
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 February 2018
Sec. Immunological Tolerance and Regulation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00354
This article is part of the Research Topic Clinical Therapeutic Tolerance: First-in-Human Data: Proceedings of the 4th Newcastle Therapeutic Tolerance Workshop View all 13 articles
 Niloufar Safinia1,2†
Niloufar Safinia1,2† Nathali Grageda1†
Nathali Grageda1† Cristiano Scottà1
Cristiano Scottà1 Sarah Thirkell1
Sarah Thirkell1 Laura J. Fry3
Laura J. Fry3 Trishan Vaikunthanathan4
Trishan Vaikunthanathan4 Robert I. Lechler1
Robert I. Lechler1 Giovanna Lombardi1*
Giovanna Lombardi1*
Solid organ transplantation is the treatment of choice for patients with end-stage organ dysfunction. Despite improvements in short-term outcome, long-term outcome is suboptimal due to the increased morbidity and mortality associated with the toxicity of immunosuppressive regimens and chronic rejection (1–5). As such, the attention of the transplant community has focused on the development of novel therapeutic strategies to achieve allograft tolerance, a state whereby the immune system of the recipient can be re-educated to accept the allograft, averting the need for long-term immunosuppression. Indeed, reports of “operational” tolerance, whereby the recipient is off all immunosuppressive drugs and maintaining good graft function, is well documented in the literature for both liver and kidney transplantations (6–8). However, this phenomenon is rare and in the setting of liver transplantation has been shown to occur late after transplantation, with the majority of patients maintained on life-long immunosupression to prevent allograft rejection (9). As such, significant research has focused on immune regulation in the context of organ transplantation with regulatory T cells (Tregs) identified as cells holding considerable promise in this endeavor. This review will provide a brief introduction to human Tregs, their phenotypic and functional characterization and focuses on our experience to date at the clinical translation of Treg immunotherapy in the setting of solid organ transplantation.
Tregs are characterized by the expression of CD4 and CD25 molecules and more specifically the transcription factor FOXP3, a master control gene underpinning Treg development and function. More recently, the inverse expression of the α-chain of IL-7R, CD127, combined with the expression of CD4 and CD25 has been shown to demarcate a pure population of Tregs demonstrating stability and optimal function (10, 11). Tregs are far from homogenous and over the years have been characterized into several subsets, most of which have been associated with various facets of Treg function (12).
Treg subsets can firstly be stratified according to their origin: thymus-derived and peripherally derived Tregs (tTregs and pTregs, respectively). While there have been phenotypic markers which have suggested differentiation between the two subsets, such as Helios and Neuropilin-1, to date these are still contentious (13–16). The only way to reliably differentiate tTregs and pTregs has been to interrogate the Treg-specific demethylated region (TSDR). Here, tTregs display a fully demethylated TSDR, whereas pTregs are only partially demethylated (17–20). Furthermore, Tregs have also been delineated on account of their antigen naivety in relation to their differential expression of CD45RA and FOXP3. The seminal work of Miyara et al., described the heterogeneity of the Treg population in three phenotypically and functionally distinct subpopulations: Population I (P1) naive or resting Tregs (CD45RA+FOXP3Lo); population II (P2) effector Tregs (CD45RA−FOXP3Hi), both of which are suppressive in vitro; and population III (P3) the “non-suppressive,” cytokine secreting non-Tregs (CD45RA−FOXP3Lo) (21). However, we and others have previously shown that population III is indeed suppressive, and within this population, identified a subpopulation of Tregs that express the C-type lectin CD161 and produce the proinflammatory cytokine, IL-17 (22–24).
In support of the heterogeneity of the Treg compartment, using a new technology, the single-cell mass cytometry (cytometry by time-of-flight) we have conducted an in-depth characterization of Tregs, further demonstrating the true extent of their heterogeneity, with 22 different clusters identified (25). In the clinical setting and through utilizing the same principles and technology, Kordasti et al. identified the Treg subset that predicted response to immunosuppressive therapy in patients with aplastic anemia (26). Additionally, we have recently extended the analysis of T helper-like subpopulations of Tregs, demonstrating that Th2-like Tregs are enriched in the tumor sites (27). There is no doubt that the future will see the discovery of many more markers heralding Treg purity. For an in-depth review into Treg phenotype subsets the reader is directed to the following reviews (12, 28, 29).
Tregs are defined by their immunoregulatory suppressive qualities. However, no one mechanism defines Tregs. Instead, it is believed that several mechanisms behave in concert, which promote immune regulation. All T lymphocytes rely on IL-2 for their survival and proliferation. By their expression of the interleukin 2 receptor, CD25, Tregs deplete stores of IL-2, curbing the survival of surrounding T lymphocytes (28). In a more active mechanism of suppression, the Treg surface molecule, cytotoxic T lymphocyte antigen 4 (CTLA-4), is known to bind the costimulatory molecules CD80/86 with a higher affinity than its proinflammatory competitor CD28, expressed on conventional T effector cells, thus preventing T effector activation. This negative costimulatory molecule has also been proposed to upregulate indoleamine 2, 3-dioxygenase expression on dendritic cells, responsible for the catabolism of tryptophan, which in turn suppresses immune responses through the generation of the immunosuppressive molecules, in particular kynurenine (30). Further in-depth investigation into the mechanism of action of CTLA-4 has revealed that CD80/86 ligands on antigen-presenting cells (APCs) are captured by a process of trans-endocytosis, in turn impairing further T cell activation (31–33). Deficiencies in CTLA-4 have been associated with lymphoproliferative disorders and the development of severe T-cell-mediated autoimmune diseases, which is why CTLA-4 is recognized as a key molecular target governing Treg-mediated suppression (33, 34).
The ectoenzyme, CD39, is abundantly expressed on Tregs, its expression allows Tregs to hydrolyze the proinflammatory danger signal, adenosine triphosphate, to the anti-inflammatory mediator, adenosine, which following interaction with the adenosine A2A receptor, has been reported to have immunosuppressive and anti-proliferative effects (35). CD39+ Tregs have also been found to suppress the release of IL-17, alongside IFN-γ and IL-2, from Th17 cells, while CD39− T cells had an increased propensity to produce IL-17 (36). Furthermore, the reconstitution of positively selected CD39-null mouse models of colitis with soluble apyrase, a mediator with enzymatic activity identical to CD39, reversed their increased susceptibility to develop auto-immune diseases and prevented a Th-1 skewed immune response. Further studies by Gibson et al., have highlighted the importance of CD39 expression for Treg mechanism of action in a T cell transfer model of colitis (37). Additionally, we have established a further mechanism by which Tregs function through the release of exosomes, expressing ectoenzyme CD73, which regulate target cells through the purinergic generation of adenosine (38).
Reports have also suggested a cytotoxic role of Tregs in depleting T effector numbers through a perforin-dependent and granzyme-dependent manner (39). Similarly, there have been reports of Tregs expressing Galectin-9, which following binding with the T-cell immunoglobulin and mucin-domain containing-3 receptor CD44 on effector T cells, has been shown to induce apoptosis (40). More recently, this mechanism of suppression has been proposed to be limited to pTregs (41). It has also been postulated that Tregs have the capacity for cytokine-mediated suppression involving the regulatory cytokines: IL-10 (42), TGF-β (43, 44), and more recently IL-35 (45).
Clinical trials of Treg therapy in transplantation are focused on tipping the balance of immune homeostasis in favor of regulation. However, in order for this to occur the in vivo Treg pool needs to be expanded significantly in order to drive immune tolerance (46). As such, there has been huge interest in either the in vivo expansion of these cells or their adoptive transfer. Here, we focus on the prerequisites that need to be fulfilled in order to permit the adoptive transfer of these cells.
Firstly, Tregs need to be isolated from the peripheral blood and this is by no means an easy feat. As mentioned earlier, Tregs are highly heterogenous and as such, the debate of which population of Tregs would serve as optimal cell product in adoptive transfers is a highly discussed topic. Isolation to date has largely been governed by magnetic bead isolation, a process whereby depletion of CD8+ T cells and positive selection of CD25+ T cells using magnetic beads allows isolation of Tregs (47, 48). This process results in the separation of a consistent number of cells from whole blood using an approach that has been utilized by us in the two clinical trials aimed at testing the safety of expanded autologous Tregs in patients receiving either kidney (ONE Study, NCT02129881) or liver (ThRIL, NCT02166177) transplants. However, this process does not allow for enrichment based on multiple parameters and isolating Tregs based upon CD25+ expression alone results in contaminating T effector cells which express low levels of CD25 molecules. Methods to overcome this have been explored in Treg expansion. One such strategy is the ex vivo expansion of these cells in the presence of the mTOR-inhibitor, rapamycin (RAPA) (49).
We and others have shown that RAPA is an ideal treatment strategy for preferential expansion of Tregs (50–52). RAPA confers a proliferative advantage to Tregs by affecting the Akt/mTor pathway. Indeed, the molecular signal controlled by this pathway is not essential for Tregs, but crucial for the activation and proliferation of conventional T cells (51, 53). Using this approach, we have shown that large-scale expansion of functionally potent Tregs is possible when starting from a population of cells with low purity (48). In our studies, another potential candidate drug for the expansion of Tregs is the vitamin A metabolite, all-trans retinoic acid (ATRA). Some studies have shown that ATRA can be a good treatment for the induction of adaptive Tregs (54) or very pure Tregs (55). Our data showed that ATRA favors the ex vivo expansion of a population of highly suppressive Tregs from magnetic bead purified Tregs, although these cells produce a significant amount of IL-17 and IFN-γ following stimulation. However, further characterization of Tregs cultured with ATRA has shown the expansion of FOXP3+CD161+ Tregs (51), which is encouraging based on our previous work reporting that CD161 identifies a specific sub-population of IL-17-producing FOXP3+ Tregs with a strong capacity to suppress conventional T cell proliferation (22). In contrast, Tregs expanded in the presence of RAPA show decreased expression of CD161, as well as reduced production of pro-inflammatory cytokines. The combined treatment of Tregs with RAPA and ATRA demonstrated that the suppressive function and stability of Tregs is maintained (decreased CD161 expression and lack of IL-17 production).
This analysis was extended to the subtypes of Tregs and while P2 Tregs did not expand in vitro, P1 Tregs expanded well in the presence of RAPA, ATRA or the combination of the two drugs. In contrast, RAPA (alone or in combination with ATRA) strongly reduced P3 Treg proliferation, while the treatment with ATRA alone showed a negligible inhibitory effect on the expansion of this subset (51). In support of this observation, we have unpublished data showing that when P1 and P3 were cultured together at 1:1 ratio, the presence of RAPA, but not ATRA, gave a much stronger proliferative advantage to P1 on P3 Tregs. At the end of culture, P1 could overgrow P3 Tregs. Additional experiments showed that RAPA treatment could inhibit the production of proinflammatory cytokines in the P3 subset and negatively affect the expansion of FOXP3+CD161+ Tregs (51). Finally, we have also demonstrated that these treatments can influence the migratory ability of expanded Tregs. Our findings showed that the treatment of Tregs with RAPA led to the expression of skin-homing receptors cutaneous lymphocyte-associated antigen and CCR4 (51), as well as CXCR3, a chemokine receptor which enables homing to the liver (48). Instead, Treg cultures in the presence of ATRA resulted in a high percentage of cells coexpressing gut-homing receptors such as CCR9 and α4β7 (51).
One of the most difficult and time-consuming aspects of translating the extensive research carried out in the laboratory to the clinical setting is the transfer of research protocols into a GMP unit. This process, referred to as technical transfer, requires careful management to ensure the resulting product maintains a consistently high level of quality, while achieving the aim of the researcher. In this pursuit, a manufacturing process was developed with the aim of creating a Treg Investigational Medicinal Product (IMP) that could be used in the ONE Study (NCT02129881) or ThRIL (NCT02166177) clinical trials.
There are many factors to consider during the technical transfer (Figure 1), and important milestones include: the sharing of technical information from the research department to the GMP facility, process development, preparation of the product specifications, process validation and finally authorization of the process by the Medicines and Healthcare Products Regulatory Agency (MHRA) through the submission of a Clinical Trials Authorization (CTA). The process initially starts with the sharing of a detailed description of all process raw materials, manufacturing methods, equipment used, specifications, and test methods from the transferring research department to the GMP unit. It is important to ensure that all raw materials, reagents and consumables that were used in the research laboratory are GMP-compatible. During the technical transfer stage of these Treg trials, different GMP-compatible reagents and culture conditions were compared, ensuring that the final protocol resulted in phenotypically stable Tregs that could be isolated and expanded consistently while maintaining their suppressive function (Table 1).
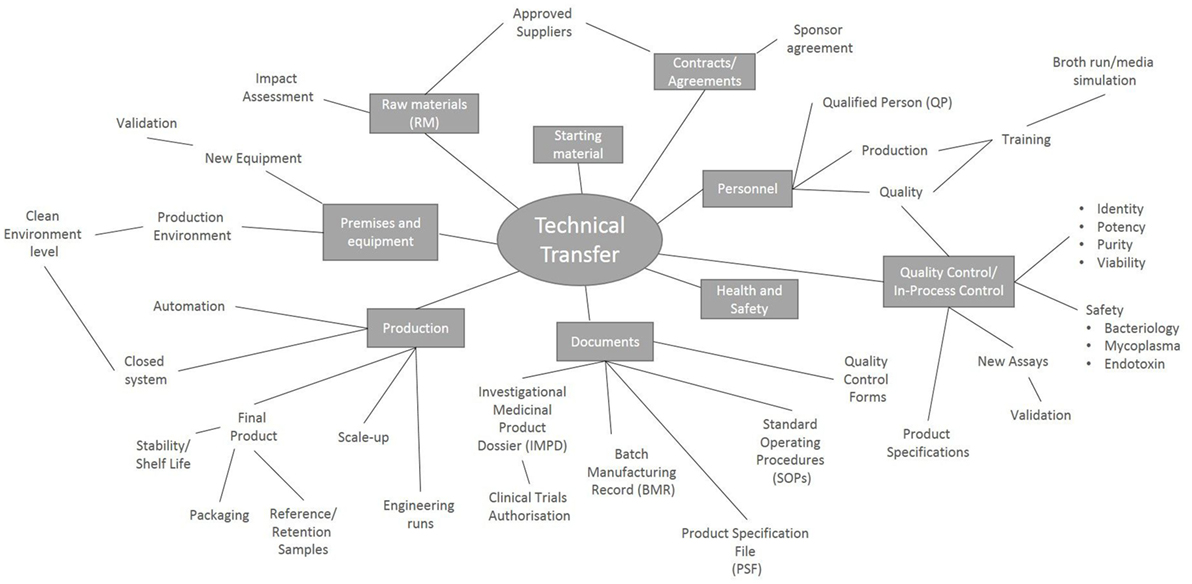
Figure 1. Key points to consider during a technical transfer process. Schematic representation of the processes involved during the transfer of a manufacturing process from the research laboratory into a Good Manufacturing Process unit.
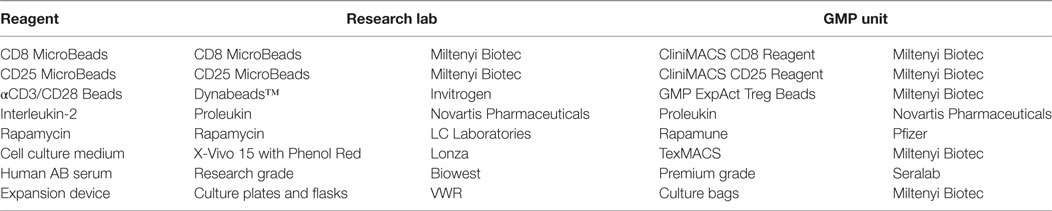
Table 1. Comparison of research- and GMP-grade reagents.
The transfer of the Treg process into the GMP unit also provided the opportunity to optimize the manufacturing method and introduce automated processes to increase the levels of consistency between batches. The original research process included a peripheral blood mononuclear cells (PBMCs) isolation using density gradient centrifugation with Ficoll® Paque Plus (GE Healthcare, Switzerland) and 50 ml centrifuge tubes. In order to close out and automate this process, the SEPAX system (Biosafe, Switzerland) was adopted. The SEPAX device is an automated cell separation system which processes within closed single-use disposable tubing sets. The maximum blood input volume to run the NeatCell Programme (SEPAX density gradient cell separation using Ficoll® Paque Plus) is 120 ml, therefore the SEPAX volume reduction programme (SmartRedux) was introduced to ensure the full initial blood sample could be processed. The data generated from these initial runs identified that the volume reduction step alone was sufficient, as the erythrocytes did not interfere with the CliniMACS Plus (Miltenyi Biotec, Germany) magnetic cell selections.
The specifications of the final product are set by the research team and from this information, a product specification file (PSF) can be written. The PSF contains, or refers to files containing, all the information required to draft the standard operating procedures, batch manufacturing records, and the quality control assay forms. These documents ensure standardization and traceability of the process for every batch manufactured.
With these documents in place, scale-up runs can be performed allowing the manufacture process to be tested within the GMP unit and ensuring enough Tregs could be generated to produce the dose required for the patients, perform all QC assays and to create the required reference and retention samples. During the scale-up runs, the opportunity to further close out the system and minimize the risk of contamination was taken. For example, Tregs were cultured using plates and T-flasks in the research laboratory. During the scale-up process, this method was adapted to utilize closed-system culture bags. However, it was found that transferring the process into bags reduced the expansion rate of the Tregs. In spite of this, the required numbers of Tregs were still achieved and so this closed system modification was incorporated into the final manufacturing process.
With the manufacturing methods finalized during the scale-up runs and following the validation of the QC assays, the entire process was validated by performing six engineering runs. These engineering runs provided documented evidence that the GMP unit could routinely and consistently produce the Treg product that met the required specifications.
The data, from both the research experiments and GMP unit, was compiled to create the IMP Dossier (IMDP). This document contains information on the quality, manufacture and control of the IMP. This is one of the essential documents, along with a EudraCT number and approval from an ethics committee, that form the CTA submission to the MHRA.
The ONE Study (NCT02129881) initiated in 2014, a dose-escalation phase I/II trial carried out under a large EU consortium. The aim of The ONE Study was to evaluate different regulatory cells in kidney transplant recipients allowing the direct comparison of the safety, practicality and therapeutic effect of each cell type. In the UK, in collaboration with The University of Oxford, our group manufactured sixteen batches of polyclonal Tregs, of which 12 were certified for administration. A dose escalation design was implemented with four escalating doses of 1 × 106, 3 × 106, 6 × 106, and 10 × 106/kg, with the last patient being treated with the maximum dose in January 2016. Four of the batches could not be certified and dosed, three of which were due to insufficient cell numbers to formulate the dose and one batch failing the release criteria due to bacterial contamination. High levels of variability were observed in the expansion capability of the Tregs obtained from different patients. Due to this, a substantial amendment was made to the IMPD allowing flexibility in setting a date for the harvesting of Treg culture. This amendment reduced the risk of failing batches during the manufacture of an expensive and time-consuming product.
The last patient in the ThRIL trial (NCT02166177) was dosed in July 2017. This was a Phase I/IIa clinical trial of Treg immunotherapy in the setting of liver transplantation initiated at King’s College London. Here, the safety, tolerability and efficacy of polyclonally expanded Tregs in combination with depletion of alloreactive T cells (ATG) and short-term immunosuppression was assessed. For this trial, patients awaiting a liver transplant were recruited with the aim of administering the cell therapy 2 months after transplantation. Out of an initial 23 patients enrolled, 7 of these patients received a transplant and the manufacture of the Treg IMP was initiated. Of these seven batches, three were completed and certified by a qualified person for administration. Two patients were removed during manufacturing, one due to proteinuria and the other due to death unrelated to the transplant. The other two batches did not meet the specifications (cell number and purity). The complications in patient retention lead to a substantial amendment to the MHRA to allow the recruitment of patients 6-month posttransplantation rather than while they were on the waiting list. This delay in recruitment was made as the transplanted patients were deemed to be more stable, and hence more amenable for cell therapy application. After this amendment was approved, a further six batches of the Treg IMP were manufactured and these patients were successfully treated.
Although these two trials are still in the patient follow-up stage, no serious adverse events have been observed suggesting that polyclonal Treg therapies are safe. We are now in the preparation stages of Phase II clinical trials in renal, liver, islet and heart transplant patients.
While the polyclonal expansion of Tregs using anti-CD3/antiCD28 beads is relatively straightforward and readily translatable into the clinic, extensive data from preclinical animal studies have demonstrated that the adoptive transfer of Tregs with direct or indirect allospecificity are superior to polyclonal Tregs at reducing graft rejection (56–58). However, the expansion of antigen-specific Tregs presents an additional set of parameters that need to be addressed, including the origin of APCs and the dose required. Initial studies by Taylor et al., used allogeneic splenocytes to enrich for murine allo-specific Tregs, which were more efficient at reducing graft versus host disease in a murine model compared to anti-CD3 stimulated Tregs (59). More recently, we have shown in a murine transplant model that Tregs need to have both direct and indirect allospecificities to induce indefinite survival of heart transplants (57). Furthermore, using a humanized transplant mouse model, in which immunodeficient mice were reconstituted with PBMCs, we show that human Tregs with direct allospecificity significantly reduced alloimmune-mediated injury of human skin grafts, when compared with polyclonal Tregs. As part of the ONE Study Consortium, Tregs with direct allospecificity are currently being evaluated at two different sites in the USA (NCT02244801 and NCT02091232). GMP regulations are far more rigorous in Europe compared to USA and the investigators have been able to use a standard cell sorter in their manufacturing process to purify Tregs, which is not possible in Europe. Few patients have been treated to date, but the completion of the trials will undoubtedly be very informative with regards to the safety of allospecific Tregs and provide possible clues on how well they compare against treatment with polyclonal Tregs.
In recent years, the transduction of chimeric antigen receptors (CAR) on T cells has shown great promise in the field of cancer cell therapy, particularly for B cell lymphomas where there is a clear target antigen such as the B lymphocyte antigens, CD20, and CD19. This has paved the way for its potential use in Tregs. We and others have shown that the expression of CAR in Tregs can potentially be used for the treatment of xeno-GVHD and allo-graft rejection (60–62). In a human skin xenograft transplant model, the adoptive transfer of CAR Tregs were more effective at alleviating the alloimmune-mediated skin injury caused by transferring allogeneic PBMCs compared to polyclonal Tregs. Recently, the US Food and Drug administration approved two CAR T cell therapies, the first one, Kymriah (Tisagenlecleucel) was developed for the treatment of patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia and Yescarta (axicabtagene ciloleucel) for the treatment of adults with refractory large B cell lymphomas. These approvals signify an important development and no doubt will pave the way for continued commercialization of cell therapies.
A key breakthrough in the translational potential of Treg cell therapy was the demonstration that human Tregs could be successfully isolated and expanded ex vivo while maintaining immunoregulatory function. This has enabled the application of these cells in the clinic, leading to Treg adoptive transfer in phase I clinical trials of bone marrow transplantation and type I diabetes (63–66) and more recently in the setting of solid organ transplantation (67, 68). The success of these trials is reliant on a highly reproducible process for the sustained manufacture of autologous patient-derived Tregs. In the setting of solid organ transplantation, we are faced with the challenge of a more targeted approach to suppress the immune system, and as such efforts have focused on the expansion of allo-antigen specific Tregs for cell therapy application. Here, we have highlighted our experience to date. However, whether a generalized effect of immunosuppression by the adoptive transfer of polyclonal Tregs could potentially be diminished by more targeted Treg therapy requires further investigation. The major drawback of the two phase I clinical trials completed by us is that the isolation technique for regulatory T cells relies on first generation magnetic bead isolation. The inability of this technique to select cells based on stricter criteria (CD25hi) or multiple parameters (low expression of CD127) has led to the development of a GMP compliant FACS cell sorter (MACSquant Tyto cell sorter, Miltenyi Biotech). The validation of the MACSQuant Tyto and its GMP accreditation has now meant that we can isolate Tregs based on several markers, further enhancing the purity and quality of the infused product. The high level of purity achieved with the MACSQuant Tyto will allow us to generate donor-specific Tregs either by using donor-derived APC or by transduction with CAR. Finally, the selection of the most favorable Treg population, which will give the best therapeutic advantage, will likely be further enhanced by the advent of new technologies.
With scientific knowledge and technology rapidly advancing in the field, the future of Treg cell therapy is set to only progress further. As such, our ultimate aim of immune tolerance in transplantation is soon to become a reality.
GL, NS, and NG contributed to the conception, design, writing, and revision of the manuscript. ST, CS, LF, TV, and RL contributed to the writing, and revision of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank all the research subjects and patients who are currently taking part in the ONE study and ThRIL. A special thank you to the GMP scientists, head of GMP production and head of GMP quality at Guy’s Hospital. This research is supported by the King’s Health Partners Research and Development Challenge Fund, Guy’s and St Thomas’ Charity (grant no. R1405170), British Heart Foundation, ONE Study Consortium, the European Cooperation in Science and Technology (COST; part of the EU Framework Programme Horizon 2020) for the AFACTT project (Action to Focus and Accelerate Cell-based Tolerance inducing Therapies; BM1305), the Medical Research Council (MRC), within the MRC Centre for Transplantation, King’s College London, UK—MRC grant no. MR/J006742/1 and the National Institute for Health Research (NIHR) Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
1. Feng S. Long-term management of immunosuppression after pediatric liver transplantation: is minimization or withdrawal desirable or possible or both? Curr Opin Organ Transplant (2008) 13(5):506–12. doi:10.1097/MOT.0b013e328310b0f7
2. Ojo AO, Held PJ, Port FK, Wolfe RA, Leichtman AB, Young EW, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med (2003) 349(10):931–40. doi:10.1056/NEJMoa021744
3. Textor SC, Taler SJ, Canzanello VJ, Schwartz L, Augustine JE. Posttransplantation hypertension related to calcineurin inhibitors. Liver Transpl (2000) 6(5):521–30. doi:10.1053/jlts.2000.9737
4. Penn I. Cancers complicating organ transplantation. N Engl J Med (1990) 323(25):1767–9. doi:10.1056/NEJM199012203232510
5. Katabathina V, Menias CO, Pickhardt P, Lubner M, Prasad SR. Complications of immunosuppressive therapy in solid organ transplantation. Radiol Clin North Am (2016) 54(2):303–19. doi:10.1016/j.rcl.2015.09.009
6. Benitez C, Londono MC, Miquel R, Manzia TM, Abraldes JG, Lozano JJ, et al. Prospective multicenter clinical trial of immunosuppressive drug withdrawal in stable adult liver transplant recipients. Hepatology (2013) 58(5):1824–35. doi:10.1002/hep.26426
7. Liu XQ, Hu ZQ, Pei YF, Tao R. Clinical operational tolerance in liver transplantation: state-of-the-art perspective and future prospects. Hepatobiliary Pancreat Dis Int (2013) 12(1):12–33. doi:10.1016/S1499-3872(13)60002-8
8. Sagoo P, Perucha E, Sawitzki B, Tomiuk S, Stephens DA, Miqueu P, et al. Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans. J Clin Invest (2010) 120(6):1848–61. doi:10.1172/JCI39922
9. Sanchez-Fueyo A. Hot-topic debate on tolerance: immunosuppression withdrawal. Liver Transpl (2011) 17(Suppl 3):S69–73. doi:10.1002/lt.22421
10. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi:10.1126/scitranslmed.aad4134
11. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203(7):1701–11. doi:10.1084/jem.20060772
12. Povoleri GA, Scotta C, Nova-Lamperti EA, John S, Lombardi G, Afzali B. Thymic versus induced regulatory T cells – who regulates the regulators? Front Immunol (2013) 4:169. doi:10.3389/fimmu.2013.00169
13. Getnet D, Grosso JF, Goldberg MV, Harris TJ, Yen HR, Bruno TC, et al. A role for the transcription factor Helios in human CD4(+)CD25(+) regulatory T cells. Mol Immunol (2010) 47(7–8):1595–600. doi:10.1016/j.molimm.2010.02.001
14. Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, et al. Differences in expression level of Helios and neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+Foxp3+ regulatory T cells. PLoS One (2015) 10(10):e0141161. doi:10.1371/journal.pone.0141161
15. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med (2012) 209(10):1723–42,S1. doi:10.1084/jem.20120914
16. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med (2012) 209(10):S1–19. doi:10.1084/jem.20120822
17. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol (2008) 38(6):1654–63. doi:10.1002/eji.200838105
18. Polansky JK, Schreiber L, Thelemann C, Ludwig L, Kruger M, Baumgrass R, et al. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J Mol Med (2010) 88(10):1029–40. doi:10.1007/s00109-010-0642-1
19. Baron U, Floess S, Wieczorek G, Baumann K, Grutzkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol (2007) 37(9):2378–89. doi:10.1002/eji.200737594
20. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463(7282):808–12. doi:10.1038/nature08750
21. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30(6):899–911. doi:10.1016/j.immuni.2009.03.019
22. Afzali B, Mitchell PJ, Edozie FC, Povoleri GA, Dowson SE, Demandt L, et al. CD161 expression characterizes a subpopulation of human regulatory T cells that produces IL-17 in a STAT3-dependent manner. Eur J Immunol (2013) 43(8):2043–54. doi:10.1002/eji.201243296
23. Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F, et al. Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol (2010) 125(1):222–30.e1–4. doi:10.1016/j.jaci.2009.10.012
24. Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol (2010) 40(8):2174–81. doi:10.1002/eji.200940257
25. Mason GM, Lowe K, Melchiotti R, Ellis R, de Rinaldis E, Peakman M, et al. Phenotypic complexity of the human regulatory T cell compartment revealed by mass cytometry. J Immunol (2015) 195(5):2030–7. doi:10.4049/jimmunol.1500703
26. Kordasti S, Costantini B, Seidl T, Perez Abellan P, Martinez Llordella M, McLornan D, et al. Deep phenotyping of Tregs identifies an immune signature for idiopathic aplastic anemia and predicts response to treatment. Blood (2016) 128(9):1193–205. doi:10.1182/blood-2016-03-703702
27. Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N, et al. An Atlas of human regulatory T helper-like cells reveals features of Th2-like Tregs that support a tumorigenic environment. Cell Rep (2017) 20(3):757–70. doi:10.1016/j.celrep.2017.06.079
28. Vaikunthanathan T, Safinia N, Boardman D, Lechler RI, Lombardi G. Regulatory T cells: tolerance induction in solid organ transplantation. Clin Exp Immunol (2017) 189(2):197–210. doi:10.1111/cei.12978
29. Edozie FC, Nova-Lamperti EA, Povoleri GA, Scotta C, John S, Lombardi G, et al. Regulatory T-cell therapy in the induction of transplant tolerance: the issue of subpopulations. Transplantation (2014) 98(4):370–9. doi:10.1097/TP.0000000000000243
30. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4(10):762–74. doi:10.1038/nri1457
31. Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med (2000) 192(2):295–302. doi:10.1084/jem.192.2.295
32. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332(6029):600–3. doi:10.1126/science.1202947
33. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med (2000) 192(2):303–10. doi:10.1084/jem.192.2.303
34. Bachmann MF, Kohler G, Ecabert B, Mak TW, Kopf M. Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol (1999) 163(3):1128–31.
35. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi:10.1084/jem.20062512
36. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O’Farrelly C, et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol (2009) 183(11):7602–10. doi:10.4049/jimmunol.0901881
37. Gibson DJ, Elliott L, McDermott E, Tosetto M, Keegan D, Byrne K, et al. Heightened expression of CD39 by regulatory T lymphocytes is associated with therapeutic remission in inflammatory bowel disease. Inflamm Bowel Dis (2015) 21(12):2806–14. doi:10.1097/MIB.0000000000000566
38. Smyth LA, Ratnasothy K, Tsang JY, Boardman D, Warley A, Lechler R, et al. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol (2013) 43(9):2430–40. doi:10.1002/eji.201242909
39. Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood (2004) 104(9):2840–8. doi:10.1182/blood-2004-03-0859
40. Wang F, Wan L, Zhang C, Zheng X, Li J, Chen ZK. Tim-3-Galectin-9 pathway involves the suppression induced by CD4+CD25+ regulatory T cells. Immunobiology (2009) 214(5):342–9. doi:10.1016/j.imbio.2008.10.007
41. Wu C, Thalhamer T, Franca RF, Xiao S, Wang C, Hotta C, et al. Galectin-9-CD44 interaction enhances stability and function of adaptive regulatory T cells. Immunity (2014) 41(2):270–82. doi:10.1016/j.immuni.2014.06.011
42. Hara M, Kingsley CI, Niimi M, Read S, Turvey SE, Bushell AR, et al. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol (2001) 166(6):3789–96. doi:10.4049/jimmunol.166.6.3789
43. Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, et al. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med (2005) 201(5):737–46. doi:10.1084/jem.20040685
44. Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J Exp Med (1996) 183(6):2669–74. doi:10.1084/jem.183.6.2669
45. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450(7169):566–9. doi:10.1038/nature06306
46. Tang Q, Lee K. Regulatory T-cell therapy for transplantation: how many cells do we need? Curr Opin Organ Transplant (2012) 17(4):349–54. doi:10.1097/MOT.0b013e328355a992
47. Afzali B, Edozie FC, Fazekasova H, Scotta C, Mitchell PJ, Canavan JB, et al. Comparison of regulatory T cells in hemodialysis patients and healthy controls: implications for cell therapy in transplantation. Clin J Am Soc Nephrol (2013) 8(8):1396–405. doi:10.2215/CJN.12931212
48. Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget (2016) 7(7):7563–77. doi:10.18632/oncotarget.6927
49. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood (2005) 105(12):4743–8. doi:10.1182/blood-2004-10-3932
50. Golovina TN, Mikheeva T, Brusko TM, Blazar BR, Bluestone JA, Riley JL. Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS One (2011) 6(1):e15868. doi:10.1371/journal.pone.0015868
51. Scotta C, Esposito M, Fazekasova H, Fanelli G, Edozie FC, Ali N, et al. Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4(+)CD25(+)FOXP3(+) T regulatory cell subpopulations. Haematologica (2013) 98(8):1291–9. doi:10.3324/haematol.2012.074088
52. Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol (2007) 178(1):320–9. doi:10.4049/jimmunol.178.1.320
53. Peter C, Waldmann H, Cobbold SP. mTOR signalling and metabolic regulation of T cell differentiation. Curr Opin Immunol (2010) 22(5):655–61. doi:10.1016/j.coi.2010.08.010
54. Mucida D, Pino-Lagos K, Kim G, Nowak E, Benson MJ, Kronenberg M, et al. Retinoic acid can directly promote TGF-beta-mediated Foxp3(+) Treg cell conversion of naive T cells. Immunity (2009) 30(4):471–2; author reply 2–3. doi:10.1016/j.immuni.2009.03.008
55. Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, et al. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A (2014) 111(33):E3432–40. doi:10.1073/pnas.1408780111
56. Tsang JY, Tanriver Y, Jiang S, Leung E, Ratnasothy K, Lombardi G, et al. Indefinite mouse heart allograft survival in recipient treated with CD4(+)CD25(+) regulatory T cells with indirect allospecificity and short term immunosuppression. Transpl Immunol (2009) 21(4):203–9. doi:10.1016/j.trim.2009.05.003
57. Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant (2013) 13(11):3010–20. doi:10.1111/ajt.12433
58. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med (2011) 3(83):83ra42. doi:10.1126/scitranslmed.3002076
59. Taylor PA, Friedman TM, Korngold R, Noelle RJ, Blazar BR. Tolerance induction of alloreactive T cells via ex vivo blockade of the CD40:CD40L costimulatory pathway results in the generation of a potent immune regulatory cell. Blood (2002) 99(12):4601–9. doi:10.1182/blood.V99.12.4601
60. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant (2017) 17(4):931–43. doi:10.1111/ajt.14185
61. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126(4):1413–24. doi:10.1172/JCI82771
62. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant (2017) 17(4):917–30. doi:10.1111/ajt.14175
63. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117(3):1061–70. doi:10.1182/blood-2010-07-293795
64. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117(14):3921–8. doi:10.1182/blood-2010-10-311894
65. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets – results of one year follow-up. Clin Immunol (2014) 153(1):23–30. doi:10.1016/j.clim.2014.03.016
66. Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol (2009) 133(1):22–6. doi:10.1016/j.clim.2009.06.001
67. Skaro A, LeFever A, Mathew J, Gallon L, Hie J, Hansen C, et al. Results of a phase 1 trial of Treg adoptive cell transfer (TRACT) in de novo living donor kidney transplant recipients. Am J Transplant (2016) 16(Suppl 3). Available from: http://atcmeetingabstracts.com/abstract/results-of-a-phase-1-trial-of-treg-adoptive-cell-transfer-tract-in-de-novo-living-donor-kidney-transplant-recipients/
68. Chandran S, Tang Q, Sarwal M, Laszik Z, Putnam A, Sigdel T, et al. Infusion of polyclonal Tregs modulates subclinical kidney transplant inflammation: final report of TASK pilot trial. Am J Transplant (2017) 17(Suppl 3). Available from: http://atcmeetingabstracts.com/abstract/infusion-of-polyclonal-tregs-modulates-subclinical-kidney-transplant-inflammation-final-report-of-task-pilot-trial/
Keywords: transplantation, regulatory T cells, clinical trials, good manufacturing practice, cell therapy, technical transfer
Citation: Safinia N, Grageda N, Scottà C, Thirkell S, Fry LJ, Vaikunthanathan T, Lechler RI and Lombardi G (2018) Cell Therapy in Organ Transplantation: Our Experience on the Clinical Translation of Regulatory T Cells. Front. Immunol. 9:354. doi: 10.3389/fimmu.2018.00354
Received: 01 December 2017; Accepted: 07 February 2018;
Published: 26 February 2018
Edited by:
Andrew L. Mellor, Newcastle University, United KingdomReviewed by:
Nick David Jones, University of Birmingham, United KingdomCopyright: © 2018 Safinia, Grageda, Scottà, Thirkell, Fry, Vaikunthanathan, Lechler and Lombardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanna Lombardi, Z2lvdmFubmEubG9tYmFyZGlAa2NsLmFjLnVr
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.