 Benjamin J. Wolf
Benjamin J. Wolf Jiyoung Elizabeth Choi
Jiyoung Elizabeth Choi Mark A. Exley
Mark A. Exley
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 March 2018
Sec. Cancer Immunity and Immunotherapy
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00384
This article is part of the Research Topic NKT Cells in Cancer Immunotherapy View all 18 articles
iNKT cells are a subset of innate-like T cells that utilize an invariant TCR alpha chain complexed with a limited repertoire of TCR beta chains to recognize specific lipid antigens presented by CD1d molecules. Because iNKT cells have an invariant TCR, they can be easily identified and targeted in both humans and mice via standard reagents, making this a population of T cells that has been well characterized. iNKT cells are some of the first cells to respond during an infection. By making different types of cytokines in response to different infection stimuli, iNKT cells help determine what kind of immune response then develops. It has been shown that iNKT cells are some of the first cells to respond during infection with a pathogen and the type of cytokines that iNKT cells make help determine the type of immune response that develops in various situations. Indeed, along with immunity to pathogens, pre-clinical mouse studies have clearly demonstrated that iNKT cells play a critical role in tumor immunosurveillance. They can mediate anti-tumor immunity by direct recognition of tumor cells that express CD1d, and/or via targeting CD1d found on cells within the tumor microenvironment. Multiple groups are now working on manipulating iNKT cells for clinical benefit within the context of cancer and have demonstrated that targeting iNKT cells can have a therapeutic benefit in patients. In this review, we briefly introduce iNKT cells, then discuss preclinical data on roles of iNKT cells and clinical trials that have targeted iNKT cells in cancer patients. We finally discuss how future trials could be modified to further increase the efficacy of iNKT cell therapies, in particular CAR-iNKT and rTCR-iNKT cells.
T cells utilize their unique T cell receptor (TCR) αβ or γδ chain pairs to recognize the universe of antigens. Although many TCRs with extensive somatic V-D-J gene rearrangements recognize peptide antigens within the context of MHC molecules, this is not the only type of antigen that can be recognized. T cells can also utilize near-germline V-J TCR rearrangements to recognize vitamin metabolites, small phosphoantigens, and lipid antigens presented within various highly conserved and non-polymorphic MHC-I like molecules (1–3). Collectively, these non-peptide-recognizing T cells are called “innate-like” T cells and make up a significant proportion of the mammalian T cell compartment (1–4). Importantly, new research is suggesting that these innate-like T cells have important roles in regulating immune reactions not only to pathogens but also to tumors, making them potentially exploitable T cell populations for immunotherapy (1–6). One of the best characterized innate-like T cell subsets that is being leveraged in immuno-oncology are Natural Killer T (NKT) cells, which recognize lipid antigens bound within the antigen presentation molecule CD1d. The best-characterized subset of NKT cells is “invariant” or “iNKT” cells (1–6). Mouse and human iNKT cells are sufficiently conserved that they can respond to each other’s CD1d (7).
NKT cells are a heterogenous population of innate-like CD1d-restricted T cells, the best known of which are invariant NKT (iNKT) cells (1–9). iNKT cells utilize a near-germline TCRα rearrangement (Vα24-Jα18 in humans and Vα14-Jα18 in mice) combined with a limited TCRβ repertoire (1–4). iNKT cells were originally named because of expression of NK1.1 (CD161C) in some mouse strains (CD161A in humans) but this does not accurately define iNKT cells (5, 6). Instead, iNKT cells are functionally defined by their ability to respond to the lipid antigen α-galactosylceramide (α-GalCer) when bound within CD1d molecules and/or by utilizing monoclonal antibodies against the human invariant TCRα chain (3–8). By utilizing (imperfect) co-expression of NK and T cell markers, absence in CD1d or Jα18 KO mice, CD1d tetramers loaded with α-GalCer, or other methods, iNKTs were discovered to make up a significant proportion of T cells within the mouse liver (~20−50%) and adipose tissue (~10−25%) and are present in significant numbers (~0.5−2%) within the murine thymus, spleen, blood, and bone marrow (3–6). Within humans, iNKT cells are represented at similar frequencies to mice in adipose tissue, but are much less frequent in the liver and other organs where “non-invariant” or “diverse” NKT cells predominate (6, 8, 9). In human peripheral blood, iNKT cells range between undetectable to over 1% of circulating T cells in rare individuals, with a median percentage of approximately 0.05% (6, 8, 9).
Unlike peptide-MHC restricted T cells, which emerge from the thymus “naïve,” iNKT cells leave the thymus fully matured and able to perform their effector functions without priming (3–6). Within the periphery, iNKT cells respond to lipid antigen and/or cytokine (e.g., IL-12/18) exposure by rapid secretion of multiple cytokines (3–6). Depending on how the iNKT cells are activated, this can include both regulatory cytokines (e.g., IL-4, IL-10, by analogy with Th2, Treg, etc., especially from NKT2, NKT10) (3–6) and/or pro-inflammatory cytokines (e.g., IL-2, IL-17, TNFα, and/or IFNγ, particularly NKT1 or NKT17) (3–6). Since iNKT cells respond rapidly and without the need for priming, they are some of the first cells within an immune response to be activated and therefore act as a “bridge” between the innate and adaptive immune systems. Indeed, iNKT cell activation via TCR engagement or IL-12 or both causes iNKT cells to upregulate IL-12 receptor [which is already basally expressed at a higher level than in NK cells (6, 8)] and CD40L, while also inducing maturation and production of IL-12 in dendritic cells (DCs). This IL-12 release then in turn greatly increases IFNγ production by iNKT cells, leading to a positive feedback loop for Th1 immunity (Figure 1) (3–6, 8, 9). Additionally, this maturation of DCs leads to trans-activation of NK cells and increased MHC class I and II antigen presentation to T cells as well as direct cognate B cell “help,” allowing for both innate and adaptive immune responses to be established (3–6).
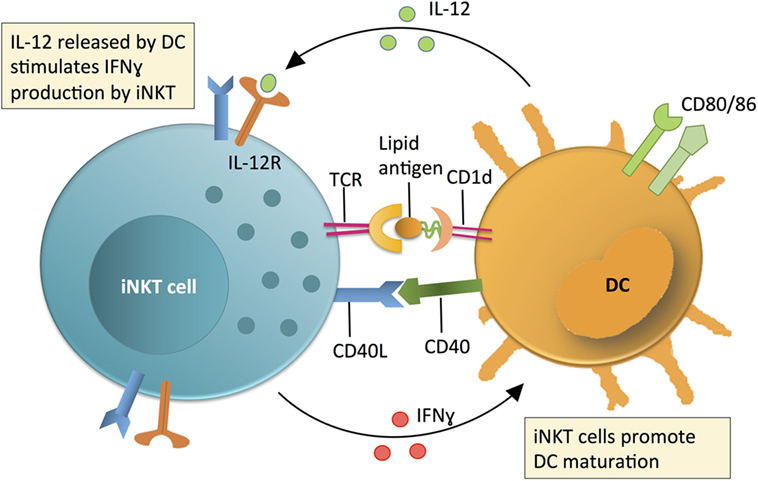
Figure 1. Invariant NKT (iNKT) cell contribution to initiating immune reactions via a positive feedback loop with dendritic cells (DCs). The contribution of iNKT cells to immune surveillance is linked to DC maturation. DCs produce IL-12 and present lipid antigens on CD1d (an MHC class 1 homolog) to stimulate iNKT cell production of IFNγ. The relative contribution of IL-12 or lipid-CD1d to iNKT cell activation is variable and context dependent. Activated iNKT cells produce IFNγ and co-stimulate DCs via CD40L-CD40 interactions to promote DCs to produce IL-12. This IL-12 then further activates iNKT cells in a positive feedback loop. Following activation of iNKT cells, iNKT cell-produced IFNγ and other iNKT-DC interactions (e.g., CD40L-CD40) mature DCs and promote production of IL-12, which further activates iNKT cells in a positive feedback loop.
Invariant NKT cells can be further subdivided into additional subsets based on anatomical location or by surface activation markers and transcription factors (1–6). The key master transcription factor of at least iNKT cell development and present in most mature iNKT populations is PLZF (4). Unlike most MHC-restricted T cells that are either CD4 + or CD8α +, iNKT cells in mice are either CD4 + or CD4/CD8αβ double negative (DN) (3–6). In humans, a minor population of iNKT cells (typically 1–5%) can instead express CD8αβ (3, 10). Additionally, CD8αα homodimers are expressed by other activated human T cells, although at lower levels on activated CD4 + T cell subsets lacking CD8β (1, 2, 11). In general, CD4 + iNKT cells are able to express more Th2-related cytokines like IL-4, although they can express as much Th1 cytokines at the same time (3–9). Human CD8αβ + and DN iNKT cells are biased toward a Th1-related phenotype, more cytotoxic and preferentially make IFNγ. In both cases, these are plastic definitions and CD4 + iNKT cells can make IFNγ and DN iNKT cells can make IL-4, at least partially depending on the stimuli given (3–6, 8, 9). Mouse iNKT cells are less clearly biased, although liver iNKT cells have greater anti-tumor activity than other organ iNKT cells (12). Indeed, within different organs, different iNKT phenotypes tend to dominate. Relatively Th1-like iNKT cells tend to be enriched within the spleen and liver, while Th2-like iNKT cells are associated with the lungs and intestine (3–6). There are also Th17-like iNKT cells that express cytokines like IL-17 and are enriched within the lungs, intestine, lymph nodes, and skin (13). Finally, a subset has recently been described in adipose tissue. Adipose iNKT tend to make anti-inflammatory cytokines like IL-10 and unlike other mature iNKT cells, lack PLZF (14, 15).
Because iNKT cells can rapidly produce IFNγ, IL-4, or both, they have been found to play a role in various diseases by establishing a Th1- or Th2-based immune response. In bacterial and viral infections, iNKT cells typically help in early control of the pathogen by establishing a productive Th1 response (1–6, 9, 13, 16). In both mouse and human studies, roles for iNKT cells have been described in diseases associated with excessive Th1 responses like type 1 diabetes (9) and chronic obstructive pulmonary disease (17, 18). Roles have also been described for iNKT cells helping to suppress Th1 responses and drive tolerogenic responses to grafts. As an example, following hematopoietic stem cell transfer, the presence of iNKT cells is predictive for survival with a reduction in graft versus host disease (GvHD) in patients and preclinical models (8, 9, 19–21).
Within the context of cancer, the frequency and/or function of iNKT cells (either within the tumor or in circulation) can be selectively and highly correlative with overall survival. In human studies, this has been demonstrated in prostate cancer, medulloblastoma and neuroblastoma, melanoma, colon, lung, breast, and head and neck squamous cell carcinomas (8, 9, 22–37). The largest numbers of patient samples and/or longest follow-ups were analyzed for tumor in neuroblastoma and circulating in head and neck squamous cell carcinomas, respectively (26, 37). Consistent with reduced numbers, proliferative response defects of iNKT cells have been noted in cancer patient iNKT cells (23, 28, 37). Decreased numbers of circulating iNKT cells can be accompanied by decreases in IFNγ production and a concurrent increase in IL-4 production (22, 24, 25, 35). Importantly, all of these defects including the shift toward an iNKT cell Th2 phenotype can be reversed in vitro. Activation via 2 strong stimuli, such as α-GalCer and IL-12, increases iNKT cell IFNγ production, promotes tumor rejection, and protects from development of metastasis in multiple mouse models and enhances cancer patient iNKT Th1 responses in vitro (8, 22, 24, 25, 35). However, such stimuli do not reverse iNKT defects individually (particularly in advanced disease) (8, 22, 24, 25, 35). Additionally, injection of α-GalCer-pulsed DCs (particularly mature DC) can provide a strong anti-tumor effect (31, 34, 35).
While the human data is correlative, the role for iNKT cells in providing tumor surveillance has been well-characterized in mouse models. Examples of iNKT-mediated tumor clearance were demonstrated by the lab of Taniguchi et al. (31, 38) as well as those of Smyth and Godfrey (9, 12). iNKT cells were found to be essential for anti-tumor responses induced by α-GalCer (12, 30, 38). Treatment with carcinogen or transfer of carcinogen-induced tumor cell lines in mice lacking iNKT cells (via TCR Jα18 deletion, Jα18-KO) caused tumors to appear at a much higher frequency than in wild-type (WT) mice (39). Additionally, transfer of iNKT cells into Jα18-KO mice was sufficient to cause protection against tumors to a level like WT mice, unless the iNKT cells came from an IFNγ KO mouse (39). Together, these and other results show that even in the absence of exogenous antigens like α-GalCer, iNKT cells can establish a Th1 response to some tumors and can contribute to tumor clearance (8, 9, 29, 32, 39). Further support for iNKT cell-mediated tumor surveillance was obtained with the spontaneous prostate cancer mouse strain: transgenic adenocarcinoma of the mouse prostate (TRAMP). By back-crossing Jα18-KO to TRAMP mice, Bellone et al. suggested that lack of iNKT cells led to accelerated tumor generation and quicker mortality than was detected in WT TRAMP mice (39), consistent with earlier human in vitro data (22). However, more recently, a caveat of studies using the original Jα18 KO mice (38) has come to light, most notably the inability of these mice to express TCR Jα regions past Jα19 (40). This impacts the TCR repertoire of conventional T cells, which could also impact in vivo immune responses, so new Jα18 KO mice have been developed that do not share this defect (41, 42).
While some CD1d-expressing tumors can probably cause Th1-biased iNKT cell activation, progressive chronic tumor cell growth can also apparently directly cause Th2-biased iNKT cell activation. By utilizing the same TRAMP prostate cancer model as a source of primary prostate tumors, we demonstrated that CD1d-expressing prostate tumor cells can directly activate iNKT cells, but biased them toward making Th2 cytokines (43). While addition of α-GalCer or IL-12 can usually help bias an iNKT cell toward a Th1 phenotype, neither of these stimuli on their own were enough to reverse the tumor cell driven Th2 bias in iNKT cells. However, pulsing the tumor cells with α-GalCer and adding IL-12 at the same time synergized to allow for IFNγ production to occur (43).
In both the models described above and in humans, activation of iNKT cells and tumor rejection can occur in one of two ways (Figure 2). The first is that iNKT cells directly recognize and kill CD1d-expressing tumor cells. This can occur in a significant portion of lymphomas, early myeloma, myeloid leukemias, medulloblastoma, and prostate cancers (24, 35, 36, 43, 44). The second is by activation of iNKT cells by other CD1d-expressing cells in the tumor microenvironment (TME) (29, 32). In this indirect system, iNKT cell activation by CD1d-expressing TME cells leads to trans-activation of NK cells and/or killing of immunosuppressive cells like tumor-associated Macrophages (TAMs) (29, 32, 45). When we directly tested the ability of iNKT cells to respond to CD1d-expressing prostate tumor cells from TRAMP mice in vitro, we found that they did not elicit a Th1 phenotype that would be indicative of killing (43). However, Bellone et al. found that iNKT cells do help delay tumor growth within intact TRAMP mice over periods of months (39). The differences in these two studies may include that within the intact mouse there is also a role for iNKT cells in killing CD1d-expressing TAMs independent of any direct anti-tumor interactions (29, 32). Therefore, relieving some of the immunosuppression within the primary tumor by killing TAMs may be a key role for iNKT cells in vivo. However, in progressive clinical cancer, TAMs can apparently overwhelm iNKT cells (29, 32). Reversing these as well as tumor cell-driven iNKT defects is the goal of the groups working on clinical trials targeting iNKT cells worldwide.
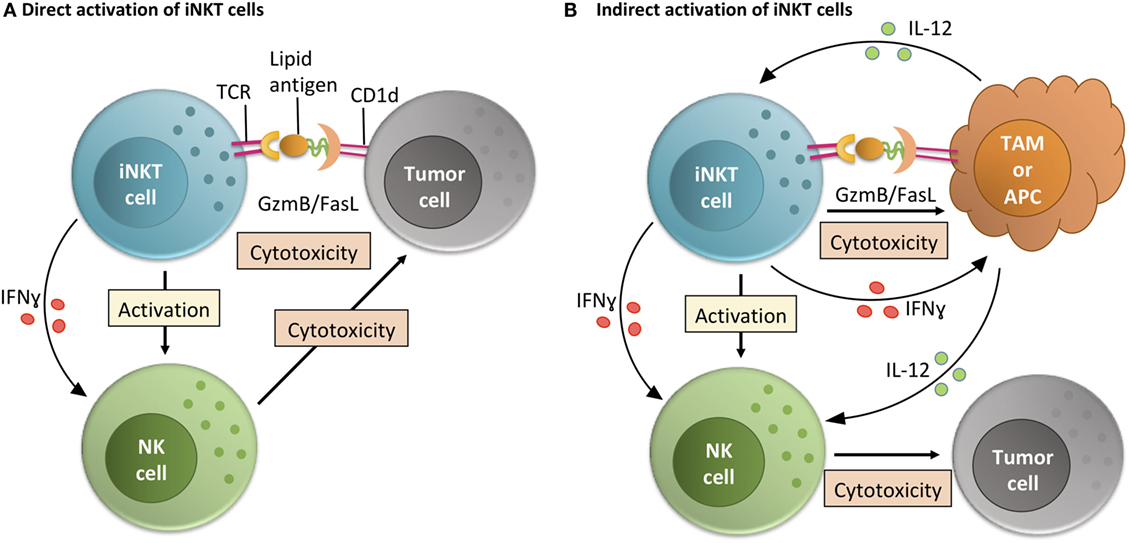
Figure 2. Invariant NKT (iNKT) cell activity within the tumor microenvironment (TME). iNKT cells can function within the TME via direct or indirect interactions with tumor cells. (A) In the direct activation pathway, iNKT cells recognize lipid-CD1d complexes on the surface of tumor cells and then directly mediate killing of tumor cells. By making inflammatory cytokines like IFNγ, this also helps TME-resident NK cells perform their anti-tumor cell effector functions (24, 35, 36, 43, 44). (B) In the indirect activation pathway, iNKT cells recognize lipid-CD1d complexes on the surface of TME-resident antigen presenting cells (APCs) or tissue-associated macrophages (TAMs) (29). This interaction leads to iNKT cell-mediated killing of immunosuppressive TAMs, leading to a less immunosuppressive environment where tumor-infiltrating NK cells can better perform their functions. Alternately, if the CD1d is on the surface of a TME-resident APC, the iNKT cell can activate that APC and stimulate production of IL-12, helping resident effector cells like NK cells overcome the immunosuppressive state of the tumor (32).
Pre-clinical murine models have shown similar defects in iNKT cells as have been seen in humans and demonstrated that iNKT stimulation in vivo or adoptive transfer can induce strong antitumor immune responses (38, 43, 46–51). This has been shown to be the case for stimulation of iNKT cells via α-GalCer infusion and when α-GalCer has been loaded on DCs (46–51). Additionally, as iNKT cells play a key role in generating a positive feedback loop for IL-12 production by DCs, low and moderate-dose IL-12 therapy in animal models is also dependent on iNKT cells (46, 50, 51). Either stimulation causes iNKT cells to rapidly produce a strong cytokine response, including large amounts of IFN-γ that stimulates NK cells, B cells, and that also enhances the generation of classical cytotoxic T cell responses (39, 49, 51). Strong antitumor immune responses to α-GalCer and/or IL-12 have been observed in most murine models, including colon carcinoma, lymphomas, sarcoma, melanoma, prostate, and lung carcinoma (39, 41, 46–51). Together, these observations indicate that restoration of iNKT cell function in humans with cancer may stimulate potent antitumor immune responses.
The pre-clinical antitumor effects of α-GalCer stimulated a phase 1 clinical study in advanced-stage cancer patients (52). Administration of α-GalCer was not accompanied by dose limiting toxicity. In this phase 1 study, as in other analyses (9, 28, 29, 37), circulating iNKT cell numbers were found to be decreased in cancer patients (52). The relevance of the decreased size of the iNKT cell pool was demonstrated in the same trial as immunological responses to α-GalCer administration (increases in GM-CSF and TNF-α) were only observed in those patients with higher iNKT cell levels comparable to healthy controls (52).
This initial clinical study and preclinical studies outlined above implied that antitumor effects of α-GalCer in cancer patients would be limited by both qualitative and quantitative defects in iNKT cells, necessitating the evaluation of alternative approaches to exploit this natural antitumor system. In mice, administration of α-GalCer-loaded DC resulted in a more powerful antitumor immune response (48, 49). Several phase 1 clinical studies have used α-GalCer-loaded immature or matured monocyte-derived dendritic cells (Mo-DC) or other monocyte-derived antigen-presenting cell (APC) preparations leading to clinically relevant antitumor responses (53–58).
In the first published autologous Mo-DC transfer clinical study, Nieda et al. investigated the transfer of purified α-GalCer-pulsed immature Mo-DC in a variety of different malignancies (53). They found that adoptive transfer of α-GalCer-pulsed Mo-DC led to minor systemic side effects in 9 of 12 patients such as fever, malaise, lethargy, and headache (53). These side effects were temporary and expected when eliciting an immune response by activating iNKT cells. Several patients experienced temporary exacerbation of tumor symptoms that were interpreted as inflammatory responses to the tumor (e.g., enlargement of tumor deposits or associated lymph nodes, bone pain, and respiratory symptoms in subjects with pulmonary metastases) (53). These exacerbated tumor symptoms had a strong temporal and reproducible relationship in terms of timing and nature with treatment cycles, were transient (generally lasting 1 to 3 days), and were absent outside of the study period. In four of the patients, there were decreases in tumor markers, and in one patient, there was extensive tumor necrosis (53). This study was also important in that it was the first to provide clinical interventional data for the role of iNKT cells as the “bridge” between the innate and adaptive immune systems in humans, as has been seen in multiple human in vitro and murine in vivo studies (39, 43, 46–51). In this clinical trial, activation of human iNKT cells in vivo by adoptive transfer of α-GalCer-loaded Mo-DC reproducibly initiated an activation program wherein iNKT cell activation led to subsequent activation of B cells, T cells, NK cells, and increased serum levels of IL-12 and IFN-γ (53).
Ishikawa et al. investigated the effects of adoptive transfer of autologous cell preparations that were enriched for α-GalCer-pulsed DCs in 11 patients with recurrent lung cancer or advanced non-small cell lung cancer (54). No serious adverse events were reported. Importantly, in several patients, an increase in the circulating number of iNKT cells was also detected. Notably, as reported previously, immunological responses were restricted to patients having “normal” pretreatment iNKT cell numbers. No patients exhibited complete or partial responses in this study, but two patients had stable disease (54).
Chang et al. performed a clinical trial where five cancer patients were treated with α-GalCer-pulsed mature Mo-DC (55). The trial was focused on evaluating the number and phenotype of iNKT cells following stimulation via DC transfer. A more than 100-fold expansion of circulating iNKT cell numbers was observed in all five patients, and this expansion was sustained for up to 6 months post-vaccination (55). Additionally, the data suggested a boost in adaptive T cell immunity, as it was accompanied by an increase in antigen-specific memory CD8 + T cells (55). In this study, no more than grade 1 toxicity was observed, and although one patient developed rheumatoid factor and transient positive anti-nuclear antibody at follow up, no clinical evidence of autoimmunity was observed (55).
In addition to these trials above, several subsequent trials have used APC (i.e., adherent monocytic cells treated with GM-CSF and IL-2) loaded with α-GalCer and have shown increasing effectiveness as dose and targeting have been improved, particularly so far in lung cancer and head and neck cancers (56–58). Specifically in a lung cancer trial, patients who had circulating iNKT able to produce IFNγ had a threefold longer lifespan (57).
Another (and complementary) approach to α-GalCer-based treatments involves the adoptive transfer of activated iNKT cells to restore iNKT cell numbers and potentially iNKT cell function in cancer patients. This approach has been tested in preclinical models of melanoma and lung cancer and shown to be more effective compared to the i.v. administration of α-GalCer (50). Trials of iNKT-enriched PBMC have supported direct use of iNKT with evidence for immunological and objective clinical responses (59–62).
The first of these adoptive iNKT cell therapies targeted six patients with non-small cell lung cancer (59). To grow out iNKT cells, bulk PBMCs were stimulated two to three times via addition of α-GalCer to the cultured cells. These iNKT cell-enriched products were then infused back into the patient, and the iNKT cell numbers, persistence, and phenotype were measured. In most patients, there was a transient but not long-term increase in iNKT cell number within the blood, and this coincided with the ability to detect IFNγ production ex vivo via α-GalCer stimulation of PBMCs. Only minor adverse effects were seen in this first trial, demonstrating that adoptive cell therapy of iNKT cells is likely to be safe. In this study, no partial or complete responses were seen (59).
The next adoptive iNKT cell-based therapy studies combined autologous iNKT cell-enriched product with in vivo boosting. In a Phase I and subsequent Phase II study, the trial group first treated head and neck squamous cell carcinoma (HNSCC) patients with two doses of α-GalCer-loaded DCs followed by an iNKT cell infusion (60). In the Phase I trial, three patients showed partial responses, four had stable disease, and one had progressive disease (60). Of the eight patients, only one had grade 3 adverse events and that patient also had a partial response: a fistula formed within the tumor apparently due to rapid tumor killing (60). In the follow-up Phase II trial for 10 patients with HNSCC, patients were first given nasal submucosal administration of α-GalCer loaded DCs followed by iNKT cell infusion directly into the tumor-feeding arteries, so that iNKT cells were more likely to end up in the tumor site (61). Adverse events were minimal and limited to grade 2 or below, five patients had a partial response, and five patients had stable disease (61). iNKT cell numbers within the tumor and in the peripheral blood were measured, and while iNKT cell numbers in the blood did increase in 9 of 10 patients post-treatment, this did not correlate with outcome. Instead, a high number of tumor-infiltrating iNKT cells correlated with an objective response of patients (61).
With clinical colleagues at Harvard Cancer Center, we performed a Phase I clinical trial of autologous purified [with the iNKTCR mAb 6B11 (62)] and expanded iNKT cells in nine melanoma cancer patients (62). In our study, iNKT cells were isolated from PBMCs with a protocol based on a monoclonal antibody that specifically recognizes the invariant TCR of iNKT cells and then expanded in vitro with plate bound anti-CD3 antibody (62–64). Compared to previous studies using α-GalCer stimulated PBMCs as a source of iNKT cells (59–61), this study transferred in generally higher purity and/or larger numbers of iNKT cells (3 doses at up to 250 million iNKT cells per dose). Since iNKT cells are activated via interaction with CD1d on APC, after the first three patients had no significant toxicities, subsequent patients were pre-treated with GM-CSF to enhance DC functions before iNKT infusion cycles 2 and 3. Like in the other studies, we noted a transient increase in circulating iNKT cell numbers following adoptive cell transfer and increased activation of other T cells and myeloid cells in some patients, and toxicities were minor and readily treatable (Grade 1 & 2 only) (62). In terms of responses at the end of the study, three patients had no evidence of disease or stable disease, three eventually progressed and responded to subsequent treatment, and three died of disease (one removed from study after infusions, two at 2 or more years post-treatment). Overall, our trial confirms that iNKT cell adoptive therapy is safe and well-tolerated, but modified treatment regimens are likely required to demonstrate efficacy. These could include further conditioning with stimulations like α-GalCer (on APC or free) and/or IL-12 in vivo.
Current clinical trials with either α-GalCer-loaded Mo-DCs or adoptive transfer of iNKT cells have produced partial and complete responses, but few if any cures in late stage patients. In contrast, T cells expressing chimeric antigen receptors (CAR-T) targeting surface proteins like CD19 have shown complete response rates of up to 90% in specific diseases such as B-ALL, leading to the first approvals for these treatments (65). Additional T cell therapies are utilizing recombinant TCR (rTCR-T) expressing T cells to be able to target peptides from tumor-associated intracellular proteins within the context of HLA molecules and are reporting similar complete response rates in myeloma (66) and solid malignancies.
One of the major drawbacks of CAR-T/rTCR-T cell therapy is a very high rate of serious adverse effects, including cytokine release syndrome (CRS) and lethal neurotoxicity (65, 67). Other risks include antigen selection issues (e.g., off-tumor on-target) and GvHD caused by TCR mispairing in rTCR-expressing cells and via allogeneic cell therapy (67). Interestingly, while GvHD is a common concern for both CAR-T and rTCR-T, iNKT cells have been shown in pre-clinical models to suppress, not cause GvHD and are associated with reduced GvHD in the clinic (19–21, 68, 69), making for a potentially safer therapeutic approach. In light of both the promises and drawbacks of CAR-T and rTCR-T cells, there is growing interest in utilizing iNKT cells as an ideal platform for CAR or rTCR therapies (CAR-iNKT or CAR-rTCR).
One of the main benefits of considering iNKT cells as an ideal vector for CAR/rTCR therapies is that iNKT cells have an endogenous TCR that is confirmed to have intrinsic anti-tumor capabilities (Figure 3). While the “random” endogenous TCRs on a bulk polyclonal T cell preparations are unlikely to contribute to anti-tumor effects, it is likely that iNKT cells could utilize both their endogenous TCR and their CAR/rTCR to target the tumor with two different targeting moieties. As mentioned previously, this could be by direct CD1d targeting either on the surface of the tumor cell or a bystander tumor-promoting myeloid cell, or via removing immunosuppression by killing CD1d + TAMs. Since strong TCR signaling cascades (as evidenced by potent iNKT cell antigens like α-GalCer) help cause Th1-based iNKT cell responses, having a second TCR signaling pathway engaged within the iNKT cell may help ensure that the iNKT cells remain Th1-biased in vivo. Another advantage for CAR-iNKT or rTCR-iNKT cell therapies is that iNKT cells naturally migrate into non-lymphoid tissues (70), suggesting that they would be ideal cells to target non-lymphoid tumors. Indeed, PLZF expression seems to drive innate T cells tissue homing in general (4, 71). iNKT cells are known to respond to tissue chemokines CCL2 (72) and CCL20 (45). While suggestive of intrinsic benefits of iNKT cells, these points remain to be formally tested in the context of CARs in the clinic.
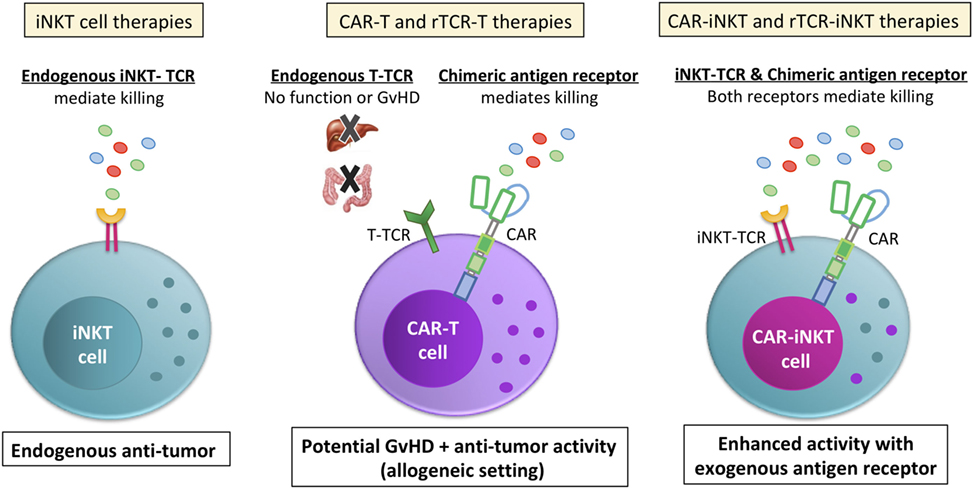
Figure 3. Invariant NKT (iNKT) cells are a viable cell vector for emerging CAR and rTCR therapies. Unlike bulk T cell preparations that are currently used for chimeric antigen receptor (CAR) or recombinant TCR (rTCR) T cell therapies, the endogenous iNKT-TCR has anti-tumor function (left side). In contrast, the bulk T cell endogenous TCRs do not contribute to the anti-tumor function of the cell (middle picture). Instead, the bulk T cell endogenous TCR is a liability, endowing the cells with the potential for graft versus host disease (GvHD) or other off-target effects. While GvHD is a common concern for both CAR-T and rTCR-T allogeneic therapy and at least a possibility due to mispairing for autologous rTCR-iNKT, iNKT cells are shown in pre-clinical models to suppress, not cause GvHD during allogeneic transfer, making for a safer therapeutic approach. Additionally, iNKT cells expressing a CAR or rTCR (right picture) would be endowed with two different anti-tumor receptors. This would allow iNKT cells to target the tumor directly with the CAR or rTCR while its endogenous iNKT-TCR would be able to target the tumor microenvironment and/or the tumor itself, depending on if the tumor expressed CD1d. Either way, CAR-iNKT and rTCR-iNKT therapies would have an endogenous TCR that contributes to tumor clearance instead of be a hindrance.
The Metelitsa group has pioneered CAR-iNKT cells for tumor therapies. In pre-clinical models, they have tested human iNKT cells purified with the iNKTCR mAb 6B11 for their ability to express GD2 CARs (against neuroblastomas) and CD19 CARs (against B cell lymphomas) (73, 74). Importantly, they demonstrated that iNKT cells could stably express either CAR construct, and that the CAR-iNKT cells kill relevant antigen-expressing tumor cell lines in vitro. With GD2 CAR-iNKT, this included killing of both GD2 + CD1d− cells and GD2− CD1d + cells, demonstrating that the endogenous iNKT TCR was still functional within GD2 CAR-iNKT cells (73). Importantly, CAR-iNKT cell homing and killing in vivo of either the solid tumor xenograft model GD2-expressing neuroblastoma or liquid xenograft B cell lymphoma was greatly increased over non-transduced iNKT cells, leading to substantially increased survival of CAR-iNKT treated mice (73).
As tumor homing and GvHD are concerns for both CAR-T and rTCR-T, they further measured if CAR-iNKT cells had better trafficking to the tumor and what effect placing the CAR into bulk T cells or iNKT cells had on GvHD. CAR-iNKT cells homed to the tumor at an even higher frequency than CAR-T cells, providing evidence that iNKT cells do indeed have better tumor homing than bulk T cells (73). To model GvHD within the context of a xenogeneic cell transfer, CAR-iNKT or CAR-T cells were transferred into humanized mice and monitored for GvHD. As would be expected when transferring in xenogeneic T cells, CAR-T cells caused severe GvHD in the livers and lungs of the mice. In contrast, CAR-iNKT cells did not cause GvHD of these organs during xenogeneic transfer (73), consistent with the GvHD suppressing activities of iNKT described above. Finally, additional work suggested that in GD2 CARs, expression of both the CD28 and 4-1BB costimulatory domains led to longer CAR-iNKT persistence and increased survival of mice compared to single CD28 or 4-1BB costimulatory domains (73). As well as the Metelitsa group, Karadimitris et al. reported in a review otherwise of potential myeloma treatments that CD19-CAR iNKT had promising preclinical anti-tumor activity in their hands also (75). Both groups are planning clinical trials in the near future.
Many groups are looking at transducing subsets of T cells for CAR/rTCR therapies, and some studies have suggested that CD62L+ (central memory) T cells are superior to other T cell subsets (76). In the Metelitsa group CD19 CAR-iNKT study, Tian et al. separated CD62L+ and CD62L− CAR-iNKT cells and measured their persistence and anti-tumor ability (74). CD62L+ CAR-iNKT cells had superior proliferation, in vivo persistence, and antitumor activity as compared to the CD62L− CAR-iNKT cells (74), suggesting that even with iNKT cells, it may be worthwhile to target a defined subset for CAR-iNKT or rTCR-iNKT therapies. Interestingly, the majority of iNKT express CD62L until they are repeatedly stimulated in vitro (74). While these CAR-iNKT preclinical studies are extremely valuable, critical issues should be generalized within future studies. First, multiple in vivo comparisons of tumor killing and survival of CAR-iNKT to bulk T cell CAR-T cells should be performed. Second, as in vivo administration of α-GalCer is well tolerated in humans, it should be determined if α-GalCer administration could help either expand CAR-iNKT in vivo and/or cause an additive/synergistic increase in anti-tumor activity. Planned clinical studies will begin to address these issues in the near future.
Jiang et al. have provided the first evidence of iNKT cells being able to express a second recombinant TCR (77). In this study, a HLA class I-restricted TCR (TCR-Vα9 TCR-Vβ5) against the Mycobacterium tuberculosis (Mtb) 38-kDa protein was cloned and expressed in iNKT cells. Using autologous 38-kDa protein pulsed Mo-DCs as APC, they confirmed that only rTCR-expressing iNKT cells recognized and killed these cells. The relative killing efficiency of α-GalCer-pulsed Mo-DCs was similar to the killing of 38-kDa pulsed Mo-DCs, suggesting that the endogenous iNKT TCR was still fully functional (77). However, it was not determined if the recombinant TCR and the endogenous TCR could both signal at the same time to cause additive or synergistic effects. Nor was it determined if expression of the recombinant TCR came at the expense of some endogenous iNKT-TCR, as could happen due to competition for CD3 complexes. Finally, as it has been reported that the anti-Mtb activity of iNKT cells is due to production of GM-CSF and not production of IFNγ or infected cell lysis (78), it is unclear what additional role(s) rTCR-iNKT cells would play during Mtb infection. Future studies are needed to determine if expressing this rTCR in iNKT cells skews their function in vivo during Mtb infection, either by helping trafficking of iNKT cells to the site of infection or otherwise. Clearly, iNKT expressing anti-tumor rTCR could also gain augmented activity, as is currently being addressed by some groups.
iNKT cells provide a novel alternative to standard T cells in cancer immunotherapy, as described above. Their tissue (and therefore also tumor) tropism, inherent direct and indirect anti-tumor activities and our ability to manipulate them in vitro and in vivo (e.g., with α-GalCer, analogs thereof, or the iNKTCR mAb 6B11) combined with reversible defects in cancer patients suggest that they can be exploited to treat a range of solid and hematological malignancies.
An important potential caveat in exploiting iNKT cells has been the observation that repeated stimulation of mouse iNKT cells (though less so with human iNKT) with α-GalCer can lead to an anergic-like state (3–6, 8, 79–81). Interestingly, this state can be reversed by PD-1/PD-L1 blockade (79), commonly now used in the clinic to overcome conventional anti-tumor responses. Furthermore, it may reflect a polarization to IL-10 producing “NKT10” that have been found in mice and man (81). Another promising approach in general, which may also overcome NKT cell anergy/polarization, is differential use of co-stimulation alongside direct invariant TCR stimulation (82).
There may well be more total “non-invariant” diverse CD1d-restricted NKT cells in the body than iNKT and their ability to make Th2 cytokines appears to impair tumor immunity (30), whereas such NKT making IFNγ stratifies with cancer patient survival (83), as does iNKT (24, 25, 54). However, non-invariant CD1d-restricted NKT cell manipulation is much more challenging and their understanding lags far behind iNKT cells. Finally, unlike other innate lymphocytes like NK and γδ T cells, iNKT are also relatively rare, so substantially increasing their numbers should be safe and is both very feasible (as described above) and has more potential to change the milieu (the other populations at ~5% of total lymphocytes probably cannot be increased more than ~10-fold without concomitant loss of conventional T cells). The next few years should provide an opportunity for iNKT cells to “put up or shut up”!
BW, JC, and ME contributed to the writing and production of this manuscript. JC designed the figures with editorial input from BW and ME.
BW, JC, and ME were employed by Agenus Inc., a company developing immuno-therapies for cancer, at the time of writing this manuscript.
We would like to thank colleagues for discussions and apologize for any references omitted for space reasons.
BW, JC, and ME were employed by Agenus Inc. at the time of writing this manuscript.
1. Kronenberg M. When less is more: T lymphocyte populations with restricted antigen receptor diversity. J Immunol (2014) 193(3):975–6. doi:10.4049/jimmunol.1401491
2. Van Rhijn I, Moody DB. Donor unrestricted T cells: a shared human T cell response. J Immunol (2015) 195(5):1927–32. doi:10.4049/jimmunol.1500943
3. Salio M, Silk JD, Yvonne Jones E, Cerundolo V. Biology of CD1- and MR1-restricted T cells. Annu Rev Immunol (2014) 32:323–66. doi:10.1146/annurev-immunol-032713-120243
4. Constantinides MG, Bendelac A. Transcriptional regulation of the NKT cell lineage. Curr Opin Immunol (2013) 25(2):161–7. doi:10.1016/j.coi.2013.01.003
5. Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI. Recognition of CD1d-restricted antigens by natural killer T cells. Nat Rev Immunol (2012) 12(12):845–57. doi:10.1038/nri3328
6. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13(2):101–17. doi:10.1038/nri3369
7. Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med (1998) 188:1521–8. doi:10.1084/jem.188.8.1521
8. Exley MA, Lynch L, Varghese B, Nowak M, Alatrakchi N, Balk S. Developing understanding of the roles of CD1d-restricted T cell subsets in cancer: reversing tumor-induced defects. Clin Immunol (2011) 140:184–95. doi:10.1016/j.clim.2011.04.017
9. Berzins SP, Smyth MJ, Baxter AG. Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol (2011) 11:131–42. doi:10.1038/nri2904
10. Lee PT, Benlagha K, Teyton L, Bendelac A. Distinct functional lineages of human V(alpha)24 natural killer T cells. J Exp Med (2002) 195:637–41. doi:10.1084/jem.20011908
11. Park Y, Moon SJ, Lee SW. Lineage re-commitment of CD4CD8αα intraepithelial lymphocytes in the gut. BMB Rep (2016) 49(1):11–7. doi:10.5483/BMBRep.2016.49.1.242
12. Crowe NY, Coquet JM, Berzins SP, Kyparissoudis K, Keating R, Pellicci DG, et al. Differential antitumor immunity mediated by NKT cell subsets in vivo. J Exp Med (2005) 202(9):1279–88. doi:10.1084/jem.20050953
13. Monteiro M, Graca L. iNKT cells: innate lymphocytes with a diverse response. Crit Rev Immunol (2014) 34(1):81–90. doi:10.1615/CritRevImmunol.2014010088
14. Lynch L, O’Shea D, Winter DC, Geoghegan J, Doherty DG, O’Farrelly C. Invariant NKT cells and CD1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol (2009) 39(7):1893–901. doi:10.1002/eji.200939349
15. Exley MA, Hand L, O’Shea D, Lynch L. Interplay between the immune system and adipose tissue in obesity. J Endocrinol (2014) 223(2):R41–8. doi:10.1530/JOE-13-0516
16. Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol (2003) 4:1230–7. doi:10.1038/ni1002
17. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med (2008) 14(6):633–40. doi:10.1038/nm1770
18. Nembrini C, Marsland BJ, Kopf M. IL-17-producing T cells in lung immunity and inflammation. J Allergy Clin Immunol (2009) 123(5):986–94. doi:10.1016/j.jaci.2009.03.033
19. Kohrt HE, Pillai AB, Lowsky R, Strober S. NKT cells, Treg, and their interactions in bone marrow transplantation. Eur J Immunol (2010) 40(7):1862–9. doi:10.1002/eji.201040394
20. Karadimitris A, Chaidos A. The role of invariant NKT cells in allogeneic hematopoietic stem cell transplantation. Crit Rev Immunol (2012) 32(2):157–71. doi:10.1615/CritRevImmunol.v32.i2.40
21. Guan P, Bassiri H, Patel NP, Nichols KE, Das R. Invariant natural killer T cells in hematopoietic stem cell transplantation: killer choice for natural suppression. Bone Marrow Transplant (2016) 51(5):629–37. doi:10.1038/bmt.2015.335
22. Tahir M, Cheng O, Shaulov A, Bubley G, Balk S, Exley M. Loss of IFN-gamma production by invariant NK T cells in advanced cancer. J Immunol (2001) 167:4046–50. doi:10.4049/jimmunol.167.7.4046
23. Yanagisawa K, Seino K, Ishikawa Y, Nozue M, Todoroki T, Fukao K. Impaired proliferative response of V alpha 24 NKT cells from cancer patients against alpha-galactosylceramide. J Immunol (2002) 168(12):6494–9. doi:10.4049/jimmunol.168.12.6494
24. Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med (2003) 197:1667–76. doi:10.1084/jem.20021650
25. Fujii S, Shimizu K, Klimek V, Geller MD, Nimer SD, Dhodapkar MV. Severe and selective deficiency of interferon-gamma-producing invariant natural killer T cells in patients with myelodysplastic syndromes. Br J Haematol (2003) 122(4):617–22. doi:10.1046/j.1365-2141.2003.04465.x
26. Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi Z, Asgharzadeh S, et al. Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J Exp Med (2004) 199(9):1213–21. doi:10.1084/jem.20031462
27. Tachibana T, Onodera H, Tsuruyama T, Mori A, Nagayama S, Hiai H, et al. Increased intratumor Valpha24-positive natural killer T cells: a prognostic factor for primary colorectal carcinomas. Clin Cancer Res (2005) 11(20):7322–7. doi:10.1158/1078-0432.CCR-05-0877
28. Molling JW, Kolgen W, van der Vliet HJ, Boomsma MF, Kruizenga H, Smorenburg CH, et al. Peripheral blood IFN-gamma-secreting Valpha24+Vbeta11+ NKT cell numbers are decreased in cancer patients independent of tumor type or tumor load. Int J Cancer (2005) 116:87–93. doi:10.1002/ijc.20998
29. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest (2009) 119(6):1524–36. doi:10.1172/JCI37869
30. Berzofsky JA, Terabe M. The contrasting roles of NKT cells in tumor immunity. Curr Mol Med (2009) 9(6):667–72. doi:10.2174/156652409788970706
31. Taniguchi M, Tashiro T, Dashtsoodol N, Hongo N, Watarai H. The specialized iNKT cell system recognizes glycolipid antigens and bridges the innate and acquired immune systems with potential applications for cancer therapy. Int Immunol (2010) 22(1):1–6. doi:10.1093/intimm/dxp104
32. Metelitsa LS. Anti-tumor potential of type-I NKT cells against CD1d-positive and CD1d-negative tumors in humans. Clin Immunol (2011) 140(2):119–29. doi:10.1016/j.clim.2010.10.005
33. Schneiders FL, Scheper RJ, von Blomberg BM, Woltman AM, Janssen HL, van den Eertwegh AJ, et al. Clinical experience with α-galactosylceramide (KRN7000) in patients with advanced cancer and chronic hepatitis B/C infection. Clin Immunol (2010) 140:130–41. doi:10.1016/j.clim.2010.11.010
34. Motohashi S, Okamoto Y, Yoshino I, Nakayama T. Anti-tumor immune responses induced by iNKT cell-based immunotherapy for lung cancer and head and neck cancer. Clin Immunol (2011) 140:167–76. doi:10.1016/j.clim.2011.01.009
35. Dhodapkar MV, Richter J. Harnessing natural killer T (NKT) cells in human myeloma: progress and challenges. Clin Immunol (2011) 140:160–6. doi:10.1016/j.clim.2010.12.010
36. Liu D, Song L, Brawley VS, Robison N, Wei J, Gao X, et al. Medulloblastoma expresses CD1d and can be targeted for immunotherapy with NKT cells. Clin Immunol (2013) 149(1):55–64. doi:10.1016/j.clim.2013.06.005
37. Schneiders FL, de Bruin RC, van den Eertwegh AJ, Scheper RJ, Leemans CR, Brakenhoff RH, et al. Circulating invariant natural killer T-cell numbers predict outcome in head and neck squamous cell carcinoma: updated analysis with 10-year follow-up. J Clin Oncol (2012) 30(5):567–70. doi:10.1200/JCO.2011.38.8819
38. Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science (1997) 278:1623–6. doi:10.1126/science.278.5343.1623
39. Bellone M, Ceccon M, Grioni M, Jachetti E, Calcinotto A, Napolitano A, et al. iNKT cells control mouse spontaneous carcinoma independently of tumor-specific cytotoxic T cells. PLoS One (2010) 5(1):e8646. doi:10.1371/journal.pone.0008646
40. Bedel R, Matsuda JL, Brigl M, White J, Kappler J, Marrack P, et al. Lower TCR repertoire diversity in Traj18-deficient mice. Nat Immunol (2012) 13(8):705–6. doi:10.1038/ni.2347
41. Dashtsoodol N, Shigeura T, Ozawa R, Harada M, Kojo S, Watanabe T, et al. Generation of novel Traj18-deficient mice lacking Vα14 natural killer T cells with an undisturbed T cell receptor α-chain repertoire. PLoS One (2016) 11(4):e0153347. doi:10.1371/journal.pone.0153347
42. Zhang J, Bedel R, Krovi SH, Tuttle KD, Zhang B, Gross J, et al. Mutation of the Traj18 gene segment using TALENs to generate natural killer T cell deficient mice. Sci Rep (2016) 6:27375. doi:10.1038/srep27375
43. Nowak M, Arredouani MS, Tun-Kyi A, Schmidt-Wolf I, Sanda MG, Balk SP, et al. Defective NKT cell activation by CD1d+ TRAMP prostate tumor cells is corrected by interleukin-12 with alpha- galactosylceramide. PLoS One (2010) 5(6):e11311. doi:10.1371/journal.pone.0011311
44. Chaudhry MS, Karadimitris A. Role and regulation of CD1d in normal and pathological B cells. J Immunol (2014) 193(10):4761–8. doi:10.4049/jimmunol.1401805
45. Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, et al. IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J Clin Invest (2012) 122(6):2221–33. doi:10.1172/JCI59535
46. Kawano T, Nakayama T, Kamada N, Kaneko Y, Harada M, Ogura N, et al. Antitumor cytotoxicity mediated by ligand-activated human V alpha24 NKT cells. Cancer Res (1999) 59:5102–5.
47. Nakagawa R, Motoki K, Ueno H, Iijima R, Nakamura H, Kobayashi E, et al. Treatment of hepatic metastasis of the colon26 adenocarcinoma with an α-galactosylceramide, KRN7000. Cancer Res (1998) 58:1202–7.
48. Toura I, Kawano T, Akutsu Y, Nakayama T, Ochiai T, Taniguchi M. Cutting edge: inhibition of experimental tumor metastasis by dendritic cells pulsed with alpha-galactosylceramide. J Immunol (1999) 163:2387–91.
49. Nishimura T, Kitamura H, Iwakabe K, Yahata T, Ohta A, Sato M, et al. The interface between innate and acquired immunity: glycolipid antigen presentation by CD1d-expressing dendritic cells to NKT cells induces the differentiation of antigen-specific cytotoxic T lymphocytes. Int Immunol (2000) 12:987–94. doi:10.1093/intimm/12.7.987
50. Shin T, Nakayama T, Akutsu Y, Motohashi S, Shibata Y, Harada M, et al. Inhibition of tumor metastasis by adoptive transfer of IL-12-activated Vα14 NKT cells. Int J Cancer (2001) 91:523–8. doi:10.1002/1097-0215(20010215)91:4<523::AID-IJC1087>3.0.CO;2-L
51. Smyth MJ, Crowe NY, Pellicci DG, Kyparissoudis K, Kelly JM, Takeda K, et al. Sequential production of interferon-γ by NK1.1+ T cells and natural killer cells is essential for the antimetastatic effect of α-galactosylceramide. Blood (2002) 99:1259–66. doi:10.1182/blood.V99.4.1259
52. Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res (2002) 8:3702–9.
53. Nieda M, Okai M, Tazbirkova A, Lin H, Yamaura A, Ide K, et al. Therapeutic activation of Valpha24+Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood (2004) 103:383–9. doi:10.1182/blood-2003-04-1155
54. Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, et al. A phase 1 study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res (2005) 11:1910–7. doi:10.1158/1078-0432.CCR-04-1453
55. Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosylceramide loaded mature dendritic cells in cancer patients. J Exp Med (2005) 201:1503–17. doi:10.1084/jem.20042592
56. Uchida T, Horiguchi S, Tanaka Y, Yamamoto H, Kunii N, Motohashi S, et al. Phase I study of alpha-galactosylceramide-pulsed antigen presenting cells administration to the nasal submucosa in unresectable or recurrent head and neck cancer. Cancer Immunol Immunother (2008) 57(3):337–45. doi:10.1007/s00262-007-0373-5
57. Motohashi S, Nagato K, Kunii N, Yamamoto H, Yamasaki K, Okita K, et al. A phase I-II study of alpha-galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J Immunol (2009) 182(4):2492–501. doi:10.4049/jimmunol.0800126
58. Nagato K, Motohashi S, Ishibashi F, Okita K, Yamasaki K, Moriya Y, et al. Accumulation of activated invariant natural killer T cells in the tumor microenvironment after α-galactosylceramide-pulsed antigen presenting cells. J Clin Immunol (2012) 32(5):1071–81. doi:10.1007/s10875-012-9697-9
59. Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res (2006) 12:6079–86. doi:10.1158/1078-0432.CCR-06-0114
60. Kunii N, Horiguchi S, Motohashi S, Yamamoto H, Ueno N, Yamamoto S, et al. Combination therapy of in vitro-expanded natural killer T cells and alpha-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma. Cancer Sci (2009) 100(6):1092–8. doi:10.1111/j.1349-7006.2009.01135.x
61. Yamasaki K, Horiguchi S, Kurosaki M, Kunii N, Nagato K, Hanaoka H, et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin Immunol (2011) 138(3):255–65. doi:10.1016/j.clim.2010.11.014
62. Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, et al. Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: a phase I clinical trial. Clin Cancer Res (2017) 23(14):3510–9. doi:10.1158/1078-0432.CCR-16-0600
63. Exley MA, Hou R, Shaulov A, Tonti E, Dellabona P, Casorati G, et al. Selective activation, expansion, and monitoring of human iNKT cells with a mAb specific for TCR alpha-chain CDR3 loop. Eur J Immunol (2008) 20:1756–1756. doi:10.1002/eji.200737389
64. Exley MA, Wilson SB, Balk SP. Isolation and functional use of human NKT cells. Curr Protoc Immunol (2017) 119:1–14. doi:10.1002/cpim.33
65. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy – assessment and management of toxicities. Nat Rev Clin Oncol (2018) 15(1):47–62. doi:10.1038/nrclinonc.2017.148
66. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med (2015) 21(8):914–21. doi:10.1038/nm.3910
67. Bedoya F, Frigault MJ, Maus MV. The flipside of the power of engineered T cells: observed and potential toxicities of genetically modified T cells as therapy. Mol Ther (2017) 25(2):314–20. doi:10.1016/j.ymthe.2016.11.011
68. Pillai AB, George TI, Dutt S, Teo P, Strober S. Host NKT cells can prevent graft-versus-host disease and permit graft antitumor activity after bone marrow transplantation. J Immunol (2007) 178(10):6242–51. doi:10.4049/jimmunol.178.10.6242
69. Dellabona P, Casorati G, de Lalla C, Montagna D, Locatelli F. On the use of donor-derived iNKT cells for adoptive immunotherapy to prevent leukemia recurrence in pediatric recipients of HLA haploidentical HSCT for hematological malignancies. Clin Immunol (2011) 140(2):152–9. doi:10.1016/j.clim.2010.11.015
70. Slauenwhite D, Johnston B. Regulation of NKT cell localization in homeostasis and infection. Front Immunol (2015) 6:255. doi:10.3389/fimmu.2015.00255
71. Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity (2008) 29(3):391–403. doi:10.1016/j.immuni.2008.07.011
72. Song L, Ara T, Wu HW, Woo CW, Reynolds CP, Seeger RC, et al. Oncogene MYCN regulates localization of NKT cells to the site of disease in neuroblastoma. J Clin Invest (2007) 117(9):2702–12. doi:10.1172/JCI30751
73. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood (2014) 124(18):2824–33. doi:10.1182/blood-2013-11-541235
74. Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J Clin Invest (2016) 126(6):2341–55. doi:10.1172/JCI83476
75. Rotolo A, Caputo V, Karadimitris A. The prospects and promise of chimeric antigen receptor immunotherapy in multiple myeloma. Br J Haematol (2016) 173(3):350–64. doi:10.1111/bjh.13976
76. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30(2):492–500. doi:10.1038/leu.2015.247
77. Jiang ZM, Luo W, Wen Q, Liu SD, Hao PP, Zhou CY, et al. Development of genetically engineered iNKT cells expressing TCRs specific for the M. tuberculosis 38-kDa antigen. J Transl Med (2015) 13:141. doi:10.1186/s12967-015-0502-4
78. Rothchild AC, Jayaraman P, Nunes-Alves C, Behar SM. iNKT cell production of GM-CSF controls Mycobacterium tuberculosis. PLoS Pathog (2014) 10(1):e1003805. doi:10.1371/journal.ppat.1003805
79. Parekh VV, Lalani S, Kim S, Halder R, Azuma M, Yagita H, et al. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol (2009) 182(5):2816–26. doi:10.4049/jimmunol.0803648
80. Kojo S, Elly C, Harada Y, Langdon WY, Kronenberg M, Liu YC. Mechanisms of NKT cell anergy induction involve Cbl-b-promoted monoubiquitination of CARMA1. Proc Natl Acad Sci U S A (2009) 106(42):17847–51. doi:10.1073/pnas.0904078106
81. Sag D, Krause P, Hedrick CC, Kronenberg M, Wingender G. IL-10-producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest (2014) 124(9):3725–40. doi:10.1172/JCI72308
82. van den Heuvel MJ, Garg N, Van Kaer L, Haeryfar SM. NKT cell costimulation: experimental progress and therapeutic promise. Trends Mol Med (2011) 17(2):65–77. doi:10.1016/j.molmed.2010.10.007
Keywords: NKT cells, CD1d, iNKT cells, cancer immunotherapy, monoclonal antibody
Citation: Wolf BJ, Choi JE and Exley MA (2018) Novel Approaches to Exploiting Invariant NKT Cells in Cancer Immunotherapy. Front. Immunol. 9:384. doi: 10.3389/fimmu.2018.00384
Received: 02 November 2017; Accepted: 12 February 2018;
Published: 02 March 2018
Edited by:
Weiming Yuan, University of Southern California, United StatesReviewed by:
S. M. Mansour Haeryfar, University of Western Ontario, CanadaCopyright: © 2018 Wolf, Choi and Exley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark A. Exley, bWFyay5leGxleUBhZ2VudXNiaW8uY29t
†Present address: Benjamin J. Wolf, Compass Therapeutics, Cambridge, MA, United States;
Jiyoung Elizabeth Choi, Novartis Institute of Biological Research, Cambridge, MA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.