
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 22 December 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01864
This article is part of the Research TopicReassessing Twenty Years of Vaccine Development Against TuberculosisView all 9 articles
 Roland Lang*
Roland Lang* Judith Schick
Judith Schick
Successful immune control of Mycobacterium tuberculosis (MTB) requires robust CD4+ T cell responses, with IFNγs as the key cytokine promoting killing of intracellular mycobacteria by macrophages. By contrast, helminth infections typically direct the immune system toward a type 2 response, characterized by high levels of the cytokines IL-4 and IL-10, which can antagonize IFNγ production and its biological effects. In many countries with high burden of tuberculosis, helminth infections are endemic and have been associated with increased risk to develop tuberculosis or to inhibit vaccination-induced immunity. Mechanistically, regulation of the antimycobacterial immune response by helminths has been mostly been attributed to the T cell compartment. Here, we review the current status of the literature on the impact of helminths on vaccine-induced and natural immunity to MTB with a focus on the alterations enforced on the capacity of macrophages to function as sensors of mycobacteria and effector cells to control their replication.
The innate immune system detects incoming mycobacteria during phagocytosis by alveolar macrophages in the lung. The hydrophobic mycobacterial cell wall contains a large number of lipids, glycolipids, and lipoglycans that act as pathogen-associated molecular patterns (PAMPs), which are recognized by several classes of pattern recognition receptors (PRRs) [for review, see Ref. (1)]. Due to the intracellular lifestyle of MTB, which persists and replicates in the phagosome, endosomal PRR have ample opportunity to interact with mycobacterial ligands released into this compartment, e.g., DNA and RNA. With increasing time spent in its host cell, mycobacterial products and even the bugs themselves can enter the cytosol (2), where yet other PRRs sense the presence of intruding microbes.
This initial interaction between macrophages and MTB is crucial: if the macrophage is able to kill MTB at this stage, no infection occurs and there is no need to call in adaptive immunity (Figure 1 “innate resistance”). Based on studies on transmission of MTB to household contacts measuring tuberculin skin test or quantiferon responses, this may be the situation in more than 50% of all exposures (3–5). However, since it is difficult to determine the true exposure of household contacts of patients with open tuberculosis to infectious aerosol, the percentage of innate resistance to MTB in humans could also be considerably lower (6). On the other hand, the finding that tuberculin skin test negativity in humans is linked to a chromosomal region overlapping the TNF1 locus provides evidence for genetic control of innate resistance to MTB infection (7). Clearly, the factors determining the initial fate of mycobacteria after ingestion by alveolar macrophages are very incompletely understood, and may range from cytokines such as TNF to antimicrobial peptides, the autophagy machinery and control of phagosomal maturation (8). Since all these macrophage functional processes are under the influence of signaling emanating from PRR, it makes sense to assume that the recognition of MTB by different PRR contributes to the initial decision if ingested bacilli survive or are killed. If the mycobacteria manage to establish an intracellular niche in the macrophage, the nature of the innate response (mostly the composition of chemokines and cytokines secreted) depends on PRR pathways and determines the type of adaptive immunity and the swiftness of a protective response characterized by robust Th1 and Th17 T cells.
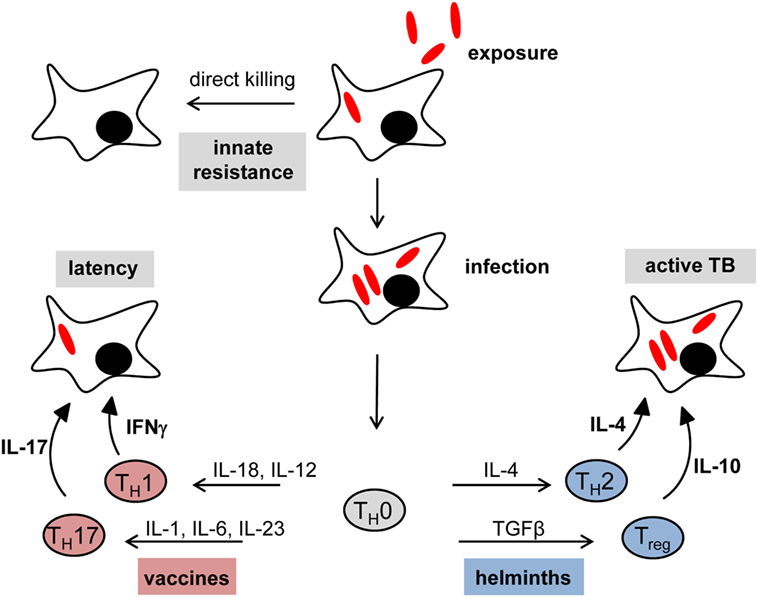
Figure 1. Immune checkpoints in tuberculosis: impact of vaccination and helminth infection. In many cases, the initial exposure to mycobacteria results in direct killing by alveolar macrophages without the need for an adaptive immune response (innate resistance). Failure of initial innate control mechanisms leads to primary infection. The set of chemokines and cytokines produced by innate immune cells are crucial for the shaping of an effective adaptive immune response. The Th1 key cytokine IFNγ is necessary to establish and maintain latent infection. Moreover, IL-17 was found to be important for vaccination-induced protection against tuberculosis. However, concomitant helminth infection shifts the immune system toward a T helper type 2 (Th2)/regulatory T cells (Treg) response rather than a protective Th1/Th17 immune status, which leads to a higher risk to develop active disease and interferes with successful vaccination responses.
Toll-like receptors (TLR) have been most intensively studied for their role in the response to mycobacteria. TLR2 and TLR4 bind to mycobacterial cell wall components lipoarabinomannan (LAM) and phosphatidylinositol mannosides (PIM), and lipomannan, respectively (9–12). The 19-kDa lipopeptide of MTB is also a TLR2 ligand (12). The endosomal TLR7 and TLR8 (the later only in humans, but not in mice) sense single-stranded RNA (13), while CpG-rich DNA was initially purified as the immunostimulatory principle of Bacille Calmette–Guerin (BCG) treatment and later explained by activation of TLR9 (14). Independent of their localization on the cell surface or in the phagosome, TLR2, TLR7/8, and TLR9 require the adapter protein Myd88 to activate gene expression. Myd88-dependent signaling is essential for host defense against experimental MTB infection in mice; however, as even the triple knockout of TLR2, TLR4, and TLR9 in mice does not increase mycobacterial load (15), the phenotype of Myd88−/− mice is likely due to a lack of IL-1 receptor signaling rather than TLR activation (16–18).
More recently, several C-type lectins receptor (CLR) have been identified as receptors for PAMPs present in the mycobacterial cell wall [reviewed in Ref. (19)]. Dectin-1 is triggered by an unknown mycobacterial ligand and induces IL-12 production from DC (20) and collaborates with TLR2 in induction of cytokine gene expression (21). The genes for the CLR Mincle (Clec4e), Mcl (Clec4d), and Dectin-2 (Clec4n) are located in close vicinity in the genome and are referred to as the Dectin-2 family (22). Mincle is the receptor for the so-called mycobacterial cord factor trehalose-6,6′-dimycolate (TDM), the most abundant glycolipid in the cell wall of MTB (23, 24). Mincle can form dimers with the related CLR Mcl (25), and both receptors mutually enhance their cell surface protein levels (26). Dectin-2 binds mannose-rich fungal ligands (27, 28), but also the TLR2 ligand mannose-capped LAM (29). Most recently, DCAR (Clec4b1), another FcRγ-coupled CLR, was revealed to bind to PIM of the cell wall, to induce MCP-1 expression by macrophages, and to induce Th1 responses (30). All of these CLR signal via the Syk-Card9-Bcl10-Malt1 pathway to activate NFκB and upregulate expression of multiple chemokines, cytokines and inflammatory mediators causing inflammation and directing developing adaptive immune responses (31, 32). Knockout mice for Card9 are highly susceptible to challenge with MTB, with increased bacterial burden, uncontrolled granulocytic inflammation, and early death (33). The phenotype of mice deficient in individual CLR in MTB infection is much more moderate, as demonstrated for Mincle (34–36), Mcl (37), or shows no difference as in the case of Dectin-1 (38). Thus, similar to the case of TLR, there appears to be considerable redundancy of individual CLR for recognition of mycobacteria, but the combined ablation of the CLR response in Card9-deficient mice is detrimental and suggests an important function of this class of receptors in antimycobacterial defense.
In addition to TLR and CLR localized to the plasma membrane or the phagosome, NOD2 is a cytosolic sensor of mycobacterial muramyl dipeptide, which induces autophagy and activates NFκB-dependent gene expression (39). The ESX-1 secretion system of MTB generates phagosomal perforation and leakage of mycobacterial DNA and RNA. Binding of cytosolic DNA by the sensor cGAS leads to production of cyclic dinucleotides binding and activating STING, which triggers IFNβ expression in an IRF3-dependent manner (40, 41). The activation of the AIM2-NLRP3 inflammasome by cytosolic DNA is an additional pathway triggered by the intracellular pathogen MTB, and causes production of IL-1β (42, 43).
The single most important immunological molecule in defense against mycobacterial infection is the cytokine IFNγ (Figure 1 “latency”). Produced mostly by Th1 CD4+ T cells but also CD8+ T cells, NK cells and NKT cells, it acts on many different cell types by activating the Jak2-Stat1 signaling pathway that drives the expression of antimicrobial genes such as Nos2 (producing NO), IFNγ-induced GTPase-binding proteins of the 65 and 47 kDa families, bactericidal peptides (e.g., cathelicidin), and many more (44). The inability to upregulate IFNγ expression (e.g., deficiency in the cytokines IL-12 or IL-18, or their receptors), produce it, or respond to it (e.g., deficiency in the IFNγ receptor chains, Jak2 or Stat1), leads to high susceptibility to experimental mycobacterial infection in mouse models and in humans (45) (Figure 1 “active TB”).
While IFNγ clearly is necessary for a successful host response to infection, several studies have shown that it is not sufficient, as increased levels of IFNγ were indicative of disease progression rather than protection in mice and humans (46, 47). Beyond a simple lack of correlation between IFNγ levels and protection in MTB infection, there is even evidence that IFNγ production has to be tightly controlled to prevent damage to the host, particularly the lung tissue, during infection (48).
Despite the nearly 100 years of vaccination with the Mycobacterium bovis strain BCG it is still very incompletely understood which vaccine-induced immune responses can be used as biomarkers for protective immunity (49). This applies to IFNγ, which is essentially required for successful immunity, but it is becoming increasingly clear that there is no linear relationship between vaccine-induced IFNγ and immune protection (48). More recently, IL-17-producing Th17 cells have been described as key players in vaccine-induced protection (50). Interestingly, the recombinant BCG vaccine VPM02 (ΔureC; hly+), which is more effective than the parental BCG in protecting against pulmonary tuberculosis in animal models, induces a balanced Th1/Th17 response and caused earlier recruitment of CD4+ T cells into the lungs after challenge infection (51). In the last years, there has been an increasing appreciation of the important role played by the route of immunization: Aguilo et al. demonstrated that BCG delivered intranasally protects DBA/2 mice against pulmonary tuberculosis much better than after subcutaneous immunization (52). An alternative strategy to improved BCG or novel recombinant MTB live vaccines is the development of subunit vaccines using recombinant MTB proteins. To be effective in generating robust antigen-specific CD4+ T cell responses, these protein vaccines require a carrier (e.g., liposomes) and adjuvants that activate antigen-presenting cells of the innate immune system for provision of costimulation and cytokines directing the desired Th cell differentiation signals. Here, the TLR9 ligands CpG ODN and IC31 are potent inducers of Th1 cells by triggering IL-12 release from DC and macrophages, and have therefore been used experimentally and in clinical studies (53–55). In contrast, ligands for certain CLR are potent inducers of Th17 differentiation: the Dectin-1 ligand Curdlan, a β-glucan, and the Mincle ligand TDB, an analog of the mycobacterial cord factor TDM, trigger production of IL-6, IL-23, and IL-1 from DC and macrophages, thereby providing robust differentiation signals toward a Th17 bias (24, 56, 57). The TDB-containing liposomal adjuvant CAF01 has been used in several experimental infection models, induces long-lived protective CD4 memory T cells in mice, and also appears to be effective in humans (58–61).
Globally, more than two billion people are infected with parasitic helminths. Intestinal nematodes (Ascaris lumbricoides, hookworms, Trichuris trichiura, Strongyloides stercoralis), filaria (Wuchereria bancrofti, Onchocerca volvulus, Loa loa), and trematodes (Schistosoma mansoni) are the most frequent and important human worm infections (62). Similar to tuberculosis, helminth infections are often chronic and not acutely life-threatening. Helminths have complex life cycles and various developmental stages, and elicit distinct host immune reactions. Despite significant differences between specific helminth infections, the immune response to helminths is generally characterized by a T helper type 2 (Th2) pattern with high levels of the cytokines IL-4, IL-5, IL-9, IL-10, and IL-13, as well as eosinophilia, goblet and mast cell hyperplasia, and IgE-biased antibody isotype switching (62). Thus, helminth infection and tuberculosis share the chronic nature, but represent two extremes of immunological bias and immune effector mechanisms. Since tuberculosis and helminth infections are coinciding in many parts of the world, most notably in Africa, South America, and Asia, the possibility that concomitant helminth infection affects antimycobacterial immunity has been investigated by epidemiological studies.
The efficacy of the BCG vaccine shows large geographic variation, with reduced protection in African and Asian countries, where worm infections are more prevalent than in Europe and North America (63, 64). Deworming of helminth-infected vaccinees before administration of BCG led to increased IFNγ and IL-12 production, yet decreased TGFβ levels, suggesting that intestinal helminths impair the development of a Th1 response to BCG (65). In the mouse model, S. mansoni infection decreased protective efficacy of BCG against MTB challenge and the capacity to produce IFNγ by splenocytes (66). As BCG vaccination is usually done in neonates, effects of maternal helminth infection on vaccination responses in the offspring have been investigated in mice and humans. Maternal infection with helminths had a strong negative effect on induction of Th1 immunity in BCG-vaccinated offspring (67). In another study, hookworm-infected mothers had reduced IFNγ responses to MTB proteins, but their children showed rather increased IFNγ production (68). In addition, anthelmintic treatment during pregnancy did not alter the neonatal responses to vaccination including BCG (69). Thus, the literature is at present contradictory whether helminth coinfection is indeed a strong factor in the suboptimal response to BCG and whether anthelmintic treatment can overcome this hurdle.
Patients with active tuberculosis were found to be more often coinfected with helminths compared to controls (70, 71). In addition, coinfection with helminths was associated with more advanced disease in tuberculosis patients, coupled to reduced IFNγ but increased production of IL-10 (72). On the other hand, a recent study showed that asymptomatic helminth infection is indeed associated with type 2 immunity in tuberculosis patients, but led to a lower frequency of smear-positive sputum samples, i.e., reduced the risk to develop open cavitary disease (73). Reflecting this ambiguous state of the literature on the impact of helminth infection on the course of active tuberculosis, a treatment study with albendazole showed no change in the clinical course after 2 months of treatment (74).
Macrophages encounter worm eggs and larval stages in tissues and therefore directly interact with helminths and their products. A number of studies have investigated how different helminth species and their products modulate macrophage activation state and responsiveness to other microbial stimuli. While these studies employed different helminths or helminth-derived products and a variety of readout parameters of macrophage activation, they in general described inhibitory or modulating effects on macrophages. ES-62, a secreted protein of the filarial nematode Acanthocheilonema viteae inhibits the production of IL-12, TNF, and IL-6 in response to LPS/IFNγ (75). Secreted filarial cystatins were found to be taken up by macrophages and to induce expression of IL-10, which was sensitive to inhibition of MAPK and was regulated by the phosphatase DUSP1 (76). Microfilariae of Brugia malayi induced a regulatory phenotype in human monocytes characterized by expression of IL-10 and PD-L1 (77). A Fasciola hepatica fatty acid binding protein exerted suppression of cytokine release and MAPK activation in response to the TLR4 ligand, and was found to bind to the co-receptor for LPS, CD14 (78). Similarly, the excretory/secretory products of the tapeworms Hymenolepis diminuta and Spirometra erinaceieuropaei inhibited the production of inflammatory cytokines such as TNF by macrophages (79, 80). Recently, Aira et al. observed that antigens prepared from H. diminuta and Trichuris muris inhibit phagolysosomal maturation and antigen presentation to CD4+ T cells in macrophages infected with MTB, with a direct negative effect on the control of mycobacterial survival in macrophages (81). It should be noted that inhibition of macrophage activation is not uniformly observed upon contact with helminths, e.g., antigens from S. mansoni showed the opposite effect, enhancing mycobacterial control and decreasing IL-10 production (81). While inhibiting production of TNF and IL-6 by TLR-triggered dendritic cells, schistosomal egg antigens (SEA) activated the Nlrp3 inflammasome and IL-1β production by activating the Dectin-2-FcRγ-Syk pathway (82).
A reduced Th1 and Th17 immunity to mycobacterial antigens in patients with latent or active tuberculosis by coinfection with helminths was described in several studies (72, 83–85). By contrast, the frequency of Treg was consistently increased in coinfected patients (73, 86), together with the production of IL-10 (73), which also contributed to inhibition of Th1 differentiation (84). In vitro exposure of human PBMC to SEA increased the expression of IL-10 and IL-4 by CD4+ T cells, which in turn caused a block in phagolysosomal maturation in mycobacteria-infected macrophages (87). The Treg/Th2-biased immune deviation in coinfected patients was corrected by anthelmintic treatment, which restored Th1 cells and diminished Treg numbers (86), decreased the levels of IL-10 (74), or reduced Th2 but increased Th1/Th17 cytokines (88). These data indicate that the bias of antimycobacterial immunity toward Th2/Treg in helminth coinfection is reversible upon treatment.
Experimental infections with filaria, schistosomes (89, 90), and the hookworm Nippostrongylus brasiliensis (91) caused a Th2-biased response to mycobacterial infection and, in part, higher bacterial burden. Potian et al. demonstrated that Nippostrongylus infection caused transiently increased mycobacterial burden through the activity of IL-4R-positive alternatively activated macrophages in the lung (91). While this study clearly demonstrated that the transfer of wild-type macrophages into IL-4R-deficient mice was sufficient to increase mycobacterial growth, the mechanistic basis has not been clarified in detail yet. One possible mechanism is upregulation of Arginase-1 expression, which is strongly induced by Stat6 signaling in response to IL-4/IL-13 signals in M2 macrophages and competes with iNOS for Arginine as a substrate (Figure 2B). Conditional Arginase-1 knockout mice lacking the enzyme in hematopoietic cells more efficiently clear MTB after pulmonary infection, correlating with enhanced NO production in the lung (92). However, in a coinfection model with MTB and S. mansoni, deletion of Arginase-1 did not alter the mycobacterial burden in coinfected mice, although it reversed lung pathology (93). Of note, infection with N. brasiliensis can also promote pulmonary Th1 cell responses and activation of alveolar macrophages, which was associated with enhanced control of early infection in a M. bovis BCG infection model (94). While these data appear to contradict those of Potian et al. (91), both studies agree in observing a reduced mycobacterial burden early in coinfection of N. brasiliensis and mycobacteria. Hence, timing of coinfection appears to be decisive for the impact of helminths on antimycobacterial immune responses.
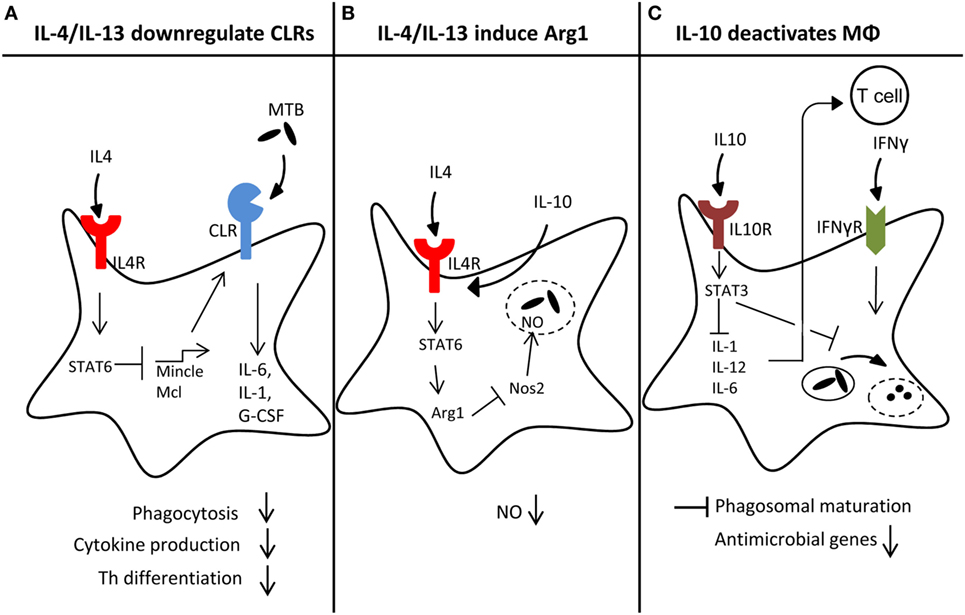
Figure 2. Macrophage reprogramming by helminth-induced type 2 responses. Helminth-induced type 2 responses can act on macrophages on different levels, thereby impeding antimicrobial responses toward Mycobacterium tuberculosis (MTB): (A) high levels of IL-4/IL-13 downregulate C-type lectin receptor (CLR) expression in a Stat6-dependent manner, which may lead to reduced amounts of pro-inflammatory cytokines like IL-6, IL-1, or G-CSF, impaired phagocytosis and Th1/Th17 differentiation. (B) Simultaneously, IL-4/IL-13 signaling through Stat6 strongly induces Arginase1 (Arg1), which counteracts Nos2 through substrate depletion, leading to reduced NO production and less effective killing of MTB inside the phagolysosome. (C) The anti-inflammatory cytokine IL-10, produced during helminth infection, can delay phagosomal maturation. Moreover, IL10R signaling through STAT3 inhibits IL-1, IL-12, or IL-6 expression, which in turn inhibits Th1 differentiation.
Babu et al. hypothesized that helminth infection may modulate immune responses by diminishing TLR expression and indeed identified reduced levels of TLR2 and TLR9 in PBMC of patients with filarial infection (95, 96). Cytokine responses to TLR2 and TLR9 ligands were decreased, suggesting that the impaired receptor expression was functionally relevant. Interestingly, anthelmintic treatment restored responsiveness to TLR ligands, indicating the reversibility of macrophage inhibition by filarial infection (95).
CLR are often expressed in cell type-specific and stimulus-dependent manners. Indeed, several CLR are used as markers for certain DC or macrophage cell types, e.g., Clec9a as marker for CD8+ DC. The cord factor receptor Mincle is strongly induced by stimulation of macrophages with the TLR ligands LPS or CpG but also by its ligand TDM itself (97, 98). Interestingly, mRNA expression of Mincle is high in monocytes and neutrophils in both mouse and human. However, during differentiation of human monocytes to DC in vitro in culture with GM-CSF plus IL-4, pronounced downregulation of Mincle mRNA was observed (99) and could be attributed to the action of IL-4 (100). This effect was also observed for the other Dectin-2 family CLR Mcl and Dectin-2, and was confirmed in mouse macrophages and DC at the mRNA and protein level by flow cytometry (100). By contrast, Dectin-1 expression was not affected negatively by IL-4. Both IL-4 and IL-13 impair expression of Mincle, and do so in a Stat6-dependent manner. Interestingly, the downregulation of Mincle and Mcl was associated with a reduced production of TNF and G-CSF when macrophages were stimulated with the Mincle/Mcl ligand TDM, but not when LPS was used (100). The functional consequence of this targeting of Dectin-2 family CLR expression by IL-4/IL-13 has not been addressed to date, but since Mincle/Mcl bind TDM and Dectin-2 has been identified as a PRR for mannose-capped LAM (29), the lack of receptors for recognition of two major mycobacterial PAMPs can be expected to diminish the response of macrophages to encounter with MTB (Figure 2A).
IL-10 by several cell types is upregulated during helminth infection (see also above in Section “Direct Regulation of Macrophage Function by Helminths”). Through its powerful macrophage deactivation properties, it inhibits the production of many cytokines important for development of Th1/Th17 responses, such as IL-12, IL-6, IL-1, and IL-23 (101) (Figure 2C). In addition, IL-10 more directly impairs killing of mycobacteria in macrophages by delaying phagosome maturation (102) and inhibiting the expression of IFNγ-induced antimycobacterial effector molecules, such as iNOS and ROS (103). Overexpression of IL-10 in mice makes them more susceptible to mycobacterial infection (104, 105), whereas its deletion or blockade of its receptor lead to enhanced immune control of MTB (106).
To date, the information about changes in the expression of TLR and CLR during helminth infection is patchy at best, with the observations on TLR2 and TLR9 made in human patients with filarial disease and the effect of IL-4/IL-13 on Dectin-2 family CLR expression in human and mouse macrophages and DC treated in vitro. A comprehensive expression analysis of different cell types (monocytes/macrophages, granulocytes, DC) from humans infected with helminths or not, or before and after anthelmintic treatment, should be performed. For comparison, the same type of expression analysis should be done using myeloid cells isolated from mice infected with helminths experimentally. This expression analysis should also be extended to other PRR families, including cytosolic sensors such as NOD proteins, cGAS, and STING.
Reduced expression of receptors for mycobacteria on myeloid cells may impair the antimycobacterial response at several levels. First, cell surface-localized CLR and TLR can be involved in phagocytosis and their absence on the membrane can lead to suboptimal uptake of mycobacteria. While Mincle and Dectin-2 appear to be dispensable for phagocytosis of mycobacteria, Mcl-deficient mice have a delayed clearance of MTB from the airways after experimental infection (37). A lack of TLR-induced signaling during phagocytosis can alter the phagosome maturation process and the capacity for antigen presentation (107, 108), which is also likely in case of CLR deficiency (109, 110). On the other hand, mycobacterial cord factor delays phagosomal acidification in a Mincle-dependent process (111). With regard to adaptive immune responses, the production of Th1- and Th17-inducing cytokines in response to contact with mycobacteria requires TLR-Myd88 and CLR-Syk-Card9 signaling, respectively (23, 24); thus, downregulated expression may alter the quality of the Th cell response toward Th2/Treg and thereby allow mycobacterial growth (Figure 2A). Finally, the capacity of macrophages to kill mycobacteria directly and without further activation by T cells is likely dependent on the strength of activation by the different PRR pathways triggered by MTB. Thus, innate resistance to MTB can be hypothesized to be compromised when the macrophages encountering MTB, such as alveolar macrophages, are exposed to type 2 environment and downregulate receptor expression. On the other hand, to demonstrate that diminished cytokine production and killing of mycobacteria is indeed due to abrogated expression of CLR or TLR, and not to other effects of IL-4-Stat6 signaling, will require to reconstitute expression of these PRR, e.g., by retroviral transduction.
Negative effects of concurrent helminth infection on the immune response to vaccination with BCG have been described repeatedly in mice and humans (65, 66). However, it is unclear by which mechanism helminth-induced type 2 immunity interferes with Th1 response induction by BCG (66). Several studies have demonstrated that helminths can inhibit DC activation and costimulatory molecule expression (112, 113). Helminth infection promotes increased induction of Treg (114, 115) and/or IL-10 production (116), leading to suppression of Th1/Th17 immune responses. Downregulation of TLR and CLR by helminths through a type 2 response may be an alternative mechanism: lack of activating signaling reduces production of cytokines driving Th1/Th17 polarization of immune response. New vaccination strategies using recombinant MTB antigens together with different adjuvants as booster vaccines after BCG priming are currently developed. Among the adjuvants employed in these experimental and preclinical vaccines are the TLR ligands IC31 (TLR9) and the CLR ligands TDB (Mincle/Mcl). Since expression of these receptors is downregulated in patients with helminth coinfection (95, 96) or in vitro by type 2 cytokines (100), the adjuvanticity of these vaccines would appear to be particularly vulnerable due to diminished innate activation in helminth-infected patients. To determine whether helminths impair the success of adjuvanted subunit vaccines via downregulation of selected PRRs, studies in mouse models should be conducted comparing the efficacy of different adjuvants acting through different innate receptors subject to regulation by helminths or not (e.g., Dectin-1/Curdlan). These studies may also identify optimal adjuvants for use in populations with high rates of helminth infection. Priming of macrophages with the TLR4 ligand LPS can overcome the inhibitory effect of IL-4 on Dectin-2 family CLR expression (100). Thus, the combination of several adjuvants should also be tested because it may circumvent the impaired receptor expression imposed by helminth-induced type 2 responses.
The connection between helminth infection and the immune response to tuberculosis and the BCG vaccine has triggered attempts to use anthelmintic treatment (“deworming”) to enhance protective natural or vaccine-induced antimycobacterial immunity. Elias et al. observed that Albendazole increased the Th1 (IFNγ/IL-12) response in adult helminth-infected BCG vaccinees (65, 117). Since most BCG doses are given to neonates, the finding that maternal helminth infection impairs the vaccine-induced IFNγ response after birth was highly relevant (67, 118) and suggested that treatment of mothers before birth should enhance the immune response to BCG in their offspring; however, this was not confirmed in a larger study performed in Uganda (69). Together, the literature is not conclusive yet and larger studies in different settings will be required. Studies into the effect of anthelmintic treatment will also be required for the newly developed live and subunit TB vaccines, depending on how strongly they are affected by coinfection with helminths.
A better understanding of the regulation of protective immunity to TB by helminths at the molecular and cellular level may also contribute to more specific modulation by targeting specific components of the type 2 response. Identification of key factors that suppress Th1/Th17 immunity may allow to specifically antagonize them during vaccination or tuberculostatic treatment without abrogating more beneficial effects of worm-induced immune regulation. It will therefore be important to dissect the contribution of different cell types (e.g., Th2, ILC2, different myeloid cells), cytokines (IL-4/IL-13, IL-10, TGFβ, but also IL-33), and mediators (e.g., Nos2, Arg1), to impaired antimycobacterial resistance and immunity.
It is now evident that helminth infestations can influence the host response to MTB at multiple levels, from the initial encounter between macrophage and bacilli, to the type of adaptive T cell immunity, and to the development of immunopathology. At the same time, many open questions remain to be answered, both at the clinical–epidemiological level (e.g., regarding the benefit of anthelmintic treatment) and at the fundamental level of immune regulation during coinfection. Clearly, to obtain a mechanistic understanding of observations made in patient cohorts and hypotheses derived from them need to be tested in experimental animal and in vitro models. Vice versa, it will be exciting to determine whether regulatory effects of type 2 cytokines on macrophages and other cell types identified in laboratory studies hold true also in cohorts of coinfected patients or in vaccination studies in humans.
RL and JS conceived and wrote the manuscript and prepared the figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by funding from the Deutsche Forschungsgemeinschaft (SFB 796, TP B6, and GRK 1660, TP A2) to RL. The authors acknowledge support by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universitat Erlangen-Nurnberg (FAU) within the funding programme Open Access Publishing.
1. Mishra A, Akhtar S, Jagannath C, Khan A. Pattern recognition receptors and coordinated cellular pathways involved in tuberculosis immunopathogenesis: emerging concepts and perspectives. Mol Immunol (2017) 87:240–8. doi:10.1016/j.molimm.2017.05.001
2. van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell (2007) 129:1287–98. doi:10.1016/j.cell.2007.05.059
3. Verhagen LM, Hermans PW, Warris A, de Groot R, Maes M, Villalba JA, et al. Helminths and skewed cytokine profiles increase tuberculin skin test positivity in Warao Amerindians. Tuberculosis (Edinb) (2012) 92:505–12. doi:10.1016/j.tube.2012.07.004
4. Kasambira TS, Shah M, Adrian PV, Holshouser M, Madhi SA, Chaisson RE, et al. QuantiFERON-TB gold in-tube for the detection of Mycobacterium tuberculosis infection in children with household tuberculosis contact. Int J Tuberc Lung Dis (2011) 15:628–34. doi:10.5588/ijtld.10.0555
5. Marks SM, Taylor Z, Qualls NL, Shrestha-Kuwahara RJ, Wilce MA, Nguyen CH. Outcomes of contact investigations of infectious tuberculosis patients. Am J Respir Crit Care Med (2000) 162:2033–8. doi:10.1164/ajrccm.162.6.2004022
6. Turner RD, Chiu C, Churchyard GJ, Esmail H, Lewinsohn DM, Gandhi NR, et al. Tuberculosis infectiousness and host susceptibility. J Infect Dis (2017) 216:S636–43. doi:10.1093/infdis/jix361
7. Cobat A, Poirier C, Hoal E, Boland-Auge A, de La Rocque F, Corrard F, et al. Tuberculin skin test negativity is under tight genetic control of chromosomal region 11p14-15 in settings with different tuberculosis endemicities. J Infect Dis (2015) 211:317–21. doi:10.1093/infdis/jiu446
8. Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discov (2017). doi:10.1038/nrd.2017.162
9. Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A (1999) 96:14459–63. doi:10.1073/pnas.96.25.14459
10. Gilleron M, Quesniaux VF, Puzo G. Acylation state of the phosphatidylinositol hexamannosides from Mycobacterium bovis Bacillus Calmette Guerin and Mycobacterium tuberculosis H37Rv and its implication in toll-like receptor response. J Biol Chem (2003) 278:29880–9. doi:10.1074/jbc.M303446200
11. Doz E, Rose S, Nigou J, Gilleron M, Puzo G, Erard F, et al. Acylation determines the toll-like receptor (TLR)-dependent positive versus TLR2-, mannose receptor-, and SIGNR1-independent negative regulation of pro-inflammatory cytokines by mycobacterial lipomannan. J Biol Chem (2007) 282:26014–25. doi:10.1074/jbc.M702690200
12. Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, et al. Cutting edge: role of toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol (2002) 169:10–4. doi:10.4049/jimmunol.169.1.10
13. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (2004) 303:1526–9. doi:10.1126/science.1093620
14. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature (2000) 408:740–5. doi:10.1038/35047123
15. Holscher C, Reiling N, Schaible UE, Holscher A, Bathmann C, Korbel D, et al. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, -4 and -9. Eur J Immunol (2008) 38:680–94. doi:10.1002/eji.200736458
16. Scanga CA, Bafica A, Feng CG, Cheever AW, Hieny S, Sher A. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect Immun (2004) 72:2400–4. doi:10.1128/IAI.72.4.2400-2404.2004
17. Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest (2004) 114:1790–9. doi:10.1172/JCI200421027
18. Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol (2007) 179:1178–89. doi:10.4049/jimmunol.179.2.1178
19. Ishikawa E, Mori D, Yamasaki S. Recognition of mycobacterial lipids by immune receptors. Trends Immunol (2017) 38:66–76. doi:10.1016/j.it.2016.10.009
20. Rothfuchs AG, Bafica A, Feng CG, Egen JG, Williams DL, Brown GD, et al. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol (2007) 179:3463–71. doi:10.4049/jimmunol.179.6.3463
21. Yadav M, Schorey JS. The beta-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood (2006) 108:3168–75. doi:10.1182/blood-2006-05-024406
22. Kerscher B, Willment JA, Brown GD. The dectin-2 family of C-type lectin-like receptors: an update. Int Immunol (2013) 25:271–7. doi:10.1093/intimm/dxt006
23. Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin mincle. J Exp Med (2009) 206:2879–88. doi:10.1084/jem.20091750
24. Schoenen H, Bodendorfer B, Hitchens K, Manzanero S, Werninghaus K, Nimmerjahn F, et al. Cutting edge: mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol (2010) 184:2756–60. doi:10.4049/jimmunol.0904013
25. Lobato-Pascual A, Saether PC, Fossum S, Dissen E, Daws MR. Mincle, the receptor for mycobacterial cord factor, forms a functional receptor complex with MCL and FcepsilonRI-gamma. Eur J Immunol (2013) 43:3167–74. doi:10.1002/eji.201343752
26. Miyake Y, Oh-Hora M, Yamasaki S. C-type lectin receptor MCL facilitates mincle expression and signaling through complex formation. J Immunol (2015) 194:5366–74. doi:10.4049/jimmunol.1402429
27. Sato K, Yang XL, Yudate T, Chung JS, Wu J, Luby-Phelps K, et al. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J Biol Chem (2006) 281:38854–66. doi:10.1074/jbc.M606542200
28. Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity (2010) 32:681–91. doi:10.1016/j.immuni.2010.05.001
29. Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, et al. Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity (2014) 41:402–13. doi:10.1016/j.immuni.2014.08.005
30. Toyonaga K, Torigoe S, Motomura Y, Kamichi T, Hayashi JM, Morita YS, et al. C-type lectin receptor DCAR recognizes mycobacterial phosphatidyl-inositol mannosides to promote a Th1 response during infection. Immunity (2016) 45:1245–57. doi:10.1016/j.immuni.2016.10.012
31. Ostrop J, Lang R. Contact, collaboration, and conflict: signal integration of Syk-coupled C-type lectin receptors. J Immunol (2017) 198:1403–14. doi:10.4049/jimmunol.1601665
32. Geijtenbeek TB, Gringhuis SI. C-type lectin receptors in the control of T helper cell differentiation. Nat Rev Immunol (2016) 16:433–48. doi:10.1038/nri.2016.55
33. Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, et al. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med (2010) 207:777–92. doi:10.1084/jem.20090067
34. Behler F, Steinwede K, Balboa L, Ueberberg B, Maus R, Kirchhof G, et al. Role of mincle in alveolar macrophage-dependent innate immunity against mycobacterial infections in mice. J Immunol (2012) 189:3121–9. doi:10.4049/jimmunol.1201399
35. Heitmann L, Schoenen H, Ehlers S, Lang R, Holscher C. Mincle is not essential for controlling Mycobacterium tuberculosis infection. Immunobiology (2013) 218:506–16. doi:10.1016/j.imbio.2012.06.005
36. Lee WB, Kang JS, Yan JJ, Lee MS, Jeon BY, Cho SN, et al. Neutrophils promote mycobacterial trehalose dimycolate-induced lung inflammation via the mincle pathway. PLoS Pathog (2012) 8:e1002614. doi:10.1371/journal.ppat.1002614
37. Wilson GJ, Marakalala MJ, Hoving JC, van Laarhoven A, Drummond RA, Kerscher B, et al. The C-type lectin receptor CLECSF8/CLEC4D is a key component of anti-mycobacterial immunity. Cell Host Microbe (2015) 17:252–9. doi:10.1016/j.chom.2015.01.004
38. Marakalala MJ, Graham LM, Brown GD. The role of Syk/CARD9-coupled C-type lectin receptors in immunity to Mycobacterium tuberculosis infections. Clin Dev Immunol (2010) 2010:567571. doi:10.1155/2010/567571
39. Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, de Jong DJ, Langenberg DM, et al. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog (2005) 1:279–85. doi:10.1371/journal.ppat.0010034
40. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe (2015) 17:799–810. doi:10.1016/j.chom.2015.05.003
41. Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe (2012) 11:469–80. doi:10.1016/j.chom.2012.03.007
42. Shah S, Bohsali A, Ahlbrand SE, Srinivasan L, Rathinam VA, Vogel SN, et al. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-beta and AIM2 inflammasome-dependent IL-1beta production via its ESX-1 secretion system. J Immunol (2013) 191:3514–8. doi:10.4049/jimmunol.1301331
43. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol (2010) 12:1046–63. doi:10.1111/j.1462-5822.2010.01450.x
44. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-{gamma}: an overview of signals, mechanisms, and functions. J Leukoc Biol (2003) 75:163–89. doi:10.1189/jlb.0603252
45. Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol (2014) 26:454–70. doi:10.1016/j.smim.2014.09.008
46. Abebe F, Mustafa T, Nerland AH, Bjune GA. Cytokine profile during latent and slowly progressive primary tuberculosis: a possible role for interleukin-15 in mediating clinical disease. Clin Exp Immunol (2006) 143:180–92. doi:10.1111/j.1365-2249.2005.02976.x
47. Doherty M, Wallis RS, Zumla A; WHO-Tropical Disease Research/European Commission Joint Expert Consultation Group. Biomarkers for tuberculosis disease status and diagnosis. Curr Opin Pulm Med (2009) 15:181–7. doi:10.1097/MCP.0b013e328326f42c
48. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T cell-derived IFN-gamma plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog (2016) 12:e1005667. doi:10.1371/journal.ppat.1005667
49. Weiner J III, Kaufmann SH. Recent advances towards tuberculosis control: vaccines and biomarkers. J Intern Med (2014) 275:467–80. doi:10.1111/joim.12212
50. Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4(+) T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol (2007) 8:369–77. doi:10.1038/ni1449
51. Desel C, Werninghaus K, Ritter M, Jozefowski K, Wenzel J, Russkamp N, et al. The Mincle-activating adjuvant TDB induces MyD88-dependent Th1 and Th17 responses through IL-1R signaling. PLoS One (2013) 8:e53531. doi:10.1371/journal.pone.0053531
52. Aguilo N, Alvarez-Arguedas S, Uranga S, Marinova D, Monzon M, Badiola J, et al. Pulmonary but not subcutaneous delivery of BCG vaccine confers protection to tuberculosis-susceptible mice by an interleukin 17-dependent mechanism. J Infect Dis (2016) 213:831–9. doi:10.1093/infdis/jiv503
53. Agger EM, Rosenkrands I, Olsen AW, Hatch G, Williams A, Kritsch C, et al. Protective immunity to tuberculosis with Ag85B-ESAT-6 in a synthetic cationic adjuvant system IC31. Vaccine (2006) 24:5452–60. doi:10.1016/j.vaccine.2006.03.072
54. van Dissel JT, Arend SM, Prins C, Bang P, Tingskov PN, Lingnau K, et al. Ag85B-ESAT-6 adjuvanted with IC31 promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in naive human volunteers. Vaccine (2010) 28:3571–81. doi:10.1016/j.vaccine.2010.02.094
55. van Dissel JT, Soonawala D, Joosten SA, Prins C, Arend SM, Bang P, et al. Ag85B-ESAT-6 adjuvanted with IC31(R) promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in volunteers with previous BCG vaccination or tuberculosis infection. Vaccine (2011) 29:2100–9. doi:10.1016/j.vaccine.2010.12.135
56. LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol (2007) 8:630–8. doi:10.1038/ni1460
57. Werninghaus K, Babiak A, Gross O, Holscher C, Dietrich H, Agger EM, et al. Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRgamma-Syk-Card9-dependent innate immune activation. J Exp Med (2009) 206:89–97. doi:10.1084/jem.20081445
58. Agger EM, Rosenkrands I, Hansen J, Brahimi K, Vandahl BS, Aagaard C, et al. Cationic liposomes formulated with synthetic mycobacterial cordfactor (CAF01): a versatile adjuvant for vaccines with different immunological requirements. PLoS One (2008) 3:e3116. doi:10.1371/journal.pone.0003116
59. Lindenstrom T, Agger EM, Korsholm KS, Darrah PA, Aagaard C, Seder RA, et al. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J Immunol (2009) 182:8047–55. doi:10.4049/jimmunol.0801592
60. Lindenstrom T, Woodworth J, Dietrich J, Aagaard C, Andersen P, Agger EM. Vaccine-induced th17 cells are maintained long-term postvaccination as a distinct and phenotypically stable memory subset. Infect Immun (2012) 80:3533–44. doi:10.1128/IAI.00550-12
61. van Dissel JT, Joosten SA, Hoff ST, Soonawala D, Prins C, Hokey DA, et al. A novel liposomal adjuvant system, CAF01, promotes long-lived Mycobacterium tuberculosis-specific T-cell responses in human. Vaccine (2014) 32:7098–107. doi:10.1016/j.vaccine.2014.10.036
62. Babu S, Nutman TB. Helminth-tuberculosis co-infection: an immunologic perspective. Trends Immunol (2016) 37:597–607. doi:10.1016/j.it.2016.07.005
63. Roy A, Eisenhut M, Harris RJ, Rodrigues LC, Sridhar S, Habermann S, et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: systematic review and meta-analysis. BMJ (2014) 349:g4643. doi:10.1136/bmj.g4643
64. Colditz GA, Brewer TF, Berkey CS, Wilson ME, Burdick E, Fineberg HV, et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA (1994) 271:698–702. doi:10.1001/jama.1994.03510330076038
65. Elias D, Wolday D, Akuffo H, Petros B, Bronner U, Britton S. Effect of deworming on human T cell responses to mycobacterial antigens in helminth-exposed individuals before and after bacille Calmette-Guerin (BCG) vaccination. Clin Exp Immunol (2001) 123:219–25. doi:10.1046/j.1365-2249.2001.01446.x
66. Elias D, Akuffo H, Pawlowski A, Haile M, Schon T, Britton S. Schistosoma mansoni infection reduces the protective efficacy of BCG vaccination against virulent Mycobacterium tuberculosis. Vaccine (2005) 23:1326–34. doi:10.1016/j.vaccine.2004.09.038
67. Malhotra I, Mungai P, Wamachi A, Kioko J, Ouma JH, Kazura JW, et al. Helminth- and Bacillus Calmette-Guerin-induced immunity in children sensitized in utero to filariasis and schistosomiasis. J Immunol (1999) 162:6843–8.
68. Elliott AM, Namujju PB, Mawa PA, Quigley MA, Nampijja M, Nkurunziza PM, et al. A randomised controlled trial of the effects of albendazole in pregnancy on maternal responses to mycobacterial antigens and infant responses to Bacille Calmette-Guerin (BCG) immunisation [ISRCTN32849447]. BMC Infect Dis (2005) 5:115. doi:10.1186/1471-2334-5-115
69. Webb EL, Mawa PA, Ndibazza J, Kizito D, Namatovu A, Kyosiimire-Lugemwa J, et al. Effect of single-dose anthelmintic treatment during pregnancy on an infant’s response to immunisation and on susceptibility to infectious diseases in infancy: a randomised, double-blind, placebo-controlled trial. Lancet (2011) 377:52–62. doi:10.1016/S0140-6736(10)61457-2
70. Elias D, Mengistu G, Akuffo H, Britton S. Are intestinal helminths risk factors for developing active tuberculosis? Trop Med Int Health (2006) 11:551–8. doi:10.1111/j.1365-3156.2006.01578.x
71. Tristao-Sa R, Ribeiro-Rodrigues R, Johnson LT, Pereira FE, Dietze R. Intestinal nematodes and pulmonary tuberculosis. Rev Soc Bras Med Trop (2002) 35:533–5. doi:10.1590/S0037-86822002000500020
72. Resende Co T, Hirsch CS, Toossi Z, Dietze R, Ribeiro-Rodrigues R. Intestinal helminth co-infection has a negative impact on both anti-Mycobacterium tuberculosis immunity and clinical response to tuberculosis therapy. Clin Exp Immunol (2007) 147:45–52. doi:10.1111/j.1365-2249.2006.03247.x
73. Abate E, Belayneh M, Idh J, Diro E, Elias D, Britton S, et al. Asymptomatic helminth infection in active tuberculosis is associated with increased regulatory and Th-2 responses and a lower sputum smear positivity. PLoS Negl Trop Dis (2015) 9:e0003994. doi:10.1371/journal.pntd.0003994
74. Abate E, Elias D, Getachew A, Alemu S, Diro E, Britton S, et al. Effects of albendazole on the clinical outcome and immunological responses in helminth co-infected tuberculosis patients: a double blind randomised clinical trial. Int J Parasitol (2015) 45:133–40. doi:10.1016/j.ijpara.2014.09.006
75. Goodridge HS, Wilson EH, Harnett W, Campbell CC, Harnett MM, Liew FY. Modulation of macrophage cytokine production by ES-62, a secreted product of the filarial nematode Acanthocheilonema viteae. J Immunol (2001) 167:940–5. doi:10.4049/jimmunol.167.2.940
76. Klotz C, Ziegler T, Figueiredo AS, Rausch S, Hepworth MR, Obsivac N, et al. A helminth immunomodulator exploits host signaling events to regulate cytokine production in macrophages. PLoS Pathog (2011) 7:e1001248. doi:10.1371/journal.ppat.1001248
77. O’Regan NL, Steinfelder S, Venugopal G, Rao GB, Lucius R, Srikantam A, et al. Brugia malayi microfilariae induce a regulatory monocyte/macrophage phenotype that suppresses innate and adaptive immune responses. PLoS Negl Trop Dis (2014) 8:e3206. doi:10.1371/journal.pntd.0003206
78. Martin I, Caban-Hernandez K, Figueroa-Santiago O, Espino AM. Fasciola hepatica fatty acid binding protein inhibits TLR4 activation and suppresses the inflammatory cytokines induced by lipopolysaccharide in vitro and in vivo. J Immunol (2015) 194:3924–36. doi:10.4049/jimmunol.1401182
79. Dirgahayu P, Fukumoto S, Miura K, Hirai K. Excretory/secretory products from plerocercoids of Spirometra erinaceieuropaei suppress the TNF-alpha gene expression by reducing phosphorylation of ERK1/2 and p38 MAPK in macrophages. Int J Parasitol (2002) 32:1155–62. doi:10.1016/S0020-7519(02)00090-5
80. Johnston MJ, Wang A, Catarino ME, Ball L, Phan VC, MacDonald JA, et al. Extracts of the rat tapeworm, Hymenolepis diminuta, suppress macrophage activation in vitro and alleviate chemically induced colitis in mice. Infect Immun (2010) 78:1364–75. doi:10.1128/IAI.01349-08
81. Aira N, Andersson AM, Singh SK, McKay DM, Blomgran R. Species dependent impact of helminth-derived antigens on human macrophages infected with Mycobacterium tuberculosis: direct effect on the innate anti-mycobacterial response. PLoS Negl Trop Dis (2017) 11:e0005390. doi:10.1371/journal.pntd.0005390
82. Ritter M, Gross O, Kays S, Ruland J, Nimmerjahn F, Saijo S, et al. Schistosoma mansoni triggers dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc Natl Acad Sci U S A (2010) 107:20459–64. doi:10.1073/pnas.1010337107
83. Babu S, Bhat SQ, Kumar NP, Jayantasri S, Rukmani S, Kumaran P, et al. Human type 1 and 17 responses in latent tuberculosis are modulated by coincident filarial infection through cytotoxic T lymphocyte antigen-4 and programmed death-1. J Infect Dis (2009) 200:288–98. doi:10.1086/599797
84. George PJ, Anuradha R, Kumar NP, Sridhar R, Banurekha VV, Nutman TB, et al. Helminth infections coincident with active pulmonary tuberculosis inhibit mono- and multifunctional CD4+ and CD8+ T cell responses in a process dependent on IL-10. PLoS Pathog (2014) 10:e1004375. doi:10.1371/journal.ppat.1004375
85. George PJ, Anuradha R, Kumaran PP, Chandrasekaran V, Nutman TB, Babu S. Modulation of mycobacterial-specific Th1 and Th17 cells in latent tuberculosis by coincident hookworm infection. J Immunol (2013) 190:5161–8. doi:10.4049/jimmunol.1203311
86. Toulza F, Tsang L, Ottenhoff TH, Brown M, Dockrell HM. Mycobacterium tuberculosis-specific CD4+ T-cell response is increased, and Treg cells decreased, in anthelmintic-treated patients with latent TB. Eur J Immunol (2016) 46:752–61. doi:10.1002/eji.201545843
87. DiNardo AR, Mace EM, Lesteberg K, Cirillo JD, Mandalakas AM, Graviss EA, et al. Schistosome soluble egg antigen decreases Mycobacterium tuberculosis-specific CD4+ T-cell effector function with concomitant arrest of macrophage phago-lysosome maturation. J Infect Dis (2016) 214:479–88. doi:10.1093/infdis/jiw156
88. Anuradha R, Munisankar S, Bhootra Y, Dolla C, Kumaran P, Nutman TB, et al. Anthelmintic therapy modifies the systemic and mycobacterial antigen-stimulated cytokine profile in helminth-latent Mycobacterium tuberculosis coinfection. Infect Immun (2017) 85(4):e973–916. doi:10.1128/IAI.00973-16
89. Elias D, Akuffo H, Thors C, Pawlowski A, Britton S. Low dose chronic Schistosoma mansoni infection increases susceptibility to Mycobacterium bovis BCG infection in mice. Clin Exp Immunol (2005) 139:398–404. doi:10.1111/j.1365-2249.2004.02719.x
90. Frantz FG, Rosada RS, Peres-Buzalaf C, Perusso FR, Rodrigues V, Ramos SG, et al. Helminth coinfection does not affect therapeutic effect of a DNA vaccine in mice harboring tuberculosis. PLoS Negl Trop Dis (2010) 4:e700. doi:10.1371/journal.pntd.0000700
91. Potian JA, Rafi W, Bhatt K, McBride A, Gause WC, Salgame P. Preexisting helminth infection induces inhibition of innate pulmonary anti-tuberculosis defense by engaging the IL-4 receptor pathway. J Exp Med (2011) 208:1863–74. doi:10.1084/jem.20091473
92. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol (2008) 9:1399–406. doi:10.1038/ni.1671
93. Monin L, Griffiths KL, Lam WY, Gopal R, Kang DD, Ahmed M, et al. Helminth-induced arginase-1 exacerbates lung inflammation and disease severity in tuberculosis. J Clin Invest (2015) 125:4699–713. doi:10.1172/JCI77378
94. du Plessis N, Kleynhans L, Thiart L, van Helden PD, Brombacher F, Horsnell WG, et al. Acute helminth infection enhances early macrophage mediated control of mycobacterial infection. Mucosal Immunol (2013) 6:931–41. doi:10.1038/mi.2012.131
95. Babu S, Bhat SQ, Kumar NP, Anuradha R, Kumaran P, Gopi PG, et al. Attenuation of toll-like receptor expression and function in latent tuberculosis by coexistent filarial infection with restoration following antifilarial chemotherapy. PLoS Negl Trop Dis (2009) 3:e489. doi:10.1371/journal.pntd.0000489
96. Babu S, Blauvelt CP, Kumaraswami V, Nutman TB. Diminished expression and function of TLR in lymphatic filariasis: a novel mechanism of immune dysregulation. J Immunol (2005) 175:1170–6. doi:10.4049/jimmunol.175.2.1170
97. Schoenen H, Huber A, Sonda N, Zimmermann S, Jantsch J, Lepenies B, et al. Differential control of Mincle-dependent cord factor recognition and macrophage responses by the transcription factors C/EBPbeta and HIF1alpha. J Immunol (2014) 193:3664–75. doi:10.4049/jimmunol.1301593
98. Matsumoto M, Tanaka T, Kaisho T, Sanjo H, Copeland NG, Gilbert DJ, et al. A novel LPS-inducible C-type lectin is a transcriptional target of NF-IL6 in macrophages. J Immunol (1999) 163:5039–48.
99. Ostrop J, Jozefowski K, Zimmermann S, Hofmann K, Strasser E, Lepenies B, et al. Contribution of MINCLE-SYK signaling to activation of primary human APCs by mycobacterial cord factor and the novel adjuvant TDB. J Immunol (2015) 195:2417–28. doi:10.4049/jimmunol.1500102
100. Hupfer T, Schick J, Jozefowski K, Voehringer D, Ostrop J, Lang R. Stat6-dependent inhibition of Mincle expression in mouse and human antigen-presenting cells by the Th2 cytokine IL-4. Front Immunol (2016) 7:423. doi:10.3389/fimmu.2016.00423
101. Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol (2002) 169:2253–63. doi:10.4049/jimmunol.169.5.2253
102. O’Leary S, O’Sullivan MP, Keane J. IL-10 blocks phagosome maturation in Mycobacterium tuberculosis-infected human macrophages. Am J Respir Cell Mol Biol (2011) 45:172–80. doi:10.1165/rcmb.2010-0319OC
103. Redpath S, Ghazal P, Gascoigne NR. Hijacking and exploitation of IL-10 by intracellular pathogens. Trends Microbiol (2001) 9:86–92. doi:10.1016/S0966-842X(00)01919-3
104. Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J Immunol (2002) 168:3402–11. doi:10.4049/jimmunol.168.7.3402
105. Schreiber T, Ehlers S, Heitmann L, Rausch A, Mages J, Murray PJ, et al. Autocrine IL-10 induces hallmarks of alternative activation in macrophages and suppresses antituberculosis effector mechanisms without compromising T cell immunity. J Immunol (2009) 183:1301–12. doi:10.4049/jimmunol.0803567
106. Redford PS, Boonstra A, Read S, Pitt J, Graham C, Stavropoulos E, et al. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur J Immunol (2010) 40:2200–10. doi:10.1002/eji.201040433
107. Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science (2004) 304:1014–8. doi:10.1126/science.1096158
108. Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol (2006) 7:1029–35. doi:10.1038/ni1006-1029
109. Khan NS, Kasperkovitz PV, Timmons AK, Mansour MK, Tam JM, Seward MW, et al. Dectin-1 controls TLR9 trafficking to phagosomes containing beta-1,3 glucan. J Immunol (2016) 196:2249–61. doi:10.4049/jimmunol.1401545
110. Mansour MK, Tam JM, Khan NS, Seward M, Davids PJ, Puranam S, et al. Dectin-1 activation controls maturation of beta-1,3-glucan-containing phagosomes. J Biol Chem (2013) 288:16043–54. doi:10.1074/jbc.M113.473223
111. Patin EC, Willcocks S, Orr S, Ward TH, Lang R, Schaible UE. Mincle-mediated anti-inflammatory IL-10 response counter-regulates IL-12 in vitro. Innate Immun (2016) 22(3):181–5. doi:10.1177/1753425916636671
112. Segura M, Su Z, Piccirillo C, Stevenson MM. Impairment of dendritic cell function by excretory-secretory products: a potential mechanism for nematode-induced immunosuppression. Eur J Immunol (2007) 37:1887–904. doi:10.1002/eji.200636553
113. Everts B, Hussaarts L, Driessen NN, Meevissen MH, Schramm G, van der Ham AJ, et al. Schistosome-derived omega-1 drives Th2 polarization by suppressing protein synthesis following internalization by the mannose receptor. J Exp Med (2012) 209(1753–1767):S1751. doi:10.1084/jem.20111381
114. Walsh KP, Brady MT, Finlay CM, Boon L, Mills KH. Infection with a helminth parasite attenuates autoimmunity through TGF-beta-mediated suppression of Th17 and Th1 responses. J Immunol (2009) 183:1577–86. doi:10.4049/jimmunol.0803803
115. Grainger JR, Smith KA, Hewitson JP, McSorley HJ, Harcus Y, Filbey KJ, et al. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J Exp Med (2010) 207:2331–41. doi:10.1084/jem.20101074
116. Hartmann W, Haben I, Fleischer B, Breloer M. Pathogenic nematodes suppress humoral responses to third-party antigens in vivo by IL-10-mediated interference with Th cell function. J Immunol (2011) 187:4088–99. doi:10.4049/jimmunol.1004136
117. Elias D, Britton S, Aseffa A, Engers H, Akuffo H. Poor immunogenicity of BCG in helminth infected population is associated with increased in vitro TGF-beta production. Vaccine (2008) 26:3897–902. doi:10.1016/j.vaccine.2008.04.083
Keywords: helminth infection, tuberculosis, vaccination, type 2 immune response, macrophage, IL-4, IL-10
Citation: Lang R and Schick J (2017) Review: Impact of Helminth Infection on Antimycobacterial Immunity—A Focus on the Macrophage. Front. Immunol. 8:1864. doi: 10.3389/fimmu.2017.01864
Received: 11 August 2017; Accepted: 08 December 2017;
Published: 22 December 2017
Edited by:
Ulrich Emil Schaible, Forschungszentrum Borstel (LG), GermanyReviewed by:
Subash Babu, International Centers for Excellence in Research (NIH), IndiaCopyright: © 2017 Lang and Schick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roland Lang, cm9sYW5kLmxhbmdAdWstZXJsYW5nZW4uZGU=
ORCID ID: orcid.org/0000-0003-0502-3677
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.