 Erica J. Brodie
Erica J. Brodie Simona Infantino
Simona Infantino Michael S. Y. Low
Michael S. Y. Low David M. Tarlinton
David M. Tarlinton
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 March 2018
Sec. B Cell Biology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00401
This article is part of the Research Topic Essential Kinases and Transcriptional Regulators and their Roles in Autoimmunity View all 10 articles
Systemic lupus erythematosus (SLE) is a progressive autoimmune disease characterized by increased sensitivity to self-antigens, auto-antibody production, and systemic inflammation. B cells have been implicated in disease progression and as such represent an attractive therapeutic target. Lyn is a Src family tyrosine kinase that plays a major role in regulating signaling pathways within B cells as well as other hematopoietic cells. Its role in initiating negative signaling cascades is especially critical as exemplified by Lyn−/− mice developing an SLE-like disease with plasma cell hyperplasia, underscoring the importance of tightly regulating signaling within B cells. This review highlights recent advances in our understanding of the function of the Src family tyrosine kinase Lyn in B lymphocytes and its contribution to positive and negative signaling pathways that are dysregulated in autoimmunity.
Systemic lupus erythematosus is a heterogeneous autoimmune disease with multiple clinical manifestations including auto-antibodies, dermatological rashes and ulcers, inflammatory markers, hematological deficiencies, arthritis, renal dysfunction, and neurological disorders (1). The incidence of SLE is approximately 1:1,500; however, the prevalence varies significantly with gender, ethnicity, and age (2). The progression of SLE symptoms is mediated by a dysregulation of innate (e.g., dendritic cells, mast cells, and macrophages) and adaptive immune cells (i.e., B and T lymphocytes) (3). Accumulating evidence emphasizes the contribution of B cells in mediating the development of autoimmunity, particularly through the breakdown in tolerance to self-antigens, the secretion of inflammatory cytokines, and the generation of auto-reactive antibodies that result in immune complex deposition in organs such as the kidneys (4–6). In keeping with this, numerous mutations affecting tyrosine kinases have been implicated in autoimmune disease progression (7), highlighting a critical requirement for the stringent regulation of intracellular signaling cascades in immune cells.
This review will focus on intracellular signaling within B cells, specifically on the role of Lyn in regulating these pathways and its contribution to the progression of autoimmune disease.
Lyn (Lck/yes-related novel tyrosine kinase) is a Src family, non-receptor tyrosine kinase found predominantly in myeloid cells and B lymphocytes (8), but it is also detectable in cell types outside of the hematopoietic compartment (9). Lyn is located on chromosome 4 A1 in mice and 8q12 in humans (9). In mice, an additional lyn gene encoding exons 1–10 is present within the genome but is not transcribed (10). Alternate splicing of exon 2 results in the translation of two different isoforms of Lyn, annotated as Lyn A (56 kDa) and Lyn B (53 kDa), that differ by an insertion within the N-terminal variable domain (11, 12). Functional analysis of the individual isoforms indicates a similar capacity to phosphorylate substrate proteins (12–14); however, both isoforms are required for normal activation and regulation of internal signaling (13, 14).
Lyn shares architectural and sequence homology with the other SFK members present in hematopoietic cells (e.g., Src, Fyn, Yes, Blk, and Hck) (15). The conserved domain organization of SFK members includes an N-terminal/unique domain, Src homology (SH) 3, SH2 and the catalytic/kinase domain. The N-terminal unique domain (SH4) contains sites for myristoylation and palmitoylation that promote localization and interaction with the cellular membrane and also integration into lipid rafts (16–18). Downstream of the N-terminus, SH3 and SH2 domains regulate the conformational and functional state of Lyn (19). Phosphorylation of the inhibitory tyrosine (Y529 in murine Src, Y508 in Lyn) on the C-terminus leads to a closed and inactive conformation via the binding and “latching” of the SH2 domain, which is further stabilized by the interaction with the SH3 domain (20, 21).
Dephosphorylation of the C-terminal tyrosine by phosphatases or competitive binding for the SH2 domain by an interacting protein relieves the inhibitory conformation imposed by SH3/2 domains, which then promotes phosphorylation of the activating tyrosine (Y416 in Src, Y397 in Lyn) within the kinase domain (19, 21, 22). Phosphorylation of Y397, either in cis or in trans, changes the conformation of the activation loop, permitting substrate binding and kinase activity (19, 22, 23). Kinase activity is mediated by interactions between the ATP binding loop (G loop) and the ATP hydrolyzing loop (catalytic loop) that facilitate optimal positioning and hydrolysis of the λ-phosphate group from ATP, respectively (20, 24, 25). The λ-phosphate group is then transferred to a protein substrate that is positioned adjacent to the catalytic loop (25). Dephosphorylation of the activating tyrosine or phosphorylation of the inhibitory tyrosine by Csk (c-Src kinase) decreases the kinase activity of Lyn and other SFKs (26).
Activation of Lyn relies on membrane-bound receptor-type phosphatases such as CD45 and CD148 to dephosphorylate the C-terminal inhibitory tyrosine (27–29). After activation, Lyn binds and phosphorylates substrate proteins that possess tyrosine residues flanked predominantly by acidic residues (30–32). The affinity for particular substrates is further regulated by the phosphorylation of the SH2 domain (33). Key substrates of Lyn include proximal membrane-bound cellular surface receptors containing immunoreceptor tyrosine activating motifs (ITAMs) or immunoreceptor tyrosine inhibitory motifs (ITIMs) within their cytoplasmic tails (34, 35). Phosphorylation of these ITAMs and ITIMs leads to the recruitment and activation of other kinases, phosphatases, and adaptor proteins that enhance or inhibit downstream signaling.
The B cell receptor (BCR) comprises the membrane-bound immunoglobulin (Ig) and the heterodimeric signaling subunit Ig-α/Ig-β (CD79α/β) that contain ITAMs within their cytoplasmic tails (36). Antigen binding to the BCR induces a variety of signaling cascades that are initiated by the proximal kinase Lyn (Figure 1), which phosphorylates Ig-α/Ig-β ITAMs thereby creating docking sites for the recruitment and activation of Syk (37–39). Activated Syk leads to the phosphorylation and activation of downstream molecules such as the adaptor protein SH2 domain-containing leukocyte protein of 65 kDa (SLP-65) (also known as BLNK or BASH), Btk, and PLCγ2 (40). Upon phosphorylation, SLP-65 organizes a signalosome that promotes calcium (Ca2+) flux and the differentiation of developing B cells (41). Phosphorylated SLP-65 also allows the recruitment of Bruton’s tyrosine kinase (Btk), Vav Guanine Nucleotide Exchange Factor 1 (Vav1), and Growth Factor Receptor Bound Protein 2 (Grb2) (42).
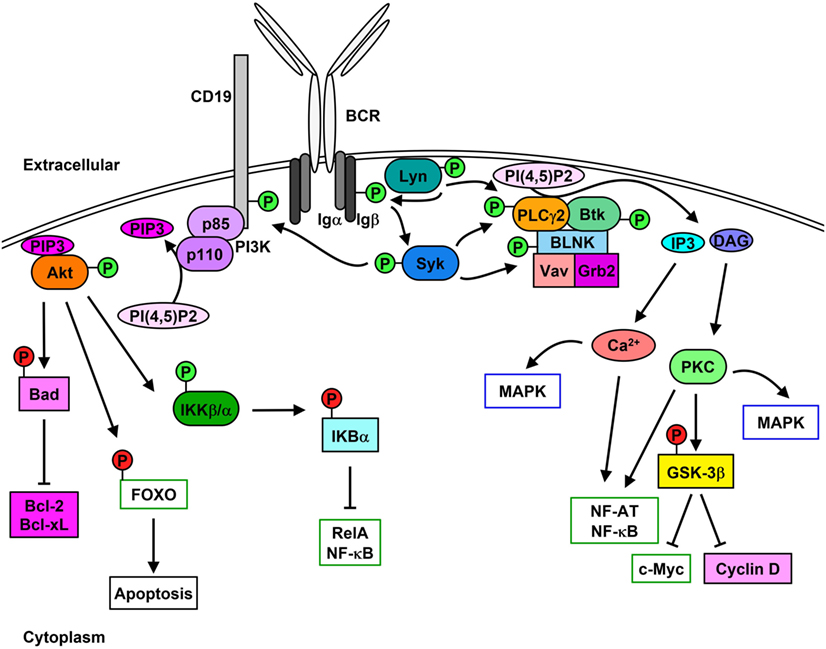
Figure 1. Lyn initiates signaling cascades following B cell receptor (BCR) cross-linking. Stimulation of the BCR leads to the activation [green (P)] of Lyn and Syk which results in Igα/β phosphorylation. PLCγ2 activity leads to the hydrolysis of PI(4,5)P2 to IP3 and diacylglycerol (DAG), which stimulates Ca2+ mobilization as well as protein kinase C (PKC) and MAPK activation and cell-cycle progression. Phosphorylation of CD19 leads to the membrane recruitment of phosphatidylinositol 3-kinase (PI3K) and activation of Akt, which phosphorylates downstream proteins leading to their inhibition [red (P)] and up-regulation of pro-survival signaling.
Phosphorylation of the non-T-cell activation linker (NTAL) by Lyn leads to the recruitment of Grb2, murine son of sevenless homolog (mSOS), and GRB2-Associated Binding Protein 1 (Gab1) (43). Phosphorylation of Gab1 promotes complex formation between SH2 Domain-Containing Transforming Protein 1 (Shc), non-receptor SH2-containing tyrosine phosphatase 2 (SHP-2), and p85 subunit of phosphatidylinositol 3-kinase (PI3K) (44). Syk-mediated phosphorylation of Shc promotes the interaction between Grb2 and mSOS, which activates Ras-MAPK pathways (45), while interaction between Shc and Gab1 leads to the activation of PI3K pathway (46, 47).
Syk-mediated phosphorylation of CD19 enables the recruitment and activation of p85, which also promotes membrane localization of PI3K (48, 49). Syk also phosphorylates E3 ubiquitin-protein ligase Cbl, which permits the interaction with p85 and activation of PI3K (50). Similarly, Syk and Btk phosphorylate B cell phosphoinositide 3-kinase adapter protein 1 (BCAP), which localizes p85 to glycolipid-enriched microdomains (GEMs) after anti-IgM stimulation (51). PI3K catalyzes the generation of phosphatidyl inositol 3,4,5-trisphosphate (PIP3) from phosphatidyl inositol 4,5-trisphosphate (PI(4,5)P2), which in turn is used as a docking site to activate other effector proteins (52). PIP3 is essential for plasma membrane translocation of Akt, placing it in proximity to 3-phosphoinositide-dependent protein kinase 1 (PDK1) and rapamycin-insensitive companion of mammalian target of rapamycin (Rictor)/mechanistic target of rapamycin (mTOR2) allowing threonine and serine phosphorylation of Akt, respectively (53, 54). Akt phosphorylation and activation following BCR cross-linking leads to the inhibition of the pro-apoptotic protein Bad and activation of the pro-survival proteins Bcl-2 and Bcl-XL (55). Additionally, Akt phosphorylates the transcription factor Forkhead box protein O (FOXO) thereby inducing its exclusion from the nucleus and preventing apoptosis while promoting cell proliferation (56, 57). Akt also activates alpha and beta subunits of the IκB kinase (IKKα/β) which phosphorylates IκBα and the p65 nuclear factor kappaB (NF-κB) subunit/RelA (58). IκBα phosphorylation leads to its proteasome-mediated degradation and exposure of the nuclear-localization sequence of RelA, which leads to its translocation and up-regulation of pro-proliferation and pro-survival proteins such as c-Myc and Bcl-2 family proteins (59, 60).
Lyn, Syk, Btk, and Blk can also phosphorylate and enhance the activation of phospholipase C gamma 2 (PLCγ2), which hydrolyzes PI(4,5)P2 to create inositol 3,4,5-trisphosphate (IP3) and diacylglycerol (DAG), stimulating Ca2+ mobilization and protein kinase C (PKC), respectively (61–64). PLCγ2 phosphorylation also stimulates the activation of MAPK pathways and nuclear location of NF-κB and nuclear factor of activated T cells (NF-AT) (65–67). Additionally, PKC activation leads to the inhibition of glycogen synthase kinase 3 beta (GSK-3β), which promotes the accumulation of beta-catenin in the nucleus and thus up-regulates the expression of c-Myc and cyclin D (68).
B cell scaffold protein with ankyrin repeats 1 (BANK1) interacts with PLCγ2, which is mediated by B-lymphocyte kinase (Blk) (69). BANK1 promotes the Lyn-mediated phosphorylation of the inositol trisphosphate receptor (IP3R) located on the endoplasmic reticulum which mediates Ca2+ flux (70).
The role of Lyn in generating the positive signaling cascade in B cells is not essential and can be compensated for the other SFKs (71, 72). However, the role of Lyn in the initiation of negative feedback loops that not only regulate downstream signaling molecules but also limit the activity of SFKs is unique (35, 73). In addition to the phosphorylation of ITAMs after BCR stimulation, Lyn phosphorylates ITIMs contained within receptors including CD22, FcγRIIB, PIR-B, PD-1, CD66a (CEACAM1), CD5, and CD72 (35, 74–79), which act as docking sites for the binding and activation of non-receptor SH2-containing tyrosine phosphatases 1 and 2 (SHP-1 and SHP-2) and, in the case of FcγRIIB, phosphatidylinositol phosphatase 5 (SHIP-1) (76, 80–86). SHP-1 has been demonstrated to dephosphorylate and, therefore, down-regulate the activity of Btk, Syk, and Lyn, leading to the inhibition of signaling cascades (85, 87–89). Similarly, SHP-2 has been shown to dephosphorylate Ig-β, Syk, and PLCγ2 following BCR cross-linking (76). However, SHP-2 has also been implicated in enhancing various signaling pathways (90).
Upon BCR activation (Figure 2), SHIP-1 dephosphorylates PIP3 to P(3,4)P2, which decreases PI3K- and Akt-mediated signaling (91). The decrease of available PIP3 also leads to the down-regulation of Btk activity and subsequent reduction of PLCγ2-mediated Ca2+ mobilization (92). Activated SHIP-1 recruits downstream of tyrosine kinase 1 (Dok1/p62dok), which down-regulates MAPK signaling pathways (93). FcγRIIB phosphorylation leads to the Dok3-mediated recruitment of SHIP-1 into the Grb2/Shc/mSOS complex, which inhibits SFK-dependent activation of Syk and decreases NF-κB and MAPK signaling pathways (94–97). A more direct role of Lyn in negative signaling is the phosphorylation of the Csk-binding protein/phosphoprotein associated with glyco-sphingolipid microdomains (Cbp/PAG), which activates Csk to down-regulate the activity of Lyn and other active SFKs (98).
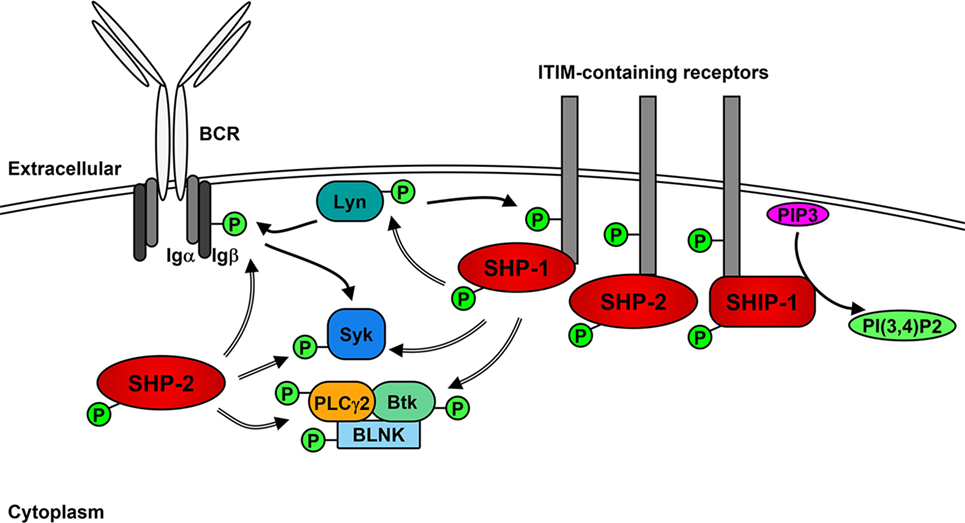
Figure 2. Lyn activates negative receptors following B cell receptor (BCR) cross-linking. In addition to initiating positive signaling, Lyn phosphorylates receptors containing immunoreceptor tyrosine inhibitory motifs (ITIMs), leading to the recruitment and activation of phosphatases SHP-1, SHP-2, and SHIP-1. SHP-1 and -2 dephosphorylate (=> arrows) and inhibit the activity of Lyn, Syk PLCγ2 and Btk leading to the down-regulation of positive signaling events. SHIP-1 dephosphorylates PIP3 and prevents activation of Akt signaling pathways.
Lyn also mediates internalization of the BCR, which acts to dampen signaling by the BCR (99) and may also play a role in the dephosphorylation of BCAP, which down-regulates PI3K activity (51). Similar to the regulation of active Src (100), the kinase activity of Lyn also promotes its own ubiquitination and subsequent degradation in B cells, most likely via the activity of Csk and suppressor of cytokine signaling (SOCS) proteins (101–104).
Consistent with the important role for negative signaling in the hematopoietic compartment, mice lacking Lyn (Lyn−/−) progressively develop symptoms of autoimmunity that are comparable to SLE in humans (105). Lyn−/− mice produce high titers of antinuclear antibodies (ANA) and develop splenomegaly, systemic inflammation, antibody complex deposition in the kidneys and glomerulonephritis (105). B cells from Lyn−/− mice display hyper-phosphorylated Akt, MEK1/2, Erk1/2, and JNK compared to wild-type (WT) B cells after BCR stimulation, indicating enhanced positive signaling in the absence of negative feedback inhibition (23, 72, 106). In keeping with this, there is a reduced phosphorylation of FcγRIIB, CD22, SHP-1, and SHIP-1 (73, 74).
The activation of Lyn-dependent inhibitory signaling in mature B cells is essential for maintaining B cell tolerance (107), including B cell anergy (108). In Lyn-deficient mice, the absence of inhibitory signaling in mature B cells increases their sensitivity to antigen stimulation and this population of now auto-reactive cells is selectively depleted via clonal deletion (109). As such, Lyn−/− mice show a significant reduction in naive, mature B cells in the periphery compared to WT mice despite similar frequencies of newly formed, immature B cells (105). The naive B cells that do persist in the periphery of Lyn−/− mice are, however, hyper-responsive to anti-IgM stimulation, show delayed but increased Ca2+ mobilization and express markers of activation (107, 109–111). Additionally, antibody secreting B cells (plasma cells) persist in lymphoid tissues at 10-times normal frequency (105, 111). The myeloid compartment is also expanded, and T cells display markers of activation (101, 112). Increased serum levels of IL-6, IFN-γ, and BAFF, primarily produced by B cells, T cells, and myeloid cells respectively, promote the activation and proliferation of immune cells, further driving autoimmunity (112, 113).
Components of the autoimmune phenotype of Lyn−/− mice are B cell intrinsic. This has been demonstrated using a B-cell-specific deletion of Lyn (Lynfl/fl mb-1Cre), which recapitulates the generation of hyper-responsive B cells, auto-antibodies and the development of glomerulonephritis (114). This phenotype may be dependent on signaling through toll-like receptor (TLR) adaptor protein myeloid differentiation primary response gene 88 (MyD88), as B-cell-specific deletion of MyD88 from Lyn−/− B cells ameliorated auto-antibody production, T cell activation, myeloid expansion, and the development of glomerulonephritis (114). Additionally, global deletion of MyD88 from Lyn−/− mice attenuated autoimmune disease development, which was considered to be due to reduced production of inflammatory cytokines IL-6 and IL-12 by Lyn−/− dendritic cells (115, 116). This result was supported by Lyn being found to negatively regulate TLR-MyD88-IRF5-dependent expression of Type 1 IFNs in dendritic cells and by the double knock out of Lyn and IRF5 (Lyn−/− IRF5−/−) in mice ameliorating SLE-like pathology (117). Perhaps surprisingly, the persistence of plasma cells was found to be independent of autoimmune disease and intrinsic to the Lyn−/− hematopoietic compartment (118). Similarly, mice double deficient in Lyn and IL-6 retain the plasma cell hyperplasia but show a dramatic reduction in kidney damage, splenomegaly, and the production of ANAs (112). As in B cells, Lyn controls intracellular signaling intensities within plasma cells in response to stimulation by cytokines thought to play a critical role in plasma cell survival (118).
The deletion of MyD88 in Lyn−/− mice also decreased spontaneous germinal center (GC) formation thus implicating GC reactions in the generation of pathogenic ANA in Lyn−/− mice (119). In line with this, removing T cells (TCRβ/δ−/−) or deleting the adaptor protein SAP (SAP−/−), thereby preventing T cell B cell interactions required for GC formation, leads to the reduction of IgG auto-antibodies in Lyn−/− mice (119). Similarly, deleting IL-21, a key regulator of GC responses and plasma cell formation (120), in Lyn−/− mice leads to reduced IgG ANAs but does not alleviate IgM ANA, plasmacytosis, or glomerulonephritis (121), suggesting that IgM and/or IgA can mediate the development of autoimmunity (122). In keeping with this, the autoimmune disease in the sanroque lupus mouse model also develops in the absence of germinal centers and depends on IgM (123).
While Lyn can act as a positive or negative regulator of signaling cascades, its role as a negative regulator is critical to the development of the phenotype seen in Lyn−/− mice. Indeed, the fact that dysregulation of other modulators of negative signaling, which are themselves targets of Lyn, also leads to autoimmunity (e.g., CD22, FcγRIIB, SHP-1, and SHIP-1) (124–128) helps define the pathways involved. This is also apparent in the compounding autoimmune disease phenotype in mice heterozygous in Lyn and SHP-1 (Lyn+/−, Mev+/−) (129). Finally, the importance of Lyn-regulated pathways in hematopoietic cells other than B cells is revealed by co-deletion of Btk, a key intermediate of several positive signaling pathways, from Lyn−/− mice. This alleviates symptoms of autoimmunity and the production of auto-antibodies, but B cells remain hyper-responsive to anti-IgM stimulation as measured by Ca2+ mobilization and the phosphorylation of Erk1/2 and Akt (130, 131). Consistent with this, mice deficient in Lyn and p110δ, a PI3K isoform, also show a reduction in inflammation, splenomegaly, T cell activation, ANA production, and glomerulonephritis, while hyper-phosphorylation of Akt and Erk1/2 compared to control mice after BCR cross-linking remained (132), indicating a unique requirement for Lyn in regulating these signaling responses, but one that is insufficient by itself to permit development of disease.
Lyn’s enzymatic activity appears to be critical to its function, as mice expressing Lyn with no or impaired kinase activity still develop autoimmune disease, albeit with delayed onset and reduced severity (23, 24). B cells from LynMld4 or kinase dead Lyn (LynKD) mice, harboring a mutation within the activation loop, display similar signaling kinetics to Lyn−/− B cells, with hyper-Ca2+ mobilization, hyper-phosphorylated Erk1/2, Akt, and JNK and reduced phosphorylation of Syk, SHIP-1, and SHP-1 after BCR stimulation (23). Expansion of the myeloid compartment, splenomegaly, and the production of IgG anti-dsDNA antibodies were significantly reduced in LynKD compared to Lyn−/− mice, but evidence of immune complex deposition in the kidneys remained (23). In contrast are LynWeeB mice, with a mutation in the G-loop of Lyn that leaves partial kinase activity, conferring B cell signaling kinetics that are intermediate between WT and Lyn−/− (24). Stimulation of LynWeeB B cells results in a partial decrease in the phosphorylation of Syk, Btk, PLCγ2, SHIP-1, and CD22 and a slight increase in the phosphorylation of Erk1/2, Akt, and JNK compared to WT B cells (24). However, in older LynWeeB mice, splenomegaly, anti-dsDNA antibodies, and glomerulonephritis were comparable to those in Lyn−/− mice (24), indicating that the partial positive signaling permitted by LynWeeB requires greater negative signaling to counterbalance its activity and prevent the development of autoimmunity. This is exemplified in mice expressing a mutant form of constitutively active Lyn (LynY508F or Lynup/up), as they develop an autoimmune disease with an increased rate of mortality (male-specific) compared to Lyn−/− mice (102). Lynup/up B cells display constitutive phosphorylation of proteins involved in positive (Syk, PLCγ2) and negative (CD22, SHP-1, SHIP-1, FcγRIIB) signaling pathways, which is further increased after BCR stimulation (102). Despite the increased phosphorylation of ITIM-containing negative regulators, Lynup/up B cells display enhanced Ca2+ mobilization compared even to Lyn−/− B cells, indicating that the increased positive signaling further outweighs the inhibitory signaling capacity within these cells (102). Thus, in the hematopoietic compartment, Lyn activity controls both negative and positive signaling and its dysregulation in mice is responsible for the breakdown of tolerance in B cells and progressive development of autoimmunity.
Systemic lupus erythematosus is a heterogeneous disease with varied clinical presentations and manifestations (1). It is therefore not surprising that its cause is multifactorial, with numerous genetic and environmental factors contributing to pathogenesis (1, 133). As such, a single gene defect in mice such as Lyn−/− is unlikely to replicate the complex phenotype seen within SLE in humans. Despite this, a reduction in Lyn expression, via increased turnover or reduced transcription, and altered sub-cellular localization are reported in patients with SLE (134–136). However, a significant susceptibility association of the Lyn locus with SLE has only been determined in a single case–control association study (137). Interestingly, proteins involved in the positive (Blk, BANK1) and negative (PTPN22/PEP, Csk, FcγRIIA, FcγRIIB, FcγRIIIA, FcγRIIIB, SOCS1) signaling pathways in B cells have all been linked to SLE via genome-wide association studies (GWAS) and this highlights the importance of regulating these signaling cascades to avoid disease onset or progression (138–143). Similarly, Blimp1 and Ets1, transcription factors involved in regulating plasma cell development, are also linked with SLE (141, 144). Mice deficient in Ets1 show enhanced generation of plasma cells and auto-antibodies, which are symptoms of autoimmunity (145). Recently, Lyn was linked with regulating Ets1 expression, via the activation of CD22 and SHP-1 reducing the Btk-dependent down-regulation of Ets1 (146, 147).
Lyn−/− mice are one of the numerous mouse models reported to develop an SLE-like illness, with none being a perfect recapitulation of the spectrum of human disease (148). Regardless, the Lyn−/− mouse model has been and remains a useful tool in dissecting the critical role that B cell signaling pathways play in the development of autoimmunity. Indeed, other models in conjunction with Lyn−/− mice have confirmed the importance of B cell dysregulation to the development of SLE-like autoimmunity. Deleting B cells in the MRL/lpr mouse model, for example, significantly decreases disease progression and mortality compared to mice that remain B cell replete (149). Furthermore, comparison of Lyn−/− with other mouse models has identified several potential novel therapies for patients with SLE such as inhibitors of BTK and HDAC (150, 151). Further determination of the molecular pathways responsible for B cell dysregulation in SLE-like autoimmunity will likely assist in the design of new treatments to ameliorate disease severity.
The role of Lyn in B cells involves fine-tuning of BCR signaling, balancing positive and negative signals to maintain tolerance to self-antigens while permitting responsiveness to foreign antigens. The Lyn-mediated phosphorylation of ITIM-containing negative receptors and subsequent activation of the inhibitory phosphatases, SHP-1 and SHIP-1, that leads to the down-regulation of BCR-mediated signaling cascades and inhibition of SFK activity represents a critical component in B cell signaling that prevents the development of autoimmunity.
Although defects in Lyn−/− mice are not an identical model for human SLE, the investigation of Lyn and the pathways it modulates have highlighted the delicate balance inherent in B cell kinase signaling cascades and the devastating consequences that can occur when they are dysregulated. Numerous SLE susceptibility genes identified through GWAS are also linked with other autoimmune diseases indicating the involvement of shared pathways that ultimately lead to the loss of tolerance (152). Therefore, future experiments examining genomic regulation or global phospho-proteomics in models of SLE could be useful in identifying all the components of the intracellular pathways involved and through that, potential therapeutic targets.
EB, SI, and ML drafted the article. DT contributed to writing and provided critical review. All authors were involved in the revising process and approve the manuscript for submission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with one of the authors [ML].
Work within DT laboratory is supported by a Distinguished Innovator Award from the Alliance for Lupus Research (formerly Lupus Research Institute). ML is funded by a CRB Blackburn scholarship (GNT1075151) jointly from the National Health and Medical Research Council (NHMRC) Australia and Royal Australasian College of Physicians. DT is also a NHMRC Research Fellow.
1. Shaikh MF, Jordan N, D’Cruz DP. Systemic lupus erythematosus. Clin Med (2017) 17(1):78–83. doi:10.7861/clinmedicine.17-1-78
2. Lim SS, Bayakly AR, Helmick CG, Gordon C, Easley KA, Drenkard C. The incidence and prevalence of systemic lupus erythematosus, 2002–2004: the georgia lupus registry. Arthritis Rheumatol (2014) 66(2):357–68. doi:10.1002/art.38239
3. Mohan C, Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat Rev Nephrol (2015) 11:329. doi:10.1038/nrneph.2015.33
4. Cambier JC. Autoimmunity risk alleles: hotspots in B cell regulatory signaling pathways. J Clin Invest (2013) 123(5):1928–31. doi:10.1172/JCI69289
5. Morawski PA, Bolland S. Expanding the B cell-centric view of systemic lupus erythematosus. Trends Immunol (2017) 38(5):373–82. doi:10.1016/j.it.2017.02.001
6. Sanz I. Rationale for B cell targeting in SLE. Semin Immunopathol (2014) 36(3):365–75. doi:10.1007/s00281-014-0430-z
7. Shao W-H, Cohen PL. The role of tyrosine kinases in systemic lupus erythematosus and their potential as therapeutic targets. Expert Rev Clin Immunol (2014) 10(5):573–82. doi:10.1586/1744666X.2014.893827
8. Yamanashi Y, Mori S, Yoshida M, Kishimoto T, Inoue K, Yamamoto T, et al. Selective expression of a protein-tyrosine kinase, p56lyn, in hematopoietic cells and association with production of human T-cell lymphotropic virus type I. Proc Natl Acad Sci U S A (1989) 86(17):6538–42. doi:10.1073/pnas.86.17.6538
9. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science (2015) 347(6220):1260419. doi:10.1126/science.1260419
10. Hibbs ML, Stanley E, Maglitto R, Dunn AR. Identification of a duplication of the mouse Lyn gene. Gene (1995) 156(2):175–81. doi:10.1016/0378-1119(95)00022-X
11. Stanley E, Ralph S, McEwen S, Boulet I, Holtzman DA, Lock P, et al. Alternatively spliced murine lyn mRNAs encode distinct proteins. Mol Cell Biol (1991) 11(7):3399–406. doi:10.1128/mcb.11.7.3399
12. Yi TL, Bolen JB, Ihle JN. Hematopoietic cells express two forms of lyn kinase differing by 21 amino acids in the amino terminus. Mol Cell Biol (1991) 11(5):2391–8. doi:10.1128/mcb.11.5.2391
13. Alvarez-Errico D, Yamashita Y, Suzuki R, Odom S, Furumoto Y, Yamashita T, et al. Functional analysis of lyn kinase A and B isoforms reveals redundant and distinct roles in FcεRI-dependent mast cell activation. J Immunol (2010) 184(9):5000–8. doi:10.4049/jimmunol.0904064
14. Yamanashi Y, Miyasaka M, Takeuchi M, Ilic D, Mizuguchi J, Yamamoto T. Differential responses of p56lyn and p53lyn, products of alternatively spliced lyn mRNA, on stimulation of B-cell antigen receptor. Cell Regul (1991) 2(12):979–87.
15. Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene (2004) 23(48):7918–27. doi:10.1038/sj.onc.1208081
16. Oneyama C, Iino T, Saito K, Suzuki K, Ogawa A, Okada M. Transforming potential of Src family kinases is limited by the cholesterol-enriched membrane microdomain. Mol Cell Biol (2009) 29(24):6462–72. doi:10.1128/MCB.00941-09
17. Patwardhan P, Resh MD. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol Cell Biol (2010) 30(17):4094–107. doi:10.1128/MCB.00246-10
18. Sato I, Obata Y, Kasahara K, Nakayama Y, Fukumoto Y, Yamasaki T, et al. Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J Cell Sci (2009) 122(7):965–75. doi:10.1242/jcs.034843
19. Williams NK, Lucet IS, Klinken SP, Ingley E, Rossjohn J. Crystal structures of the Lyn protein tyrosine kinase domain in its apo- and inhibitor-bound state. J Biol Chem (2009) 284(1):284–91. doi:10.1074/jbc.M807850200
20. Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell (1999) 3(5):629–38. doi:10.1016/S1097-2765(00)80356-1
21. Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, et al. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure (2005) 13(6):861–71. doi:10.1016/j.str.2005.03.012
22. Breitenlechner CB, Kairies NA, Honold K, Scheiblich S, Koll H, Greiter E, et al. Crystal structures of active Src kinase domain complexes. J Mol Biol (2005) 353(2):222–31. doi:10.1016/j.jmb.2005.08.023
23. Verhagen AM, Wallace ME, Goradia A, Jones SA, Croom HA, Metcalf D, et al. A kinase-dead allele of Lyn attenuates autoimmune disease normally associated with Lyn deficiency. J Immunol (2009) 182(4):2020–9. doi:10.4049/jimmunol.0803127
24. Barouch-Bentov R, Che J, Lee CC, Yang Y, Herman A, Jia Y, et al. A conserved salt bridge in the G loop of multiple protein kinases is important for catalysis and for in vivo Lyn function. Mol Cell (2009) 33(1):43–52. doi:10.1016/j.molcel.2008.12.024
25. Roskoski R Jr. Src protein–tyrosine kinase structure and regulation. Biochem Biophys Res Commun (2004) 324(4):1155–64. doi:10.1016/j.bbrc.2004.09.171
26. Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H. CSK: a protein-tyrosine kinase involved in regulation of src family kinases. J Biol Chem (1991) 266(36):24249–52.
27. Zhu JW, Brdicka T, Katsumoto TR, Lin J, Weiss A. Structurally distinct phosphatases CD45 and CD148 both regulate B cell and macrophage immunoreceptor signaling. Immunity (2008) 28(2):183–96. doi:10.1016/j.immuni.2007.11.024
28. Zikherman J, Doan K, Parameswaran R, Raschke W, Weiss A. Quantitative differences in CD45 expression unmask functions for CD45 in B-cell development, tolerance, and survival. Proc Natl Acad Sci U S A (2012) 109(1):E3–12. doi:10.1073/pnas.1117374108
29. Skrzypczynska KM, Zhu JW, Weiss A. Positive regulation of Lyn kinase by CD148 is required for B cell receptor signaling in B1 but not B2 B cells. Immunity (2016) 45(6):1232–44. doi:10.1016/j.immuni.2016.10.013
30. Ritz A, Shakhnarovich G, Salomon AR, Raphael BJ. Discovery of phosphorylation motif mixtures in phosphoproteomics data. Bioinformatics (2009) 25(1):14–21. doi:10.1093/bioinformatics/btn569
31. Schmitz R, Baumann G, Gram H. Catalytic Specificity of phosphotyrosine kinases Blk, Lyn, c-Src and Syk as assessed by phage display. J Mol Biol (1996) 260(5):664–77. doi:10.1006/jmbi.1996.0429
32. Amano M, Hamaguchi T, Shohag MH, Kozawa K, Kato K, Zhang X, et al. Kinase-interacting substrate screening is a novel method to identify kinase substrates. J Cell Biol (2015) 209(6):895. doi:10.1083/jcb.201412008
33. Jin LL, Wybenga-Groot LE, Tong J, Taylor P, Minden MD, Trudel S, et al. Tyrosine phosphorylation of the Lyn Src Homology 2 (SH2) domain modulates its binding affinity and specificity. Mol Cell Proteomics (2015) 14(3):695–706. doi:10.1074/mcp.M114.044404
34. Johnson SA, Pleiman CM, Pao L, Schneringer J, Hippen K, Cambier JC. Phosphorylated immunoreceptor signaling motifs (ITAMs) exhibit unique abilities to bind and activate Lyn and Syk tyrosine kinases. J Immunol (1995) 155(10):4596–603.
35. Nishizumi H, Horikawa K, Mlinaric-Rascan I, Yamamoto TA. Double-edged kinase Lyn: a positive and negative regulator for antigen receptor-mediated signals. J Exp Med (1998) 187(8):1343–8. doi:10.1084/jem.187.8.1343
37. Yamanashi Y, Kakiuchi T, Mizuguchi J, Yamamoto T, Toyoshima K. Association of B cell antigen receptor with protein tyrosine kinase Lyn. Science (1991) 251(4990):192–4. doi:10.1126/science.1702903
38. Pao LI, Famiglietti SJ, Cambier JC. Asymmetrical phosphorylation and function of immunoreceptor tyrosine-based activation motif tyrosines in B cell antigen receptor signal transduction. J Immunol (1998) 160(7):3305–14.
39. Stepanek O, Draber P, Drobek A, Horejsi V, Brdicka T. Nonredundant roles of Src-family kinases and Syk in the initiation of B-cell antigen receptor signaling. J Immunol (2013) 190(4):1807–18. doi:10.4049/jimmunol.1202401
40. Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol (2002) 2:945. doi:10.1038/nri955
41. Chiu CW, Dalton M, Ishiai M, Kurosaki T, Chan AC. BLNK: molecular scaffolding through ‘cis’-mediated organization of signaling proteins. EMBO J (2002) 21(23):6461–72. doi:10.1093/emboj/cdf658
42. Geahlen RL. Syk and pTyr’d: signaling through the B cell antigen receptor. Biochim Biophys Acta (2009) 1793(7):1115–27. doi:10.1016/j.bbamcr.2009.03.004
43. Brdička T, Imrich M, Angelisová P, Brdičková N, Horváth O, Špička J, et al. Non-T cell activation linker (NTAL). J Exp Med (2002) 196(12):1617. doi:10.1084/jem.20021405
44. Ingham RJ, Holgado-Madruga M, Siu C, Wong AJ, Gold MR. The Gab1 protein is a docking site for multiple proteins involved in signaling by the B cell antigen receptor. J Biol Chem (1998) 273(46):30630–7. doi:10.1074/jbc.273.46.30630
45. Lai K-MV, Pawson T. The ShcA phosphotyrosine docking protein sensitizes cardiovascular signaling in the mouse embryo. Genes Dev (2000) 14(9):1132–45. doi:10.1101/gad.14.9.1132
46. Ravichandran KS. Signaling via Shc family adapter proteins. Oncogene (2001) 20:6322. doi:10.1038/sj.onc.1204776
47. Ingham RJ, Santos L, Dang-Lawson M, Holgado-Madruga M, Dudek P, Maroun CR, et al. The Gab1 docking protein links the B cell antigen receptor to the phosphatidylinositol 3-kinase/Akt signaling pathway and to the SHP2 tyrosine phosphatase. J Biol Chem (2001) 276(15):12257–65. doi:10.1074/jbc.M010590200
48. Yokozeki T, Adler K, Lankar D, Bonnerot C. B cell receptor-mediated Syk-independent activation of phosphatidylinositol 3-kinase, Ras, and mitogen-activated protein kinase pathways. J Immunol (2003) 171(3):1328. doi:10.4049/jimmunol.171.3.1328
49. Otero DC, Omori SA, Rickert RC. CD19-dependent activation of Akt kinase in B-lymphocytes. J Biol Chem (2001) 276(2):1474–8. doi:10.1074/jbc.M003918200
50. Beitz LO, Fruman DA, Kurosaki T, Cantley LC, Scharenberg AM. SYK is upstream of phosphoinositide 3-kinase in B cell receptor signaling. J Biol Chem (1999) 274(46):32662–6. doi:10.1074/jbc.274.46.32662
51. Okada T, Maeda A, Iwamatsu A, Gotoh K, Kurosaki T. BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity (2000) 13(6):817–27. doi:10.1016/S1074-7613(00)00079-0
52. Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol (2012) 13:195. doi:10.1038/nrm3290
53. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307(5712):1098. doi:10.1126/science.1106148
54. Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJC, Frech M, et al. Role of translocation in the activation and function of protein kinase B. J Biol Chem (1997) 272(50):31515–24. doi:10.1074/jbc.272.50.31515
55. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell (1997) 91(2):231–41. doi:10.1016/S0092-8674(00)80405-5
56. Werner M, Hobeika E, Jumaa H. Role of PI3K in the generation and survival of B cells. Immunol Rev (2010) 237(1):55–71. doi:10.1111/j.1600-065X.2010.00934.x
57. Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, et al. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell (2008) 32(2):221–31. doi:10.1016/j.molcel.2008.09.013
58. Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int J Cancer (2009) 125(12):2863–70. doi:10.1002/ijc.24748
59. Grumont RJ, Strasser A, Gerondakis S. B cell growth is controlled by phosphatidylinosotol 3-kinase-dependent induction of Rel/NF-κB regulated c-myc transcription. Mol Cell (2002) 10(6):1283–94. doi:10.1016/S1097-2765(02)00779-7
60. Sen R. Control of B lymphocyte apoptosis by the transcription factor NF-κB. Immunity (2006) 25(6):871–83. doi:10.1016/j.immuni.2006.12.003
61. Li Z, Wahl MI, Eguinoa A, Stephens LR, Hawkins PT, Witte ON. Phosphatidylinositol 3-kinase-γ activates Bruton’s tyrosine kinase in concert with Src family kinases. Proc Natl Acad Sci U S A (1997) 94(25):13820–5. doi:10.1073/pnas.94.25.13820
62. Ishiai M, Kurosaki M, Pappu R, Okawa K, Ronko I, Fu C, et al. BLNK required for coupling Syk to PLCγ2 and Rac1-JNK in B cells. Immunity (1999) 10(1):117–25. doi:10.1016/S1074-7613(00)80012-6
63. Engels N, Wollscheid B, Wienands J. Association of SLP-65/BLNK with the B cell antigen receptor through a non-ITAM tyrosine of Ig-α. Eur J Immunol (2001) 31(7):2126–34. doi:10.1002/1521-4141(200107)31:7<2126:AID-IMMU2126>3.0.CO;2-O
64. Kabak S, Skaggs BJ, Gold MR, Affolter M, West KL, Foster MS, et al. The direct recruitment of BLNK to immunoglobulin α couples the B-cell antigen receptor to distal signaling pathways. Mol Cell Biol (2002) 22(8):2524–35. doi:10.1128/MCB.22.8.2524-2535.2002
65. Antony P, Petro JB, Carlesso G, Shinners NP, Lowe J, Khan WN. B-cell antigen receptor activates transcription factors NFAT (nuclear factor of activated T-cells) and NF-κB (nuclear factor κB) via a mechanism that involves diacylglycerol. Biochem Soc Trans (2004) 32(1):113–5. doi:10.1042/bst0320113
66. Petro JB, Khan WN. Phospholipase C-γ2 couples Bruton’s tyrosine kinase to the NF-κB signaling pathway in B lymphocytes. J Biol Chem (2001) 276(3):1715–9. doi:10.1074/jbc.M009137200
67. Hashimoto A, Okada H, Jiang A, Kurosaki M, Greenberg S, Clark EA, et al. Involvement of guanosine triphosphatases and phospholipase C-γ2 in extracellular signal–regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J Exp Med (1998) 188(7):1287–95. doi:10.1084/jem.188.7.1287
68. Christian SL, Sims PV, Gold MR. The B cell antigen receptor regulates the transcriptional activator β-catenin via protein kinase C-mediated inhibition of glycogen synthase kinase-3. J Immunol (2002) 169(2):758–69. doi:10.4049/jimmunol.169.2.758
69. Bernal-Quirós M, Wu Y-Y, Alarcón-Riquelme ME, Castillejo-López C. BANK1 and BLK act through phospholipase C gamma 2 in B-cell signaling. PLoS One (2013) 8(3):e59842. doi:10.1371/journal.pone.0059842
70. Yokoyama K, Su IH, Tezuka T, Yasuda T, Mikoshiba K, Tarakhovsky A, et al. BANK regulates BCR-induced calcium mobilization by promoting tyrosine phosphorylation of IP(3) receptor. EMBO J (2002) 21(1–2):83–92. doi:10.1093/emboj/21.1.83
71. Saijo K, Schmedt C, Su IH, Karasuyama H, Lowell CA, Reth M, et al. Essential role of Src-family protein tyrosine kinases in NF-κB activation during B cell development. Nat Immunol (2003) 4:274. doi:10.1038/ni893
72. Chan VWF, Meng F, Soriano P, DeFranco AL, Lowell CA. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity (1997) 7(1):69–81. doi:10.1016/S1074-7613(00)80511-7
73. Smith KGC, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of the B cell by CD22: a requirement for Lyn. J Exp Med (1998) 187(5):807–11. doi:10.1084/jem.187.5.807
74. Chan VWF, Lowell CA, DeFranco AL. Defective negative regulation of antigen receptor signaling in Lyn-deficient B lymphocytes. Curr Biol (1998) 8(10):545–53. doi:10.1016/S0960-9822(98)70223-4
75. Ho LH, Uehara T, Chen C-C, Kubagawa H, Cooper MD. Constitutive tyrosine phosphorylation of the inhibitory paired Ig-like receptor PIR-B. Proc Natl Acad Sci U S A (1999) 96(26):15086–90. doi:10.1073/pnas.96.26.15086
76. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A (2001) 98(24):13866–71. doi:10.1073/pnas.231486598
77. Adachi T, Flaswinkel H, Yakura H, Reth M, Tsubata T. Cutting edge: the B cell surface protein CD72 recruits the tyrosine phosphatase SHP-1 upon tyrosine phosphorylation. J Immunol (1998) 160(10):4662.
78. Skubitz KM, Campbell KD, Ahmed K, Skubitz AP. CD66 family members are associated with tyrosine kinase activity in human neutrophils. J Immunol (1995) 155(11):5382.
79. Ochi H, Watanabe T. Negative regulation of B cell receptor-mediated signaling in B-1 cells through CD5 and Ly49 co-receptors via Lyn kinase activity. Int Immunol (2000) 12(10):1417–23. doi:10.1093/intimm/12.10.1417
80. Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, et al. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity (1998) 8(4):497–508. doi:10.1016/S1074-7613(00)80554-3
81. Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor FcγRIIB. Nature (1996) 383(6597):263–6. doi:10.1038/383263a0
82. Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell (1997) 90(2):293–301. doi:10.1016/S0092-8674(00)80337-2
83. D’Ambrosio D, Hippen K, Minskoff S, Mellman I, Pani G, Siminovitch K, et al. Recruitment and activation of PTP1C in negative regulation of antigen receptor signaling by Fc gamma RIIB1. Science (1995) 268(5208):293–7. doi:10.1126/science.7716523
84. Doody GM, Justement LB, Delibrias CC, Matthews RJ, Lin J, Thomas ML, et al. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science (1995) 269(5221):242. doi:10.1126/science.7618087
85. Maeda A, Scharenberg AM, Tsukada S, Bolen JB, Kinet J-P, Kurosaki T. Paired immunoglobulin-like receptor B (PIR-B) inhibits BCR-induced activation of Syk and Btk by SHP-1. Oncogene (1999) 18:2291. doi:10.1038/sj.onc.1202552
86. Chen T, Zimmermann W, Parker J, Chen I, Maeda A, Bolland S. Biliary glycoprotein (BGPa, CD66a, CEACAM1) mediates inhibitory signals. J Leukoc Biol (2001) 70(2):335–40. doi:10.1189/jlb.70.2.335
87. Zhang J, Somani A-K, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol (2000) 12(4):361–78. doi:10.1006/smim.2000.0223
88. Yang W, McKenna SD, Jiao H, Tabrizi M, Lynes MA, Shultz LD, et al. SHP-1 deficiency in B-lineage cells is associated with heightened lyn protein expression and increased lyn kinase activity. Exp Hematol (1998) 26(12):1126–32.
89. Mizuno K, Tagawa Y, Mitomo K, Arimura Y, Hatano N, Katagiri T, et al. Src homology region 2 (SH2) domain-containing phosphatase-1 dephosphorylates B cell linker protein/SH2 domain leukocyte protein of 65 kDa and selectively regulates c-Jun NH2-terminal kinase activation in B cells. J Immunol (2000) 165(3):1344. doi:10.4049/jimmunol.165.3.1344
90. Tamir I, Dal Porto JM, Cambier JC. Cytoplasmic protein tyrosine phosphatases SHP-1 and SHP-2: regulators of B cell signal transduction. Curr Opin Immunol (2000) 12(3):307–15. doi:10.1016/S0952-7915(00)00092-3
91. Aman MJ, Lamkin TD, Okada H, Kurosaki T, Ravichandran KS. The inositol phosphatase SHIP inhibits Akt/PKB activation in B cells. J Biol Chem (1998) 273(51):33922–8. doi:10.1074/jbc.273.51.33922
92. Bolland S, Pearse RN, Kurosaki T, Ravetch JV. SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity (1998) 8(4):509–16. doi:10.1016/S1074-7613(00)80555-5
93. Zhao M, Schmitz AAP, Qin Y, Di Cristofano A, Pandolfi PP, Van Aelst L. Phosphoinositide 3-kinase-dependent membrane recruitment of P62(dok) is essential for its negative effect on mitogen-activated protein (Map) kinase activation. J Exp Med (2001) 194(3):265–74. doi:10.1084/jem.194.3.265
94. Stork B, Neumann K, Goldbeck I, Alers S, Kähne T, Naumann M, et al. Subcellular localization of Grb2 by the adaptor protein Dok-3 restricts the intensity of Ca(2+) signaling in B cells. EMBO J (2007) 26(4):1140–9. doi:10.1038/sj.emboj.7601557
95. Honma M, Higuchi O, Shirakata M, Yasuda T, Shibuya H, Iemura S-I, et al. Dok-3 sequesters Grb2 and inhibits the Ras-Erk pathway downstream of protein-tyrosine kinases. Genes Cells (2006) 11(2):143–51. doi:10.1111/j.1365-2443.2006.00926.x
96. Robson JD, Davidson D, Veillette A. Inhibition of the Jun N-terminal protein kinase pathway by SHIP-1, a lipid phosphatase that interacts with the adaptor molecule Dok-3. Mol Cell Biol (2004) 24(6):2332–43. doi:10.1128/MCB.24.6.2332-2343.2004
97. Lemay S, Davidson D, Latour S, Veillette A. Dok-3, a novel adapter molecule involved in the negative regulation of immunoreceptor signaling. Mol Cell Biol (2000) 20(8):2743–54. doi:10.1128/MCB.20.8.2743-2754.2000
98. Xu Y, Huntington ND, Harder KW, Nandurkar H, Hibbs ML, Tarlinton DM. Phosphatidylinositol-3 kinase activity in B cells is negatively regulated by Lyn tyrosine kinase. Immunol Cell Biol (2012) 90:903. doi:10.1038/icb.2012.31
99. Ma H, Yankee TM, Hu J, Asai DJ, Harrison ML, Geahlen RL. Visualization of Syk-antigen receptor interactions using green fluorescent protein: differential roles for Syk and Lyn in the regulation of receptor capping and internalization. J Immunol (2001) 166(3):1507. doi:10.4049/jimmunol.166.3.1507
100. Hakak Y, Martin GS. Ubiquitin-dependent degradation of active Src. Curr Biol (1999) 9(18):S1–1042. doi:10.1016/S0960-9822(99)80453-9
101. Harder KW, Parsons LM, Armes J, Evans N, Kountouri N, Clark R, et al. Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity (2001) 15(4):603–15. doi:10.1016/S1074-7613(01)00208-4
102. Hibbs ML, Harder KW, Armes J, Kountouri N, Quilici C, Casagranda F, et al. Sustained activation of Lyn tyrosine kinase in vivo leads to autoimmunity. J Exp Med (2002) 196(12):1593–604. doi:10.1084/jem.20020515
103. Ingley E, Schneider JR, Payne CJ, McCarthy DJ, Harder KW, Hibbs ML, et al. Csk-binding protein mediates sequential enzymatic down-regulation and degradation of Lyn in erythropoietin-stimulated cells. J Biol Chem (2006) 281(42):31920–9. doi:10.1074/jbc.M602637200
104. Ingley E. Csk-binding protein can regulate Lyn signals controlling cell morphology. Int J Biochem Cell Biol (2009) 41(6):1332–43. doi:10.1016/j.biocel.2008.12.001
105. Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, et al. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell (1995) 83(2):301–11. doi:10.1016/0092-8674(95)90171-X
106. Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, et al. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice. Immunity (1995) 3(5):549–60. doi:10.1016/1074-7613(95)90126-4
107. Gross AJ, Lyandres JR, Panigrahi AK, Prak ETL, DeFranco AL. Developmental acquisition of the Lyn–CD22–SHP-1 inhibitory pathway promotes B cell tolerance. J Immunol (2009) 182(9):5382–92. doi:10.4049/jimmunol.0803941
108. Getahun A, Beavers NA, Larson SR, Shlomchik MJ, Cambier JC. Continuous inhibitory signaling by both SHP-1 and SHIP-1 pathways is required to maintain unresponsiveness of anergic B cells. J Exp Med (2016) 213(5):751–69. doi:10.1084/jem.20150537
109. Gross AJ, Proekt I, DeFranco AL. Elevated BCR signaling and decreased survival of Lyn-deficient transitional and follicular B cells. Eur J Immunol (2011) 41(12):3645–55. doi:10.1002/eji.201141708
110. Takata M, Sabe H, Hata A, Inazu T, Homma Y, Nukada T, et al. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J (1994) 13(6):1341–9.
111. Wang J, Koizumi T, Watanabe T. Altered antigen receptor signaling and impaired Fas-mediated apoptosis of B cells in Lyn-deficient mice. J Exp Med (1996) 184(3):831–8. doi:10.1084/jem.184.3.831
112. Tsantikos E, Oracki SA, Quilici C, Anderson GP, Tarlinton DM, Hibbs ML. Autoimmune disease in Lyn-deficient mice is dependent on an inflammatory environment established by IL-6. J Immunol (2010) 184(3):1348–60. doi:10.4049/jimmunol.0901878
113. Scapini P, Hu Y, Chu C-L, Migone T-S, DeFranco AL, Cassatella MA, et al. Myeloid cells, BAFF, and IFN-γ establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J Exp Med (2010) 207(8):1757–73. doi:10.1084/jem.20100086
114. Lamagna C, Hu Y, DeFranco AL, Lowell CA. B cell-specific loss of Lyn kinase leads to autoimmunity. J Immunol (2014) 192(3):919–28. doi:10.4049/jimmunol.1301979
115. Silver KL, Crockford TL, Bouriez-Jones T, Milling S, Lambe T, Cornall RJ. MyD88-dependent autoimmune disease in Lyn-deficient mice. Eur J Immunol (2007) 37(10):2734–43. doi:10.1002/eji.200737293
116. Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA. Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci U S A (2013) 110(35):E3311–20. doi:10.1073/pnas.1300617110
117. Ban T, Sato GR, Nishiyama A, Akiyama A, Takasuna M, Umehara M, et al. Lyn kinase suppresses the transcriptional activity of IRF5 in the TLR-MyD88 pathway to restrain the development of autoimmunity. Immunity (2016) 45(2):319–32. doi:10.1016/j.immuni.2016.07.015
118. Infantino S, Jones SA, Walker JA, Maxwell MJ, Light A, O’Donnell K, et al. The tyrosine kinase Lyn limits the cytokine responsiveness of plasma cells to restrict their accumulation in mice. Sci Signal (2014) 7(338):ra77. doi:10.1126/scisignal.2005105
119. Hua Z, Gross AJ, Lamagna C, Ramos-Hernández NM, Scapini P, Ji M, et al. Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J Imunol (2014) 192(3):875–85. doi:10.4049/jimmunol.1300683
120. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell–intrinsic mechanism. J Exp Med (2010) 207(2):365–78. doi:10.1084/jem.20091777
121. Gutierrez T, Mayeux JM, Ortega SB, Karandikar NJ, Li Q-Z, Rakheja D, et al. IL-21 promotes the production of anti-DNA IgG but is dispensable for kidney damage in lyn−/− mice. Eur J Immunol (2013) 43(2):382–93. doi:10.1002/eji.201142095
122. Oracki SA, Tsantikos E, Quilici C, Light A, Schmidt T, Lew AM, et al. CTLA4Ig alters the course of autoimmune disease development in Lyn−/− mice. J Immunol (2010) 184(2):757–63. doi:10.4049/jimmunol.0804349
123. Chevrier S, Kratina T, Emslie D, Karnowski A, Corcoran LM. Germinal center-independent, IgM-mediated autoimmunity in sanroque mice lacking Obf1. Immunol Cell Biol (2013) 92:12. doi:10.1038/icb.2013.71
124. Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, et al. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell (1993) 73(7):1445–54. doi:10.1016/0092-8674(93)90369-2
125. O’Keefe TL, Williams GT, Davies SL, Neuberger MS. Hyperresponsive B cells in CD22-deficient mice. Science (1996) 274(5288):798–801. doi:10.1126/science.274.5288.798
126. Jellusova J, Wellmann U, Amann K, Winkler TH, Nitschke L. CD22 × Siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J Immunol (2010) 184(7):3618–27. doi:10.4049/jimmunol.0902711
127. Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature (1996) 379:346. doi:10.1038/379346a0
128. Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev (1998) 12(11):1610–20. doi:10.1101/gad.12.11.1610
129. Tsantikos E, Maxwell MJ, Kountouri N, Harder KW, Tarlinton DM, Hibbs ML. Genetic interdependence of Lyn and negative regulators of B cell receptor signaling in autoimmune disease development. J Immunol (2012) 189(4):1726. doi:10.4049/jimmunol.1103427
130. Takeshita H, Taniuchi I, Kato J, Watanabe T. Abrogation of autoimmune disease in Lyn-deficient mice by the mutation of the Btk gene. Int Immunol (1998) 10(4):435–44. doi:10.1093/intimm/10.4.435
131. Whyburn LR, Halcomb KE, Contreras CM, Lowell CA, Witte ON, Satterthwaite AB. Reduced dosage of Bruton’s tyrosine kinase uncouples B cell hyperresponsiveness from autoimmunity in Lyn-/- mice. J Immunol (2003) 171(4):1850–8. doi:10.4049/jimmunol.171.4.1850
132. Maxwell MJ, Tsantikos E, Kong AM, Vanhaesebroeck B, Tarlinton DM, Hibbs ML. Attenuation of phosphoinositide 3-kinase δ signaling restrains autoimmune disease. J Autoimmun (2012) 38(4):381–91. doi:10.1016/j.jaut.2012.04.001
133. Hiraki LT, Silverman ED. Genomics of systemic lupus erythematosus: insights gained by studying monogenic young-onset systemic lupus erythematosus. Rheum Dis Clin North Am (2017) 43(3):415–34. doi:10.1016/j.rdc.2017.04.005
134. Flores-Borja F, Kabouridis PS, Jury EC, Isenberg DA, Mageed RA. Decreased Lyn expression and translocation to lipid raft signaling domains in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum (2005) 52(12):3955–65. doi:10.1002/art.21416
135. Liossis S-NC, Solomou EE, Dimopoulos M-A, Panayiotidis P, Mavrikakis MM, Sfikakis PP. B-cell kinase Lyn deficiency in patients with systemic lupus erythematosus. J Investig Med (2001) 49(2):157–65. doi:10.2310/6650.2001.34042
136. Liu Y, Dong J, Mu R, Gao Y, Tan X, Li Y, et al. MicroRNA-30a promotes B cell hyperactivity in patients with systemic lupus erythematosus by direct interaction with Lyn. Arthritis Rheum (2013) 65(6):1603–11. doi:10.1002/art.37912
137. Lu R, Vidal GS, Kelly JA, Delgado-Vega AM, Kim-Howard X, Macwana SR, et al. Genetic associations of LYN with systemic lupus erythematosus. Genes Immun (2009) 10(5):397–403. doi:10.1038/gene.2009.19
138. Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, et al. The autoimmune disease–associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet (2011) 43:902. doi:10.1038/ng.904
139. Manjarrez-Orduño N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, et al. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B cell signaling and activation. Nat Genet (2012) 44(11):1227–30. doi:10.1038/ng.2439
140. Kozyrev SV, Abelson A-K, Wojcik J, Zaghlool A, Linga Reddy MVP, Sanchez E, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet (2008) 40:211. doi:10.1038/ng.79
141. Yang W, Shen N, Ye D-Q, Liu Q, Zhang Y, Qian X-X, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet (2010) 6(2):e1000841. doi:10.1371/journal.pgen.1000841
142. Saeed M. Lupus pathobiology based on genomics. Immunogenetics (2017) 69(1):1–12. doi:10.1007/s00251-016-0961-7
143. International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN)Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet (2008) 40:204. doi:10.1038/ng.81
144. Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet (2009) 41(11):1228–33. doi:10.1038/ng.468
145. Wang D, John SA, Clements JL, Percy DH, Barton KP, Garrett-Sinha LA. Ets-1 deficiency leads to altered B cell differentiation, hyperresponsiveness to TLR9 and autoimmune disease. Int Immunol (2005) 17(9):1179–91. doi:10.1093/intimm/dxh295
146. Luo W, Mayeux J, Gutierrez T, Russell L, Getahun A, Müller J, et al. A balance between B cell receptor and inhibitory receptor signaling controls plasma cell differentiation by maintaining optimal Ets1 levels. J Immunol (2014) 193(2):909. doi:10.4049/jimmunol.1400666
147. Mayeux J, Skaug B, Luo W, Russell LM, John S, Saelee P, et al. Genetic interaction between Lyn, Ets1, and Btk in the control of antibody levels. J Immunol (2015) 195(5):1955–63. doi:10.4049/jimmunol.1500165
148. Li W, Titov AA, Morel L. An update on lupus animal models. Curr Opin Rheumatol (2017) 29(5):434–41. doi:10.1097/bor.0000000000000412
149. Chan OTM, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med (1999) 189(10):1639–48. doi:10.1084/jem.189.10.1639
150. Waibel M, Christiansen AJ, Hibbs ML, Shortt J, Jones SA, Simpson I, et al. Manipulation of B-cell responses with histone deacetylase inhibitors. Nat Commun (2015) 6:6838. doi:10.1038/ncomms7838
151. Mina-Osorio P, LaStant J, Keirstead N, Whittard T, Ayala J, Stefanova S, et al. Suppression of glomerulonephritis in lupus-prone NZB × NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum (2013) 65(9):2380–91. doi:10.1002/art.38047
Keywords: Lyn, systemic lupus erythematosus, cell signaling, SFK, B cell, B-cell receptor, autoimmunity, lupus
Citation: Brodie EJ, Infantino S, Low MSY and Tarlinton DM (2018) Lyn, Lupus, and (B) Lymphocytes, a Lesson on the Critical Balance of Kinase Signaling in Immunity. Front. Immunol. 9:401. doi: 10.3389/fimmu.2018.00401
Received: 08 December 2017; Accepted: 13 February 2018;
Published: 01 March 2018
Edited by:
Marc Pellegrini, Walter and Eliza Hall Institute of Medical Research, AustraliaReviewed by:
Aaron James Marshall, University of Manitoba, CanadaCopyright: © 2018 Brodie, Infantino, Low and Tarlinton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David M. Tarlinton, ZGF2aWQudGFybGludG9uQG1vbmFzaC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.