 Soumaya Kouidhi
Soumaya Kouidhi Farhat Ben Ayed3
Farhat Ben Ayed3 Amel Benammar Elgaaied
Amel Benammar Elgaaied- 1Laboratory BVBGR, LR11ES31, Higher Institute of Biotechnology of Sidi Thabet (ISBST), Department of Biotechnology, University of Manouba, Sidi Thabet, Tunisia
- 2Laboratory of Genetics, Immunology and Human Pathology, Faculty of Sciences of Tunis, Department of Biology, University Tunis El Manar, Tunis, Tunisia
- 3Association Tunisienne de Lutte contre le Cancer (ATCC), Tunis, Tunisia
Currently, a marked number of clinical trials on cancer treatment have revealed the success of immunomodulatory therapies based on immune checkpoint inhibitors that activate tumor-specific T cells. However, the therapeutic efficacy of cancer immunotherapies is only restricted to a small fraction of patients. A deeper understanding of key mechanisms generating an immunosuppressive tumor microenvironment (TME) remains a major challenge for more effective antitumor immunity. There is a growing evidence that the TME supports inappropriate metabolic reprogramming that dampens T cell function, and therefore impacts the antitumor immune response and tumor progression. Notably, the immunosuppressive TME is characterized by a lack of crucial carbon sources critical for T cell function and increased inhibitory signals. Here, we summarize the basics of intrinsic and extrinsic metabolic remodeling and metabolic checkpoints underlying the competition between cancer and infiltrating immune cells for nutrients and metabolites. Intriguingly, the upregulation of tumor programmed death-L1 and cytotoxic T lymphocyte-associated antigen 4 alters the metabolic programme of T cells and drives their exhaustion. In this context, targeting both tumor and T cell metabolism can beneficially enhance or temper immunity in an inhospitable microenvironment and markedly improve the success of immunotherapies.
Introduction
Over the past decades, huge efforts have focused on refinement of conventional cancer therapeutic strategies of chemotherapy, radiation, surgery, or targeted therapies. Although all these advances have displayed clear improvement of clinical outcomes for many types of cancers (1–3), their therapeutic efficacy remains unsatisfactory. Since the cells and the molecules of the immune system are a fundamental component of the tumor microenvironment (TME), cancer immunotherapy has emerged as a powerful new therapeutic approach to boost antitumor immunity response (4). Collectively, the immunotherapy principle consists in the modulation of the immune cells activity, predominantly T cells, using adoptive cell transfer, chimeric-antigen receptor T-cells, or monoclonal antibodies (mAbs) (5, 6). The “Checkpoint blockade” that utilizes mAbs specific to cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and the programmed cell death protein 1 pathway (PD-1/PD-L1), is arising as a newer strategy used to fight cancer and one of the most promising immunotherapies (7, 8). Indeed, encouraging results demonstrate unprecedented responses in patients with several types of metastatic tumors that were previously resistant to available treatment options (9–11). While these clinical successes have dramatically harnessed host antitumor immunity and clinical outcomes for patients, there are several limitations for immunotherapy (12). In fact, this approach is confronting a highly immunosuppressive TME and low immunogenicity of cancer cells (13). Moreover, despite the success of immunotherapy, mechanisms that govern anticancer immunity and their relevant biomarkers are still being elucidated. Therefore, the development of new methods to overcome such challenge and to improve the efficacy of this therapy is needed in cancer therapy.
Tumor-infiltrating lymphocytes (TIL) reflect tumor biology and prognostic significance. However, they are challenged with a hostile microenvironment that dampens their function and produces antitumor effects (14). Nevertheless, in the setting of malignancy, multiple mechanisms of immune suppression may exist that prevent effective antitumor immunity (15, 16). Along with negative immunologic regulators called “immune checkpoints,” TIL function is also negatively impacted by a variety of “metabolic checkpoints” (17). Increasing evidence suggests that the deregulation of energy metabolism plays a pivotal role in the inhibition of the antitumor immune response and thereby in tumor progression and metastasis (18). Under a suppressive microenvironment, TIL operate with a metabolic disadvantage since they are subjected to a lack of crucial carbon sources and increased inhibitory signals (19). This may be mainly due to the competition between T cells and tumor cells with deregulated metabolic activities, for limiting nutrients (20). Rapidly dividing tumor cells exhibit complex and dynamic metabolic reprogramming and highly glycolytic level, a phenomenon called the “Warburg effect” and recognized as one of the hallmarks of cancer (21, 22). Thus, tumor cells impede T cell access to nutrients necessary for their activation and generate high levels of lactate. The resulting nutrient scarceness and metabolic waste products accumulation in the TME lead to TIL metabolic switch that impairs their appropriate proliferation and function (23).
Collectively, the cancer cell energetics dictates the metabolic landscape of the TME. Abnormal metabolic activities of cancer cells lead to intratumoral heterogeneity and immunosuppression that could be responsible for the failure of immunotherapy (24, 25). Therefore, a deeper understanding of the metabolic challenges within the TME and their impacts on metabolic fitness of immune cells might contribute the discovery of novel promising approaches to rewire metabolic fitness of TILs that boost existing immunotherapies.
Overlapping Metabolic Profiles of Cancer Cells and T Lymphocytes
Metabolism Impacts T Cell Fate and Activation
T cells fate and activation is closely linked to metabolic reprogramming to acquire effector functions (26). Briefly, Naive CD4+ T cells can differentiate into T helper (Th) subsets or into regulatory T cells Treg, while CD8+ T cells differentiate into effector cytotoxic T lymphocytes (CTLs). Importantly, each T cell functional subset utilizes a distinct metabolic program (27, 28).
Highly proliferative cells increase glucose uptake and undergo upregulated aerobic glycolysis, a critical metabolic pathway for activated T cells (29). In parallel to glucose metabolism, T cell activation also enhances mitochondrial biogenesis and oxidative phosphorylation (OXPHOS) and drives mitochondrial membrane hyperpolarization, amino acid uptake, and glutaminolysis (30). There are several signaling pathways that govern the metabolic reprogramming of activated T cells. The critical checkpoint pathways known to regulate the metabolic switch are mammalian target of rapamycin (mTOR) (31) and adenosine monophosphate-activated protein (AMPK) pathways (32). The phosphoinositide-3-kinase (PI3-kinase)-Akt-mTOR pathway is a central integrator of T cell metabolism to sense and require nutrient availability in order to support high glycolytic rate in proliferating T cells (33). Notably, both activated mTOR complexes mTORC1 and mTORC2 play a role in driving glycolysis (34). Additionally, glycolysis activation is concomitant with the pentose phosphate pathway (PPP) upregulation, necessary to build-up of biochemical intermediates that are necessary for nucleotide, amino acid and fatty acid synthesis. Hypoxia-inducible factor-1α (HIF1α) is a master transcription factor enhanced by mTORC activity, which is monitoring and promoting glycolytic enzymes expression (35).
Also, in response to metabolic stress, AMPK inhibits mTOR signaling and increases catabolic metabolism (36). This results in glycolysis suppression and upregulation of oxidative metabolism and mitochondrial complex 1 activity. AMPK activation promotes generation of Treg, Th1, and Th17 subsets (37).
Metabolism Impacts Tumor Proliferation and Progression
Cancer progression has been recognized for a long time as consequence of multiple genetic events that imply activation of oncogenes and function loss of specific tumor suppressor genes (38, 39). Increasing data point out that this is directly linked to an altered tumor metabolism. Cancer cells exhibit increased glycolysis despite the presence of oxygen, because they must divide rapidly to ensure malignant transformation and tumor development (40, 41). This phenomenon of metabolic reprogramming called “the Warburg effect,” has been recognized as one of the 10 hallmarks of cancer (42). The rate of glycolysis is largely faster than OXPHOS, providing competitive advantages to cancer cells to consume more glucose than surrounding slow-dividing cells and to grow under hypoxia and nutrient deprivation conditions over the TME (43, 44).
Furthermore, glycolysis is an effective metabolic pathway for highly proliferative cancer cells to supply nucleotide, lipid, and amino acid synthesis (45). For instance, increased levels of the glycolysis intermediates provide essential precursors for pivotal anabolic pathways such as the PPP and the serine pathway (46).
It is well established that hypoxia is as a key process supporting glycolysis in tumorigenesis (47). HIF-1α, a transcription factor induced by hypoxia, induces glucose transport by increasing expression of glucose transporters 1–3 along with the transcription of pyruvate dehydrogenase kinase (48). As a result, the tricarboxylic acid cycle is inhibited and several glycolytic enzymes activities are enhanced, including hexokinase 2 (HK2) (49) and lactate dehydrogenase A that converts pyruvate to lactate (50, 51). Therefore, intensive aerobic glycolysis generates high rate of lactate. For instance, the accumulation of lactate in TME results in acidic pH that promotes tumor progression and metastasis and contributes to cancer therapy resistance (52).
While aerobic glycolysis is considered as a key feature in cancer metabolism, clear evidence suggest that mitochondrial metabolism remains functional in most glycolytic cancer cells.
Although most cancer cells rely on aerobic glycolysis, it is clear that a tumor displays considerable heterogeneity in metabolic phenotypes (53). Such intratumorally metabolic heterogeneity may be critical for the failure of therapeutic effects. In fact, recent data has shown that cancer stem-like cells (CSCs) exhibit a distinct metabolism from the rest of tumor cells (54). This CSC metabolism depends on mitochondria function (55, 56). Moreover, the particular metabolic phenotype of CSCs may probably render them resistant to conventional antitumor therapies and explain minimal residual disease (57). Interestingly, encouraging results showed that targeting CSC metabolism (by inhibiting mitochondrial biogenesis) could be an attractive approach to reduce drug resistance (58, 59).
T Cell Impaired Function Under Hostile TME
Metabolic Interplay between Cancer Cells and TIL in TME
The tumor tissue consists of complex sets of cell populations including tumor cells, endothelial cells, T cells, natural killer (NK) cells, macrophages, dendritic cells, fibroblasts, and adipocytes. Regarding its genetic and metabolic diversity, this intricate network of cells contributes to the intratumoral heterogeneity. Tumors exhibit a metabolic shift and shape the TME in such a way to support cancer proliferation and metastasis (17, 60). Yet, this milieu is very hostile for T cells to mediate their antitumor effects because of hypoxia, reduced pH and acidosis, inhibitory signals, competition for nutrients, and waste products accumulation (61, 62).
It is well known that tumor cells like effector T cells (Teff), exhibit intensive aerobic glycolysis that improve their metabolic fitness and provide cell-extrinsic advantage, resulting in competition for vital metabolites such as glucose and amino acids. Therefore, tumor-infiltrating T cells are exposed to nutrient depletion in TME and become dysfunctional (62, 63). Nutrient competition has emerged as one of the major axis of tumor immunosuppression due to the anergy and exhaustion of TILs. Indeed, resources scarceness alters T cell activation and antitumor effector functions tumors through several ways (64). Rapidly dividing tumor cells impede T cell access to glucose essential for T cell metabolic switch and activation. Therefore, glucose depletion enhances AMPK pathways and decreases mTORC1 activity, glycolytic capacity, interferon-γ (IFN-γ) production, and cytolytic activity of T cells (65). This may favor Treg subsets instead of Teff and promote tumor progression. Furthermore, decreased levels of amino acids critical for efficient T cell activation and proliferative responses, can modulate the activity of TILs. Glutamine, arginine, and tryptophan deficiency in TME is immunosuppressive and dampens the proliferation of Teff subset (66).
Moreover, it has been recognized that in addition to consumption of key nutrients, tumors produce large amounts of waste products: lactate, arginine and tryptophan by-products, and phosphoenolpyruvate, that impair T cell metabolism and function and confer worse prognosis for patients (67, 68).
Lactate accumulation due to the use of aerobic glycolysis by cancer cells has been described in TME, accompanied by consequent low pH and acidification of the milieu. In mouse models, lactate levels negatively correlate with markers of T cell activation in melanoma (69). The tumor-derived lactate has positive effects on promoting survival, migration and invasion of cancer cells (70). However, lactate negatively impacts T-cell proliferation and function (71). Such acidic condition increases the expression of proangiogenic factors IL-8 and VEGF, both important involved in cancer metastasis (72). Yet, lactate inhibits the phosphatidylinositol-3 kinase (PI3K)/Akt/mTOR pathway and thus glycolytic metabolism in T cells by abolishing their cytokine production (73). Lactate also impairs the migration of T cells by reducing the chemokine receptors expression. Added to that, lactate has been demonstrated to be preferentially utilized by Tregs since they prefer oxidative metabolism, resulting in T-cell polarization toward a Treg phenotype. Excess of lactate may also regulate macrophage polarization and represses NK cells functions through a restriction of IFN-γ, IL-10, and TGF-beta (74, 75). Hence, the acidic TME has been contemplated as an attractive target for cancer therapy. Interesting results showed that buffering the tumor pH with bicarbonate improved immunotherapy outcomes.
Proliferative cancer cells create a state of tryptophan deprivation in the TME because of their increased demand for tryptophan (76). Indoleamine 2,3-dioxygenase (IDO) is a pivotal enzyme involved in tryptophan catabolism. IDO is also the first enzyme involved in the production of nicotinamid adenine nucleotide. Upregulation of IDO has been demonstrated to be correlated with an increased malignancy (77). In such context, cancer cells express high levels of IDO that deplete tryptophan availability in the TME and consequently impede T cell responses. In addition to its role in cancer cells, expression of IDO has been shown in other cells: endothelial, tumor-associated macrophages, and dendritic cells and was associated with suppression of antitumor Teff response. IDO contributes to tryptophan deprivation and degradation to kynurenine (78). Accumulation of kynurenine in TME has been described in several tumors leading to immunosuppression (79). Moreover, kynurenine is endogenously able to promote Treg cells and to reduce proliferation of Teff (40). Currently, several trials targeting IDO in combination with checkpoint inhibition are under investigation (80).
Crosstalk between Immunologic Checkpoints and T Cell Metabolism
Immune checkpoint regulators are critical to coordinate effective and efficient immune response, to maintain self-tolerance and to prevent the onset of autoimmunity (81). Nevertheless, T cell effector function is correlated with the expression patterns of coinhibitory and costimulatory immune checkpoint receptors (82). The most described checkpoint proteins playing a central role in maintain immune self-tolerance belong to the TNFR superfamily (83) and B7 family (84).
Tumors can evade immune surveillance through defective immune-checkpoint signaling pathways (81, 85). It is now clear that under tumoral context, aberrantly expressed inhibitory checkpoint proteins are described to disrupt antitumor immune response. CTLA-4 and PD1 are critical coinhibitory receptors highly expressed in T cells under TME (86). Moreover, PD-1 ligands PD-L1 and PD-L2 are upregulated by cancer cells and thus disrupt T cells mediated antitumor response (87). Accordingly, immune checkpoints ligand–receptor interactions were proven to be effective targets to enhance antitumor immunity moving immunotherapy into a new era (88). In fact, immune checkpoints blocking antibodies have achieved an outstanding benefit in cancer treatment enabling patients to produce an effective and durable antitumor response. Currently, three checkpoint inhibitors are approved for the treatment of advanced melanomas: ipilimumab, a CTLA-4-specific mAb (89), and pembrolizumab and nivolumab, which are PD-1-specific mAbs (11). Furthermore, remarkable clinical effectiveness has been reported in other cancers such as, ovarian (90) non–small cell lung carcinoma (91), breast (92), prostate (93), and lymphoma (94).
Although the effectiveness of the immune checkpoint blockade in enhancing antitumor immunity by reducing the number and/or the suppressive activity of Tregs and by restoring the activity of Teff has been reported, little is known about mechanisms underlying T-cell activation. Recent evidence suggest that both checkpoint ligation and inhibition may directly modify metabolism of T cells and cancer cells and alter their metabolic feature. Emerging data have shown that PD-1 binding to its ligands impairs the metabolic phenotype of TIL, by inhibiting glycolysis and upregulating fatty acid oxidation (FAO) (95, 96). CTLA-4 ligation to B7 inhibits glycolysis without augmenting FAO, which suggests that CTLA-4 would not affect the metabolic profile of non-stimulated cells (95). Hence, this abrogation of energy generation impacts antitumor response and leads to reduced cytokine secretion and Teff exhaustion (97). Moreover, immune checkpoints also have an impact on cancer cell metabolic reprogramming. Ligation of PD-L1 directly upregulate glycolysis in cancer cells by promoting glucose uptake and production of lactate (98). Hence, signaling through PD-L1benefits cancer cell metabolism, leading to their expansion and survival (61).
Interestingly, the immune checkpoint blockade appears to differentially impact the metabolic profile in TME by favoring T cell activation and in contrast inhibiting cancer cells. Blocking PD-1 and PD-L1 may reduce glycolysis level in cancer cells by inhibiting mTOR pathway (61). Consequently cancer glucose uptake and lactate secretion decrease which restore glucose availability in TME. Besides, the immune checkpoint blockade has a benefit on T cell metabolism and function. A melanoma mice model study showed that tumor treatment with immune checkpoint inhibitors increases glucose rates in TME and enhances T-cell glycolysis and cytotoxic function (99).
In conclusion, clear evidences demonstrated that tumor cell metabolism deeply affects TME differentiation and functions. By modulating tumor cell metabolism, one can control nutrient availability for T cells, thus promoting either their antitumor or immunosuppressive functions.
Targeting Metabolism for Efficient Immunotherapy
Targeting Glucose Metabolism
In tumors, T cell activation and proliferation could be impaired by metabolic disruption, therefore cell metabolism becomes an attractive target to restore anti tumor immunity and to develop anticancer therapy (100). However, in tumoral context, it is wise to consider the overlapping metabolic requirements of tumor and immune cells.
Several drugs have been proposed to target tumor glucose metabolism for cancer treatment. For instance, inhibition of glycolytic enzymes that catalyze several steps of glucose metabolism has been known to support anticancer effects (101). 2-Deoxyglucose (2DG) is a non-metabolizable glucose analog and inhibitor of HK used to shut down glycolysis since the first steps. Despite the safety of this drug in cancer patients and its efficiency beyond glycolysis inhibition in cancer cells (102–104), 2DG has also been shown to impair the metabolism of T cells, which results in decreased secretion of cytokines and reduced T cell antitumor function that may be critical for therapeutic success (105). Dichloroacetate (DCA) is another drug targeting cancer cell metabolism which showed conflicting results. DCA is a metabolic disruptor inducing a shift from glycolysis to OXPHOS and inhibiting growth of tumor cells in vitro (106, 107) and in mouse models (108). Similar to 2DG, DCA is not specific to tumor cell metabolism, therefore, it mediates the same metabolic shift in T cells, favoring Treg formation (109).
The TME is particularly immunosuppressive because of lactic acid production in the extracellular milieu that may stand against the therapeutic efficacy (110). To overcome the “Warburg effect” in cancer cells, some therapeutic approaches target lactate with lactate dehydrogenase (LDH) and monocarboxylate transporter (MCT) inhibitors or oral bicarbonate supplementation to tamper the acidic microenvironment (111). Importantly, the inhibition of LDH, the enzyme that catalyzes the conversion of pyruvate into lactate, shows impaired glycolysis and growth arrest in cancer cells (51, 112). Moreover, lactate blockade improves the response to 5-fluorouracil treatment in colorectal cancer (113). However, LDH inhibition demonstrates contradictory results in proliferating T cells response. While it has been reported that deletion of LDH using small-molecule FX11 or Galloflavin ameliorates lactate levels (114, 115), other studies demonstrate that such inhibition leads to a decrease in T cells IFN-γ production (116). Therefore, the differential impact of LDH inhibitors on cancer and immune cells should be considered when administrated for tumor therapy.
Beside the inhibition of the enzyme LDH, the lactate transporters MCT-1–4 may also be targeted to avoid acidic milieu (117). MCT of the SLC16A gene family influences substrate availability, the metabolic path of lactate and pH balance within the tumor (118). Recent studies have described new MCT disruptors, thalidomide, lenalidomide, and pomalidomide that act on cancer cells to impair the CD147–MCT-1 ligation (119, 120). In addition, the treatment with lenalidomide has been reported to enhance IL-2 and IFN-γ secretion in T cells (121), suggesting that lenalidomide could suppress tumor cell proliferation while favoring T cells activation. Although these drugs cause a loss of cell surface expression of MCT-1, the efficacy may be limited as cancer cells express not only MCT-1 but also MCT-4. Further, AZD3965 another lactate transporter inhibitor, is currently in phase I clinical trials for advanced solid tumors and diffuse large B cell lymphomas (http://www.clinicaltrials.gov/ct2/show/NCT01791595). AZD3965 is targeting MCT-1/MCT-2. Yet, the inhibitory effect has also been observed in T cells (122). Recently, the effect of diclofenac, a non-steroidal anti-inflammatory drug, has been investigated on lactate transport and secretion. Diclofenac has been reported to reduce tumor growth, the number of infiltrating Tregs and the lactate rate in the microenvironment in glioma model (123, 124). Therefore, this result raises the possibility that the application of diclofenac should be feasible to improve the efficacy of immunotherapies.
Further, lactic acid production and resulting low-pH TME are shown to dampen CTLs proliferation and cytotoxic response (125–127). Hence, neutralization of TME may have a meaningful impact on improving the efficacy and outcomes of anticancer immunotherapy therapeutics (128). Emerging data show that buffering lactic acid with bicarbonate or proton pump inhibitor, Esomeprazole improves the pH of TME (129, 130). More importantly, neutralization of TME pH improves outcomes in CTLs and in NK cell mediated anticancer as well. Notably, buffering TME with oral bicarbonate inhibits tumor growth when combined with anti-PD-1 immunotherapy in a melanoma model, and improves survival when combined with adoptive T-cell transfer (131). Altogether, these data indicate that targeting TME acidification by buffering provide a new perspective for immunotherapy outcomes.
The PI3K-AKT-mTOR is an important pathway well known to play a critical role in cancer and immune cell metabolism (31, 132). Further, this pathway has been extensively studied in various cancers showing inappropriate activation supporting tumor growth and survival. Over the last decades, several therapies were developed against mTOR signaling in several solid malignancies (133, 134). Analogs of rapamycin, a drug that inhibits the mTOR signaling, have been approved for the treatment of breast (135), renal (136), and pancreatic cancers (137). An increasing number of studies have reported that inhibition of the mTOR pathway suppresses the glycolytic metabolism and sensitizes tumor cells to chemotherapy (138, 139). Yet, it has been reported that rapamycin can mediate opposite effects on T cells since it broadens Tregs and cytotoxic memory T cells but at the same time decreases Teff proliferation (140). Interestingly, recent evidence suggest that treatment with rapamycin combined with immunotherapy augments cytotoxic and memory T-cell functions in glioblastoma cancer (141). Therefore, rapamycin could be an attractive adjuvant to be used in combination with immunotherapy.
Besides glycolysis, OXPHOS is also a possible target structure in cancer cells. Several reports have described the potential effects of metformin, which is commonly used to treat type II diabetes, as an anticancer drug. Indeed, a large number of retrospective clinical studies and randomized control trials show that metformin prevents tumor growth and improves clinical prognosis in various cancers including lung and prostate cancers (142, 143).
Interestingly, those effects seem to be partially immune-mediated as metformin improved T cell function in vivo (144). Further, metformin has been proposed as a treatment for melanomas due to the limitations of current therapies (145). Metformin is known to target the mitochondrial respiratory complex I and to activate AMPK pathway signal transduction (146, 147). Several reports have demonstrated that AMPK plays pleiotropic and conflicting effects at the interface of cellular metabolism and function (37). In fact, activated AMPK may engender both antitumor and protumor effects in a manner not yet understood (148, 149). Notably, activated AMPK pathway impedes mTOR signaling, and shuts down glycolytic gene expression leading to antiproliferative effects in cancer (150, 151). However, AMPK activation on another side helps cancer cells accommodation to metabolic stresses, which raises their survival (152). Metformin’s AMPK activating effects could also impact T cells behavior mainly by enhancing memory T cells (105, 153) and Treg expansion (154). Therefore, metformin treatment may improve secondary responses. Yet it could favor immunosuppressive Treg cells in TME.
Targeting Amino Acid Catabolism
In the context of the TME, cancer cells require a continuous and high rate of supply of energy to take advantage of their metabolic reprogramming and to avoid immune surveillance. In fact, cancer cells create a state of nutrient deprivation for the T cells and redirect glucose and amino acids for their own advantage. It is well known that l-arginine, tryptophan and glutamine are fundamental in tumor progression and immunity (155). Therefore, targeting theses amino acids in cancer therapy becomes a promising strategy for the development of novel therapeutic agents (156). In fact, many clinical trials are actually testing specific drugs inhibiting amino acid metabolism in cancer cells. Depletion of arginine was assessed using ADI-PEG20 inhibitor (157). It can inhibit cell proliferation in vitro and tumor growth in vivo and decrease Treg accumulation (158). However, it would be more pertinent to prevent amino acid depletion by tumor cells or myeloid cells rather than decreasing amino acid rates in the TME. This approach is currently tested in a clinical trial with CB-1158, an ARG inhibitor, in combination with checkpoint therapy (159).
Furthermore, increasing evidence suggest that tryptophan is critical in supporting oncogenic signature and in maintaining the immunosuppressive phenotype in several cancers (160). Interestingly, it has been reported that the silencing of IDO boosted antitumor immunity in metastatic liver tumor model (161), improved cytotoxic T cell function and decreased Treg numbers (162). Accordingly, it is well established that IDO is a key target of drug discovery in cancer immunotherapy (80, 163). Imatinib is another drug displaying improved anti tumor immunity by activating T effector cells and suppressing Tregs, in a manner dependent on IDO pathway (164). For instance, a current clinical trial is assessing the combination between imatinib and anti-CTLA4 approach in GIST (165).
Glutamine is considered as a critical amino acid for cancer cell metabolism as well as for rapidly dividing T cells. To overcome the high glutamine consumption rates of cancer cells, several therapeutic agents targeting glutamine metabolism have been explored in preclinical studies (166). Three compounds were assessed as glutamine analogs, 6-diazo-5-oxo-L-norleucine, azaserine, and acivicin. These agents showed impaired activity of enzymes utilizing glutamine in many tumor models (167, 168). Moreover, testing glutamine transporter inhibitors gamma-l-glutamyl-p-nitroanilide and benzylserine [H-Ser(Bzl)-OH], showed reduced glutamine uptake and cell growth in lung and prostate cancers (169, 170). Yet, glutamine plays also a key role in normal Teff. Therefore, it is conceivable to consider better tumor-targeting options under the TME.
Concluding Remarks
Cancer immunotherapy provides successful and powerful opportunity in cancer treatment. However, it is important to get comprehensive understanding of mechanisms leading to reduced antitumor immunity under hostile TME. Importantly, TILs have to surpass not only immune checkpoints but also a wide range of metabolic checkpoints that fate their energetic behavior defects and dampen their function. In fact, cancer cells upregulate nutrients uptake and waste metabolites production to generate an immunosuppressive TME that allows their evasion and growth, and that dictates immune cell fate (Figure 1A). Increasing emerging data point out the modulation of cellular metabolism, using combinational approaches of metabolic disruptors with immune checkpoint blockade (Figure 1B). However, a special attention should be devoted to target specific tumor site, in order to avoid systemic toxicity and innumerable other side effects. In summary, by operating through distinct and complementary mechanisms, these new therapeutic strategies might reinvigorate TILs by restoring their metabolic properties and improving the efficacy of immunotherapies.
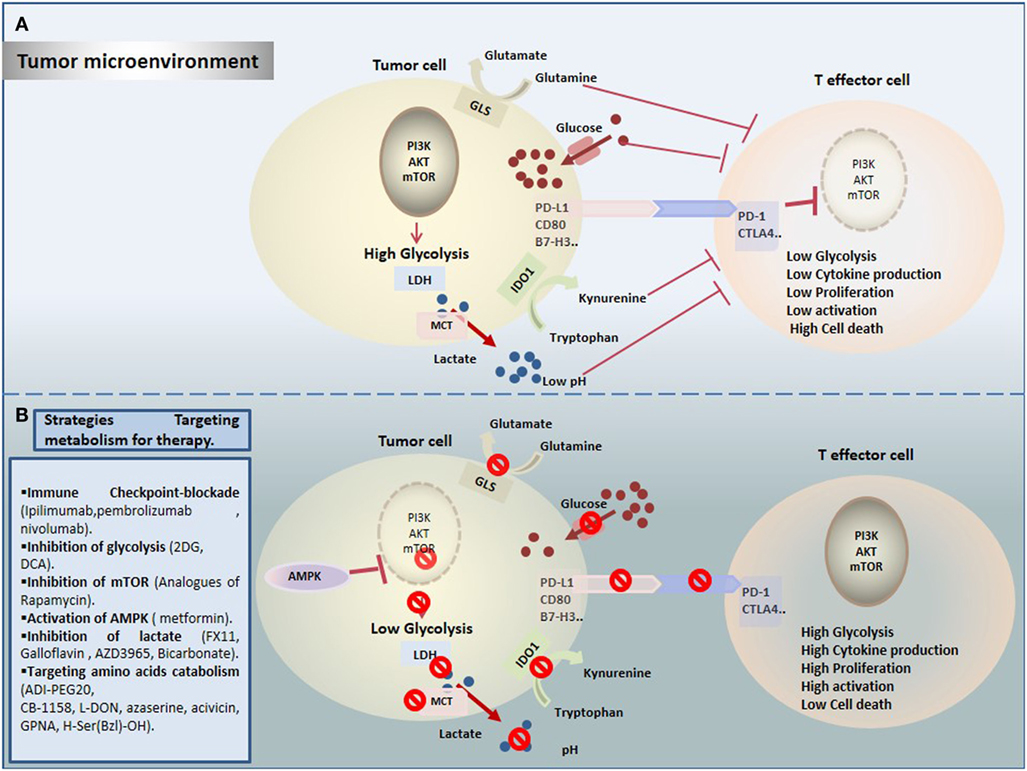
Figure 1. Therapeutic targeting cell metabolism in the tumor microenvironment (TME). (A) Tumor cells create a hostile TME that affects metabolic fitness of T cells through multiple ways. T cells are challenged by different immunologic and metabolic checkpoints: Glucose and amino acid depletion, high acidity and lactate, and upregulation of immune checkpoints influence T cell metabolism to suppress glycolysis thereby reducing their activation and proliferation. (B) Currently, several novel promising approaches are proposed to rewire metabolic fitness of T cells in the TME and to boost existing immunotherapies.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
2DG, 2-deoxyglucose; ACT, adoptive cell transfer; AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate; CAR, chimeric-antigen receptor; CTLs, cytolytic T cells; CTLA-4, cytotoxic T-lymphocyte antigen; FAO, fatty acid oxidation; HIF, hypoxia-inducible factor; IDO, indoleamine 2,3-dioxygenase; IFN-γ, interferon-γ; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; mTOR, mammalian target of rapamycin; OXPHOS, oxidative phosphorylation; PD-1, programmed death-1; PD-L1, programmed death ligand-1; PI3K, phosphatidylinositol-3 kinase; PPP, pentose phosphate pathway; TCA, tricarboxylic acid; Teff, effector T cells; Th, helper T cells; TIL, tumor-infiltrating lymphocytes; TME, Tumor microenvironment; Treg, regulatory T cells.
References
1. Palumbo MO, Kavan P, Miller WH, Panasci L, Assouline S, Johnson N, et al. Systemic cancer therapy: achievements and challenges that lie ahead. Front Pharmacol (2013) 4:57. doi:10.3389/fphar.2013.00057
2. Chabner BA, Roberts TG. Timeline: chemotherapy and the war on cancer. Nat Rev Cancer (2005) 5(1):65–72. doi:10.1038/nrc1529
3. DeVita VT, Chu E. A history of cancer chemotherapy. Cancer Res (2008) 68(21):8643–53. doi:10.1158/0008-5472.CAN-07-6611
4. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer (2011) 11(11):805–12. doi:10.1038/nrc3153
5. Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O, Samara P. Harnessing the immune system to improve cancer therapy. Ann Transl Med (2016) 4(14):261. doi:10.21037/atm.2016.04.01
6. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi:10.1158/1078-0432.CCR-11-0116
7. Zavala VA, Kalergis AM. New clinical advances in immunotherapy for the treatment of solid tumours. Immunology (2015) 145(2):182–201. doi:10.1111/imm.12459
8. Schwab CL, English DP, Roque DM, Pasternak M, Santin AD. Past, present and future targets for immunotherapy in ovarian cancer. Immunotherapy (2014) 6(12):1279–93. doi:10.2217/imt.14.90
9. Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther (2015) 37(4):764–82. doi:10.1016/j.clinthera.2015.02.018
10. Lee JH, Lyle M, Menzies AM, Chan MM, Lo S, Clements A, et al. Metastasis-specific patterns of response and progression with anti-PD-1 treatment in metastatic melanoma. Pigment Cell Melanoma Res (2017). doi:10.1111/pcmr.12675
11. Ivashko IN, Kolesar JM. Pembrolizumab and nivolumab: PD-1 inhibitors for advanced melanoma. Am J Health Syst Pharm (2016) 73(4):193–201. doi:10.2146/ajhp140768
12. Hwang SJE, Fernández-Peñas P. Adverse reactions to biologics: melanoma (Ipilimumab, Nivolumab, Pembrolizumab). Curr Probl Dermatol (2018) 53:82–92. doi:10.1159/000478081
13. Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab (2017) 26(1):94–109. doi:10.1016/j.cmet.2017.06.016
14. Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity (2013) 39(1):11–26. doi:10.1016/j.immuni.2013.07.008
15. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi:10.1016/j.immuni.2013.07.012
16. Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity (2013) 39(1):61–73. doi:10.1016/j.immuni.2013.07.005
17. Scharping NE, Delgoffe GM. Tumor microenvironment metabolism: a new checkpoint for anti-tumor immunity. Vaccines (Basel) (2016) 4(4):46. doi:10.3390/vaccines4040046
18. Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol (2017) 14(1):11–31. doi:10.1038/nrclinonc.2016.60
19. Kouidhi S, Elgaaied AB, Chouaib S. Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front Immunol (2017) 8:270. doi:10.3389/fimmu.2017.00270
20. Eikawa S, Udono H. [Metabolic competition in tumor microenvironment]. Gan To Kagaku Ryoho (2017) 44(11):972–6.
21. Chiavarina B, Whitaker-Menezes D, Martinez-Outschoorn UE, Witkiewicz AK, Birbe R, Howell A, et al. Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol Ther (2011) 12(12):1101–13. doi:10.4161/cbt.12.12.18703
22. Kareva I, Hahnfeldt P. The emerging «Hallmarks» of metabolic reprogramming and immune evasion: distinct or linked? Cancer Res (2013) 73(9):2737–42. doi:10.1158/0008-5472.CAN-12-3696
23. Ruffell B, DeNardo DG, Affara NI, Coussens LM. Lymphocytes in cancer development: polarization towards pro-tumor immunity. Cytokine Growth Factor Rev (2010) 21(1):3–10. doi:10.1016/j.cytogfr.2009.11.002
24. Diaz-Cano SJ. Tumor heterogeneity: mechanisms and bases for a reliable application of molecular marker design. Int J Mol Sci (2012) 13(2):1951–2011. doi:10.3390/ijms13021951
25. Kleppe M, Levine RL. Tumor heterogeneity confounds and illuminates: assessing the implications. Nat Med (2014) 20(4):342–4. doi:10.1038/nm.3522
26. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13(10):907. doi:10.1038/ni.2386
27. Delgoffe GM, Powell JD. Feeding an army: the metabolism of T cells in activation, anergy, and exhaustion. Mol Immunol (2015) 68(2 Pt 100):492–6. doi:10.1016/j.molimm.2015.07.026
28. Bantug GR, Galluzzi L, Kroemer G, Hess C. The spectrum of T cell meta-bolism in health and disease. Nat Rev Immunol (2017) 8(1):19–34. doi:10.1038/nri.2017.99
29. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol (2015) 6:1. doi:10.3389/fimmu.2015.00001
30. Araujo L, Khim P, Mkhikian H, Mortales C-L, Demetriou M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. eLife (2017). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5257256/
31. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 168(6):960–76. doi:10.1016/j.cell.2017.02.004
32. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42(1):41–54. doi:10.1016/j.immuni.2014.12.030
33. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249(1):43–58. doi:10.1111/j.1600-065X.2012.01152.x
34. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity (2016) 45(3):540–54. doi:10.1016/j.immuni.2016.08.017
35. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer (2008) 8(11):851. doi:10.1038/nrc2501
36. Gwinn DM, Shaw RJ. AMPK control of mTOR signaling and growth. (2010) 28:49–75. doi:10.1016/S1874-6047(10)28003-4
37. Ma EH, Poffenberger MC, Wong AH-T, Jones RG. The role of AMPK in T cell metabolism and function. Curr Opin Immunol (2017) 46:45–52. doi:10.1016/j.coi.2017.04.004
38. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. The Molecular Basis of Cancer-Cell Behavior. (2002). Available from: https://www.ncbi.nlm.nih.gov/books/NBK26902/
39. Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Proto-Oncogenes and Tumor-Suppressor Genes. (2000). Available from: https://www.ncbi.nlm.nih.gov/books/NBK21662/
40. Routy J-P, Routy B, Graziani GM, Mehraj V. The kynurenine pathway is a double-edged sword in immune-privileged sites and in cancer: implications for immunotherapy. Int J Tryptophan Res (2016) 9:67–77. doi:10.4137/IJTR.S38355
41. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi:10.1126/science.1160809
42. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell (2012) 21(3):297–308. doi:10.1016/j.ccr.2012.02.014
43. Singh D, Arora R, Kaur P, Singh B, Mannan R, Arora S. Overexpression of hypoxia-inducible factor and metabolic pathways: possible targets of cancer. Cell Biosci (2017) 7:62. doi:10.1186/s13578-017-0190-2
44. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science (2009) 325(5947):1555–9. doi:10.1126/science.1174229
45. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2(5). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4928883/
46. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi:10.1146/annurev-cellbio-092910-154237
47. Guillaumond F, Leca J, Olivares O, Lavaut M-N, Vidal N, Berthezène P, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A (2013) 110(10):3919–24. doi:10.1073/pnas.1219555110
48. Zhao H, Jiang H, Li Z, Zhuang Y, Liu Y, Zhou S, et al. 2-Methoxyestradiol enhances radiosensitivity in radioresistant melanoma MDA-MB-435R cells by regulating glycolysis via HIF-1α/PDK1 axis. Int J Oncol (2017) 50(5):1531–40. doi:10.3892/ijo.2017.3924
49. Nakashima RA, Paggi MG, Scott LJ, Pedersen PL. Purification and characterization of a bindable form of mitochondrial bound hexokinase from the highly glycolytic AS-30D rat hepatoma cell line. Cancer Res (1988) 48(4):913–9.
50. Parra-Bonilla G, Alvarez DF, Al-Mehdi A-B, Alexeyev M, Stevens T. Critical role for lactate dehydrogenase A in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. Am J Physiol Lung Cell Mol Physiol (2010) 299(4):L513–22. doi:10.1152/ajplung.00274.2009
51. Fiume L, Manerba M, Vettraino M, Di Stefano G. Impairment of aerobic glycolysis by inhibitors of lactic dehydrogenase hinders the growth of human hepatocellular carcinoma cell lines. Pharmacology (2010) 86(3):157–62. doi:10.1159/000317519
52. Hirschhaeuser F, Sattler UGA, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res (2011) 71(22):6921–5. doi:10.1158/0008-5472.CAN-11-1457
53. Gentric G, Mieulet V, Mechta-Grigoriou F. Heterogeneity in cancer metabolism: new concepts in an old field. Antioxid Redox Signal (2017) 26(9):462–85. doi:10.1089/ars.2016.6750
54. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer (2016) 114(12):1305–12. doi:10.1038/bjc.2016.152
55. Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res (2016) 18. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4879746/
56. De Luca A, Fiorillo M, Peiris-Pagès M, Ozsvari B, Smith DL, Sanchez-Alvarez R, et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget (2015) 6(17):14777–95. doi:10.18632/oncotarget.4401
57. Vermeersch KA, Wang L, McDonald JF, Styczynski MP. Distinct metabolic responses of an ovarian cancer stem cell line. BMC Syst Biol (2014) 18(8):134. doi:10.1186/s12918-014-0134-y
58. Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond) (2012) 7(4):597–615. doi:10.2217/nnm.12.22
59. Deshmukh A, Deshpande K, Arfuso F, Newsholme P, Dharmarajan A. Cancer stem cell metabolism: a potential target for cancer therapy. Mol Cancer (2016) 15. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5101698/
60. Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris J-H, Noman MZ, et al. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol (2015) 6:482. doi:10.3389/fimmu.2015.00482
61. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi:10.1016/j.cell.2015.08.016
62. Beckermann KE, Dudzinski SO, Rathmell JC. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev (2017) 35(Suppl C):7–14. doi:10.1016/j.cytogfr.2017.04.003
63. Boldajipour B, Nelson A, Krummel MF. Tumor-infiltrating lymphocytes are dynamically desensitized to antigen but are maintained by homeostatic cytokine. JCI Insight (2016) 1(20):e89289. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5135268/
64. Delgoffe GM. Filling the tank: keeping antitumor T cells metabolically fit for the long haul. Cancer Immunol Res (2016) 4(12):1001–6. doi:10.1158/2326-6066.CIR-16-0244
65. Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism. Cancer Biol Ther (2014) 15(2):156–69. doi:10.4161/cbt.26726
66. Nakaya M, Xiao Y, Zhou X, Chang J-H, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40(5):692–705. doi:10.1016/j.immuni.2014.04.007
67. Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi:10.1016/j.cell.2015.08.012
68. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol (2017) 8:248. doi:10.3389/fimmu.2017.00248
69. Gao C, Yan X, Wang B, Yu L, Han J, Li D, et al. Jolkinolide B induces apoptosis and inhibits tumor growth in mouse melanoma B16F10 cells by altering glycolysis. Sci Rep (2016) 6. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5086858/
70. Wang J, Wang H, Liu A, Fang C, Hao J, Wang Z. Lactate dehydrogenase A negatively regulated by miRNAs promotes aerobic glycolysis and is increased in colorectal cancer. Oncotarget (2015) 6(23):19456–68. doi:10.18632/oncotarget.3318
71. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi:10.1182/blood-2006-07-035972
72. Ruan G-X, Kazlauskas A. Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J Biol Chem (2013) 288(29):21161–72. doi:10.1074/jbc.M113.474619
73. Chen JL-Y, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, et al. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet (2008) 4(12):e1000293. doi:10.1371/journal.pgen.1000293
74. Scott KEN, Cleveland JL. Lactate wreaks havoc on tumor-infiltrating T and NK cells. Cell Metab (2016) 24(5):649–50. doi:10.1016/j.cmet.2016.10.015
75. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab (2016) 24(5):657–71. doi:10.1016/j.cmet.2016.08.011
76. Timosenko E, Ghadbane H, Silk JD, Shepherd D, Gileadi U, Howson LJ, et al. Nutritional stress induced by tryptophan-degrading enzymes results in ATF4-dependent reprogramming of the amino acid transporter profile in tumor cells. Cancer Res (2016) 76(21):6193–204. doi:10.1158/0008-5472.CAN-15-3502
77. Silk JD, Lakhal S, Laynes R, Vallius L, Karydis I, Marcea C, et al. IDO induces expression of a novel tryptophan transporter in mouse and human tumor cells. J Immunol (2011) 187(4):1617–25. doi:10.4049/jimmunol.1000815
78. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res (2012) 72(21):5435–40. doi:10.1158/0008-5472.CAN-12-0569
79. Katz JB, Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev (2008) 222:206–21. doi:10.1111/j.1600-065X.2008.00610.x
80. Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol (2015) 5:673. doi:10.3389/fimmu.2014.00673
81. Lim S, Phillips JB, da Silva LM, Zhou M, Fodstad O, Owen LB, et al. Interplay between immune checkpoint proteins and cellular metabolism. Cancer Res (2017) 77(6):1245–9. doi:10.1158/0008-5472.CAN-16-1647
82. Cogdill AP, Andrews MC, Wargo JA. Hallmarks of response to immune checkpoint blockade. Br J Cancer (2017) 117(1):1–7. doi:10.1038/bjc.2017.136
83. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell (2001) 104(4):487–501. doi:10.1016/S0092-8674(01)00237-9
84. Collins M, Ling V, Carreno BM. The B7 family of immune-regulatory ligands. Genome Biol (2005) 6(6):223. doi:10.1186/gb-2005-6-6-223
85. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res (2015) 21(4):687–92. doi:10.1158/1078-0432.CCR-14-1860
86. Dong Y, Sun Q, Zhang X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget (2016) 8(2):2171–86. doi:10.18632/oncotarget.13895
87. Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother (2005) 54(4):307–14. doi:10.1007/s00262-004-0593-x
88. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol (2017) 8:561. doi:10.3389/fphar.2017.00561
89. Sachpekidis C, Hassel JC, Dimitrakopoulou-Strauss A. Metastatic melanoma response to combination therapy with ipilimumab and vemurafenib. Hell J Nucl Med (2017) 20(3):251–3. doi:10.1967/s002449910611
90. Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res (2013) 73(23):6900–12. doi:10.1158/0008-5472.CAN-13-1550
91. Ramos-Esquivel A, van der Laat A, Rojas-Vigott R, Juárez M, Corrales-Rodríguez L. Anti-PD-1/anti-PD-L1 immunotherapy versus docetaxel for previously treated advanced non-small cell lung cancer: a systematic review and meta-analysis of randomised clinical trials. ESMO Open (2017) 2(3):e000236. doi:10.1136/esmoopen-2017-000236
92. Jia H, Truica CI, Wang B, Wang Y, Ren X, Harvey HA, et al. Immunotherapy for triple-negative breast cancer: existing challenges and exciting prospects. Drug Resist Updat (2017) 32:1–15. doi:10.1016/j.drup.2017.07.002
93. Thompson RH, Allison JP, Kwon ED. Anti-cytotoxic T lymphocyte antigen-4 (CTLA-4) immunotherapy for the treatment of prostate cancer. Urol Oncol (2006) 24(5):442–7. doi:10.1016/j.urolonc.2005.08.011
94. Montico B, Lapenta C, Ravo M, Martorelli D, Muraro E, Zeng B, et al. Exploiting a new strategy to induce immunogenic cell death to improve dendritic cell-based vaccines for lymphoma immunotherapy. Oncoimmunology (2017) 6(11):e1356964. doi:10.1080/2162402X.2017.1356964
95. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4389235/
96. Qorraj M, Böttcher M, Mougiakakos D. PD-L1/PD-1: new kid on the «immune metabolic» block. Oncotarget (2017) 8(43):73364–5. doi:10.18632/oncotarget.20639
97. Saunders PA, Hendrycks VR, Lidinsky WA, Woods ML. PD-L2:PD-1 involvement in T cell proliferation, cytokine production, and integrin-mediated adhesion. Eur J Immunol (2005) 35(12):3561–9. doi:10.1002/eji.200526347
98. Wangpaichitr M, Kandemir H, Li YY, Wu C, Nguyen D, Feun LG, et al. Relationship of metabolic alterations and PD-L1 expression in cisplatin resistant lung cancer. Cell Dev Biol (2017) 6(2):183. doi:10.4172/2168-9296.1000183
99. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol (2016) 17(1):95–103. doi:10.1038/ni.3313
100. Obre E, Rossignol R. Emerging concepts in bioenergetics and cancer research: metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int J Biochem Cell Biol (2015) 59:167–81. doi:10.1016/j.biocel.2014.12.008
101. Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle (2010) 9(10):1960–71. doi:10.4161/cc.9.10.11601
102. Gu L, Yi Z, Zhang Y, Ma Z, Zhu Y, Gao J. Low dose of 2-deoxy-D-glucose kills acute lymphoblastic leukemia cells and reverses glucocorticoid resistance via N-linked glycosylation inhibition under normoxia. Oncotarget (2017) 8(19):30978–91. doi:10.18632/oncotarget.16046
103. Mühlenberg T, Grunewald S, Treckmann J, Podleska L, Schuler M, Fletcher JA, et al. Inhibition of KIT-glycosylation by 2-deoxyglucose abrogates KIT-signaling and combination with ABT-263 synergistically induces apoptosis in gastrointestinal stromal tumor. PLoS One (2015) 10(3). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4364009/
104. Stein M, Lin H, Jeyamohan C, Dvorzhinski D, Gounder M, Bray K, et al. Targetingtumor metabolismwith 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate (2010) 70(13):1388–94. doi:10.1002/pros.21172
105. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi:10.1172/JCI69589
106. Vella S, Conti M, Tasso R, Cancedda R, Pagano A. Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells. Int J Cancer (2012) 130(7):1484–93. doi:10.1002/ijc.26173
107. Tong J, Xie G, He J, Li J, Pan F, Liang H. Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. J Biomed Biotechnol (2011) 2011:740564. doi:10.1155/2011/740564
108. Zheng M-F, Shen S, Huang W. DCA increases the antitumor effects of capecitabine in a mouse B16 melanoma allograft and a human non-small cell lung cancer A549 xenograft. Cancer Chemother Pharmacol (2013) 72(5):1031–41. doi:10.1007/s00280-013-2281-z
109. Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Antoniadis N, Liakopoulos V, et al. Dichloroacetate at therapeutic concentration alters glucose metabolism and induces regulatory T-cell differentiation in alloreactive human lymphocytes. J Basic Clin Physiol Pharmacol (2013) 24(4):271–6. doi:10.1515/jbcpp-2013-0001
110. McCarty MF, Whitaker J. Manipulating tumor acidification as a cancer treatment strategy. Altern Med Rev (2010) 15(3):264–72.
111. Peppicelli S, Bianchini F, Calorini L. Extracellular acidity, a «reappreciated» trait of tumor environment driving malignancy: perspectives in diagnosis and therapy. Cancer Metastasis Rev (2014) 33(2–3):823–32. doi:10.1007/s10555-014-9506-4
112. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A (2010) 107(5):2037–42. doi:10.1073/pnas.0914433107
113. Li X, Zhao H, Zhou X, Song L. Inhibition of lactate dehydrogenase A by microRNA-34a resensitizes colon cancer cells to 5-fluorouracil. Mol Med Rep (2015) 11(1):577–82. doi:10.3892/mmr.2014.2726
114. Granchi C, Paterni I, Rani R, Minutolo F. Small-molecule inhibitors of human LDH5. Future Med Chem (2013) 5(16):1967–91. doi:10.4155/fmc.13.151
115. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest (2013) 123(9):3685–92. doi:10.1172/JCI69741
116. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. Cancer acidity: an ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol (2017) 43(Suppl C):74–89. doi:10.1016/j.semcancer.2017.03.001
117. Hong CS, Graham NA, Gu W, Espindola Camacho C, Mah V, Maresh EL, et al. MCT1 modulates cancer cell pyruvate export and growth of tumors that co-express MCT1 and MCT4. Cell Rep (2016) 14(7):1590–601. doi:10.1016/j.celrep.2016.01.057
118. Kennedy KM, Dewhirst MW. Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol (2010) 6(1):127. doi:10.2217/fon.09.145
119. Pinheiro C, Reis RM, Ricardo S, Longatto-Filho A, Schmitt F, Baltazar F. Expression of monocarboxylate transporters 1, 2, and 4 in human tumours and their association with CD147 and CD44. J Biomed Biotechnol (2010). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2863082/
120. Eichner R, Heider M, Fernández-Sáiz V, van Bebber F, Garz A-K, Lemeer S, et al. Immunomodulatory drugs disrupt the cereblon-CD147-MCT1 axis to exert antitumor activity and teratogenicity. Nat Med (2016) 22(7):735–43. doi:10.1038/nm.4128
121. Görgün G, Calabrese E, Soydan E, Hideshima T, Perrone G, Bandi M, et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood (2010) 116(17):3227–37. doi:10.1182/blood-2010-04-279893
122. Bola BM, Chadwick AL, Michopoulos F, Blount KG, Telfer BA, Williams KJ, et al. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol Cancer Ther (2014) 13(12):2805–16. doi:10.1158/1535-7163.MCT-13-1091
123. Chirasani SR, Leukel P, Gottfried E, Hochrein J, Stadler K, Neumann B, et al. Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model. Int J Cancer (2013) 132(4):843–53. doi:10.1002/ijc.27712
124. Pantziarka P, Sukhatme V, Bouche G, Meheus L, Sukhatme VP. Repurposing Drugs in Oncology (ReDO)—diclofenac as an anti-cancer agent. Ecancermedicalscience (2016) 10. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4720497/
125. Nakagawa Y, Negishi Y, Shimizu M, Takahashi M, Ichikawa M, Takahashi H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol Lett (2015) 167(2):72–86. doi:10.1016/j.imlet.2015.07.003
126. Bosticardo M, Ariotti S, Losana G, Bernabei P, Forni G, Novelli F. Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur J Immunol (2001) 31(9):2829–38. doi:10.1002/1521-4141(200109)31:9<2829::AID-IMMU2829>3.0.CO;2-U
127. Romero-Garcia S, Moreno-Altamirano MMB, Prado-Garcia H, Sánchez-García FJ. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol (2016) 7:52. doi:10.3389/fimmu.2016.00052
128. Choi SYC, Collins CC, Gout PW, Wang Y. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol (2013) 230(4):350–5. doi:10.1002/path.4218
129. Fais S. Proton pump inhibitor-induced tumour cell death by inhibition of a detoxification mechanism. J Intern Med (2010) 267(5):515–25. doi:10.1111/j.1365-2796.2010.02225.x
130. Koltai T. Cancer: fundamentals behind pH targeting and the double-edged approach. Onco Targets Ther (2016) 9:6343–60. doi:10.2147/OTT.S115438
131. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res (2016) 76(6):1381–90. doi:10.1158/0008-5472.CAN-15-1743
132. Zeng H. mTOR signaling in immune cells and its implications for cancer immunotherapy. Cancer Lett (2017) 1(408):182–9. doi:10.1016/j.canlet.2017.08.038
133. Wang X-Y, Zhang X-H, Peng L, Liu Z, Yang Y-X, He Z-X, et al. Bardoxolone methyl (CDDO-Me or RTA402) induces cell cycle arrest, apoptosis and autophagy via PI3K/Akt/mTOR and p38 MAPK/Erk1/2 signaling pathways in K562 cells. Am J Transl Res (2017) 9(10):4652–72.
134. Liu W, Wang X, Liu Z, Wang Y, Yin B, Yu P, et al. SGK1 inhibition induces autophagy-dependent apoptosis via the mTOR-Foxo3a pathway. Br J Cancer (2017) 117(8):1139–53. doi:10.1038/bjc.2017.293
135. Woo S-U, Sangai T, Akcakanat A, Chen H, Wei C, Meric-Bernstam F. Vertical inhibition of the PI3K/Akt/mTOR pathway is synergistic in breast cancer. Oncogenesis (2017) 6(10):e385. doi:10.1038/oncsis.2017.86
136. Deng F, Ma Y-X, Liang L, Zhang P, Feng J. The pro-apoptosis effect of sinomenine in renal carcinoma via inducing autophagy through inactivating PI3K/AKT/mTOR pathway. Biomed Pharmacother (2017) 13(97):1269–74. doi:10.1016/j.biopha.2017.11.064
137. Schmidt KM, Hellerbrand C, Ruemmele P, Michalski CW, Kong B, Kroemer A, et al. Inhibition of mTORC2 component RICTOR impairs tumor growth in pancreatic cancer models. Oncotarget (2017) 8(15):24491–505. doi:10.18632/oncotarget.15524
138. Chi K-H, Wang Y-S, Huang Y-C, Chiang H-C, Chi M-S, Chi C-H, et al. Simultaneous activation and inhibition of autophagy sensitizes cancer cells to chemotherapy. Oncotarget (2016) 7(36):58075–88. doi:10.18632/oncotarget.10873
139. Huang S, Yang ZJ, Yu C, Sinicrope FA. Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J Biol Chem (2011) 286(46):40002–12. doi:10.1074/jbc.M111.297432
140. Chi H. Regulation and function of mTOR signalling in T cell fate decision. Nat Rev Immunol (2012) 12(5):325–38. doi:10.1038/nri3198
141. Mineharu Y, Kamran N, Lowenstein PR, Castro MG. Blockade of mTOR signaling via rapamycin combined with immunotherapy augments antiglioma cytotoxic and memory T-cell functions. Mol Cancer Ther (2014) 13(12):3024–36. doi:10.1158/1535-7163.MCT-14-0400
142. Whitburn J, Edwards CM, Sooriakumaran P. Metformin and prostate cancer: a new role for an old drug. Curr Urol Rep (2017) 18(6). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5405102/
143. Sakoda LC, Ferrara A, Achacoso NS, Peng T, Ehrlich SF, Quesenberry CP, et al. Metformin use and lung cancer risk in patients with diabetes. Cancer Prev Res (Phila) (2015) 8(2):174–9. doi:10.1158/1940-6207.CAPR-14-0291
144. Watanabe M, Eikawa S, Shien K, Yamamoto H, Shien T, Soh J, et al. Abstract 5592: metformin improves immune function of exhausted peripheral CD8+ T cells derived from cancer patients. Cancer Res (2017) 77(13 Suppl):5592–5592. doi:10.1158/1538-7445.AM2017-5592
145. Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis (2011) 2(9):e199. doi:10.1038/cddis.2011.86
146. Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia (2013) 56(9):1898–906. doi:10.1007/s00125-013-2991-0
147. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) (2012) 122(6):253–70. doi:10.1042/CS20110386
148. Fonseca EAI, Oliveira MA, Eichler R, Akamine EH, Carvalho MHC, Tostes RC, et al. Antitumor effect of metformin is mediated by AMPK and FOXO3a. FASEB J (2012) 26(1 Suppl):1038.10. doi:10.1096/fasebj.26.1_supplement.1038.10
149. Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol (2010) 6(3):457–70. doi:10.2217/fon.09.174
150. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab (2013) 17(1):113–24. doi:10.1016/j.cmet.2012.12.001
151. Dandapani M, Hardie DG. AMPK: opposing the metabolic changes in both tumour cells and inflammatory cells? Biochem Soc Trans (2013) 41(2):687–93. doi:10.1042/BST20120351
152. Zhao Y, Hu X, Liu Y, Dong S, Wen Z, He W, et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer (2017) 16. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5390360/
153. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi:10.1038/nature08097
154. Lee S-Y, Lee SH, Yang E-J, Kim E-K, Kim J-K, Shin D-Y, et al. Metformin ameliorates inflammatory bowel disease by suppression of the stat3 signaling pathway and regulation of the between Th17/Treg balance. PLoS One (2015) 10(9):e0135858. doi:10.1371/journal.pone.0135858
155. Lukey MJ, Katt WP, Cerione RA. Targeting amino acid metabolism for cancer therapy. Drug Discov Today (2017) 22(5):796–804. doi:10.1016/j.drudis.2016.12.003
156. Ananieva E. Targeting amino acid metabolism in cancer growth and anti-tumor immune response. World J Biol Chem (2015) 6(4):281–9. doi:10.4331/wjbc.v6.i4.281
157. Kremer JC, Prudner BC, Lange SES, Bean GR, Schultze MB, Brashears CB, et al. Arginine deprivation inhibits the warburg effect and upregulates glutamine anaplerosis and serine biosynthesis in ASS1-deficient cancers. Cell Rep (2017) 18(4):991–1004. doi:10.1016/j.celrep.2016.12.077
158. Brin E, Wu K, Lu H-T, He Y, Dai Z, He W. PEGylated arginine deiminase can modulate tumor immune microenvironment by affecting immune checkpoint expression, decreasing regulatory T cell accumulation and inducing tumor T cell infiltration. Oncotarget (2017) 8(35):58948–63. doi:10.18632/oncotarget.19564
159. Arginase inhibitor INCB001158 as a single agent and in combination with immune checkpoint therapy in patients with advanced/metastatic solid tumors. ClinicalTrials gov (2017). Available from: https://clinicaltrials.gov/ct2/show/NCT02903914
160. van Baren N, Van den Eynde BJ. Tryptophan-degrading enzymes in tumoral immune resistance. Front Immunol (2015) 6:34. doi:10.3389/fimmu.2015.00034
161. Huang T-T, Yen M-C, Lin C-C, Weng T-Y, Chen Y-L, Lin C-M, et al. Skin delivery of short hairpin RNA of indoleamine 2,3 dioxygenase induces antitumor immunity against orthotopic and metastatic liver cancer. Cancer Sci (2011) 102(12):2214–20. doi:10.1111/j.1349-7006.2011.02094.x
162. Yen M-C, Lin C-C, Chen Y-L, Huang S-S, Yang H-J, Chang C-P, et al. A novel cancer therapy by skin delivery of indoleamine 2,3-dioxygenase siRNA. Clin Cancer Res (2009) 15(2):641–9. doi:10.1158/1078-0432.CCR-08-1988
163. Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular pathways: targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res (2015) 21(24):5427–33. doi:10.1158/1078-0432.CCR-15-0420
164. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates anti-tumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med (2011) 17(9):1094–100. doi:10.1038/nm.2438
165. Reilley MJ, Bailey A, Subbiah V, Janku F, Naing A, Falchook G, et al. Phase I clinical trial of combination imatinib and ipilimumab in patients with advanced malignancies. J Immunother Cancer (2017) 5:35. doi:10.1186/s40425-017-0238-1
166. Altman BJ, Stine ZE, Dang CV. From krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(10):619–34. doi:10.1038/nrc.2016.71
167. Lynch G, Kemeny N, Casper E. Phase II evaluation of DON (6-diazo-5-oxo-L-norleucine) in patients with advanced colorectal carcinoma. Am J Clin Oncol (1982) 5(5):541–3. doi:10.1097/00000421-198210000-00014
168. Ahluwalia GS, Grem JL, Hao Z, Cooney DA. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther (1990) 46(2):243–71. doi:10.1016/0163-7258(90)90094-I
169. van der Mijn JC, Panka DJ, Geissler AK, Verheul HM, Mier JW. Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab (2016) 4. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4944519/
Keywords: T-lymphocyte metabolism, tumor cell metabolism, tumor microenvironment, immunotherapy, immune checkpoints, metabolic checkpoints
Citation: Kouidhi S, Ben Ayed F and Benammar Elgaaied A (2018) Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front. Immunol. 9:353. doi: 10.3389/fimmu.2018.00353
Received: 30 November 2017; Accepted: 07 February 2018;
Published: 23 February 2018
Edited by:
Salem Chouaib, Institut Gustave Roussy, FranceReviewed by:
Graham Robert Leggatt, The University of Queensland, AustraliaViktor Umansky, Deutsches Krebsforschungszentrum (DKFZ), Germany
Copyright: © 2018 Kouidhi, Ben Ayed and Benammar Elgaaied. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Soumaya Kouidhi, c291bWF5YWtvdWlkaGlAZ21haWwuY29t