 Cynthia Louis
Cynthia Louis Chris Burns2
Chris Burns2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 06 March 2018
Sec. B Cell Biology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00434
This article is part of the Research TopicEssential Kinases and Transcriptional Regulators and their Roles in AutoimmunityView all 10 articles
The pathogenesis of autoimmune diseases, such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) is driven by genetic predisposition and environmental triggers that lead to dysregulated immune responses. These include the generation of pathogenic autoantibodies and aberrant production of inflammatory cytokines. Current therapies for RA and other autoimmune diseases reduce inflammation by targeting inflammatory mediators, most of which are innate response cytokines, resulting in generalized immunosuppression. Overall, this strategy has been very successful, but not all patients respond, responses can diminish over time and numerous side effects can occur. Therapies that target the germinal center (GC) reaction and/or antibody-secreting plasma cells (PC) potentially provide a novel approach. TANK-binding kinase 1 (TBK1) is an IKK-related serine/threonine kinase best characterized for its involvement in innate antiviral responses through the induction of type I interferons. TBK1 is also gaining attention for its roles in humoral immune responses. In this review, we discuss the role of TBK1 in immunological pathways involved in the development and maintenance of antibody responses, with particular emphasis on its potential relevance in the pathogenesis of humoral autoimmunity. First, we review the role of TBK1 in the induction of type I IFNs. Second, we highlight how TBK1 mediates inducible T cell co-stimulator signaling to the GC T follicular B helper population. Third, we discuss emerging evidence on the contribution of TBK1 to autophagic pathways and the potential implications for immune cell function. Finally, we discuss the therapeutic potential of TBK1 inhibition in autoimmunity.
TANK-binding kinase 1 (TBK1) is an IKK-related serine/threonine kinase best known for the induction of antiviral type I interferons (IFN-Is) in innate immunity. However, a growing body of evidence highlights the relevance of TBK1 for other responses. In this review, we discuss our present understanding of the role of TBK1 in nucleic acid sensing pathways, antibody responses, and autophagy. We conclude by speculating how these diverse TBK1-regulated responses could potentially culminate in the induction, promotion, and maintenance of autoimmunity, as well as how pharmacological modulation of TBK1 could represent an alternative treatment strategy, particularly in the context of humorally mediated autoimmunity.
TANK-binding kinase 1 is an IKK-related serine/threonine kinase, best known for the induction of innate antiviral type I IFNs. However, TBK1 potentially has much broader functions, which we discuss in this review (Figure 1). TBK1 is ubiquitously expressed in both hematopoietic and non-hematopoietic compartments. Germline deletion of TBK1 is embryonically lethal in mice (1), highlighting its homeostatic functions during development. Through biochemical studies, TBK1 was shown to be activated by double stranded (ds)-RNA (via TLR3-TRIF), LPS (via TLR4-TRIF), viral RNA (via RIG-I-MAVS), and dsDNA (via cGAS-STING) in innate immune signaling pathways (2, 3). TRIF (TIR-domain-containing adapter-inducing IFN β), MAVS (mitochondrial antiviral-signaling), and STING (stimulator of IFN genes) are innate immune adaptor proteins that transduce signal downstream of their corresponding sensors to the activation of interferon regulatory factor 3 (IRF3). Mechanistically, TBK1 activation is thought to occur via trans-autoactivation, in response to adaptor proteins that shuttle TBK1 to specific signaling complexes and direct subcellular localizations, such as to the ER-Golgi compartments (4–7). Activated TBK1 then phosphorylates IRF3 and induces the production of type I IFN-Is (8–12). Other TBK1 substrates include AKT (13, 14) and PLK1, which are involved in TLR activation or oncogenicity of cancer cells (15). Closely related to TBK1, IKKε shares 60% homology and is initially thought to participate also in IFN-Is induction (8, 9). Subsequent studies show that IKKε is dispensable for IFN-I responses (16). IKKε is abundantly expressed in T cells and have been shown to regulate a number of T cell responses (17–19).
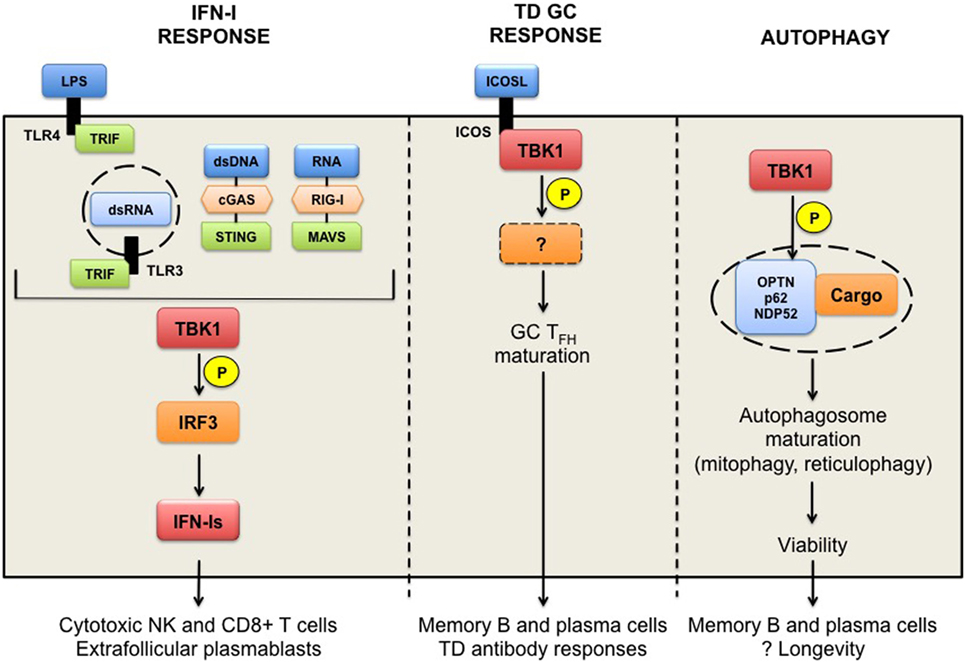
Figure 1. TANK-binding kinase 1 (TBK1) in humoral responses. TBK1 functions downstream of TLR3/4-TRIF and DNA receptor cGAS-STING pathways leading to the activation of the transcription factor interferon regulatory factor 3 and the production of interferons (IFN-Is). Chronic IFN-Is prime cytotoxic functions promote the survival of NK and CD8+ T cells, presumed to have pathogenic roles in autoimmunity, as well as the formation of extrafollicular plasmablasts. TBK1 is also implicated in the inducible T cell co-stimulator (ICOS) signaling pathway in T follicular B helper (TFH) cells to thymus-dependent (TD) antigens. TBK1 is recruited to and activated upon ICOS engagement to ICOS ligand, and promotes the maturation of pre-TFH to germinal center (GC) TFH cells. TBK1 targets downstream of ICOS signaling remain to be determined. TBK1-driven ICOS signaling is necessary for the generation of GC-derived memory B and plasma cells, and TD antibody responses. Finally, TBK1 can promote autophagy through the phosphorylation of autophagy receptors proteins (optineurin, p62, or NDP52), which sequester ubiquitinated cargo (damaged or redundant organelles). Mitophagy in memory B cells and reticulophagy in plasma cells are required for their longevity in vivo. TBK1 involvement in these processes remains to be determined.
Interferons are a family of cytokines with potent antiviral priming effects, but are also associated with humorally mediated autoimmune diseases, most notably systemic lupus erythematosus (SLE) (20–22). Recently, TBK1 was also shown to associate with the inducible T cell co-stimulator (ICOS) in CD4+ T follicular B helper (TFH) cells that support efficient antibody responses (23). However, the downstream target(s) of TBK1 in ICOS signaling have not yet been identified. Finally, TBK1 is also implicated in promoting autophagy by phosphorylating autophagy receptor proteins, including optineurin (OPTN), SQSTM1/p62, and NDP52 (24, 25). TBK1-mediated regulation of autophagy is currently under evaluation because TBK1 haploinsufficiency is a major risk factor in neurodegenerative diseases (26, 27). Autophagy is thought to protect senescent neuronal cells from the accumulation of defective or redundant organelles. TBK1 may likewise physiologically protect long-lived immune cells through autophagy (28). Although no particular TBK1 genetic variants have to date been directly linked to the development of autoimmune diseases, the diverse functions of TBK1 may contribute to one or more aspects of autoimmunity, which is the focus of this review.
TANK-binding kinase 1 has a well described role in activating the transcription factor IRF3 to induce type I IFNs production (8–12). IFN-Is are a family of cytokines with pleiotropic functions that have potent antiviral and antimicrobial effects against some intracellular bacteria, but are also implicated in pathogenesis of SLE (SLE, discussed below). TBK1 is ubiquitously expressed in both hematopoietic and non-hematopoietic compartments and it is activated by sensor-adaptor pairs, including TLR3/4-TRIF, RIG-I-MAVS, or cGAS-STING, in response to LPS, dsRNA, virus infection, and cytoplasmic DNA, respectively (2, 3). Consequently, TBK1−/− mouse embryonic fibroblasts (MEFs) have impaired production of IFN-Is (IFN-α and IFN-β) and IFN-inducible chemokines (CCL5 and CXCL10), among other genes, following activation with synthetic dsRNA (poly I:C) or viruses, or LPS (8, 10).
Molecular characterization of the sequence of events leading to TBK1 induction, IRF3 activation, and IFN-Is production have mainly been performed in cell lines and MEFs, in response to LPS stimulation or in the context of antiviral responses (2, 3, 8, 10). Elucidating roles for TBK1 in more complex biological settings in vivo has been challenging due to the embryonic lethality of germline TBK1-deficiency in mice. This is thought to be due to TNF-α-induced hepatocyte apoptosis and can be rescued by combined loss of TNF (i.e., TBK1−/− TNF−/− mice are viable) (1). Subsequently, TBK1 has been suggested to control cell survival through PAI-2/serpinB2 and transglutaminase 2 in the TNF-activated anti-apoptotic response (29).
High levels IFN-α or induction of IFN-stimulated genes (i.e., the “IFN signature”) is a remarkably consistent feature of SLE and is associated with high titers of affinity-matured autoantibodies and worse disease outcome (20, 21, 22). A similar IFN signature and correlation with high levels of autoantibodies and disease activity is also found in some patients with RA and primary Sjogren’s syndrome (30, 31) consistent with a pathogenic role for IFN-α in autoimmunity. Consequently, the possibility of targeting TBK1-dependent IFN-Is induction has received attention as a treatment strategy (32).
Among members of the IFN-I family in humans and mice, IFN-α and IFN-β are the best characterized and most broadly expressed. They signal through a shared, ubiquitously expressed heterodimeric receptor (IFNAR), and prime a rapid antiviral response that acts directly or indirectly on many cell types, including NK cells, T cells, B cells, DCs, and macrophages (33–35). IFNAR signaling mediates early attrition of existing memory CD8+ T cells in response to viral infections, which is thought to permit a more vigorous, diverse, and efficient T cell response emanating from the naïve T cell pool (36). In later stages, IFNAR signaling in activated cytotoxic CD8+ T cells (CTLs) (37) and NK cells (38) is important for long-term survival against perforin-mediated cytotoxicity, thereby preventing rapid elimination in vivo and sustaining antiviral immunity. IFNAR signaling is also required for optimal NK cell effector function through upregulation of granzyme B (38). IFN-Is is gaining attention in anti-cancer therapy, where it is generally considered pro-cytotoxic for CTLs and presumably NK cells. This is exemplified by the observations that IFNAR downregulation in CTLs endows colorectal cancers with an immune-privileged niche that promotes aggressive tumorigenesis, associated with poor prognosis, and lessens the response to immunotherapy. Conversely, IFNAR expression suppresses tumor growth and improves the efficacy of combined anti-cancer chimeric antigen receptor T cell transfer and PD-1 inhibition (39). Targeted intratumoral delivery of IFN-I-inducing (i.e., interferogenic) cyclic dinucleotide GMP-adenosine monophosphate (AMP), which activates the STING-TBK1 pathway and IFN-Is production in endothelial cells, has been shown to control tumor growth by boosting antitumor CD8+ T responses in murine models of melanoma and colon cancer (40).
Emerging evidence also implicates dysregulated NK cells and CD8+ T cells in SLE and potentially RA (41). Despite an overall reduction in circulating NK cell number in lupus patients and lupus mouse models, presumably owing to activation-induced death of these cells, NK cells with an activated phenotype infiltrate the kidneys of pre-disease lupus mice and may contribute to tissue injury by releasing cytotoxic granules (42). Another study showed that SLE patients have an expanded population of CTLs, which may contribute to tissue damage (43). Further studies are needed to determine whether IFN-Is contribute to the activation of these human effector cells.
Persistent IFN-Is exposure, particularly IFN-α, has long been implicated in immune dysfunction and autoimmune diseases, through a number of mechanisms. Some patients treated with IFN-α therapy develop autoimmunity, including RA and lupus-like autoimmune syndrome (44, 45). Chronic IFN-α overexpression in vivo induces rapid and lethal lupus, with immune complex glomerulonephritis in NZB/W lupus-prone mice (46). Such excess IFN-α can also induce sustained B cell proliferation in vivo, accompanied by uncontrolled production of proliferating, short-lived, autoantibody-secreting plasmablasts in secondary lymphoid organs of NZB/W mice (47). pDC-derived IFN-Is have been shown to increase the translocation of marginal zone B cells to the follicular region of the spleen, which disrupt the ability of marginal zone macrophages to clear apoptotic cells and promote the loss of immune tolerance to apoptotic cell-derived antigens in SLE (48). IFN-Is also promotes affinity maturation of antibodies by activating DCs to produce IL-6 (49). The severity of lupus-related pathology is attenuated with IFNAR-deficiency or IFNAR-blocking antibody in several murine lupus models (50–52).
While the involvement of IFN-Is-IFNAR signaling is a consistent feature of murine lupus models, there is less consensus in RA. In contrast to the association of IFN-α with humoral autoimmunity, IFN-β has homeostatic and anti-inflammatory functions. In RA synovium, IFN-β reduced the secretion of RA-associated proinflammatory mediators, including IL-6, TNF-α, matrix metalloproteinases, and prostaglandin E2 (53). IFN-β also primes an anti-inflammatory phenotype of endothelial cells by upregulating the expression of CD73, an ecto-5′-nucleotidase that produces anti-inflammatory adenosine from AMP, at least in neuroinflammation (54). Other studies have shown that exogenous IFN-β can inhibit autoimmune collagen-induced arthritis (CIA) (55, 56). In contrast, IFN-β deficient mice develop prolonged CIA, with a higher incidence relative to control mice (57). IFN-β delivery has been used therapeutically in multiple sclerosis (58) and has been considered for RA (59). Monoclonal antibody therapies inhibiting IFN-Is signaling or depleting of IFN-overproducing plasmacytoid DCs (pDCs) are under evaluation for the treatment of SLE (60, 61). The opposing roles of IFN-α and IFN-β clearly require careful consideration in relation to these potential IFN-Is-targeted therapies in autoimmunity.
The prevailing concept in SLE and murine lupus models is that immune complexes containing autoantibodies bound to self-DNA and RNA can act as interferogenic stimuli, following Fc receptor-mediated internalization and activation of endosomal TLR7 and TLR9 in pDCs (62). TLR7- or TLR9-mediated induction of IFN-Is, however, does not require TBK1. For instance, TBK1 is not required for IFN-I production in the TLR7-dependent pristane-induced lupus model (51). TLR9 ligand (CpG-B) induces IFN-Is production by B cells and DCs through IRF3, but independently of TBK1. Autocrine IFNAR signaling in B cells is required for enhanced IgM and IgG2a autoantibody production and these are dominant autoantibody isotypes in murine lupus (63).
Using viable TBK1−/− TNF−/− mice, Ishii and colleagues demonstrated functional distinctions between TBK1 signaling in hematopoietic and non-hematopoietic cells for the induction of Ag-specific responses in a plasmid-DNA immunization model (64). TBK1−/− TNF−/− mice had no difference in total serum IgG1 and IgG2a, suggesting normal B cell function. However, TBK1−/− TNF−/− mice had completely abrogated primary and secondary antigen-specific IgG responses upon vaccination with plasmid-DNA, relative to wild type, TNF−/− TBK1+/+, Myd88−/−, or TRIF−/− mice. Mechanistically, the DNA component of the plasmid-DNA vaccine was shown to activate DCs in a TBK1- and IFN-I-dependent manner, but this occurred independently of the CpG DNA sensor, TLR9 (64). Along the same lines, alum and hydroxypropyl-β-cyclodextrin adjuvants have been shown to induce cell death and DNA release as part of their immunogenic properties and TBK1−/− TNF−/− mice immunized with these adjuvants had reduced levels of antigen-specific IgG1 responses (65, 66). Ishikawa and colleagues subsequently demonstrated that intracellular DNA induced DC activation and IFN-Is production through the cGAS-STING-TBK1 pathway (67).
As mentioned, TBK1-dependent IFN-Is responses are activated by cytoplasmic nucleic acids. In the autoimmune context, pathogenic TBK1-mediated IFN-Is responses can be caused by aberrant self-DNA that leads to chronic IRF3 activation, such as is the case in TREX1 deficiency (68). TREX1 is an endoplasmic reticulum (ER)-associated 3′–5′ exonuclease, which degrades cytoplasmic viral DNA before sensing occurs. TREX1 is also required to clear endogenous retroelements and genomic DNA. TREX1 deficiency in patients and murine models causes lupus-like autoimmune manifestations. TREX1-deficient mice develop aberrant interferogenic responses and features of lupus owing to the cytoplasmic accumulation of endogenous nucleic acids and chronic activation of the TBK1-dependent DNA-sensing pathway (68–70). Mutations in TREX1 are associated with human autoimmune disorders, including Aicardi–Goutières syndrome (71), familial chilblain lupus (72), and SLE (73). An inhibitor of TBK1 was effective in treating TREX1−/− mice (74).
In summary, TBK1 is an important signaling kinase for the induction of IFN-Is in response to a number of ligands that activate TLR3, TLR4, and the STING pathways. TBK1 may be less relevant in other IFN-Is induction pathways, including TLR7 and TLR9. Mutations leading to aberrant activation of TBK1 and IFN-Is overproduction can contribute to lupus. Limiting pathogenic IFN-Is production through TBK1 inhibition may alleviate lupus. However, TBK1-driven responses other than IFN-Is induction may also contribute to humoral autoimmunity and are discussed in the next section.
Antibody-mediated autoimmune diseases share underlying immune mechanism(s). High-affinity autoantibodies arise from a GC reaction occurring in the B cell follicles (75, 76). The GC is a specialized structure in secondary lymphoid tissues, where B cells undergo iterative rounds of somatic hypermutation in Ig variable (V) gene segments, class switching and affinity selection, as well as post-translational modifications. Normally, the GC reaction is transient (self-terminating) (77) and only B cells expressing affinity-matured, class-switched antibodies specific for the antigen exit GCs, and survive as long-lived memory B cells and/or antibody-secreting plasma cells (78–80). However, the GC reaction can persist and give rise to antibody-mediated autoimmunity (81).
Intrinsic B cell defects can directly contribute to the development of spontaneous GCs, breakdown of B cell tolerance and humoral autoimmunity, such as Tlr7 gene duplication (80) or WAS (Wiskott–Aldrich syndrome) protein deficiency (82, 83) in lupus. However, CD4+ TFH cells provide another essential cellular component regulating GC B cells. TFH cells are required for the generation of high-affinity antibodies by promoting the GC reaction, including B cell clonal proliferation, affinity selection and the development of high-affinity antibody-producing cells (75, 84, 85). TFH cells are characterized by the expression of chemokine receptor CXCR5, which facilitates migration and proximity to follicular B cells. Here, they provide cognate help to B cells via stable interactions such as SAP (SLAM-associated protein), costimulatory molecules such as CD40L, and cytokines such as IL-21, IL-4, and IFN-γ (86–89). While TFH cells are critical for an optimal GC reaction and subsequent generation of protective antibodies following immunization, abnormal development and/or function of TFH have also been implicated in loss of tolerance and the development of humoral autoimmunity.
An increased TFH population in the GC as well as GC numbers may contribute to aberrant positive selection and autoantibody formation in SLE (90). This is exemplified in Sanroque mice, in which exaggerated TFH generation occurs in a cell-intrinsic manner and leads to spontaneous GC formation, and lupus-like pathology (91, 92). Furthermore, adoptively transferred Roquinsan/san TFH cells are able to induce spontaneous GC B cell expansion and GC formation in naïve recipient mice (92). Similarly, an enlarged TFH population accompanies increased GC size and more productive humoral responses in immunization models (77). Conversely, mice with conditional Bcl6 deficiency in T cells (Bcl6 is a transcriptional repressor that regulates both TFH and GC B cell differentiation) have impairment of TFH development, GC reactions, and antibody responses (93, 94). CD4 transgenic autoreactive T cells deficient in SAP (SLAM-associated protein, which mediates stable T-B interactions critical for GC formation), failed to mount GC reactions, develop IgG1 autoantibodies, and autoantibody-mediated arthritis (95, 96). Thus, the size of the TFH population is directly coupled with GC function and ensuing humoral responses. Abnormal TFH accumulation may also contribute to the production of pathogenic autoantibodies through enhanced positive selection of self-reactive B cells.
In clinical settings, the frequency of TFH-like cells is increased in the peripheral blood of RA patients and correlates with higher elevated levels of anti-CCP (cyclic citrullinated peptide) autoantibodies, as well as disease activity (97, 98). Conversely, treatment responsive, new onset RA patients have a reduced frequency of circulating TFH, which is accompanied by a decrease in anti-CCP antibody (98). SLE patients also demonstrate a similar expansion of TFH-like cells, which correlates with disease activity, frequency of circulating plasmablasts, and anti-double-stranded DNA antibody positivity (99). TFH expansion and its association with autoantibody responses have also been noted in other humoral autoimmune syndromes, including type 1 diabetes (100) and primary Sjogren’s syndrome (101, 102). Given the robust correlation between TFH numbers and high-affinity autoantibody levels, manipulation of the differentiation program and plasticity of TFH cells may provide new therapeutic options in autoimmune diseases, such as SLE and RA.
Among many determinants of optimal humoral immunity, ICOS has been consistently associated with GC reactions and the induction of GC-dependent thymus-dependent (TD) antibody responses. ICOS is a critical coreceptor, distinct from CD28, on activated or antigen-experienced T cells (103, 104) and is highly expressed on TFH (105). Through interaction with ICOS ligand (ICOSL) on antigen-presenting cells (DCs and B cells), ICOS delivers robust costimulatory signals that promote TFH positioning and thus supports GC function (106). ICOSL−/− mice mount comparable antigen-specific IgM and IgG3 responses, but have reduced IgG1 and IgG2a production upon immunization with thymus-independent antigens (107). ICOS−/− or ICOSL−/− mice have defective production of class-switched antibodies against TD antigens (particularly IgG1, IgG2a, and IgG2b isotypes, but not IgM), along with reduced number and size of GCs and a lack of B cell memory (107–110). Additionally, mice with a tyrosine-to-phenylalanine point mutation at residue 181 in the cytoplasmic tail of ICOS have abrogated TFH generation, GC reactions, antibody class switching, and antibody affinity maturation (111). ICOS deficiency or antibody-mediated depletion of ICOS-expressing CD4+ T cells in SLE1 lupus mice results in diminished pathogenic TFH expansion, inhibited plasma cell generation, and a reduction in class-switched IgG autoantibodies (112). ICOSL−/− or B cell-specific ICOSL−/− mice have markedly inhibited development of proteoglycan-induced arthritis, with notable reductions in TFH and GC B cells, IL-21 production, and proteoglycan-specific IgG antibody responses (113).
Blockade of the ICOSL pathway ameliorates autoimmune CIA, the K/BxN spontaneous arthritis model, and the SLE (NZB/NZW) F1 mouse model, with marked reductions in disease manifestations, numbers of TFH and GC B cells, and pathogenic, class-switched, high-affinity autoantibodies (113–115). Interestingly, inhibition of CIA was observed even when ICOSL blocking antibody was given after the onset of disease (114). Sanroque mice have excessive ICOS activation due to genetic mutation of a RING-type ligase that represses ICOS (91). These mice develop spontaneous GC in the absence of foreign antigen, increased numbers of TFH cells, spontaneous autoantibodies, including antinuclear antibodies, and lupus-like manifestations, such as glomerulonephritis. Intriguingly, although ICOS or ICOSL deficiency in Sanroque mice substantially reduced autoantibody production, it did not result in complete inhibition of autoantibody production (91). This observation suggests a contribution from residual GC-independent extra-follicular pathway (85, 113). Thus, ICOS/ICOSL signaling drives optimal GC-dependent TD antibody responses and inhibition of this pathway abrogates the GC reaction, autoantibody responses, and disease features in humorally mediated autoimmune disease models. Indeed, therapies targeting ICOS/ICOSL are under evaluation in early phase clinical trials of SLE (116).
Similar to other CD4+ T cell subsets (Th1, Th2, Th17, and Treg cells), TFH development is a multi-step process which involves initial priming of naïve CD4+ T cells by dendritic cells in the T cell zone, followed by expansion and differentiation that are regulated through signaling pathways activated downstream of cytokines and cell surface molecules. Subsequent activation of lineage-defining transcription factors (T-bet for Th1, GATA3 for Th2, RORγt for Th17, FoxP3 for Treg cells, and Bcl6 for TFH) promotes T cell differentiation (93, 94, 117).
T follicular B helper development can be separated into two stages—(i) naive to Bcl6+ pre-TFH and (ii) pre-TFH to mature GC TFH. Pre-TFH development follows DC priming in vivo, through an ICOS costimulation signal and the phosphoinositide-3 kinase (PI3K) pathway. The ICOS-PI3K pathway instructs TFH differentiation via induction of Bcl6 and the subsequent Bcl6-dependent expression of CXCR5 on pre-TFH (23, 105). The activation ICOS-PI3K signaling alone is, however, insufficient to drive full GC TFH maturation and the GC reaction (23, 111, 118). For final differentiation of nascent TFH into GC TFH, pre-TFH cells require a second costimulatory signal through ICOS (119, 120). This has been demonstrated by the inability of T cell-selective deletion of PI3K components to fully recapitulate the phenotype of CD4+ T cells from ICOS−/− mice (111, 121).
A recent report identified TBK1 as a unique signaling kinase in the ICOS pathway (23). In this study, Pedros and colleagues identified a conserved TRAF-like motif in the cytoplasmic tail of ICOS (iProx motif), which mediated TBK1 recruitment and activation following a combination of strong TCR and ICOS signals. These authors showed that by deleting the iProx motif on ICOS specifically in CD4+ T cells, TBK1 failed to associate with ICOS. T cells modified in this way displayed severely impaired differentiation into GC TFH and TD antibody responses, despite generating pre-TFH cells. A similar effect was obtained by CD4+ T cell-specific TBK1 depletion. In a series of reconstitution experiments, transducing ICOS and TBK1 constructs into ICOS−/− TCR transgenic CD4+ T cells, it was shown that intact ICOS is required for the generation of both nascent and final GC TFH populations, while TBK1 controls progression from the pre-TFH to mature GC TFH phenotype (23).
Although the downstream mediators of ICOS-TBK1 signaling in TFH have not been identified, FoxO1 is a potential candidate because ICOS signaling instructs the TFH program via AKT-mediated FoxO1 phosphorylation (119). AKT has been shown to be a TBK1 substrate in some settings (13, 14, 122). FoxO1 is a transcription factor that, in its active unphosphorylated state, represses TFH programming. FoxO1 phosphorylation results in its transient inactivation and cytoplasmic translocation from the nucleus (119). FoxO1 inactivation also reduces FoxO1-dependent KLF2 expression, together with expression of KLF2-dependent chemokine receptors, necessary for optimal repositioning of TFH in the GC (120). FoxO1 inactivation, specifically in CD4+ T cells (Foxo1fl/fl:CD4-Cre mice) caused defective Tregs and systemic autoimmunity, characterized by accumulation of the TFH population, with exaggerated Bcl6 induction and GC formation, and production of anti-DNA antibodies (123). While FoxO1-sufficient CD4+ T cells give rise to effector T cells, pre-TFH, and GC TFH upon immunization, FoxO1−/− CD4+ T cells generate pre-TFH cells with higher expression of TFH-defining markers (Bcl6, CXCR5, and PD-1) and lowered T cell zone chemokine receptors (CD62L, PSGL1) (119). ICOS-driven FoxO1 inactivation thus alters the chemokine receptor profile of pre-TFH, facilitating migration from the T cell zone toward the B cell follicles (119). Conversely, ICOS/ICOSL blockade results in the relocation of fully developed TFH back to the T cell zone. This relocation reverses their phenotype toward non-TFH effector T cells, with a consequent reduction in antigen-specific GC B cells, as well as serum antigen-specific IgG1 and IgG2a responses, indicating collapse of the GC response (120). This study also concluded that ICOS is not required for TFH survival or expression of TFH-related transcription factors, but rather, it regulates the expression of TFH homing markers. Changes in TFH transcription factors are thus likely to be a secondary effect upon failure to maintain the positioning of pre-TFH and impaired costimulatory signals from follicular B cells (120).
Although TBK1 has been identified as an ICOS-specific signaling kinase required for full maturation of GC TFH, the role of TBK1-mediated FoxO1 regulation in this process has not been elucidated. One study using conditional TBK1−/− in CD4+ T cells (Tbk1fl/fl:CD4-Cre mice) and stimulation with TCR and CD28, suggested that basal TBK1 is required for constitutive AKT turnover to prevent hyperactivation of AKT upon T cell activation (124). These authors also reported a marked increase in IFN-γ production and activation markers in CD4+ T cells derived from Tbk1fl/fl:CD4-Cre mice, indicating the propensity of these cells to become a Th1-like population in the absence of basal TBK1 (124). Interestingly, ICOS−/− or ICOSL−/− mice also exhibit enhanced Th1 responses in secondary lymphoid tissues with marked elevation of IFN-γ in the context of infection (125–127).
Given the role of TBK1 in ICOS signaling in GC TFH and downstream GC-driven antibody responses, TBK1 inhibition may curtail humoral autoimmunity through an ICOS-driven GC pathway. Importantly, targeting ICOS/ICOSL and/or TBK1 may not result in generalized immunosuppression, but rather reverse cell fate decisions in TFH. Understanding how TBK1 signals in TFH, how it affects cell fate decisions in T helper cell polarization, positioning and migratory pathways may provide new therapeutic strategies, especially for antibody-mediated autoimmunity.
The functional effects of TBK1 extend beyond innate immune signaling. Autophagy is a conserved homeostatic process in eukaryotic cells involving sequestration and lysosomal degradation of cytoplasmic contents, including damaged or surplus organelles (mitophagy for mitochondria, pexophagy for peroxisomes, ribophagy for ribosomes, reticulophagy for endoplasmic reticulum), cytotoxic macromolecular aggregates (aggrephagy), and intracellular microorganisms (xenophagy) (128–131). Autophagy will not be discussed in detail as it has been extensively reviewed elsewhere (128–131). Instead, we discuss reports which have implicated TBK1 in autophagic processes and how these translate to immune responses.
TANK-binding kinase 1-mediated regulation of autophagy has been described in the context of antimicrobial defense (xenophagy), in which intracytoplasmic pathogens are sequestered into autophagosomes and targeted for lytic, lysosomal degradation. TBK1 and its homolog IKKε have been identified as binding partners of the autophagy receptor protein NDP52 that recognizes polyubiquitylated Salmonella enterica in human cells. However, only TBK1 is required for xenophagy of S. enterica (132, 133) and mycobacteria (134). Canonical IKKs initiate autophagy, while TBK1 knockdown suppresses the maturation of autophagosomes into autolysosomes. Mechanistically, TBK1 phosphorylates autophagy receptor proteins, including NDP52, OPTN on Ser177, and p62 (also known as SQSTM1) at Ser403 (located at the ubiquitin-associated/UBA domain) (132–134). Phosphorylation increases the affinity of LC3-binding autophagy adaptors for K48- and K63-ubiquitinated cytoplasmic bacteria, as well as polyubiquitinated protein aggregates (135), and it promotes autophagic clearance (132–134). Knockdown or pharmacological inhibition of TBK1 impairs autophagic killing of S. enterica or M. tuberculosis (133, 134). Mice deficient in autophagic proteins (Atg3, Atg5, Atg7, Atg9, and Atg16L1) have embryonic lethality (129). Viable conditional autophagy knockout mice often have impaired pathogen clearance, reduced survival, and severe tissue injury due to enhanced inflammasome and cytokine responses, and in some cases, enhanced Th17 responses (136–138). In the same study, TBK1 was shown to be important for delivery of the lysosomal hydrolase cathepsin D to the autophagolysosomal compartment (134). TBK1, therefore, appears to play an essential role in late autophagic flux.
Autophagy is increasingly appreciated for its role in maintaining cell homeostasis through clearance or normal turnover of cytoplasmic contents or defective cellular organelles, including mitochondria (mitophagy). Mice deficient for Atg5 specifically in neural cells develop progressive decline in motor function in the absence of any disease-associated mutant proteins, accompanied by the accumulation of cytoplasmic inclusion bodies in neurons (139). Damaged mitochondria are detrimental to cellular homeostasis and efficient removal through autophagy is crucial for cell survival, particularly for senescent cells, such as neurons, which cannot dilute cytotoxic contents through cell division (140). Mitochondrial damage induces concomitant PINK1-PARKIN-mediated poly-ubiquitylation of damaged mitochondria and also activates TBK1. In turn, TBK1 can phosphorylate autophagy receptors (OPTN, SQSTM1/p62, and NDP52), thereby enhancing the ability of these receptors to associate with ubiquitinated cargo (e.g., ubiquitin-tagged, depolarized mitochondria) and autophagic membranes (24, 25, 28, 141). This post-translational modification creates a signal amplification loop that recruits and retains autophagy receptor/TBK1/ubiquitinated cargo complexes, thereby promoting mitophagy. Separate mutations that disrupt TBK1’s association with OPTN, or of OPTN with ubiquitin, abolish the translocation and activation of TBK1, and, therefore, impair mitophagy (25).
Exome sequencing identifies TBK1 as a neurodegenerative disease gene in amyotropic lateral sclerosis (ALS) and frontotemporal dementia (26, 27). Further, it was shown that mutations of TBK1 at the C-terminal TBK1 coiled-coil domain, resulted in TBK1’s dissociation from OPTN, while preserving its kinase activity (located at the N-terminal ubiquitin-like domain) (27). These studies provide a potential mechanistic basis for TBK’s involvement in ALS. Mutations linking OPTN to impaired autophagy and neurodegenerative diseases have also been characterized (142). Thus, although it has not been directly demonstrated, TBK1-regulated autophagy appears to maintain cellular homeostasis of long-lived neuronal cells through mitophagy.
TANK-binding kinase 1-regulated autophagy may also be important for regulation of immune cells. As mentioned above, mice deficient in autophagic proteins (Atg3, Atg5, Atg7, Atg9, and Atg16L1) have neonatal lethality, as do TBK1−/− mice (1, 129). In contrast, STING−/− mice are viable, but have impaired TBK1-dependent IFN-I responses to cytoplasmic DNA (67). Autophagy allows dynamic changes necessary for proper mammalian development through the recycling and provision of macromolecules and clearance of apoptotic bodies. Conditional Atg7fl/fl:Vav-Cre mice (i.e., hematopoietic cell-specific deletion of Atg7) overcomes embryonic lethality, but these mice are anemic and lymphopenic, linking autophagy to erythropoiesis and lymphopoiesis (129). Similarly, T cell-specific deletion of Atg5 or Atg7 (Atg5fl/fl:Lck-Cre or Atg7fl/fl:Lck-Cre mice) or innate lymphoid cell (ILC)-specific deletion of Atg5 (Atg5fl/fl:Nkp46-Cre) reduces peripheral T cells and ILC subpopulations, respectively (143, 144). Maturation of naïve T cells depends on autophagy to reduce mitochondrial and ER contents through mitophagy and reticulophagy, respectively (143, 145, 146). Defective autophagy in T cells results in accumulated mitochondrial biomass, disturbed Ca2+ homeostasis, higher levels of reactive oxygen species (superoxide), and enhanced susceptibility to apoptosis (143, 145, 146). In contrast to lymphopenia, the myeloid compartment in Atg7fl/fl:Vav-Cre mice is expanded (147), implicating autophagy in the balance between lymphopoiesis and myelopoiesis. It was also recently shown that autophagy is required for full granulopoiesis (148). Autophagy regulates cellular differentiation and activation by accommodating metabolic adaptation, which can occur in parallel and independently of transcriptional regulators. Neutrophils from Atg7fl/fl:Vav-Cre or Atg7fl/fl:Cebpa-Cre (granulocyte-macrophage progenitor-specific deletion of Atg7) mice are numerically expanded, but are unable to complete maturation and, therefore, are functionally defective. In this case, autophagy-mediated lipolysis (lipophagy) provides free fatty acids to support a mitochondrial respiration pathway essential for neutrophil differentiation (148). Autophagy regulates cytosolic processing of antigen for presentation on MHCII in DCs. The absence of Atg5 in DCs results in failure to mount full Th1 cell immunity to viral infection (149). TBK1 has also been associated with metabolic adaptation of DC after TLR stimulation, whereby TBK1 phosphorylates AKT for the glycolysis which is necessary for DC activation. shRNA-mediated TBK1 knockdown in DCs results in a blunted glycolytic shift and reduced ability of these DCs to prime antigen-specific T cells in vitro (14).
In summary, separate lines of evidence have linked autophagy to cell metabolism, TBK1 to autophagy, and TBK1 to metabolism in immune cell development and activation. Further studies of TBK1’s role in cellular autophagy and metabolism in various immune contexts could allow manipulation of immune function—either for protection against pathogens or rewiring toward tolerance in autoimmunity.
In contrast to the maturation defect of neutrophils in the absence of autophagy, macrophages derived from Atg5fl/fl:LysM-Cre mice display a heightened proinflammatory phenotype. These mice develop greater hepatitis on a high fat diet and low dose LPS and also spontaneous uveitis (150, 151). Autophagy-deficient macrophages activate the NLRP3 inflammasome and develop IL-1β-mediated inflammation (151–153). Similar to macrophages, Atg16L1-deficient DCs have heightened activation in graft-versus-host disease (154). Loss of function polymorphisms of Atg16L1 have been associated with age-dependent development of inflammatory bowel disease (Crohn’s disease) owing to impaired clearance of ileal pathogens or endogenous protein aggregates, and chronic elevation of inflammatory cytokine responses (155, 156). Interestingly, DC-specific deletion of TBK1 (Tbk1fl/fl:CD11c-Cre mice) also display age-related cellular hyperactivation, with marked upregulation of costimulatory molecules on DCs, T cell activation, and autoimmune features (splenomegaly, lymphadenopathy, and tissue infiltration with lymphocytes) (157). These Tbk1fl/fl:CD11c-Cre mice have an increased frequency of activated IFN-γ-producing CD4+ and CD8+ T cells, while Tregs remain comparable to TBK1-sufficient mice. Consequently, these mice are more sensitive to EAE and mount more robust antitumor immunity against poorly immunogenic B16F10 melanoma cells (157).
Enhanced macrophage or DC activation in the absence of TBK1 may be due to impaired autophagy, which normally limits age-related inflammasome activation. Of note, the age-dependent hyperinflammatory status of autophagy- or TBK1-deficient macrophages and DCs in Atg conditional knockout mouse models resembles aging macrophages. These cells shift from an anti-inflammatory to a proinflammatory phenotype, with an age-related reduction in autophagic activity and sensing of endogenous damage-associated molecular patterns (DAMPs) (158, 159). Thus, it is tempting to speculate that TBK1 maintains cellular autophagy and sustains immune cell longevity and homeostasis. Hallmarks of accelerated immune cell aging with chronic TBK1 deficiency are also notable in the Tbk1fl/fl:CD19-Cre mice (B cell-specific ablation of TBK1) (160). These mice have normal B cell populations in the spleen and bone marrow, but develop age-related dysregulation of the non-canonical NF-κB pathway, uncontrolled production of IgA, increased levels of autoantibody antinuclear antigen and anti-dsDNA, with nephropathy-like disease (160). In this study, it was concluded that steady state TBK1 negatively regulates IgA class switching in B cells by attenuating noncanonical NF-κB signaling. This effect was thought to be due to TBK1-mediated phosphorylation and degradation of NF-κB-inducing kinase, downstream of BAFF or APRIL signaling (160). Intriguingly, it is possible that Tbk1fl/fl:CD19-Cre mice phenocopy the aging B cell repertoire because the B1 population (responsible for IgA responses) and autoantibody production are enhanced by age (161). While chronic deficiency of autophagy or TBK1 results in amplified endogenous inflammation to DAMPs in myeloid cells and abnormalities in the B cell repertoire, inhibition of autophagy may be exploited to target long-lived autoimmune populations (discussed below) through acceleration of immune cell aging.
As discussed above, autophagy is cytoprotective in senescent cells, such as neurons. This cytoprotective function also appears to apply in long-lived immune cells. Mice with B cell-specific deletion of Atg5 or Atg7 (Atg5fl/fl:CD19-Cre or Atg7fl/fl:CD19-Cre mice) have mostly normal B cell development, but are unable to maintain long-lived humoral antibody responses owing to the failure to maintain long-lived plasma cells and memory B cells, respectively (162, 163). In both studies, plasma cells and memory B cells arising from immunization and GC were shown to upregulate components of the autophagic machinery. In plasma cells, autophagy is thought to maintain longevity by reticulophagy (autophagic clearance of redundant endoplasmic reticulum) to limit excessive antibody synthesis and conserve energy balance (162). Further studies are needed to investigate whether TBK1 is involved in reticulophagy and physiological adaptation of plasma cells.
Autophagy also supports the lifespan of quiescent, antigen-experienced, long-lived, GC-derived memory B cells through mitophagy. Atg7fl/fl:CD19-Cre mice mount normal primary antibody responses and have normal differentiation of post-GC memory B cells, but fail to generate secondary antibody responses to influenza virus due to spontaneous death of memory B cells (163). Memory B cells from Atg7fl/fl:CD19-Cre mice are unable to efficiently remove damaged mitochondria, resulting in accumulation of reactive oxygen species, lipid peroxidation, and oxidative stress-induced death. Interestingly, these mice also develop enhanced Th17 responses to viral infection, possibly as a compensatory mechanism (163). Whether TBK1 also regulates mitophagy and, therefore, the survival of memory B cells requires further investigation. Mitophagy was shown to support the generation of LCMV-induced memory CD8+ T cells (164, 165) and MCMV-induced memory NK cells (166). These studies highlight that autophagy is not apparently required for germline T cell development, nor for T cell activation and proliferation, but is important for established effector T cells to generate a pool of memory cells (165, 166). Autophagy is dynamically induced at various stages of immune cell development, activation, and differentiation, but plays a particular role in the formation and maintenance of long-lived immune populations, including memory B cells and senescent plasma cells. Because long-lived plasma cells or plasma cells deriving from memory B cells can drive persistent autoimmune disease (167), abrogation of autophagy through TBK1 inhibition might reduce resistance to autonomous cell aging and death, and diminish pathogenic autoantibody responses.
In summary, a number of studies demonstrate significant overlap between TBK1 and autophagy, most notably in antimicrobial xenophagy and maintenance of neuronal cell health. Similar to autophagy, TBK1 has a complex role in immune cells. It is known that autophagy is dynamically regulated to accommodate rapid metabolic adaptation and organelle turnover associated with cell development, differentiation, activation, and longevity. TBK1 regulates autophagy through post-translational modifications of autophagy adaptor/receptor proteins required for the maturation of autophagosomes. TBK1’s involvement in other types of organelle autophagy and metabolic signaling pathways in immune lineage cells and immune responses is of great interest. Autophagy supports the extended lifespan of cells, such as neurons, immune memory populations, and long-lived plasma cells. Targeting autophagy through the inhibition of TBK1 may provide a novel approach for treating humoral autoimmune diseases. Inhibition of autophagy or TBK1 may favor the generation of short-lived effector cells, rather than long-lived memory populations. Both autophagy and TBK1 have been shown to regulate the delicate balance between cellular adaptation for efficient immune response and aging-associated autoinflammation. Studies exploring how, when, and where TBK1 facilitates autophagy in distinct immune lineages will inform potential modulation of protective or pathogenic immune responses.
In this review, we discuss the remarkable functional diversity of TBK1 in the context of humoral autoimmunity. These pathways are summarized in the Figure 1. TBK1 is required for IFN-Is production in the context of sensing viral or aberrant cytoplasmic nucleic acids. Overactive TBK1 can precipitate IFN-Is and lupus, such as is the case for TREX1 deficiency. Recent literature reports that TBK1 is associated with humoral antibody responses via its recruitment to and activation of ICOS in the CD4+ TFH population in GCs of lymph nodes. ICOS is required for full maturation of the GC TFH population, the GC reaction, GC-mediated generation of affinity-matured long-lived plasma cell and memory B cells, and productive GC-derived antibody responses. Thus, TBK1 inhibition may be useful in pathogenic autoantibody responses mediated by GC. Finally, we highlight the similarity of TBK1 deficiency to that of autophagy-deficiency. Therapeutic TBK1 inhibition may therefore lead to premature aging and/or death of pathogenic immune cells, such as long-lived plasma cells, and memory B cells in autoimmune diseases.
All authors contributed to the assembly and revision of this review manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Reid Charitable Trusts, National Health and Medical Research Council of Australia Clinical Practitioner Fellowship (1023407), Program Grant (1016647, 1113577) and Development Grant (1055374) and IRIISS (9000220), Victorian Government Operational Infrastructure Support, Australian Cancer Research Foundation and Dyson Bequest Funding (Dunn Fellowship to CB).
1. Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J (2000) 19(18):4976–85. doi:10.1093/emboj/19.18.4976
2. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347(6227):aaa2630. doi:10.1126/science.aaa2630
3. Li X, Yang M, Yu Z, Tang S, Wang L, Cao X, et al. The tyrosine kinase Src promotes phosphorylation of the kinase TBK1 to facilitate type I interferon production after viral infection. Sci Signal (2017) 10(460):eaae0435. doi:10.1126/scisignal.aae0435
4. Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, et al. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U S A (2012) 109(24):9378–83. doi:10.1073/pnas.1121552109
5. Larabi A, Devos JM, Ng SL, Nanao MH, Round A, Maniatis T, et al. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep (2013) 3(3):734–46. doi:10.1016/j.celrep.2013.01.034
6. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe (2015) 18(2):157–68. doi:10.1016/j.chom.2015.07.001
7. Pourcelot M, Zemirli N, Silva Da Costa L, Loyant R, Garcin D, Vitour D, et al. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol (2016) 14:69. doi:10.1186/s12915-016-0292-z
8. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol (2003) 4(5):491–6. doi:10.1038/ni921
9. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science (2003) 300(5622):1148–51. doi:10.1126/science.1081315
10. Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med (2004) 199(12):1641–50. doi:10.1084/jem.20040520
11. McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A (2004) 101(1):233–8. doi:10.1073/pnas.2237236100
12. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal (2012) 5(214):ra20. doi:10.1126/scisignal.2002521
13. Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, et al. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci U S A (2011) 108(16):6474–9. doi:10.1073/pnas.1016132108
14. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15(4):323–32. doi:10.1038/ni.2833
15. Kim JY, Welsh EA, Oguz U, Fang B, Bai Y, Kinose F, et al. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc Natl Acad Sci U S A (2013) 110(30):12414–9. doi:10.1073/pnas.1220674110
16. Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science (2007) 315(5816):1274–8. doi:10.1126/science.1136567
17. Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol (2011) 12(9):844–52. doi:10.1038/ni.2080
18. Gulen MF, Bulek K, Xiao H, Yu M, Gao J, Sun L, et al. Inactivation of the enzyme GSK3alpha by the kinase IKKi promotes AKT-mTOR signaling pathway that mediates interleukin-1-induced Th17 cell maintenance. Immunity (2012) 37(5):800–12. doi:10.1016/j.immuni.2012.08.019
19. Zhang J, Feng H, Zhao J, Feldman ER, Chen SY, Yuan W, et al. IkappaB kinase epsilon is an NFATc1 kinase that inhibits T cell immune response. Cell Rep (2016) 16(2):405–18. doi:10.1016/j.celrep.2016.05.083
20. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A (2003) 100(5):2610–5. doi:10.1073/pnas.0337679100
21. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197(6):711–23. doi:10.1084/jem.20021553
22. Ronnblom L, Alm GV. Systemic lupus erythematosus and the type I interferon system. Arthritis Res Ther (2003) 5(2):68–75. doi:10.1186/ar625
23. Pedros C, Zhang Y, Hu JK, Choi YS, Canonigo-Balancio AJ, Yates JR III, et al. A TRAF-like motif of the inducible costimulator ICOS controls development of germinal center TFH cells via the kinase TBK1. Nat Immunol (2016) 17(7):825–33. doi:10.1038/ni.3463
24. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell (2015) 60(1):7–20. doi:10.1016/j.molcel.2015.08.016
25. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A (2016) 113(15):4039–44. doi:10.1073/pnas.1523926113
26. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science (2015) 347(6229):1436–41. doi:10.1126/science.aaa3650
27. Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci (2015) 18(5):631–6. doi:10.1038/nn.4000
28. Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A (2016) 113(24):E3349–58. doi:10.1073/pnas.1523810113
29. Delhase M, Kim SY, Lee H, Naiki-Ito A, Chen Y, Ahn ER, et al. TANK-binding kinase 1 (TBK1) controls cell survival through PAI-2/serpinB2 and transglutaminase 2. Proc Natl Acad Sci U S A (2012) 109(4):E177–86. doi:10.1073/pnas.1119296109
30. Nordmark G, Eloranta ML, Ronnblom L. Primary Sjogren’s syndrome and the type I interferon system. Curr Pharm Biotechnol (2012) 13(10):2054–62. doi:10.2174/138920112802273290
31. Lubbers J, Brink M, van de Stadt LA, Vosslamber S, Wesseling JG, van Schaardenburg D, et al. The type I IFN signature as a biomarker of preclinical rheumatoid arthritis. Ann Rheum Dis (2013) 72(5):776–80. doi:10.1136/annrheumdis-2012-202753
32. Hasan M, Yan N. Therapeutic potential of targeting TBK1 in autoimmune diseases and interferonopathies. Pharmacol Res (2016) 111:336–42. doi:10.1016/j.phrs.2016.04.008
33. Cervantes-Barragan L, Kalinke U, Zust R, Konig M, Reizis B, Lopez-Macias C, et al. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J Immunol (2009) 182(2):1099–106. doi:10.4049/jimmunol.182.2.1099
34. Prchal M, Pilz A, Simma O, Lingnau K, von Gabain A, Strobl B, et al. Type I interferons as mediators of immune adjuvants for T- and B cell-dependent acquired immunity. Vaccine (2009) 27(Suppl 6):G17–20. doi:10.1016/j.vaccine.2009.10.016
35. Zust R, Toh YX, Valdes I, Cerny D, Heinrich J, Hermida L, et al. Type I interferon signals in macrophages and dendritic cells control dengue virus infection: implications for a new mouse model to test dengue vaccines. J Virol (2014) 88(13):7276–85. doi:10.1128/JVI.03827-13
36. Bahl K, Kim SK, Calcagno C, Ghersi D, Puzone R, Celada F, et al. IFN-induced attrition of CD8 T cells in the presence or absence of cognate antigen during the early stages of viral infections. J Immunol (2006) 176(7):4284–95. doi:10.4049/jimmunol.176.7.4284
37. Xu HC, Grusdat M, Pandyra AA, Polz R, Huang J, Sharma P, et al. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity (2014) 40(6):949–60. doi:10.1016/j.immuni.2014.05.004
38. Madera S, Rapp M, Firth MA, Beilke JN, Lanier LL, Sun JC. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J Exp Med (2016) 213(2):225–33. doi:10.1084/jem.20150712
39. Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, et al. Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell (2017) 31(2):194–207. doi:10.1016/j.ccell.2017.01.004
40. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A (2015) 112(50):15408–13. doi:10.1073/pnas.1512832112
41. Morawski PA, Bolland S. Expanding the B cell-centric view of systemic lupus erythematosus. Trends Immunol (2017) 38(5):373–82. doi:10.1016/j.it.2017.02.001
42. Huang Z, Fu B, Zheng SG, Li X, Sun R, Tian Z, et al. Involvement of CD226+ NK cells in immunopathogenesis of systemic lupus erythematosus. J Immunol (2011) 186(6):3421–31. doi:10.4049/jimmunol.1000569
43. Kim JS, Cho BA, Sim JH, Shah K, Woo CM, Lee EB, et al. IL-7Ralphalow memory CD8+ T cells are significantly elevated in patients with systemic lupus erythematosus. Rheumatology (Oxford) (2012) 51(9):1587–94. doi:10.1093/rheumatology/kes100
44. Passos de Souza E, Evangelista Segundo PT, Jose FF, Lemaire D, Santiago M. Rheumatoid arthritis induced by alpha-interferon therapy. Clin Rheumatol (2001) 20(4):297–9. doi:10.1007/PL00011206
45. Gota C, Calabrese L. Induction of clinical autoimmune disease by therapeutic interferon-alpha. Autoimmunity (2003) 36(8):511–8. doi:10.1080/08916930310001605873
46. Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S. IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black x New Zealand White) F1 but not in BALB/c mice. J Immunol (2005) 174(5):2499–506. doi:10.4049/jimmunol.174.5.2499
47. Mathian A, Gallegos M, Pascual V, Banchereau J, Koutouzov S. Interferon-alpha induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZBxNZW)F1 mice but not in BALB/c mice. Eur J Immunol (2011) 41(3):863–72. doi:10.1002/eji.201040649
48. Li H, Fu YX, Wu Q, Zhou Y, Crossman DK, Yang P, et al. Interferon-induced mechanosensing defects impede apoptotic cell clearance in lupus. J Clin Invest (2015) 125(7):2877–90. doi:10.1172/JCI81059
49. Cucak H, Yrlid U, Reizis B, Kalinke U, Johansson-Lindbom B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity (2009) 31(3):491–501. doi:10.1016/j.immuni.2009.07.005
50. Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Lyons R, Sobel E, et al. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum (2007) 56(11):3770–83. doi:10.1002/art.23023
51. Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med (2008) 205(13):2995–3006. doi:10.1084/jem.20080462
52. Baccala R, Gonzalez-Quintial R, Schreiber RD, Lawson BR, Kono DH, Theofilopoulos AN. Anti-IFN-alpha/beta receptor antibody treatment ameliorates disease in lupus-predisposed mice. J Immunol (2012) 189(12):5976–84. doi:10.4049/jimmunol.1201477
53. Smeets TJ, Dayer JM, Kraan MC, Versendaal J, Chicheportiche R, Breedveld FC, et al. The effects of interferon-beta treatment of synovial inflammation and expression of metalloproteinases in patients with rheumatoid arthritis. Arthritis Rheum (2000) 43(2):270–4. doi:10.1002/1529-0131(200002)43:2<270::AID-ANR5>3.0.CO;2-H
54. Airas L, Niemela J, Yegutkin G, Jalkanen S. Mechanism of action of IFN-beta in the treatment of multiple sclerosis: a special reference to CD73 and adenosine. Ann N Y Acad Sci (2007) 1110:641–8. doi:10.1196/annals.1423.067
55. Triantaphyllopoulos KA, Williams RO, Tailor H, Chernajovsky Y. Amelioration of collagen-induced arthritis and suppression of interferon-gamma, interleukin-12, and tumor necrosis factor alpha production by interferon-beta gene therapy. Arthritis Rheum (1999) 42(1):90–9. doi:10.1002/1529-0131(199901)42:1<90::AID-ANR12>3.0.CO;2-A
56. van Holten J, Reedquist K, Sattonet-Roche P, Smeets TJ, Plater-Zyberk C, Vervoordeldonk MJ, et al. Treatment with recombinant interferon-beta reduces inflammation and slows cartilage destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res Ther (2004) 6(3):R239–49. doi:10.1186/ar1165
57. Treschow AP, Teige I, Nandakumar KS, Holmdahl R, Issazadeh-Navikas S. Stromal cells and osteoclasts are responsible for exacerbated collagen-induced arthritis in interferon-beta-deficient mice. Arthritis Rheum (2005) 52(12):3739–48. doi:10.1002/art.21496
58. Manfredonia F, Pasquali L, Dardano A, Iudice A, Murri L, Monzani F. Review of the clinical evidence for interferon beta 1a (Rebif) in the treatment of multiple sclerosis. Neuropsychiatr Dis Treat (2008) 4(2):321–36. doi:10.2147/NDT.S476
59. Vervoordeldonk MJ, Aalbers CJ, Tak PP. Interferon beta for rheumatoid arthritis: new clothes for an old kid on the block. Ann Rheum Dis (2009) 68(2):157–8. doi:10.1136/ard.2008.097899
60. Oon S, Huynh H, Tai TY, Ng M, Monaghan K, Biondo M, et al. A cytotoxic anti-IL-3Ralpha antibody targets key cells and cytokines implicated in systemic lupus erythematosus. JCI Insight (2016) 1(6):e86131. doi:10.1172/jci.insight.86131
61. Oon S, Wilson NJ, Wicks I. Targeted therapeutics in SLE: emerging strategies to modulate the interferon pathway. Clin Transl Immunology (2016) 5(5):e79. doi:10.1038/cti.2016.26
62. Ronnblom L, Alm GV, Eloranta ML. Type I interferon and lupus. Curr Opin Rheumatol (2009) 21(5):471–7. doi:10.1097/BOR.0b013e32832e089e
63. Oganesyan G, Saha SK, Pietras EM, Guo B, Miyahira AK, Zarnegar B, et al. IRF3-dependent type I interferon response in B cells regulates CpG-mediated antibody production. J Biol Chem (2008) 283(2):802–8. doi:10.1074/jbc.M704755200
64. Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature (2008) 451(7179):725–9. doi:10.1038/nature06537
65. Marichal T, Ohata K, Bedoret D, Mesnil C, Sabatel C, Kobiyama K, et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med (2011) 17(8):996–1002. doi:10.1038/nm.2403
66. Onishi M, Ozasa K, Kobiyama K, Ohata K, Kitano M, Taniguchi K, et al. Hydroxypropyl-beta-cyclodextrin spikes local inflammation that induces Th2 cell and T follicular helper cell responses to the coadministered antigen. J Immunol (2015) 194(6):2673–82. doi:10.4049/jimmunol.1402027
67. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461(7265):788–92. doi:10.1038/nature08476
68. Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell (2008) 134(4):587–98. doi:10.1016/j.cell.2008.06.032
69. Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell (2007) 131(5):873–86. doi:10.1016/j.cell.2007.10.017
70. Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr, Barber GN, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity (2012) 36(1):120–31. doi:10.1016/j.immuni.2011.11.018
71. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet (2006) 38(8):917–20. doi:10.1038/ng1845
72. Gunther C, Berndt N, Wolf C, Lee-Kirsch MA. Familial chilblain lupus due to a novel mutation in the exonuclease III domain of 3’ repair exonuclease 1 (TREX1). JAMA Dermatol (2015) 151(4):426–31. doi:10.1001/jamadermatol.2014.3438
73. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet (2007) 39(9):1065–7. doi:10.1038/ng2091
74. Hasan M, Dobbs N, Khan S, White MA, Wakeland EK, Li QZ, et al. Cutting edge: inhibiting TBK1 by compound II ameliorates autoimmune disease in mice. J Immunol (2015) 195(10):4573–7. doi:10.4049/jimmunol.1500162
75. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi:10.1146/annurev-immunol-020711-075032
76. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol (2015) 15(3):137–48. doi:10.1038/nri3804
77. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity (2013) 38(3):596–605. doi:10.1016/j.immuni.2012.11.020
78. Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, et al. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med (2006) 203(11):2419–24. doi:10.1084/jem.20061254
79. Suan D, Sundling C, Brink R. Plasma cell and memory B cell differentiation from the germinal center. Curr Opin Immunol (2017) 45:97–102. doi:10.1016/j.coi.2017.03.006
80. Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, et al. Germinal-center development of memory B cells driven by IL-9 from follicular helper T cells. Nat Immunol (2017) 18(8):921–30. doi:10.1038/ni.3788
81. Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol (2009) 9(12):845–57. doi:10.1038/nri2637
82. Becker-Herman S, Meyer-Bahlburg A, Schwartz MA, Jackson SW, Hudkins KL, Liu C, et al. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J Exp Med (2011) 208(10):2033–42. doi:10.1084/jem.20110200
83. Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B cell signalling in autoimmunity. Nat Rev Immunol (2017) 17(7):421–36. doi:10.1038/nri.2017.24
84. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi:10.1016/j.immuni.2014.10.004
85. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol (2015) 16(2):142–52. doi:10.1038/ni.3054
86. Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, et al. Mice deficient for the CD40 ligand. Immunity (1994) 1(5):423–31. doi:10.1016/1074-7613(94)90073-6
87. King IL, Mohrs M. IL-4-producing CD4+ T cells in reactive lymph nodes during helminth infection are T follicular helper cells. J Exp Med (2009) 206(5):1001–7. doi:10.1084/jem.20090313
88. Reinhardt RL, Liang HE, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol (2009) 10(4):385–93. doi:10.1038/ni.1715
89. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med (2010) 207(2):365–78. doi:10.1084/jem.20091777
90. Woods M, Zou YR, Davidson A. Defects in germinal center selection in SLE. Front Immunol (2015) 6:425. doi:10.3389/fimmu.2015.00425
91. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435(7041):452–8. doi:10.1038/nature03555
92. Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med (2009) 206(3):561–76. doi:10.1084/jem.20081886
93. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi:10.1126/science.1175870
94. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325(5943):1001–5. doi:10.1126/science.1176676
95. Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, et al. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity (2010) 32(2):253–65. doi:10.1016/j.immuni.2010.01.010
96. Chevalier N, Macia L, Tan JK, Mason LJ, Robert R, Thorburn AN, et al. The role of follicular helper T cell molecules and environmental influences in autoantibody production and progression to inflammatory arthritis in mice. Arthritis Rheumatol (2016) 68(4):1026–38. doi:10.1002/art.39481
97. Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol (2012) 2012:827480. doi:10.1155/2012/827480
98. Wang J, Shan Y, Jiang Z, Feng J, Li C, Ma L, et al. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin Exp Immunol (2013) 174(2):212–20. doi:10.1111/cei.12162
99. Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol (2015) 67(4):988–99. doi:10.1002/art.39020
100. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest (2015) 125(1):292–303. doi:10.1172/JCI76238
101. Szabo K, Papp G, Barath S, Gyimesi E, Szanto A, Zeher M. Follicular helper T cells may play an important role in the severity of primary Sjogren’s syndrome. Clin Immunol (2013) 147(2):95–104. doi:10.1016/j.clim.2013.02.024
102. Verstappen GM, Kroese FG, Meiners PM, Corneth OB, Huitema MG, Haacke EA, et al. B cell depletion therapy normalizes circulating follicular Th cells in primary Sjogren syndrome. J Rheumatol (2017) 44(1):49–58. doi:10.3899/jrheum.160313
103. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi:10.1038/16717
104. Lohning M, Hutloff A, Kallinich T, Mages HW, Bonhagen K, Radbruch A, et al. Expression of ICOS in vivo defines CD4+ effector T cells with high inflammatory potential and a strong bias for secretion of interleukin 10. J Exp Med (2003) 197(2):181–93. doi:10.1084/jem.20020632
105. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34(6):932–46. doi:10.1016/j.immuni.2011.03.023
106. Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol (2016) 17(10):1197–205. doi:10.1038/ni.3554
107. Wong SC, Oh E, Ng CH, Lam KP. Impaired germinal center formation and recall T-cell-dependent immune responses in mice lacking the costimulatory ligand B7-H2. Blood (2003) 102(4):1381–8. doi:10.1182/blood-2002-08-2416
108. Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature (2001) 409(6816):97–101. doi:10.1038/35051100
109. McAdam AJ, Greenwald RJ, Levin MA, Chernova T, Malenkovich N, Ling V, et al. ICOS is critical for CD40-mediated antibody class switching. Nature (2001) 409(6816):102–5. doi:10.1038/35051107
110. Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, et al. ICOS is essential for effective T-helper-cell responses. Nature (2001) 409(6816):105–9. doi:10.1038/35051113
111. Gigoux M, Shang J, Pak Y, Xu M, Choe J, Mak TW, et al. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc Natl Acad Sci U S A (2009) 106(48):20371–6. doi:10.1073/pnas.0911573106
112. Mittereder N, Kuta E, Bhat G, Dacosta K, Cheng LI, Herbst R, et al. Loss of immune tolerance is controlled by ICOS in Sle1 mice. J Immunol (2016) 197(2):491–503. doi:10.4049/jimmunol.1502241
113. Hamel KM, Cao Y, Olalekan SA, Finnegan A. B cell-specific expression of inducible costimulator ligand is necessary for the induction of arthritis in mice. Arthritis Rheumatol (2014) 66(1):60–7. doi:10.1002/art.38207
114. Iwai H, Kozono Y, Hirose S, Akiba H, Yagita H, Okumura K, et al. Amelioration of collagen-induced arthritis by blockade of inducible costimulator-B7 homologous protein costimulation. J Immunol (2002) 169(8):4332–9. doi:10.4049/jimmunol.169.8.4332
115. Hu YL, Metz DP, Chung J, Siu G, Zhang M. B7RP-1 blockade ameliorates autoimmunity through regulation of follicular helper T cells. J Immunol (2009) 182(3):1421–8. doi:10.4049/jimmunol.182.3.1421
116. Sullivan BA, Tsuji W, Kivitz A, Peng J, Arnold GE, Boedigheimer MJ, et al. Inducible T-cell co-stimulator ligand (ICOSL) blockade leads to selective inhibition of anti-KLH IgG responses in subjects with systemic lupus erythematosus. Lupus Sci Med (2016) 3(1):e000146. doi:10.1136/lupus-2016-000146
117. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31(3):457–68. doi:10.1016/j.immuni.2009.07.002
118. Dodson LF, Boomer JS, Deppong CM, Shah DD, Sim J, Bricker TL, et al. Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol Cell Biol (2009) 29(13):3710–21. doi:10.1128/MCB.01869-08
119. Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai CY, Emslie E, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity (2015) 42(2):239–51. doi:10.1016/j.immuni.2015.01.017
120. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz LJ, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Kruppel-like factor 2. J Exp Med (2015) 212(2):217–33. doi:10.1084/jem.20141432
121. Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LM, et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol (2010) 185(7):4042–52. doi:10.4049/jimmunol.1001730
122. Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell (2011) 41(4):458–70. doi:10.1016/j.molcel.2011.01.019
123. Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch’en IL, Stockmann C, et al. Foxo transcription factors control regulatory T cell development and function. Immunity (2010) 33(6):890–904. doi:10.1016/j.immuni.2010.12.002
124. Yu J, Zhou X, Chang M, Nakaya M, Chang JH, Xiao Y, et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat Commun (2015) 6:6074. doi:10.1038/ncomms7074
125. Kadkhoda K, Wang S, Joyee AG, Fan Y, Yang J, Yang X. Th1 cytokine responses fail to effectively control chlamydia lung infection in ICOS ligand knockout mice. J Immunol (2010) 184(7):3780–8. doi:10.4049/jimmunol.0901384
126. Nouailles G, Day TA, Kuhlmann S, Loewe D, Dorhoi A, Gamradt P, et al. Impact of inducible co-stimulatory molecule (ICOS) on T-cell responses and protection against Mycobacterium tuberculosis infection. Eur J Immunol (2011) 41(4):981–91. doi:10.1002/eji.201040608
127. Wikenheiser DJ, Ghosh D, Kennedy B, Stumhofer JS. The costimulatory molecule ICOS regulates host Th1 and follicular Th cell differentiation in response to Plasmodium chabaudi AS infection. J Immunol (2016) 196(2):778–91. doi:10.4049/jimmunol.1403206
128. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469(7330):323–35. doi:10.1038/nature09782
129. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol (2011) 27:107–32. doi:10.1146/annurev-cellbio-092910-154005
130. Deretic V. Autophagy: an emerging immunological paradigm. J Immunol (2012) 189(1):15–20. doi:10.4049/jimmunol.1102108
131. Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell (2014) 54(2):224–33. doi:10.1016/j.molcel.2014.03.009
132. Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol (2009) 10(11):1215–21. doi:10.1038/ni.1800
133. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (2011) 333(6039):228–33. doi:10.1126/science.1205405
134. Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity (2012) 37(2):223–34. doi:10.1016/j.immuni.2012.04.015
135. Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell (2011) 44(2):279–89. doi:10.1016/j.molcel.2011.07.039
136. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A (2012) 109(46):E3168–76. doi:10.1073/pnas.1210500109
137. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13(10):722–37. doi:10.1038/nri3532
138. Pu Q, Gan C, Li R, Li Y, Tan S, Li X, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in Pseudomonas sepsis. J Immunol (2017) 198(8):3205–13. doi:10.4049/jimmunol.1601196
139. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature (2006) 441(7095):885–9. doi:10.1038/nature04724
140. Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci (2015) 16(6):345–57. doi:10.1038/nrn3961
141. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature (2015) 524(7565):309–14. doi:10.1038/nature14893
142. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature (2010) 465(7295):223–6. doi:10.1038/nature08971
143. Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol (2009) 182(7):4046–55. doi:10.4049/jimmunol.0801143
144. O’Sullivan TE, Geary CD, Weizman OE, Geiger TL, Rapp M, Dorn GW II, et al. Atg5 is essential for the development and survival of innate lymphocytes. Cell Rep (2016) 15(9):1910–9. doi:10.1016/j.celrep.2016.04.082
145. Jia W, He YW. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol (2011) 186(9):5313–22. doi:10.4049/jimmunol.1002404
146. Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol (2011) 186(3):1564–74. doi:10.4049/jimmunol.1001822
147. Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med (2011) 208(3):455–67. doi:10.1084/jem.20101145
148. Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, et al. Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity (2017) 47(3):466–480e465. doi:10.1016/j.immuni.2017.08.005
149. Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity (2010) 32(2):227–39. doi:10.1016/j.immuni.2009.12.006
150. Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy (2015) 11(2):271–84. doi:10.1080/15548627.2015.1009787
151. Santeford A, Wiley LA, Park S, Bamba S, Nakamura R, Gdoura A. Impaired autophagy in macrophages promotes inflammatory eye disease. Autophagy (2016) 12(10):1876–85. doi:10.1080/15548627.2016.1207857
152. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J (2011) 30(23):4701–11. doi:10.1038/emboj.2011.398
153. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi:10.1038/ni.1980
154. Hubbard-Lucey VM, Shono Y, Maurer K, West ML, Singer NV, Ziegler CG, et al. Autophagy gene Atg16L1 prevents lethal T cell alloreactivity mediated by dendritic cells. Immunity (2014) 41(4):579–91. doi:10.1016/j.immuni.2014.09.011
155. Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature (2014) 506(7489):456–62. doi:10.1038/nature13044
156. Tschurtschenthaler M, Adolph TE, Ashcroft JW, Niederreiter L, Bharti R, Saveljeva S, et al. Defective ATG16L1-mediated removal of IRE1alpha drives Crohn’s disease-like ileitis. J Exp Med (2017) 214(2):401–22. doi:10.1084/jem.20160791
157. Xiao Y, Zou Q, Xie X, Liu T, Li HS, Jie Z, et al. The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J Exp Med (2017) 214(5):1493–507. doi:10.1084/jem.20161524
158. Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab (2013) 18(4):519–32. doi:10.1016/j.cmet.2013.09.010
159. Becker L, Nguyen L, Gill J, Kulkarni S, Pasricha PJ, Habtezion A. Age-dependent shift in macrophage polarisation causes inflammation-mediated degeneration of enteric nervous system. Gut (2017). doi:10.1136/gutjnl-2016-312940
160. Jin J, Xiao Y, Chang JH, Yu J, Hu H, Starr R, et al. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat Immunol (2012) 13(11):1101–9. doi:10.1038/ni.2423
161. Johnson SA, Rozzo SJ, Cambier JC. Aging-dependent exclusion of antigen-inexperienced cells from the peripheral B cell repertoire. J Immunol (2002) 168(10):5014–23. doi:10.4049/jimmunol.168.10.5014
162. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol (2013) 14(3):298–305. doi:10.1038/ni.2524
163. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med (2014) 20(5):503–10. doi:10.1038/nm.3521
164. Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife (2014) 3:1–21. doi:10.7554/eLife.03706
165. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol (2014) 15(12):1152–61. doi:10.1038/ni.3025
166. O’Sullivan TE, Johnson LR, Kang HH, Sun JC. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity (2015) 43(2):331–42. doi:10.1016/j.immuni.2015.07.012
Keywords: TANK-binding kinase 1, type 1 interferons, germinal center, autophagy, humoral immunity, autoimmunity
Citation: Louis C, Burns C and Wicks I (2018) TANK-Binding Kinase 1-Dependent Responses in Health and Autoimmunity. Front. Immunol. 9:434. doi: 10.3389/fimmu.2018.00434
Received: 08 December 2017; Accepted: 19 February 2018;
Published: 06 March 2018
Edited by:
Raffi Gugasyan, Burnet Institute, AustraliaCopyright: © 2018 Louis, Burns and Wicks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cynthia Louis, bG91aXMuY0B3ZWhpLmVkdS5hdQ==;
Ian Wicks, d2lja3NAd2VoaS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.