 Alessandro Poggi
Alessandro Poggi Serena Varesano
Serena Varesano Maria Raffaella Zocchi
Maria Raffaella Zocchi- 1Molecular Oncology and Angiogenesis Unit, Policlinico San Martino, Genoa, Italy
- 2Division of Immunology, Transplants and Infectious Diseases, San Raffaele Scientific Institute, Milan, Italy
Experimental evidence indicates that mesenchymal stromal cells (MSCs) may regulate tumor microenvironment (TME). It is conceivable that the interaction with MSC can influence neoplastic cell functional behavior, remodeling TME and generating a tumor cell niche that supports tissue neovascularization, tumor invasion and metastasization. In addition, MSC can release transforming growth factor-beta that is involved in the epithelial–mesenchymal transition of carcinoma cells; this transition is essential to give rise to aggressive tumor cells and favor cancer progression. Also, MSC can both affect the anti-tumor immune response and limit drug availability surrounding tumor cells, thus creating a sort of barrier. This mechanism, in principle, should limit tumor expansion but, on the contrary, often leads to the impairment of the immune system-mediated recognition of tumor cells. Furthermore, the cross-talk between MSC and anti-tumor lymphocytes of the innate and adaptive arms of the immune system strongly drives TME to become immunosuppressive. Indeed, MSC can trigger the generation of several types of regulatory cells which block immune response and eventually impair the elimination of tumor cells. Based on these considerations, it should be possible to favor the anti-tumor immune response acting on TME. First, we will review the molecular mechanisms involved in MSC-mediated regulation of immune response. Second, we will focus on the experimental data supporting that it is possible to convert TME from immunosuppressive to immunostimulant, specifically targeting MSC.
Introduction
Mesenchymal stromal cells (MSCs) are a key component of solid tumor microenvironment (TME) (1–4). They include fibroblasts, myofibroblasts, pericytes, vascular or lymphatic endothelial cells, and undifferentiated mesenchymal stem cells. These cells produce the large part of the extracellular matrix and are involved in the homeostasis of tissues in different organs. There is experimental evidence that MSC can be influenced by tumor cells and, in turn, regulate tumor cell growth and expansion (1–4). In many instances, MSCs are driven by tumor cells to modify the extracellular matrix components, allowing tumor cell adaptation to the surrounding microenvironment and eventually metastasization (1–4). In healthy tissues, MSCs represent the network on which epithelial cells, blood, and lymphatic vasculature are organized and polarized. After receiving a danger signal, induced by biological, chemical, or physical injury, MSCs respond to maintain tissue homeostasis, favoring the repair of the tissue and reconstituting the healthy condition. During this process, MSCs come across the innate and adaptive arms of the immune system. This interaction should be highly regulated to avoid, on one hand, uncomplete repair and, on the other hand, an inefficient shut down of immune response leading to chronic inflammation (1–4). During this process, microenvironment is plenty of stimuli that, when out of control, can favor the overwhelming growth of epithelial cells with genetic alterations that are the basis of oncogenesis (1–4). Thus, the generation of a neoplasia can be dependent on the response of MSC to pathogenetic signals and to the cross-talk with immune and epithelial cells. Indeed, MSC can show immunosuppressive properties that are necessary during wound healing and repair process, but this feature is a drawback when a tumor is growing within the damaged tissue (1–4). Herein, we will briefly review the main features of MSC, from phenotype to functional properties, to clarify the molecular mechanisms whereby these cells can become immunosuppressive. Then, we will focus on the possible ways to modify MSC behavior and commute the TME from immunosuppressive to immunostimulant.
MSC: Phenotypic and Functional Characteristics
To talk about a cell type and its functional features, it is important to define their phenotypic and functional characteristics to avoid confusion among the different reports found in the literature (1–4). To simplify, a very comprehensive definition of MSC is that they are cells of mesodermal origin that are neither epithelial cells nor leukocytes (1–4). The term “MSCs” have been coined by the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy (5, 6). These “MSCs” are defined as multipotent mesenchymal cells that can be found in several different tissues (1–6) and can differentiate, under appropriate culture conditions, into adipocytes, osteoblasts, and chondrocytes (5–10). It has been shown that adipocytes and osteoblasts can be obtained from cultures of fibroblast-like cells from skin biopsies (11). Thus, it is possible that the cultures set up to select fibroblasts contain residual stem cells that in turn differentiate to other stromal cells, such as adipocytes, chondrocytes, and osteoblasts (5–11). This implies that MSC is not a synonym of mesenchymal stem cell. Also, MSC can include fibroblasts, endothelial cells, pericytes, and mesenchymal stem cells (1–4); in turn, mesenchymal stem cells are precursors of osteoblasts, chondrocytes, and adipocytes, which can be considered as MSC. On this basis, the different cell types can be distinguished for their differentiation potential and preferential production of a given component of extracellular matrix, related to the grade of differentiation (1, 3). It is not clear whether all these kinds of cells can de-differentiate to give rise to different members of MSC, in other words, what is the degree of plasticity of a differentiated MSC (Figure 1). It is conceivable that the tissue microenvironment of a given organ leads a stem cell to differentiate into a given MSC with peculiar functional properties (1–4). If this is the case, any kind of cell derived from mesenchymal stem cells should share some phenotypic and functional characteristics (Figure 1). Although several phenotypic characteristics and functional activities of MSC have been well reviewed recently (1–10), we will briefly summarize the most relevant phenotypes, found in MSC cultured in vitro, related to their function in TME.
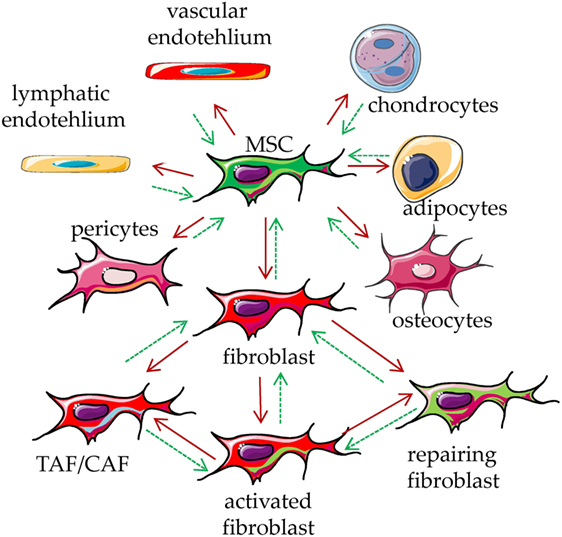
Figure 1. Mesenchymal stromal cell (MSC) plasticity. MSCs are present in every tissue, where they represent a key component characterized by the ability to differentiate into several types of mesodermal cells, including osteocytes, adipocytes, chondrocytes, endothelial cells, pericytes, and fibroblasts (red arrows). It is not clear whether all these kinds of cells can in turn de-differentiate back to MSC (green dotted arrows). The function of these cells is to maintain the homeostasis of the tissue/organ where they are present, regulating the production of the extracellular matrix components. Upon stimulation with physical, chemical, or biological stimuli, they participate in the reconstitution of the equilibrium among cellular and matrix components of a given tissue, leading to damage repair. They can be considered as sensor of the tissue conditions which can coordinate the molecular mechanisms that maintain tissue integrity. Upon influence of microenvironment, fibroblasts can lead to tumor-associated/carcinoma-associated fibroblasts (TAF/CAF), activated fibroblast and fibroblast involved in repair of the tissue.
Collectively, MSC can be identified as cells that grow adherent to plastic, with elongated-diamond (fibroblast-like) shape, expressing a definite set of markers, including CD73, CD90, and CD105, but lacking the typical hematopoietic lineage and non-lineage-specific markers, such as CD34, CD45, CD14, CD11b, CD31, CD79, CD19, and HLA-DR (2–4). In some instances, some MSC cultures show peculiar markers, such as the fibroblast activation protein (FAP) found in tumor-associated fibroblasts (1–3), but it is hard to identify subpopulations of MSC on the basis of the bimodal expression of a given antigen. In other words, it is difficult to define a MSC-specific marker, as occurs in the case of CD4+ or CD8+ lymphocytes. Indeed, although distinct fibroblast subpopulations have been reported, based on the different intensity of expression of some cell surface molecules (1–4), it is not easy to distinguish these markers by immunofluorescence. In addition, MSC can produce a variety of cytokines, chemokines, and factors, such as basic fibroblast growth factor, heparin epidermal growth factor, insulin-like growth factor (IGF) 1, keratinocyte growth factor, platelet-derived growth factor-β chain (PDGF-β), vascular endothelial growth factor (VEGF), and angiopoietins, involved in tissue repair (1–3). Indeed, the main function of MSC is thought to be the repair of injuries: this process is triggered by both differentiation of MSC in specialized tissue elements, producing peculiar extracellular matrix proteins, and regeneration of the tissue and vessel architecture (1–4). In this context, the immunosuppressive properties of MSC have been demonstrated for differentiated mesenchymal stem cells (12–15) and fibroblasts (11). Unfortunately, several MSC properties have been discovered after ex vivo expansion upon culture in vitro, so that the resulting cell population may represent a selected subset of MSC. This can also explain why findings reported from different laboratories may be conflicting (16, 17). Another relevant point to be considered is the culture ratio between MSC and tumor cells or leukocytes. Several reports have shown that the maximal inhibiting effect exerted by MSC on lymphocyte functions is achieved at MSC-lymphocyte ratios ranging from 1:1 to 1:10 (16–28). While it is possible that these ratios can be found also in situ, it is evident that in ex vivo conventional cultures the microenvironment does not dynamically change as it occurs in vivo. Indeed, in the large majority of reports, the time points chosen to analyze an inhibiting effect were set up after several days of co-culture (16–30). This implies that the vitro culture microenvironment is composed of metabolites and factors not necessarily present in situ; indeed, in vivo, blood and lymphatic vessels are involved in the clearance and renewal of the tissue milieu (31). Experimental evidence has been reported to support that MSC can display immunosuppressive behavior in vivo (32–38). However, a direct demonstration of the immunosuppression exerted by MSC is far from to be demonstrated and even the potential relevance of these cells for regenerative medicine is not unequivocally proven (32).
To summarize, MSCs are present in both healthy and neoplastic tissues as undifferentiated and differentiated cells that maintain the homeostasis with a strong relevance in regulating epithelial cells growth and immune response.
MSC and Carcinoma-Associated Fibroblasts
Mesenchymal stromal cells present in solid tumors are fibroblasts that are called carcinoma (or tumor)-associated fibroblasts (CAF or TAF) (1–4). These cells display characteristics different from MSC of healthy tissues, conceivably related to the surrounding milieu (1–4). Several factors produced by MSC, such as hepatocyte growth factor (HGF), IGF1, and FGF, in TME can interact with surface receptors on tumor cells influencing their growth (1–4). In addition, pro-angiogenic factors, such as VEGF and PDGF, produced by MSC can favor tumor cell growth indirectly, promoting the tumor niche neovascularization (1–4). Thus, it is evident the possibility of blocking tumor cell growth by inhibiting the VEGF and/or the PDGF signaling axis (39–41). Of course, also tumor and immune cells, including tumor-associated macrophages and tumor-infiltrating lymphocytes (of both the innate and the adaptive arm of the immune system) can produce these factors; thus, the block of angiogenesis can hit several components of the TME, besides MSC. MSCs are also able to release TGF-β; this cytokine can exert several opposite effects on tumor cells, depending on the type and stage of tumor (42). Indeed, TGF-β can act as a tumor promoter as well as a tumor suppressor (42); furthermore, this cytokine is a relevant factor in epithelial–mesenchymal transition (EMT), a phase of tumor life which is considered essential for the generation of cancer metastasis (42). Recently, molecular mechanisms underlining the cross-talk between MSC and carcinoma cells have been deeply reviewed (1–4, 43–47). It is of note that, besides the direct MSC–tumor cell interactions, exosomes released by MSC can contain factors, such as micro RNA (47–56), that may drive either solid tumor cell apoptosis or tumor growth and spreading.
MSC as Regulators of Immune Response
There is experimental evidence that MSC, mainly the MSC from bone marrow, can suppress immune responses in vivo (1–4, 10, 23, 24). In particular, the ability of MSC to reduce graft-versus-host disease (GVHD) has been reported (32–38). In vitro experiments have shed a light on which leukocyte populations MSC can regulate (1–4). MSC can act on both the innate arm and the adaptive arm of the immune system, blocking the expression and function of activating surface receptors on effector cells, impairing the maturation of antigen-presenting cells (APC) and favoring the expansion of regulatory cells (1–4, 12, 26, 57–67). This evidence derives from experiments where, in well-defined settings, different cells of the immune system are cocultured with a feeder layer of MSC and triggered by a given stimulus (12, 26, 68–72). Usually, such stimuli can induce proliferation, secretion of pro-inflammatory cytokines, or acquisition of a potent cytolytic potential. Upon coculture with MSC, both lymphocytes and APC are impaired in the acquisition of functional features essential to evoke a “normal” immune response (12, 26). Indeed, APC do not differentiate adequately to permit a full response to antigen-dependent or -independent stimuli (12, 26) and do not express high amounts of accessory molecules, such as CD80 and CD86, essential to deliver an optimal second signal. On the other hand, T lymphocytes express low levels of receptors, including CD25, typical of an activation state and do not respond to IL2 (12, 22, 23). The generation, in cocultures with MSC, of T cells with regulatory activities is an additional mean through which MSC can indirectly deliver an inhibiting signal to immune response (57, 58). Several papers have pointed out that different types of MSC can exert different degrees of inhibition of immune responses (1–4). In addition, differentiated MSC can still act as potent regulators of immunity (12, 72, 73). However, depending on the type of fully differentiated mesenchymal cells, pro-stimulating or pro-inhibiting effects have been described. For instance, it has been shown that mature adipocytes can trigger T cell proliferation and both HLA-DR and HLA-I appeared to be involved (74–76). Indeed, mature adipocytes express low levels of HLA-G, a surface structure responsible for the MSC-mediated T cell inhibition (76). It is of note that the ability of adipocytes to stimulate T cells was related with a stronger expression of HLA-DR and of the master transcriptional regulator CIITA factor, compared to de-differentiated adipocytes (76). On the other hand, fully differentiated chondrocytes can inhibit T cell proliferation triggered through the CD3–CD28 activating receptors, impairing CD25 expression. More importantly, chondrocytes can affect the differentiation of monocytes to dendritic cells (12). All these effects can in turn amplify each other, thus making the immunoregulatory activity of MSC really strong (Figure 2).
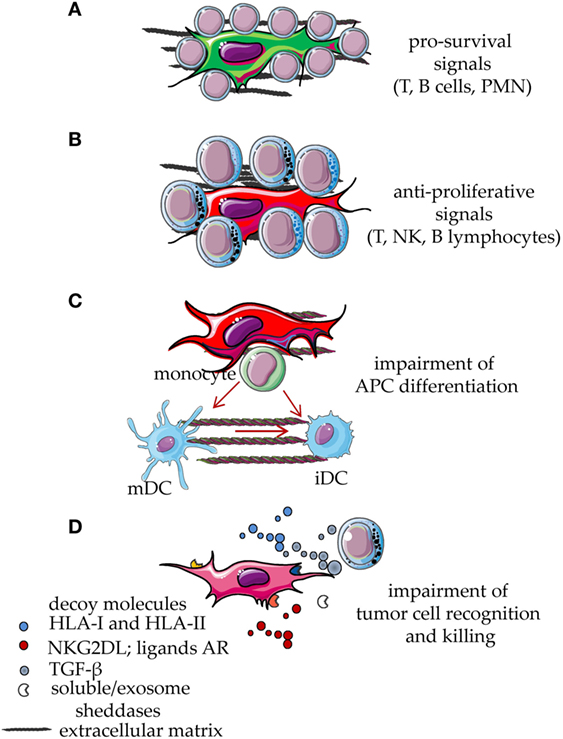
Figure 2. Functional behavior of mesenchymal stromal cells (MSCs) interacting with cells of the immune system. MSCs display several functional abilities during the interaction with cell of the immune system. Resting leukocytes are typically supported by MSC through direct cell-to-cell contact involving different types of receptor on the leukocyte membrane and the corresponding ligands expressed on MSC or on differentiated MSC. Some soluble factors and interleukins are also involved, such as stromal-derived factor 1, IL-6, and IL-15. Due to this interaction, leukocytes receive an anti-apoptotic signal which leads to their survival (A). This effect is also involved in the maintenance of the neoplastic counterpart of T, B lymphocytes and myeloid cells. After signals inducing proliferation, cytokine release, or activation of cytolytic machinery, MSCs exert a potent inhibitory effect that reduces leukocyte proliferation and effector functions (B). In addition, activated MSCs interfere with the differentiation of monocytes to immature DC (iDC) or mature DC (mDC), thus blocking the generation of professional antigen-presenting cells (APC) (C). The release via microvescicles, exosomes or in soluble form, of decoy molecules such as HLA-I, ligands for NKG2D or other activating receptors involved in tumor cell recognition and killing, hampers the anti-tumor activity of T and NK lymphocytes (D). Furthermore, MSC can release TGF-β, sheddases, such as metalloproteinases, and a disintegrin and metalloproteinase members, which can induce the release of decoy receptors from MSC, tumor cells, and bystander cells in the microenvironment. TGF-β can inhibit tumor cell recognition reducing the activation-induced increase of NKG2D expression on anti-tumor effector lymphocytes (D). All these events eventually lead to the impairment of both innate and adaptive immune responses.
Molecular Mechanisms of the Immunoregulation Mediated by MSC
Mesenchymal stromal cells regulate immune response by different means (1–4), shared with other components of the TME, such as myeloid-derived suppressor cells (MDSC), tumor cells, and infiltrating Treg lymphocytes (1–4, 77–84). Indeed, indoleamine 2,3 dioxygenase (IDO), hemeoxygenase (HO), arginase 1 and 2 (ARG1 and ARG2), nitric oxidase synthase 2 (NOS2), HGF, TGF-β, IL10, prostaglandin E2 (PGE2), and adenosine are all factors involved in the MSC-mediated regulation of innate and adaptive immunity (1–4, 23, 85–92) (Figure 3). It is of note that several of these factors are upregulated by inflammatory stimuli, such as IFN-γ (69). IDO and PGE2, are strongly induced upon inflammation, conceivably to switch off the inflammatory response to danger signals. In the TME, IDO- and PGE2-mediated immunosuppression can be the marker of a physiological response triggered to favor tissue repair, but undesired because it favors also tumor cell growth. Indeed, IDO induces kynurenine synthesis that can strongly inhibit both the innate and the adaptive immune response (93–98). Furthermore, TGF-β is not only relevant for tumor cell growth but can also directly inhibit the function of anti-tumor effector cells. This cytokine downregulates, at the surface of natural killer (NK) cells, CD8+ cytolytic T cells and γδ T cells, the expression of the NKG2D activating receptor, which in turn cannot interact with the NKG2D ligands expressed by tumor cells. These events would limit the immunosurveillance to stress signals mediated by the growing tumor (1–4, 25). In addition, TGF-β is a critical factor to generate conventional CD4+CD25high + Treg and regulatory γδ T cells (42, 99–104). Moreover, TAF expressing α-smooth muscle actin (SMA) can convert arginine in ornithine through the involvement of ARG2; this leads to the inhibition of TIL functional activities, especially in hypoxic conditions (88). PGE2 derived from NK–MSC cocultures can impair the IL-2-dependent upregulation of activating NK-cell receptors, such as members of the natural cytotoxicity receptors and DNAM-1, thus inhibiting melanoma cell recognition (20). Adenosine is an additional factor involved in MSC-mediated immunosuppression. Indeed, the ecto-5′-nucleotidase activity of CD73 expressed on MSC can catalyze the hydrolysis of the extracellular adenosine monophosphate (AMP) to adenosine. This metabolite can influence the activity of adenylyl cyclase, the synthesis of cyclic AMP and the function of PKA exerting potent immunosuppressive effects (90–92).
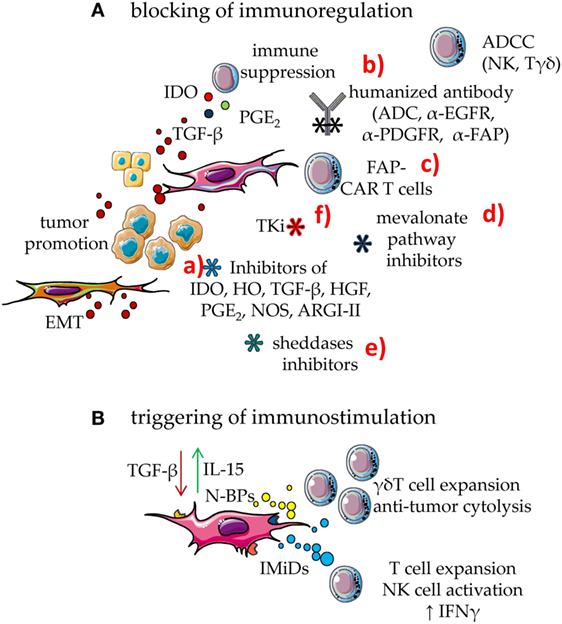
Figure 3. Means to enhance the immune response in the tumor microenvironment (TME). To counteract the mesenchymal stromal cell (MSC)-mediated downregulation of immune response, two main approaches can be utilized: (A) blocking of immunosuppressive effect; (B) triggering MSC to be immunostimulant rather than immunosuppressive. (A) MSC can downregulate immune response through several soluble factors such as indoleamine 2,3 dioxygenase (IDO) prostaglandin E2 and TGF-β. In turn, TGF-β from MSC, tumor cells, and bystander cells in TME can support tumor cell growth and dissemination. This latter event is linked to epithelial–mesenchymal transition (EMT) that triggers the generation of metastasis. The blockade of MSC immunosuppression can be obtained by several means: (a) drugs that inhibit the activity or the generation of molecules involved in immunosuppression such as inhibitors of IDO, HO, TGF-β, hepatocyte growth factor (HGF), PGE2, NOS, and ARGI–II; (b) antibodies directed either to MSC growth receptors, as the epidermal growth factor and platelet-derived growth factor (PDGF) or to the fibroblast activation protein (FAP). It is of note that some of these receptors are shared by tumor cells; thus, human or humanized antibodies-based therapy can target both MSC and cancer cells. These antibodies act inhibiting the effect of a given growth factor but also impairing the function of the target molecule. In addition, they trigger complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity (ADCC) elicited by Fcγ receptor-expressing cells, including natural killer (NK) cells and γδ T cells. These antibodies can be a portion of antibody–drug conjugates (ADC), which join the antibody-mediated effect to that of a cytotoxic drug, leading to a strong inhibition of tumor cell growth or MSC-mediated functions. (c) cytotoxic T cells equipped with chimeric antigen receptors (CARs) specific for FAP (FAP-CAR T cells) that can recognize FAP+ cells; (d) drugs affecting the mevalonate pathway that is essential for both MSC and tumor cell metabolism; unfortunately, mevalonate is relevant also for the development of an optimal immune response; they should therefore be used carefully; (e) inhibitors of sheddases, as matrix metalloproteinase and a disintegrin and metalloproteinases, which can inhibit tumor cell growth limiting the generation of growth factors in a suitable form to trigger proliferation; furthermore, these inhibitors should impair the generation of decoy molecules, reducing the competition between membrane and soluble ligands for activating receptors on effector lymphocytes; (f) tyrosine kinase inhibitors (TKi) which block the activity of MSC besides hindering tumor cell growth. (B) Immunomodulatory drugs (IMiDs), among which thalidomide, pomalidomide, lenalidomide, and avadomide can trigger the innate and the adaptive immune responses, besides hampering angiogenesis in the tumor. Aminobiphosphonates (N-BPs), such as zoledronic acid, can interfere with the mevalonate pathway strongly enhancing the production of isopentenyl pyrophosphate (IPP) and dimethyl allyl pyrophosphate (DMPP). These small pyrophosphates can trigger the expansion of γδ T cells of the Vδ2 subset, a cell population with potent anti-tumoral capabilities. Furthermore, Vδ2+ T cells express the FcγR involved in ADCC, reinforcing the anti-tumor effect of human/humanized antibodies.
Targeting MSC with Anti-Tumor Drugs
Tyrosine kinase inhibitors (TKi) are recent drugs that block the signaling cascade that follows the interaction of a growth factor with its specific receptor (105–107). It is not surprising that some TKi can affect MSC as well (Figure 3). Indeed, MSCs bear at the cell surface several receptors that can be considered as targets for tumor cell therapy with TKi. In particular, the expression on MSC of PDGFR-β and EGFR is well established; the effects of TKi such as imatinib, nilotinib, or gefitinib in vitro have pointed out that these drugs can affect both MSC proliferation and differentiation (108–122). These effects have been recently reviewed in very detail (122). It is clear from all these findings that, as expected, TKi can exert a strong inhibition on MSC growth and function, but their effects on MSC-mediated immunosuppression have not been studied. It is conceivable that the inhibition of MSC proliferation leads to the inhibition of MSC responsiveness to TME signals, but this is not determined yet. However, it has been recently shown that the encapsulated TKi sunitinib can work synergistically with vaccine therapy in an advanced mouse melanoma model, leading to the remodeling of TAF, collagen, and vessels of the tumor. Furthermore, TKi can induce a shift from Th2 to Th1 pattern of TIL, accompanied by an increment of these lymphocytes and a decrease of MDSC (123).
Targeting MSC Antigens to Modulate TME
It is now evident that the immune system can have a significant role in limiting and controlling tumor cell growth (124–131). Indeed, both adoptive and immune check point inhibitor immunotherapies are based on the possibility of triggering, either passively or actively, the specific anti-tumor immune response (124–131). A third possibility of adoptive immunotherapy is the administration of tumor vaccines; however, tumor vaccination has led to contrasting results in clinical practice (132–138). In this setting, it is attractive to target not only tumor cells but also different components of the TME (40, 41, 139–162). Indeed, specific vaccines to tumor endothelial cells or blockers of the VEGF signaling have been used in preclinical studies, and clinical trials are ongoing (40, 41). MSC can become a target for anti-tumor vaccines as well (141–162). For instance, the strong production of collagen type I by MSC can interfere with the uptake of anti-tumor drugs (149, 150); thus the targeting of MSC and the inhibition of extracellular matrix components can render more sensitive tumor cells to chemotherapy. Furthermore, antigens shared by tumor cells and TAF can be good targets for a vaccine. The fibroblast activation protein (FAP), a member of the serine protease family, can be expressed by TAF at higher levels than on resident fibroblast of healthy tissue. In addition, FAP can be also expressed by tumor cells; this would imply that an immune-based therapy focused on FAP can beat both tumor cells and TAF (140, 143–162). Indeed, it has been shown, in a murine model, that FAP+ tumor cells can be used as a vaccine, leading to reduced vascular dissemination and elimination of different tumors. In the same model, tumor-infiltrating CD8+ T cells increased and a net decrease of intratumor TAF, accompanied by a reduced recruitment of cells with immunosuppressive phenotype, was found in treated animals (144). In this context, the use of the humanized anti-FAP monoclonal antibody sibrotuzumab has been proposed in non-small cell lung and colorectal cancer (CRC), but the pilot study in CRC did not reach the minimal requirements for the continuation of the trial (163–166). However, FAP has been considered as a target for redirected T cells or chimeric antigen receptor (CAR) T cells (158, 159, 162) (Figure 3). It has been reported that transfer of murine T cells transduced with FAP-CAR construct can affect tumor cell growth increasing the CD8+ T cell response. Also, the administration of anti-fibrotic agents, in several murine tumor models (E-G7 lymphoma, LLC1 Lewis lung cancer, or B16F1 melanoma) induced a strong increment of CD8+ T cells, NK activity, and humoral immunity and a sharp decrease of MDSC, Treg, stromal-derived factor 1, TGF-β, and PGE2 (162).
Can MSC Counteract Cancer Development and Growth?
Taking together the findings reported, it appears clear that MSC as TAF should be a mean by which tumor cells are facilitated in their growth and spreading. Thus, the higher is the content of TAF in a given tumor, the faster will be the expansion of that tumor. TAF elimination leads to an enhancement of immune response and, at the same time, to a lower support of tumor cell growth. By contrast, recent evidence in pancreatic ductal adenocarcinoma (PDAC) indicates, that the depletion of αSMA+ myofibroblast, in a murine model can trigger tumor cell expansion and paradoxically accelerate disease progression (167). In addition, this depletion led to an increment of regulatory T cells without affecting NK cell infiltration. This was accompanied by a strong remodeling of the extracellular matrix composition and the therapy with CTLA-4 immune check point inhibitors could rescue the detrimental effect due to myofibroblast depletion. Furthermore, it appeared that the lower was the number of αSMA+ myofibroblast in human PDAC, the worse was the prognosis of patients (167). How to explain this unexpected effect? The simplest explanation is that the reaction due to αSMA+ myofibroblast represents a tool by which healthy MSC try to repair tissue and limit the expansion of PDAC, as suggested for other malign tumors (168–183). This phenomenon is known as desmoplastic reaction, which serves to repair tissue injury (175–182). It is conceivable that, at the onset of tumor growth, fibroblasts may function also as a physical barrier to tumor expansion. During tumor growth, due to the presence of subclones and/or cancer stem cells, this barrier can be modified by reciprocal cross-talk between tumor components and MSC. An additional explanation is that within αSMA+ myofibroblast are present subsets of cells with different functional behaviors, with either positive or negative effects on tumor cell growth. After depletion of all αSMA+ myofibroblast, these populations are lost and PDAC can grow without any brake (168, 173, 175, 177, 183). In such TME, immune system can receive misleading information with conflicting, undesired outcomes. Recently, it has been shown that NK cells can recognize and eliminate pancreatic stellate cells, bona fide myofibroblasts (171); this would suggest that innate immunity, in this case, can favor rather than inhibit tumor cell expansion by limiting stromal reaction.
Research Gaps and Future Developments
At present, targeting MSC is complicated by the fact that a specific marker of these cells is missing (1–4). Indeed, MSCs have the property to differentiate and it is not clear whether there is also an intrinsic de-differentiation potential (1–4); these functional/plastic properties can impair the efficacy of a drug specific for a given MSC subpopulation. In addition, from data obtained in PDAC, it is clear that MSC can aid the host against cancer evolution. Finally, MSCs are present in each tissue and represent the key cell involved in the maintenance of the structural architecture of the whole body. Thus, therapeutic targeting of MSC should be made very carefully.
Targeting MSC with Antibodies
All the above reported matters render the targeting of MSC not as specific as desired and possibly accompanied by relevant drawbacks. By contrast, tumor cell targeting can be more specific, since the marker used as target is more expressed in tumor cells than in their healthy counterpart. For instance, in Hodgkin lymphoma and non-Hodgkin lymphomas (NHL), tumor targeting can be really efficient (184–187). Indeed, in these instances, administration of therapeutic antibodies to CD30 or CD20 molecules can spare the healthy counterpart of B cells, because the target antigen is not expressed or is expressed at low levels. Also, B lymphocyte precursors can substitute the bystander healthy B cells damaged by target therapy (184–187). An additional relevant question is whether therapies aimed to eliminate cancer cells have also an effect on MSC. Indeed, humanized monoclonal antibodies (huAb), directed to receptors involved in the proliferation of tumor cells, including EGFR or Her2b, may hit MSCs that share these molecules at the cell surface (Figure 3). MSC targeting might be useful, on the one hand, but the availability of the therapeutic huAb can be reduced. Moreover, it is conceivable that anti-EGFR and/or anti-HerB2 huAb can affect MSC-tumor cell cross-talk due to the signal delivered upon huAb/receptor interaction (188, 189). The study of this interaction can shed new light on the reported unexpected effects observed with huAb therapy in some type of cancers, among which is CRC (190–193). As reported above, targeting FAP+ TAF, or αSMA+ myofibroblast has elicited unexpected drawbacks, since these cells can also function as negative regulators of cancer cell growth (167). The definition of subsets of MSC, myofibroblasts and even TAF, using a specific marker is a prerequisite to selectively hit the population that can favor the tumor cell growth and inhibit anti-tumor immune cell response. In this context, besides FAP, CD73, and CD105 (90, 91, 141–162), the finding that fibroblasts present in scar tissue and basal cell carcinoma express gremlin1, the secreted bone morphogenetic protein antagonist, would suggest that this can be a specific molecular target to distinguish TAF from healthy fibroblasts (194).
Interference with EMT and Role of MSC
It is well known that EMT is a key step of the spreading of cancer cells far from the primary tumor (100, 101, 104, 195) (Figure 3). TGF-β plays a relevant role in EMT (101, 104, 194, 196–204); thus, it is conceivable that the blockade of TGF-β production by MSC can impair EMT (197). Some evidence is reported on the prometastatic effect of CAF in different types of cancer (198, 199, 202, 203). It is of note that EMT can be also triggered by anti-EGFR huAb therapy in squamous cell carcinoma of the head and neck. Indeed, it has been reported that cetuximab therapy can induce modifications in the expression of genes and proteins implicated both in EMT and in the extracellular matrix production by CAF (201). Importantly, upregulation of CXCL12, ASPN, and OLFM3, factors secreted by CAF, has been observed; CXCL12, through the interaction with its receptor CXCR4, can lead to CXCL12 and TGFβ production and concur to myelofibrosis (205). One can speculate that EGFR signaling can drive TME to generate therapy resistance involving CAF. Targeting CAF to reduce production and release of TGFβ, CXCL12, and matrix metalloproteinases (MPP) can limit cancer cell spreading favored by TGFβ and MPP and the anti-apoptotic effect of CXCL12 on tumor cells. Unfortunately, the clinical use of inhibitors of TGFβ and MPP is far from to be well established, although the interference with CXCR4/CXCL12 axis, using AMD3100 or huAb, is already applied in several clinical trials (204–210).
Targeting Immunosuppressive Molecular Mechanisms of MSC with Inhibitory Drugs
The interference with MSC-mediated immunosuppressive molecular mechanisms, obtained using specific inhibitory drugs, is an additional mean by which the immune escape favored by tumor MSC can be avoided (1–4). In this context, all the inhibitors already used in therapeutic schemes to block IDO, HO, ARGI and II, NOS2, PGE2, and TGF-β activity can be employed to reduce MSC influence on tumor cell growth (211–217) (Figure 3). In this context, the immune check point inhibitors anti-PD1 and/or PDL-1 huAb can have an important role (132, 137, 218–220). Indeed, it has been shown that PD1 is involved in MSC immunoregulation of T and B cell proliferation (18, 221, 222). The striking therapeutic effect observed upon blockade of PD1–PDL-1 with huAb can be dependent not only on the direct effect on tumor cell–effector lymphocyte interaction, but also on the switch off of the inhibiting signal elicited by PD1–PDL-1 binding during lymphocyte–MSC interaction. PDL-1 expression is upregulated on MSC by IFNγ and this cytokine can upregulate IDO as well (223); this suggests that the combination of IDO and immune check point inhibitors can concur to overcome TME immunosuppression (224). Some drugs, such as hydroxy-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors, can influence both immunosuppressive effects and cancer pro-survival signals delivered by MSC (28, 225) (Figure 3). Furthermore, it is clear that mevalonate, the metabolic product of the HMG-CoA reductase activity, is a key molecule for tumor cell fate (226). However, limiting mevalonate production can influence the functional behavior of macrophages and lead to regulatory T cell expansion, thus favoring tumor cell spreading (227). In addition, anti-tumor effector cell-mediated lytic activity is strongly reduced by HMG-CoA reductase inhibition (228–232). This can be related to the decrease of cholesterol content in lymphocyte membrane that limits the formation of rafts; these rafts are essential in the delivery of the activating signals that lead to granzyme and perforin release, upon effector–target interaction (232, 233), and consequent target cell killing. Thus, it is relevant to design inhibitors of mevalonate pathway that can be delivered specifically to MSC in order to limit tumor cell growth sparing immune surveillance.
Drugs to Transform MSC from Immunosuppressive to Immunostimulant
Another approach to downregulate the inhibitory effect of MSC on immune system is to convert their behavior from immunosuppressive to immunostimulant. Recently, it has been demonstrated, both in NHL and CRC, that priming of MSC, derived from lymph nodes or colon mucosa, with the aminobisphosphonate (N-BP) zoledronic acid can trigger Vδ2 T cell proliferation (25, 234, 235). In NHL, zoledronate-pulsed MSC are impaired in the secretion of TGF-β, whereas there is an increment in the production of IL-15 (234) (N-BPs in Figure 3). It should be defined whether priming with zoledronate can favor the expansion of other anti-tumor effector cells that are inhibited by MSC and whether MSC can become a target of Vδ2 T cells. If this is the case, the specific delivery of zoledronic acid to the lymph node TME would trigger anti-tumor immunity. It is well known that N-BPs have a strong tropism to bone (236); for this reason they are commonly used to treat neoplasias primarily localized in the bone, such as multiple myeloma, or bone metastases of different carcinomas (237–239). In these instances, N-BPs have a dual effect: support the deposition of bone matrix to repair the osteolytic damage induced by tumor cells and trigger γδ T cell-mediated anti-tumor immune response (237–241). When tumors are localized in other tissues, a major issue for the administration of N-BPs is to efficiently target the tumor outside the bone. It can be hypothesized that the generation of antibody–drug conjugates (ADC) (242), made of huAb linked to N-BPs, can be a good tool to deliver N-BPs to a specific tumor site. So far, ADC have been developed with huAb specific for a tumor marker linked to cytotoxic drugs, the specificity of the antibody being the key parameter to maximize anti-tumor effect. It is conceivable that also the linkage of immunostimulant drugs to huAb specific for tumor cells and MSC can combine the specifity for the target with the triggering of anti-tumor γδ T cell immune response.
Immunomodulatory drugs (IMiDs), from the first described thalidomide to the recent reported avadomide (CC-122) (Figure 3), can affect both directly and indirectly tumor cell growth (243–251). Indeed, it has been reported that IMiDs can impair cereblon, a ubiquitin ligase constitutive in every cell type but crucial for cancer cell survival, causing mis-regulation of developmental signaling molecules and generation of reactive oxygen species, which in turn kill tumor cells. Furthermore, IMiDs inhibit tumor neoangiogenesis leading to the reduction of tumor cell growth. IMiDs can also modulate NK cell number and function, besides co-stimulate T cell proliferation; these effects have led to their use in multiple myeloma and several types of lymphomas. IMiDs administration has been proven to be effective in clinical trials, because these compounds can hit different components of the TME, including MSC (124, 139, 239, 243–248). It is conceivable that a progressively larger application to several kinds of solid tumors, since these drugs have shown remarkable effects in CRC and sarcomas (249–272). Importantly, in the bone marrow microenvironment, IMiDs inhibit the production of IL-6, essential for myeloma cell growth, by regulating SOCS1 (273). In addition, these compounds affect osteoblast differentiation, indicating that bone anabolic therapeutics are needed in myeloma to counteract the negative effect on bone metabolism of IMiD exposure. In this instance, the use of N-BPs can favor the deposition of bone matrix, thus limiting the damage induced by IMiDs.
Drugs to Interfere with the Generation of Decoy Receptors from MSC and Tumor Cells
MSC can release the MHC-class-I related molecules MIC-A, MIC-B, and the UL16-binding proteins (ULBPs) into TME, through the enzymatic activity of members of the a disintegrin and metalloproteinases (ADAMs) family (274–279). These released NKG2D-L can function as decoy ligands blocking the NKG2D-mediated recognition of cancer cells that usually express them on the cell membrane (277–281) (Figure 3). It is reasonable that ADAM10 and ADAM17 in MSC can act on such stressed molecules expressed not only by MSC but also by other cells present in TME. In addition, ADAMs can be released in exosomes and microvescicles by MSC, thus spreading their enzymatic activity. This would imply that ADAMs inhibitors can reduce the MSC-mediated release of stress molecules, allowing cancer cell recognition by immune cells and eventually leading to an increment of tumor cell killing (280). In this context, it is becoming evident that the analysis of MSC secretome is highly relevant to understand the physiological and pathological behavior of these cells (282). The targeting of ADAMs inhibitors to TME could be achieved again, using ADC which recognize MSC and/or tumor cells. Importantly, the delivery to MSC of drugs, such as N-BPs and ADAMs inhibitors, either alone or in combination with huAb as ADC, can take advantage of nanotechnology (283–285). Nanovectors can be artificially built with different morphology and physico-chemical properties (283–285). The choice of these parameters is relevant to design the optimal combination and obtain the maximal effect (283–285).
The New Frontier of Three-Dimensional (3D) Models: To Study the Interactions among MSC, Tumor Cell, and the Immune System
The study of the functional cross-talk among MSC, tumor cells, and the immune system can be more reliable using 3D models instead of classical in vitro culture systems (272, 286–299). Indeed, in these 3D models, the control of cell culture conditions and the regulation of biomechanical stimuli can give relevant insight on how biophysical cues can influence stromal cell phenotype and function; this can clarify how these modifications impact on tumor drug sensitivity. In addition, the cross-talk of tumor and stromal cells with immune cells can be studied in detail, varying the experimental conditions in a setting that reproduces tissue architecture; this can spare time, limit the costs of animal experimentation and reduce the environmental impact of animal breeding farms (290, 295, 297, 299). These culture systems, validated by the EU Reference Laboratories (EURL-ECVAM) as preclinical models, are reproducible 3D culture microenvironments useful for studying pharmaceuticals or biological pathways (300, 301). Among them, the hydrogels of matrix components, such as collagen, fibronectin, or cell derivates such as Matrigel or amorphic scaffold have been used (286, 293, 295). More recently, in multiple myeloma a model that recapitulates the interactions among MSC, myeloma cells, endothelial cells, and bone remodeling has been set up in order to analyze dynamically the cross-talk among all these cell populations (273). Indeed, this 3D model uses silk protein-based scaffolds that allow active cell attachment and growth on the scaffolds, rather than passive encapsulation in 3D hydrogel cultures. This represents a unique model to analyze under mechanical stress, similar to the bone tissue, the interactions of cancer cells and bone in a 3D microenvironment. The interaction among tumor cells, anti-tumor lymphocytes and MSC can be achieved in different 3D experimental setting as tumor spheroids, organoids and 3D on-chip cell cultures (291, 297, 301, 302). The 3D models where metabolic microenvironment is dynamically changed are essential to confirm the findings obtained in the murine system regarding the role of PDL-1 blocking in tumor metabolism (303, 304). Infact, in a mouse sarcoma model, it has been shown that glucose consumption by tumors can metabollically inhibit T cell responses, impairing glycolytic activity and IFNγ production. More importantly, anti-PDL-1 antibodies can block tumor glucose utilization favoring T cell glycolysis and IFNγ release (304). To validate these findings and further analyze the mechanisms of regulation of metabolism of immune cells humanized mice can be employed (305). However, these mice are engrafted with human hematopoietic stem cells and, for this reason, should be immunodeficient. Although this model can aid in mimicking the pathophysiological conditions of human beings, it is evident that the large majority of TME is composed of murine cells. On the contrary, organoids of tumors from patients’ specimens can be obtained and analyzed in detail (306–309). For instance, it has been recently shown that human intestinal organoids can be generated and used, not only for research purposes but even to treat intestinal injury (310). In addition, bioprinting techniques have led to the biofabrication of accurate models that can recreate the biophysical and biochemical characteristics of a given tissue (292). Thus, in the near future, the cross-talk among the different components of the TME will be analyzed using more and more precise 3D models and organoids from a given patient to test the sensitivity to selected targeted therapy (289, 294, 302, 306, 311–313).
Concluding Remarks
It is now clear that MSC represent a key player in regulating TME through direct cell-to-cell interactions, producing several cytokines and releasing exosomes (314–322). The secretome of MSC can play an important role in immunosuppression (319, 320): its modification with drugs can represent a new tool for drug delivery and cell-free regeneration after tumor injury (314–318, 321, 322). Because of the lack of specific markers that identify subsets of MSC, the specific targeting of these cells appears to be difficult, to achieve selective inhibition of immunosuppression. Furthermore, it is still to be elucidated whether different subsets of MSC, due to their plasticity, can represent functional subsets of cancer-associated fibroblasts (323–328). This would imply that a specific marker for the immunosuppressive MSC will be still elusive for a long time. Nevertheless, it is conceivable that drug combination therapies of cancer, which limit, on the one hand, tumor cell proliferation and, on the other hand, trigger immune responses, which already involve MSC. The in situ analysis of MSC functional features, together with their study in 3D tumor culture systems, would allow to clarify the existence in humans of MSC subsets and to assess the effects of drug treatment in order to choose the right combination of therapeutic means for each patient.
Author Contributions
AP, SV, and MZ wrote, edited, and revised the paper. AP takes primary responsibility of the manuscript content.
Conflict of Interest Statement
The authors declare no conflict of interest and the founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Acknowledgments
This work has been partially supported by grants from AIRC to AP (IG15483) and AIRC to MZ (IG17074) and from 5xmille 2012 Ministero della Salute, 5xmille 2013 Ministero della Salute, 5xmille 2014 Ministero della Salute, and 5xmille 2015 Ministero della Salute to AP. Figures were produced using Servier Medical Art: www.servier.com.
References
1. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16:582–98. doi:10.1038/nrc.2016.73
2. Poggi A, Musso A, Dapino I, Zocchi MR. Mechanisms of tumor escape from immune system: role of mesenchymal stromal cells. Immunol Lett (2014) 159:55–72. doi:10.1016/j.imlet.2014.03.001
3. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol (2015) 15:669–82. doi:10.1038/nri3902
4. Poggi A, Giuliani M. Mesenchymal stromal cells can regulate the immune response in the tumor microenvironment. Vaccines (Basel) (2016) 4. doi:10.3390/vaccines4040041
5. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy (2006) 8:315–7. doi:10.1080/14653240600855905
6. Horwitz EM, Le Blanc K, Dominici M, Mueller I, Slaper-Cortenbach I, Marini FC, et al. Clarification of the nomenclature for MSC: the International Society for Cellular Therapy position statement. Cytotherapy (2005) 7:393–5. doi:10.1080/14653240500319234
7. Friedenstein AJ, Chailakhyan RK, Gerasimov UV. Bone marrow osteogenic stem cells: in vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet (1987) 20:263–72.
9. Raffaghello L, Dazzi F. Classification and biology of tumour associated stromal cells. Immunol Lett (2015) 168:175–82. doi:10.1016/j.imlet.2015.06.016
10. Haniffa MA, Collin MP, Buckley CD, Dazzi F. Mesenchymal stem cells: the fibroblasts’ new clothes? Haematologica (2009) 94:258–63. doi:10.3324/haematol.13699
11. Jones S, Horwood N, Cope A, Dazzi F. The antiproliferative effect of mesenchymal stem cells is a fundamental property shared by all stromal cells. J Immunol (2007) 179:2824–31. doi:10.4049/jimmunol.179.5.2824
12. Pereira RC, Martinelli D, Cancedda R, Gentili C, Poggi A. Human articular chondrocytes regulate immune response by affecting directly T cell proliferation and indirectly inhibiting monocyte differentiation to professional antigen-presenting cells. Front Immunol (2016) 7:415. doi:10.3389/fimmu.2016.00415
13. Adkisson HD, Milliman C, Zhang X, Mauch K, Maziarz RT, Streeter PR. Immune evasion by neocartilage-derived chondrocytes: implications for biologic repair of joint articular cartilage. Stem Cell Res (2010) 4:57–68. doi:10.1016/j.scr.2009.09.004
14. Bocelli-Tyndall C, Barbero A, Candrian C, Ceredig R, Tyndall A, Martin I. Human articular chondrocytes suppress in vitro proliferation of anti-CD3 activated peripheral blood mononuclear cells. J Cell Physiol (2006) 209:732–4. doi:10.1002/jcp.20789
15. Lohan P, Treacy O, Lynch K, Barry F, Murphy M, Griffin MD, et al. Culture expanded primary chondrocytes have potent immunomodulatory properties and do not induce an allogeneic immune response. Osteoarthritis Cartilage (2016) 24:521–33. doi:10.1016/j.joca.2015.10.005
16. Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, et al. Human mesenchymal stem cells modulate B-cell functions. Blood (2006) 107:367–72. doi:10.1182/blood-2005-07-2657
17. Traggiai E, Volpi S, Schena F, Gattorno M, Ferlito F, Moretta L, et al. Bone marrow-derived mesenchymal stem cells induce both polyclonal expansion and differentiation of B cells isolated from healthy donors and systemic lupus erythematosus patients. Stem Cells (2008) 26:562–9. doi:10.1634/stemcells.2007-0528
18. Augello A, Tasso R, Negrini SM, Amateis A, Indiveri F, Cancedda R, et al. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol (2005) 35:1482–90. doi:10.1002/eji.200425405
19. Li T, Yi S, Liu W, Jia C, Wang G, Hua X, et al. Colorectal carcinoma-derived fibroblasts modulate natural killer cell phenotype and antitumor cytotoxicity. Med Oncol (2013) 30:663. doi:10.1007/s12032-013-0663-z
20. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A (2009) 106:20847–52. doi:10.1073/pnas.0906481106
21. Pradier A, Passweg J, Villard J, Kindler V. Human bone marrow stromal cells and skin fibroblasts inhibit natural killer cell proliferation and cytotoxic activity. Cell Transplant (2011) 20:681–91. doi:10.3727/096368910X536545
22. Haniffa MA, Wang XN, Holtick U, Rae M, Isaacs JD, Dickinson AM, et al. Adult human fibroblasts are potent immunoregulatory cells and functionally equivalent to mesenchymal stem cells. J Immunol (2007) 179:1595–604. doi:10.4049/jimmunol.179.3.1595
23. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood (2002) 99:3838–43. doi:10.1182/blood.V99.10.3838
24. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood (2005) 105:1815–22. doi:10.1182/blood-2004-04-1559
25. Zocchi MR, Catellani S, Canevali P, Tavella S, Garuti A, Villaggio B, et al. High ERp5/ADAM10 expression in lymph node microenvironment and impaired NKG2D-ligands recognition in Hodgkin lymphomas. Blood (2012) 119:1479–89. doi:10.1182/blood-2011-07-370841
26. Spaggiari GM, Capobianco A, Becchetti S, Mingari MC, Moretta L. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood (2006) 107:1484–90. doi:10.1182/blood-2005-07-2775
27. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood (2008) 111:1327–33. doi:10.1182/blood-2007-02-074997
28. Musso A, Zocchi MR, Poggi A. Relevance of the mevalonate biosynthetic pathway in the regulation of bone marrow mesenchymal stromal cell-mediated effects on T-cell proliferation and B-cell survival. Haematologica (2011) 96:16–23. doi:10.3324/haematol.2010.031633
29. Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood (2004) 103:4619–21. doi:10.1182/blood-2003-11-3909
30. Najar M, Rouas R, Raicevic G, Boufker HI, Lewalle P, Meuleman N, et al. Mesenchymal stromal cells promote or suppress the proliferation of T lymphocytes from cord blood and peripheral blood: the importance of low cell ratio and role of interleukin-6. Cytotherapy (2009) 11:570–83. doi:10.1080/14653240903079377
31. Wittkowske C, Reilly GC, Lacroix D, Perrault CM. In vitro bone cell models: impact of fluid shear stress on bone formation. Front Bioeng Biotechnol (2016) 4:87. doi:10.3389/fbioe.2016.00087
32. Parekkadan B, Milwid JM. Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng (2010) 12:87–117. doi:10.1146/annurev-bioeng-070909-105309
33. Koç ON, Gerson SL, Cooper BW, Dyhouse SM, Haynesworth SE, Caplan AI, et al. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol (2000) 18:307–16. doi:10.1200/JCO.2000.18.2.307
34. Lee ST, Jang JH, Cheong JW, Kim JS, Maemg HY, Hahn JS, et al. Treatment of high-risk acute myelogenous leukaemia by myeloablative chemoradiotherapy followed by co-infusion of T cell-depleted haematopoietic stem cells and culture-expanded marrow mesenchymal stem cells from a related donor with one fully mismatched human leucocyte antigen haplotype. Br J Haematol (2002) 118:1128–31. doi:10.1046/j.1365-2141.2002.03767.x
35. Kim N, Im KI, Lim JY, Jeon EJ, Nam YS, Kim EJ, et al. Mesenchymal stem cells for the treatment and prevention of graft-versus-host disease: experiments and practice. Ann Hematol (2013) 92:1295–308. doi:10.1007/s00277-013-1796-z
36. Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY, et al. Isolated allogeneic bone-marrow derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci U S A (2002) 99:8932–7. doi:10.1073/pnas.132252399
37. Le Blanc K. Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy (2003) 5:485–9. doi:10.1080/14653240310003611
38. Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hussan M, Uzunel M, et al. Treatment of severe acute graft-versus host disease with third party haploidentical mesenchymal stem cells. Lancet (2004) 363:1439–41. doi:10.1016/S0140-6736(04)16104-7
39. Shaked Y. Balancing efficacy of and host immune responses to cancer therapy: the yin and yang effects. Nat Rev Clin Oncol (2016) 13(10):611–26. doi:10.1038/nrclinonc.2016.57
40. Zhao Y, Adjei AA. Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. Oncologist (2015) 20:660–73. doi:10.1634/theoncologist.2014-0465
41. Martin-Broto J, Hindi N. Targeted treatments of sarcomas and connective tumors beside gastrointestinal stromal tumor. Curr Opin Oncol (2016) 28:338–44. doi:10.1097/CCO.0000000000000302
42. Yang L. TGFbeta, a potent regulator of tumor microenvironment and host immune response, implication for therapy. Curr Mol Med (2010) 10:374–80. doi:10.2174/156652410791317039
43. Lazennec G, Jorgensen C. Concise review: adult multipotent stromal cells and cancer: risk or benefit? Stem Cells (2008) 26:1387–94. doi:10.1634/stemcells.2007-1006
44. Yang Y, Bucan V, Baehre H, von der Ohe J, Otte A, Hass R. Acquisition of newtumor cell properties by MSC-derived exosomes. Int J Oncol (2015) 47:244–52. doi:10.3892/ijo.2015.3001
45. Yang Y, Otte A, Hass R. Human mesenchymalstroma/stem cells exchange membrane proteins and alter functionality during interaction with different tumor cell lines. Stem Cells Dev (2015) 24:1205–22. doi:10.1089/scd.2014.0413
46. Shi S, Zhang Q, Xia Y, You B, Shan Y, Bao L, et al. Mesenchymal stem cell-derived exosomes facilitate nasopharyngeal carcinoma progression. Am J Cancer Res (2016) 6:459–72.
47. Melzer C, Yang Y, Hass R. Interaction of MSC with tumor cells. Cell Commun Signal (2016) 14:20. doi:10.1186/s12964-016-0143-0
48. Nargesi AA, Lerman LO, Eirin A. Mesenchymal stem cell-derived extracellular vesicles for renal repair. Curr Gene Ther (2017) 17:29–42. doi:10.2174/1566523217666170412110724
49. Zhang X, Tu H, Yang Y, Fang L, Wu Q, Li J. Mesenchymal stem cell-derived extracellular vesicles: roles in tumor growth, progression, and drug resistance. Stem Cells Int (2017) 2017:1758139. doi:10.1155/2017/1758139
50. Clark EA, Kalomoiris S, Nolta JA, Fierro FA. Concise review: microRNA function in multipotent mesenchymal stromal cells. Stem Cells (2014) 32:1074–82. doi:10.1002/stem.1623
51. Yang F, Ning Z, Ma L, Liu W, Shao C, Shu Y, et al. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol Cancer (2017) 16:148. doi:10.1186/s12943-017-0718-4
52. Schoepp M, Ströse AJ, Haier J. Dysregulation of miRNA expression in cancer associated fibroblasts (CAFs) and its consequences on the tumor microenvironment. Cancers (Basel) (2017) 24:9. doi:10.3390/cancers9060054
53. Sansone P, Berishaj M, Rajasekhar VK, Ceccarelli C, Chang Q, Strillacci A, et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res (2017) 77:1927–41. doi:10.1158/0008-5472
54. Donnarumma E, Fiore D, Nappa M, Roscigno G, Adamo A, Iaboni M, et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget (2017) 8:19592–608. doi:10.18632/oncotarget
55. Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene (2017) 36:1770–8. doi:10.1038/onc.2016.353
56. Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, et al. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci (2015) 106:1362–9. doi:10.1111/cas.12747
57. Prevosto C, Zancolli M, Canevali P, Zocchi MR, Poggi A. Generation of CD4+ or CD8+ regulatory T cells upon mesenchymal stem cell-lymphocyte interaction. Haematologica (2007) 92:881–8. doi:10.3324/haematol.11240
58. Maccario R, Podestà M, Moretta A, Cometa A, Comoli P, Montagna D, et al. Interaction of human mesenchymal stem cells with cells involved in alloantigen-specific immune response favors the differentiation of CD4+ T-cell subsets expressing a regulatory/suppressive phenotype. Haematologica (2005) 90:516–25. doi:10.1016/j.bbmt.2005.08.039
59. Poggi A, Prevosto C, Massaro AM, Negrini S, Urbani S, Pierri I, et al. Interaction between human NK cells and bone marrow stromal cells induces NK cell triggering: role of NKp30 and NKG2D receptors. J Immunol (2005) 175:6352–60. doi:10.4049/jimmunol.175.10.6352
60. Najar M, Raicevic G, Fayyad-Kazan H, De Bruyn C, Bron D, Toungouz M, et al. Immune-related antigens, surface molecules and regulatory factors in human-derived mesenchymal stromal cells: the expression and impact of inflammatory priming. Stem Cell Rev (2012) 8:1188–98. doi:10.1007/s12015-012-9408-1
61. Luz-Crawford P, Kurte M, Bravo-Alegría J, Contreras R, Nova-Lamperti E, Tejedor G, et al. Mesenchymal stem cells generate a CD4+CD25+Foxp3+ regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res Ther (2013) 4:65. doi:10.1186/scrt216
62. Zhou Y, Singh AK, Hoyt RF Jr, Wang S, Yu Z, Hunt T, et al. Regulatory T cells enhance mesenchymal stem cell survival and proliferation following autologous cotransplantation in ischemic myocardium. J Thorac Cardiovasc Surg (2014) 148:1131–7. doi:10.1016/j.jtcvs.2014.06.029
63. Reinders ME, Hoogduijn MJ. NK cells and MSCs: possible implications for MSC therapy in renal transplantation. J Stem Cell Res Ther (2014) 4:1000166. doi:10.4172/2157-7633.1000166
64. Fu B, Tian Z, Wei H. Subsets of human natural killer cells and their regulatory effects. Immunology (2014) 141:483–9. doi:10.1111/imm.12224
65. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol (2012) 30:221–41. doi:10.1146/annurev-immunol-020711-074934
66. Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev (2008) 224:201–14. doi:10.1111/j.1600-065X.2008.00661.x
67. Peng Y, Chen X, Liu Q, Zhang X, Huang K, Liu L, et al. Mesenchymal stromal cells infusions improve refractory chronic graft versus host disease through an increase of CD5+ regulatory B cells producing interleukin 10. Leukemia (2015) 29:636–46. doi:10.1038/leu.2014.225
68. Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol (2012) 12:383–96. doi:10.1038/nri3209
69. Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A, et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells (2006) 24:386–98. doi:10.1634/stemcells.2005-0008
70. Prasanna SJ, Gopalakrishnan D, Shankar SR, Vasandan AB. Pro-inflammatory cytokines, IFN-gamma and TNFalpha, influence immune properties of human bone marrow and Wharton jelly mesenchymal stem cells differentially. PLoS One (2010) 5. doi:10.1371/journal.pone.0009016
71. Ryan JM, Barry F, Murphy JM, Mahon BP. Interferon-gamma does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin Exp Immunol (2007) 149:353–63. doi:10.1111/j.1365-2249.2007.03422.x
72. Kronsteiner B, Wolbank S, Peterbauer A, Hackl C, Redl H, van Griensven M, et al. Human mesenchymal stem cells from adipose tissue and amnion influence T-cells depending on stimulation method and presence of other immune cells. Stem Cells Dev (2011) 20:2115–26. doi:10.1089/scd.2011.0031
73. Mazanet MM, Hughes CC. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol (2002) 169:3581–8. doi:10.4049/jimmunol.169.7.3581
74. Poloni A, Maurizi G, Leoni P, Serrani F, Mancini S, Frontini A, et al. Human dedifferentiated adipocytes show similar properties to bone marrow-derived mesenchymal stem cells. Stem Cells (2012) 30:965–74. doi:10.1002/stem.1067
75. Poloni A, Maurizi G, Ciarlantini M, Medici M, Mattiucci D, Mancini S, et al. Interaction between human mature adipocytes and lymphocytes induces T-cell proliferation. Cytotherapy (2015) 17:1292–301. doi:10.1016/j.jcyt.2015.06.007
76. Maurizi G, Della Guardia L, Maurizi A, Poloni A. Adipocytes properties and crosstalk with immune system in obesity-related inflammation. J Cell Physiol (2018) 233:88–97. doi:10.1002/jcp.25855
77. Wang Z, Till B, Gao Q. Chemotherapeutic agent-mediated elimination of myeloid-derived suppressor cells. Oncoimmunology (2017) 16:6. doi:10.1080/2162402X.2017.1331807
78. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol (2017) 7:93. doi:10.3389/fimmu.2017.00093
79. Zhang H, Maric I, DiPrima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood (2013) 122:1105–13. doi:10.1182/blood-2012-08-449413
80. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37:208–20. doi:10.1016/j.it.2016.01.004
81. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol (2017) 17:703–17. doi:10.1038/nri.2017.75
82. Maccalli C, Parmiani G, Ferrone S. Immunomodulating and immunoresistance properties of cancer-initiating cells: implications for the clinical success of immunotherapy. Immunol Invest (2017) 46:221–38. doi:10.1080/08820139.2017.1280051
83. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting from immunosurveillance to tumour escape. Nat Immunol (2002) 3:991–8. doi:10.1038/ni1102-991
84. Von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol (2005) 6:338–43. doi:10.1038/ni1180
85. Mougiakakos D, Jitschin R, Johansson CC, Okita R, Kiessling R, Le Blanc K. The impact of inflammatory licensing on heme oxygenase-1-mediated induction of regulatory T cells by human mesenchymal stem cells. Blood (2011) 117:4826–35. doi:10.1182/blood-2010-12-324038
86. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoural immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9:1269–74. doi:10.1038/nm934
87. Vig M, Srivastava S, Kandpal U, Sade H, Lewis V, Sarin A, et al. Inducible oxid nitric synthase in T cells regulates T cell death and immune memory. J Clin Invest (2004) 113:1734–42. doi:10.1172/JCI20225
88. Ino Y, Yamazaki-Itoh R, Oguro S, Shimada K, Kosuge T, Zavada J, et al. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS One (2013) 8:e55146. doi:10.1371/journal.pone.0055146
89. Barnas JL, Simpson-Abelson MR, Brooks SP, Kelleher RJ Jr, Bankert RB. Reciprocal functional modulation of the activation of T lymphocytes and fibroblasts derived from human solid tumors. J Immunol (2010) 185:2681–92. doi:10.4049/jimmunol.1000896
90. Allard D, Allard B, Gaudreau PO, Chrobak P, Stagg J. CD73-adenosine: a next-generation target in immuno-oncology. Immunotherapy (2016) 8:145–63. doi:10.2217/imt.15.106
91. Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discov (2014) 4:879–88. doi:10.1158/2159-8290.CD-14-0341
92. Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol (2016) 7:109. doi:10.3389/fimmu.2016.00109
93. Frumento G, Piazza T, Di Carlo E, Ferrini S. Targeting tumor-related immunosuppression for cancer immunotherapy. Endocr Metab Immune Disord Drug Targets (2006) 6:233–7. doi:10.2174/187153006778250019
94. Xu H, Zhang GX, Ciric B, Rostami A. IDO: a double-edged sword for T(H)1/T(H)2 regulation. Immunol Lett (2008) 121:1–6. doi:10.1016/j.imlet.2008.08.008
95. Harden JL, Egilmez NK. Indoleamine 2,3-dioxygenase and dendritic cell tolerogenicity. Immunol Invest (2012) 41:738–64. doi:10.3109/08820139.2012.676122
96. Nguyen NT, Nakahama T, Le DH, Van Son L, Chu HH, Kishimoto T. Aryl hydrocarbon receptor and kynurenine: recent advances in autoimmune disease research. Front Immunol (2014) 5:551. doi:10.3389/fimmu.2014.00551
97. Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol (2015) 5:673. doi:10.3389/fimmu.2014.00673
98. Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Mellor AL, Munn DH, Blazar BR. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood (2009) 114:5062–70. doi:10.1182/blood-2009-06-227587
99. Paul S, Lal G. Regulatory and effector functions of gamma-delta (γδ) T cells and their therapeutic potential in adoptive cellular therapy for cancer. Int J Cancer (2016) 139:976–85. doi:10.1002/ijc.30109
100. Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol (2010) 31:220–7. doi:10.1016/j.it.2010.04.002
101. Calon A, Tauriello DV, Batlle E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol (2014) 15:22. doi:10.1016/j.semcancer.2013.12.008
102. Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol (2006) 24:99–146. doi:10.1146/annurev.immunol.24.021605.090737
103. Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell (2005) 8:369–80. doi:10.1016/j.ccr.2005.10.012
104. Trapani JA. The dual adverse effects of TGF-beta secretion on tumor progression. Cancer Cell (2005) 8:349–50. doi:10.1016/j.ccr.2005.10.018
105. Russo A, Franchina T, Ricciardi GRR, Smiroldo V, Picciotto M, Zanghì M, et al. Third generation EGFR TKIs in EGFR-mutated NSCLC: where are we now and where are we going. Crit Rev Oncol Hematol (2017) 117:38–47. doi:10.1016/j.critrevonc.2017.07.003
106. Tong CWS, Wu WKK, Loong HHF, Cho WCS, To KKW. Drug combination approach to overcome resistance to EGFR tyrosine kinase inhibitors in lung cancer. Cancer Lett (2017) 405:100–10. doi:10.1016/j.canlet.2017.07.023
107. Hughes A, Yong ASM. Immune effector recovery in chronic myeloid leukemia and treatment-free remission. Front Immunol (2017) 8:469. doi:10.3389/fimmu.2017.00469
108. Fierro F, Illmer T, Jing D, Schleyer E, Ehninger G, Boxberger S, et al. Inhibition of platelet-derived growth factor receptor beta by imatinibmesylate suppresses proliferation and alters differentiation of human mesenchymal stem cells in vitro. Cell Prolif (2007) 40:355–66. doi:10.1111/j.1365-2184.2007.00438.x
109. O’Sullivan S, Naot D, Callon K, Porteous F, Horne A, Wattie D, et al. Imatinib promotes osteoblast differentiation by inhibiting PDGFR signaling and inhibits osteoclastogenesis by both direct and stromal cell-dependent mechanisms. J Bone Miner Res (2007) 22:1679–89. doi:10.1359/jbmr.070719
110. Tibullo D, Giallongo C, La Cava P, Berretta S, Stagno F, Chiarenza A, et al. Effects of imatinib mesylate in osteoblastogenesis. Exp Hematol (2009) 37:461–8. doi:10.1016/j.exphem.2008.12.008
111. Fitter S, Vandyke K, Gronthos S, Zannettino AC. Suppression of PDGF-induced PI3 kinase activity by imatinib promotes adipogenesis and adiponectin secretion. J Mol Endocrinol (2012) 48:229–40. doi:10.1530/JME-12-0003
112. Jönsson S, Hjorth-Hansen H, Olsson B, Wadenvik H, Sundan A, Standal T. Imatinib inhibits proliferation of human mesenchymal stem cells and promotes early but not late osteoblast differentiation in vitro. J Bone Miner Metab (2012) 30:119–23. doi:10.1007/s00774-011-0323-3
113. Id Boufker H, Lagneaux L, Najar M, Piccart M, Ghanem G, Body JJ, et al. The Src inhibitor dasatinib accelerates the differentiation of human bone marrow-derived mesenchymal stromal cells into osteoblasts. BMC Cancer (2010) 10:298. doi:10.1186/1471-2407-10-298
114. Mallampati S, Leng X, Ma H, Zeng J, Li J, Wang H, et al. Tyrosine kinase inhibitors induce mesenchymal stem cell-mediated resistance in BCR-ABL+ acute lymphoblastic leukemia. Blood (2015) 125:2968–73. doi:10.1182/blood-2014-05-576421
115. Borghese C, Cattaruzza L, Pivetta E, Normanno N, De Luca A, Mazzucato M, et al. Gefitinib inhibits the cross-talk between mesenchymal stem cells and prostate cancer cells leading to tumor cell proliferation and inhibition of docetaxel activity. J Cell Biochem (2013) 114:1135–44. doi:10.1002/jcb.24456
116. Liu X, Qin J, Luo Q, Bi Y, Zhu G, Jiang W, et al. Cross-talk between EGF and BMP9 signalling pathways regulates the osteogenic differentiation of mesenchymal stem cells. J Cell Mol Med (2013) 17:1160–72. doi:10.1111/jcmm.12097
117. Liu L, Wang Y, Wu K, Fu S, Zhang L, Xu Y, et al. The tyrosine kinase inhibitor nilotinib inhibits proliferation and osteoblast differentiation of human mesenchymal stromal cells. Bone (2011) 49:1117–8. doi:10.1016/j.bone.2011.07.026
118. Tibullo D, Barbagallo I, Giallongo C, La Cava P, Branca A, Conticello C, et al. Effects of second-generation tyrosine kinase inhibitors towards osteogenic differentiation of human mesenchymal cells of healthy donors. Hematol Oncol (2012) 30:27–33. doi:10.1002/hon.988
119. Jönsson S, Hjorth-Hansen H, Olsson B, Wadenvik H, Sundan A, Standal T. Second-generation TKI dasatinib inhibits proliferation of mesenchymal stem cells and osteoblast differentiation in vitro. Leukemia (2010) 24:1357–9. doi:10.1038/leu.2010.84
120. Borriello A, Caldarelli I, Basile MA, Bencivenga D, Tramontano A, Perrotta S, et al. The tyrosine kinase inhibitor dasatinib induces a marked adipogenic differentiation of human multipotent mesenchymal stromal cells. PLoS One (2011) 6:e28555. doi:10.1371/journal.pone.0028555
121. Normanno N, De Luca A, Aldinucci D, Maiello MR, Mancino M, D’Antonio A. Gefitinib inhibits the ability of human bone marrow stromal cells to induce osteoclast differentiation: implications for the pathogenesis and treatment of bone metastasis. Endocr Relat Cancer (2005) 12:471–82. doi:10.1677/erc.1.00956
122. Borriello A, Caldarelli I, Bencivenga D, Stampone E, Perrotta S, Oliva A, et al. Tyrosine kinase inhibitors and mesenchymal stromal cells: effects on self-renewal, commitment and functions. Oncotarget (2017) 8:5540–65. doi:10.18632/oncotarget.12649
123. Huo M, Zhao Y, Satterlee AB, Wang Y, Xu Y, Huang L. Tumor-targeted delivery of sunitinib base enhances vaccine therapy for advanced melanoma by remodeling the tumor microenvironment. J Control Release (2017) 245:81–94. doi:10.1016/j.jconrel.2016.11.013
124. Larocca A, Mina R, Gay F, Bringhen S, Boccadoro M. Emerging drugs and combinations to treat multiple myeloma. Oncotarget (2017) 8:60656–72. doi:10.18632/oncotarget.19269
125. Khoja L, Day D, Wei-Wu Chen T, Siu LL, Hansen AR. Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol (2017) 28:2377–85. doi:10.1093/annonc/mdx286
126. Kim BJ, Kim JH, Kim HS. Survival benefit of immune checkpoint inhibitors according to the histology in non-small-cell lung cancer: a meta-analysis and review. Oncotarget (2017) 8:51779–85. doi:10.18632/oncotarget.17213
127. Datta J, Berk E, Cintolo JA, Xu S, Roses RE, Czerniecki BJ. Rationale for a multimodality strategy to enhance the efficacy of dendritic cell-based cancer immunotherapy. Front Immunol (2015) 6:271. doi:10.3389/fimmu.2015.00271
128. Zhao H, Bo C, Kang Y, Li H. What else can CD39 tell us? Front Immunol (2017) 8:727. doi:10.3389/fimmu.2017.00727
129. Keller HR, Zhang X, Li L, Schaider H, Wells JW. Overcoming resistance to targeted therapy with immunotherapy and combination therapy for metastatic melanoma. Oncotarget (2017) 8:75675–86. doi:10.18632/oncotarget.18523
130. Bethune MT, Joglekar AV. Personalized T cell-mediated cancer immunotherapy: progress and challenges. Curr Opin Biotechnol (2017) 48:142–52. doi:10.1016/j.copbio.2017.03.024
131. Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: recent innovations and future developments. Front Immunol (2017) 8:631. doi:10.3389/fimmu.2017.00631
132. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi:10.1038/nrc3239
133. Chitadze G, Lettau M, Luecke S, Wang T, Janssen O, Fürst D, et al. NKG2D- and T-cell receptor-dependent lysis of malignant glioma cell lines by human γδ T cells: modulation by temozolomide and A disintegrin and metalloproteases 10 and 17 inhibitors. Oncoimmunology (2015) 5. doi:10.1080/2162402X.2015.1093276
135. MadorskyRowdo FP, Baron A, Urrutia M, Mordoh J. Immunotherapy in cancer: a combat between tumors and the immune system; you win some, you lose some. Front Immunol (2015) 6:127. doi:10.3389/fimmu.2015.00127
136. Pizzurro GA, Barrio MM. Dendritic cell-based vaccine efficacy: aiming for hot spots. Front Immunol (2015) 6:91. doi:10.3389/fimmu.2015.00091
137. Ansell SM. Targeting immune checkpoints in lymphoma. Curr Opin Hematol (2015) 22:337–42. doi:10.1097/MOH.0000000000000158
138. Westdorp H, Fennemann FL, Weren RD, Bisseling TM, Ligtenberg MJ, Figdor CG, et al. Opportunities for immunotherapy in microsatellite instable colorectal cancer. Cancer Immunol Immunother (2016) 65:1249–59. doi:10.1007/s00262-016-1832-7
139. Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood (2014) 124:1873–9. doi:10.1182/blood-2014-05-578732
140. Zhang Y, Ertl HC. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget (2016) 7:23282–99. doi:10.18632/oncotarget.7818
141. Meng M, Wang W, Yan J, Tan J, Liao L, Shi J, et al. Immunization of stromal cell targeting fibroblast activation protein providing immunotherapy to breast cancer mouse model. Tumour Biol (2016) 37:10317–27. doi:10.1007/s13277-016-4825-4
142. Li X, Wang Y, Zhao Y, Yang H, Tong A, Zhao C, et al. Immunotherapy of tumor with vaccine based on basic fibroblast growth factor-activated fibroblasts. J Cancer Res Clin Oncol (2014) 140:271–80. doi:10.1007/s00432-013-1547-5
143. Lee J, Fassnacht M, Nair S, Boczkowski D, Gilboa E. Tumor immunotherapy targeting fibroblast activation protein, a product expressed in tumor-associated fibroblasts. Cancer Res (2005) 65:11156–63. doi:10.1158/0008-5472.CAN-05-2805
144. Fassnacht M, Lee J, Milazzo C, Boczkowski D, Su Z, Nair S, et al. Induction of CD4(+) and CD8(+) T-cell responses to the human stromal antigen, fibroblast activation protein: implication for cancer immunotherapy. Clin Cancer Res (2005) 11:5566–71. doi:10.1158/1078-0432.CCR-05-0699
145. Fang J, Xiao L, Joo KI, Liu Y, Zhang C, Liu S, et al. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. Int J Cancer (2015) 138:1013–23. doi:10.1002/ijc.29831
146. Gottschalk S, Yu F, Ji M, Kakarla S, Song XT. A vaccine that co-targets tumor cells and cancer associated fibroblasts results in enhanced antitumor activity by inducing antigen spreading. PLoS One (2013) 8:e82658. doi:10.1371/journal.pone.0082658
147. Jones B, Adams S, Miller GT, Jesson MI, Watanabe T, Wallner BP. Hematopoietic stimulation by a dipeptidyl peptidase inhibitor reveals a novel regulatory mechanism and therapeutic treatment for blood cell deficiencies. Blood (2003) 102:1641–8. doi:10.1182/blood-2003-01-0208
148. Wolf BB, Quan C, Tran T, Wiesmann C, Sutherlin D. On the edge of validation – cancer protease fibroblast activation protein. Mini Rev Med Chem (2008) 8:719–27. doi:10.2174/138955708784567449
149. Puré E. The road to integrative cancer therapies: emergence of a tumor-associated fibroblast protease as a potential therapeutic target in cancer. Expert Opin Ther Targets (2009) 13:967–73. doi:10.1517/14728220903103841
150. Jackson KW, Christiansen VJ, Yadav VR, Silasi-Mansat R, Lupu F, Awasthi V, et al. Suppression of tumor growth in mice by rationally designed pseudopeptide inhibitors of fibroblast activation protein and prolyloligopeptidase. Neoplasia (2015) 17:43–54. doi:10.1016/j.neo.2014.11.002
151. Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res (2004) 64:5471–80. doi:10.1158/0008-5472.CAN-04-0447
152. Wen Y, Wang CT, Ma TT, Li ZY, Zhou LN, Mu B, et al. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci (2010) 101:2325–32. doi:10.1111/j.1349-7006.2010.01695.x
153. Lu C, Tilan JU, Everhart L, Czarnecka M, Soldin SJ, Mendu DR, et al. Dipeptidyl peptidases as survival factors in Ewing sarcoma family of tumors: implications for tumor biology and therapy. J Biol Chem (2011) 286:27494–505. doi:10.1074/jbc.M111.224089
154. Petrausch U, Schuberth PC, Hagedorn C, Soltermann A, Tomaszek S, Stahel R, et al. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer (2012) 12:615. doi:10.1186/1471-2407-12-615
155. Hamson EJ, Keane FM, Tholen S, Schilling O, Gorrell MD. Understanding fibroblast activation protein (FAP): substrates, activities, expression and targeting for cancer therapy. Proteomics Clin Appl (2014) 8:454–63. doi:10.1002/prca.201300095
156. Teichgräber V, Monasterio C, Chaitanya K, Boger R, Gordon K, Dieterle T, et al. Specific inhibition of fibroblast activation protein (FAP)-alpha prevents tumor progression in vitro. Adv Med Sci (2015) 60:264–72. doi:10.1016/j.advms.2015.04.006
157. Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, et al. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci (2015) 106:134–42. doi:10.1111/cas.12584
158. Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest (2006) 116:1955–62. doi:10.1172/JCI26532
159. Chen M, Xiang R, Wen Y, Xu G, Wang C, Luo S, et al. A whole-cell tumor vaccine modified to express fibroblast activation protein induces antitumor immunity against both tumor cells and cancer-associated fibroblasts. Sci Rep (2015) 5:14421. doi:10.1038/srep14421
160. Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One (2009) 4:e7965. doi:10.1371/journal.pone.0007965
161. Narunsky L, Oren R, Bochner F, Neeman M. Imaging aspects of the tumor stroma with therapeutic implications. Pharmacol Ther (2014) 141:192–208. doi:10.1016/j.pharmthera.2013.10.003
162. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2013) 2:154–66. doi:10.1158/2326-6066.CIR-13-0027
163. Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jäger D, Renner C, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie (2003) 26:44–8. doi:10.1159/000069863
164. Cheng JD, Weiner LM. Tumors and their microenvironments: tilling the soil. Commentary re: A. M. Scott et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 9: 1639–1647, 2003. Clin Cancer Res (2003) 9:1590–5.
165. Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res (2003) 9:1639–47.
166. Liu R, Li H, Liu L, Yu J, Ren X. Fibroblast activation protein: a potential therapeutic target in cancer. Cancer Biol Ther (2012) 13:123–9. doi:10.4161/cbt.13.3.18696
167. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2015) 28:831–3. doi:10.1016/j.ccell.2015.11.002
168. Tjomsland V, Aasrum M, Christoffersen T, Gladhaug IP. Functional heterogeneity in tumor-derived human pancreatic stellate cells: differential expression of HGF and implications for mitogenic signaling and migration in pancreatic cancer cells. Oncotarget (2017) 8:71672–84. doi:10.18632/oncotarget.17800
169. Muzumdar MD, Chen PY, Dorans KJ, Chung KM, Bhutkar A, Hong E, et al. Survival of pancreatic cancer cells lacking KRAS function. Nat Commun (2017) 8:1090. doi:10.1038/s41467-017-00942-5
170. Liang C, Shi S, Meng Q, Liang D, Ji S, Zhang B, et al. Do anti-stroma therapies improve extrinsic resistance to increase the efficacy of gemcitabine in pancreatic cancer? Cell Mol Life Sci (2017). doi:10.1007/s00018-017-2678-7
171. Van Audenaerde JRM, De Waele J, Marcq E, Van Loenhout J, Lion E, Van den Bergh JMJ, et al. Interleukin-15 stimulates natural killer cell-mediated killing of both human pancreatic cancer and stellate cells. Oncotarget (2017) 8:56968–79. doi:10.18632/oncotarget.18185
172. Evan GI, Hah N, Littlewood TD, Sodir NM, Campos T, Downes M, et al. Re-engineering the pancreas tumor microenvironment: a “regenerative program” hacked. Clin Cancer Res (2017) 23:1647–55. doi:10.1158/1078-0432.CCR-16-3275
173. Drifka CR, Loeffler AG, Esquibel CR, Weber SM, Eliceiri KW, Kao WJ. Human pancreatic stellate cells modulate 3D collagen alignment to promote the migration of pancreatic ductal adenocarcinoma cells. Biomed Microdevices (2016) 18:105. doi:10.1007/s10544-016-0128-1
174. Ware MJ, Keshishian V, Law JJ, Ho JC, Favela CA, Rees P, et al. Generation of an in vitro 3D PDACstroma rich spheroid model. Biomaterials (2016) 108:129–42. doi:10.1016/j.biomaterials.2016.08.041
175. Nielsen MF, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol (2016) 22:2678–700. doi:10.3748/wjg.v22.i9.2678
176. Ariza-Prota MA, Pando-Sandoval A, Fole-Vázquez D, Casan P. Desmoplastic small round cell tumor of the lung: a case report and literature review. Respir Med Case Rep (2015) 16:112–6. doi:10.1016/j.rmcr.2015.08.010
177. Chirumbolo S, Bjørklund G. The sesquiterpene α-bisabolol in the adipocyte-cancer desmoplastic crosstalk: does it have an action on epithelial-mesenchymal transition mechanisms? Int J Clin Oncol (2017) 22:222–8. doi:10.1007/s10147-016-1072-z
178. Ueno H, Kanemitsu Y, Sekine S, Ishiguro M, Ito E, Hashiguchi Y, et al. Desmoplastic pattern at the tumor front defines poor-prognosis subtypes of colorectal cancer. Am J Surg Pathol (2017) 41:1506–12. doi:10.1097/PAS.0000000000000946
179. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer (2006) 6:392–401. doi:10.1038/nrc1877
180. Ayala G, Tuxhorn JA, Wheeler TM, Frolov A, Scardino PT, Ohori M, et al. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin Cancer Res (2003) 9:4792–801.
181. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
182. Troester MA, Lee MH, Carter M, Fan C, Cowan DW, Perez ER, et al. Activation of host wound responses in breast cancer microenvironment. Clin Cancer Res (2009) 15:7020–8. doi:10.1158/1078-0432.CCR-09-1126
183. Herreros-Villanueva M, Hijona E, Cosme A, Bujanda L. Spontaneous regression of pancreatic cancer: real or a misdiagnosis? World J Gastroenterol (2012) 18:2902–8. doi:10.3748/wjg.v18.i23.2902
184. Eichenauer DA, Engert A. The evolving role of targeted drugs in the treatment of Hodgkin lymphoma. Expert Rev Hematol (2017) 10:775–82. doi:10.1080/17474086.2017.1350167
185. Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest (2017) 127:3462–71. doi:10.1172/JCI94306
186. Marshall MJE, Stopforth RJ, Cragg MS. Therapeutic antibodies: what have we learnt from targeting CD20 and where are we going? Front Immunol (2017) 8:1245. doi:10.3389/fimmu.2017.01245
187. Zappasodi R, de Braud F, Di Nicola M. Lymphoma Immunotherapy: current status. Front Immunol (2015) 6:448. doi:10.3389/fimmu.2015.00448
188. Venè R, Tosetti F, Minghelli S, Poggi A, Ferrari N, Benelli R. Celecoxib increases EGF signaling in colon tumor associated fibroblasts, modulating EGFR expression and degradation. Oncotarget (2015) 6:12310–25. doi:10.18632/oncotarget.3678
189. Koledova Z, Zhang X, Streuli C, Clarke RB, Klein OD, Werb Z, et al. SPRY1 regulates mammary epithelial morphogenesis by modulating EGFR-dependent stromal paracrine signaling and ECM remodeling. Proc Natl Acad Sci U S A (2016) 113:E5731–40. doi:10.1073/pnas.1611532113
190. Liang YH, Lin YL, Liau JY, Tsai JH, Liang JT, Lin BR, et al. Cetuximab might be detrimental to metastatic colorectal cancer patients with KRAS codon 12 mutations. Anticancer Res (2015) 35:4207–14.
191. Bokemeyer C, Köhne CH, Ciardiello F, Lenz HJ, Heinemann V, Klinkhardt U, et al. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur J Cancer (2015) 51:1243–52. doi:10.1016/j.ejca.2015.04.007
192. Ochenduszko SL, Krzemieniecki K. Targeted therapy in advanced colorectal cancer: more data, more questions. Anticancer Drugs (2010) 21:737–48. doi:10.1097/CAD.0b013e32833cfc99
193. Punt CJ, Tol J. More is less – combining targeted therapies in metastatic colorectal cancer. Nat Rev Clin Oncol (2009) 6:731–3. doi:10.1038/nrclinonc.2009.168
194. Sneddon JB, Zhen HH, Montgomery K, van de Rijn M, Tward AD, West R, et al. Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc Natl Acad Sci U S A (2006) 103(40):14842–7. doi:10.1073/pnas.0606857103
195. Assi J, Srivastava G, Matta A, Chang MC, Walfish PG, Ralhan R. Transglutaminase 2 overexpression in tumor stroma identifies invasive ductal carcinomas of breast at high risk of recurrence. PLoS One (2013) 8:e74437. doi:10.1371/journal.pone.0074437
196. Kim S, Lee J, Jeon M, Nam SJ, Lee JE. Elevated TGF-β1 and -β2 expression accelerates the epithelial to mesenchymal transition in triple-negative breast cancer cells. Cytokine (2015) 75:151–8. doi:10.1016/j.cyto.2015.05.020
197. Ide M, Jinnin M, Tomizawa Y, Wang Z, Kajihara I, Fukushima S, et al. Transforming growth factor β-inhibitor Repsox downregulates collagen expression of scleroderma dermal fibroblasts and prevents bleomycin-induced mice skin fibrosis. Exp Dermatol (2017) 26:1139–43. doi:10.1111/exd.13366
198. Shan T, Chen S, Chen X, Lin WR, Li W, Ma J, et al. Prometastatic mechanisms of CAF-mediated EMT regulation in pancreatic cancer cells. Int J Oncol (2016) 50:121–8. doi:10.3892/ijo.2016.3779
199. Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer (2014) 110:724–32. doi:10.1038/bjc.2013.768
200. Flechsig P, Dadrich M, Bickelhaupt S, Jenne J, Hauser K, Timke C, et al. LY2109761 attenuates radiation-induced pulmonary murine fibrosis via reversal of TGF-β and BMP associated proinflammatory and proangiogenic signals. Clin Cancer Res (2012) 18:3616–27. doi:10.1158/1078-0432.CCR-11-2855
201. Schmitz S, Bindea G, Albu RI, Mlecnik B, Machiels JP. Cetuximab promotes epithelial to mesenchymal transition and cancer associated fibroblasts in patients with head and neck cancer. Oncotarget (2015) 6:34288–99. doi:10.18632/oncotarget.5924
202. Alsina-Sanchis E, Figueras A, Lahiguera Á, Vidal A, Casanovas O, Graupera M, et al. The TGFβ pathway stimulates ovarian cancer cell proliferation by increasing IGF1R levels. Int J Cancer (2016) 139:1894–903. doi:10.1002/ijc.30233
203. Frassanito MA, De Veirman K, Desantis V, Di Marzo L, Vergara D, Ruggieri S, et al. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia (2016) 30:640–8. doi:10.1038/leu.2015.289
204. Nemunaitis J, Dillman RO, Schwarzenberger PO, Senzer N, Cunningham C, Cutler J, et al. Phase II study of belagenpumatucel-L, a transforming growth factor β-2 antisense gene-modified allogeneic tumor cell vaccine in nonsmall-cell lung cancer. J Clin Oncol (2006) 24:4721–30. doi:10.1200/JCO.2005.05.5335
205. Dutta A, Hutchison RE, Mohi G. Hmga2 promotes the development of myelofibrosis in Jak2V617F knockin mice by enhancing TGF-β1 and Cxcl12 pathways. Blood (2017) 130:920–32. doi:10.1182/blood-2016-12-757344
206. Levin M, Udi Y, Solomonov I, Sagi I. Next generation matrix metalloproteinase inhibitors – novel strategies bring new prospects. Biochim Biophys Acta (2017) 1864:1927–39. doi:10.1016/j.bbamcr.2017.06.009
207. Pawig L, Klasen C, Weber C, Bernhagen J, Noels H. Diversity and inter-connections in the CXCR4 chemokine receptor/ligand family: molecular perspectives. Front Immunol (2015) 6:429. doi:10.3389/fimmu.2015.00429
208. Scala S. Molecular pathways: targeting the CXCR4-CXCL12 axis – untapped potential in the tumor microenvironment. Clin Cancer Res (2015) 21:4278–85. doi:10.1158/1078-0432.CCR-14-0914
209. Schroeder MA, Rettig MP, Lopez S, Christ S, Fiala M, Eades W, et al. Mobilization of allogeneic peripheral blood stem cell donors with intravenous plerixafor mobilizes a unique graft. Blood (2017) 129:2680–92. doi:10.1182/blood-2016-09-739722
210. Kashyap MK, Amaya-Chanaga CI, Kumar D, Simmons B, Huser N, Gu Y, et al. Targeting the CXCR4 pathway using a novel anti-CXCR4 IgG1 antibody (PF-06747143) in chronic lymphocytic leukemia. J Hematol Oncol (2017) 10:112. doi:10.1186/s13045-017-0435-x
211. Zulfiqar B, Mahroo A, Nasir K, Farooq RK, Jalal N, Rashid MU, et al. Nanomedicine and cancer immunotherapy: focus on indoleamine 2,3-dioxygenase inhibitors. Onco Targets Ther (2017) 10:463–76. doi:10.2147/OTT.S119362
212. Cheng CC, Guan SS, Yang HJ, Chang CC, Luo TY, Chang J, et al. Blocking heme oxygenase-1 by zinc protoporphyrin reduces tumor hypoxia-mediated VEGF release and inhibits tumor angiogenesis as a potential therapeutic agent against colorectal cancer. J Biomed Sci (2016) 23:18. doi:10.1186/s12929-016-0219-6
213. Caldwell RB, Toque HA, Narayanan SP, Caldwell RW. Arginase: an old enzyme with new tricks. Trends Pharmacol Sci (2015) 36:395–405. doi:10.1016/j.tips.2015.03.006
214. Vannini F, Kashfi K, Nath N. The dual role of iNOS in cancer. Redox Biol (2015) 6:334–43. doi:10.1016/j.redox.2015.08.009
215. Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol (2013) 35:123–37. doi:10.1007/s00281-012-0342-8
216. Norberg JK, Sells E, Chang HH, Alla SR, Zhang S, Meuillet EJ. Targeting inflammation: multiple innovative ways to reduce prostaglandin E2. Pharm Pat Anal (2013) 2:265–88. doi:10.4155/ppa.12.90
217. Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov (2012) 11:790–811. doi:10.1038/nrd3810
218. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer (2012) 12:278–87. doi:10.1038/nrc3236
219. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer (2012) 12:237–51. doi:10.1038/nrc3237
220. Henricks LM, Schellens JH, Huitema AD, Beijnen JH. The use of combinations of monoclonal antibodies in clinical oncology. Cancer Treat Rev (2015) 41:859–67. doi:10.1016/j.ctrv.2015.10.008
221. Liu O, Xu J, Ding G, Liu D, Fan Z, Zhang C, et al. Periodontal ligament stem cells regulate B lymphocyte function via programmed cell death protein 1. Stem Cells (2013) 31:1371–82. doi:10.1002/stem.1387
222. Chinnadurai R, Copland IB, Patel SR, Galipeau J. IDO-independent suppression of T cell effector function by IFN-γ-licensed human mesenchymal stromal cells. J Immunol (2014) 192:1491–501. doi:10.4049/jimmunol.1301828
223. Davies LC, Heldring N, Kadri N, Le Blanc K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cells (2017) 35:766–76. doi:10.1002/stem.2509
224. De Felice F, Musio D, Cassese R, Gravina GL, Tombolini V. New approaches in glioblastoma multiforme: the potential role of immune- check point inhibitors. Curr Cancer Drug Targets (2017) 17:282–9. doi:10.2174/1568009616666160813183738
225. Park A, Barrera-Ramirez J, Ranasinghe I, Pilon S, Sy R, Fergusson D, et al. Use of statins to augment progenitor cell function in preclinical and clinical studies of regenerative therapy: a systematic review. Stem Cell Rev (2016) 12:327–39. doi:10.1007/s12015-016-9647-7
226. Gruenbacher G, Thurnher M. Mevalonate metabolism in cancer. Cancer Lett (2015) 356:192–6. doi:10.1016/j.canlet.2014.01.013
227. Comito G, Pons Segura C, Taddei ML, Lanciotti M, Serni S, Morandi A, et al. Zoledronic acid impairs stromal reactivity by inhibiting M2-macrophages polarization and prostate cancer-associated fibroblasts. Oncotarget (2017) 8:118–32. doi:10.18632/oncotarget.9497
228. Blanco-Colio LM, Muñoz-García B, Martín-Ventura JL, Lorz C, Díaz C, Hernández G, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors decrease Fas ligand expression and cytotoxicity in activated human T lymphocytes. Circulation (2003) 108:1506–13. doi:10.1161/01.CIR.0000089086.48617.2B
229. Katznelson S, Wang XM, Chia D, Ozawa M, Zhong HP, Hirata M, et al. The inhibitory effects of pravastatin on natural killer cell activity in vivo and on cytotoxic T lymphocyte activity in vitro. J Heart Lung Transplant (1998) 17:335–40.
230. Pierce SK. Lipid rafts and B-cell activation. Nat Rev Immunol (2002) 2:96–105. doi:10.1038/nri726
231. Varshney P, Yadav V, Saini N. Lipid rafts in immune signalling: current progress and future perspective. Immunology (2016) 149:13–24. doi:10.1111/imm.12617
232. Poggi A, Boero S, Musso A, Zocchi MR. Selective role of mevalonate pathway in regulating perforin but not FasL and TNFalpha release in human natural killer cells. PLoS One (2013) 8:e62932. doi:10.1371/journal.pone.0062932
233. Raemer PC, Kohl K, Watzl C. Statins inhibit NK-cell cytotoxicity by interfering with LFA-1-mediated conjugate formation. Eur J Immunol (2009) 39:1456–65. doi:10.1002/eji.200838863
234. Musso A, Catellani S, Canevali P, Tavella S, Venè R, Boero S, et al. Aminobisphosphonates prevent the inhibitory effects exerted by lymph node stromal cells on anti-tumor Vδ 2 T lymphocytes in non-Hodgkin lymphomas. Haematologica (2014) 99:131–9. doi:10.3324/haematol.2013.097311
235. Zocchi MR, Costa D, Venè R, Tosetti F, Ferrari N, Minghelli S, et al. Zoledronate can induce colorectal cancer microenvironment expressing BTN3A1 to stimulate effector γδ T cells with antitumor activity. Oncoimmunology (2017) 6:e1278099. doi:10.1080/2162402X.2016.1278099
236. Dhillon S. Zoledronic acid (Reclast®, Aclasta®): a review in osteoporosis. Drugs (2016) 76:1683–97. doi:10.1007/s40265-016-0662-4
237. Himelstein AL, Foster JC, Khatcheressian JL, Roberts JD, Seisler DK, Novotny PJ, et al. Effect of longer-interval vs standard dosing of zoledronic acid on skeletal events in patients with bone metastases: a randomized clinical trial. JAMA (2017) 317:48–58. doi:10.1001/jama.2016.19425
238. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Adjuvant bisphosphonate treatment in early breast cancer: meta-analyses of individual patient data from randomised trials. Lancet (2015) 386:1353–61. doi:10.1016/S0140-6736(15)60908-4
239. Mhaskar R, Redzepovic J, Wheatley K, Clark OA, Miladinovic B, Glasmacher A, et al. Bisphosphonates in multiple myeloma: a network meta-analysis. Cochrane Database Syst Rev (2012) 5:CD003188. doi:10.1002/14651858.CD003188.pub3
240. Lo Presti E, Dieli F, Meraviglia S. Tumor-infiltrating γδ T lymphocytes: pathogenic role, clinical significance, and differential programing in the tumor microenvironment. Front Immunol (2014) 5:607. doi:10.3389/fimmu.2014.00607
241. Poggi A, Zocchi MR. γδ T lymphocytes as a first line of immune defense: old and new ways of antigen recognition and implications for cancer immunotherapy. Front Immunol (2014) 5:575. doi:10.3389/fimmu.2014.00575
242. Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov (2017) 16:315–37. doi:10.1038/nrd.2016.268
243. Camicia R, Winkler HC, Hassa PO. Novel drug targets for personalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: a comprehensive review. Mol Cancer (2015) 14:207. doi:10.1186/s12943-015-0474-2
244. Nowakowski GS. Evolution: IMiDs to PPMs, revolution in DLBCL? Blood (2015) 126:698–700. doi:10.1182/blood-2015-06-649483
245. Jacques V, Czarnik AW, Judge TM, Van der Ploeg LH, DeWitt SH. Differentiation of antiinflammatory and antitumorigenic properties of stabilized enantiomers of thalidomide analogs. Proc Natl Acad Sci U S A (2015) 112:E1471–9. doi:10.1073/pnas.1417832112
246. Cubillos-Zapata C, Cordoba R, Avendaño-Ortiz J, Arribas-Jiménez C, Hernández-Jiménez E, Toledano V, et al. CC-122 immunomodulatory effects in refractory patients with diffuse large B-cell lymphoma. Oncoimmunology (2016) 5:e1231290. doi:10.1080/2162402X.2016.1231290
247. Hagner PR, Man HW, Fontanillo C, Wang M, Couto S, Breider M, et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood (2015) 126:779–89. doi:10.1182/blood-2015-02-628669
248. Engelhardt M, Wäsch R, Reinhardt H, Kleber M. Pomalidomide. Recent Results Cancer Res (2014) 201:359–72. doi:10.1007/978-3-642-54490-3_22
249. Zeidner JF, Foster MC. Immunomodulatory drugs: IMiDs in acute myeloid leukemia (AML). Curr Drug Targets (2015) 18:304–14. doi:10.2174/1389450116666150304104315
250. Gamerith G, Auer T, Amann A, Putzer D, Schenk B, Kircher B, et al. Increase in antibody-dependent cellular cytotoxicity (ADCC) in a patient with advanced colorectal carcinoma carrying a KRAS mutation under lenalidomide therapy. Cancer Biol Ther (2014) 15:266–70. doi:10.4161/cbt.27327
251. Liu WM, Henry JY, Meyer B, Bartlett JB, Dalgleish AG, Galustian C. Inhibition of metastatic potential in colorectal carcinoma in vivo and in vitro using immunomodulatory drugs (IMiDs). Br J Cancer (2009) 101:803–12. doi:10.1038/sj.bjc.6605206
252. Pourcher V, Desnoyer A, Assoumou L, Lebbe C, Curjol A, Marcelin AG, et al. Phase II trial of lenalidomide in HIV-infected patients with previously treated Kaposi’s sarcoma: results of the ANRS 154 lenakap trial. AIDS Res Hum Retroviruses (2017) 33:1–10. doi:10.1089/AID.2016.0069
253. Said R, Kakadiaris E, Piha-Paul S, Fu S, Falchook G, Janku F, et al. Phase I clinical trial of lenalidomide in combination with bevacizumab in patients with advanced cancer. Cancer Chemother Pharmacol (2016) 77:1097–102. doi:10.1007/s00280-016-3000-3
254. Steff M, Joly V, Di Lucca J, Feldman J, Burg S, Sarda-Mantel L, et al. Clinical activity of lenalidomide in visceral human immunodeficiency virus related Kaposi sarcoma. JAMA Dermatol (2013) 149:1319–22. doi:10.1001/jamadermatol.2013.5751
255. Wu L, Parton A, Lu L, Adams M, Schafer P, Bartlett JB. Lenalidomide enhances antibody-dependent cellular cytotoxicity of solid tumor cells in vitro: influence of host immune and tumor markers. Cancer Immunol Immunother (2011) 60:61–73. doi:10.1007/s00262-010-0919-9
256. Rubegni P, Sbano P, De Aloe G, Flori ML, Fimiani M. Thalidomide in the treatment of Kaposi’s sarcoma. Dermatology (2007) 215:240–4. doi:10.1159/000106583
257. Hu S, Yuan L, Yan H, Li Z. Design, synthesis and biological evaluation of lenalidomide derivatives as tumor angiogenesis inhibitor. Bioorg Med Chem Lett (2017) 27:4075–81. doi:10.1016/j.bmcl.2017.07.046
258. Ullenhag GJ, Mozaffari F, Broberg M, Mellstedt H, Liljefors M. Clinical and immune effects of lenalidomide in combination with gemcitabine in patients with advanced pancreatic cancer. PLoS One (2017) 12:e0169736. doi:10.1371/journal.pone.0169736
259. Zuo W, Zhu X, Yang J, Mei Z, Deng M, Lin Q, et al. Bortezomib combined with lenalidomide as the first-line treatment for the rare synchronous occurrence of multiple myeloma and pulmonary adenocarcinoma: a case report. Medicine (Baltimore) (2017) 96:e5787. doi:10.1097/MD.0000000000005787
260. Qu Z, Jiang C, Wu J, Ding Y. Lenalidomide induces apoptosis and inhibits angiogenesis via caspase-3 and VEGF in hepatocellular carcinoma cells. Mol Med Rep (2016) 14:4781–6. doi:10.3892/mmr.2016.5797
261. Shahda S, Loehrer PJ, Clark RS, Spittler AJ, Althouse SK, Chiorean EG. Phase I study of lenalidomide and sorafenib in patients with advanced hepatocellular carcinoma. Oncologist (2016) 21:664–5. doi:10.1634/theoncologist.2016-0071
262. Mizutani S, Kuroda J, Sasaki N, Kiyota M, Tatekawa S, Tsukamoto T, et al. Quadruple cancers of non-producing multiple myeloma, cholangiocellular carcinoma, and two different thyroid cancers. Intern Med (2016) 55:1183–6. doi:10.2169/internalmedicine.55.5319
263. Parnell K, Ahmed M, Smalligan RD, Nadesan S. Extramedullary plasmacytoma mimicking colon carcinoma: an unusual presentation and review of the literature. BMJ Case Rep (2015) 2015:bcr2015210973. doi:10.1136/bcr-2015-210973
264. Huang YT, Cheng CC, Chiu TH, Lai PC. Therapeutic potential of thalidomide for gemcitabine-resistant bladder cancer. Int J Oncol (2015) 47:1711–24. doi:10.3892/ijo.2015.3155
265. Ullenhag GJ, Rossmann E, Liljefors M. A phase I dose-escalation study of lenalidomide in combination with gemcitabine in patients with advanced pancreatic cancer. PLoS One (2015) 10:e0121197. doi:10.1371/journal.pone.0121197
266. Bilen MA, Fu S, Falchook GS, Ng CS, Wheler JJ, Abdelrahim M, et al. Phase I trial of valproic acid and lenalidomide in patients with advanced cancer. Cancer Chemother Pharmacol (2015) 75:869–74. doi:10.1007/s00280-015-2695-x
267. Selle F, Sevin E, Ray-Coquard I, Mari V, Berton-Rigaud D, Favier L, et al. A phase II study of lenalidomide in platinum-sensitive recurrent ovarian carcinoma. Ann Oncol (2014) 25:2191–6. doi:10.1093/annonc/mdu392
268. Ou DL, Chang CJ, Jeng YM, Lin YJ, Lin ZZ, Gandhi AK, et al. Potential synergistic anti-tumor activity between lenalidomide and sorafenib in hepatocellular carcinoma. J Gastroenterol Hepatol (2014) 29:2021–31. doi:10.1111/jgh.12708
269. Agarwal N, Apolo AB, Tsao CK, Lee KM, Godbold JH, Soto R, et al. Phase Ib/II trial of gemcitabine, cisplatin, and lenalidomide as first-line therapy in patients with metastatic urothelial carcinoma. Oncologist (2014) 19:915–6. doi:10.1634/theoncologist.2014-0153
270. Jian W, Levitt JM, Lerner SP, Sonpavde G. The preclinical activity of lenalidomide in indolent urothelial carcinoma. Anticancer Res (2014) 34:3383–9.
271. Rini B, Redman B, Garcia JA, Burris HA III, Li S, Fandi A, et al. A phase I/II study of lenalidomide in combination with sunitinib in patients with advanced or metastatic renal cell carcinoma. Ann Oncol (2014) 25:1794–9. doi:10.1093/annonc/mdu212
272. Jakubikova J, Cholujova D, Hideshima T, Gronesova P, Soltysova A, Harada T, et al. A novel 3D mesenchymal stem cell model of the multiple myeloma bone marrow niche: biologic and clinical applications. Oncotarget (2016) 7:77326–41. doi:10.18632/oncotarget.12643
273. Görgün G, Calabrese E, Soydan E, Hideshima T, Perrone G, Bandi M, et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood (2010) 116:3227–37. doi:10.1182/blood-2010-04-279893
274. Bolomsky A, Schreder M, Meißner T, Hose D, Ludwig H, Pfeifer S, et al. Immunomodulatory drugs thalidomide and lenalidomide affect osteoblast differentiation of human bone marrow stromal cells in vitro. Exp Hematol (2014) 42:516–25. doi:10.1016/j.exphem.2014.03.005
275. Mullooly M, McGowan PM, Crown J, Duffy MJ. The ADAMs family of proteases as targets for the treatment of cancer. Cancer Biol Ther (2016) 17:870–80. doi:10.1080/15384047.2016.1177684
276. Murphy G. The ADAMs: signalling scissors in the tumor microenvironment. Nat Rev Cancer (2008) 8:929–41. doi:10.1038/nrc2459
277. Moss ML, Stoeck A, Yan W, Dempsey PJ. ADAM10 as a target for anti-cancer therapy. Curr Pharm Biotechnol (2008) 9:2–8. doi:10.2174/138920108783497613
278. Waldhauer I, Steinle A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res (2006) 66:2520–6. doi:10.1158/0008-5472.CAN-05-2520
279. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res (2008) 68:6368–76. doi:10.1158/0008-5472.CAN-07-6768
280. Zocchi MR, Camodeca C, Nuti E, Rossello A, Venè R, Tosetti F, et al. ADAM10 new selective inhibitors reduce NKG2D ligand release sensitizing Hodgkin lymphoma cells to NKG2D-mediated killing. Oncoimmunology (2015) 5:e1123367. doi:10.1080/2162402X.2015.1123367
281. Camodeca C, Nuti E, Tepshi L, Boero S, Tuccinardi T, Stura EA, et al. Discovery of a new selective inhibitor of A disintegrin and metalloprotease 10 (ADAM-10) able to reduce the shedding of NKG2D ligands in Hodgkin’s lymphoma cell models. Eur J Med Chem (2016) 111:193–201. doi:10.1016/j.ejmech.2016.01.053
282. Crivelli B, Chlapanidas T, Perteghella S, Lucarelli E, Pascucci L, Brini AT, et al. Italian mesenchymal stem cell group (GISM). J Control Release (2017) 262:104–17. doi:10.1016/j.jconrel.2017.07.023
283. Xu J, Zhou X, Li Y, Tian Y. Cancer nanotechnology: recent trends and developments in strategies for targeting cancer cells to improve cancer imaging and treatment. Curr Drug Metab (2017) 18:266–79. doi:10.2174/1389200218666170116113103
284. Williams DS, Pijpers IAB, Ridolfo R, van Hest JCM. Controlling the morphology of copolymeric vectors for next generation nanomedicine. J Control Release (2017) 259:29–39. doi:10.1016/j.jconrel.2017.02.030
285. Campani V, De Rosa G, Misso G, Zarone MR, Grimaldi A. Lipid nanoparticles to deliver miRNA in cancer. Curr Pharm Biotechnol (2016) 17:741–9. doi:10.2174/138920101708160517234941
286. Santoro M, Menegaz BA, Lamhamedi-Cherradi SE, Molina ER, Wu D, Priebe W, et al. Modeling stroma-induced drug resistance in a tissue-engineered tumor model of ewing sarcoma. Tissue Eng Part A (2017) 23:80–9. doi:10.1089/ten.TEA.2016.0369
287. McAndrews KM, McGrail DJ, Ravikumar N, Dawson MR. Mesenchymal stem cells induce directional migration of invasive breast cancer cells through TGF-β. Sci Rep (2015) 5:16941. doi:10.1038/srep16941
288. Reagan MR, Mishima Y, Glavey SV, Zhang Y, Manier S, Lu ZN, et al. Investigating osteogenic differentiation in multiple myeloma using a novel 3D bone marrow niche model. Blood (2014) 124:3250–9. doi:10.1182/blood-2014-02-558007
289. Kuen J, Darowski D, Kluge T, Majety M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS One (2017) 12:e0182039. doi:10.1371/journal.pone.0182039
290. Pavesi A, Tan AT, Koh S, Chia A, Colombo M, Antonecchia E, et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight (2017) 2:89762. doi:10.1172/jci.insight.89762
291. Feng H, Ou BC, Zhao JK, Yin S, Lu AG, Oechsle E, et al. Homogeneous pancreatic cancer spheroids mimic growth pattern of circulating tumor cell clusters and macrometastases: displaying heterogeneity and crater-like structure on inner layer. J Cancer Res Clin Oncol (2017) 143:1787. doi:10.1007/s00432-017-2456-9
292. Zhang YS, Duchamp M, Oklu R, Ellisen LW, Langer R, Khademhosseini A. Bioprinting the cancer microenvironment. ACS Biomater Sci Eng (2016) 2:1710–21. doi:10.1021/acsbiomaterials.6b00246
293. Fong EL, Harrington DA, Farach-Carson MC, Yu H. Heralding a new paradigm in 3D tumor modeling. Biomaterials (2016) 108:197–213. doi:10.1016/j.biomaterials.2016.08.052
294. Bartucci M, Ferrari AC, Kim IY, Ploss A, Yarmush M, Sabaawy HE. Personalized medicine approaches in prostate cancer employing patient derived 3D organoids and humanized mice. Front Cell Dev Biol (2016) 4:64. doi:10.3389/fcell.2016.00064
295. Alonso-Nocelo M, Abuín C, López-López R, de la Fuente M. Development and characterization of a three-dimensional co-culture model of tumor T cell infiltration. Biofabrication (2016) 8:025002. doi:10.1088/1758-5090/8/2/025002
296. Adjei IM, Blanka S. Modulation of the tumor microenvironment for cancer treatment: a biomaterials approach. J Funct Biomater (2015) 6:81–103. doi:10.3390/jfb6010081
297. Christakou AE, Ohlin M, Önfelt B, Wiklund M. Ultrasonic three-dimensional on-chip cell culture for dynamic studies of tumor immune surveillance by natural killer cells. Lab Chip (2015) 15:3222–31. doi:10.1039/c5lc00436e
298. Kuhl S, Voss E, Scherer A, Lusche DF, Wessels D, Soll DR. 4D tumorigenesis model for quantitating coalescence, directed cell motility and chemotaxis, identifying unique cell behaviors, and testing anticancer drugs. Methods Mol Biol (2016) 1407:229–50. doi:10.1007/978-1-4939-3480-5_18
299. Nyga A, Neves J, Stamati K, Loizidou M, Emberton M, Cheema U. The next level of 3D tumour models: immunocompetence. Drug Discov Today (2016) 21:1421–8. doi:10.1016/j.drudis.2016.04.010
300. Zdzisińska B, Roliński J, Piersiak T, Kandefer-Szerszeń M. A comparison of cytokine production in 2-dimensional and 3-dimensional cultures of bone marrow stromal cells of multiple myeloma patients in response to RPMI8226 myeloma cells. Folia Histochem Cytobiol (2009) 47:69–74. doi:10.2478/v10042-009-0015-1
301. Kirshner J, Thulien KJ, Martin LD, Debes Marun C, Reiman T, Belch AR, et al. A unique three-dimensional model for evaluating the impact of therapy on multiple myeloma. Blood (2008) 112:2935–45. doi:10.1182/blood-2008-02-142430
302. Aljitawi OS, Li D, Xiao Y, Zhang D, Ramachandran K, Stehno-Bittel L, et al. A novel three-dimensional stromal-based model for in vitro chemotherapy sensitivity testing of leukemia cells. Leuk Lymphoma (2014) 55:378–91. doi:10.3109/10428194.2013.793323
303. Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med (2015) 212:1345–60. doi:10.1084/jem.20151159
304. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi:10.1016/j.cell.2015.08.016
305. Drake AC, Chen Q, Chen J. Engineering humanized mice for improved hematopoietic reconstitution. Cell Mol Immunol (2012) 9:215–24. doi:10.1038/cmi.2012.6
306. Han HW, Hsu SH. Chitosan-hyaluronan based 3D co-culture platform for studying the crosstalk of lung cancer cells and mesenchymal stem cells. Acta Biomater (2016) 42:157–67. doi:10.1016/j.actbio.2016.06.014
307. Marinkovic M, Block TJ, Rakian R, Li Q, Wang E, Reilly MA, et al. One size does not fit all: developing a cell-specific niche for in vitro study of cell behavior. Matrix Biol (2016) 52-54:426–41. doi:10.1016/j.matbio.2016.01.004
308. Shah SB, Singh A. Cellular self-assembly and biomaterials-based organoid models of development and diseases. Acta Biomater (2017) 53:29–45. doi:10.1016/j.actbio.2017.01.075
309. Chatzinikolaidou M, Rekstyte S, Danilevicius P, Pontikoglou C, Papadaki H, Farsari M, et al. Adhesion and growth of human bone marrow mesenchymal stem cells on precise-geometry 3D organic-inorganic composite scaffolds for bone repair. Mater Sci Eng C Mater Biol Appl (2015) 48:301–9. doi:10.1016/j.msec.2014.12.007
310. Cruz-Acuña R, Quirós M, Farkas AE, Dedhia PH, Huang S, Siuda D, et al. Synthetic hydrogels for human intestinal organoid generation and colonic wound repair. Nat Cell Biol (2017) 19:1326–35. doi:10.1038/ncb3632
311. Shimony N, Avrahami I, Gorodetsky R, Elkin G, Tzukert K, Zangi L, et al. A 3D rotary renal and mesenchymal stem cell culture model unveils cell death mechanisms induced by matrix deficiency and low shear stress. Nephrol Dial Transplant (2008) 23:2071–80. doi:10.1093/ndt/gfn062
312. Ramadan Q, Ting FC. In vitro micro-physiological immune-competent model of the human skin. Lab Chip (2016) 16:1899–908. doi:10.1039/c6lc00229c
313. Kim SH, Chi M, Yi B, Kim SH, Oh S, Kim Y, et al. Three-dimensional intestinal villi epithelium enhances protection of human intestinal cells from bacterial infection by inducing mucin expression. Integr Biol (Camb) (2014) 6:1122–31. doi:10.1039/c4ib00157e
314. Kim MS, Haney MJ, Zhao Y, Yuan D, Deygen I, Klyachko NL, et al. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: in vitro and in vivo evaluations. Nanomedicine (2017) S1549-9634:30178–30178. doi:10.1016/j.nano.2017.09.011
315. Pascucci L, Coccè V, Bonomi A, Ami D, Ceccarelli P, Ciusani E, et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release (2014) 192:262–70. doi:10.1016/j.jconrel.2014.07.042
316. Kusuzaki K, Matsubara T, Murata H, Logozzi M, Iessi E, Di Raimo R, et al. Natural extracellular nanovesicles and photodynamic molecules: is there a future for drug delivery? J Enzyme Inhib Med Chem (2017) 32:908–16. doi:10.1080/14756366.2017.1335310
317. Wang Y, Yu D, Liu Z, Zhou F, Dai J, Wu B, et al. Exosomes from embryonic mesenchymal stem cells alleviate osteoarthritis through balancing synthesis and degradation of cartilage extracellular matrix. Stem Cell Res Ther (2017) 8:189. doi:10.1186/s13287-017-0632-0
318. Konala VB, Mamidi MK, Bhonde R, Das AK, Pochampally R, Pal R. The current landscape of the mesenchymal stromal cell secretome: a new paradigm for cell-free regeneration. Cytotherapy (2016) 18:13–24. doi:10.1016/j.jcyt.2015.10.008
319. Pachler K, Ketterl N, Desgeorges A, Dunai ZA, Laner-Plamberger S, Streif D, et al. An in vitro potency assay for monitoring the immunomodulatory potential of stromal cell-derived extracellular vesicles. Int J Mol Sci (2017) 18. doi:10.3390/ijms18071413
320. Bruno S, Deregibus MC, Camussi G. The secretome of mesenchymal stromal cells: role of extracellular vesicles in immunomodulation. Immunol Lett (2015) 168:154–8. doi:10.1016/j.imlet.2015.06.007
321. Zimmerlin L, Park TS, Zambidis ET, Donnenberg VS, Donnenberg AD. Mesenchymal stem cell secretome and regenerative therapy after cancer. Biochimie (2013) 95:2235–45. doi:10.1016/j.biochi.2013.05.010
322. Lou G, Song X, Yang F, Wu S, Wang J, Chen Z, et al. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J Hematol Oncol (2015) 8:122. doi:10.1186/s13045-015-0220-7
323. Hass R, Otte A. Mesenchymal stem cells as all-round supporters in a normal and neoplastic microenvironment. Cell Commun Signal (2012) 10:26. doi:10.1186/1478-811X-10-26
324. Orenstein JM. The “myofibroblast” that is omnipresent in pathology and key to the EMT concepts does not actually exist, since normal fibroblasts contain stress fibril organelles (SMA bundles with dense bodies) variably detected by TEM and IHC: conclusions by a diagnostic pathologist with decades of ultrastructural experience. Ultrastruct Pathol (2014) 38:387–98. doi:10.3109/01913123.2014.940231
325. Wang L, Cao L, Wang H, Liu B, Zhang Q, Meng Z, et al. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget (2017) 8:76116–28. doi:10.18632/oncotarget.18814
326. Poggi A, Zocchi MR. Modulating mesenchymal stromal cell function with cholesterol synthesis inhibitors. Curr Med Chem (2011) 18:5196–205. doi:10.2174/092986711798184244
327. Mushahary D, Spittler A, Kasper C, Weber V, Charwat V. Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytometry (2017) 93:19–31. doi:10.1002/cyto.a.23242
Keywords: mesenchymal stromal cells, carcinoma-associated fibroblast, tumor-associated fibroblast, tumor microenvironment, immunosuppression
Citation: Poggi A, Varesano S and Zocchi MR (2018) How to Hit Mesenchymal Stromal Cells and Make the Tumor Microenvironment Immunostimulant Rather Than Immunosuppressive. Front. Immunol. 9:262. doi: 10.3389/fimmu.2018.00262
Received: 28 November 2017; Accepted: 30 January 2018;
Published: 19 February 2018
Edited by:
Salem Chouaib, Institut Gustave Roussy, FranceCopyright: © 2018 Poggi, Varesano and Zocchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Poggi, YWxlc3NhbmRyby5wb2dnaUBoc2FubWFydGluby5pdA==