 Shiyang Li
Shiyang Li John W. Bostick
John W. Bostick Liang Zhou
Liang Zhou- 1Department of Infectious Diseases and Immunology, College of Veterinary Medicine, University of Florida, Gainesville, FL, United States
- 2Department of Chemical and Biological Engineering, Northwestern University, Evanston, IL, United States
With striking similarity to their adaptive T helper cell counterparts, innate lymphoid cells (ILCs) represent an emerging family of cell types that express signature transcription factors, including T-bet+ Eomes+ natural killer cells, T-bet+ Eomes− group 1 ILCs, GATA3+ group 2 ILCs, RORγt+ group 3 ILCs, and newly identified Id3+ regulatory ILC. ILCs are abundantly present in barrier tissues of the host (e.g., the lung, gut, and skin) at the interface of host–environment interactions. Active research has been conducted to elucidate molecular mechanisms underlying the development and function of ILCs. The aryl hydrocarbon receptor (Ahr) is a ligand-dependent transcription factor, best known to mediate the effects of xenobiotic environmental toxins and endogenous microbial and dietary metabolites. Here, we review recent progresses regarding Ahr function in ILCs. We focus on the Ahr-mediated cross talk between ILCs and other immune/non-immune cells in host tissues especially in the gut. We discuss the molecular mechanisms of the action of Ahr expression and activity in regulation of ILCs in immunity and inflammation, and the interaction between Ahr and other pathways/transcription factors in ILC development and function with their implication in disease.
Introduction
Innate lymphoid cells (ILCs) are newly identified cell populations, which mirror helper T cells, such as Th1, Th2, and Th17 cells, by expressing similar transcription factors and cytokines (1–3). ILCs are divided into group 1 ILCs (ILC1) (T-bet+), group 2 ILCs (ILC2) (GATA3+), and group 3 ILCs (ILC3) (RORγt+) (1). To join the group, a new type of ILC that express the transcription factor Id3 and exhibit regulatory function [known as regulatory ILC (ILCreg)] have also recently been identified (4). Notably, natural killer (NK) cells have been defined as distinct population from ILC1, based on eomesdermin (Eomes) expression and a distinct progenitor from other ILCs (5). ILCs are predominantly locate at the mucosal barriers and participate in various biological processes, such as control of pathogenic infection, progression of autoimmune disease, as well as development of cancer (2, 6, 7). Different from adaptive immune cells, ILCs lack the antigen stimulation step and respond quickly under certain contexts of disease (8). The aryl hydrocarbon receptor (Ahr) is a ligand-dependent transcriptional factor, which can sense environmental and endogenous compounds generated by commensal, dietary, or cellular metabolism (9–11). Ahr has been studied in the development and/or function of various immune/non-immune cells (11) and recently found to be key regulator of ILC3 (12–14). There are many extensive reviews on Ahr in other immune cells. In this review, we focus our efforts on summarizing the recent progresses on decoding Ahr physiological functions in the development and function of ILCs, as well as Ahr-mediated cross talk between ILCs and other immune/non-immune cells in host tissues, especially in the gut. We discuss the molecular regulation of Ahr expression and activity in ILCs, and the interaction between Ahr and other pathways/transcription factors in ILC development and function. We also identify areas that need further study, especially the role of Ahr in group 1 and group 2 ILCs.
Description and Function of ILCs
Innate lymphoid cells share the same progenitor, common lymphoid progenitors (CLPs), as adaptive immune cells, including T and B cells (15). CLPs differentiate toward unique direction to α-lymphoid precursor, and then common helper innate lymphoid progenitor (CHILP), to become ILCs, including NK cells, ILC1, ILC2, ILC3, and ILCreg (5, 16). The lineage-defining transcription factors, key regulators, stimuli, and effector molecules are summarized in Table 1.
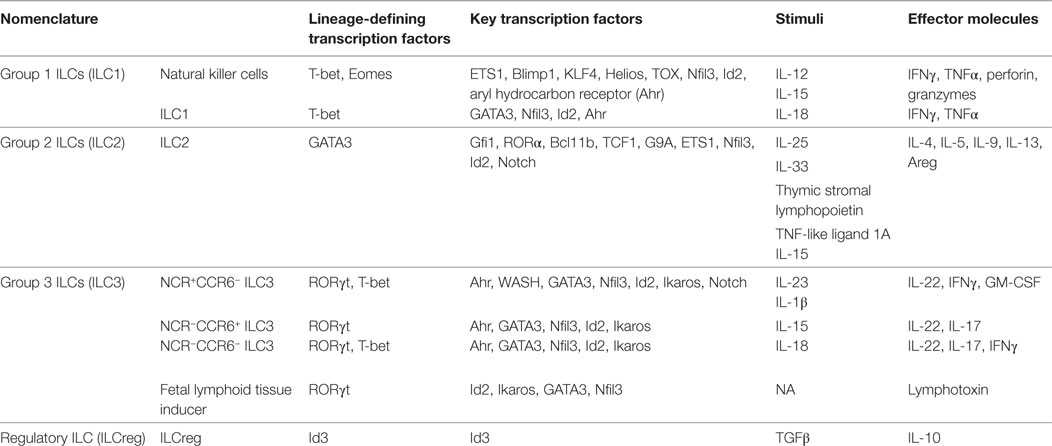
Table 1. Features of innate lymphoid cell (ILC) subsets.
Group 1 ILC
While NK cells are predominantly circulating in the blood and secondary lymphoid organs such as the lymph nodes and spleen, NK cells are also found in some non-lymphoid tissues such as the liver, uterus, and lung (17). Closely related in function to NK cells, ILC1 are present in various non-lymphoid tissues, including intestine, liver, salivary glands, and the female reproductive tract (18). The development and function of ILC1 depend on T-bet, while the requirement of T-bet by NK cells appears to be complicated since deletion of T-bet reduces the numbers of NK cells in liver, spleen, and peripheral blood (19, 20), but not in bone marrow and intestine (5, 19, 20). The transcription factor, Eomes, distinguishes NK cells from ILC1 and is indispensable for the development of NK cells (5). Recent studies indicate that NK cells and ILC1 derive from different progenitors, which further separate NK cells from ILC1 (5). Although developmentally identified as two distinct populations, NK cells and ILC1 can be stimulated by IL-12, IL-15, or IL-18 to produce interferon γ (IFNγ) and tumor necrosis factor (TNF) (18), which are critical for the immune response to control intracellular pathogens, viruses, and tumors (5, 21, 22). NK cells have the ability to secrete granzyme and perforin to promote cytotoxic function, which imparts NK cells tumor suppression activity, distinct from ILC1 (23). Different from intestinal lamina proprial ILC1 that express T-bet but not Eomes, intraepithelial ILC1 have been shown to express both T-bet and Eomes, and produce granzyme and perforin; however, lack of the requirement of IL-15 signals for their maintenance distinguishes intraepithelial ILC1 from NK cells (24).
Group 2 ILC
Group 2 ILCs have been identified to localize in various lymphoid/non-lymphoid tissues, including intestine, lung, adipose tissue, spleen, nasal tissue, and skin, while immature ILC2 are also reported in bone marrow (25–27). The development and function of ILC2 require GATA3, RORα, Gfi1, TCF1, Bcl11b, and Notch signaling, of which GATA3 acts as the defining marker of ILC2 (26, 28–34). Upon stimulation with IL-25, IL-33, or thymic stromal lymphopoietin (TSLP), ILC2 can produce IL-5, IL-13, and IL-4, similar to Th2 cells, which contribute to the control of helminth infection and pathology of allergic inflammation (25, 35–39). ILC2 can also express IL-9 to promote the epithelial cell maintenance in the lung (40, 41). Amphiregulin is an effector molecule produced by ILC2 to participate in the tissue repair in the gut (42). Additionally, ILC2 have been shown to promote the beiging of white adipose tissue to control obesity through the production of methionine-enkephalin peptides (43, 44).
Group 3 ILC
Group 3 ILCs are mainly found in gastrointestinal tract, while few ILC3 are present in other tissues (45, 46). ILC3 are heterogeneous, and can be divided, based on the expression of the natural cytotoxicity receptor (NCR or NKp46/NKp44) and chemokine receptor 6 (CCR6), into three major groups: NCR+CCR6− ILC3, NCR−CCR6+ ILC3, and NCR−CCR6− ILC3 (47). It should be noted that the above discussion is on ILC3 after birth. Fetal ILC3, also known as lymphoid tissue inducer (LTi) cells, which express RORγt, function in the formation of secondary lymphoid organs, such as lymph nodes and gut-associated lymphoid tissue (48–50). Postnatal CCR6+ ILC3 found in the gut and other lymphoid organs are known as LTi-like cells (51). While RORγt is the common transcription factor that is required for the development, maintenance, and function of all ILC3 (52), NCR+ ILC3 also appear to depend on T-bet for development and function (53). When stimulated, all three subsets of ILC3 produce IL-22, while NCR+ ILC3, relying on T-bet, can express IFNγ (53). In addition, ILC3 can also secret IL-17A and GM-CSF (51, 54). GATA3 is required for development of all IL-7Rα-expressing ILCs (55). Although GATA3 expression is high in ILC2, it is also expressed at a lower level in ILC1 and ILC3 and required for their maintenance (5, 56). It has been shown that GATA3 is important for ILC3 function to produce IL-22 (47). ILC3 are involved in clearance of bacterial and fungal infection, control of enteric virus infection, and maintenance of microbiota (57–62), while recent studies suggest that GM-CSF, as well as IL-22, expressed by ILC3 participate in ILC-driven colitis (63–65). After birth, ILC3 are also required for the development of cryptopatches and isolated lymphoid follicles (ILFs) in the gut through expression of lymphotoxin and CCR6 (66–69).
Regulatory ILC
In addition to ILCs discussed above, a new ILC subset, with the ability to suppress ILC1 and ILC3 to promote the resolution of intestinal inflammation, has been identified recently in mice (4). Although further work is needed to confirm the existence and function of this cell type, ILCreg, mainly populate in the gut, develop from CHILP in bone marrow, and require transcription factor Id3 for their development. The regulatory function of ILCreg is mediated by IL-10. TGFβ1 is required for the expansion of ILCreg during inflammation (4). In human, the regulatory ILC (ILCreg) are also reported in the context of cancer recently (70), to suppress the expansion of tumor-associated T cells. Different from the mouse ILCreg that do not express other ILC signature transcription factors, the human ILCreg, present in the tumor tissue, express high levels of Eomes, T-bet, GATA3, RORα, and Ahr, suggesting an overlapping transcriptional profile of the human ILCreg and other ILC subsets.
Ahr Structure and Activation
Aryl hydrocarbon receptor is a ligand-dependent transcription factor and belongs to Per-Arnt-Sim (PAS) superfamily (71, 72). Various Ahr ligands have been identified, including environmental pollutants such as dioxins, and multiple physiologic ligands generated by microbiota, diet, and host metabolism (73–76). Without ligand binding, Ahr localizes in the cytoplasm, and this inactive status is maintained by interacting with 90-kDa heat shock protein (HSP90) (77). Ahr also interacts with Ahr-interacting protein (AIP) which protects Ahr from degradation (78), as well as p23 (79). Upon ligand activation, the conformation of Ahr is changed, leading to the release of Ahr from the protein complex and the translocation of Ahr into the nucleus, where Ahr interacts with Ahr nuclear translocator (ARNT) through PAS-A domain and bHLH domain (80) and acts as a transcription factor targeting dioxin response element (DRE)-containing genes, which are prototypically cytochrome P450 family, like Cyp1a1, but also include genes involved in other important biological processes (13, 81). Several partners of Ahr have been identified, such as RORγt, sterol regulatory element binding transcription factor 1, LXR, NF-κB (13, 82, 83). The involvement of ARNT in these reported interactions remains to be determined.
Aryl hydrocarbon receptor was initially identified as the sensor for 2,3,7,8-tetracholrodibenzo-p-dioxin (TCDD) (84). Later, a variety of Ahr ligands were identified from different physiological sources, such as tryptophan (Trp) metabolism and microbiota. The metabolism of Trp generates Ahr ligands through catalysis by indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO) to kynurenine (Kyn), which acts as an Ahr ligand (76, 85, 86). Independent of IDO/TDO, Trp can also be metabolized by the tryptamine and serotonin pathway, of which the metabolites can act as Ahr agonist (87, 88). Notably, Trp can be photo-oxidized by ultraviolet light or metabolized by other pathways to 6-formylindolo[3,2-b]carbazole (FICZ), which has been proven as a physiologically relevant Ahr agonist (89, 90). Of note, a higher concentration of Kyn, at micromolar concentration, compared to nanomolar of TCDD or FICZ, is required for Ahr activation.
In addition to cellular metabolism, commensal bacteria can catalyze Trp into Ahr ligands as well (74, 91). Lactobacilli expand when the energy source switches from sugar to Trp, and produce indole-3-aldehyde which acts as Ahr ligand to promote IL-22 production by ILC3 (74). Consequently, the Ahr-IL-22 axis provides resistance to the fungus Candida albicans and protection from dextran sulfate sodium (DSS)-induced colitis. In accordance with the importance of Trp in mice, recent research suggests that dysregulation of commensal bacteria that use Trp to generate Ahr ligands may correlate with the pathogenesis of human inflammatory bowel disease (IBD) (92). Besides the Ahr ligands generated by cellular metabolism or commensal bacteria, bacterial pigment factors such as the phenazines from Pseudomonas aeruginosa and the naphthoquinone phthiocol from Mycobacterium tuberculosis can also act as ligands for Ahr, and contribute to the antibacterial response through activation of the Ahr pathway (93).
Ahr Expression in ILCs
Aryl hydrocarbon receptor is thought to be expressed ubiquitously in various organs and cell types, including immune cells, such as Th17 cells, IL-17-producing γδ T cells, Treg cells, CD8αα IEL lymphocytes, B cells, Langerhans cells, monocytes, and splenic dendritic cells (DCs) (94–100). However, the expression of Ahr in ILCs, at both mRNA and protein level, remains to be clarified. Genome-wide transcription analysis of different ILC populations, which is available at IMMGEN.ORG, has shown that Ahr mRNA is detectable among ILCs (101). It has been reported that cytokine stimulation, including IL-2, IL-12, or IL-15, can enhance Ahr expression in splenic NK cells (102, 103). In addition, the transcription factor, Distal-Less Homeobox 3 is found to enhance Ahr transcription in NK cells, while its function remains to be determined (104).
We and other groups have reported the expression of Ahr in ILC3. Differential levels of Ahr were observed in different subsets of ILC3 (13, 37, 41). NCR+ ILC3 express higher Ahr than the other two subsets of ILC3, which lack NCR on the surface (13). How Ahr expression is regulated in ILCs has been a subject of active research. Recent study has shown that in NCR+ ILC3, Wiskott-Aldrich syndrome protein and SCAR homolog (WASH) activates Ahr expression by recruiting AT-Rich Interaction Domain 1A (Arid1a) to the Ahr promoter, and thus maintains NCR+ ILC3 in the gut (105).
Although further investigation on Ahr expression, especially at the protein level, needs to be conducted, the public data at IMMGEN.ORG appears to show that the special microenvironment of the gut correlates with the high Ahr transcriptional expression, since lower Ahr expression is observed in spleen or liver NK cells or ILC1. In a Cyp1a1 (a target gene of Ahr) reporter mouse, Ahr was shown mainly active in the gut in homeostatic conditions (106). A recent paper using a mouse model in which GFP was knocked into the endogenous locus of Ahr showed that among Tregs in various tissues, gut Treg cells express the highest amounts of Ahr, suggesting a tissue adaptation of Ahr expression (107). Identification of the gut specific factors, such as cytokines/metabolites and transcription factors that facilitate Ahr expression will provide insights into the regulation of Ahr expression in ILCs, and potentially be translated into clinical manipulation of the Ahr pathway. To get a molecular understanding on the regulation of Ahr expression, it is of importance to analyze chromatin status of the Ahr locus and Ahr interactions with key transcription factors in different ILC populations.
Involvement of Ahr in ILC Function and Regulation
Ahr and NK Cells/ILC1
In tumor, Ahr promotes NK cell cytotoxicity and its production of IFNγ (103). During T. gondii infection, Ahr is also required for maximal IL-10 production by NK cells (102). It has also been shown that Ahr maintains liver-resident CD49a+ cells by regulating cytokine-induced cell death (108). Notably, CD49a is considered as a marker for ILC1 in the liver, instead of NK cells (18). Therefore, these data may suggest that Ahr is required for liver ILC1 maintenance (108).
So far, the studies on Ahr in NK cells or ILC1 have been predominantly focused in the liver or spleen. The function of Ahr in the gut ILC1 and NK cells still remains to be elucidated, given that the gastrointestinal tract is another site for these two cell populations, especially for ILC1 (5).
Ahr and ILC2
Currently, limited knowledge is available on the function of Ahr in ILC2. IFNγ has recently been shown to inhibit ILC2 activation (109, 110). In addition, IFNγ can induce Ido1 mRNA and Ido protein expression in some cell types (111, 112). Given that Ido1 is able to catalyze Trp to Kyn, which acts as a ligand for Ahr, it is tempting to speculate that Ahr ligands, such as Kyn, might suppress ILC2 function but additional works are needed to test this hypothesis. TNF-like ligand 1A (TL1A) has been shown to promote expansion and function of ILC2 in the gut (113). RNA-seq data reveal that TL1A enhances Ahr expression in the presence of IL-33 and IL-25 in human ILC2 (114). Thus, the function of Ahr in ILC2 and in in vivo models of ILC2-driven pathology remains to be investigated.
Ahr and ILC3
Aryl hydrocarbon receptor has been relatively well studied in gut ILC3. Although it is dispensable for fetal LTi development, Ahr is essential for the maintenance and IL-22 production of ILC3 (12, 13, 45). Although the precise mechanisms by which Ahr regulates the homeostasis of ILC3 still remain to be determined, it has been described that Ahr can regulate survival and/or proliferation of ILC3 (Figure 1). First, it is reported that Ahr is important for the survival of ILC3 by promoting the expression of anti-apoptotic proteins, such as Bcl-2. Ahr upregulates IL-7 receptor (IL-7R) in ILC3, in line with the role of IL-7/IL-7R signaling pathway in the supporting the survival of ILC3 (13). Second, it has been shown that Ahr-deficient ILC3 have reduced Ki67 expression, indicating that decreased proliferation may lead to the defective expansion of ILC3. Furthermore, Ahr can regulate the expression of Kit through binding to DRE at the promoter of Kit locus, suggesting direct regulation of Kit expression by Ahr at the transcriptional level (12). Finally, Ahr supports the development of ILC3 presumably through promoting the transcription of Notch 1 and Notch 2, although defects in Notch signaling have more effect on NCR-expressing ILC3 than NCR-negative ILC3 (45). By regulating the maintenance and function of ILC3, Ahr is critical for the clearance of Citrobacter rodentium, a murine pathogen that models human enterohemorrhagic Escherichia coli and enteropathogenic E. coli infections in the gut (12, 13, 64), as well as for the pathology of anti-CD40-incuced colitis (64).
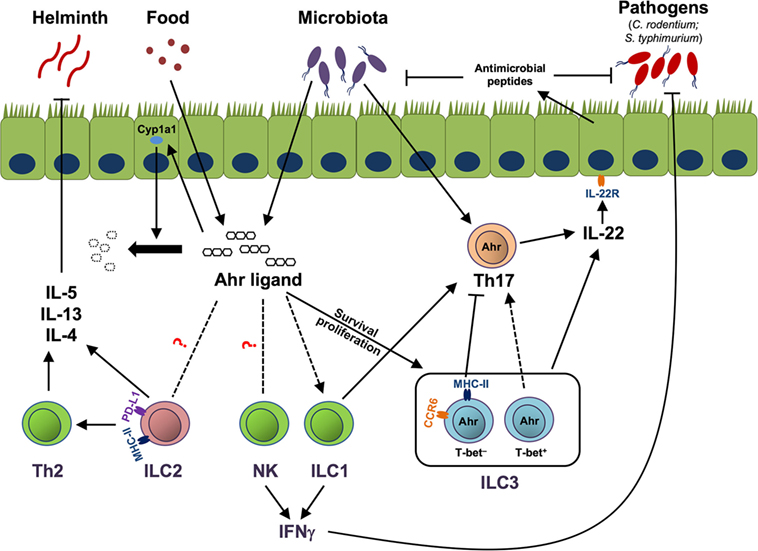
Figure 1. Aryl hydrocarbon receptor (Ahr)-mediated cross talk between innate lymphoid cells (ILCs) and immune/non-immune cells. Ahr ligands derived from the diet or microbiota activate Ahr to promote group 3 ILCs (ILC3) homeostasis by enhancing the survival or proliferation of ILC3. MHC-II+ ILC3, which are mainly CCR6+, suppress pathogenic Th17 response to commensal bacteria, while T-bet+ ILC3, together with group 1 ILC (ILC1), promote Th17 cells. Both Th17 cells and ILC3 can produce IL-22 to control commensal/pathogenic bacteria through facilitating the production of antimicrobial peptides by epithelial cells. Ahr ligand could potentially regulate natural killer (NK) cells, ILC1, and group 2 ILCs (ILC2) through the Ahr pathway in the gut. NK cells and ILC1 can help the host to clear pathogens, like Salmonella typhimurium, by production of effector cytokine IFNγ. ILC2, through expression of MHC class II (MHC-II) and programmed death ligand 1 (PD-L1), enhance Th2 cells. ILC2 and Th2 cells protect the host from helminth infection by secreting type 2 cytokines, including IL-5, IL-13, and IL-4. Ahr ligand enhances Cyp1a1 expression in gut epithelial cells, and as a feedback negative control loop, Cyp1a1 degrades Ahr ligand to prevent overt Ahr-mediated immune responses. Solid lines and arrows depict known regulation. Dotted lines and arrows depict to-be-determined regulation in the gut.
Ahr in ILC Plasticity
The plasticity of ILCs has been observed in both human and mouse systems under steady state or certain disease models, while the mechanism that drives the plasticity of ILCs is still not well understood. The conversion of ILC3 to ILC1 is characterized by the loss of RORγt and gain of T-bet expression to become exILC3 (115, 116). These exILC3 stop the production of IL-22, and begin to secrete IFNγ. IL-15 and IL-12 can lead to downregulation of RORγt, and enhance IFNγ expression (116). In support, IL-12 has been shown to participate in the transition of ILC3 to ILC1 in humans (115, 117). There is an increase of ILC1 and decrease in ILC3 in the intestines of patients with Crohn’s disease, suggesting the ILC3-derived ILC1 might contribute to the pathology of human IBD (115, 117). Human ILC1 can also convert to ILC3 in the presence of IL-2, IL-23, and IL-1β, and retinoic acid can accelerate this process which may depend on the receptors for retinoic acid (117). Although Ahr has been shown to prevent the differentiation of human ILC3 to NK cells (118), it is of interest to determine whether Ahr participates in the transition of ILC3 to ILC1. Of note, microbiota has been shown to maintain the RORγt expression by ILC3 through IL-7 signaling in the gut (116). Since commensal bacteria have the ability to produce Ahr ligands, Ahr might receive the signals from microbiota to maintain ILC3 through upregulating IL-7R.
The plasticity of ILC2 has recently been reported (31, 119–121). Gfi1, a key transcription factor for ILC2 development and function, appears to sustain ILC2, as deletion of Gfi1 in ILC2 leads to upregulation of RORγt and IL-17 production by these ILC2 (29). Similarly, Bcl11b, a recently defined transcription factor for ILC2, maintains the stability of ILC2 by suppressing RORγt and Ahr expression (31). ILC2 were found to convert to IFNγ-producing ILC1 in the lung by IL-12 and IL-18 (119–121). The conversion is dependent on T-bet expression, and enhanced by IL-1β through induction of the IL-12 receptor alpha (Il12rα). As the frequency of ILC1 shows positive correlation with disease severity in patients with chronic obstructive pulmonary disease or chronic rhinosinusitis with nasal polyps (119, 121), the plasticity of ILC2 could be a therapeutic target for these respiratory diseases. Whether Ahr plays any role in ILC2 conversion to ILC1 needs to be established.
Ahr-Mediated Modulation of the Cross Talk Between ILCs and Other Cells
Cross Talk with Innate Immune Cells
Innate immune cells, such as dendritic cells (DCs) and mononuclear phagocytes (MNPs), have been shown to interact with ILCs. Both CX3CR1+ MNPs and CD103+ DCs can induce IL-22 production by ILC3, while CX3CR1+ MNPs can also recruit ILC3 to the gut through CXCL16–CXCR6 pathway (122–124). Ahr controls the differentiation and function of DCs by arresting the differentiation of progenitors, as well as regulating antigen presentation in DCs (125–128). In addition, recent work has shown that Ahr controls differentiation of monocyte to monocyte-derived macrophages in the human system (129). Thus, it is possible that Ahr controls ILC3 through regulating these innate immune cells. On the other hand, innate immune cells can be attracted by ILC-secreted cytokines. IL-5 and IL-13, produced by ILC2, can recruit eosinophils which are essential for the clearance of helminth infections (25). ILC3 can secrete IL-17A, which is proved to attract neutrophils into the intestine (130, 131). Thus, lack of ILC3 in Ahr-deficient mice may account for the resistance of anti-CD40 colitis (64).
Cross Talk with Adaptive Immune Cells
The absence of Ahr in ILC3 leads to defects in the IL-22-producing ability of ILC3. The impaired IL-22 production in the gut of Ahr-deficient mice causes a decrease in antimicrobial peptide production by gut epithelial cells (62, 132), leading to increased segmented filamentous bacteria (SFB) which has been established to induce Th17 cells in the gut (133, 134). However, recent papers also show that SFB induced IL-22 production by ILC3 can induce epithelial production of Serum Amyloid A, which in turn promotes Th17 cells (135, 136). Thus, the role of ILC3-derived IL-22 in regulating Th17 cells will require further investigation into the underlying molecular mechanisms, which are most likely indirect given the lack of expression IL-22R by immune cells. By supporting ILC3 homeostasis, Ahr controls cryptopatches formation, and consequently the genesis of ILFs in the gut (12, 137). As ILFs have been recognized as a site for the production of intestinal IgA responses (138), it is possible that Ahr contributes to B cell responses via the regulation of ILC3, in addition to its B cell-intrinsic roles (96, 97, 139).
Recent research showed that ILC2 are critical for memory Th2 cell responses, as impaired Th2 cells are found in sensitized mice, which lack ILC2 (140). During helminth infection, ILC2 have been shown to express the checkpoint molecule Programmed Death Ligand 1, through which ILC2 support Th2 polarization, and effective Th2 dependent-anti-helminth response (141). Additionally, ILC2, through producing IL-9, can sustain the proliferation of ILC2 and activation of Treg cells in arthritis, by which promote the resolution of inflammation (142). It is of interest to note that, with the resistance to IL-7-induced downregulation of IL-7R, ILCs limit the availability of IL-7 for T cells, thus controlling the homeostasis of T cells (143). Given that Ahr deficiency leads to reduction of ILC3, it remains to be determined whether enhanced T cell proliferation and Th17 cell differentiation observed in Ahr knockout mice are caused by increased IL-7 that is made available to T cells.
In addition to the cross talk between ILCs and adaptive immune cells through cytokines, ILCs interact with T cells through the expression of MHC class II (MHC-II) molecules on the surface. The MHC-II-mediated interaction between ILCs and T cells controls the activation or anergy of T cells (Figure 1). For example, ILC2, via MHC-II and co-stimulatory molecules, CD80 and CD86, interact with and activate T cells (144). Different from ILC2, ILC3 expressing MHC-II but not the co-stimulatory molecules CD80 and CD86, induce T cell apoptosis and tolerance in the gut (145, 146). However, ILC3 express CD30 ligand and OX40 ligand, which may contribute to the maintenance of CD4+ T cell memory (147). Although there is no direct evidence indicating whether Ahr regulates MHC-II or co-stimulatory molecule expression by ILC2 and ILC3, Ahr may mediate the cross talk between ILCs and T cells, at least through regulating ILC numbers (Figure 1). A recent study reveals that NCR-expressing ILCs, including ILC1 and NCR+ ILC3, support Th17 cells in inflamed central nervous system (148), which raises intriguing questions that whether similar event is evident in the gut, and how the host keeps the balance between the induction of Th17 cells by NCR+ ILCs, and the inhibition of Th17 cells by CCR6+ ILC3 through MHC-II expression (Figure 1).
Cross Talk with Epithelial Cells
The cross talk between gut epithelial cells and ILC3 has been recently investigated. Over-expression of Cyp1a1, a target gene of Ahr, in epithelial cells consumes Ahr ligands in the gut, which consequently leads to the decrease of gut ILC3 (106) (Figure 1). These findings raise the possibility that activation of Ahr may not only promote gut ILC3 in a cell-intrinsic manner, but also maintain the ILC3 at a physiological level through controlling the availability of Ahr ligands in the gut. On the other hand, ILC3, via expression of IL-22 and lymphotoxin, regulate the fucosylation of epithelial cells which is critical for the host to control Salmonella typhimurium infection (149). In addition, ILC3, via producing IL-22, promote the expansion of intestinal stem cell, and consequently promote the regeneration of intestinal epithelium after gut injury (150, 151).
Cross Talk with Commensals
Aryl hydrocarbon receptor appears to mediate the interaction of ILC3 and microbiota. The absence of caspase recruitment domain family member 9 (CARD9) results in alteration of microbiota, and the altered microbiota fail to metabolize Trp into Ahr ligands, leading to decreased ILC3 and IL-22 production, and increased susceptibility of the host to colitis (92). Accordingly, Ahr ligands are found decreased in the microbiota of IBD patients, especially in the individuals with IBD-associated single-nucleotide polymorphism within CARD9 (rs10781499), suggesting microbiota–Ahr ligand axis may be a therapeutic target of colitis in humans (92). Although the cross talk between ILCs and microbiota remains to be further explored, genome-wide analysis at the transcriptional level of ILCs has been conducted using RNA-seq by comparing specific pathogen-free mice to those with microbiota depletion (152). Marked numbers of transcripts change significantly in all ILCs upon antibiotics treatment, but the expression profile is generally maintained. Intriguingly, depletion of microbiota shows more effects on the gene expression of ILC1 and ILC2 than that of ILC3. Given the important role of Ahr in ILC3 and Ahr could sense ligands generated by commensals, for example, Lactobacillus reuteri (74, 91), these findings may suggest ligands from other sources (e.g., diet) could activate the Ahr pathway in the absence of microbiota.
Regulation of ILCs by ILCreg
With the minimal Ahr expression in mouse ILCreg at least under the steady state (4), it remains to be determined whether Ahr plays a role in ILCreg. In contrast to the mouse ILCreg, human ILCreg in cancer that suppress T cell expansion appear to express high level of Ahr, indicating potential role of Ahr in this population (70). The mouse ILCreg have been shown to regulate ILC1 and ILC3 (4), it is unclear whether ILCreg can suppress ILC2.
ILC-Nervous System Interaction
The nervous system has been shown to affect ILCs. Glial cells in the gut, through secreting neurotrophic factors that bind to the neuroregulatory receptor rearranged during transfection (RET) on ILC3, promote the expression of IL-22, and consequently decrease the susceptibility to intestinal inflammation and infection (153). Recent studies demonstrate that among various hematopoietic cells, ILC2 uniquely express the neuropeptide neuromedin U (NMU) receptor 1 (NMUR1), which makes them respond to NMU (154–156). The activation of ILC2 by NMU leads to enhanced cell expansion and type 2 cytokine production, which promote the clearance of helminth in the gut. It remains to be determined that whether Ahr modulates ILC responses to neuromediators.
Cooperation of Ahr and Partners in Regulating ILCs
Aryl hydrocarbon receptor has been studied for decades, and some interacting proteins, like HSP90 and AIP, have been well documented. However, only a few partners of Ahr have been functionally implicated in ILCs. In Th17 and IL-17-producing γδ T cells, Ahr regulates IL-22 expression while the molecular mechanism of action of Ahr is unclear (94, 95). However, Ahr has been shown to interact with RORγt in an overexpression system to promote IL-22 expression (13). RORγt is required for the recruitment of Ahr to the Il22 locus, as Ahr alone fails to bind to the Il22 locus. In contrast to the Il22 locus, Ahr is recruited to the Cyp1a1 locus independent of RORγt. These data raise a question regarding how Ahr, by cooperating with other transcription factors (e.g., RORγt), regulates gene expression in ILC3 and other lymphocytes (e.g., Th17 and γδ T cells). In addition to RORγt, the C2H2 zinc finger transcription factor Ikaros, a key regulator of hematopoiesis, is a binding protein of Ahr in ILC3 (157). Ikaros negatively regulates ILC3 through zinc finger 4-dependent inhibition of transcriptional activity of the Ahr by disruption of the Ahr–ARNT complex. It will be of interest to investigate whether Ikaros participates in a complex of Ahr and RORγt to regulate RORγt activity in ILC3 development and/or function. Intriguingly, Ikaros but not Ahr is required for fetal LTi cell development, demonstrating the distinct transcriptional regulation of fetal and postnatal ILC3.
As ILC3 resemble Th17 cells in regards to key transcription factor and cytokines, knowledge of the function of Ahr in Th17 cells might be adopted into ILC3 potentially. Transcription factor Musculoaponeurotic Fibrosarcoma (MAF) has been shown to be induced by TGFβ in Th17 cells to promote IL-17 production and suppress IL-22 secretion (158). Although the interaction between Ahr and MAF has been only implicated in type 1 regulatory T cells (159), the cross talk of these two proteins may provide insight into the molecular regulation of IL-22 expression in ILC3.
Aryl hydrocarbon receptor has been shown to interact with RelB, a key component of NF-κB signaling, and synergize to induce the transcription of certain genes, such as IL-6 and IL-8 in DC or macrophage (160, 161). Additionally, another component of NF-κB, RelA, binds to Ahr, and the interaction consequently promotes IL-6 transcription (162). Therefore, the interplay between Ahr and NF-κB pathway might be important for ILCs since the critical function of NF-κB has been investigated throughout various cell types.
Not limited to transcriptional function, Ahr has been reported to participate in posttranslational regulation in non-immune cells. It is described that Ahr acts as a component of cullin 4B ubiquitin ligase complex, which targets sex steroid receptors for degradation (163, 164). More investigation directed to confirm and extend this non-genomic function of Ahr in ILC and other cell types will be necessary to understand how Ahr is linked to protein degradation in different contexts.
In non-immune cells, Ahr exhibits a rhythmic expression, and its sensitivity to Ahr ligands is time-dependent (165). Reciprocally, genes associated with circadian clock and the behavioral responses of mice to circadian clock are regulated by Ahr (165). Ahr has been shown to interact with Bmal1, which forms a complex with Clock to facilitate the transcription of circadian genes (166–168). ILC2 activation and consequent eosinophil recruitment is responsive to the circadian clock, suggesting a conserved circadian mechanism in ILCs (25). Understanding of the synergetic function of Ahr and circadian signaling could improve our understanding of the basic biology of ILCs, and provide new targets of interest for regulation of ILCs.
Translational Potential of Ahr in ILCs
Changes in ILCs have been reported in the patients with IBD. IL-22-producing ILC3 decreased in the intestine of Crohn’s patients (115, 169, 170), in line with the protective role of IL-22 on the integrity of gut barrier which has been implicated several mouse models (171). Other studies also reveal that IL-22 produced by ILC3 increased in inflammatory sites of the colons in both CD and UC patients (122, 172), which might be due to a compensatory response of the host to inflammation but also might reveal the pathological aspects of ILC3, especially NCR+ ILC3 (63, 64). The MHC-II expression on ILC3 is critical to induce T cell tolerance to gut commensal bacteria and avoid overt inflammation. It has been shown that pediatric IBD patients have reduced MHC-II expression on colonic ILC3, consistent with the model that compromised ILC3 regulatory function can lead to T cell-mediated inflammation (146). It has been shown that the expression of Ahr is reduced in the gut tissues from IBD patients compared to healthy controls (173). Accordingly, treatment of Ahr ligand ameliorated the pathology of several mouse colitis models, including 2,4,6-trinitrobenzenesulfonic acid (TNBS)-, DSS-, and T cell transfer-induced colitis, in which IL-22 is required (173, 174). Considering the role of Ahr in the maintenance of gut ILC3 and IL-22 production by ILC3, Ahr pathway could be potentially manipulated to regulate gut inflammation by increasing ILC3 in the gut of IBD patients. However, given the different functions between NCR+ ILC3 and NCR− ILC3, special considerations are needed while targeting the Ahr pathway in IBD.
Type 2 immunity has been considered to mediate ulcerative colitis in human, which has been modeled by oxazolone-induced colitis in mice (175). A known Ahr ligand, 3,3’-Diindolylmethane, has been found to alleviate oxazolone-induced colitis, probably through inhibition of Th2/Th17 cells and induction of Treg cells (176). Since ILC2 express large amounts of type 2 cytokines, this population could potentially play a pathogenic role in ulcerative colitis (177). Despite the reduced expression of Ahr in IBD, the role of Ahr in ILC2 and disease pathogenesis remains to be determined. In addition, it will be of interest to investigate the balance between ILC2 (or type 2 immunity) and ILC3 in colitis. IL-33, a cytokine that acts on ILC2 and Th2 to promote the cytokine production, increased in IBD patients and in experimental colitis models of mice, including TNBS and DSS model (178). Ablation of IL-33-ST2 pathway relieves experimental colitis in mice. Of note, IL-33 and soluble ST2 have been shown increased in the colons of IBD patients (179), in line with the proinflammatory role of type 2 immunity. Thus, the functions of ILC2 and ILC3 in colitis could be dissected into two phases as ILC2 initiate the pathology via IL-13 (177), while ILC3, probably through IL-22, facilitate the tissue repair in the later phase of disease. However, IL-22 could also participate in the gut inflammation, highlighting its “double-edged sword” nature (65, 180). A recent study reveals that IL-33 stimulates ILC2 to secrete amphiregulin to promote tissue repair in experimental colitis (42), suggesting ILC2 at different stage of the disease and/or some subset of ILC2 (i.e., amphiregulin+ ILC2) may have protective function in the resolution of colitis as well.
Allergic asthma is a chronic inflammatory disease, in which type 2 cytokines, IL-4, IL-5, and IL-13 are associated with the pathology (181). These type 2 cytokines are required for IgE response, recruitment of eosinophils, and mucus production. ILC2 have been implicated in asthma, since ILC2 produce large amounts of IL-5 and IL-13, as well as IL-4 under certain context, in response to IL-33, IL-25, and TSLP (182). Additionally, recent study showed that ILC2 increase in the airways of severe asthma patients, suggesting ILC2 may contribute to airway inflammation in mouse and human (183). Although the function of Ahr in ILC2 remains to be determined, several Ahr ligands have been reported to suppress allergic airway inflammation in different mouse models, through suppressing type 2 cytokines, IL-4 and IL-5, production, eosinophilia, and specific IgE expression (184–186). Thus, study of the role of Ahr in ILC2 would provide another potential target for clinical intervention in airway inflammation, like asthma. Although type 2 cytokines have been well documented in asthma, elevated IL-17 has been noticed clinically (187). Given that IL-25 can induce a population of lung ILC2 with IL-17-producing ability, the potential role of this special ILC2 subset in the pathology of asthma in humans needs to be studied in the future.
Both pro- and antitumor action of Ahr has been implicated (188), and the potential function of Ahr in ILC-mediated tumor immunology remains largely unknown. Ahr has been demonstrated to promote the antitumor activity of NK cells (103). IL-22, mainly produced by ILC3 under the steady state, has been shown to associate with increased risk in colon cancer (189). Accordingly, IL-22-producing ILC3 are found to promote an experimental cancer model in mice (190). Therefore, understanding of the precise function of Ahr in ILCs in cancer needs to be carefully studied.
Concluding Remarks
The tissue microenvironment may be involved in regulating the differentiation, homeostasis, and function of ILCs. Thus, the expression and activity of Ahr in ILCs from different organs under the steady state need to be carefully considered when designing therapeutics to target Ahr. Furthermore, it will be of great interest to investigate whether the Ahr level/activity in ILCs can be changed under different contexts, like in infection, inflammation, and/or cancers.
Cell-intrinsic role of Ahr in ILCs has to be determined given the broad expression of Ahr in other cell types. The molecular mechanism by which Ahr regulates the development or homeostasis of ILCs remains to be explored. Mechanistic insights of Ahr expression and/or activity in various ILC subsets or any given ILCs in different tissues are important for designing targeted strategy to modulate the Ahr function pharmacologically. It is of interest to investigate whether various ILCs have different sensitivity to Ahr ligand, or unique machinery to uptake Ahr ligand. Furthermore, single cell-omics studies involving RNA-seq and ATAC-seq analyses, together with ChIP-seq analysis of Ahr, will delineate the functional pattern and role of Ahr in regulating transcriptional landscape of ILCs. Identification of Ahr-binding partners in ILCs will provide insights into the mechanism by which Ahr cooperates with other factors to differentially regulate gene expression. These molecular findings could uncover more specific and effective therapeutic targets on the Ahr pathway, in cell-type/tissue-specific manner, in disease treatment and prevention.
Author Contributions
SL wrote the manuscript with JB’s contribution. LZ supervised the research and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MC and handling editor declared their shared affiliation.
Acknowledgments
The authors thank the entire LZ laboratory their help and suggestions. The work was supported by the National Institutes of Health (AI132391 and DK105562 to LZ), and by a Cancer Research Institute Investigator Award (LZ). LZ is a Pew Scholar in Biomedical Sciences, supported by the Pew Charitable Trusts, and an Investigator in the Pathogenesis of Infectious Disease, supported by Burroughs Wellcome Fund.
References
1. Ebbo M, Crinier A, Vely F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol (2017) 17(11):665–78. doi:10.1038/nri.2017.86
2. Klose CSN, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17(7):765–74. doi:10.1038/ni.3489
3. Fang DF, Zhu JF. Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J Exp Med (2017) 214(7):1861–76. doi:10.1084/jem.20170494
4. Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell (2017) 171(1):201–16.e18. doi:10.1016/j.cell.2017.07.027
5. Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157(2):340–56. doi:10.1016/j.cell.2014.03.030
6. Mattner J, Wirtz S. Friend or foe? The ambiguous role of innate lymphoid cells in cancer development. Trends Immunol (2017) 38(1):29–38. doi:10.1016/j.it.2016.10.004
7. Bostick JW, Zhou L. Innate lymphoid cells in intestinal immunity and inflammation. Cell Mol Life Sci (2016) 73(2):237–52. doi:10.1007/s00018-015-2055-3
8. Shih HY, Sciume G, Mikami Y, Guo LY, Sun HW, Brooks SR, et al. Developmental acquisition of regulomes underlies innate lymphoid cell functionality. Cell (2016) 165(5):1120–33. doi:10.1016/j.cell.2016.04.029
9. Zhou L. AHR function in lymphocytes: emerging concepts. Trends Immunol (2016) 37(1):17–31. doi:10.1016/j.it.2015.11.007
10. Cella M, Colonna M. Aryl hydrocarbon receptor: linking environment to immunity. Semin Immunol (2015) 27(5):310–4. doi:10.1016/j.smim.2015.10.002
11. Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol (2014) 32:403–32. doi:10.1146/annurev-immunol-032713-120245
12. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (2011) 334(6062):1561–5. doi:10.1126/science.1214914
13. Qiu J, Heller JJ, Guo XH, Chen ZME, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity (2012) 36(1):92–104. doi:10.1016/j.immuni.2011.11.011
14. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol (2012) 13(2):144–51. doi:10.1038/ni.2187
15. Eberl G, Di Santo JP, Vivier E. The brave new world of innate lymphoid cells. Nat Immunol (2015) 16(1):1–5. doi:10.1038/ni.3059
16. Yu X, Wang Y, Deng M, Li Y, Ruhn KA, Zhang CC, et al. The basic leucine zipper transcription factor NFIL3 directs the development of a common innate lymphoid cell precursor. Elife (2014) 3:e04406. doi:10.7554/eLife.04406
17. Yu JH, Freud AG, Caligiuri MA. Location and cellular stages of natural killer cell development. Trends Immunol (2013) 34(12):573–82. doi:10.1016/j.it.2013.07.005
18. Fuchs A. ILC1s in tissue inflammation and infection. Front Immunol (2016) 7:104. doi:10.3389/fimmu.2016.00104
19. Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity (2004) 20(4):477–94. doi:10.1016/S1074-7613(04)00076-7
20. van Helden MJ, Goossens S, Daussy C, Mathieu AL, Faure F, Marcais A, et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J Exp Med (2015) 212(12):2015–25. doi:10.1084/jem.20150809
21. Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, et al. Cancer immunosurveillance by tissue-resident innate lymphoid cells and innate-like T cells. Cell (2016) 164(3):365–77. doi:10.1016/j.cell.2016.01.002
22. Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol (2001) 1(1):41–9. doi:10.1038/35095564
23. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol (2016) 17(7):758–64. doi:10.1038/ni.3482
24. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity (2013) 38(4):769–81. doi:10.1016/j.immuni.2013.02.010
25. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature (2013) 502(7470):245. doi:10.1038/nature12526
26. Halim TYF, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity (2012) 37(3):463–74. doi:10.1016/j.immuni.2012.06.012
27. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464(7293):1367–70. doi:10.1038/nature08900
28. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity (2012) 37(4):634–48. doi:10.1016/j.immuni.2012.06.020
29. Spooner CJ, Lesch J, Yan D, Khan AA, Abbas A, Ramirez-Carrozzi V, et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol (2013) 14(12):1229–36. doi:10.1038/ni.2743
30. Yang Q, Monticelli LA, Saenz SA, Chi AWS, Sonnenberg GF, Tang JB, et al. T cell factor 1 is required for group 2 innate lymphoid cell generation. Immunity (2013) 38(4):694–704. doi:10.1016/j.immuni.2012.12.003
31. Califano D, Cho JJ, Uddin MN, Lorentsen KJ, Yang Q, Bhandoola A, et al. Transcription factor Bcl11b controls identity and function of mature type 2 innate lymphoid cells. Immunity (2015) 43(2):354–68. doi:10.1016/j.immuni.2015.07.005
32. Yu Y, Wang C, Clare S, Wang JX, Lee SC, Brandt C, et al. The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J Exp Med (2015) 212(6):865–74. doi:10.1084/jem.20142318
33. Walker JA, Oliphant CJ, Englezakis A, Yu Y, Clare S, Rodewald HR, et al. Bcl11b is essential for group 2 innate lymphoid cell development. J Exp Med (2015) 212(6):875–82. doi:10.1084/jem.20142224
34. Zhang KN, Xu XY, Pasha MA, Siebel CW, Costello A, Haczku A, et al. Cutting edge: notch signaling promotes the plasticity of group-2 innate lymphoid cells. J Immunol (2017) 198(5):1798–803. doi:10.4049/jimmunol.1601421
35. Salimi M, Barlow JL, Saunders SP, Xue LZ, Gutowska-Owsiak D, Wang XW, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med (2013) 210(13):2939–50. doi:10.1084/jem.20130351
36. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature (2016) 529(7585):221–5. doi:10.1038/nature16161
37. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12(11):1045–54. doi:10.1031/ni.2131
38. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol (2011) 12(11):1055–62. doi:10.1038/ni.2104
39. Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med (2013) 5(170):170ra16. doi:10.1126/scitranslmed.3005374
40. Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med (2013) 210(13):2951–65. doi:10.1084/jem.20130071
41. Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang HE, Locksley RM. Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol (2016) 9(1):275–86. doi:10.1038/mi.2015.59
42. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A (2015) 112(34):10762–7. doi:10.1073/pnas.1509070112
43. Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell (2015) 160(1–2):74–87. doi:10.1016/j.cell.2014.12.011
44. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature (2015) 519(7542):242–6. doi:10.1038/nature14115
45. Hazenberg MD, Spits H. Human innate lymphoid cells. Blood (2014) 124(5):700–9. doi:10.1182/blood-2013-11-427781
46. Gasteiger G, Fan XY, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350(6263):981–5. doi:10.1126/science.aac9593
47. Zhong C, Cui KR, Wilhelm C, Hu GQ, Mao KR, Belkaid Y, et al. Group 3 innate lymphoid cells continuously require the transcription factor GATA-3 after commitment. Nat Immunol (2016) 17(2):169–78. doi:10.1038/ni.3318
48. Kim MY, Kim KS, McConnell F, Lane P. Lymphoid tissue inducer cells: architects of CD4 immune responses in mice and men. Clin Exp Immunol (2009) 157(1):20–6. doi:10.1111/j.1365-2249.2009.03932.x
49. Cherrier M, Sawa S, Eberl G. Notch, Id2, and RORgammat sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J Exp Med (2012) 209(4):729–40. doi:10.1084/jem.20111594
50. Ivanov II, Diehl GE, Littman DR. Lymphoid tissue inducer cells in intestinal immunity. Curr Top Microbiol Immunol (2006) 308:59–82. doi:10.1007/3-540-30657-9_3
51. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med (2009) 206(1):35–41. doi:10.1084/jem.20072713
52. Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi YW, Littman DR. An essential function for the nuclear receptor ROR gamma t in the generation of fetal lymphoid tissue inducer cells. Nat Immunol (2004) 5(1):64–73. doi:10.1038/ni1022
53. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature (2013) 494(7436):261–5. doi:10.1038/nature11813
54. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science (2014) 343(6178):1477. doi:10.1126/Science.1249288
55. Yagi RJ, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, et al. The transcription factor GATA3 is critical for the development of all IL-7R alpha-expressing innate lymphoid cells. Immunity (2014) 40(3):378–88. doi:10.1016/j.immuni.2014.01.012
56. Serafini N, Klein Wolterink RG, Satoh-Takayama N, Xu W, Vosshenrich CA, Hendriks RW, et al. Gata3 drives development of RORgammat+ group 3 innate lymphoid cells. J Exp Med (2014) 211(2):199–208. doi:10.1084/jem.20131038
57. Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity (2009) 31(1):15–23. doi:10.1016/j.immuni.2009.06.008
58. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity (2011) 34(1):122–34. doi:10.1016/j.immuni.2010.12.009
59. Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol (2013) 190(2):521–5. doi:10.4049/jimmunol.1202924
60. Hernandez PP, Mahlakoiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat Immunol (2015) 16(7):698–707. doi:10.1038/ni.3180
61. Powell N, Walker AW, Stolarczyk E, Canavan JB, Gokmen MR, Marks E, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity (2012) 37(4):674–84. doi:10.1016/j.immuni.2012.09.008
62. Qiu J, Guo XH, Chen ZME, He L, Sonnenberg GF, Artis D, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity (2013) 39(2):386–99. doi:10.1016/j.immuni.2013.08.002
63. Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife (2016) 5:e10066. doi:10.7554/eLife.10066
64. Song C, Lee JS, Gilfillan S, Robinette ML, Newberry RD, Stappenbeck TS, et al. Unique and redundant functions of NKp46(+) ILC3s in models of intestinal inflammation. J Exp Med (2015) 212(11):1869–82. doi:10.1084/jem.20151403
65. Eken A, Singh AK, Treuting PM, Oukka M. IL-23R(+) innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol (2014) 7(1):143–54. doi:10.1038/mi.2013.33
66. Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature (2008) 456(7221):507–10. doi:10.1038/nature07450
67. Kanamori Y, Ishimaru K, Nanno M, Maki K, Ikuta K, Nariuchi H, et al. Identification of novel lymphoid tissues in murine intestinal mucosa where clusters of c-kit(+) IL-7R(+) Thy1(+) lympho-hemopoietic progenitors develop. J Exp Med (1996) 184(4):1449–59. doi:10.1084/jem.184.4.1449
68. Taylor RT, Lugering A, Newell KA, Williams IR. Intestinal cryptopatch formation in mice requires lymphotoxin alpha and the lymphotoxin beta receptor. J Immunol (2004) 173(12):7183–9. doi:10.4049/jimmunol.173.12.7183
69. Lugering A, Ross M, Sieker M, Heidemann J, Williams IR, Domschke W, et al. CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clin Exp Immunol (2010) 160(3):440–9. doi:10.1111/j.1365-2249.2010.04103.x
70. Crome SQ, Nguyen LT, Lopez-Verges S, Yang SY, Martin B, Yam JY, et al. A distinct innate lymphoid cell population regulates tumor-associated T cells. Nat Med (2017) 23(3):368–75. doi:10.1038/nm.4278
71. Ema M, Sogawa K, Watanabe N, Chujoh Y, Matsushita N, Gotoh O, et al. cDNA cloning and structure of mouse putative Ah receptor. Biochem Biophys Res Commun (1992) 184(1):246–53. doi:10.1016/0006-291X(92)91185-S
72. Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci U S A (1992) 89(17):8185–9. doi:10.1073/pnas.89.17.8185
73. Marshall NB, Kerkvliet NI. Dioxin and immune regulation emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann N Y Acad Sci (2010) 1183:25–37. doi:10.1111/j.1749-6632.2009.05125.x
74. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39(2):372–85. doi:10.1016/j.immuni.2013.08.003
75. Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci U S A (1991) 88(21):9543–7. doi:10.1073/pnas.88.21.9543
76. Mezrich JD, Fechner JH, Zhang XJ, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (2010) 185(6):3190–8. doi:10.4049/jimmunol.0903670
77. Pongratz I, Mason GG, Poellinger L. Dual roles of the 90-kDa heat shock protein hsp90 in modulating functional activities of the dioxin receptor. Evidence that the dioxin receptor functionally belongs to a subclass of nuclear receptors which require hsp90 both for ligand binding activity and repression of intrinsic DNA binding activity. J Biol Chem (1992) 267(19):13728–34.
78. Lees MJ, Peet DJ, Whitelaw ML. Defining the role for XAP2 in stabilization of the dioxin receptor. J Biol Chem (2003) 278(38):35878–88. doi:10.1074/jbc.M302430200
79. Nair SC, Toran EJ, Rimerman RA, Hjermstad S, Smithgall TE, Smith DF. A pathway of multi-chaperone interactions common to diverse regulatory proteins: estrogen receptor, Fes tyrosine kinase, heat shock transcription factor HSF1, and the aryl hydrocarbon receptor. Cell Stress Chaperones (1996) 1(4):237–50. doi:10.1379/1466-1268(1996)001<0237:APOMCI>2.3.CO;2
80. Fukunaga BN, Probst MR, Reisz-Porszasz S, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor. J Biol Chem (1995) 270(49):29270–8. doi:10.1074/jbc.270.49.29270
81. Furman DP, Oshchepkova EA, Oshchepkov DY, Shamanina MY, Mordvinov VA. Promoters of the genes encoding the transcription factors regulating the cytokine gene expression in macrophages contain putative binding sites for aryl hydrocarbon receptor. Comput Biol Chem (2009) 33(6):465–8. doi:10.1016/j.compbiolchem.2009.10.004
82. Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene (2000) 19(48):5498–506. doi:10.1038/sj.onc.1203945
83. Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, et al. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest (2011) 121(2):658–70. doi:10.1172/JCI42974
84. Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol (1982) 22:517–54. doi:10.1146/annurev.pa.22.040182.002505
85. Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov (2002) 1(8):609–20. doi:10.1038/nrd870
86. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478(7368):197–203. doi:10.1038/nature10491
87. Heath-Pagliuso S, Rogers WJ, Tullis K, Seidel SD, Cenijn PH, Brouwer A, et al. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry (1998) 37(33):11508–15. doi:10.1021/bi980087p
88. Bittinger MA, Nguyen LP, Bradfield CA. Aspartate aminotransferase generates proagonists of the aryl hydrocarbon receptor. Mol Pharmacol (2003) 64(3):550–6. doi:10.1124/mol.64.3.550
89. Rannug A, Rannug U, Rosenkranz HS, Winqvist L, Westerholm R, Agurell E, et al. Certain photooxidized derivatives of tryptophan bind with very high-affinity to the Ah receptor and are likely to be endogenous signal substances. J Biol Chem (1987) 262(32):15422–7.
90. Esser C, Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev (2015) 67(2):259–79. doi:10.1124/pr.114.009001
91. Cervantes-Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, Merriman J, et al. Lactobacillus reuteri induces gut intraepithelial CD4+CD8alphaalpha+ T cells. Science (2017) 357(6353):806–10. doi:10.1126/science.aah5825
92. Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med (2016) 22(6):598. doi:10.1038/nm.4102
93. Moura-Alves P, Fae K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature (2014) 512(7515):387–92. doi:10.1038/nature13684
94. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature (2008) 453(7191):106–9. doi:10.1038/nature06881
95. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity (2009) 31(2):321–30. doi:10.1016/j.immuni.2009.06.020
96. Vaidyanathan B, Chaudhry A, Yewdell WT, Angeletti D, Yen WF, Wheatley AK, et al. The aryl hydrocarbon receptor controls cell-fate decisions in B cells. J Exp Med (2017) 214(1):197–208. doi:10.1084/jem.20160789
97. Villa M, Gialitakis M, Tolaini M, Ahlfors H, Henderson CJ, Wolf CR, et al. Aryl hydrocarbon receptor is required for optimal B-cell proliferation. EMBO J (2017) 36(1):116–28. doi:10.15252/embj.201695027
98. Platzer B, Richter S, Kneidinger D, Waltenberger D, Woisetschlager M, Strobl H. Aryl hydrocarbon receptor activation inhibits in vitro differentiation of human monocytes and Langerhans dendritic cells. J Immunol (2009) 183(1):66–74. doi:10.4049/jimmunol.0802997
99. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A (2010) 107(46):19961–6. doi:10.1073/pnas.1014465107
100. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell (2011) 147(3):629–40. doi:10.1016/j.cell.2011.09.025
101. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang YM, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol (2015) 16(3):306–17. doi:10.1038/ni.3094
102. Wagage S, John B, Krock BL, Hall AO, Randall LM, Karp CL, et al. The aryl hydrocarbon receptor promotes IL-10 production by NK cells. J Immunol (2014) 192(4):1661–70. doi:10.4049/jimmunol.1300497
103. Shin JH, Zhang LH, Murillo-Sauca O, Kim J, Kohrt HEK, Bui JD, et al. Modulation of natural killer cell antitumor activity by the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A (2013) 110(30):12391–6. doi:10.1073/pnas.1302856110
104. Shin JH, Haggadone MD, Sunwoo JB. Transcription factor Dlx3 induces aryl hydrocarbon receptor promoter activity. Biochem Biophys Rep (2016) 7:353–60. doi:10.1016/j.bbrep.2016.06.023
105. Xia P, Liu J, Wang S, Ye B, Du Y, Xiong Z, et al. WASH maintains NKp46+ ILC3 cells by promoting AHR expression. Nat Commun (2017) 8:15685. doi:10.1038/ncomms15685
106. Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, Potocnik AJ, et al. Feedback control of AHR signalling regulates intestinal immunity. Nature (2017) 542(7640):242–5. doi:10.1038/nature21080
107. Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S, et al. The aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep (2017) 21(8):2277–90. doi:10.1016/j.celrep.2017.10.114
108. Zhang LH, Shin JH, Haggadone MD, Sunwoo JB. The aryl hydrocarbon receptor is required for the maintenance of liver-resident natural killer cells. J Exp Med (2016) 213(11):2249–57. doi:10.1084/jem.20151998
109. Duerr CU, McCarthy CDA, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol (2016) 17(1):65. doi:10.1038/ni.3308
110. Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and interferon-gamma counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity (2015) 43(1):161–74. doi:10.1016/j.immuni.2015.05.019
111. Badawy AA. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int J Tryptophan Res (2017) 10:1178646917691938. doi:10.1177/1178646917691938
112. Jurgens B, Hainz U, Fuchs D, Felzmann T, Heitger A. Interferon-gamma-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood (2009) 114(15):3235–43. doi:10.1182/blood-2008-12-195073
113. Yu X, Pappu R, Ramirez-Carrozzi V, Ota N, Caplazi P, Zhang J, et al. TNF superfamily member TL1A elicits type 2 innate lymphoid cells at mucosal barriers. Mucosal Immunol (2014) 7(3):730–40. doi:10.1038/mi.2013.92
114. Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L, et al. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med (2016) 213(4):569–83. doi:10.1084/jem.20151750
115. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14(3):221–9. doi:10.1038/ni.2534
116. Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity (2010) 33(5):736–51. doi:10.1016/j.immuni.2010.10.017
117. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity (2015) 43(1):146–60. doi:10.1016/j.immuni.2015.06.019
118. Hughes T, Briercheck EL, Freud AG, Trotta R, McClory S, Scoville SD, et al. The transcription factor AHR prevents the differentiation of a stage 3 innate lymphoid cell subset to natural killer cells. Cell Rep (2014) 8(1):150–62. doi:10.1016/j.celrep.2014.05.042
119. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1 beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol (2016) 17(6):636. doi:10.1038/ni.3444
120. Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity (vol 17, pg 646, 2016). Nat Immunol (2016) 17(8):1005. doi:10.1038/ni.3447
121. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs (vol 17, pg 626, 2016). Nat Immunol (2016) 17(8):1005. doi:10.1038/ni.3443
122. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med (2014) 211(8):1571–83. doi:10.1084/jem.20140678
123. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med (2013) 210(6):1117–24. doi:10.1084/jem.20121588
124. Satoh-Takayama N, Serafini N, Verrier T, Rekiki A, Renauld JC, Frankel G, et al. The chemokine receptor CXCR6 controls the functional topography of interleukin-22 producing intestinal innate lymphoid cells. Immunity (2014) 41(5):776–88. doi:10.1016/j.immuni.2014.10.007
125. Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science (2010) 329(5997):1345–8. doi:10.1126/science.1191536
126. Thordardottir S, Hangalapura BN, Hutten T, Cossu M, Spanholtz J, Schaap N, et al. The aryl hydrocarbon receptor antagonist StemRegenin 1 promotes human plasmacytoid and myeloid dendritic cell development from CD34+ hematopoietic progenitor cells. Stem Cells Dev (2014) 23(9):955–67. doi:10.1089/scd.2013.0521
127. Bankoti J, Rase B, Simones T, Shepherd DM. Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicol Appl Pharmacol (2010) 246(1–2):18–28. doi:10.1016/j.taap.2010.03.013
128. Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A (2010) 107(48):20768–73. doi:10.1073/pnas.1009201107
129. Goudot C, Coillard A, Villani AC, Gueguen P, Cros A, Sarkizova S, et al. Aryl hydrocarbon receptor controls monocyte differentiation into dendritic cells versus macrophages. Immunity (2017) 47(3):582. doi:10.1016/j.immuni.2017.08.016
130. Flannigan KL, Ngo VL, Geem D, Harusato A, Hirota SA, Parkos CA, et al. IL-17A-mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol (2017) 10(3):673–84. doi:10.1038/mi.2016.80
131. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol (2014) 32:659–702. doi:10.1146/annurev-immunol-032713-120145
132. Shih VFS, Cox J, Kljavin NM, Dengler HS, Reichelt M, Kumar P, et al. Homeostatic IL-23 receptor signaling limits Th17 response through IL-22-mediated containment of commensal microbiota. Proc Natl Acad Sci U S A (2014) 111(38):13942–7. doi:10.1073/pnas.1323852111
133. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139(3):485–98. doi:10.1016/j.cell.2009.09.033
134. Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity (2014) 40(4):594–607. doi:10.1016/j.immuni.2014.03.005
135. Sano T, Huang WD, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell (2015) 163(2):381–93. doi:10.1016/j.cell.2015.08.061
136. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell (2015) 163(2):367–80. doi:10.1016/j.cell.2015.08.058
137. Pabst O, Herbrand H, Worbs T, Friedrichsen M, Yan S, Hoffmann MW, et al. Cryptopatches and isolated lymphoid follicles: dynamic lymphoid tissues dispensable for the generation of intraepithelial lymphocytes. Eur J Immunol (2005) 35(1):98–107. doi:10.1002/eji.200425432
138. Tsuji M, Suzuki K, Kitamura H, Maruya M, Kinoshita K, Ivanov II, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity (2008) 29(2):261–71. doi:10.1016/j.immuni.2008.05.014
139. Li J, Bhattacharya S, Zhou J, Phadnis-Moghe AS, Crawford RB, Kaminski NE. Aryl hydrocarbon receptor activation suppresses EBF1 and PAX5 and impairs human B lymphopoiesis. J Immunol (2017) 199(10):3504–15. doi:10.4049/jimmunol.1700289
140. Halim TY, Hwang YY, Scanlon ST, Zaghouani H, Garbi N, Fallon PG, et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat Immunol (2016) 17(1):57–64. doi:10.1038/ni.3294
141. Schwartz C, Khan AR, Floudas A, Saunders SP, Hams E, Rodewald HR, et al. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J Exp Med (2017) 214(9):2507–21. doi:10.1084/jem.20170051
142. Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med (2017) 23(8):938–44. doi:10.1038/nm.4373
143. Martin CE, Spasova DS, Frimpong-Boateng K, Kim HO, Lee M, Kim KS, et al. Interleukin-7 availability is maintained by a hematopoietic cytokine sink comprising innate lymphoid cells and T cells. Immunity (2017) 47(1):171. doi:10.1016/j.immuni.2017.07.005
144. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 Immunity and promotes parasitic helminth expulsion. Immunity (2014) 41(2):283–95. doi:10.1016/j.immuni.2014.06.016
145. Hepworth MR, Monticelli LA, Fung TC, Ziegler CGK, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4(+) T-cell responses to intestinal commensal bacteria. Nature (2013) 498(7452):113. doi:10.1038/nature12240
146. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science (2015) 348(6238):1031–5. doi:10.1126/science.aaa4812
147. Kim MY, Gaspal FMC, Wiggett HE, McConnell FM, Gulbranson-Judge A, Raykundalia C, et al. CD4(+)CD3(-) accessory cells costimulate primed CD4 T cells through OX40 and CD30 at sites where T cells collaborate with B cells. Immunity (2003) 18(5):643–54. doi:10.1016/S1074-7613(03)00110-9
148. Kwong B, Rua R, Gao Y, Flickinger J Jr, Wang Y, Kruhlak MJ, et al. T-bet-dependent NKp46+ innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat Immunol (2017) 18(10):1117–27. doi:10.1038/ni.3816
149. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science (2014) 345(6202):1310. doi:10.1126/Science.1254009
150. Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature (2015) 528(7583):560–4. doi:10.1038/nature16460
151. Aparicio-Domingo P, Romera-Hernandez M, Karrich JJ, Cornelissen F, Papazian N, Lindenbergh-Kortleve DJ, et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J Exp Med (2015) 212(11):1783–91. doi:10.1084/jem.20150318
152. Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell (2016) 166(5):1231. doi:10.1016/j.cell.2016.07.043
153. Ibiza S, Garcia-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature (2016) 535(7612):440–3. doi:10.1038/nature18644
154. Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature (2017) 549:282–6. doi:10.1038/nature23676
155. Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature (2017) 549(7671):277–81. doi:10.1038/nature23469
156. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature (2017) 549(7672):351–6. doi:10.1038/nature24029
157. Li S, Heller JJ, Bostick JW, Lee A, Schjerven H, Kastner P, et al. Ikaros inhibits group 3 innate lymphoid cell development and function by suppressing the aryl hydrocarbon receptor pathway. Immunity (2016) 45(1):185–97. doi:10.1016/j.immuni.2016.06.027
158. Rutz S, Noubade R, Eidenschenk C, Ota N, Zeng W, Zheng Y, et al. Transcription factor c-Maf mediates the TGF-beta-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol (2011) 12(12):1238–45. doi:10.1038/ni.2134
159. Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol (2010) 11(9):854–61. doi:10.1038/ni.1912
160. Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol (2007) 21(12):2941–55. doi:10.1210/me.2007-0211
161. Vogel CF, Wu D, Goth SR, Baek J, Lollies A, Domhardt R, et al. Aryl hydrocarbon receptor signaling regulates NF-kappaB RelB activation during dendritic-cell differentiation. Immunol Cell Biol (2013) 91(9):568–75. doi:10.1038/icb.2013.43
162. Chen PH, Chang H, Chang JT, Lin P. Aryl hydrocarbon receptor in association with RelA modulates IL-6 expression in non-smoking lung cancer. Oncogene (2012) 31(20):2555–65. doi:10.1038/onc.2011.438
163. Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature (2007) 446(7135):562–6. doi:10.1038/nature05683
164. Luecke-Johansson S, Gralla M, Rundqvist H, Ho JC, Johnson RS, Gradin K, et al. A molecular mechanism to switch the aryl hydrocarbon receptor from a transcription factor to an E3 ubiquitin ligase. Mol Cell Biol (2017) 37(13). doi:10.1128/MCB.00630-16
165. Jaeger C, Tischkau SA. Role of aryl hydrocarbon receptor in circadian clock disruption and metabolic dysfunction. Environ Health Insights (2016) 10:133–41. doi:10.4137/EHI.S38343
166. Tischkau SA, Jaeger CD, Krager SL. Circadian clock disruption in the mouse ovary in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Lett (2011) 201(2):116–22. doi:10.1016/j.toxlet.2010.12.013
167. Xu CX, Wang C, Krager SL, Bottum KM, Tischkau SA. Aryl hydrocarbon receptor activation attenuates Per1 gene induction and influences circadian clock resetting. Toxicol Sci (2013) 132(2):368–78. doi:10.1093/toxsci/kfs345
168. Xu CX, Krager SL, Liao DF, Tischkau SA. Disruption of CLOCK-BMAL1 transcriptional activity is responsible for aryl hydrocarbon receptor-mediated regulation of period1 gene. Toxicol Sci (2010) 115(1):98–108. doi:10.1093/toxsci/kfq022
169. Takayama T, Kamada N, Chinen H, Okamoto S, Kitazume MT, Chang J, et al. Imbalance of NKp44(+)NKp46(-) and NKp44(-)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology (2010) 139(3):882–92, 892.e1–3. doi:10.1053/j.gastro.2010.05.040
170. Mizuno S, Mikami Y, Kamada N, Handa T, Hayashi A, Sato T, et al. Cross-talk between RORgammat+ innate lymphoid cells and intestinal macrophages induces mucosal IL-22 production in Crohn’s disease. Inflamm Bowel Dis (2014) 20(8):1426–34. doi:10.1097/MIB.0000000000000105
171. Rutz S, Wang X, Ouyang W. The IL-20 subfamily of cytokines – from host defence to tissue homeostasis. Nat Rev Immunol (2014) 14(12):783–95. doi:10.1038/nri3766
172. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208(6):1127–33. doi:10.1084/jem.20101712
173. Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology (2011) 141(1):237–48, 248.e1. doi:10.1053/j.gastro.2011.04.007
174. Benson JM, Shepherd DM. Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease. Toxicol Sci (2011) 120(1):68–78. doi:10.1093/toxsci/kfq360
175. Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity (2002) 17(5):629–38. doi:10.1016/S1074-7613(02)00453-3
176. Huang Z, Jiang YC, Yang Y, Shao J, Sun XL, Chen JN, et al. 3,3 ’-Diindolylmethane alleviates oxazolone-induced colitis through Th2/Th17 suppression and Treg induction. Mol Immunol (2013) 53(4):335–44. doi:10.1016/j.molimm.2012.09.007
177. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol (2014) 14(5):329–42. doi:10.1038/nri3661
178. Sedhom MAK, Pichery M, Murdoch JR, Foligne B, Ortega N, Normand S, et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut (2013) 62(12):1714–23. doi:10.1136/gutjnl-2011-301785
179. Pastorelli L, Garg RR, Hoang SB, Spina L, Mattioli B, Scarpa M, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A (2010) 107(17):8017–22. doi:10.1073/pnas.0912678107
180. Zenewicz LA, Flavell RA. Recent advances in IL-22 biology. Int Immunol (2011) 23(3):159–63. doi:10.1093/intimm/dxr001
181. Fahy JV. Type 2 inflammation in asthma – present in most, absent in many (vol 15, pg 57, 2015). Nat Rev Immunol (2015) 15(2):130. doi:10.1038/nri3807
182. McKenzie AN. Type-2 innate lymphoid cells in asthma and allergy. Ann Am Thorac Soc (2014) 11(Suppl 5):S263–70. doi:10.1513/AnnalsATS.201403-097AW
183. Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O’Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol (2016) 137(1):75–86.e8. doi:10.1016/j.jaci.2015.05.037
184. Li XM, Peng J, Gu W, Guo XJ. TCDD-induced activation of aryl hydrocarbon receptor inhibits Th17 polarization and regulates non-eosinophilic airway inflammation in asthma. PLoS One (2016) 11(3):e0150551. doi:10.1371/journal.pone.0150551
185. Thatcher TH, Williams MA, Pollock SJ, McCarthy CE, Lacy SH, Phipps RP, et al. Endogenous ligands of the aryl hydrocarbon receptor regulate lung dendritic cell function. Immunology (2016) 147(1):41–54. doi:10.1111/imm.12540
186. Luebke RW, Copeland CB, Daniels M, Lambert AL, Gilmour MI. Suppression of allergic immune responses to house dust mite (HDM) in rats exposed to 2,3,7,8-TCDD. Toxicol Sci (2001) 62(1):71–9. doi:10.1093/toxsci/62.1.71
187. Wang YH, Wills-Karp M. The potential role of interleukin-17 in severe asthma. Curr Allergy Asthma Rep (2011) 11(5):388–94. doi:10.1007/s11882-011-0210-y
188. Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer (2014) 14(12):801–14. doi:10.1038/nrc3846
189. Triantafillidis JK, Nasioulas G, Kosmidis PA. Colorectal cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res (2009) 29(7):2727–37.
Keywords: aryl hydrocarbon receptor, innate lymphoid cell, mucosal immunity, gut, microbiota and immunity
Citation: Li S, Bostick JW and Zhou L (2018) Regulation of Innate Lymphoid Cells by Aryl Hydrocarbon Receptor. Front. Immunol. 8:1909. doi: 10.3389/fimmu.2017.01909
Received: 14 September 2017; Accepted: 14 December 2017;
Published: 05 January 2018
Edited by:
Marina Cella, Washington University in St. Louis, United StatesReviewed by:
John B. Sunwoo, Stanford University, United StatesMarco Colonna, Washington University in St. Louis, United States
Copyright: © 2018 Li, Bostick and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Zhou, bGlhbmd6aG91NDk3QHVmbC5lZHU=