 Loni Berkowitz1,2
Loni Berkowitz1,2 Bárbara M. Schultz1
Bárbara M. Schultz1 Geraldyne A. Salazar1
Geraldyne A. Salazar1 Catalina Pardo-Roa1
Catalina Pardo-Roa1 Valentina P. Sebastián1
Valentina P. Sebastián1 Manuel M. Álvarez-Lobos2*
Manuel M. Álvarez-Lobos2* Susan M. Bueno1*
Susan M. Bueno1*
- 1Millennium Institute on Immunology and Immunotherapy, Departamento de Genética Molecular y Microbiología, Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Departamento de Gastroenterología, Facultad de Medicina, Pontificia Universidad Católica de Chile, Santiago, Chile
Cigarette smoking is a major risk factor for gastrointestinal disorders, such as peptic ulcer, Crohn’s disease (CD), and several cancers. The mechanisms proposed to explain the role of smoking in these disorders include mucosal damage, changes in gut irrigation, and impaired mucosal immune response. Paradoxically, cigarette smoking is a protective factor for the development and progression of ulcerative colitis (UC). UC and CD represent the two most important conditions of inflammatory bowel diseases, and share several clinical features. The opposite effects of smoking on these two conditions have been a topic of great interest in the last 30 years, and has not yet been clarified. In this review, we summarize the most important and well-understood effects of smoking in the gastrointestinal tract; and particularly, in intestinal inflammation, discussing available studies that have addressed the causes that would explain the opposite effects of smoking in CD and UC.
Introduction
Cigarette smoking is a major risk factor for the development of vascular diseases, such as atherosclerosis and pulmonary hypertension (1). Moreover, smoking-related diseases are the leading cause of preventable deaths and respiratory dysfunctions worldwide. Not only respiratory and cardiovascular diseases contribute to the morbidity and mortality associated with smoking but also other conditions such as multiple inflammatory and carcinogenic disorders (2). The underlying cause of these diseases is the presence of a large number of toxic components in cigarette smoke, whose impact at the respiratory and vascular level has been widely studied (1). Nevertheless, the pathways that are affected by cigarette smoke within the gastrointestinal tract, especially in inflammatory bowel disease (IBD), have not been clarified.
This review will focus on the impact of cigarette smoking on the gastrointestinal system, focusing on recent studies that have addressed its opposite effects on the two major forms of IBD: Crohn’s disease (CD) and ulcerative colitis (UC).
General Harmful Effects of Cigarette Smoking
The main component of cigarette, tobacco, is a natural product with a complex molecular composition. During smoking, this complex biomass is subjected to high temperatures and varying oxygen concentrations, producing an incomplete combustion that generates more than 7,000 toxic compounds. Cigarette smoke can be divided into mainstream and sidestream smoke. Mainstream smoke is the fraction that the smoker inhales directly, and is composed of a particulate phase and a gas phase (3). Low molecular weight components, such as carbon monoxide (CO) and light aldehydes, are the main substances present in the gas phase, which immediately enters the pulmonary circulation (4). On the other hand, compounds such as nicotine, polycyclic aromatic compounds, nitrosamines, and heavy metals are predominantly found in the particulate phase, which is absorbed by mucous membranes, skin, alveoli, and the gastrointestinal system (5).
It has been shown that chronic inhalation of cigarette smoke alters cell proliferation, endothelial function (1), and immune response (6). To date, more than 60 carcinogenic and mutagenic compounds have been identified in cigarette smoke, most of them in the particulate phase. Among the most dangerous compounds are some polycyclic aromatic hydrocarbons and N-nitrosamines (7). In addition, cigarette smoke has high levels of reactive oxygen species, peroxynitrite, peroxynitrate, free radicals, and reactive organic compounds that produce oxidative stress and modulate nitric oxide-dependent endothelial function (4). In fact, both smokers and animals exposed to cigarette smoke have markers of systemic oxidative stress (8), which can negatively affect susceptible tissues, such as the gastrointestinal tract.
Effects of Cigarette Smoke on the Gastrointestinal Tract
In addition to the respiratory and cardiovascular effects produced by cigarette smoke, several studies suggest that smoking is also harmful to the gastrointestinal tract, as summarized below (Table 1). Cigarette smoke contains between 1014 and 1016 free radicals per puff (9), both in the gas phase and in the particulate phase. The active chemicals include aldehydes, quinones, benzo(a)pyrene, epoxides, and peroxides (3), which can induce the production of reactive oxygen species. If these are not neutralized by antioxidants, oxidative stress occurs, which causes tissue damage. Accordingly, it has been shown that cigarette smoking increases the incidence and relapse of peptic ulcer disease (10), and has been associated with IBD, CD, and the development of esophageal, stomach, liver, pancreatic, and colon cancer (5).

Table 1. Effects of cigarette smoke on the gastrointestinal tract.
The intestinal effects produced by cigarette smoking could be partially related to the large amounts of particulate matter ingested by the smoker. It has been described that the amount of nicotine in gastric fluid is 10 times higher than in arterial blood and 80 times higher than in venous blood (23). Nevertheless, the gastrointestinal tract can also be affected by circulating components. Evidence suggests that chronic cigarette smoking (people who have smoked for more than 2 years) could increase the secretion of gastric acid and lower the stomach pH (5). In agreement with this idea, some studies have demonstrated a positive correlation between smoking and the probability of Helicobacter pylori infection, as well as with disease progression (24). In addition, chronic cigarette smoking seems to modify mucus production by the gastric (5) and intestinal mucosa (12), and to alter mucosal repair in the gut (5). Moreover, the vasoconstrictor and procoagulant characteristics of cigarette smoke can have different effects at the gastrointestinal level. For example, it has been described that chronic cigarette smoking alters microcirculation and significantly reduces blood flow to the gastrointestinal mucosa (25), which may favor the development of inflammatory diseases.
The role of cigarette smoking in intestinal inflammation has been extensively studied due to the contradictory effects observed in patients with IBD. IBD is a chronic disorder that usually begins in early adulthood, and whose symptoms include recurrent diarrhea, abdominal pain, and the presence of blood in stool (26). The two main disorders of IBD are CD (26) and UC (27). Although CD and UC share several characteristics, they differ in terms of clinical, endoscopic, and histological features (27). They also differ in their associated risk factors, such as cigarette smoking and some susceptibility loci. CD is characterized by an inflammatory process that can affect any portion of the gastrointestinal tract, from the mouth to the perianal area, in a discontinuous and transmural fashion. Depending on the location of the inflammation, CD has traditionally been classified into ileal, colonic, ileocolonic, and upper gastrointestinal phenotype (26). The most frequent phenotype among CD patients is the ileocolonic disease (26). Colonic inflammation manifests with more symptoms, whereas ileal inflammation seems to progress more rapidly toward transmural lesions, such as fistulas or stenosis (28).
Conversely, UC is a chronic and idiopathic inflammatory disorder of the colonic mucosa that begins in the rectum and usually extends proximally in a continuous manner through the entire colon or through a defined area. However, some patients with proctitis or left-sided colitis may have a cecal inflammation patch. Bloody diarrhea is the characteristic symptom of UC. The clinical course is unpredictable and is marked by alternating periods of exacerbation and remission (27).
In recent years, there has been a substantial improvement in the understanding of the pathophysiology of gastrointestinal inflammatory diseases; however, their etiological pathways remain unclear, and the incidence of CD and UC has increased markedly throughout the world. Unfortunately, there is still no cure for IBD (29). Several implicated environmental factors have been studied, cigarette smoking being the most widely described. Interestingly, although cigarette smoking increases the risk of CD, it is currently the epidemiological factor most associated with a lower incidence of UC (30). These contradictory effects are not fully understood; however, they seem to depend on etiological differences between both disorders, as well as on site-dependent impacts, which will be discussed in this review.
IBD: Dissimilarities between CD and UC
In recent years, three main factors have been identified that contribute to IBD pathogenesis: genetic factors, the host’s immune system, environmental factors, and gut microbiota (31). Some of the environmental factors that could affect the composition of intestinal microbiota and produce alterations in the immune system are changes in lifestyle, such as taking antibiotics, Western-style diet, and cigarette smoking. All of these could explain the increase in the incidence of CD and UC (32). It seems that in both disorders, genetically susceptible individuals have an inappropriate mucosal immune response against their gut microbiota, which leads to an aberrant inflammation response in the digestive tract (31). It has been shown that innate cells such as neutrophils, monocytes, macrophages, and dendritic cells, as well as non-immune cells such as epithelial and stromal cells, contribute to IBD pathogenesis by producing large amounts of cytokines (33). Moreover, it was found that mucosal dendritic cells express high levels of toll-like receptor (TLR) in both CD and UC, which can induce proinflammatory responses upon stimulation by enteric microbiota (33).
Crohn’s disease has been linked to more than 140 genetic susceptibility loci. About 30% of these loci are shared with UC, and about 50% of them are also shared with at least another immune-mediated disease (34). Moreover, predictive models based on genetic analysis can distinguish between colonic and ileal CD (35). Shared loci are enriched in genes involved in primary immunodeficiencies, T-cell function, and modulation of cytokine production (31). For instance, variants of IL23R and HLA are associated with both colonic CD and UC (31, 35). Remarkably, the strongest associations with extensive UC are with variants of the ancestral 8.1 HLA haplotype, which is a known recessive risk for primary sclerosing cholangitis, another inflammatory disease (35). On the other hand, CD seems to be more related to alterations in the immune response and effector functions involved in bacterial clearance. An example is the association between ileal CD and NOD2 variants (35). NOD2 encodes the primary receptor for muramyl dipeptide (MDP) and is essential for bacterial recognition. A loss of NOD2 function is a key event in the pathogenesis of ileal CD, since defective NOD2 leads to an increase in inflammation due to impaired bacterial clearance (36). In addition, a genome-wide association study identified an association between CD and variants of ATG16L1 and IRGM, two genes involved in autophagy (31). The susceptibility loci ATG16L1 directly affect the function of Paneth cells, which exhibit defects in the exocytosis of their granules (37) and affect, in the case of some specific mutations, their response to endoplasmic reticulum stress (38). The defects described above lead to a poor antimicrobial response, which allows the persistence of bacteria in the intestine lining (36, 38). This favors the activation of the immune response against the commensal microbiota. The identification of these genetic predispositions has allowed a deeper understanding of the aberrant response generated in CD and UC. However, genetic factors account for only about 20% of heritability in IBD (34).
The impaired immune response in susceptible individuals who develop IBD is essential to perpetuate intestinal inflammation. However, CD and UC have different adaptive immune responses. In CD, infiltrating CD4+ T cells produce a large number of Th1/Th17-associated pro-inflammatory cytokines such as IFN-γ, IL-17A, and TNF-α (26); and the secretion of these cytokines changes during the course of the disease. In the first stage, T cells secrete Th1 cytokines and TNF-α; in the transition stage, Th17 immune response begins with the secretion of IL-6 and IL-23; and finally, during the establishment of the disease, the combination of the Th1 and Th17 immune response is perpetuated, without TNF-α production (39). In UC patients, on the other hand, CD4+ T cells secrete Th2-associated proinflammatory cytokines, such as IL-4, IL-13, and IL-14 (27). IL-13 secretion promotes apoptosis of epithelial cells and alteration of their tight junctions (40, 41).
NKT cells are also an important part of the immune response in UC pathology: they secrete IL-13, which causes damage in the epithelium, and this consequently generates a positive feedback that increases NKT cell function (42). Interestingly, UC mucosa is specifically populated by type-II NKT cells that respond to a sulfatide autoantigen by producing IL-13. This suggests that this form of IBD is an autoimmune condition in which a self-glycolipid activates lamina propria NKT cells that mediate epithelial cell damage (43). However, one study shows that IL-13 is not related to UC immune response, because neutralization of this cytokine does not prevent or ameliorate the disease (44). More research is needed to solve this discrepancy.
It has been also suggested that humoral immunity has a more relevant etiological role in UC than in CD. Some studies have reported that intestinal damage and extra-intestinal manifestations present in patients with UC could be caused by an adaptive immune response mediated by antibodies that target autoantigens (45–47). Conversely, CD patients develop antibodies against bacterial antigens, suggesting a humoral response triggered primarily by impaired intestinal barrier function and, therefore, an increased interaction with the gut microbiota (48). In fact, the fundamental role of the disruption of the epithelium and the microbiota in the pathogenesis of CD has been widely described, both in human and animal models (48). Alterations in microbiota composition have been observed in both CD and UC. In CD, it has been reported an increase in Bacteroidetes, Proteobacteria, Enterobacteriaceae, Pasteurellaceae, Vellonellaceae, and Fusobacteriaceae, and a reduction in Firmicutes, Erysipelotrichales, and Clostridiales (33). In fact, patients suffering from ileal CD show a distinct change in their microbiota, characterized by a reduction of Faecalibacterium and Roseburia and an increase of Enterobacteriaceae and Ruminococcus gnavus (49). In UC, a change in the gut microbiota has also been reported, but less dramatic than that reported for CD. Here, sequence analysis of 16S rDNA revealed a reduction of Firmicutes and Bacteroidetes and an increase of Actinobacteria and Proteobacteria (33).
Opposite Effects of Cigarette Smoking on CD and UC
Epidemiology
Currently, it is widely accepted that cigarette smoking confers protection against UC (29). A meta-analysis confirmed that the risk of suffering UC is significantly lower in people who smoke, compared with people who have never smoked [OR 0.58 (0.45–0.75), (50)]. In addition, hospitalization and relapse rates, along with the need for more potent drugs and colectomy, are significantly lower in patients who smoke (30).
However, the protective effect of cigarette smoking in UC is temporary, since the relative risk of developing the disease increases after smoking cessation, compared with patients who have never smoked (OR 1.64 [1.36–1.98]) (51). In addition, cigarette smoking has been reported to increase the activity of the disease (52). Although the effects of smoking are not the same as those of nicotine, there is clinical evidence that suggests that nicotine and/or its metabolites, such as cotinine, are responsible for the beneficial effect of smoking in patients with active UC.
Nicotine has been used as a therapeutic agent in UC patients, and its administration has been in the form of chewing gum, transdermal patches, and nicotine-based enemas (53, 54). These studies indicated that chewing nicotine gum results in maximum levels of nicotine in the blood, similarly as those observed during smoking (55, 56). Moreover, a clinical trial with UC patients comparing the use of transdermal nicotine patches to placebo patches suggested that the direct use of nicotine is more effective in controlling mild-to-moderate colitis manifestations (57, 58). Although transdermal nicotine may be effective for UC, it has limited use due to its side effects such as acute pancreatitis, headache, and nausea (58). However, topical administration of nicotine into the colon by enema or administration of delayed-release oral capsule formulations decreases side effects, endoscopic characteristics, and histological damage in UC patients (58). Contradictorily, a recent study indicates that nicotine enemas do not provide benefits for the treatment of active UC; therefore, the benefits of these treatments are not yet conclusive (54).
On the other hand, early smoking significantly increases the risk of CD [OR 2.0 (1.65–2.47), (51)]. In addition, CD patients who smoke have a worse course of illness and quality of life, and are more likely to develop complications, have a higher rate of hospitalization, show a worse response to treatments, and have a greater need for surgery (59). Interestingly, there is a clear association between cigarette smoking and the site of inflammation in these patients, which mainly involves the ileum (ileal phenotype) (60). These associations have been confirmed in animal models (15). In addition, clinical and surgical recurrence rates are significantly higher in these patients, compared with non-smoking patients. In fact, 70% of CD patients who smoke and underwent ileal resection suffered macroscopic lesions at the site of anastomosis within the first year after surgery, as compared with only 35% of non-smoking patients (61).
As in UC, the impact of cigarette smoking on CD is temporary, and smoking cessation improves the course of the disease. In fact, it has been estimated that after 2 years of smoking cessation, the activity of the disease and the therapeutic requirement of former smokers are the same as those of patients who have never smoked (62, 63).
Proposed Mechanisms of Action
The pathways by which cigarette smoking exerts opposite effects in CD and UC are hard to establish, due to the large number of components in the smoke and the diverse cellular and tissue functions that may be affected, such as the mucosal barrier, gut microbiota, immune system, microvasculature, and others. Several studies have described the effect of cigarette smoking on the immune system. The first tissues that interact directly with cigarette smoke are the respiratory and gastrointestinal tract, where it can affect the local immune response. In agreement with this idea, it has been described that cigarette smoking is a risk factor for Mycobacterium tuberculosis (Mtb) infection and disease (64). One of the explanations for this susceptibility lies in an abnormal innate immune response. Smokers have significantly more alveolar macrophages (AM) than non-smokers and former smokers. In addition, AM from smokers cannot control intracellular Mtb and secrete less TNF-α, IFN-γ, and IL-1β after infection, which explains the susceptibility of smokers to Mtb infection and disease (65).
Neutrophils are involved in the protection and elimination of bacterial infections in lungs, due to their ability to form neutrophil extracellular traps (NETosis). In a recent study, human blood neutrophils were stimulated with phorbol ester (PMA for phorbol-12-myristate-13-acetate, a compound known for its ability to stimulate NETosis) in the presence or absence of cigarette smoke condensate (CSC). In the presence of CSC, neutrophils showed an inhibition of NETosis that correlated with an attenuation of oxygen consumption, suggesting a mechanistic relationship between these events. If operative in vivo, attenuation of NETosis due to cigarette smoke may impair host immune responses and increase the risk of respiratory tract infections (66). These impairments in the innate immune response can in turn cause an ineffective adaptive immune response.
In an Mtb infection model, continuous exposure to cigarette smoke locally impaired the accumulation of antigen-presenting cells (APC) and their production of TNF-α, IL-12, and RANTES (CCL5). It also impaired the recruitment of CD4+ IFN-γ+ T cells into the lung, and weakened the formation of granuloma. On the other hand, smoking cessation was found to help restore type-1 immunity by rapidly improving lung APC functionality, enhancing the recruitment of CD4+ IFN-γ+ T cells into the lung, and promoting the granuloma formation (67). In addition, the number of Treg cells and the expression of IL-10 and Foxp3 were lower in the lungs of mice exposed to cigarette smoke than in the control group. More important, the numbers of Tregs negatively correlated with the numbers of Th17 and Tc17 cells (68). These effects on the immune system fit nicely in the pathogenesis of CD, given that Th1/Th17 responses and impairment of bacterial clearance are essential factors for the development of inflammation.
Cigarette smoke has an inhibitory effect on ulcer healing and repair of the gastrointestinal tract, possibly due to its toxic chemical components (10). It was found that the permeability of the intestinal barrier in healthy subjects is unaffected by smoking (69); however, an in vitro study with mice exposed to smoke described increased apoptosis in follicle-associated epithelium overlying Peyer’s patches. Moreover, total dendritic cells, macrophages, CD4+, and CD8+ T cells increased significantly after smoke exposure, together with an upregulation in mRNA expression of CCL9 and CCL20, two important chemokines in CD pathogenesis (19) (Figure 1). Additionally, one study demonstrated that cigarette smoking triggers colitis in mice mediated by CD4+ IFN-γ+ T cells (70). These results suggest that the injury caused in the epithelium barrier can lead to the exposure of lumen antigens, which can be recognized by immune cells whose recruitment is increased, inducing the development of CD (Figure 1).
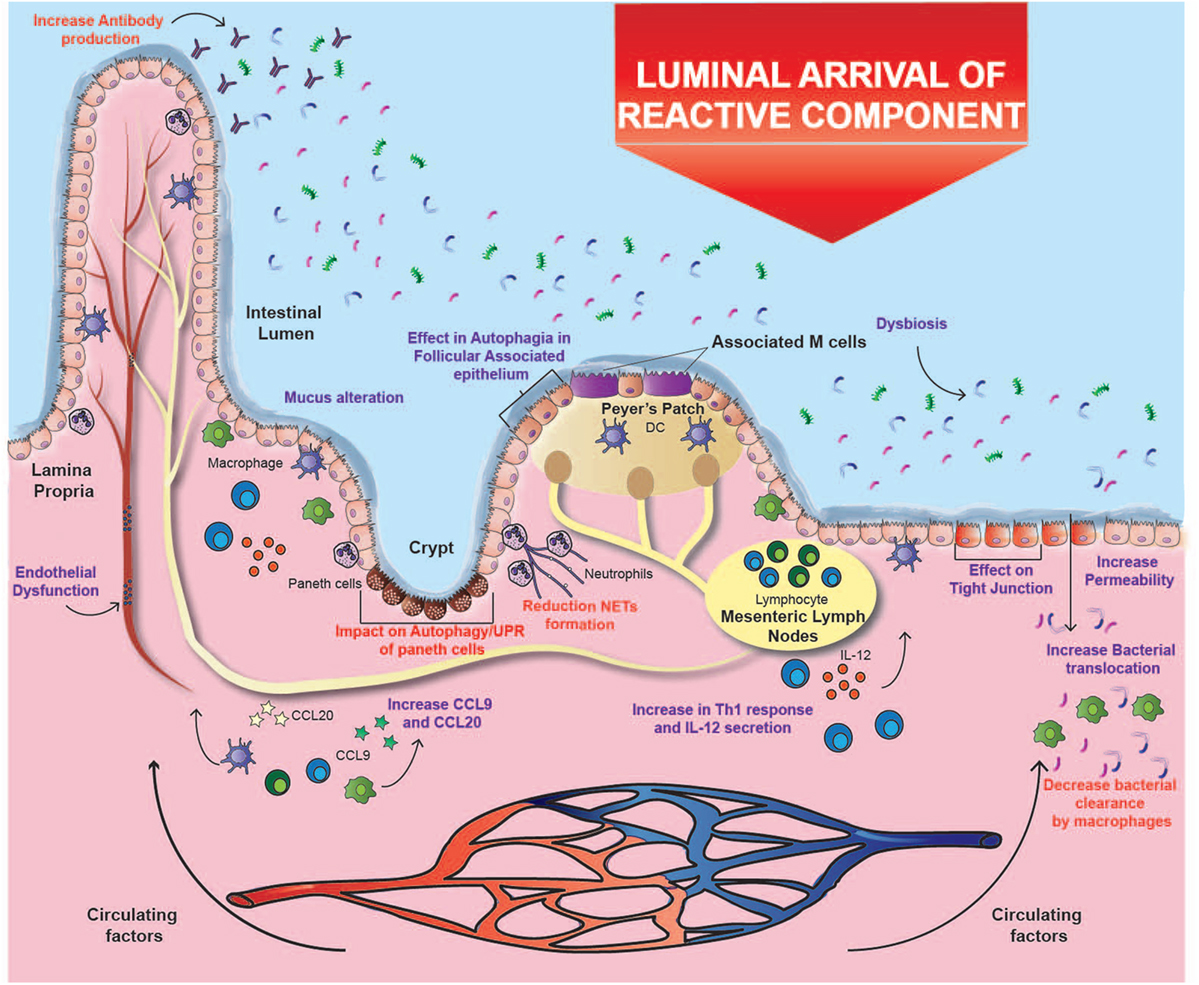
Figure 1. Effects of cigarette smoking on Crohn’s ileitis. A high percentage of particulate matter may reach the ileum, where it could alter the interaction of the intestinal mucosa with the microbiota through several mechanisms, e.g., affecting bacterial clearance, changing microbiota composition, increasing the permeability of the intestinal barrier, and disregulating immune responses. These alterations could prevail over the immunomodulatory effects of carbon monoxide and nicotine. Purple text denotes described effects and red text denotes proposed effects.
As mentioned above, it is intriguing that despite the harmful effect of cigarette smoking in the gastrointestinal tract, it confers protection against UC. In this context, several inherent pharmacological properties of cigarette smoke could explain this. First, nicotine has an immunomodulatory effect mediated by the activation of nicotinic receptors α7 (α7-nAChR) in immune cells such as macrophages and dendritic cells. More specifically, stimulation of these receptors leads to decreased production of pro-inflammatory cytokines, both in vitro and in vivo (71). Additional evidence suggests that activation of α7-nAChR receptors is key in the immunosuppressive function of CD4+ CD25+ regulatory T cells, reducing NF-κB activation and IL-2 production (71). Other studies have shown that the α7-nAChR receptor is also expressed by endothelial cells, where its activation decreases the production of chemokines and the expression of adhesion molecules in the endothelium––a mechanism that could help to modulate leukocyte migration and inflammation (72). On the other hand, CO, the main component of the gas phase of cigarette smoke, has also been associated with anti-inflammatory properties. CO prevents the maturation of dendritic cells and reduces antigen presentation. In addition, it lowers the production of pro-inflammatory cytokines and the proliferation of effector T cells, whereas it stimulates the secretion of the anti-inflammatory cytokine IL-10 (73). Furthermore, exposure to CO causes improvements in genetic and chemical murine models of UC (73, 74). However, this anti-inflammatory effect predominates in UC but not in ileal CD, which could be explained by the different expression of receptors and other molecules by immune cells residing in the different parts of the intestine that are affected in CD or UC. An example of this is the different expression of aryl hydrocarbon receptor (AhR). Normal stimulation of this receptor by dioxin, a by-product of tobacco combustion, should induce anti-inflammatory and protective responses (75). However, it has been reported that the expression of AhR is downregulated in inflamed CD tissue compared with inflamed UC tissue (75).
In contrast to the beneficial effects observed in UC, the harmful effects of cigarette smoking prevail in ileal CD. Considering that CD development may be accompanied by multifocal microinfarcts in the gastrointestinal tract (76, 77), it seems logical that cigarette smoking can worsen the disease by increasing endothelial dysfunction. Accordingly, it has been described that the amount of oxidative and highly reactive compounds of cigarette smoke can intensify vasodilation malfunction in a chronically inflamed microvasculature; resulting in ischemia, ulcers, and fibrosis (4). Indeed, oxidative stress has been widely associated with CD, especially because enhanced oxidative stress and decreased antioxidant levels have been reported in patients with active CD (78). Interestingly, Li et al. demonstrated that cigarette smoke exposure increases oxidative stress in the small intestine of rats, causing upregulation of the nicotinamide adenine dinucleotide phosphate oxidase, while the antioxidative enzyme superoxide dismutase was downregulated (8). Again, this deleterious effect seems to predominate in CD, due to the impact it would have on bacterial clearance, as explained below.
The oxidative damage caused by cigarette smoke deteriorates lipids, proteins, DNA and even organelles; which finally undergo autophagy, a system of intracellular degradation that prevents cell death (19). In parallel to autophagy, intracellular damage can activate an interconnected response known as unfolded protein response (UPR), which is induced by reticulum stress (79). Interestingly, genes involved in both pathways (e.g., ATG16L1 and XBP1) have been associated with CD (80). Indeed, murine models with alterations in both pathways develop a phenotype that closely resembles the transmural inflammation observed in patients with CD (81). Therefore, the exhaustive use of these processes in response to damage caused by cigarette compounds could help explain the increased risk of developing CD, especially in genetically susceptible individuals (19). Remarkably, these interconnected pathways seem to be essential mechanisms in the immune response against pathogens (79). Consistently, Deuring et al. demonstrated that patients with an ATG16L1 risk allele present higher levels of reticulum stress markers and an altered intestinal microbiota (38). Therefore, cigarette smoke may alter the interaction between intestinal mucosa and microbiota through several mechanisms, including the effects mentioned above in autophagy and UPR (19) (Figure 1).
In addition, nicotine can affect bacterial clearance through the reduction of macrophages due to the activation of α7-nAChR (71). In fact, it has been shown that cigarette smoking significantly modifies the composition of intestinal microbiota by favoring the presence of opportunistic pathogens such as some species of Enterobacteriaceae and the phylum Bacteroidetes (13, 82). In relation to this, one study showed that the impact of smoking on the gut microbiota is temporary, since the amount of Enterobacteriaceae and Bacteroidetes decreases a few months after smoking cessation, while the abundance of some Firmicutes bacteria increases (13). Given that it is widely accepted that the disruption of the relationship between microbiota and intestinal mucosa is one of the main contributors to the pathophysiology of CD, the impact of cigarette smoking on the microbiota can have a great impact in the onset of this disease (Figure 1).
Etiological Differences or Site-Specific Impact?
The opposite effects of cigarette smoking on ileal CD and UC could be explained by three different mechanisms. One of them is the etiological difference between both disorders. As mentioned above, the disruption of the intestinal mucosa and its interaction with a modified microbiota seems to be determinant in the etiology of CD. A murine study showed that exposure to cigarette smoke causes damage to the ileal mucosal barrier, generating greater permeability to bacteria (15) (Figure 1). Thus, it is likely that alterations of the gut microbiota caused by cigarette smoke have a more significant impact on CD. On the contrary, humoral immunity seems to play a more relevant role in the development and progression of UC (31, 46, 47). Therefore, in the intestinal mucosa, the reported immunomodulatory effects of nicotine and CO could prevail over the harmful impact of cigarette smoke in UC, but not in CD (Figure 2).
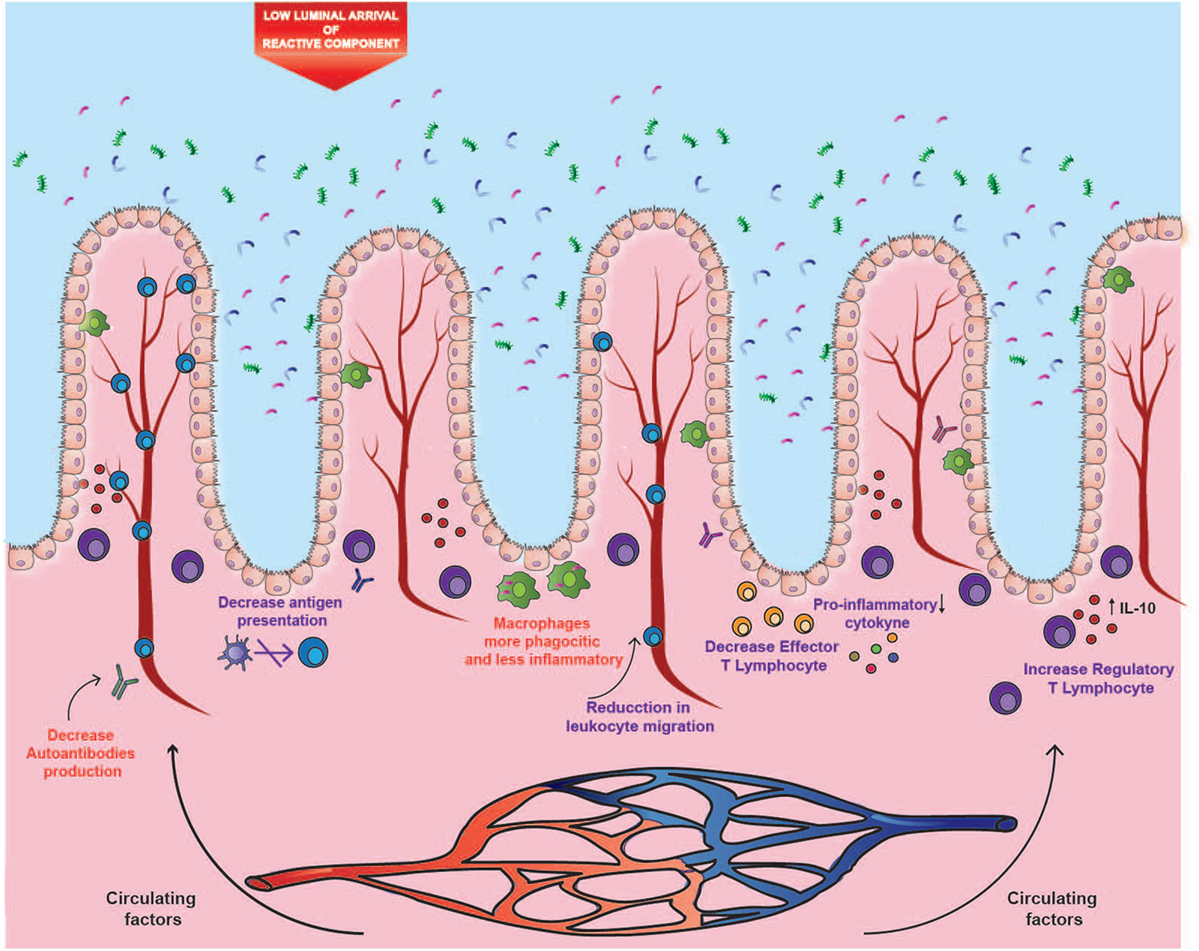
Figure 2. Impact of cigarette components on ulcerative colitis (UC). The distal segment of the large intestine could be mainly affected by the circulating components, where both carbon monoxide and nicotine could modulate the inflammatory profile, autoantibodies production, and leukocyte migration. These immunomodulatory effects could prevail in UC over the harmful effects observed in ileal Crohn’s disease, probably due to the lower arrival of luminal components and to the etiological differences between both disorders. Purple text denotes described effects and red text denotes proposed effects.
With respect to the location of inflammation, the opposite effects of cigarette smoke seem to be site-specific. In fact, murine studies have shown that exposure to cigarette smoke intensifies damage in the small intestine (15), whereas it reduces colitis (83). This site-specific effect could be given by a differential supply of cigarette compounds to the tissues, or by a tissue-dependent mechanism, thus originating the two remaining hypotheses. In this context, volatile compounds are rapidly absorbed through mucous membranes and alveoli, where they reach circulation. Conversely, a high percentage of particulate matter is swallowed, reaching high levels in saliva and gastric juices (23). In these fluids, several alkaline substances derived from tobacco accumulate because of their similar pH, and because they cannot be absorbed in their ionized form (23). Subsequently, these substances reach the small intestine, where they can be absorbed in the distal area as the pH increases, specifically in the ileum (84). As a second hypothesis, the considerable impact of cigarette smoke on the ileal mucosa can be attributed to the fact that this area suffers from a high exposure to these cigarette compounds (Figure 1). In fact, the distal segment of the large intestine can be affected mainly by the circulating components, where both CO and nicotine could modulate the inflammatory profile and leukocyte migration, and the immunomodulatory role would prevail over the impact on the mucosa (Figure 2).
Altogether, and as a third hypothesis, the harmful effects of cigarette smoking in the ileum could be tissue-dependent. In this context, some of the elements intrinsic to the ileum that could be affected by cigarette compounds are Peyer’s patches, Paneth cells, and tissue-specific microbiota (12, 19) (Figure 1). Interestingly, immune cells and Paneth cells present in Peyer’s patches are highly dependent on autophagy, and therefore could be severely affected by the reactive components of cigarette smoke (81). As a matter of fact, one study showed that mice exposed to cigarette smoke had a significant increase in autophagy levels in Peyer’s patches and in the surrounding epithelium (19), but there is still a lack of research on its actual effects on IBD patients. In fact, ileal epithelium and particularly Paneth cells are extremely sensitive to alterations in autophagy and UPR (81). As mentioned above, the impact of cigarette smoke on these pathways can lead to intestinal inflammation, impaired ileal barrier, and alterations in bacterial clearance (12, 19). Therefore, considering that there is a higher exposure to some cigarette components in that area, and that these processes are important in the pathophysiology of CD, the three hypotheses converge to help explain the increased risk of developing ileal CD in smokers.
Conclusion
Although all the mechanisms mentioned above help explain the impact of cigarette smoke on CD and UC, they fail to explain the opposite effects on intestinal inflammation. In this scenario, there are three non-exclusive possibilities: the different etiology of both diseases, the location of the disease, and the differences in the immune response that are specific for the affected tissue. In addition, the interaction between genetic factors and other environmental aspects works in a complementary and non-exclusive way to promote the different effects of cigarette smoke on intestinal inflammation. Understanding the opposite effects of cigarette smoking on CD and UC will help to better understand the causes of these diseases, and the appropriate therapeutic approach to be used.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the National Fund of Scientific and Technological Development FONDECYT (grant number 1131012 and grant number 1170964); Millennium Institute on Immunology and Immunotherapy (grant number P09/016-F); Iniciativa Científica Milenio from the Ministry of Economy and Tourism; the National Council of Scientific and Technological Research (CONICYT) (grant number 21140273, grant number 21140169, and grant number 21171014); and Escuela de Medicina, Pontificia Universidad Católica de Chile (grant number PMD-08/17).
References
1. Csordas A, Bernhard D. The biology behind the atherothrombotic effects of cigarette smoke. Nat Rev Cardiol (2013) 10:219–30. doi:10.1038/nrcardio.2013.8
2. Stämpfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol (2009) 9:377–84. doi:10.1038/nri2530
3. Marcilla A, Martínez I, Berenguer D, Gómez-Siurana A, Beltrán MI. Comparative study of the main characteristics and composition of the mainstream smoke of ten cigarette brands sold in Spain. Food Chem Toxicol (2012) 50:1317–33. doi:10.1016/j.fct.2012.01.046
4. Jaimes EA, DeMaster EG, Tian R-X, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol (2004) 24:1031–6. doi:10.1161/01.ATV.0000127083.88549.58
5. Li LF, Chan RL, Lu L, Shen J, Zhang L, Wu WK, et al. Cigarette smoking and gastrointestinal diseases: the causal relationship and underlying molecular mechanisms (review). Int J Mol Med (2014) 34:372–80. doi:10.3892/ijmm.2014.1786
6. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet (2011) 378:1015–26. doi:10.1016/S0140-6736(11)60988-4
7. World Health Organization. Tabaco. (2015). Available from: http://www.who.int/mediacentre/factsheets/fs339/es/
8. Li H, Wu Q, Xu L, Li X, Duan J, Zhan J, et al. Increased oxidative stress and disrupted small intestinal tight junctions in cigarette smoke-exposed rats. Mol Med Rep (2015) 11:4639–44. doi:10.3892/mmr.2015.3234
9. Yang S, Valvo S, Yao H, Kode A, Rajendrasozhan S, Edirisinghe I, et al. IKKα causes chromatin modification on pro-inflammatory genes by cigarette smoke in mouse lung. Am J Respir Cell Mol Biol (2008) 38:689–98. doi:10.1165/rcmb.2007-0379OC
10. Zhang L, Ren JW, Wong CC, Wu WK, Ren SX, Shen J, et al. Effects of cigarette smoke and its active components on ulcer formation and healing in the gastrointestinal mucosa. Curr Med Chem (2012) 19:63–9. doi:10.2174/092986712803413926
11. Ma L, Wang WP, Chow JY, Yuen ST, Cho CH. Reduction of EGF is associated with the delay of ulcer healing by cigarette smoking. Am J Physiol Gastrointest Liver Physiol (2000) 278:G10–7. doi:10.1152/ajpgi.2000.278.1.G10
12. Allais L, Kerckhof FM, Verschuere S, Bracke KR, De Smet R, Laukens D, et al. Chronic cigarette smoke exposure induces microbial and inflammatory shifts and mucin changes in the murine gut. Environ Microbiol (2016) 18(5):1352–63. doi:10.1111/1462-2920.12934
13. Biedermann L, Brülisauer K, Zeitz J, Frei P, Scharl M, Vavricka SR, et al. Smoking cessation alters intestinal microbiota: insights from quantitative investigations on human fecal samples using FISH. Inflamm Bowel Dis (2014) 20:1496–501. doi:10.1097/MIB.0000000000000129
14. Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol (2005) 43:3380–9. doi:10.1128/JCM.43.7.3380-3389.2005
15. Zuo L, Li Y, Wang H, Wu R, Zhu W, Zhang W, et al. Cigarette smoking is associated with intestinal barrier dysfunction in the small intestine but not in the large intestine of mice. J Crohns Colitis (2014) 8:1710–22. doi:10.1016/j.crohns.2014.08.008
16. Ma L, Chow JY, Cho CH. Cigarette smoking delays ulcer healing: role of constitutive nitric oxide synthase in rat stomach. Am J Physiol (1999) 276:G238–48.
17. Lee H-M, Kim C-W, Hwang K-A, Choi D-W, Choi K-C. Three components of cigarette smoke altered the growth and apoptosis of metastatic colon cancer cells via inducing the synthesis of reactive oxygen species and endoplasmic reticulum stress. Environ Toxicol Pharmacol (2016) 45:80–9. doi:10.1016/j.etap.2016.05.016
18. Wang H, Ma L, Li Y, Cho CH. Exposure to cigarette smoke increases apoptosis in the rat gastric mucosa through a reactive oxygen species-mediated and p53-independent pathway. Free Radic Biol Med (2000) 28:1125–31. doi:10.1016/S0891-5849(00)00207-0
19. Verschuere S, Bracke KR, Demoor T, Plantinga M, Verbrugghe P, Ferdinande L, et al. Cigarette smoking alters epithelial apoptosis and immune composition in murine GALT. Lab Invest (2011) 91:1056–67. doi:10.1038/labinvest.2011.74
20. Oberg M, Jaakkola MS, Woodward A, Peruga A, Prüss-Ustün A. Worldwide burden of disease from exposure to second-hand smoke: a retrospective analysis of data from 192 countries. Lancet (2011) 377:139–46. doi:10.1016/S0140-6736(10)61388-8
21. Ueno A, Jijon H, Traves S, Chan R, Ford K, Beck PL, et al. Opposing effects of smoking in ulcerative colitis and Crohn’s disease may be explained by differential effects on dendritic cells. Inflamm Bowel Dis (2014) 20:800–10. doi:10.1097/MIB.0000000000000018
22. Allais L, De Smet R, Verschuere S, Talavera K, Cuvelier CA, Maes T. Transient receptor potential channels in intestinal inflammation: what is the impact of cigarette smoking? Pathobiology (2017) 84:1–15. doi:10.1159/000446568
23. Lindell G, Farnebo LO, Chen D, Nexø E, Rask Madsen J, Bukhave K, et al. Acute effects of smoking during modified sham feeding in duodenal ulcer patients. An analysis of nicotine, acid secretion, gastrin, catecholamines, epidermal growth factor, prostaglandin E2, and bile acids. Scand J Gastroenterol (1993) 28:487–94. doi:10.3109/00365529309098254
24. Ogihara A, Kikuchi S, Hasegawa A, Kurosawa M, Miki K, Kaneko E, et al. Relationship between Helicobacter pylori infection and smoking and drinking habits. J Gastroenterol Hepatol (2000) 15:271–6. doi:10.1046/j.1440-1746.2000.02077.x
25. Hunsballe JM, Rittig S, Pedersen EB, Djurhuus JC. Smokeless nicotinergic stimulation of vasopressin secretion in patients with persisting nocturnal enuresis and controls. Scand J Urol Nephrol (2001) 35:117–21. doi:10.1080/003655901750170489
26. Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet (2012) 380:1590–605. doi:10.1016/S0140-6736(12)60026-9
27. Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet (2012) 380:1606–19. doi:10.1016/S0140-6736(12)60150-0
28. Oberhuber G, Stangl PC, Vogelsang H, Schober E, Herbst F, Gasche C. Significant association of strictures and internal fistula formation in Crohn’s disease. Virchows Arch (2000) 437:293–7. doi:10.1007/s004280000226
29. Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology (2011) 140:1785–94. doi:10.1053/j.gastro.2011.01.055
30. Cosnes J. What is the link between the use of tobacco and IBD? Inflamm Bowel Dis (2008) 14(Suppl 2):S14–5. doi:10.1002/ibd.20555
31. Souza HSP, de & Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol (2016) 13:13–27. doi:10.1038/nrgastro.2015.186
32. Bernstein CN, Shanahan F. Disorders of a modern lifestyle: reconciling the epidemiology of inflammatory bowel diseases. Gut (2008) 57:1185–91. doi:10.1136/gut.2007.122143
33. Park JH, Peyrin-Biroulet L, Eisenhut M, Shin J. Il IBD immunopathogenesis: a comprehensive review of inflammatory molecules. Autoimmun Rev (2017) 16:416–26. doi:10.1016/j.autrev.2017.02.013
34. Liu JZ, Anderson CA. Genetic studies of Crohn’s disease: past, present and future. Best Pract Res Clin Gastroenterol (2014) 28:373–86. doi:10.1016/j.bpg.2014.04.009
35. Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet (2016) 387:156–67. doi:10.1016/S0140-6736(15)00465-1
36. Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A (2009) 106:15813–8. doi:10.1073/pnas.0907722106
37. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene ATG16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456:259–63. doi:10.1038/nature07416
38. Deuring JJ, Fuhler GM, Konstantinov SR, Peppelenbosch MP, Kuipers EJ, de Haar C, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut (2014) 63:1081–91. doi:10.1136/gutjnl-2012-303527
39. Zorzi F, Monteleone I, Sarra M, Calabrese E, Marafini I, Cretella M, et al. Distinct profiles of effector cytokines mark the different phases of Crohn’s disease. PLoS One (2013) 8:e54562. doi:10.1371/journal.pone.0054562
40. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology (2005) 129:550–64. doi:10.1053/j.gastro.2005.05.002
41. Heller F, Fromm A, Gitter AH, Mankertz J, Schulzke J-D. Epithelial apoptosis is a prominent feature of the epithelial barrier disturbance in intestinal inflammation: effect of pro-inflammatory interleukin-13 on epithelial cell function. Mucosal Immunol (2008) 1(Suppl 1):S58–61. doi:10.1038/mi.2008.46
42. Fuss IJ, Strober W. The role of IL-13 and NK T cells in experimental and human ulcerative colitis. Mucosal Immunol (2008) 1(Suppl 1):S31–3. doi:10.1038/mi.2008.40
43. Fuss IJ, Joshi B, Yang Z, Degheidy H, Fichtner-Feigl S, de Souza H, et al. IL-13Rα2-bearing, type II NKT cells reactive to sulfatide self-antigen populate the mucosa of ulcerative colitis. Gut (2014) 63:1728–36. doi:10.1136/gutjnl-2013-305671
44. Tilg H, Kaser A. Failure of interleukin 13 blockade in ulcerative colitis. Gut (2015) 64:857–8. doi:10.1136/gutjnl-2015-309464
45. Takahashi F, Das KM. Isolation and characterization of a colonic autoantigen specifically recognized by colon tissue-bound immunoglobulin G from idiopathic ulcerative colitis. J Clin Invest (1985) 76:311–8. doi:10.1172/JCI111963
46. Duerr RH, Targan SR, Landers CJ, LaRusso NF, Lindsay KL, Wiesner RH, et al. Neutrophil cytoplasmic antibodies: a link between primary sclerosing cholangitis and ulcerative colitis. Gastroenterology (1991) 100:1385–91. doi:10.1016/0016-5085(91)90793-K
47. Biancone L, Mandal A, Yang H, Dasgupta T, Paoluzi AO, Marcheggiano A, et al. Production of immunoglobulin G and G1 antibodies to cytoskeletal protein by lamina propria cells in ulcerative colitis. Gastroenterology (1995) 109:3–12. doi:10.1016/0016-5085(95)90263-5
48. Man SM, Kaakoush NO, Mitchell HM. The role of bacteria and pattern-recognition receptors in Crohn’s disease. Nat Rev Gastroenterol Hepatol (2011) 8:152–68. doi:10.1038/nrgastro.2011.56
49. Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology (2010) 139:1844–54.e1. doi:10.1053/j.gastro.2010.08.049
50. Mahid SS, Minor KS, Soto RE, Hornung CA, Galandiuk S. Smoking and inflammatory bowel disease: a meta-analysis. Mayo Clin Proc (2006) 81:1462–71. doi:10.4065/81.11.1462
51. Calkins BM. A meta-analysis of the role of smoking in inflammatory bowel disease. Dig Dis Sci (1989) 34:1841–54. doi:10.1007/BF01536701
52. Beaugerie L, Massot N, Carbonnel F, Cattan S, Gendre JP, Cosnes J. Impact of cessation of smoking on the course of ulcerative colitis. Am J Gastroenterol (2001) 96:2113–6. doi:10.1111/j.1572-0241.2001.03944.x
53. Lunney PC, Leong RWL. Review article: ulcerative colitis, smoking and nicotine therapy. Aliment Pharmacol Ther (2012) 36:997–1008. doi:10.1111/apt.12086
54. Ingram JR, Thomas GA, Rhodes J, Green JT, Hawkes ND, Swift JL, et al. A randomized trial of nicotine enemas for active ulcerative colitis. Clin Gastroenterol Hepatol (2005) 3:1107–14. doi:10.1016/S1542-3565(05)00849-9
55. Benowitz NL, Porchet H, Sheiner L, Jacob P. Nicotine absorption and cardiovascular effects with smokeless tobacco use: comparison with cigarettes and nicotine gum. Clin Pharmacol Ther (1988) 44:23–8. doi:10.1038/clpt.1988.107
56. Lashner BA, Hanauer SB, Silverstein MD. Testing nicotine gum for ulcerative colitis patients. Experience with single-patient trials. Dig Dis Sci (1990) 35:827–32. doi:10.1007/BF01536795
57. Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, et al. Transdermal nicotine for active ulcerative colitis. N Engl J Med (1994) 330:811–5. doi:10.1056/NEJM199403243301202
58. Sandborn WJ, Tremaine WJ, Leighton JA, Lawson GM, Zins BJ, Compton RF, et al. Nicotine tartrate liquid enemas for mildly to moderately active left-sided ulcerative colitis unresponsive to first-line therapy: a pilot study. Aliment Pharmacol Ther (1997) 11:663–71. doi:10.1046/j.1365-2036.1997.00208.x
59. Cosnes J, Carbonnel F, Carrat F, Beaugerie L, Cattan S, Gendre J. Effects of current and former cigarette smoking on the clinical course of Crohn’s disease. Aliment Pharmacol Ther (1999) 13:1403–11. doi:10.1046/j.1365-2036.1999.00630.x
60. Brant SR, Picco MF, Achkar JP, Bayless TM, Kane SV, Brzezinski A, et al. Defining complex contributions of NOD2/CARD15 gene mutations, age at onset, and tobacco use on Crohn’s disease phenotypes. Inflamm Bowel Dis (2003) 9:281–9. doi:10.1097/00054725-200309000-00001
61. Yamamoto T, Keighley MR. Smoking and disease recurrence after operation for Crohn’s disease. Br J Surg (2000) 87:398–404. doi:10.1046/j.1365-2168.2000.01443.x
62. Cosnes J, Beaugerie L, Carbonnel F, Gendre JP. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology (2001) 120:1093–9. doi:10.1053/gast.2001.23231
63. Lewis CM, Whitwell SC, Forbes A, Sanderson J, Mathew CG, Marteau TM. Estimating risks of common complex diseases across genetic and environmental factors: the example of Crohn disease. J Med Genet (2007) 44:689–94. doi:10.1136/jmg.2007.051672
64. Davies PD, Yew WW, Ganguly D, Davidow AL, Reichman LB, Dheda K, et al. Smoking and tuberculosis: the epidemiological association and immunopathogenesis. Trans R Soc Trop Med Hyg (2006) 100:291–8. doi:10.1016/j.trstmh.2005.06.034
65. O’Leary SM, Coleman MM, Chew WM, Morrow C, McLaughlin AM, Gleeson LE, et al. Cigarette smoking impairs human pulmonary immunity to mycobacterium tuberculosis. Am J Respir Crit Care Med (2014) 190:1430–6. doi:10.1164/rccm.201407-1385OC
66. Bokaba RP, Anderson R, Theron AJ, Tintinger GR. Cigarette smoke condensate attenuates phorbol ester-mediated neutrophil extracellular trap formation. Afr Health Sci (2017) 17:896–904. doi:10.4314/ahs.v17i3.33
67. Shaler CR, Horvath CN, McCormick S, Jeyanathan M, Khera A, Zganiacz A, et al. Continuous and discontinuous cigarette smoke exposure differentially affects protective Th1 immunity against pulmonary tuberculosis. PLoS One (2013) 8:e59185. doi:10.1371/journal.pone.0059185
68. Duan MC, Zhang JQ, Liang Y, Liu GN, Xiao J, Tang HJ, et al. Infiltration of IL-17-producing T cells and Treg cells in a mouse model of smoke-induced emphysema. Inflammation (2016) 39:1334–44. doi:10.1007/s10753-016-0365-8
69. Suenaert P, Bulteel V, Den Hond E, Hiele M, Peeters M, Monsuur F, et al. The effects of smoking and indomethacin on small intestinal permeability. Aliment Pharmacol Ther (2000) 14:819–22. doi:10.1046/j.1365-2036.2000.00754.x
70. Lee G, Jung KH, Shin D, Lee C, Kim W, Lee S, et al. Cigarette smoking triggers colitis by IFN-γ+ CD4+ T cells. Front Immunol (2017) 8:1344. doi:10.3389/fimmu.2017.01344
71. Lakhan SE, Kirchgessner A. Anti-inflammatory effects of nicotine in obesity and ulcerative colitis. J Transl Med (2011) 9:129. doi:10.1186/1479-5876-9-129
72. Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med (2005) 201:1113–23. doi:10.1084/jem.20040463
73. Mackern-Oberti JP, Riquelme SA, Llanos C, Schmidt CB, Simon T, Anegon I, et al. Heme oxygenase-1 as a target for the design of gene and pharmaceutical therapies for autoimmune diseases. Curr Gene Ther (2014) 14:218–35. doi:10.2174/1566523214666140424150308
74. Sheikh SZ, Hegazi RA, Kobayashi T, Onyiah JC, Russo SM, Matsuoka K, et al. An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J Immunol (2011) 186:5506–13. doi:10.4049/jimmunol.1002433
75. Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology (2011) 141:237–48.e1. doi:10.1053/j.gastro.2011.04.007
76. Desreumaux P, Huet G, Zerimech F, Gambiez L, Balduyck M, Baron P, et al. Acute inflammatory intestinal vascular lesions and in situ abnormalities of the plasminogen activation system in Crohn’s disease. Eur J Gastroenterol Hepatol (1999) 11:1113–9. doi:10.1097/00042737-199910000-00007
77. Dhillon AP, Anthony A, Sim R, Wakefield AJ, Sankey EA, Hudson M, et al. Mucosal capillary thrombi in rectal biopsies. Histopathology (1992) 21:127–33. doi:10.1111/j.1365-2559.1992.tb00360.x
78. Maor I, Rainis T, Lanir A, Lavy A. Oxidative stress, inflammation and neutrophil superoxide release in patients with Crohn’s disease: distinction between active and non-active disease. Dig Dis Sci (2008) 53:2208–14. doi:10.1007/s10620-007-0141-6
79. Fritz T, Niederreiter L, Adolph T, Blumberg RS, Kaser A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut (2011) 60:1580–8. doi:10.1136/gut.2009.206466
80. Hoefkens E, Nys K, John JM, Van Steen K, Arijs I, Van der Goten J, et al. Genetic association and functional role of Crohn disease risk alleles involved in microbial sensing, autophagy, and endoplasmic reticulum (ER) stress. Autophagy (2013) 9:2046–55. doi:10.4161/auto.26337
81. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Böck J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature (2013) 503:272–6. doi:10.1038/nature12599
82. Benjamin JL, Hedin CR, Koutsoumpas A, Ng SC, McCarthy NE, Prescott NJ, et al. Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm Bowel Dis (2012) 18:1092–100. doi:10.1002/ibd.21864
83. Eliakim R, Fan QX, Babyatsky MW. Chronic nicotine administration differentially alters jejunal and colonic inflammation in interleukin-10 deficient mice. Eur J Gastroenterol Hepatol (2002) 14:607–14. doi:10.1097/00042737-200206000-00005
Keywords: cigarette smoking, gastrointestinal inflammation, inflammatory bowel disease, ulcerative colitis, Crohn’s disease
Citation: Berkowitz L, Schultz BM, Salazar GA, Pardo-Roa C, Sebastián VP, Álvarez-Lobos MM and Bueno SM (2018) Impact of Cigarette Smoking on the Gastrointestinal Tract Inflammation: Opposing Effects in Crohn’s Disease and Ulcerative Colitis. Front. Immunol. 9:74. doi: 10.3389/fimmu.2018.00074
Received: 28 October 2017; Accepted: 11 January 2018;
Published: 30 January 2018
Edited by:
Eric Cox, Ghent University, BelgiumCopyright: © 2018 Berkowitz, Schultz, Salazar, Pardo-Roa, Sebastián, Álvarez-Lobos and Bueno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel M. Álvarez-Lobos, bWFuYWx2YXJlemxAZ21haWwuY29t;
Susan M. Bueno, c2J1ZW5vQGJpby5wdWMuY2w=