
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 26 February 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00312
This article is part of the Research Topic Mediators and Organ Crosstalk in Sterile and Infectious Inflammation View all 26 articles
 Sonja Oehmcke-Hecht*
Sonja Oehmcke-Hecht* Juliane Köhler
Juliane Köhler
The name human contact system is related to its mode of action, as “contact” with artificial negatively charged surfaces triggers its activation. Today, it is generally believed that the contact system is an inflammatory response mechanism not only against artificial material but also against misfolded proteins and foreign organisms. Upon activation, the contact system is involved in at least two distinct (patho)physiologic processes:i. the trigger of the intrinsic coagulation via factor XI and ii. the cleavage of high molecular weight kininogen with release of bradykinin and antimicrobial peptides (AMPs). Bradykinin is involved in the regulation of inflammatory processes, vascular permeability, and blood pressure. Due to the release of AMPs, the contact system is regarded as a branch of the innate immune defense against microorganisms. There is an increasing list of pathogens that interact with contact factors, in addition to bacteria also fungi and viruses bind and activate the system. In spite of that, pathogens have developed their own mechanisms to activate the contact system, resulting in manipulation of this host immune response. In this up-to-date review, we summarize present research on the interaction of pathogens with the human contact system, focusing particularly on bacterial and viral mechanisms that trigger inflammation via contact system activation.
The human contact system consists of two proteases, factor XII (FXII) and plasma prekallikrein (PPK) as well as the non-enzymatic cofactor high molecular weight kininogen (HK, see Figure 1). The proteins are produced in the liver and circulate as zymogens in the blood stream or are assembled on endothelial cells, neutrophils, and platelets. When blood is exposed to foreign biological or artificial surfaces, zymogen FXII binds through and autoactivates into an enzyme. Activation is accompanied by a major conformational change in the structure of FXII (1). Classically, it is stated that FXII has to interact with negatively charged surfaces for activation, but the current paradigm is that any artificial surface has the potential for FXII autoactivation (2). HK, which is in a noncovalent complex with PPK (3), also binds to the surface, thereby exposing PPK for activation by FXII cleavage. In turn, activated plasma kallikrein (PK) cleaves and activates more FXII, forming a powerful activation feedback loop. When sufficient amounts of FXII are activated on the surface, FXII activates coagulation factor XI (FXI), leading to subsequent thrombin formation. This result—in vitro—in the formation of a fibrin clot and is used as a diagnostic coagulation test—the activated partial thromboplastin time (aPTT). However, individuals with congenital deficiencies in FXII, PPK, or HK, who show a prolonged aPTT, do not have bleeding diathesis or abnormal hemostasis, indicating that the intrinsic coagulation pathway does not contribute to physiological hemostasis (4). Moreover, contact activation in vivo always occurs under pathological conditions, such as thrombosis (5), sepsis, or ARDS (6, 7), which makes FXII a promising therapeutic target to limit thrombosis without increasing bleeding risk (8). Thus, it has been questioned whether activation of the intrinsic coagulation by FXII is really its main physiological function. Instead, it was suggested that the pro-inflammatory arm of the contact system—the kallikrein–kinin system—is more related to physiological in vivo functions (9).
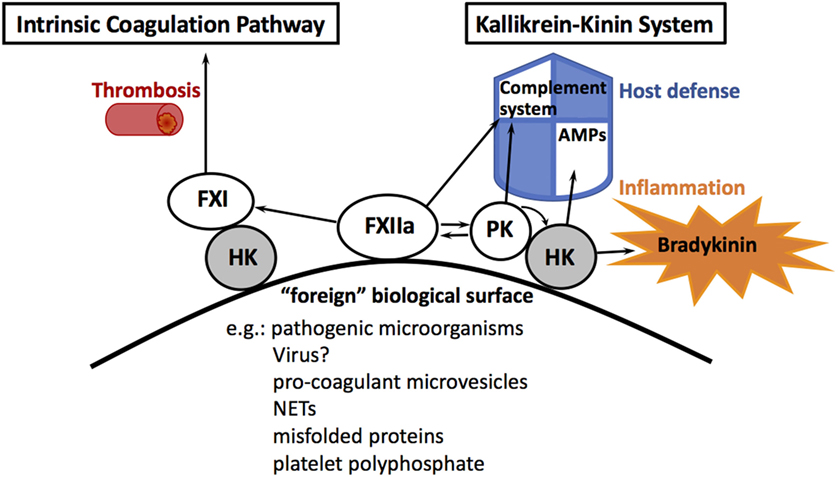
Figure 1. The human contact system. Assembly of contact system factors on foreign biological or artificial surfaces activates factor XII (FXII). FXIIa activates factor XI (FXI) that triggers the intrinsic pathway of coagulation, which is involved in thrombosis. FXII also activates plasma kallikrein (PK), which cleaves high molecular weight kininogen (HK), followed by the release of the pro-inflammatory peptide bradykinin and antimicrobial peptides (AMPs). FXII and PK contribute in vitro to complement activation.
High molecular weight kininogen is encoded by the KNG1 gene, which is alternatively spliced into two products, high and low molecular weight kininogen. High molecular weight kininogen (HK) contains six domains (D1–D6) with a range of procoagulant, pro-inflammatory, or antimicrobial functions. Low molecular weight kininogen (LK) lacks D6, wherefore it cannot bind PK or FXI and does not belong to the contact system. Upon activation by FXII, PK cleaves HK and the nonapeptide bradykinin will be released from D4 (10).
Bradykinin is one of the most potent inflammatory mediators in humans, after binding through its cell receptor B2R (11) it activates signaling pathways resulting in increased vascular permeability, vasodilation, hypotension, pain, fever. Furthermore, kinin receptors appear to be involved in autoimmune diseases (12). Although bradykinin is a short-lived mediator, it stimulates the production of superoxide radicals and nitric oxide and modulates the mobilization and release of histamine, arachidonic acid, prostaglandin E2, pro-inflammatory interleukin-1, and TNF-alpha (13).
Additionally, bradykinin is involved in activation of cellular innate immune responses, such as migration of neutrophils (14) and stimulation of alveolar macrophages (15). Exogenous bradykinin activates immature dendritic cells via B2R, thereby stimulating adaptive immunity (16). Moreover, cooperative activation of B2R and toll-like receptor 2 is responsible for an interferon-γ response in dendritic cells, linking innate and adaptive immune responses (17).
It has been proposed that neutrophils interact with the contact system to boost neutrophil extravasation by bradykinin-mediated vasodilatation (18). Moreover, PK and FXII itself trigger inflammation by causing aggregation and degranulation of human neutrophils (19, 20). FXII contributes further to inflammation by induction of pro-inflammatory cytokines from macrophages (21).
Neutrophil extracellular traps (NETs) have been shown to bind and activate contact factors (22). Released from neutrophils in response to infectious and pro-inflammatory stimuli, NETs immobilize invading pathogens within a fibrous matrix consisting of DNA, histones, and antimicrobial peptides (AMPs) (23), providing a suitable surface for contact system activation. The overall consequences of NETs release are not clear, far from being univocal. NETs may also be protective for the invading pathogen and contribute to autoimmune diseases (24).
Recent studies further reveal that also procoagulant microvesicles are equipped with a surface that allows binding and activation of contact factors and bradykinin release (25). Microvesicles are continuously shed from the membrane of every cell type examined to date. Procoagulant microvesicles are shed due to an infectious stimulus from the plasma membrane of monocytes (25, 26). The outer surface of such microvesicles is enriched in phosphatidylserine, which provides a catalytic surface for the assembly of contact and coagulation factors (27). Moreover, microvesicles can explore antimicrobial activity (28), entrap bacteria, and prevent their dissemination from the local focus of infection in an animal model of sepsis (29). Formation and release of procoagulant microvesicles follows the principles of pattern recognition, as activation of monocytes is triggered by the binding of streptococcal M1 protein to toll-like receptor 2 (30), which suggests that microvesicle release is part of the innate immune reaction.
A further link between the contact system and host defense is activation of the alternative complement pathway by FXII that triggers activation of the C1 complex (31). In the alternative complement pathway, PK can replace factor D for the activation of C3 convertase (32, 33). Simultaneous activation of the contact and complement system results in the generation of nascent molecules that have significant impact in various in inflammatory diseases including angioedema and cancer (34). Whether contact system factors trigger activation of the complement system in vivo remains to be investigated.
Due to the generation of bradykinin by PK, the cleavage product HKa is formed, which differs from HK because of conformational changes (35). HKa stimulates secretion of the cytokines TNFα, interleukin IL-1β, IL-6, and the chemokines IL-8 and MCP-1 from human mononuclear cells, all of which are known to contribute to the inflammatory process (36).
Finally, HK-derived peptides display potent antibacterial and antifungal properties (37–40), contributing pivotal components of innate immunity, as such AMPs represent a first-line defense against invading pathogens. Recently Cagliani et al. (41) published a phylogenetic analysis indicating that mammalian kininogen genes evolved adaptively, in contrast to the other contact system genes. It has been proposed that kininogen gene KNG1 has been a target of long-lasting and strong selective pressures, suggesting that kininogen plays a central role in the modulation of immune responses (41).
Taken together, the kallikrein–kinin system contributes to innate immune defense by bradykinin dependent and independent mechanisms. Activation of contact factors triggers inflammatory reactions that potentiate the host response against invading pathogens.
In order to respond to a broad range of microbes the innate immune system uses a variety of proteins, which recognize surface features of microbial pathogens that differ from human cell membranes. Although the contact system is activated in vitro by high doses of purified bacterial lipopolysaccharides (LPS) (42, 43), contact factors bound to specific proteins and virulence determinants on the bacterial surface. Over 20 bacterial species are known to bind and activate contact factors on their surface, but the bacterial binding protein and the activating mechanism is often unknown (44). However, there are certain similarities in structure and property of bacterial proteins, which interact with contact factors. Many bacterial species possess long filamentous structures known as curli, fimbriae, or pili extending from their surfaces (45). Gram-negative bacteria, such as Escherichia coli and Salmonella enterica subsp. enterica ser. Typhimurium express curli fibers that bind all contact factors (46, 47). Curli play a major role in biofilm formation (48) and as adhesins, as they bind to proteins of the extracellular matrix. Similarly, Porphyromonas gingivalis, a Gram-negative periodontal pathogen, expresses long peritrichous, filamentous components, known as fimbriae, on the bacterial surface that are implicated in binding of contact factors (49). Interestingly both, curli from E. coli or Salmonella enterica as well as fimbriae from P. gingivalis (50) belong to a class of stable, ordered proteins, characterized structurally by repeating beta-strand units and known as bacterial amyloids (51). It might be that FXII recognizes bacterial exogenous amyloid structures as a pathogen-associated molecular pattern (52). This idea is supported by studies showing that FXII binds and activates on endogenous amyloids and misfolded proteins (53, 54), proposing that FXII-dependent activation of PK is a conserved protective response that recognize and clear non-physiological or damaged host proteins in the extracellular space (55). We have recently shown that different pili of Streptococcus gallolyticus—a Gram-positive strain and endocarditis isolate—are involved in binding and activation of contact factors. The adhesin from the pilus binds FXII with high affinity, and we proposed that S. gallolyticus may trigger inflammation on the endocardium by activation of host blood coagulation and contact system activation (56). For both, the pilin adhesin Gallo2179 and the major pilin (Gallo2178) several amylogenic regions can be predicted by using “Waltz” an amyloid-prediction tool (57, 58). Nevertheless, whether these proteins are amyloids remains to be investigated.
The Gram-positive group A Streptococcus (GAS) bind HK, FXII, and FXI via their surface M protein (59), which forms fibrous hair-like structures at the bacterial surface (60), but is not part of a pilus (61). Protein FOG, a fibrinogen-binding M-like protein, and protein G, from Group G streptococci also bind HK, FXII, and FXI (62). Interestingly in this context, immunoglobulin binding domain of the streptococcal protein G also forms amyloid fibrils (63).
Moreover, several adhesins from Candida spp. have been demonstrated to bind all contact factors (64).
Factor XII binds to human proteins with amyloid-like properties by the fibronectin type I domain (54); however, the precise binding site for pathogens on FXII are unknown. In HK some interactions have been mapped to D3, D5, and D6 (41).
Thus, so far identified bacterial and fungal proteins that are bound by contact factors have several properties in common, a fibrous hair-like structure, extension from the bacterial cell surface, and function as adhesins by binding of fibrinogen, fibronectin, collagen, or laminin. It remains to be investigated to what extend bacterial amyloid proteins play a role for activation of FXII, as also Streptococcus mutants or Mycobacterium tuberculosis display amyloid fimbriae (65, 66).
Binding and local activation of contact factors at the pathogens surface triggers inflammatory reactions that support the first line in host defense against the invaders. However, eukaryotic and prokaryotic microorganisms can exploit the system and induce its activation by different mechanisms. This may promote invasive spread via bradykinin-induced vascular leakage, since inflowing nutrient-rich plasma to the infected tissue site might serve as a route for the disseminating pathogen. Microbial cysteine proteases such as SpeB from GAS (67), staphopain A and B from Staphylococcus aureus (68), gingipains from Porphyromonas gingivalis (69), and cruzipain from Trypanosoma cruzi [for a review see Ref. (70)] can directly liberate kinins from HK (see Table 1). Aeromonas sobria, a pathogen causing gastroenteritis and sepsis, secrets a serine protease that activates PK, and also directly cleaves HK as well as LK, thereby producing vascular leakage activity (71).
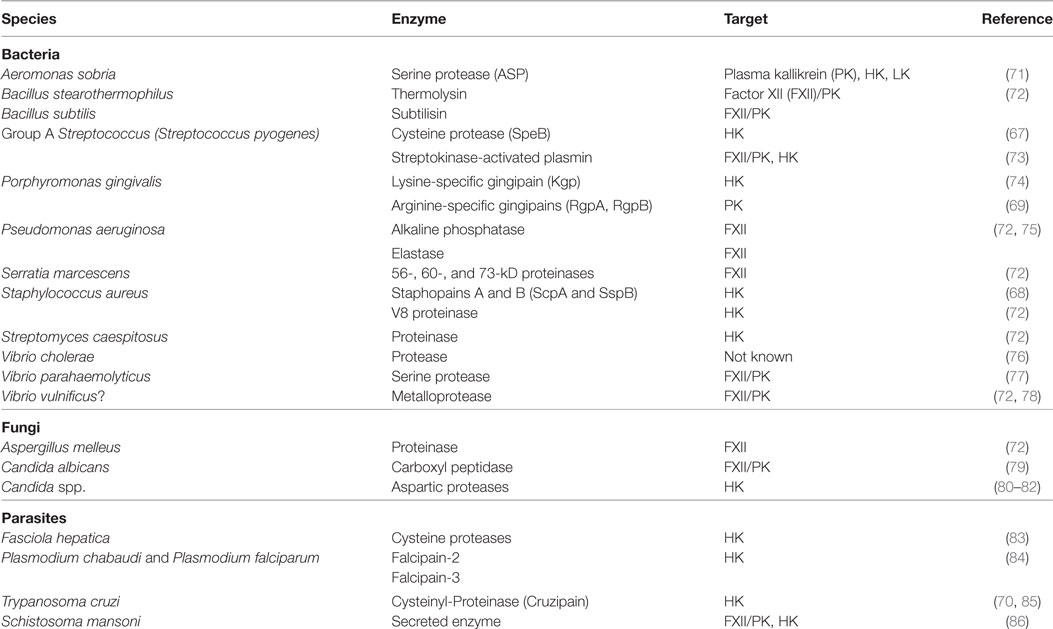
Table 1. Enzymes produced by pathogens that activate or cleave contact factors.
Plasmodium parasites, which cause malaria in the host, generate bradykinin in a different way. They process HK intracellular, probably by cysteine proteases. Thus, by releasing vasoactive peptides, derived from host HK, plasmodium is able to induce vasodilatation and endothelial cell permeability to facilitate parasite survival (84, 87).
Furthermore, extracellular bacterial or fungal proteinases generate proteolytic activity of FXII or PK, thereby producing bradykinin indirectly [see Table 1, for a review see Ref. (44, 72)]. Indirect bradykinin liberation can also be induced by activation of host proteinases, as it has been recently shown for secreted streptokinase, a GAS plasminogen activator (73). Many invasive pathogens exploit plasmin as a virulence factor to degrade fibrin clots, overcome tissue barriers, and evade peptide-derived host immune defenses (88, 89). Contact activation by streptokinase-activated plasmin could explain systemic contact activation and bradykinin liberation seen during invasive streptococcal infection (90). Moreover, dysregulation of the tightly regulated hemostasis by contact system activation may represent another virulence mechanism for streptokinase. Accordingly, the data reveal that GAS isolates from invasive infections trigger an activation of the contact system more potently than strains isolated from noninvasive infections (73).
Hence, activation of the contact system by the pathogen adds another level of complexity to the interaction between pathogen and host during infections.
Besides pro- and eukaryotes, there are few studies describing contact system activation arising from viral infections. Infection of ferrets with influenza A virus results in an increased generation of bradykinin in nasal secretion, suggesting that kinins may contribute to local symptoms of sneezing, nasal congestion, and rhinorrhea (91).
Dengue fever is a rapidly spreading mosquito-borne viral infection often manifests in severe forms. Dengue hemorrhagic fever and dengue shock syndrome can lead to life-threatening complications, including vascular permeability and hemorrhagic manifestations. Reduced serum levels of kininogen were observed in dengue fever patients, which may be due to proteolysis and generation of bradykinin to trigger inflammatory reactions (92).
Human immunodeficiency virus (HIV) progressively damage the immune system, which can lead to endothelial dysfunction and liver damage leading to coagulopathy and over time acquired immunodeficiency syndrome. It was shown that HIV-positive patient have significant prolonged prothrombin time and aPTT (93). Another study showed significant decreased PK activity, but HK concentrations were not significant different between healthy controls and HIV-positive patients (94). Similarly, in patients with HIV–hepatitis B virus co-infections, a significant decrease of PK concentration was measured (95), indicating consumption due to activation.
Hantaviruses are responsible for hemorrhagic fever with renal and pulmonary syndrome, both of which present with edema and hemorrhage. Recently Taylor et al. demonstrated that hantavirus-infected cells trigger activation of the kallikrein–kinin system, revealing a novel mechanism of hantavirus-induced vascular leakage. Incubation of contact factors FXII, PK, and HK with hantavirus-infected endothelial cells leads to an increased cleavage of HK, increased amounts of activated FXII and PK, and liberation of bradykinin. In addition, cell permeability could be avoided using inhibitors that directly block bradykinin binding, the activity of FXIIa, or the activity of PK. Furthermore, they first demonstrated a FXII binding and autoactivation on hantavirus-infected endothelial cells (96).
It has not been shown yet whether the virus will be bound and activate contact factors directly. But enveloped viruses probably provide an appropriate surface for contact activation as the viral envelop is typically derived from host cell membranes. Herpes simplex virus 1 (HSV-1) contains phosphatidylserine and tissue factor on its surface, both derived from the host cell membrane. After addition of HSV-1 to plasma, clotting was induced by the extrinsic and intrinsic pathway of coagulation (97), similarly to procoagulant microvesicles (25).
Thus, there is evidence that the contact system is involved in vascular leakage and inflammatory reactions seen in viral infections. It remains to be investigated whether contact factors bind and activate on the viral surface or on viral infected cells, and whether this interaction may protect from virus-induced disease.
Sepsis is the archetypical disease state were systemic contact activation occurs (6, 98), and multiple animal studies were done targeting the system to evaluate potential therapeutic options. In animal studies with different species pharmacological interventions that inhibit FXII, PK, or bradykinin-receptors during sepsis implicate beneficial for the host; however, human trials still lack the same confidence [for a review see Ref. (99)]. Moreover, little studies exist, revealing the role of single contact factors during microbial sepsis, using knockout animals or specific inhibitors. In a first study, FXII deficient mice were protected against hypotension induced by LPS, but coagulopathy, inflammatory responses, and lethality were not affected (100). Contrary, mice deficient in HK were resistant to LPS-induced mortality and had significantly reduced circulating LPS levels. Binding of LPS to HK induced cleavage and bradykinin release, proposing that HK—as a major LPS carrier in circulation—plays an essential role in endotoxemia (101). With regard to bacterial infection, a recent study by Stroo et al. show, that FXII deficiency in mice improved survival and reduced bacterial outgrowth, in an airway infection with the Gram-negative Klebsiella pneumoniae, but the protecting mechanism is unclear. In contrast, FXII-deficient mice did not show a protective phenotype by using Gram-positive Streptococcus pneumoniae in the same infection model (102). Thus, the consequences of activating the contact system within the infection process have yet to be established.
John Hageman was the first patient identified with FXII deficiency, and he suffered from recurrent infections all his life. Beside this case, there are no reports in the literature linking contact protein deficiencies to increased susceptibility for infections. Because there are redundancies in the immune system, it is more than likely that such deficiencies only accidentally will be diagnosed, a phenomenon also seen in complement deficiencies.
Local activation due to contact factor binding on the pathogens surface may be protective against several infections, but activation by the pathogen may trigger systemic reactions that result in detrimental effects for the host.
These findings may offer a great promise for the development of novel therapeutic approaches, potentially complementing existing antibiotic therapies. However, the different mechanisms that trigger a systemic contact activation need to be understood more in detail.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research is supported by Deutsche Forschungsgemeinschaft (OE 547/4-1) and Federal Excellence Initiative of Mecklenburg Western Pomerania and European Social Fund (ESF) Grant KoInfekt (ESF_14-BM-A55-00xx_16).
1. Samuel M, Pixley RA, Villanueva MA, Colman RW, Villanueva GB. Human factor-Xii (Hageman-factor) autoactivation by dextran sulfate – circular-dichroism, fluorescence, and ultraviolet difference spectroscopic studies. J Biol Chem (1992) 267:19691–7.
2. Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost (2016) 14:28–39. doi:10.1111/jth.13194
3. Mandle RJ, Colman RW, Kaplan AP. Identification of prekallikrein and high-molecular-weight kininogen as a complex in human-plasma. Proc Natl Acad Sci U S A (1976) 73:4179–83. doi:10.1073/pnas.73.11.4179
4. Meijers JCM. No contact, no thrombosis? Blood (2014) 123:1629–1629. doi:10.1182/blood-2014-01-549691
5. Renné T, Schmaier AH, Nickel KF, Blomback M, Maas C. In vivo roles of factor XII. Blood (2012) 120:4296–303. doi:10.1182/blood-2012-07-292094
6. Oehmcke S, Herwald H. Contact system activation in severe infectious diseases. J Mol Med-Jmm (2010) 88:121–6. doi:10.1007/s00109-009-0564-y
7. Hess R, Wujak L, Hesse C, Sewald K, Jonigk D, Warnecke G, et al. Coagulation factor XII regulates inflammatory responses in human lungs. Thromb Haemost (2017) 117:1896–907. doi:10.1160/TH16-12-0904
8. Long AT, Kenne E, Jung R, Fuchs TA, Renné T. Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost (2016) 14:427–37. doi:10.1111/jth.13235
9. Jukema BN, de Maat S, Maas C. Processing of factor XII during inflammatory reactions. Front Med (2016) 3:52. doi:10.3389/fmed.2016.00052
10. Colman RW, Schmaier AH. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood (1997) 90:3819–43.
11. Leeb-Lundberg LMF, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev (2005) 57:27–77. doi:10.1124/pr.57.1.2
12. Dutra RC. Kinin receptors: key regulators of autoimmunity. Autoimmun Rev (2017) 16:192–207. doi:10.1016/j.autrev.2016.12.011
13. Lalmanach G, Naudin C, Lecaille F, Fritz H. Kininogens: more than cysteine protease inhibitors and kinin precursors. Biochimie (2010) 92:1568–79. doi:10.1016/j.biochi.2010.03.011
14. Paegelow I, Trzeczak S, Böckmann S, Vietinghoff G. Migratory responses of polymorphonuclear leukocytes to kinin peptides. Pharmacology (2002) 66:153–61. doi:10.1159/000063797
15. Sato E, Koyama S, Nomura H, Kubo K, Sekiguchi M. Bradykinin stimulates alveolar macrophages to release neutrophil, monocyte, and eosinophil chemotactic activity. The Journal of Immunology (1996) 157:3122–9.
16. Aliberti J, Viola JPB, Vieira-de-Abreu A, Bozza PT, Sher A, Scharfstein J. Cutting edge: bradykinin induces IL-12 production by dendritic cells: a danger signal that drives Th1 polarization. The Journal of Immunology (2003) 170:5349–53. doi:10.4049/jimmunol.170.11.5349
17. Scharfstein J, Schmitz V, Svensjö E, Granato A, Monteiro AC. Kininogens coordinate adaptive immunity through the proteolytic release of bradykinin, an endogenous danger signal driving dendritic cell maturation. Scand J Immunol (2007) 66:128–36. doi:10.1111/j.1365-3083.2007.01983.x
18. Hofman Z, de Maat S, Hack CE, Maas C. Bradykinin: inflammatory product of the coagulation system. Clin Rev Allergy Immunol (2016) 51:152–61. doi:10.1007/s12016-016-8540-0
19. Wachtfogel YT, Kucich U, James HL, Scott CF, Schapira M, Zimmerman M, et al. Human-plasma kallikrein releases neutrophil elastase during blood-coagulation. J Clin Invest (1983) 72:1672–7. doi:10.1172/JCI111126
20. Wachtfogel YT, Pixley RA, Kucich U, Abrams W, Weinbaum G, Schapira M, et al. Purified plasma factor XIIa aggregates human neutrophils and causes degranulation. Blood (1986) 67:1731–7.
21. Vorlova S, Koch M, Manthey HD, Cochain C, Busch M, Chaudhari SM, et al. Coagulation factor XII induces pro-inflammatory cytokine responses in macrophages and promotes atherosclerosis in mice. Thromb Haemost (2017) 117:1–12. doi:10.1160/TH16-06-0466
22. Oehmcke S, Mörgelin M, Herwald H. Activation of the human contact system on neutrophil extracellular traps. J Innate Immun (2009) 1:225–30. doi:10.1159/000203700
23. Köckritz-Blickwede von M, Nizet V. Innate immunity turned inside-out: antimicrobial defense by phagocyte extracellular traps. J Mol Med (Berl) (2009) 87:775–83. doi:10.1007/s00109-009-0481-0
24. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol (2017) 7:461. doi:10.1038/nri.2017.105
25. Oehmcke S, Mörgelin M, Malmström J, Linder A, Chew M, Thorlacius H, et al. Stimulation of blood mononuclear cells with bacterial virulence factors leads to the release of pro-coagulant and pro-inflammatory microparticles. Cell Microbiol (2012) 14:107–19. doi:10.1111/j.1462-5822.2011.01705.x
26. Toti F, Dachary-Prigent J, Eschwège V, Freyssinet JM. Monocyte vesiculation is a possible mechanism for dissemination of membrane-associated procoagulant activities and adhesion molecules after stimulation by lipopolysaccharide. J Immunol (1994) 153:3245–55.
27. van der Poll T, Herwald H. The coagulation system and its function in early immune defense. Thromb Haemost (2014) 112:640–8. doi:10.1160/TH14-01-0053
28. Timár CI, Lorincz AM, Csépányi-Kömi R, Vályi-Nagy A, Nagy G, Buzás EI, et al. Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood (2013) 121:510–8. doi:10.1182/blood-2012-05-431114
29. Oehmcke S, Westman J, Malmström J, Mörgelin M, Olin AI, Kreikemeyer B, et al. A novel role for pro-coagulant microvesicles in the early host defense against Streptococcus pyogenes. PLoS Pathog (2013) 9:e1003529. doi:10.1371/journal.ppat.1003529
30. Påhlman LI, Mörgelin M, Eckert J, Johansson L, Russell W, Riesbeck K, et al. Streptococcal M protein: a multipotent and powerful inducer of inflammation. J Immunol (2006) 177:1221–8. doi:10.4049/jimmunol.177.2.1221
31. Ghebrehiwet B, Silverberg M, Kaplan AP. Activation of the classical pathway of complement by Hageman factor fragment. J Exp Med (1981) 153:665–76. doi:10.1084/jem.153.3.665
32. DiScipio RG. The activation of the alternative pathway C3 convertase by human plasma kallikrein. Immunology (1982) 45:587–95.
33. Irmscher S, Döring N, Halder LD, Jo EAH, Kopka I, Dunker C, et al. Kallikrein cleaves C3 and activates complement. J Innate Immun (2017). doi:10.1159/000484257
34. Ghebrehiwet B, Kaplan AP, Joseph K, Peerschke EIB. The complement and contact activation systems: partnership in pathogenesis beyond angioedema. Immunol Rev (2016) 274:281–9. doi:10.1111/imr.12469
35. Weisel JW, Nagaswami C, Woodhead JL, Cadena DeLa RA, Page JD, Colman RW. The shape of high molecular weight kininogen: organization into structural domains, changes with activation, and interactions with prekallikrein, as determined by electron microscopy. J Biol Chem (1994) 269:10100–6.
36. Khan MM, Bradford HN, Isordia-Salas I, Liu Y, Wu Y, Espinola RG, et al. High-molecular-weight kininogen fragments stimulate the secretion of cytokines and chemokines through uPAR, Mac-1, and gC1qR in monocytes. Arterioscl Throm Vas (2006) 26:2260–6. doi:10.1161/01.ATV.0000240290.70852.c0
37. Frick I-M, Åkesson P, Herwald H, Mörgelin M, Malmsten M, Nägler DK, et al. The contact system – a novel branch of innate immunity generating antibacterial peptides. EMBO J (2006) 25:5569–78. doi:10.1038/sj.emboj.7601422
38. Nordahl EA, Rydengård V, Mörgelin M, Schmidtchen A. Domain 5 of high molecular weight kininogen is antibacterial. J Biol Chem (2005) 280:34832–9. doi:10.1074/jbc.M507249200
39. Schmidtchen A, Pasupuleti M, Mörgelin M, Davoudi M, Alenfall J, Chalupka A, et al. Boosting antimicrobial peptides by hydrophobic oligopeptide end tags. J Biol Chem (2009) 284:17584–94. doi:10.1074/jbc.M109.011650
40. Sonesson A, Nordahl EA, Malmsten M, Schmidtchen A. Antifungal activities of peptides derived from domain 5 of high-molecular-weight kininogen. Int J Pept (2011) 2011:761037. doi:10.1155/2011/761037
41. Cagliani R, Forni D, Riva S, Pozzoli U, Colleoni M, Bresolin N, et al. Evolutionary analysis of the contact system indicates that kininogen evolved adaptively in mammals and in human populations. Mol Biol Evol (2013) 30:1397–408. doi:10.1093/molbev/mst054
42. Kalter ES, van Dijk WC, Timmerman A, Verhoef J, Bouma BN. Activation of purified human plasma prekallikrein triggered by cell wall fractions of Escherichia coli and Staphylococcus aureus. J Infect Dis (1983) 148:682–91. doi:10.1093/infdis/148.4.682
43. Roeise O, Bouma BN, Stadaas JO, Aasen AO. Dose dependence of endotoxin-induced activation of the plasma contact system – an invitro study. Circ Shock (1988) 26:419–30.
44. Nickel KF, Renné T. Crosstalk of the plasma contact system with bacteria. Thromb Res (2012) 130(Suppl 1):S78–83. doi:10.1016/j.thromres.2012.08.284
45. Proft T, Baker EN. Pili in Gram-negative and Gram-positive bacteria – structure, assembly and their role in disease. Cell Mol Life Sci (2009) 66:613–35. doi:10.1007/s00018-008-8477-4
46. BenNasr A, OlsenN A, Sjöbring U, Müller-Esterl W, Björck L. Assembly of human contact phase proteins and release of bradykinin at the surface of curli-expressing Escherichia coli. Mol Microbiol (1996) 20:927–35. doi:10.1111/j.1365-2958.1996.tb02534.x
47. Herwald H, Mörgelin M, Olsen A, Rhen M, Dahlbäck B, Müller-Esterl W, et al. Activation of the contact-phase system on bacterial surfaces-a clue to serious complications in infectious diseases. Nat Med (1998) 4:298–302. doi:10.1038/nm0398-298
48. Hung C, Zhou Y, Pinkner JS, Dodson KW, Crowley JR, Heuser J, et al. Escherichia coli biofilms have an organized and complex extracellular matrix structure. MBio (2013) 4:e645–713. doi:10.1128/mBio.00645-13
49. Rapala-Kozik M, Bras G, Chruscicka B, Karkowska-Kuleta J, Sroka A, Herwald H, et al. Adsorption of components of the plasma kinin-forming system on the surface of Porphyromonas gingivalis involves gingipains as the major docking platforms. Infect Immun (2011) 79:797–805. doi:10.1128/IAI.00966-10
50. Cueno ME, Nagano K, Imai K, Tamura M, Yoshimura F, Ochiai K. Ab initio modeling approach towards establishing the structure and docking orientation of the Porphyromonas gingivalis FimA. J Mol Graph Model (2015) 55:65–71. doi:10.1016/j.jmgm.2014.11.001
51. Van Gerven N, Klein RD, Hultgren SJ, Remaut H. Bacterial amyloid formation: structural insights into curli biogensis. Trends Microbiol (2015) 23:693–706. doi:10.1016/j.tim.2015.07.010
52. Gebbink MFBG, Claessen D, Bouma B, Dijkhuizen L, Wösten HAB. Amyloids – a functional coat for microorganisms. Nat Rev Micro (2005) 3:333–41. doi:10.1038/nrmicro1127
53. Zamolodchikov D, Renné T, Strickland S. The Alzheimer’s disease peptide β-amyloid promotes thrombin generation through activation of coagulation factor XII. J Thromb Haemost (2016) 14:995–1007. doi:10.1111/jth.13209
54. Maas C, Schiks B, Strangi RD, Hackeng TM, Bouma BN, Gebbink MFBG, et al. Identification of fibronectin type I domains as amyloid-binding modules on tissue-type plasminogen activator and three homologs. Amyloid (2008) 118(9):3208–18. doi:10.1080/13506120802193498
55. Maas C, Govers-Riemslag JWP, Bouma B, Schiks B, Hazenberg BPC, Lokhorst HM, et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest (2008):1–11. doi:10.1172/JCI35424
56. Isenring J, Köhler J, Nakata M, Frank M, Jans C, Renault P, et al. Streptococcus gallolyticus subsp. gallolyticus endocarditis isolate interferes with coagulation and activates the contact system. Virulence (2017) 9:1–14. doi:10.1080/21505594.2017.1393600
57. Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Methods (2010) 7:237–42. doi:10.1038/nmeth.1432
58. Oliveberg M. Waltz, an exciting new move in amyloid prediction. Nat Methods (2010) 7:187–8. doi:10.1038/nmeth0310-187
59. Herwald H, Mörgelin M, Dahlbäck B, Björck L. Interactions between surface proteins of Streptococcus pyogenes and coagulation factors modulate clotting of human plasma. J Thromb Haemost (2003) 1:284–91. doi:10.1046/j.1538-7836.2003.00105.x
60. Oehmcke S, Shannon O, Mörgelin M, Herwald H. Streptococcal M proteins and their role as virulence determinants. Clin Chim Acta (2010) 411:1172–80. doi:10.1016/j.cca.2010.04.032
61. Kreikemeyer B, Gámez G, Margarit I, Giard J-C, Hammerschmidt S, Hartke A, et al. Genomic organization, structure, regulation and pathogenic role of pilus constituents in major pathogenic streptococci and enterococci. Int J Med Microbiol (2011) 301:240–51. doi:10.1016/j.ijmm.2010.09.003
62. Wollein Waldetoft K, Svensson L, Mörgelin M, Olin AI, Nitsche-Schmitz DP, Björck L, et al. Streptococcal surface proteins activate the contact system and control its antibacterial activity. J Biol Chem (2012) 287:25010–8. doi:10.1074/jbc.M112.373217
63. Ramirez-Alvarado M, Merkel JS, Regan L. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc Natl Acad Sci U S A (2000) 97:8979–84. doi:10.1073/pnas.150091797
64. Karkowska-Kuleta J, Kedracka-Krok S, Rapala-Kozik M, Kamysz W, Bielinska S, Karafova A, et al. Molecular determinants of the interaction between human high molecular weight kininogen and Candida albicans cell wall: identification of kininogen-binding proteins on fungal cell wall and mapping the cell wall-binding regions on kininogen molecule. Peptides (2011) 32:2488–96. doi:10.1016/j.peptides.2011.10.021
65. Oli MW, Otoo HN, Crowley PJ, Heim KP, Nascimento MM, Ramsook CB, et al. Functional amyloid formation by Streptococcus mutans. Microbiology (Reading, Engl) (2012) 158:2903–16. doi:10.1099/mic.0.060855-0
66. Sawyer EB, Claessen D, Gras SL, Perrett S. Exploiting amyloid: how and why bacteria use cross-β fibrils. Biochem Soc Trans (2012) 40:728–34. doi:10.1042/BST20120013
67. Herwald H, Collin M, Müller-Esterl W, Björck L. Streptococcal cysteine proteinase releases kinins: a virulence mechanism. J Exp Med (1996) 184:665–73. doi:10.1084/jem.184.2.665
68. Imamura T, Tanase S, Szmyd G, Kozik A, Travis J, Potempa J. Induction of vascular leakage through release of bradykinin and a novel kinin by cysteine proteinases from Staphylococcus aureus. J Exp Med (2005) 201:1669–76. doi:10.1084/jem.20042041
69. Imamura T, Pike RN, Potempa J, Travis J. Pathogenesis of periodontitis: a major arginine-specific cysteine proteinase from Porphyromonas gingivalis induces vascular permeability enhancement through activation of the kallikrein/kinin pathway. J Clin Invest (1994) 94:361–7. doi:10.1172/JCI117330
70. Scharfstein J, Andrade D, Svensjö E, Oliveira AC, Nascimento CR. The kallikrein-kinin system in experimental Chagas disease: a paradigm to investigate the impact of inflammatory edema on GPCR-mediated pathways of host cell invasion by Trypanosoma cruzi. Front Immunol (2012) 3:396. doi:10.3389/fimmu.2012.00396
71. Imamura T, Kobayashi H, Khan R, Nitta H, Okamoto K. Induction of vascular leakage and blood pressure lowering through kinin release by a serine proteinase from Aeromonas sobria. The Journal of Immunology (2006) 177:8723–9. doi:10.4049/jimmunol.177.12.8723
72. Molla A, Yamamoto T, Akaike T, Miyoshi S, Maeda H. Activation of hageman factor and prekallikrein and generation of kinin by various microbial proteinases. J Biol Chem (1989) 264:10589–94.
73. Nitzsche R, Rosenheinrich M, Kreikemeyer B, Oehmcke-Hecht S. Streptococcus pyogenes triggers activation of the human contact system by streptokinase. Infect Immun (2015) 83:3035–42. doi:10.1128/IAI.00180-15
74. Scott CF, Whitaker EJ, Hammond BF, Colman RW. Purification and characterization of a potent 70-kDa thiol lysyl-proteinase (Lys-gingivain) from Porphyromonas gingivalis that cleaves kininogens and fibrinogen. J Biol Chem (1993) 268:7935–42.
75. Sakata Y, Akaike T, Suga M, Ijiri S, Ando M, Maeda H. Bradykinin generation triggered by Pseudomonas proteases facilitates invasion of the systemic circulation by Pseudomonas aeruginosa. Microbiol Immunol (1996) 40:415–23. doi:10.1111/j.1348-0421.1996.tb01088.x
76. Sakata Y, Akaike T, Khan MM, Ichinose Y, Hirayama H, Suga M, et al. Activation of bradykinin generating cascade by Vibrio cholerae protease. Immunopharmacology (1996) 33:377–9. doi:10.1016/0162-3109(96)00090-2
77. Miyoshi S-I, Watanabe H, Kawase T, Yamada H, Shinoda S. Generation of active fragments from human zymogens in the bradykinin-generating cascade by extracellular proteases from Vibrio vulnificus and V. parahaemolyticus. Toxicon (2004) 44:887–93. doi:10.1016/j.toxicon.2004.08.013
78. Miyoshi N, Miyoshi S, Sugiyama K, Suzuki Y, Furuta H, Shinoda S. Activation of the plasma kallikrein-kinin system by Vibrio vulnificus protease. Infect Immun (1987) 55:1936–9.
79. Kaminishi H, Tanaka M, Cho T, Maeda H, Hagihara Y. Activation of the plasma kallikrein-kinin system by Candida albicans proteinase. Infect Immun (1990) 58:2139–43.
80. Bras G, Bochenska O, Rapala-Kozik M, Guevara-Lora I, Faussner A, Kozik A. Extracellular aspartic protease SAP2 of Candida albicans yeast cleaves human kininogens and releases proinflammatory peptides, Met-Lys-bradykinin and des-Arg(9)-Met-Lys-bradykinin. Biol Chem (2012) 393:829–39. doi:10.1515/hsz-2012-0157
81. Bras G, Bochenska O, Rapala-Kozik M, Guevara-Lora I, Faussner A, Kamysz W, et al. Release of biologically active kinin peptides, Met-Lys-bradykinin and Leu-Met-Lys-bradykinin from human kininogens by two major secreted aspartic proteases of Candida parapsilosis. Peptides (2013) 48:114–23. doi:10.1016/j.peptides.2013.08.003
82. Kozik A, Gogol M, Bochenska O, Karkowska-Kuleta J, Wolak N, Kamysz W, et al. Kinin release from human kininogen by 10 aspartic proteases produced by pathogenic yeast Candida albicans. BMC Microbiol (2015) 15:60. doi:10.1186/s12866-015-0394-8
83. Cordova M, Jara J, Del Nery E, Hirata IY, Araújo MS, Carmona AK, et al. Characterization of two cysteine proteinases secreted by Fasciola hepatica and demonstration of their kininogenase activity. Mol Biochem Parasitol (2001) 116:109–15. doi:10.1016/S0166-6851(01)00309-7
84. Bagnaresi P, Barros NM, Assis DM, Melo PM, Fonseca RG, Juliano MA, et al. Intracellular proteolysis of kininogen by malaria parasites promotes release of active kinins. Malar J (2012) 11:156. doi:10.1186/1475-2875-11-156
85. DelNery E, Juliano MA, Lima A, Scharfstein J, Juliano L. Kininogenase activity by the major cysteinyl proteinase (cruzipain) from Trypanosoma cruzi. J Biol Chem (1997) 272:25713–8. doi:10.1074/jbc.272.41.25713
86. Carvalho WS, Lopes CT, Juliano L, Coelho PM, Cunha-Melo JR, Beraldo WT, et al. Purification and partial characterization of kininogenase activity from Schistosoma mansoni adult worms. Parasitology (1998) 117(Pt 4):311–9. doi:10.1017/S0031182098003175
87. Cotrin SS, Gouvea IE, Melo PMS, Bagnaresi P, Assis DM, Araújo MS, et al. Substrate specificity studies of the cysteine peptidases falcipain-2 and falcipain-3 from Plasmodium falciparum and demonstration of their kininogenase activity. Mol Biochem Parasitol (2013) 187:111–6. doi:10.1016/j.molbiopara.2013.01.002
88. Peetermans M, Vanassche T, Liesenborghs L, Lijnen RH, Verhamme P. Bacterial pathogens activate plasminogen to breach tissue barriers and escape from innate immunity. Crit Rev Microbiol (2016) 42:866–82. doi:10.3109/1040841X.2015.1080214
89. Nitzsche R, Köhler J, Kreikemeyer B, Oehmcke-Hecht S. Streptococcus pyogenes escapes killing from extracellular histones through plasminogen binding and activation by streptokinase. J Innate Immun (2016) 8:589–600. doi:10.1159/000448039
90. Sriskandan S, Cohen J. Kallikrein-kinin system activation in streptococcal toxic shock syndrome. Clin Infect Dis (2000) 30:961–2. doi:10.1086/313827
91. Barnett JK, Cruse LW, Proud D. Kinins are generated in nasal secretions during influenza A infections in ferrets. Am Rev Respir Dis (1990) 142:162–6. doi:10.1164/ajrccm/142.1.162
92. Ray S, Srivastava R, Tripathi K, Vaibhav V, Patankar S, Srivastava S. Serum proteome changes in dengue virus-infected patients from a dengue-endemic area of India: towards new molecular targets? OMICS (2012) 16:527–36. doi:10.1089/omi.2012.0037
93. Omoregie R, Osakue SI, Ihemeje V, Omokaro EU, Ogeferet HO. Correlation of CD4 count with platelet count, prothrombin time and activated partial thromboplastin time among HIV patients in Benin City, Nigeria. West Indian Med J (2009) 58:437–40.
94. Reis dos ML, Maeda S, Rosatelli JB, Donadi EA, Roselino A. Kininogens and kallikrein in pruritic papular eruption. Immunopharmacology (1999) 45:115–20. doi:10.1016/S0162-3109(99)00063-6
95. Sun T, Liu L, Wu A, Zhang Y, Jia X, Yin L, et al. iTRAQ based investigation of plasma proteins in HIV infected and HIV/HBV coinfected patients—C9 and KLK are related to HIV/HBV coinfection. Int J Infect Dis (2017) 63:64–71. doi:10.1016/j.ijid.2017.08.006
96. Taylor SL, Wahl-Jensen V, Copeland AM, Jahrling PB, Schmaljohn CS. Endothelial cell permeability during hantavirus infection involves factor XII-dependent increased activation of the kallikrein-kinin system. PLoS Pathog (2013) 9:e1003470. doi:10.1371/journal.ppat.1003470
97. Gershom ES, Sutherland MR, Lollar P, Pryzdial ELG. Involvement of the contact phase and intrinsic pathway in herpes simplex virus-initiated plasma coagulation. J Thromb Haemost (2010) 8:1037–43. doi:10.1111/j.1538-7836.2010.03789.x
98. Schmaier AH. Physiologic activities of the contact activation system. Thromb Res (2014) 133:S41–4. doi:10.1016/j.thromres.2014.03.018
99. Nicola H. The role of contact system in septic shock: the next target? An overview of the current evidence. J Intensive Care (2017) 5:31. doi:10.1186/s40560-017-0228-x
100. Iwaki T, Cruz-Topete D, Castellino FJ. A complete factor XII deficiency does not affect coagulopathy, inflammatory responses, and lethality, but attenuates early hypotension in endotoxemic mice. J Thromb Haemost (2008) 6:1993–5. doi:10.1111/j.1538-7836.2008.03142.x
101. Yang A, Xie Z, Wang B, Colman RW, Dai J, Wu Y. An essential role of high-molecular-weight kininogen in endotoxemia. J Exp Med (2017) 214:2649–70. doi:10.1084/jem.20161900
Keywords: contact system, bradykinin, inflammation, infection, pathogen
Citation: Oehmcke-Hecht S and Köhler J (2018) Interaction of the Human Contact System with Pathogens—An Update. Front. Immunol. 9:312. doi: 10.3389/fimmu.2018.00312
Received: 17 November 2017; Accepted: 05 February 2018;
Published: 26 February 2018
Edited by:
Christoph Thiemermann, Queen Mary University of London, United KingdomReviewed by:
Krzysztof Guzik, Jagiellonian University, PolandCopyright: © 2018 Oehmcke-Hecht and Köhler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonja Oehmcke-Hecht, c29uamEub2VobWNrZS1oZWNodEBtZWQudW5pLXJvc3RvY2suZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.