 Martin Y. Fan
Martin Y. Fan Laurence A. Turka
Laurence A. Turka- 1Center for Transplantation Sciences, Department of Surgery, Massachusetts General Hospital, Boston, MA, United States
- 2Program in Immunology, Division of Medical Sciences, Harvard Medical School, Boston, MA, United States
CD4+ Foxp3+ regulatory T cells (Tregs) are an essential component of immune homeostasis. Modulation of Treg function has been proposed as a means of treating autoimmune conditions and preventing rejection of organ transplants, although achieving this goal will require a detailed understanding of Treg signaling pathways. Signaling within Tregs is known to differ considerably from that observed in other T cell subsets. Of note, Tregs are the only cell type known to constitutively express CD25, the main ligand-binding subunit of the IL-2 receptor. The PI(3)K/Akt/mTOR cascade constitutes a major signaling pathway downstream of IL-2 and is closely tied to cellular metabolism. Due to increasing recognition of the links between cellular fuel usage and immune cell function, the interplay between IL-2 signaling and Treg metabolism represents an important space for exploration and a potential approach for immunomodulation. Here, we discuss how IL-2 may affect Treg metabolism via PI(3)K signaling, as well as the effects of altered metabolism on Treg lineage stability and suppressor function.
Introduction
Regulatory T cells (Tregs) play a key role in maintaining immune homeostasis and in preventing the onset of autoimmune diseases (1). Modulation of Treg suppressor function is being actively explored as a promising new approach to treat autoimmunity (2–4), promote transplant tolerance (5, 6), and enhance anti-tumor responses (7, 8). Although several subsets of Tregs have been described, the best characterized is defined by the expression of CD4, CD25, and the transcription factor Foxp3 (9). The majority of circulating Tregs originate from the thymus and are termed “tTregs.” Naïve CD4+ T cells may also be induced to express Foxp3 in the periphery, thereby constituting a minority “pTreg” population (10) which is required for fetal tolerance (11). Although reports do not always specify which of the two populations is examined, any findings concerning “Tregs” most likely apply primarily to tTregs since they constitute the majority of Tregs in blood and secondary lymphoid organs. The importance of Tregs in maintaining peripheral tolerance is illustrated by the fact that mice (12) or humans (13) lacking Foxp3 suffer severe systemic autoimmunity. Similar, albeit less severe, autoimmune phenotypes are observed in mice (14) or humans (15) lacking CD25. Most Tregs constitutively express CD25 in addition to Foxp3, and it is generally believed that Tregs require continuous IL-2 signals through CD25 for their survival, lineage maintenance, and suppressor function (16, 17).
It is now appreciated that cell-intrinsic metabolic pathways directly impact cellular fate and function (18). Broadly speaking, aerobic glycolysis tends to support the function of pro-inflammatory cells, while fatty acid oxidation (FAO) tends to be used by anti-inflammatory cells such as Tregs (19). However, increasing evidence shows that Tregs also utilize aerobic glycolysis to achieve full suppressor function (20, 21). These metabolic programs are controlled in large part by the PI(3)K/Akt/mTOR signaling axis (22), offering multiple pharmacologic avenues to differentially target immune subsets depending on their metabolic preferences. Given the importance of CD25 for initiating PI(3)K signaling (23), in this review, we will focus on how IL-2 may interact with metabolism and the mechanisms through which metabolism influences Treg function. We touch on the difficulty of directly evaluating the interplay between IL-2 signaling, metabolism, and Treg function using existing germline knockout models and propose a means by which this issue can be addressed.
IL-2 and Tregs
IL-2 Signaling
The IL-2 receptor is composed of three subunits: CD25, CD122, and CD132, which are, respectively, referred to as the α, β, and γc (also termed the common gamma chain) subunits (24). CD122 and CD132 are the sole mediators of downstream signaling and may form a heterodimer capable of low-affinity binding to IL-2 (16) (Figure 1). The alpha subunit CD25 does not signal, but is needed for high-affinity binding to IL-2. Most Tregs constitutively express all three subunits, while conventional CD4+ and CD8+ T cells constitutively express the CD122/CD132 dimer and only express CD25 upon activation. Conventional T cells begin producing IL-2 1 h after activation (25) and constitute the primary source of IL-2 in vivo. IL-2 activates three major signaling axes: the STAT5, PI(3)K, and MAPK/ERK pathways. STAT5 is particularly important for Treg development, as it is necessary to initiate Foxp3 expression (26).
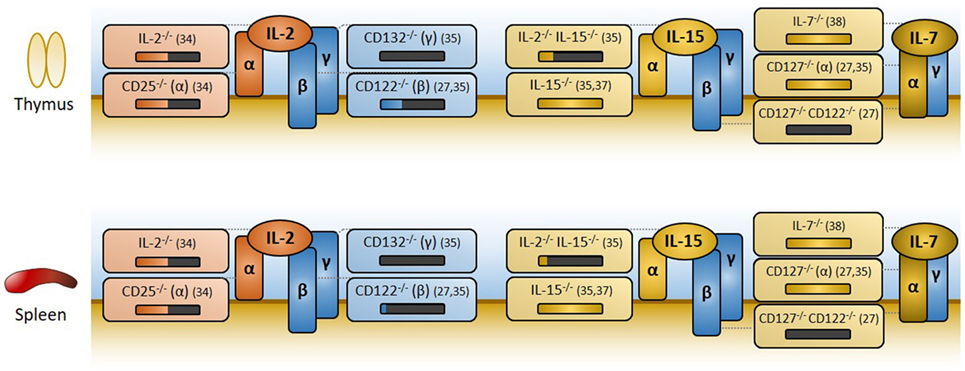
Figure 1. Overview of IL-2, IL-15, and IL-7 receptor components, and effects of knockouts on regulatory T cell (Treg) generation. The IL-2 and IL-15 receptors are trimers with common β and γc subunits (CD122 and CD132, respectively) that mediate signaling. High ligand affinity is conferred by their α subunit (CD25 for IL-2, CD215 for IL-15) which does not signal. The IL-7 receptor is a dimer of CD127 (α) and CD132 (γc). Disruption of IL-2 signaling is detrimental to Treg development and subsequent Treg representation in the periphery, as measured by the percentage of Foxp3+ cells among CD4+ cells in the thymus and spleen, respectively. In the above figure, losses in Tregs are represented visually as black bars below knockout mouse genotypes, with relevant references for each knockout provided immediately to the right. Deletion of IL-2 or CD25 (IL-2Rα) leads to an approximate 50% reduction in Foxp3+ cells. In the absence of IL-2 signaling, IL-15 or IL-7 appears to compensate, albeit imperfectly. Concomitant knockout of IL-2 and IL-15, or knockout of CD122 (the shared β subunit of both IL-2 and IL-15 receptors), exacerbates defects in Treg production. Removal of signaling through all three cytokines, whether through deletion of the common gamma chain CD132 (γc) or through the more targeted CD122/CD127 double knockout, virtually eliminates Treg development. Mice deficient in IL-15, IL-7, or CD127 (IL-7Rα) alone experience lymphopenia, but have normal percentages of Foxp3+ cells among CD4+ T cells and do not develop autoimmunity. Thus, IL-15 and IL-7 may partially compensate for Treg development in the absence of IL-2 signaling, but neither are required for Treg development when IL-2 signaling is fully functional.
The receptors for two other cytokines, IL-15 and IL-7, share subunits with the IL-2 receptor and partially compensate for losses of IL-2 or CD25 (27). The IL-15 receptor is a trimer that is strikingly similar to the IL-2 receptor, sharing the CD122 and CD132 subunits used for downstream signaling. Its alpha subunit CD215, like CD25, does not signal but instead confers high ligand affinity (28). On the other hand, the IL-7 receptor is a dimer composed of CD132 and a unique alpha subunit, CD127, which is capable of activating STAT5 (29).
IL-2 Is (Partially) Needed for Treg Development
Germline knockouts of IL-2 or its receptor components yield similar autoimmune phenotypes due to Treg deficiency (14, 30). Mice develop hemolytic anemia and colitis accompanied by thymic involution, lymph node hyperplasia, and splenomegaly, with elevated numbers of activated effector CD4+ and CD8+ T cells. Analogous findings have been reported in three clinical cases of CD25 loss (15, 31, 32), indicating that human Tregs are similarly dependent on CD25 and IL-2 signaling. These phenotypes are less severe than the scurfy phenotype resulting from Foxp3 deletion (33), most likely because the loss of IL-2 signaling also impacts effector T cells.
The fact that IL-2 and CD25 knockout mice maintain appreciable numbers of Foxp3+ cells in both thymus and spleen (34) indicates that IL-2 is not absolutely required for Treg development or subsequent survival, though it may be needed to achieve full suppressor function. The primary compensatory factor appears to be IL-15, as mice lacking both IL-2 and IL-15 are severely deficient in Foxp3+ cells (as are mice lacking either of the shared CD122 or CD132 subunits) (35, 36). In the presence of IL-2, however, IL-15 and IL-7 are dispensable for Treg development and function: IL-15−/−(37), IL-7−/− (38), and CD127−/− (27, 35) mice have normal percentages of Foxp3+ cells and do not develop autoimmunity.
Post-Developmental Roles of IL-2 in Tregs
Although IL-2 signaling is an important component of Treg development (39), its roles following development are less thoroughly explored. It is generally believed that Tregs require constitutive IL-2 signals to survive and maintain Foxp3 expression, much in the same way these signals are needed during thymic development (40, 41). The role of IL-2 in Treg suppressor function has been difficult to address due to its roles in Treg survival during development. To date, the most prominent attempt to evaluate Treg function has been a Bim−/− IL-2−/− double knockout (42), in which targeting of the pro-apoptotic protein Bim was intended to decouple the roles of CD25 in Treg survival versus suppressor function. Although this study suggested that IL-2 is needed for full suppressor function, it should be noted that all germline knockout models of IL-2 signaling components are subject to a critical confounding factor: knockout mice develop lethal autoimmunity, which by its very nature is accompanied by immune activation and widespread inflammation. For this reason, it has been difficult to study how constitutive IL-2 signaling influences Treg lineage stability and function post development, much less study its effects on Treg metabolism.
Blocking antibody approaches can be dosed to avoid inducing autoimmunity. Although they are insufficient to address the issue of Treg function, due to off-target effects on effector T cells, these studies do not support IL-2 as a survival factor for Tregs. Anti-CD25 clone 7D4, widely used in commercial Treg magnetic isolation kits (43), induces loss of CD25 for up to 2 weeks following injection, yet Tregs persist and mice fail to develop autoimmunity (44, 45). It is critical to note that this antibody is distinct from anti-CD25 clone PC61, commonly as a tool to deplete Tregs in vivo, which is now recognized to act via opsonization for phagocytosis rather than through IL-2 deprivation (46–48).
PI(3)K Signaling in Tregs
Because the role of IL-2-induced STAT5 signaling in Treg development has been reviewed extensively (16), here we focus on how lineage stability and suppressor function are influenced by metabolism in mature, post-developmental Tregs. PI(3)K catalyzes the conversion of PIP2 (PtdIns-4,5-P2) to PIP3 (PtdIns-3,4,5-P3) to permit activation of kinases with plextrin homology domains, most notably Akt. Targets of Akt include the protein translation regulator complex mTOR, which promotes cellular growth and survival (49). Thus, one major downstream effect of PI(3)K signaling is induction of aerobic glycolysis, which is increasingly emerging as a key control mechanism of Treg function (see below). The lipid phosphatase PTEN, which dephosphorylates PIP3 back into PIP2, and the protein phosphatase PHLPP, which dephosphorylates Akt, are the primary negative regulators of PI(3)K activity in T cells (50, 51). Excessive PI(3)K activity is detrimental to Tregs since loss of PTEN in mice (52, 53), loss of PHLPP in mice or in human cell culture (51), and induced Akt activation in human cell culture (54) all lead to Treg lineage instability and loss of suppressor function. Tregs may receive signals from three sources which would normally induce strong PI(3)K signaling: the TCR, CD28, and the IL-2 receptor (23). To prevent excessive PI(3)K signaling from these sources, Tregs express high levels of PTEN (55, 56) and PHLPP (51).
Treg Metabolism
Glycolysis
Following immune cell activation by antigen or inflammatory signals, aerobic glycolysis and fatty acid synthesis are rapidly induced to support cell proliferation and cytokine secretion (57). This is reflected in the metabolic profiles of relevant immune subsets: effector T cells such as Th1, Th2, and Th17 cells show increased glycolytic rates following activation, as do effector CD8+ T cells. Tregs, like memory CD8+ T cells, rely on FAO for their basal metabolism but utilize some degree of aerobic glycolysis to properly execute their suppressor functions.
Beyond mere association with immune activation, several causal links have emerged between inflammatory stimuli, glycolysis, and Tregs (Figure 2). In T cells, signals through the TCR, CD28, or IL-2 activate the PI(3)K/Akt/mTOR cascade (58), which induces expression of the glucose transporter Glut1 to facilitate increased glycolysis (59). Akt also inhibits Foxo1 and Foxo3 transcription factors which are important for Foxp3 gene expression (60–62). mTOR engages Hif-1α, which may also be independently activated through toll-like receptor signaling, to promote the expression of key glycolytic genes (63). Hif-1α may also directly bind Foxp3 and target it for proteasomal degradation (64). Reciprocally, forced Foxp3 expression is sufficient to suppress glycolysis and promote FAO in vitro (20). Treg effector molecules such as CTLA4 and PD-1 suppress glycolysis in CD4+ T cells by activating PTEN to antagonize PI(3)K signaling and subsequent glycolysis, with PD-1 also actively promoting FAO by increasing expression of CPT1A (65). These data suggest that elevated glycolysis is detrimental to Treg lineage stability and suppressor function.
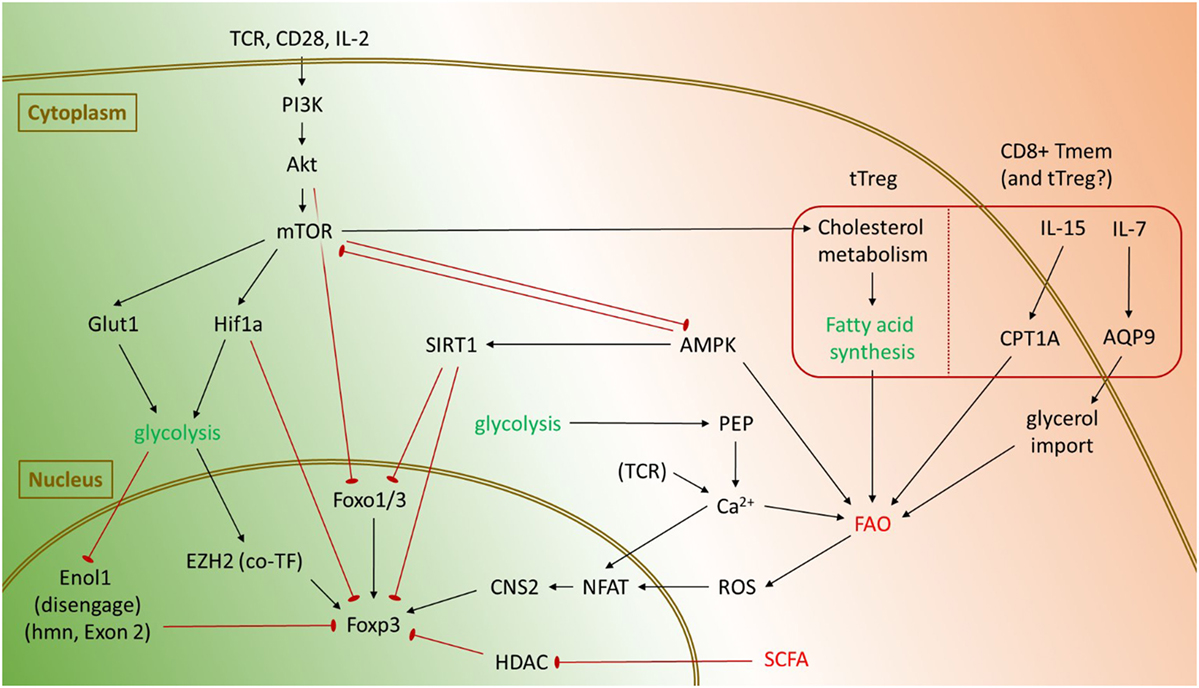
Figure 2. Pathways promoting glycolysis and fatty acid oxidation (FAO) in regulatory T cells (Tregs), and known mechanisms affecting Foxp3. Glycolysis is primarily activated in Tregs through mTOR and tends to suppress Foxp3 expression and Treg lineage stability. Activation of the PI(3)K/Akt/mTOR signaling axis inhibits Foxo transcription factors and promotes activation of Hif-1α, which can directly target Foxp3 for degradation. However, under certain conditions, glycolysis also promotes Foxp3 expression. By disengaging Enolase 1 from its nuclear role, glycolysis enables expression of the Foxp3-E2 splice isoform in humans. Glycolysis also represses microRNAs such as miR-101 and miR-26a to enable expression of EZH2, which is a cotranscription factor for Foxp3. Tregs generally rely upon FAO for their metabolic needs. In the gut, short-chain fatty acids (SCFA) inhibit histone deacetylases (HDACs) to promote Foxp3 expression and conversion of naïve CD4+ T cells into pTregs. Under certain conditions, FAO may also impinge upon Treg lineage stability. Sirt1 may repress Foxp3, either through direct deacetylation of Foxp3 or by targeting Foxo transcription factors. In CD8+ memory T cells, cytokines such as IL-7 and IL-15 promote uptake of fatty acid precursors and increased FAO, respectively. It remains to be seen whether similar processes occur in Tregs as well. Both glycolysis and FAO can also promote Foxp3 expression through an NFAT-dependent mechanism.
However, most studies showing detrimental effects of glycolysis on Tregs were performed in vitro, where T cell activation and glycolysis were driven to their maximum extent. Under certain conditions, glycolysis actually supports Foxp3 expression, promotes Treg proliferation, and potentiates suppressor function. Among in vitro induced human Tregs, the glycolytic enzyme Enolase-1 binds the Foxp3 promoter and its CNS2 regions. This represses transcription of a splice isoform containing Exon 2 (Foxp3-E2), which is needed for optimal Treg suppressor function. Engaging glycolysis forces Enolase-1 into the cytoplasm, thereby allowing transcription of Foxp3-E2 (66). Glycolysis also favors expression of the histone methyltransferase EZH2 by repressing inhibitory microRNAs such as miR-101 and miR-26a (67). EZH2 in turn binds Foxp3 to assist suppression of target genes (68), although no experiment has yet confirmed glycolysis-dependent EZH2 expression is essential for Treg lineage stability. The glycolytic metabolite phosphoenol pyruvate (PEP) can also increase Foxp3 expression through an NFAT-dependent mechanism. By inhibiting the calcium ATPase SERCA, PEP increases intracellular Ca2+ levels to promote nuclear translocation of NFAT, which facilitates interactions between the Foxp3 promoter and its CNS2 regions (69, 70).
A recent study suggests a possible resolution of these conflicting roles for glycolysis in Tregs. Using a Glut1 transgene to increase glucose uptake and glycolysis, the authors found that although elevated glycolysis boosts tTreg proliferation, it comes at the cost of their ability to execute suppressor functions (20). This suggests that for optimal Treg activity, a balance must be struck between the cell activating effects of glycolysis with its negative effects on the lineage.
Fatty Acid Oxidation
Fatty acid oxidation is generally associated with an anti-inflammatory phenotype and maintenance of Treg lineage stability. One mechanism is through simple antagonism of glycolysis: Tregs express high levels of AMPK, which simultaneously promotes FAO while inhibiting mTOR and subsequent glycolysis (71). In the gut, short-chain fatty acids are also known to inhibit mTOR (72). They have the added benefit of stabilizing pTregs by inhibiting histone deacetylases (HDACs) such as HDAC6 and HDAC9 which would otherwise inhibit Foxp3 expression (73, 74). Reactive oxygen species generated as a byproduct of oxidative phosphorylation have been shown to promote Foxp3 stability by increasing activity of the transcription factor NFAT, which binds the CNS2 enhancer of Foxp3 (70, 75). In addition, Foxp3 may experience post-transcriptional modifications such as acetylation, which prevents Foxp3 from being targeted for degradation thereby increasing its half-life (76). Foxp3 acetylation is dependent on nuclear availability of acetyl-CoA, whose supply is increased upon breakdown of fatty acids. As with glycolysis however, under certain conditions FAO may antagonize Treg lineage stability. FAO promotes an increased NAD+/NADH ratio, which elevates the activity of the deacetylase SIRT1 (77). By deacetylating Foxp3, SIRT1 promotes Foxp3 poly-ubiquitination and subsequent proteasomal degradation (78).
pTregs and tTregs diverge considerably in their execution of FAO: although pTregs generally rely upon exogenous fatty acids for their metabolic needs (79), it is uncertain whether tTregs can import exogenous fatty acids in vivo (18). While the coming years will likely clarify this issue, available literature suggests one peculiar metabolic feature among tTregs. One of the major roles for mTOR signaling in tTregs is to drive synthesis of endogenous fatty acid stores, primarily along cholesterol biosynthetic pathways (80). Whether these endogenously synthesized fatty acids are then used for energy is not known, although these specific pathways are needed for tTreg proliferation and optimal suppressor function. Memory CD8+ T cells constitute the only major T cell subset known to synthesize endogenous fatty acids for subsequent FAO in vivo (18, 81) and rely in part on IL-7 and IL-15 to regulate these processes. IL-7 induces expression of the channel protein aquaporin 9, which facilitates glycerol import for fatty acid synthesis (82). IL-15 increases FAO by stimulating mitochondrial biogenesis and elevating expression of CPT1a, a key regulator of FAO (83). Given that Tregs appear to rely on IL-7 and/or IL-15 in the absence of IL-2, we speculate that tTregs from IL-2 or CD25 knockout mice may experience a shift from glycolysis to FAO, possibly with an associated loss of suppressor function. Whether similar events might occur in pTregs is unknown, although prior literature (11) suggests a loss of suppressor function in pTregs would result in increased fetal resorption among any IL-2 or CD25 knockout mothers which reach breeding age.
Therapeutic Interventions
One of the most exciting prospects of immunometabolism is developing therapeutic interventions which can selectively target T cell subsets. Since activated effector T cells are more reliant on glycolysis than Tregs, studies have examined whether inhibiting glycolysis might improve outcomes in mouse models of autoimmunity and transplant rejection. Blocking glycolysis with 2-DG (a competitive inhibitor of hexokinase), or with dichloroacetate (an inhibitor of PDHK isoforms) reduced the severity of experimental autoimmune encephalomyelitis with associated decreases in the percentage of Th17, but not Treg, cells (63, 84). Similar outcomes were reported following inhibition of another glycolytic enzyme, acetyl-CoA carboxylase 1 (ACC1), with soraphen A or with T cell specific genetic deletion of ACC1 (79). Furthermore, treatment with metformin (an agonist of AMPK, which increases fatty acid uptake and oxidation) reduced airway inflammation and fibrosis in a murine asthma model (85). In the transplant setting, treatment with 2-DG, metformin, and a glutamine uptake inhibitor DON prolonged allograft survival in heart and skin transplants, in part by suppressing the proliferation of antigen-specific T cells and by increasing the relative frequency of Tregs (86).
Conversely, interventions that promote glycolysis enhance immune function, presumably by increasing the proliferation and function of effector T cells while inhibiting Treg function. Pharmacological blockade or genetic loss of PTEN leads to Akt-dependent inhibition of Foxo3a and subsequent loss of Foxp3 and tumor regression (87). Furthermore, increasing glycolysis through forced expression of the glucose transporter Glut1 in Tregs exacerbated pathology in an adoptive transfer model of colitis (20). Tregs recovered from this system were also found to have lower levels of Foxp3 protein.
Conclusion and Future Directions
The metabolic state of Tregs defies simple categorical explanations with regard to glycolysis and FAO. Although elevated glycolysis is generally associated with immune activation and can be detrimental to Treg lineage stability and function, controlled levels of glycolysis are necessary to sustain the same processes. The list of known links between metabolism and Treg function is far from complete, and the coming years will likely reveal other metabolic enzymes with moonlighting roles in Treg biology. In particular, the “futile cycle” approach of tTregs to FAO, and its preference for cholesterol synthesis may be a promising area of discovery.
Metabolic interventions offer a promising new approach to modulating Treg function and may be used to fine-tune therapies targeting other signaling pathways or used as a primary therapy in their own right. Of note, while there is clear potential for interplay between IL-2 signaling and immunometabolism through the PI(3)K/Akt/mTOR signaling cascade, to date no studies have specifically evaluated the effects of IL-2 signaling on Treg metabolism. In part, this is due to the inadequacies of germline knockout models to address this question. As mentioned before, such knockouts experience an autoimmune environment in which immune cells are already highly active and presumably glycolytic. It would be more appropriate to use a model in which Tregs can be inducibly made to lose IL-2 signaling while maintaining immune homeostasis. A tamoxifen-inducible CD25 knockout, with tamoxifen dosage adjusted to leave enough CD25-competent cells to prevent autoimmunity, would be well suited for this approach. Such studies would lay the framework for combination treatments in which metabolic interventions would be used with existing therapies such as CD25 blockade.
Author Contributions
MF and LT conceived of and wrote the review.
Conflict of Interest Statement
LT discloses personal or family financial interests with Third Rock Ventures, Tango Therapeutics, and Neon Therapeutics. LT has served as a consultant to Lycera, Solid Bio, UCB, and Intellia. M.F declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank B. Blazar, J. Bluestone, H. Chi, J. Rathmell, and members of the Turka laboratory for discussions.
Funding
The authors acknowledge financial support from the US National Institutes of Health (P01-HL018646 to LT, and T32-AI007529 to MF) and from the Herchel Smith Graduate Fellowship Program (MF).
References
1. Sakaguchi S. Regulatory T cells: history and perspective. Methods Mol Biol (2011) 707:3–17. doi:10.1007/978-1-61737-979-6_1
2. Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev (2008) 223:371–90. doi:10.1111/j.1600-065X.2008.00637.x
3. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev (2006) 212:8–27. doi:10.1111/j.0105-2896.2006.00427.x
4. Bluestone JA, Tang Q. How do CD4+CD25+ regulatory T cells control autoimmunity? Curr Opin Immunol (2005) 17(6):638–42. doi:10.1016/j.coi.2005.09.002
5. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117(14):3921–8. doi:10.1182/blood-2010-10-311894
6. Priyadharshini B, Turka LA. T-cell energy metabolism as a controller of cell fate in transplantation. Curr Opin Organ Transplant (2015) 20(1):21–8. doi:10.1097/MOT.0000000000000149
7. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol (2014) 27:1–7. doi:10.1016/j.coi.2013.12.005
8. Curiel TJ. Tregs and rethinking cancer immunotherapy. J Clin Invest (2007) 117(5):1167–74. doi:10.1172/JCI31202
9. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi:10.1126/science.1079490
10. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14(4):307–8. doi:10.1038/ni.2554
11. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell (2012) 150(1):29–38. doi:10.1016/j.cell.2012.05.031
12. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol (2003) 4(4):337–42. doi:10.1038/ni909
13. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi:10.1038/83713
14. Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity (1995) 3(4):521–30. doi:10.1016/1074-7613(95)90180-9
15. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol (2007) 119(2):482–7. doi:10.1016/j.jaci.2006.10.007
16. Malek TR. The biology of interleukin-2. Annu Rev Immunol (2008) 26:453–79. doi:10.1146/annurev.immunol.26.021607.090357
17. Benoist C, Mathis D. Treg cells, life history, and diversity. Cold Spring Harb Perspect Biol (2012) 4(9):a007021. doi:10.1101/cshperspect.a007021
18. Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol (2016) 17(6):618–25. doi:10.1038/ni.3466
19. O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi:10.1038/nri.2016.70
20. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol (2016) 17(12):1459–66. doi:10.1038/ni.3577
21. Tanimine N, Turka LA, Priyadharshini B. Navigating T cell immunometabolism in transplantation. Transplantation (2017) Forthcoming. doi:10.1097/TP.0000000000001951
22. Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol (2014) 15(3):155–62. doi:10.1038/nrm3757
23. Han JM, Patterson SJ, Levings MK. The role of the PI3K signaling pathway in CD4(+) T cell differentiation and function. Front Immunol (2012) 3:245. doi:10.3389/fimmu.2012.00245
24. Stauber DJ, Debler EW, Horton PA, Smith KA, Wilson IA. Crystal structure of the IL-2 signaling complex: paradigm for a heterotrimeric cytokine receptor. Proc Natl Acad Sci U S A (2006) 103(8):2788–93. doi:10.1073/pnas.0511161103
25. Sojka DK, Bruniquel D, Schwartz RH, Singh NJ. IL-2 secretion by CD4+ T cells in vivo is rapid, transient, and influenced by TCR-specific competition. J Immunol (2004) 172(10):6136–43. doi:10.4049/jimmunol.172.10.6136
26. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108(5):1571–9. doi:10.1182/blood-2006-02-004747
27. Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol (2008) 181(5):3285–90. doi:10.4049/jimmunol.181.5.3285
28. Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol (2006) 6(8):595–601. doi:10.1038/nri1901
29. Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol (2006) 24:657–79. doi:10.1146/annurev.immunol.24.021605.090727
30. Schorle H, Holtschke T, Hünig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature (1991) 352(6336):621–4. doi:10.1038/352621a0
31. Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc Natl Acad Sci U S A (1997) 94(7):3168–71. doi:10.1073/pnas.94.7.3168
32. Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Mannurita SC, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol (2013) 146(3):248–61. doi:10.1016/j.clim.2013.01.004
33. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27(1):68–73. doi:10.1038/83784
34. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol (2005) 6(11):1142–51. doi:10.1038/ni1263
35. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol (2007) 178(1):280–90. doi:10.4049/jimmunol.178.1.280
36. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol (2016) 17(11):1322–33. doi:10.1038/ni.3540
37. Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med (2000) 191(5):771–80. doi:10.1084/jem.191.5.771
38. Peffault de latour R, Dujardin HC, Mishellany F, Burlen-Defranoux O, Zuber J, Marques R, et al. Ontogeny, function, and peripheral homeostasis of regulatory T cells in the absence of interleukin-7. Blood (2006) 108(7):2300–6. doi:10.1182/blood-2006-04-017947
39. Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity (2008) 28(1):100–11. doi:10.1016/j.immuni.2007.11.021
40. Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol (2011) 11(2):119–30. doi:10.1038/nri2916
41. Lin JX, Li P, Liu D, Jin HT, He J, Ur Rasheed MA, et al. Critical role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity (2012) 36(4):586–99. doi:10.1016/j.immuni.2012.02.017
42. Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, et al. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol (2010) 185(11):6426–30. doi:10.4049/jimmunol.0903940
43. Miltenyi Biotec. CD4+CD25+ Regulatory T Cell Isolation Kit Mouse. (2006). Available from: http://www.miltenyibiotec.com/~/media/Images/Products/Import/0001100/IM0001159.ashx?force=1
44. Kohm AP, Mcmahon JS, Podojil JR, Begolka WS, DeGutes M, Kasprowicz DJ, et al. Cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J Immunol (2006) 176(6):3301–5. doi:10.4049/jimmunol.176.6.3301
45. Couper KN, Blount DG, De souza JB, Suffia I, Belkaid Y, Riley EM. Incomplete depletion and rapid regeneration of Foxp3+ regulatory T cells following anti-CD25 treatment in malaria-infected mice. J Immunol (2007) 178(7):4136–46. doi:10.4049/jimmunol.178.7.4136
46. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res (1999) 59(13):3128–33.
47. Setiady YY, Coccia JA, Park PU. In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. Eur J Immunol (2010) 40(3):780–6. doi:10.1002/eji.200939613
48. Mohr F, Fischer JC, Nikolaus M, Stemberger C, Dreher S, Verschoor A, et al. Minimally manipulated murine regulatory T cells purified by reversible Fab multimers are potent suppressors for adoptive T-cell therapy. Eur J Immunol (2017) 47(12):2153–62. doi:10.1002/eji.201747137
49. Vanhaesebroeck B, Guillermet-guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol (2010) 11(5):329–41. doi:10.1038/nrm2882
50. Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene (2008) 27(41):5527–41. doi:10.1038/onc.2008.247
51. Patterson SJ, Han JM, Garcia R, Assi K, Gao T, O’Neill A, et al. Cutting edge: PHLPP regulates the development, function, and molecular signaling pathways of regulatory T cells. J Immunol (2011) 186(10):5533–7. doi:10.4049/jimmunol.1002126
52. Huynh A, Dupage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol (2015) 16(2):188–96. doi:10.1038/ni.3077
53. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol (2015) 16(2):178–87. doi:10.1038/ni.3076
54. Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood (2007) 109(5):2014–22. doi:10.1182/blood-2006-07-035279
55. Buckler JL, Walsh PT, Porrett PM, Choi Y, Turka LA. Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J Immunol (2006) 177(7):4262–6. doi:10.4049/jimmunol.177.7.4262
56. Bensinger SJ, Walsh PT, Zhang J, Carroll M, Parsons R, Rathmell JC, et al. Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J Immunol (2004) 172(9):5287–96. doi:10.4049/jimmunol.172.9.5287
57. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342(6155):1242454. doi:10.1126/science.1242454
58. Newton RH, Turka LA. Regulation of T cell homeostasis and responses by pten. Front Immunol (2012) 3:151. doi:10.3389/fimmu.2012.00151
59. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi:10.1016/j.cmet.2014.05.004
60. Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch’en IL, Stockmann C, et al. Foxo transcription factors control regulatory T cell development and function. Immunity (2010) 33(6):890–904. doi:10.1016/j.immuni.2010.12.002
61. Ouyang W, Beckett O, Ma Q, Paik JH, Depinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol (2010) 11(7):618–27. doi:10.1038/ni.1884
62. Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature (2012) 491(7425):554–9. doi:10.1038/nature11581
63. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi:10.1084/jem.20110278
64. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi:10.1016/j.cell.2011.07.033
65. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi:10.1038/ncomms7692
66. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol (2015) 16(11):1174–84. doi:10.1038/ni.3269
67. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol (2016) 17(1):95–103. doi:10.1038/ni.3313
68. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity (2015) 42(2):227–38. doi:10.1016/j.immuni.2015.01.007
69. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi:10.1016/j.cell.2015.08.012
70. Li X, Liang Y, Leblanc M, Benner C, Zheng Y. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell (2014) 158(4):734–48. doi:10.1016/j.cell.2014.07.030
71. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi:10.4049/jimmunol.1003613
72. Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol (2015) 8(1):80–93. doi:10.1038/mi.2014.44
73. Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal (2012) 5(229):ra45. doi:10.1126/scisignal.2002873
74. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341(6145):569–73. doi:10.1126/science.1241165
75. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi:10.1016/j.immuni.2012.10.020
76. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YYJ, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115(5):965–74. doi:10.1182/blood-2009-02-207118
77. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (2009) 324(5930):1076–80. doi:10.1126/science.1164097
78. van Loosdregt J, Brunen D, Fleskens V, Pals CE, Lam EW, Coffer PJ. Rapid temporal control of Foxp3 protein degradation by sirtuin-1. PLoS One (2011) 6(4):e19047. doi:10.1371/journal.pone.0019047
79. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. doi:10.1038/nm.3704
80. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature (2013) 499(7459):485–90. doi:10.1038/nature12297
81. O’Sullivan D, Van der windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41(1):75–88. doi:10.1016/j.immuni.2014.06.005
82. Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, et al. IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell (2015) 161(4):750–61. doi:10.1016/j.cell.2015.03.021
83. Van der windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36(1):68–78. doi:10.1016/j.immuni.2011.12.007
84. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125(1):194–207. doi:10.1172/JCI76012
85. Park CS, Bang BR, Kwon HS, Moon KA, Kim TB, Lee KY, et al. Metformin reduces airway inflammation and remodeling via activation of AMP-activated protein kinase. Biochem Pharmacol (2012) 84(12):1660–70. doi:10.1016/j.bcp.2012.09.025
86. Lee CF, Lo YC, Cheng CH, Furtmuller GJ, Oh B, Andrade-Oliveira V, et al. Preventing allograft rejection by targeting immune metabolism. Cell Rep (2015) 13(4):760–70. doi:10.1016/j.celrep.2015.09.036
Keywords: regulatory T cells, CD25, IL-2, metabolism, aerobic glycolysis, fatty acid oxidation, PI(3)K
Citation: Fan MY and Turka LA (2018) Immunometabolism and PI(3)K Signaling As a Link between IL-2, Foxp3 Expression, and Suppressor Function in Regulatory T Cells. Front. Immunol. 9:69. doi: 10.3389/fimmu.2018.00069
Received: 26 November 2017; Accepted: 10 January 2018;
Published: 29 January 2018
Edited by:
Carrie L. Lucas, Yale University, United StatesReviewed by:
Megan K. Levings, University of British Columbia, CanadaJose R. Regueiro, Complutense University of Madrid, Spain
Copyright: © 2018 Fan and Turka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurence A. Turka, bHR1cmthQHBhcnRuZXJzLm9yZw==