 Miho Ushijima1
Miho Ushijima1 Takehito Uruno1,2*
Takehito Uruno1,2* Akihiko Nishikimi3
Akihiko Nishikimi3 Fumiyuki Sanematsu4
Fumiyuki Sanematsu4 Yasuhisa Kamikaseda1
Yasuhisa Kamikaseda1 Kazufumi Kunimura1
Kazufumi Kunimura1 Daiji Sakata1,2
Daiji Sakata1,2 Takaharu Okada5
Takaharu Okada5 Yoshinori Fukui1,2
Yoshinori Fukui1,2
- 1Division of Immunogenetics, Medical Institute of Bioregulation, Kyushu University, Fukuoka, Japan
- 2Research Center for Advanced Immunology, Kyushu University, Fukuoka, Japan
- 3Department of Biosciences, School of Science, Kitasato University, Sagamihara, Japan
- 4Department of Pharmacology, Faculty of Medicine, University of Miyazaki, Miyazaki, Japan
- 5Laboratory for Tissue Dynamics, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan
A hallmark of humoral immune responses is the production of antibodies. This process involves a complex cascade of molecular and cellular interactions, including recognition of specific antigen by the B cell receptor (BCR), which triggers activation of B cells and differentiation into plasma cells (PCs). Although activation of the small GTPase Rac has been implicated in BCR-mediated antigen recognition, its precise role in humoral immunity and the upstream regulator remain elusive. DOCK2 is a Rac-specific guanine nucleotide exchange factor predominantly expressed in hematopoietic cells. We found that BCR-mediated Rac activation was almost completely lost in DOCK2-deficient B cells, resulting in defects in B cell spreading over the target cell-membrane and sustained growth of BCR microclusters at the interface. When wild-type B cells were stimulated in vitro with anti-IgM F(ab′)2 antibody in the presence of IL-4 and IL-5, they differentiated efficiently into PCs. However, BCR-mediated PC differentiation was severely impaired in the case of DOCK2-deficient B cells. Similar results were obtained in vivo when DOCK2-deficient B cells expressing a defined BCR specificity were adoptively transferred into mice and challenged with the cognate antigen. In addition, by generating the conditional knockout mice, we found that DOCK2 expression in B-cell lineage is required to mount antigen-specific IgG antibody. These results highlight important role of the DOCK2–Rac axis in PC differentiation and IgG antibody responses.
Introduction
B cells play an important role in protective immunity through production of antibodies that bind to and eliminate foreign antigens. During development in the bone marrow (BM), precursor B cells undergo rearrangements of the gene encoding the B cell receptor (BCR) and differentiate into immature B cells, which migrate to the spleen to complete their development via T1 and T2 transitional stages (1, 2). Mature follicular B cells then enter secondary lymphoid tissues such as the lymph nodes (LNs) in search for cognate antigens. Specific recognition of antigen by the BCR triggers intracellular signaling cascades, leading to activation of mature B cells and differentiation into plasma cells (PCs) (3, 4). During T cell-dependent (TD) humoral immune responses, PCs are initially produced in transient extrafollicular proliferative foci, but are subsequently derived from B cells participating in the follicular germinal center (GC) reactions (5–7). Accumulating evidence indicates that low-affinity antigens fail to induce PC differentiation (8–10). However, its underlying mechanism and cellular response are poorly understood.
Although soluble antigens can activate B cells, membrane-bound antigens are more effective in promoting B cell activation and are likely to constitute the dominant form of antigens responsible for B cell stimulation in vivo (11). When a mature B cell recognizes antigens tethered on the surface of a target cell such as the follicular dendritic cell (FDC), a microcluster of BCR and its cognate antigen forms and grows at the site of the contact (4), which is surrounded by adhesion molecules, leukocyte function-associated antigen-1 (LFA-1), and intercellular adhesion molecule-1 (ICAM-1) on the surface of B cells and FDCs, respectively. This structure is known as immunological synapse (IS), and its formation involves membrane polarization and cytoskeletal reorganization (4). Previous studies have indicated that the affinity of the BCR for antigen affects the extent of antigen accumulation at the contact site (12, 13). Additionally, it is well established that intracellular signaling molecules also polarize to the IS, following a precise relative topology (4). Therefore, IS formation may be an important factor that determines the fate of antigen-specific B cells during humoral immune responses.
Rac is a member of Rho family GTPases that function as molecular switches by cycling between GDP-bound inactive and GTP-bound active states (14, 15). Rac exists in the cytosol in the GDP-bound form and is recruited to membranes, where its GDP is exchanged for GTP by the action of one or more guanine nucleotide exchange factors (GEFs) (14, 15). Once activated, Rac binds to multiple effector molecules and regulates various cellular functions including remodeling of the actin cytoskeleton. Rac is composed of three isoforms, Rac1, Rac2, and Rac3. Rac1 is ubiquitously expressed and Rac3 is highly expressed in the brain, whereas Rac2 expression is restricted largely to hematopoietic cells (15). So far, the role of Rac in B cells has been extensively analyzed using conventional Rac2 knockout (KO; Rac2–/–) mice and/or conditional KO mice lacking Rac1 expression in B cell lineage (16–18). These results have shown that Rac2 is more important than Rac1 in B cell development, B cell adhesion, and IS formation. However, the effect of loss of Rac activation on antibody production remains unknown, because genetic deletion of both Rac1 and Rac2 in B cell lineage leads to virtually complete absence of mature B cells (17).
DOCK2 is a member of the CDM family of proteins (Caenorhabditis elegans CED-5, mammals DOCK180, and Drosophila melanogaster Myoblast City) and is predominantly expressed in hematopoietic cells (19, 20). Although DOCK2 does not contain the pleckstrin homology (PH) and Dbl homology (DH) domains typically found in GEFs, DOCK2 can bind to phosphatidylinositol 3,4,5-triphosphate (PIP3) through its DOCK homology region (DHR)-1 domain and mediates the GTP–GDP exchange reaction for Rac by means of its DHR-2 domain (21–25). DOCK2 plays key roles in migration and activation of T cells, and its deficiency severely impairs humoral immune responses to TD antigens in mice and humans (26–29). However, the B cell-intrinsic role of DOCK2 in antibody production remains unknown. In this study, we found that BCR-mediated Rac activation and IS formation critically depend on DOCK2. By analyzing three different models, we demonstrate here that DOCK2 expression in B-lineage cells is required for PC differentiation and antigen-specific IgG production.
Materials and Methods
Mice
Dock2−/− mice on C57BL/6 background (CD45.2+) have been described previously (26–28). HyHEL10 mice were generated by crossing VDJ9 HyHEL10 heavy-chain knock-in mice with Vκ5 HyHEL10 light chain transgenic mice (30) and bred to congenic C57BL/6 mice carrying CD45.1+ allele. For adoptive transfer experiments, Dock2+/− or Dock2−/− HyHEL10 mice carrying CD45.1+ allele were generated. For development of DOCK2 conditional KO mice, ES cell harboring loxP-flanked exon 3 of Dock2 allele (EUCOMM consortium) were microinjected into C57BL/6 blastocysts, and the male chimeras were crossed with C57BL/6 mice to obtain Dock2lox/lox mice (for details, see Figure S1 in Supplementary Material). Dock2lox/lox mice were crossed with CD19-Cre mice (CD19-Cre+/−), in which a Cre recombinase gene is inserted heterozygously into the first exon of the CD19 (31).
Cell Preparation and In Vitro Functional Assays
B cells were purified from the peripheral LN cells with B cell Isolation kit (Miltenyi Biotec) and cultured in RPMI 1640 medium (Wako) containing 10% heat-inactivated fetal calf serum (Nichirei Biosciences), 50 µM 2-mercaptoethanol (Nacalai tesque), 2 mM l-glutamine (Life Technologies), 100 U/ml penicillin (Life Technologies), 100 µg/ml streptomycin (Life Technologies), 1 mM sodium pyruvate (Life Technologies), and MEM non-essential amino acids (Life Technologies) (designated complete RPMI medium). For proliferation assay, LN B cells (5 × 104/well) were stimulated in complete RPMI medium with the specified concentrations of anti-IgM F(ab′)2 antibody (Jackson ImmunoResearch Laboratories), anti-CD40 antibody (BD Biosciences), or lipopolysaccharide (LPS; Sigma-Aldrich) in the presence or absence of IL-4 (8 ng/ml; PeproTech) or IL-5 (10 ng/ml; PeproTech) for 48 h, and [3H]-thymidine (37 kBq) was added during the final 18 h of the culture. To assess PC differentiation in vitro, LN B cells (1 × 105/well) were stimulated in complete RPMI medium with anti-IgM F(ab′)2 antibody (33 µg/ml; Jackson ImmunoResearch Laboratories), anti-CD40 antibody (5 µg/ml; BD Biosciences) or LPS (10 µg/ml; Sigma-Aldrich) in the presence or absence of IL-4 and IL-5 (both from PeproTech; 10 ng/ml) for 96 h, as described previously (32). In some experiments, CPYPP (33), a small-molecule inhibitor of DOCK2, was added to the culture at 12.5 µM.
Flow Cytometry
The following antibodies and reagents were used at the indicated concentrations. Allophycocyanin (APC)-conjugated or phycoerythrin (PE)-conjugated anti-mouse CD45R/B220 (RA3-6B2; 2 µg/ml), fluorescein isothiocyanate (FITC)- or PE-conjugated anti-IgM (R6-60.2; 5 µg/ml), APC-anti-CD19 (1D3; 2 µg/ml), FITC-anti-IgD (11-26c.2a; 5 µg/ml), FITC-anti-CD21/CD35 (7G6; 10 µg/ml), biotinylated anti-heat stable antigen (HSA; M1/69; 0.1 µg/ml), FITC-anti-CD45.1 (A20; 2 µg/ml), PE-anti-CD38 (90; 0.7 µg/ml), PE-anti-IgG1 (A85-1; 1 µg/ml), PE-anti-CD138 (281-1; 2 µg/ml), PE-conjugated streptavidin (1 µg/ml) or PerCP-5.5cyanine-conjugated streptavidin (0.5 µg/ml) were from BD Bioscience. Biotinylated anti-GL7 (GL7; 2 µg/ml) purchased from eBioscience. Alexa Fluor 647-labeled HEL was prepared with an Alexa Fluor647 antibody-labeling kit (Invitrogen). Before staining with the antibodies, cells were incubated for 10 min on ice with anti-Fcγ III/II receptor (2.4G2; 0.5 µg/ml; BD Bioscience) to block Fc receptors. In some experiments, mice were injected intraperitoneally with 300 µl of BrdU (Invitrogen) and sacrificed 5 h later. Splenocytes were then stained with APC BrdU flow kit (Becton-Dickinson). Flow cytometric analyses were done on FACS Calibur (BD Bioscience).
ELISA
Ninety six-well polystyrene plates (Thermo 3855) were coated overnight at 4°C with OVA (0.5 µg), HEL (2 µg), or goat anti-mouse Ig (IgM + IgG + IgA, H + L; #1010-01) antibody (Southern Biotech). After the wells were blocked with 150 µl phosphate-buffered saline (PBS) containing 1% sodium casein and 0.1% Tween-20, serial dilutions of sera were added. Alkaline phosphatase-conjugated isotype-specific antibodies (IgM, IgG1, IgG2b; Southern Biotech) were used to detect bound antibody. The reactions were visualized with the substrate p-nitrophenyl phosphate (Sigma-Aldrich) and detected at 405 nm.
Plasmids and Transfection
The cDNA encoding HEL was amplified by PCR using the pET-22b HEL (amino acid residues 19–147) as a template (34). The following primers were used: 5′-ATGAGGTCTTTGCTAATCTTGGTGCTTTGCTTCCTGCCCCTGGCTGCTCTGGGGAAAGTCTTT
GGACGATGTGAG-3′ and 5′-TCACAGCCGGCAGCCTCTGA-3′. The cDNAs encoding enhanced green fluorescent protein (EGFP) and the GPI anchor domain were prepared as described previously (35). The HEL-, EGFP-, and GPI anchor-coding cDNAs were cloned into the EcoR I-Pst I, Pst I-BamH I, and BamH I-Not I sites of the pBSSK vector, respectively, which was then cloned into the Xho I-Not I site of the pBJ1 vector. The pBJ1-HEL-GFP-GPI construct was linearized with Sal I and electroporated into the baby hamster kidney (BHK) cells expressing ICAM-1-GPI (36), together with pTRE2-puro vector. Cells were cultured in the presence of puromycin (0.3 µg/ml) and clones stably expressing HEL-GFP-GPI were selected.
Pull-Down Assays and Immunoblotting
For Rac activation assays, LN B cells (1.25 × 107 per sample) were stimulated with anti-IgM F(ab′)2 antibody (33 µg/ml; Jackson ImmunoResearch Laboratories) at 37°C for the specified times. Cells were then lysed by adding 1× MLB [Mg2+ Lysis Buffer: 25 mM Hepes (pH 7.5), 150 mM NaCl, 1% lgepal CA-630, 10 mM MgCl2, 1 mM EDTA, 10% glycerol; Millipore], followed by centrifugation at 20,000 × g for 1 min at 4°C. Aliquots were saved for total cell lysate controls, and the remaining lysates were incubated with agarose beads containing the GST-fusion Rac binding domain of PAK1 (#14-325; Millipore) at 4°C for 1 h. The beads were washed twice with 1× MLB buffer and suspended in 1× SDS-PAGE sample buffer [62.5 mM Tris–HCl (pH 6.8), 0.005% bromophenol blue, 2% SDS, 10% glycerol, 100 mM dithiothreitol]. The bound proteins and the same amount of total lysates were analyzed by SDS-PAGE, and blots were probed with the anti-Rac1 (23A8; Millipore) or Rac2 (3B10-2D9; Sigma-Aldrich) antibody.
To examine expression and activation of each signaling molecule, cells were lysed on ice in 20 mM Tris–HCl buffer (pH 7.5) containing 1% Triton X-100, 150 mM NaCl, 1 mM β-glycerophosphate, 1 mM Na3VO4, and complete™ protease inhibitors (Roche). After centrifugation, the supernatants were mixed with an equal volume of 2× SDS-PAGE sample buffer (125 mM Tris–HCl, 0.01% bromophenol blue, 4% SDS, 20% glycerol, and 200 mM dithiothreitol). Samples were boiled for 5 min and analyzed by immunoblotting with the following antibodies: rabbit anti-DOCK2 (09-454; 1:1,000 dilution; Millipore); rabbit anti-Syk (1:1,000 dilution; Cell Signaling Technology), rabbit anti-phospho-Syk (Tyr323; 1:1,000 dilution; Cell Signaling Technology), rabbit anti-p44/42 MAPK (Erk1/2) (1:1,000 dilution; Cell Signaling Technology), rabbit anti- phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204; 1:1,000 dilution; Cell Signaling Technology), rabbit anti-Akt (1:1,000 dilution; Cell Signaling Technology), rabbit anti- phospho-Akt (Thr308; 1:1,000 dilution; Cell Signaling Technology), rabbit anti-BLNK (1:1,000 dilution; Cell Signaling Technology), rabbit anti- phospho-BLNK (Tyr96; 1:1,000 dilution; Cell Signaling Technology), rabbit anti-CD19 (1:1,000 dilution; Cell Signaling Technology), rabbit anti- phospho-CD19 (Tyr513; 1:1,000 dilution; Cell Signaling Technology). To analyze tyrosine phosphorylation of Vav or PLCγ2, cell extracts were incubated with protein G sepharose conjugated with anti-Vav (C-14; 1:1,000 dilution; Santa Cruz Biotechnology) or anti-PLCγ2 (Q-20; 1:1,000 dilution; Santa Cruz Biotechnology) antibody. The precipitates were subjected to immunoblotting using anti-phosphotyrosine antibody (pY99; 1:1,000 dilution; Santa Cruz Biotechnology).
Calcium Flux Assays
Lymph node B cells (1 × 106) were loaded with 3 µM Fura 2-AM (Wako Chemicals) for 30 min at 37°C. Cells were then resuspended in Hank’s buffered salt solution containing calcium and magnesium, and were stimulated with anti-IgM F(ab′)2 antibody (33 µg/ml). Fluorescence intensities were monitored at an excitation wavelength of 340 or 380 nm and emission wavelength of 510 nm using a Flex Station3 (Molecular Devices). Ionomycin (10 µM; Sigma-Aldrich) was used as a positive control.
Assays for IS Formation
To analyze IS formation, LN B cells (3 × 105) from HyHEL10 mice were stained with PKH26 (Sigma-Aldrich) or biotinylated anti-LFA-1 antibody (2D7; BD Biosciences) followed by Alexa546-conjugated streptavidin (Invitrogen) before assays. After LN B cells were incubated on a monolayer of BHK cells expressing ICAM1-GPI and HEL-GFP-GPI (designated BHK-ICAM-HEL cells) at 37°C for the specified times, cells were fixed with 4% paraformaldehyde for 12 min. All images were taken with FV3000 laser scanning confocal microscopy (Olympus).
Adoptive Transfers and Immunization
HEL was covalently conjugated to sheep red blood cell (SRBC; KOHJIN BIO) with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (Sigma-Aldrich) as described previously (37). CD45.1+ LN B cells (1 × 105 B cells from Dock2+/− HyHEL10 mice or 2 × 105 B cells from Dock2−/− HyHEL10 mice) were adoptively transferred into 6- to 7-week-old C57BL/6 mice (CD45.2+) together with 2 × 108 HEL-SRBC or SRBC alone. Mice were sacrificed at the specified time points, and spleen cells were analyzed by flow cytometry. In some experiments, mice were injected intraperitoneally with 300 µl of BrdU (Invitrogen) to assess cell proliferation.
To examine antigen-specific antibody responses, mice were immunized by intraperitoneal injection of ovalbumin (OVA; 50 µg per mouse; Sigma-Aldrich) emulsified in complete Freund’s adjuvant (CFA; Difco Laboratories). Fourteen days later, the serum levels of anti-OVA antibody were determined by ELISA.
Immunohistochemical Analyses
Freshly prepared spleens were embedded in Tissue-Tek OCT compound (Sakura Finetechnical), and frozen at −80°C. Cryosections (10 µm) were fixed with 4% (w/v) paraformaldehyde for 10 min at 37°C. After being blocked with 10% horse serum (Sigma-Aldrich), samples were stained with FITC-conjugated anti-B220 (RA3-6B2; BD Biosciences), PE-conjugated anti-CD3 (17A2; BioLegend), anti-metallophillic macrophages (MOMA1; BMA Biomedicals) followed by Alexa 647-conjugated goat anti-rat antibody (Invitrogen). All images were obtained with a laser-scanning confocal microscope (LSM510 META, Carl Zeiss).
Reverse Transcription (RT)-PCR
Total RNA was isolated using ISOGEN (Nippon Gene). After treatment with RNase-free DNase I (Life Technologies), RNA samples were reverse-transcribed with oligo(dT) primers (Life Technologies) and SuperScript III reverse transcriptase (Life Technologies) for amplification by PCR. The following PCR primers were used: Prdm1; 5′-GACTGGGTGGACATGAGAGAG-3′ and 5′-CCATCAATGAAGTGGTGGAAC-3′. Gapdh; 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′.
Homing Assays
B cells were purified from the LNs from Dock2+/+ and Dock2−/− mice and labeled with PKH26 fluorescent cell linkers (Sigma-Aldrich) or CMTMR (Life Technologies), respectively. After intravenous injection of LN B cells (1–2 × 107) into C57BL/6 mice, the ratio of transferred B cells in the white pulp was compared at 48 h later.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism. The data was initially tested with a Kolmogorov–Smirnov test for normal distribution. Parametric data were analyzed using a two-tailed unpaired Student’s t-test when two groups were compared. Nonparametric data were analyzed with a two-tailed Mann–Whitney test when two groups were compared. P-values less than 0.05 were considered significant.
Results
DOCK2 Is a Major Rac GEF Acting Downstream of BCR
Although Dock2−/− mice exhibited diminished numbers of transitional B cells and mature follicular B cells in the spleen (Figures S2B,C in Supplementary Material), LN B cells from C57BL/6 (designated Dock2+/+) mice and Dock2−/− mice showed similar IgMlowIgDhi mature phenotype due to the lack of transitional B cells (2) (Figure S2D in Supplementary Material). Therefore, to examine whether DOCK2 functions downstream of BCR, we prepared LN B cells and analyzed activation and phosphorylation of the signaling molecules. When LN B cells from Dock2+/+ mice were stimulated with anti-IgM F(ab′)2 antibody, the GTP-bound, activated Rac1 and Rac2 were readily detected at 0.5 min after stimulation (Figure 1A). However, BCR-mediated activation of Rac1 and Rac2 were reduced in Dock2−/− B cells to 4.7 and 20.9% of the wild-type (WT) levels, respectively (Figure 1A). These results indicate that DOCK2 is a major Rac GEF acting downstream of BCR. On the other hand, BCR-mediated calcium influx occurred normally even in Dock2−/− B cells (Figure 1B). In addition, we found that DOCK2 deficiency did not affect phosphorylations of other signaling molecules such as Erk, Syk, Akt, BLNK, CD19, PLCγ2, and Vav (Figures 1C,D).
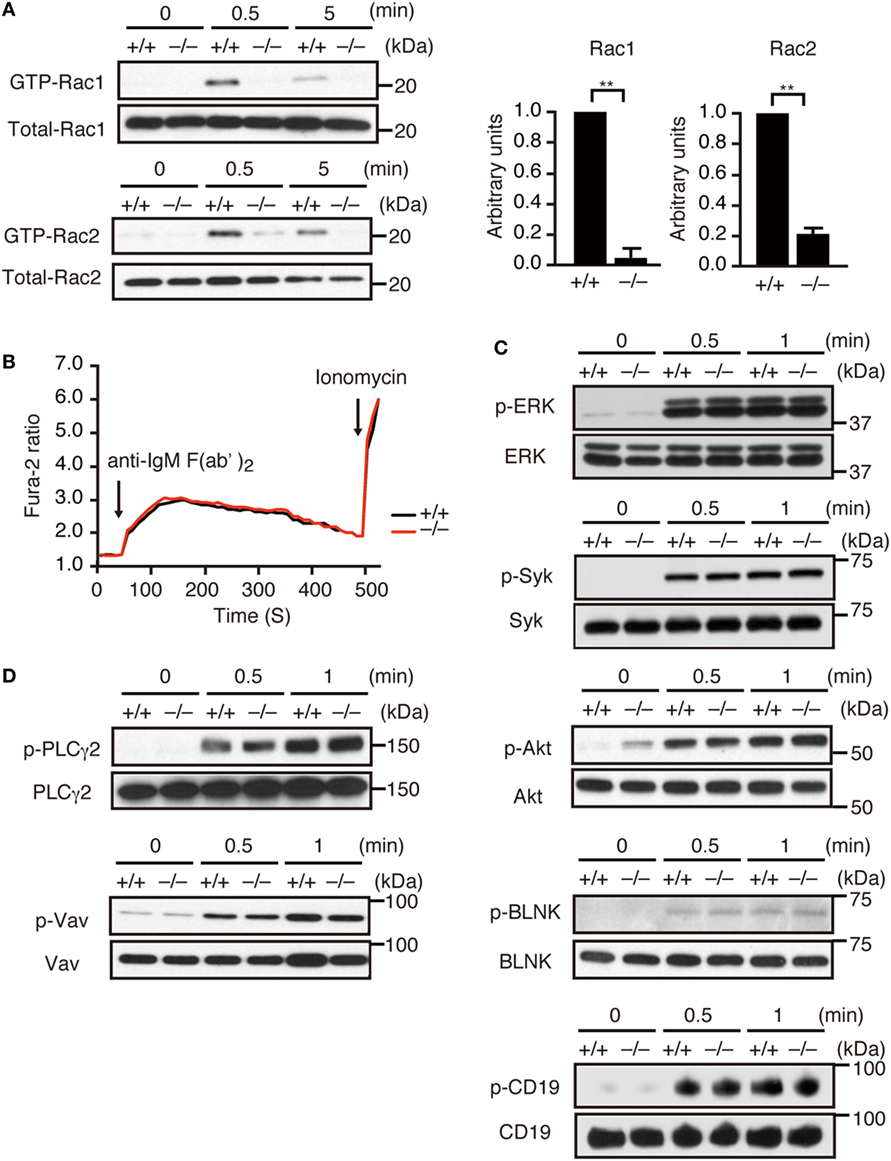
Figure 1. DOCK2 is a major Rac GEF acting downstream of B cell receptor (BCR). (A) BCR-mediated activation of Rac1 and Rac2 were compared between Dock2+/+ and Dock2−/− lymph node (LN) B cells. Results were quantified by densitometry and are expressed as the ratio of GTP-bound form to the total protein after normalization of the 0.5 min-value of Dock2+/+ samples to an arbitrary value of 1. Data for 0.5 min are indicated as the mean ± SD of five independent experiments. **p < 0.01 (two-tailed Mann–Whitney test). (B) Fura-2-loaded Dock2+/+ and Dock2−/− LN B cells were stimulated with anti-IgM F(ab′)2 antibody or ionomycin. Data are indicated as the Fura-2 ratio at 340:380 nm and are representative of three independent experiments. (C,D) Dock2+/+ and Dock2−/− LN B cells were stimulated with anti-IgM F(ab′)2 antibody and analyzed for phosphorylation of each molecule. In (C), phosphorylations of ERK, Syk, Akt, BLNK, and CD19 were analyzed using phosphorylation-specific antibodies. In (D), cell extracts were immunoprecipitated with anti-Vav or anti-PLCγ2 antibody and analyzed with anti-phosphotyrosine antibody.
DOCK2 Regulates BCR-Mediated B Cell Proliferation and PC Differentiation In Vitro
Having found that DOCK2 acts downstream of BCR, we next examined whether DOCK2 deficiency affects BCR-mediated B cell functions in vitro. Although Dock2+/+ B cells proliferated vigorously when stimulated with anti-IgM F(ab′)2 antibody in the presence or absence of IL-4/IL-5, BCR-mediated B-cell proliferation was impaired in the absence of DOCK2 (Figure 2A). When Dock2+/+ B cells were stimulated with anti-IgM F(ab′)2 antibody plus IL-4 and IL-5 for 4 days, they efficiently differentiated into CD138+ PCs (Figure 2B). However, in the case of Dock2−/− B cells, CD138+ PCs were hardly detected under the same culture condition (Figure 2B). Consistent with this, the expression of Prdm1, which encodes the transcription factor Blimp-1 important for PC differentiation (38), was readily detected in Dock2+/+ B cells, but not Dock2−/− B cells (Figure 2C). Importantly, BCR-mediated PC differentiation was impaired when Dock2+/+ B cells were treated with CPYPP (Figure 2D), a small-molecule inhibitor of DOCK2 that binds to the DOCK2 DHR-2 domain and inhibits its Rac GEF activity (33). On the other hand, DOCK2 deficiency did not affect B-cell proliferation and PC differentiation in response to CD40 ligation or LPS stimulation (Figures 2A,B). Thus, DOCK2 selectively regulates BCR-mediated B cell proliferation and PC differentiation via Rac activation.
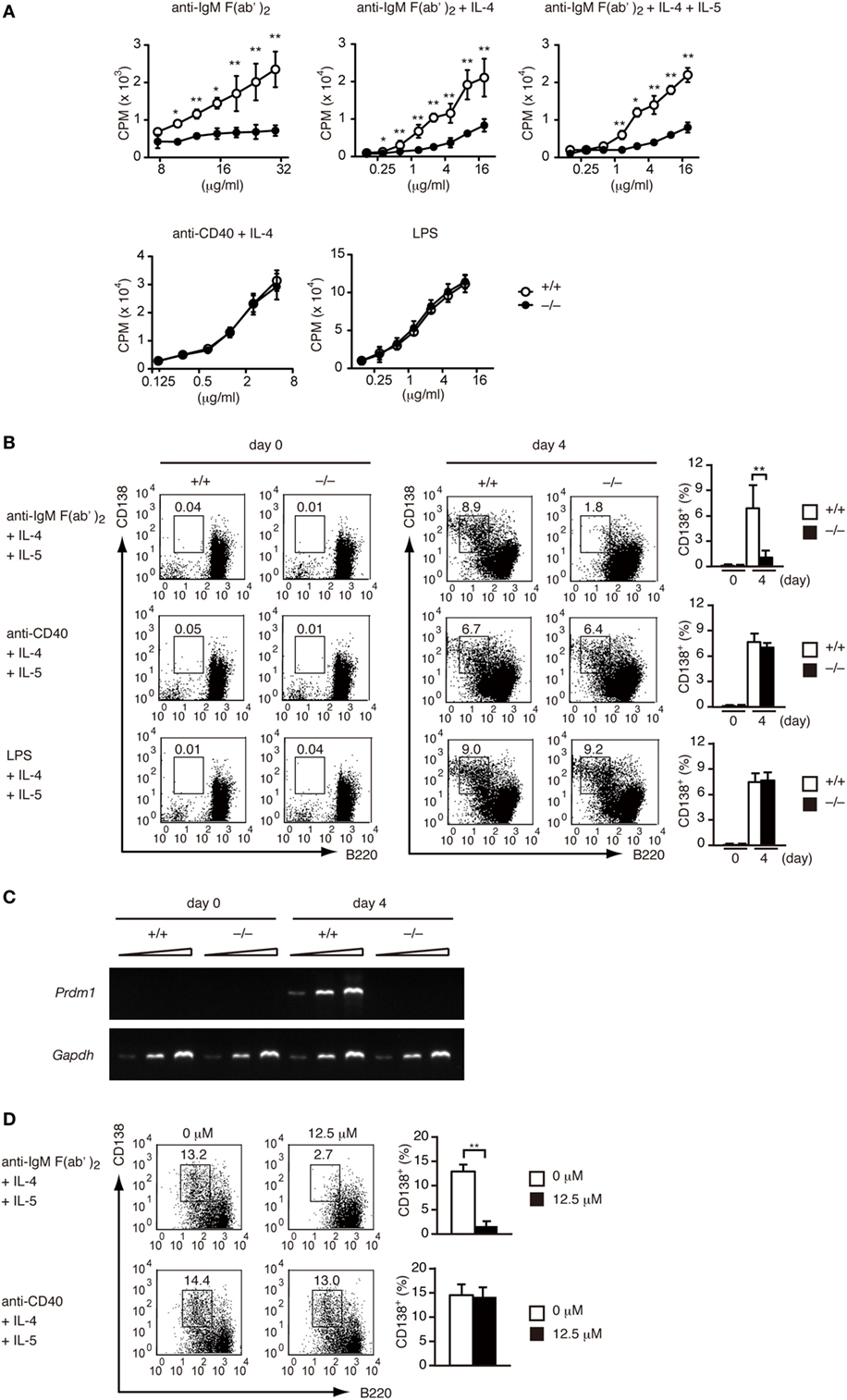
Figure 2. DOCK2 regulates B cell receptor-mediated B cell proliferation and PC differentiation in vitro. (A) Dock2+/+ and Dock2−/− LN B cells were stimulated with anti-IgM F(ab′)2 antibody, anti-CD40 antibody, or lipopolysaccharide at the indicated concentrations in the presence or absence of IL-4/IL-5, and B cell proliferation was analyzed. Data are indicated as the mean ± SD of five independent experiments. *p < 0.05; **p < 0.01 (two-tailed Mann–Whitney test). (B) Following stimulation with anti-IgM F(ab′)2 or anti-CD40 antibody in the presence of IL-4 and IL-5 for 4 days, Dock2+/+ and Dock2−/− LN B cells were analyzed for the expression of CD138 and B220 to assess PC differentiation. FACS profiles at day 0 and day 4 after stimulation are shown. Data are indicated as the mean ± SD of five independent experiments. **p < 0.01 (two-tailed unpaired Student’s t-test). (C) Following stimulation of Dock2+/+ and Dock2−/− LN B cells with anti-IgM F(ab′)2 antibody in the presence of IL-4 and IL-5, the expression of Prdm1 and Gapdh were analyzed with reverse transcription-PCR. Amplification increased by three cycles from the left to the right starting at 28 cycles for Prdm1 or 20 cycles for Gapdh. Data are representative of three independent experiments. (D) The effect of CPYPP (12.5 µM) on in vitro PC differentiation was analyzed as in (B). Data are indicated as the mean ± SD of seven independent experiments. **p < 0.01 (two-tailed Mann–Whitney test).
DOCK2 Regulates BCR-Mediated IS Formation
Although a previous study has indicated that B cell adhesion and IS formation are impaired in Rac2-deficient B cells (18), the physiological function of Rac1 and Rac2 activation in this process is not completely understood. To address this issue, we crossed Dock2−/− mice with HyHEL10 mice that express a defined anti-HEL BCR and are capable of normal Ig class-switch recombination and somatic hypermutation. Irrespective of DOCK2 expression, LN B cells from HyHEL10 mice comparably bound to HEL (Figure 3A). When PKH26-labeled LN B cells from Dock2+/− HyHEL10 mice were incubated with BHK-ICAM-HEL cells, they rapidly spread over the target membrane, where small clusters of GFP-fusion HEL were formed by 3 min within the area of interaction (Figure 3B). However, in the case of Dock2−/− HyHEL10 B cells, a spreading response was impaired with a significant reduction of the number of BCR microclusters at the site of the contact (Figures 3B–D). Similarly, LFA-1 accumulation was reduced in the case of Dock2−/− HyHEL10 B cells (Figures 3E,F). These results indicate that BCR-mediated IS formation critically depends on DOCK2.
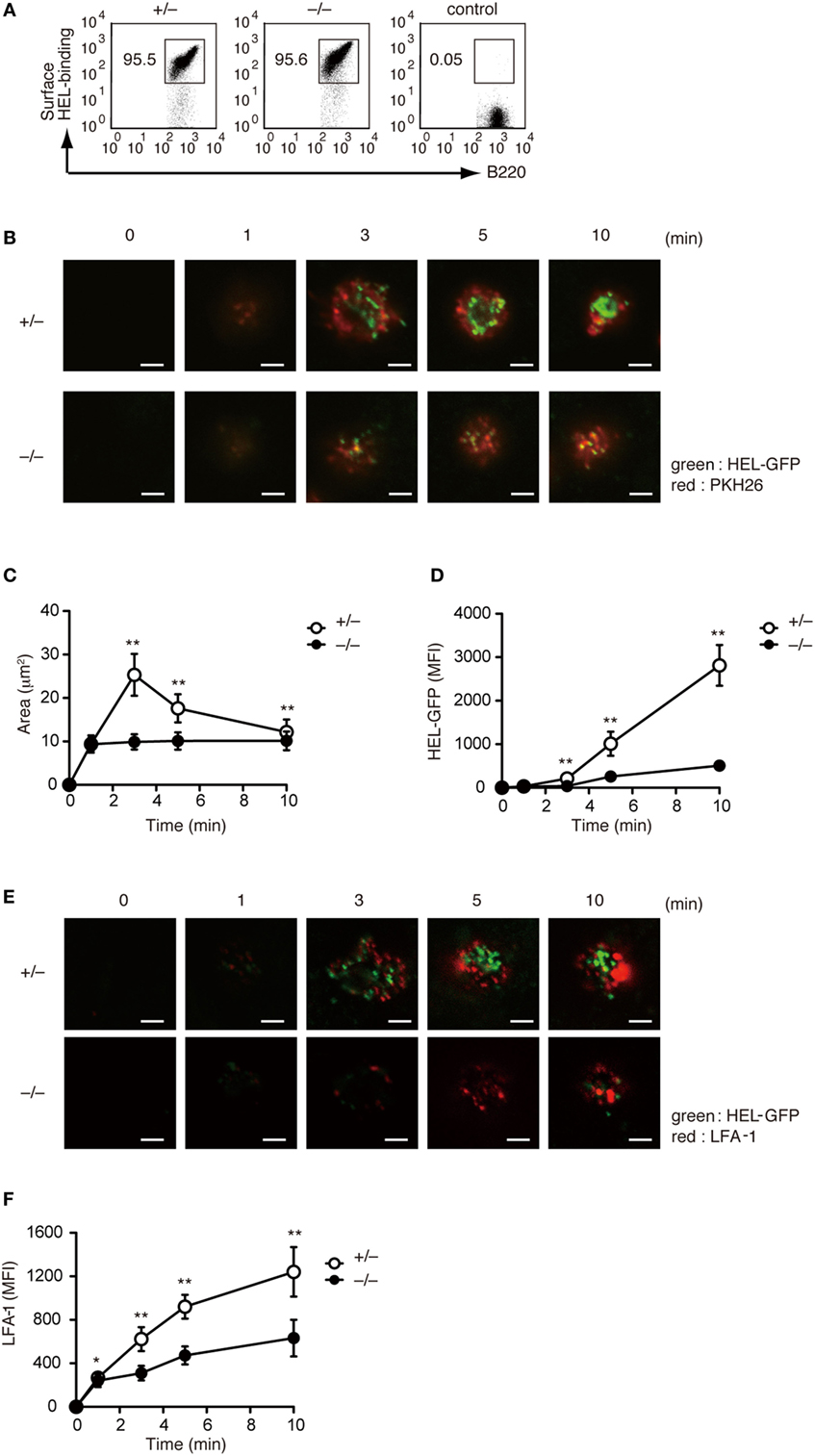
Figure 3. DOCK2 regulates immunological synapse formation. (A) FACS profiles showing comparable binding of HEL to Dock2+/− and Dock2−/− LN B cells. LN B cells from C57BL/6 mice were used as control samples. (B–D) Following incubation of Dock2+/− and Dock2−/− HyHEL10 mice with baby hamster kidney (BHK)-ICAM-HEL, the area of B cell contact (C) and the mean fluorescence intensity (MFI) of HEL-GFP (D) were compared at the indicated time points. Data are indicated as the mean ± SD of 60 cells collected from three separate experiments. **p < 0.01 (two-tailed Mann–Whitney test). (E,F) Following incubation of Dock2+/− and Dock2−/− HyHEL10 mice with BHK-ICAM-HEL, the MFI of leukocyte function-associated antigen-1 (LFA-1) (F) was compared at the indicated time points. Data are indicated as the mean ± SD of 60 cells collected from three separate experiments. *p < 0.05; **p < 0.01 (two-tailed Mann–Whitney test).
DOCK2 Is Required for Expansion of GC B Cells and Differentiation into PCs in Adoptive Transfer Model
To examine the role of DOCK2 in PC differentiation in vivo, we prepared LN CD45.1+ B cells from Dock2+/− and Dock2−/− HyHEL10 mice and adoptively transferred them into C57BL/6 mice (CD45.2) with HEL-conjugated SRBCs (Figure 4A). As DOCK2 deficiency reduces B cell homing to the secondary lymphoid organs (26, 39), we injected Dock2−/− B cells twice as much as Dock2+/− B cells to compensate the number of B cells in the lymphoid follicle (Figure S3 in Supplementary Material). In both cases, the frequencies of GL7+CD38− B cells and IgG1+ B cells to the total CD45.1+ B cells were comparable between Dock2+/− and Dock2−/− B cells at day 5 and day 6 after transfer (Figures 4B,C), indicating that DOCK2 deficiency does not affect differentiation of antigen-engaged B cells to GC B cells and class-switch recombination. However, while Dock2+/− GC B cells proliferated well from day 4 to day 5, such expansion was impaired in the case of Dock2−/− GC B cells (Figure 4D). This was further supported by analyzing BrdU incorporation (Figure 4E). More importantly, we found that B cells from Dock2−/− HyHEL10 mice failed to differentiate efficiently to CD138+ PCs (Figures 4B–D). These results indicate that DOCK2 is required for expansion of GC B cells and differentiation into PCs during TD antibody response.
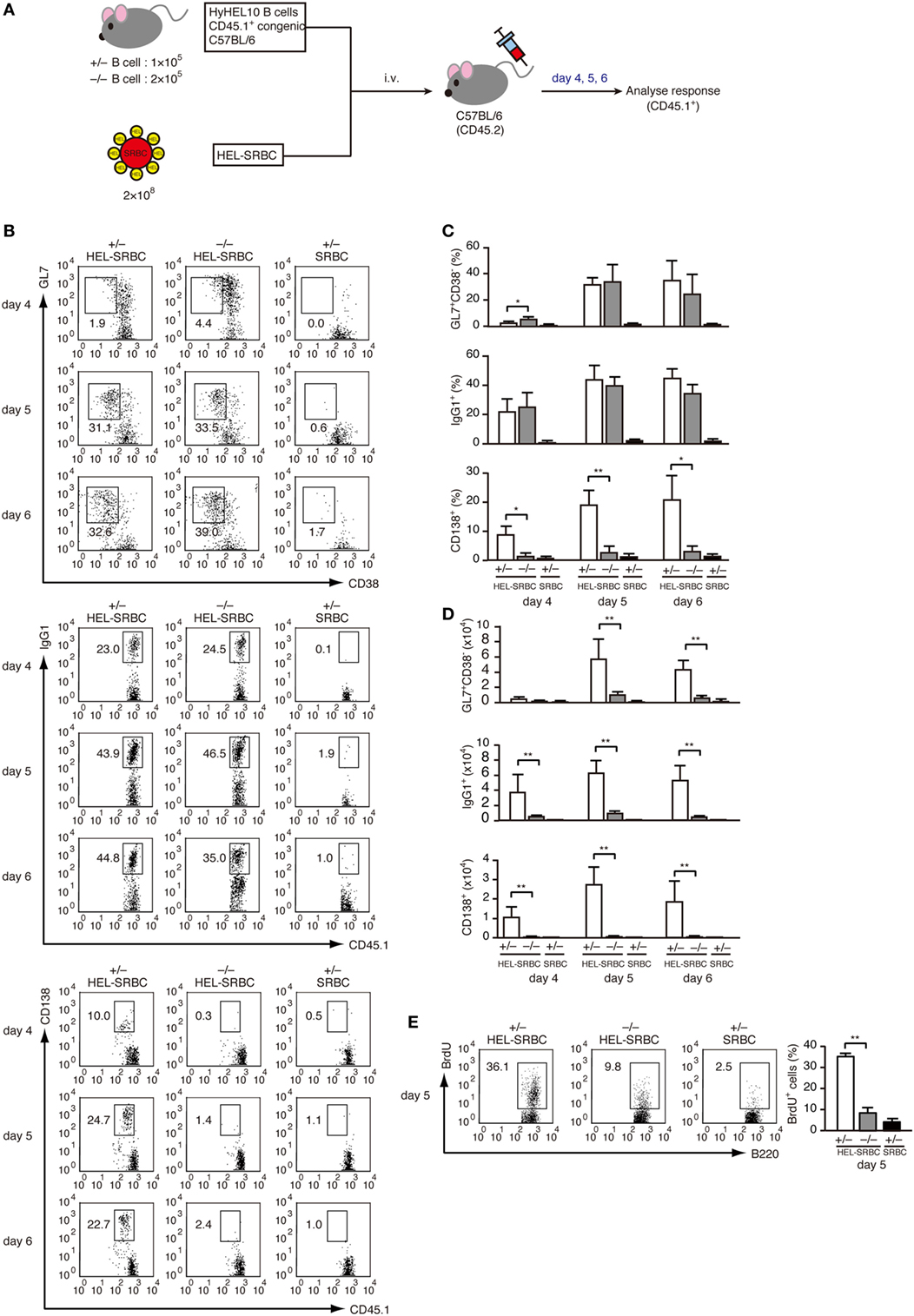
Figure 4. DOCK2 is required for germinal center (GC) B cell expansion and PC differentiation in adoptive transfer model. (A) Schematic representation of the adoptive transfer model used in this study. (B) FACS profiles indicating the expression of GL7, CD38, IgG1, and/or CD138 in CD45.1+ transferred B cells from Dock2+/− and Dock2−/− HyHEL10 mice. Data were obtained at day 4, day 5, and day 6 after transfer and are representative of five independent experiments. (C) Following adoptive transfer, the percentages of GL7+CD38− GC B cells, IgG1+ B cells, CD138+ plasma cells (PCs) in CD45.1+ transferred B cells were compared at the indicated time points between Dock2+/− and Dock2−/− HyHEL10 mice. Data are indicated as the mean ± SD of five independent experiments. *p < 0.05; **p < 0.01 (two-tailed Mann–Whitney test). (D) Following adoptive transfer, the numbers of GL7+CD38− GC B cells, IgG1+ B cells, CD138+ PCs were compared at the indicated time points between Dock2+/− and Dock2−/− HyHEL10 mice. Data are indicated as the mean ± SD of five independent experiments. **p < 0.01 (two-tailed Mann–Whitney test). (E) The percentages of BrdU+ B cells in CD45.1+ transferred B cells were compared between Dock2+/− and Dock2−/− HyHEL10 mice 5 days after adoptive transfer. Data are indicated as the mean ± SD of nine independent experiments. **p < 0.01 (two-tailed Mann–Whitney test).
Development and Characterization of Conditional KO Mice Lacking DOCK2 in a B Cell-Specific Manner
To examine the B cell intrinsic role of DOCK2 under more physiological condition, we developed conditional KO mice lacking DOCK2 in a B cell-specific manner (CD19-Cre+/− Dock2lox/lox mice). Western blot analyses revealed that DOCK2 expression was selectively deleted in B-lineage cells in these mice (Figure S4 in Supplementary Material). We first compared B cell development between CD19-Cre+/− Dock2lox/lox and CD19-Cre−/− Dock2lox/lox mice. Although the amounts of pre/pro B cells and immature B cells in the BM were unchanged between them (Figure 5A), the number of mature recirculating B cells was reduced to 44% of the control level (Figure 5A). Similarly, CD19-Cre+/− Dock2lox/lox mice had diminished numbers of transitional B cells and mature follicular B cells in the spleen (Figures 5B,C), as seen in Dock2−/− mice (Figures S2B,C in Supplementary Material). On the other hand, no phenotypic difference was found when LN B cells from CD19-Cre+/− Dock2lox/lox and CD19-Cre−/− Dock2lox/lox mice were stained for IgM and IgD, or CD21 and HSA (Figure 5D). These phenotypes were similar to those of Dock2−/− mice (Figure S2D in Supplementary Material). Consistent with the FACS data, immunohistochemical analyses of the spleen revealed that the relative size and number of B cell follicles was significantly reduced in CD19-Cre+/− Dock2lox/lox mice, compared with CD19-Cre–/– Dock2lox/lox mice (Figure 5E). However, the organization of T cells and macrophages in the white pulp was not altered in CD19-Cre+/− Dock2lox/lox mice (Figure 5E).
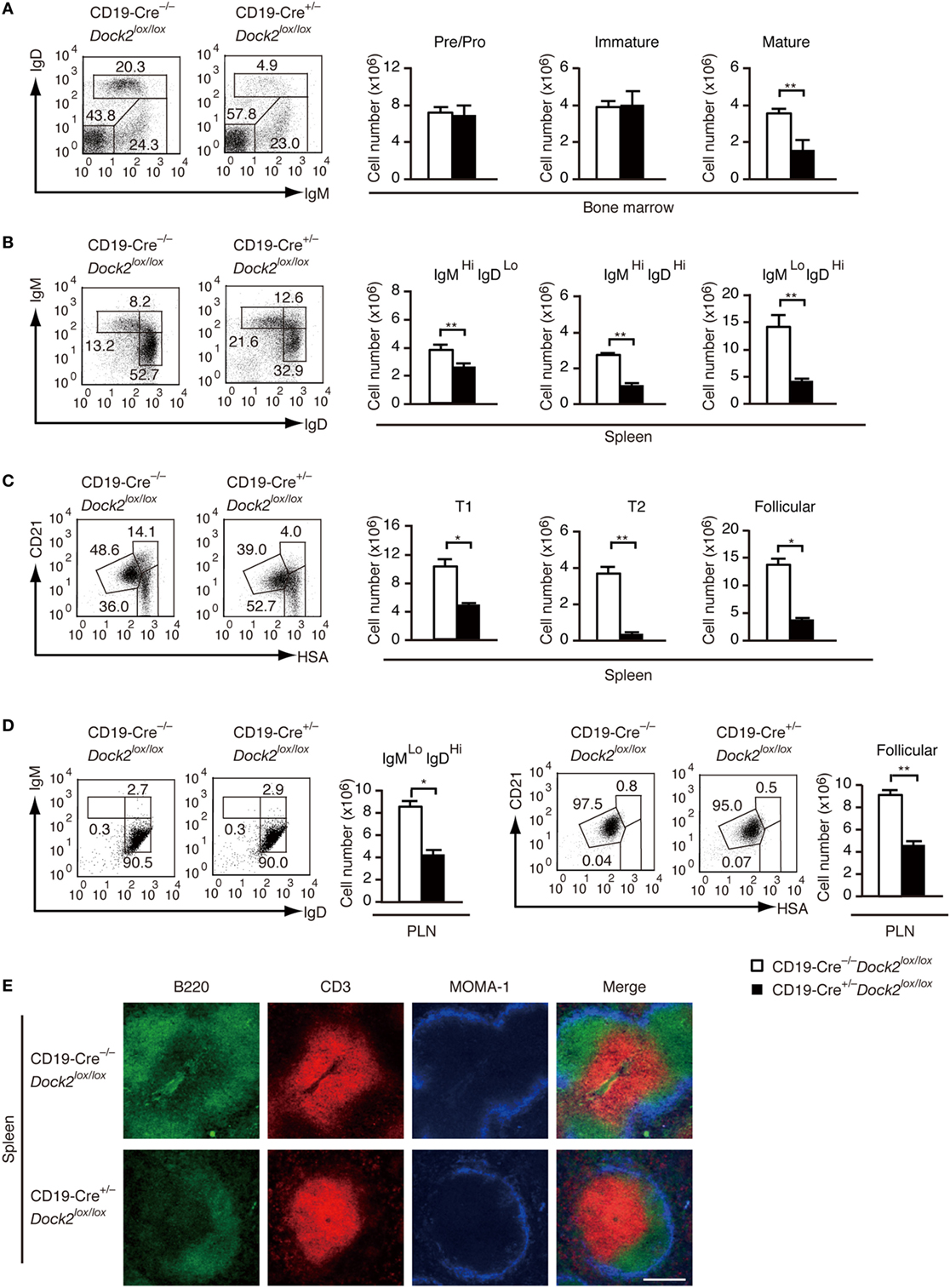
Figure 5. Characterization of CD19-Cre+/− Dock2lox/lox mice. (A) FACS profiles for expression of IgM and IgD in the CD19+ bone marrow B cells. The number of each subset of B cells was compared between CD19-Cre−/− Dock2lox/lox and CD19-Cre+/− Dock2lox/lox mice. Data are indicated as the mean ± SD of five mice. **p < 0.01 (two-tailed Mann–Whitney test). (B) FACS profiles for expression of IgM and IgD in the B220+ splenic B cells. The number of each subset of B cells was compared between CD19-Cre−/− Dock2lox/lox and CD19-Cre+/− Dock2lox/lox mice. Data are indicated as the mean ± SD of 5 mice. **p < 0.01 (two-tailed unpaired Student’s t-test). (C) FACS profiles for expression of CD21 and heat stable antigen (HSA) in the B220+ splenic B cells. The number of each subset of B cells (T1, T2, and follicular B cells) was compared between CD19-Cre–/– Dock2lox/lox and CD19-Cre+/− Dock2lox/lox mice. Data are indicated as the mean ± SD of five mice. *p < 0.05; **p < 0.01 (two-tailed Mann–Whitney test). (D) FACS profiles for expression of IgM and IgD or CD21 and HSA in the B220+ peripheral LN (PLN) B cells. The number of each subset of B cells was compared between CD19-Cre−/− Dock2lox/lox and CD19-Cre+/− Dock2lox/lox mice. Data are indicated as the mean ± SD of five mice. *p < 0.05; **p < 0.01 (two-tailed Mann–Whitney test). (E) Immunohistochemical analyses of the spleen sections from CD19-Cre−/− Dock2lox/lox and CD19-Cre+/− Dock2lox/lox mice.
A Critical Role of DOCK2 in IgG Antibody Responses In Vivo
Under basal condition, serum levels of IgG1 and IgG2b were significantly reduced in CD19-Cre+/− Dock2lox/lox mice, compared with CD19-Cre−/− Dock2lox/lox control mice (Figure 6A). We then compared TD antibody response between CD19-Cre+/− Dock2lox/lox and CD19-Cre−/− Dock2lox/lox mice. When CD19-Cre−/− Dock2lox/lox mice were injected intraperitoneally with OVA in CFA, antigen-specific IgG1 and IgG2b antibodies were readily detected at 14 days after immunization (Figure 6B). However, OVA-specific IgG antibody production was severely impaired in CD19-Cre+/− Dock2lox/lox mice (Figure 6B). These results demonstrate a critical role of DOCK2 in IgG antibody responses in vivo.
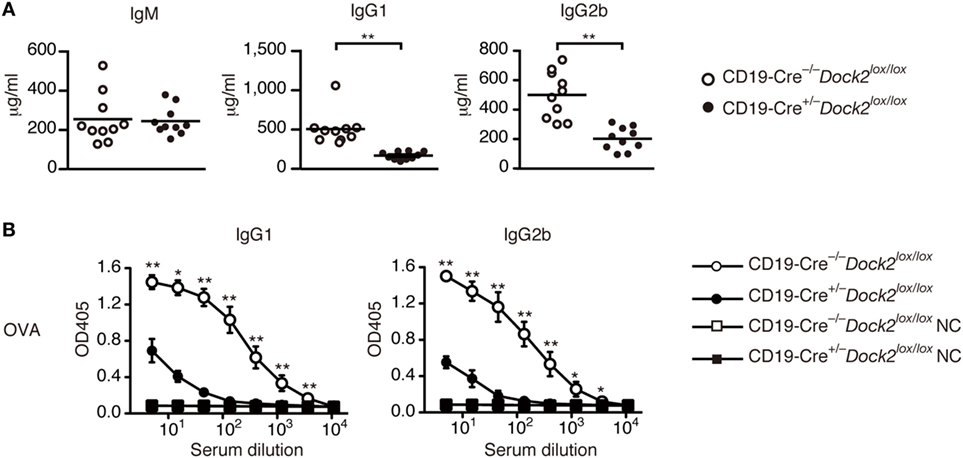
Figure 6. Defective antibody production in CD19-Cre+/− Dock2lox/lox mice. (A) Comparison of serum IgM, IgG1 and IgG2b in CD19-Cre−/− Dock2lox/lox and CD19-Cre+/− Dock2lox/lox mice under the steady state. Data are indicated as the mean ± SD of 10 mice. **p < 0.01 (two-tailed Mann–Whitney test). (B) OVA-specific antibody production was compared between CD19-Cre−/− Dock2lox/lox and CD19-Cre+/− Dock2lox/lox mice at day 14 after immunization. For negative controls, wells were coated with HEL. Data are indicated as the mean ± SD of five independent experiments. *p < 0.05; **p < 0.01 (two-tailed Mann–Whitney test).
Discussion
DOCK2 regulates B cell migration and adhesion by acting downstream of chemokine receptors (26, 39), yet, its role in BCR signaling is poorly understood. Here, we have shown that activations of Rac1 and Rac2 following BCR stimulation were markedly reduced in the absence of DOCK2. Our results thus identify DOCK2 as a key Rac GEF acting downstream of BCR. So far, the DH-type GEFs Vav proteins (Vav1, Vav2, and Vav3) have been considered to regulate B cell functions as the Rac GEFs (40, 41). Although tyrosine phosphorylation of Vav augments its Rac GEF activity (42), BCR-mediated Vav phosphorylation was unchanged between WT and Dock2−/− B cells. In addition, DOCK2 deficiency did not affect BCR-mediated calcium influx, which is defective in B cells from Vav1−/−Vav2−/− double KO mice (40, 41). The precise relationship between DOCK2 and Vav proteins in BCR signaling is currently unknown. However, recent studies have shown that Vav proteins play important roles in T cells and NK cells independently of the Rac GEF activities (43, 44). In light of this, it seems likely that Vav proteins act as adaptor molecules and regulate B cell functions via calcium mobilization.
Although Rac activation has been implicated in BCR-mediated IS formation (4, 18), its physiological relevance and the upstream signaling cascade are not completely understood. We found that antigen-driven B cell spreading and sustained growth of BCR microclusters were impaired in Dock2−/− primary B cells. As these cellular responses are abrogated by actin polymerization inhibitors (4, 45), Rac activation-induced remodeling of the actin cytoskeleton is likely to be involved. In addition, a recent study using chicken DT40 B cells revealed that the growth of BCR microclusters critically depends on PIP3, a lipid product of phosphatidylinositol 3-kinases (PI3Ks) (46). Indeed, DOCK2 binds to PIP3 through its DHR-1 domain (21, 23). Therefore, it is highly conceivable that PI3K activity is required to recruit DOCK2 to the synaptic membrane and activate Rac locally for IS formation, as seen in other lymphocytes (35, 46, 47). While DOCK2 deficiency leads to defective IS formation, it did not affect phosphorylation of major signaling molecules downstream of BCR stimulated with a soluble cross-linking antibody. The role of DOCK2 in signal transduction might be more critical in the in vivo situations where antigen concentrations are often low and the signaling induced by antigen presented on the membrane with adhesion molecules becomes more important.
In this study, we have also shown that in the absence of DOCK2, BCR-mediated PC differentiation was severely impaired in vitro and in vivo. Similar results were obtained when WT B cells were treated with CPYPP, which binds to the DOCK2 DHR-2 domain and inhibits its Rac GEF activity (33). These results indicate that DOCK2 regulates BCR-mediated PC differentiation through Rac activation. How DOCK2-Rac signaling axis regulates PC differentiation remains to be determined. However, accumulating evidence indicates that low affinity antigens fail to induce PC differentiation (8–10). As B cell spreading and growth of BCR microclusters act to increase the number of signalosomes within the membrane (4), their defects in Dock2−/− B cells may lead to the failure to amplify signaling above the threshold required for PC differentiation. Alternatively, in light of the fact that Rac has direct roles in the regulation of gene transcription (48, 49), activated Rac may be involved in the expression of Prdm1 or its related genes during PC differentiation. Also, it may be possible that DOCK2–Rac axis regulates the expression of other molecules required for survival, growth, or differentiation during PC differentiation, because it has been reported that DOCK2 deficiency affects helper T cell differentiation by modulating cytokine receptor expression (50).
Finally, we have shown that CD19-Cre+/− Dock2lox/lox conditional KO mice fail to mount antigen-specific IgG antibody upon immunization of OVA. This result is in marked contrast to a recent study showing that after treatment with tamoxifen to delete Rac1 in Rac2–/– B cells, Mb1-Cre-ERT2 Rac1lox/lox Rac2–/– mice exhibited increased IgG1 and IgG2b antibody to a TD antigen (51). As DOCK2 was deleted early during B cell development in CD19-Cre+/− Dock2lox/lox mice, B cell trafficking is also impaired in this model. On the other hand, there is a time lag between the last tamoxifen treatment and antibody measurement in Mb1-Cre-ERT2 Rac1lox/lox Rac2−/− mice. These differences may affect the outcome of antibody production to TD antigens. Alternatively, it would be possible to speculate that genetic loss of Rac and functional loss of activated Rac are not essentially the same in terms of the regulation of humoral immunity. Further analyses are needed to determine the underlying mechanisms.
Ethics Statement
Mice were maintained under specific-pathogen-free conditions in the animal facility of Kyushu University. The protocol of animal experiments was performed in accordance with the guidelines of the committee of Ethics on Animal Experiments of Kyushu University.
Author Contributions
MU, TU, AN, and FS performed functional, histological, and biochemical analyses; YK and DS performed in vivo experiments; KK contributed to histological analyses; TO provided reagents; MU, TU, and TO contributed to writing the manuscript; TU and YF conceived the project, interpreted the data, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jason G. Cyster for his kind permission to use HyHEL10 mice in this study. We also thank to Ayumi Inayoshi, Arisa Aosaka, and Linh Thi Hoai Nguyen for technical assistance.
Funding
This research was supported by the Leading Advanced Projects for Medical Innovation (LEAP) from Japan Agency for Medical Research and Development (AMED to YF; JP17gm0010001); Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT to YF) of Japan; and the Japan Society for the Promotion of Science (JSPS to YF).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2018.00243/full#supplementary-material.
References
1. Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J Exp Med (1991) 173(5):1213–25. doi:10.1084/jem.173.5.1213
2. Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, et al. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med (1999) 190(1):75–89. doi:10.1084/jem.190.1.75
3. Reth M, Wienands J. Initiation and processing of signals from the B cell antigen receptor. Annu Rev Immunol (1997) 15:453–79. doi:10.1146/annurev.immunol.15.1.453
4. Harwood NE, Batista FD. Early events in B cell activation. Annu Rev Immunol (2010) 28:185–210. doi:10.1146/annurev-immunol-030409-101216
5. MacLennan ICM, Toellner KM, Cunningham AF, Serre K, Sze DMY, Zúñiga E, et al. Extrafollicular antibody responses. Immunol Rev (2003) 194:8–18. doi:10.1034/j.1600-065X.2003.00058.x
6. Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J Exp Med (1991) 173(5):1165–75. doi:10.1084/jem.173.5.1165
7. Liu YJ, Zhang J, Lane PJL, Chan EYT, MacLennan ICM. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur J Immunol (1991) 21(12):2951–62. doi:10.1002/eji.1830211209
8. Smith KGC, Light A, Nossal GJV, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J (1997) 16(11):2996–3006. doi:10.1093/emboj/16.11.2996
9. Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, et al. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med (2006) 203(11):2419–24. doi:10.1084/jem.20061254
10. Brink R, Phan TG, Paus D, Chan TD. Visualizing the effects of antigen affinity on T-dependent B-cell differentiation. Immunol Cell Biol (2008) 86(1):31–9. doi:10.1038/sj.icb.7100143
11. Carrasco YR, Batista FD. B cell recognition of membrane-bound antigen: an exquisite way of sensing ligands. Curr Opin Immunol (2006) 18(3):286–91. doi:10.1016/j.coi.2006.03.013
12. Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity (2004) 20(5):589–99. doi:10.1016/S1074-7613(04)00105-0
13. Fleire SJ, Goldman JP, Carrasco YR, Weber M, Bray D, Batista FD. B cell ligand discrimination through a spreading and contraction response. Science (2006) 312(5774):738–41. doi:10.1126/science.1123940
14. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature (2002) 420(6916):629–35. doi:10.1038/nature01148
15. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol (2008) 9(9):690–701. doi:10.1038/nrm2476
16. Croker BA, Tarlinton DM, Cluse LA, Tuxen AJ, Light A, Yang FC, et al. The Rac2 guanosine triphosphatase regulates B lymphocyte antigen receptor responses and chemotaxis and is required for establishment of B-1a and marginal zone B lymphocytes. J Immunol (2002) 168(7):3376–86. doi:10.4049/jimmunol.168.7.3376
17. Walmsley MJ, Ooi SKT, Reynolds LF, Smith SH, Ruf S, Mathiot A, et al. Critical roles for Rac1 and Rac2 GTPases in B cell development and signaling. Science (2003) 302(5644):459–62. doi:10.1126/science.1089709
18. Arana E, Vehlow A, Harwood NE, Vigorito E, Henderson R, Turner M, et al. Activation of the small GTPase Rac2 via the B cell receptor regulates B cell adhesion and immunological-synapse formation. Immunity (2008) 28(1):88–99. doi:10.1016/j.immuni.2007.12.003
19. Reif K, Cyster JG. The CDM protein DOCK2 in lymphocyte migration. Trends Cell Biol (2002) 12(8):368–73. doi:10.1016/S0962-8924(02)02330-9
20. Nishikimi A, Kukimoto-Niino M, Yokoyama S, Fukui Y. Immune regulatory functions of DOCK family proteins in health and disease. Exp Cell Res (2013) 319(15):2343–9. doi:10.1016/j.yexcr.2013.07.024
21. Kunisaki Y, Nishikimi A, Tanaka Y, Takii R, Noda M, Inayoshi A, et al. DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J Cell Biol (2006) 174(5):647–52. doi:10.1083/jcb.200602142
22. Côté JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci (2002) 115(24):4901–13. doi:10.1242/jcs.00219
23. Nishikimi A, Fukuhara H, Su W, Hongu T, Takasuga S, Mihara H, et al. Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science (2009) 324(5925):384–7. doi:10.1126/science.1170179
24. Kulkarni K, Yang J, Zhang Z, Barford D. Multiple factors confer specific Cdc42 and Rac protein activation by dedicator of cytokinesis (DOCK) nucleotide exchange factors. J Biol Chem (2011) 286(28):25341–51. doi:10.1074/jbc.M111.236455
25. Laurin M, Côté JF. Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev (2014) 28(6):533–47. doi:10.1101/gad.236349.113
26. Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, et al. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature (2001) 412(6849):826–31. doi:10.1038/35090591
27. Sanui T, Inayoshi A, Noda M, Iwata E, Oike M, Sasazuki T, et al. DOCK2 is essential for antigen-induced translocation of TCR and lipid rafts, but not PKC-θ and LFA-1, in T cells. Immunity (2003) 19(1):119–29. doi:10.1016/S1074-7613(03)00169-9
28. Jiang H, Pan F, Erickson LM, Jang MS, Sanui T, Kunisaki Y, et al. Deletion of DOCK2, a regulator of the actin cytoskeleton in lymphocytes, suppresses cardiac allograft rejection. J Exp Med (2005) 202(8):1121–30. doi:10.1084/jem.20050911
29. Dobbs K, Domínguez Conde C, Zhang SY, Parolini S, Audry M, Chou J, et al. DOCK2 and a recessive immunodeficiency with early-onset invasive infections. N Engl J Med (2015) 372(25):2409–22. doi:10.1056/NEJMoa1413462
30. Allen CDC, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science (2007) 315(5811):528–31. doi:10.1126/science.1136736
31. Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature (1995) 376(6583):352–5. doi:10.1038/376352a0
32. Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity (2009) 31(2):259–69. doi:10.1016/j.immuni.2009.06.016
33. Nishikimi A, Uruno T, Duan X, Cao Q, Okamura Y, Saitoh T, et al. Blockade of inflammatory responses by a small-molecule inhibitor of the Rac activator DOCK2. Chem Biol (2012) 19(4):488–97. doi:10.1016/j.chembiol.2012.03.008
34. Mine S, Ueda T, Hshimoto Y, Imoto T. Improvement of the refolding yield and solubility of hen egg-white lysozyme by altering the Met residue attached to its N-terminus to Ser. Protein Eng (1997) 10(11):1333–8. doi:10.1093/protein/10.11.1333
35. Sakai Y, Tanaka Y, Yanagihara T, Watanabe M, Duan X, Terasawa M, et al. The Rac activator DOCK2 regulates natural killer cell-mediated cytotoxicity in mice through the lytic synapse formation. Blood (2013) 122(3):386–93. doi:10.1182/blood-2012-12-475897
36. Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, et al. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol (2005) 6(12):1253–62. doi:10.1038/ni1272
37. Phan TG, Gardam S, Basten A, Brink R. Altered migration, recruitment, and somatic hypermutation in the early response of marginal zone B cells to T cell-dependent antigen. J Immunol (2005) 174(8):4567–78. doi:10.4049/jimmunol.174.8.4567
38. Turner CA Jr, Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell (1994) 77(2):297–306. doi:10.1016/0092-8674(94)90321-2
39. Nombela-Arrieta C, Lacalle RA, Montoya MC, Kunisaki Y, Megías D, Marqués M, et al. Differential requirements for DOCK2 and phosphoinositide-3-kinase γ during T and B lymphocyte horming. Immunity (2004) 21(3):429–41. doi:10.1016/j.immuni.2004.07.012
40. Doody GM, Bell SE, Vigorito E, Clayton E, McAdam S, Tooze R, et al. Signal transduction through Vav-2 participates in humoral immune responses and B cell maturation. Nat Immunol (2001) 2(6):542–7. doi:10.1038/88748
41. Tedford K, Nitschke L, Girkontaite I, Charlesworth A, Chan G, Sakk V, et al. Compensation between Vav-1 and Vav-2 in B cell development and antigen receptor signaling. Nat Immunol (2001) 2(6):548–55. doi:10.1038/88756
42. Crespo P, Schuebel KE, Ostrom AA, Gutkind JS, Bustelo XR. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature (1997) 385(6612):169–72. doi:10.1038/385169a0
43. Upshaw JL, Arneson LN, Schoon RA, Dick CJ, Billadeau DD, Leibson PJ. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat Immunol (2006) 7(5):524–32. doi:10.1038/ni1325
44. Miletic AV, Graham DB, Sakata-Sogawa K, Hiroshima M, Hamann MJ, Cemerski S, et al. Vav links the T cell antigen receptor to the actin cytoskeleton and T cell activation independently of intrinsic Guanine nucleotide exchange activity. PLoS One (2009) 4(8):e6599. doi:10.1371/journal.pone.0006599
45. Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, et al. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol (2008) 9(1):63–72. doi:10.1038/ni1547
46. Wang J, Xu L, Shaheen S, Liu S, Zheng W, Sun X, et al. Growth of B cell receptor microclusters is regulated by PIP2 and PIP3 equilibrium and Dock2 recruitment and activation. Cell Rep (2017) 21(9):2541–57. doi:10.1016/j.celrep.2017.10.117
47. Le Floc’h A, Tanaka Y, Bantilan NS, Voisinne G, Altan-Bonnet G, Fukui Y, et al. Annular PIP3 accumulation controls actin architecture and modulates cytotoxicity at the immunological synapse. J Exp Med (2013) 210(12):2721–37. doi:10.1084/jem.20131324
48. Benitah SA, Valerón PF, van Aelst L, Marshall CJ, Lacal JC. Rho GTPases in human cancer: an unresolved link to upstream and downstream transcriptional regulation. Biochim Biophys Acta (2004) 1705(2):121–32. doi:10.1016/j.bbcan.2004.10.002
49. Barros P, Jordan P, Matos P. Rac1 signaling modulates BCL-6-mediated repression of gene transcription. Mol Cell Biol (2009) 29(15):4156–66. doi:10.1128/MCB.01813-08
50. Tanaka Y, Hamano S, Gotoh K, Murata Y, Kunisaki Y, Nishikimi A, et al. T helper type 2 differentiation and intracellular trafficking of the interleukin 4 receptor-alpha subunit controlled by the Rac activator Dock2. Nat Immunol (2007) 8(10):1067–75. doi:10.1038/ni1506
Keywords: Rac activation, DOCK2, B cell receptor, immunological synapse, plasma cell, antibody production
Citation: Ushijima M, Uruno T, Nishikimi A, Sanematsu F, Kamikaseda Y, Kunimura K, Sakata D, Okada T and Fukui Y (2018) The Rac Activator DOCK2 Mediates Plasma Cell Differentiation and IgG Antibody Production. Front. Immunol. 9:243. doi: 10.3389/fimmu.2018.00243
Received: 25 September 2017; Accepted: 29 January 2018;
Published: 16 February 2018
Edited by:
Deborah K. Dunn-Walters, University of Surrey, United KingdomReviewed by:
Paolo Casali, The University of Texas Health Science Center San Antonio, United StatesWenxia Song, University of Maryland, College Park, United States
Copyright: © 2018 Ushijima, Uruno, Nishikimi, Sanematsu, Kamikaseda, Kunimura, Sakata, Okada and Fukui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takehito Uruno, dXJ1bm9AYmlvcmVnLmt5dXNodS11LmFjLmpw