 Nader Rahimi
Nader Rahimi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 December 2017
Sec. Alloimmunity and Transplantation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01847
This article is part of the Research Topic Endothelial Dysfunction during Inflammation and Alloimmunity View all 11 articles
Regulated vascular permeability is an essential feature of normal physiology and its dysfunction is associated with major human diseases ranging from cancer to inflammation and ischemic heart diseases. Integrity of endothelial cells also play a prominent role in the outcome of surgical procedures and organ transplant. Endothelial barrier function and integrity are regulated by a plethora of highly specialized transmembrane receptors, including claudin family proteins, occludin, junctional adhesion molecules (JAMs), vascular endothelial (VE)-cadherin, and the newly identified immunoglobulin (Ig) and proline-rich receptor-1 (IGPR-1) through various distinct mechanisms and signaling. On the other hand, vascular endothelial growth factor (VEGF) and its tyrosine kinase receptor, VEGF receptor-2, play a central role in the destabilization of endothelial barrier function. While claudins and occludin regulate cell–cell junction via recruitment of zonula occludens (ZO), cadherins via catenin proteins, and JAMs via ZO and afadin, IGPR-1 recruits bullous pemphigoid antigen 1 [also called dystonin (DST) and SH3 protein interacting with Nck90/WISH (SH3 protein interacting with Nck)]. Endothelial barrier function is moderated by the function of transmembrane receptors and signaling events that act to defend or destabilize it. Here, I highlight recent advances that have provided new insights into endothelial barrier function and mechanisms involved. Further investigation of these mechanisms could lead to the discovery of novel therapeutic targets for human diseases associated with endothelial dysfunction.
To live and reproduce, all vertebrate animals are evolved to have a circulatory system (i.e., heart, veins, and arteries) that safeguards an uninterrupted supply of blood and oxygen to all tissues, followed by the return of the deoxygenated blood to the lungs for re-oxygenation. In addition to its emissary function, the vascular system also plays an indispensable role in hemostasis, immune surveillance, angiogenesis, and vascular permeability (1). Although they differ in function and morphologies, endothelial cells are the main constituents of blood vessels. In some organs such as the brain, endothelial cells form a strong and highly selective blood–brain barrier, but in other organs such as the kidney and pancreas, endothelial cells display selective permeability by forming highly specialized holes on the plasma membrane called fenestrae, which allows rapid exchange of solute and molecules such as hormones.
To maintain the structural and functional integrity that retains the highly dynamic barrier function of blood vessels, which permits continuous leakage of solutes and small molecules but limits extravasation of larger molecules and cells, metazoan cells are evolved to form highly specialized cell–cell junctions such as desmosomes, adherent junctions, and gap junctions. Not only do these junctions glue cells together, they also generate intracellular signaling and permit junctional remodeling in response to various external and internal cues (2). Curiously, certain viruses employ cell adhesion receptors for their entry into human cells. For example, hepatitis C virus (HCV) uses occludin and claudins to enter liver cells. Coxsackievirus and adenovirus use junctional adhesion molecule (JAM)/coxsackie and adenovirus receptor (CAR) and reoviruses uses JAM-A for their entry into cells [for review see Ref. (3)]. In many human diseases, such as cancer, diabetes, age-related macular degeneration, and chronic inflammatory conditions, this core barrier function of endothelial cells breaks down, leading to the leakage of larger molecules and blood with serious life-threatening consequences. Blood vessel leakiness also is associated with tumor-induced angiogenesis and represents a significant challenge for an effective delivery of anti-cancer drugs to the site of tumors as tumor-associated blood vessels are structurally fragile and hyperpermeable (4).
In addition to their pivotal roles in angiogenesis and inflammation, endothelial cells also play important functions in various other conditions such as surgical trauma, ischemia–reperfusion, alloimmune responses, and chemotherapy and immunosuppressant treatments (5, 6). Activated endothelial cells often upregulate expression of various growth factors, cytokines and chemokines that stimulate endothelial cell proliferation, permeability, and migration (7, 8). Furthermore, they upregulate thrombogenic molecules and specific adhesion molecules that promote thrombosis and immune cell activation (Figure 1). Endothelial cells also respond to immunosuppressant and chemotherapeutic drugs. Although the cardiotoxic effects of conventional chemotherapeutic agents are well-documented, the targeted therapeutic drugs such as the antiangiogenic are also associated with endothelial dysfunction, such as hypertension, thromboembolism, myocardial infarction, and proteinuria (9, 10). In organ transplantation, the host immune system is brought into direct contact with the endothelial cell lining of graft vessels, where the graft endothelial cells play a major role in allograft vasculopathy (i.e., allograft rejection) and in the overall long-term survival after any organ transplantation (11, 12).
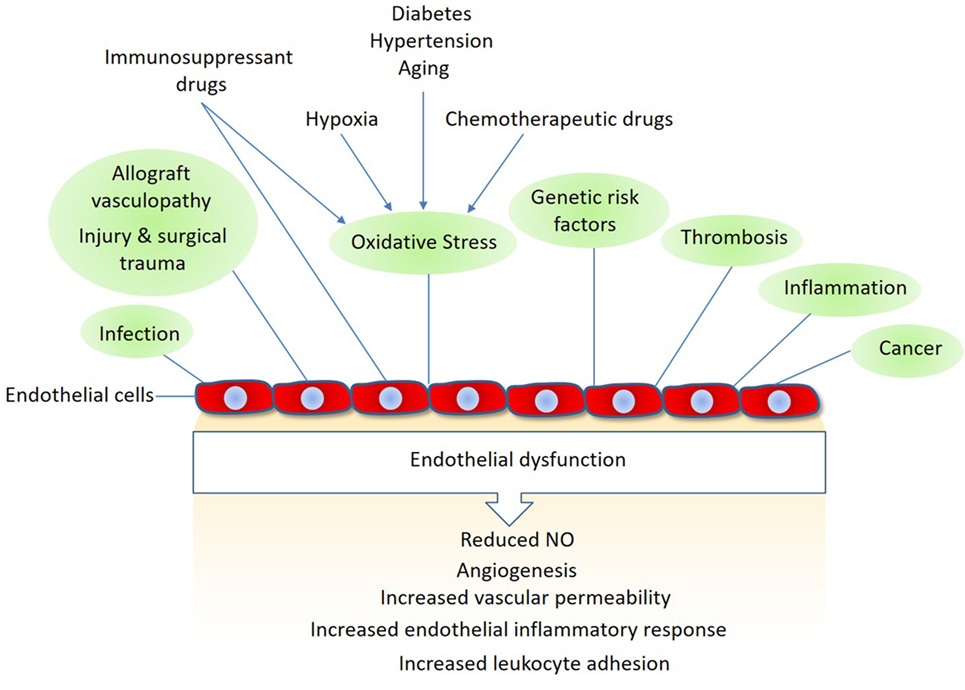
Figure 1. Mechanisms linked to endothelial dysfunction: several key mechanisms that promote endothelial dysfunction and vascular damage are shown. Also, shown are the major endothelial responses triggered by these factors Nitric oxide (NO).
Adherens junctions, gap junctions, and desmosomes are principal cell–cell junctions that provide structural integrity and create highly polarized barriers with selective paracellular permeability to solutes, macromolecules, and other cells, which is an essential element of homeostatic maintenance in endothelial and epithelial cells. Tight junctions, in particular, control monolayer permeability and play a significant role in endothelial cells that maintain rigorous barriers, whereas adherens junctions partake in multiple roles, such as establishment and maintenance of cell–cell adhesion, actin cytoskeleton remodeling, signal transduction, and transcriptional regulation. However, unlike epithelial cells, adherens and tight junctions in endothelial cells are highly interconnected. In addition to their cardinal role in the regulation of homeostatic maintenance and barrier function, proteins involved in the regulation of cell–cell junctions play major role in cellular differentiation, proliferation, migration, signal transduction, and gene expression (13, 14). Altered cell–cell junctions are also associated with the pathogenesis of various diseases, including cancers, diabetic retinopathy, and inflammation (15, 16). A plethora of cell surface receptors including claudins family proteins, occludin, JAMs, vascular endothelial (VE)-cadherin, and the recently identified immunoglobulin (Ig) and proline-rich receptor-1 (IGPR-1) are involved in cell–cell junction signaling through various means and mechanisms. While occludin through its cytoplasmic coiled-coil (CC) domain interacts ZO proteins, claudins family proteins and JAMs through their PDZ-binding motif interact with PDZ-containing proteins such as ZO. JAMs also interact with PAR3, PAR6 and AF6, which are also PDZ-containing proteins. On the other hand, VE-cadherin through its armadillo-binding domain recruits p120, catenin proteins, whereas IGPR-1 through its proline-rich motif interacts with (BPAG1 or BP230), also called dystonin (DST) and SH3 protein interacting with Nck90 (SPIN90)/WISH (SH3 protein interacting with Nck), also called NCK-interacting protein with SH3 domain (NCKIPSD) (Figure 2). Regardless of their mechanisms of recruitment of cytoplasmic-binding partners, it is clear that these receptor-interacting proteins transduce signals that are required for cell–cell junction assembly, cell morphology, and barrier function. In a way, these transmembrane receptors along with their intracellular-binding partners are the defenders of endothelial integrity and barrier function.
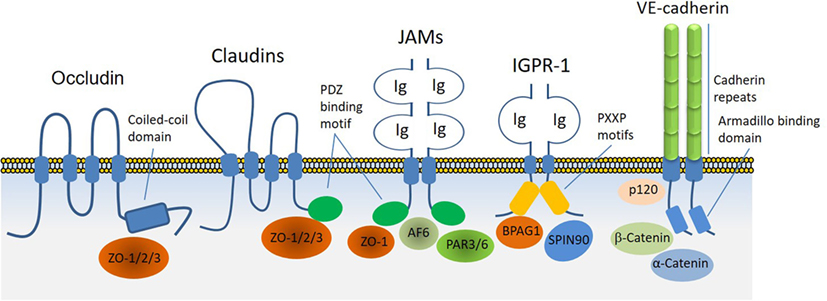
Figure 2. Transmembrane receptors involved in the endothelial cell–cell junction and their key cytoplasmic-binding partners. Claudin family proteins, occludin, junctional adhesion molecules (JAMs), vascular endothelial (VE)-cadherin an immunoglobulin (Ig) and proline-rich receptor-1 (IGPR-1) are major receptors in endothelial cells that regulate endothelial cell–cell junctions and barrier. The core mechanism associated with the function of these receptors involves with their ability to recruit specific signaling proteins that signal to strength the cell–cell junctions. Zonula occludens (ZO 1–3), catenin proteins (α-catenin, β-catenin, and γ-catenin), polarity protein-3 and 6 (PAR3/6), afadin (AF6), bullous pemphigoid antigen 1 [BPAG1 also called dystonin and SH3 protein interacting with Nck90 (SPIN90)/WISH (SH3 protein interacting with Nck)] are key substrates involved with these receptors.
Claudin family proteins are four-transmembrane type proteins and there are at least 24 claudins present in human genome (17, 18), which represents the largest family of tight junction proteins. By forming homophilic- and heterophilic-trans/cis dimerization, claudins determine the barrier properties and cell–cell interactions (19, 20). Claudins, with the exception of claudin-12, contain a PDZ-binding domain at C-terminal tail that allows them to interact with PDZ-containing scaffold proteins such as zonula occludens (ZO) (19). It appears that unlike cadherins, claudins mediate cell–cell adhesion through a calcium-independent manner (20). Claudins are expressed in both endothelial and epithelial cells, though with some degree of cell type specificity. Claudin-3, claudin-5, and claudin-12 are predominantly expressed in brain endothelial cells (21, 22), whereas renal endothelial cells express claudin-5 and claudin-15 (21, 23). Some claudins such as claudin-2 and claudin-16 specifically control paracellular ionic selectivity by forming ion channels (24, 25), while others such as claudin-8 is proposed to control paracellular Na+ permeability (26). In addition to their canonical function, some claudins also interact with other proteins. For example, claudin-1 acts as a receptor for HCV (27) and for dengue virus (28).
Similar to claudins, occludin is a four-transmembrane protein and one of the key components of tight junctions that plays a critical role in the regulation of trans-epithelial/endothelial electrical resistance (29, 30) and actin assembly (31). While the N-terminal extracellular domain is involved in the adhesive function of occludin, its C-terminal is subject to phosphorylation at several tyrosine and serine/threonine residues through multiple kinases and is also involved in the recruitment of SH3 and PDZ-containing zonula occluden (ZO) proteins, which anchor occludin to the actin fibril assembly (Figure 3). In endothelial cells, it regulate tight junction barriers in response to IFNγ and vascular endothelial growth factor (VEGF) (32–34). The barrier function of occludin is regulated by the phosphorylation of key residues at the cytoplasmic domain (19, 35). For example, phosphorylation of Ser490 was proposed to promote ubiquitination of occludin, which promotes its downregulation (36), whereas phosphorylation of occludin at different sites is associated with its barrier function (37–39). The key kinases involved in the phosphorylation of occludin are shown (Figure 3). The interaction of occludin with tight junction proteins such as ZO family proteins is also affected by phosphorylation at its CC domain (40, 41). Overall, phosphorylation of the C-terminal of occludin at serine/threonine and tyrosine sites by various kinases (Figure 3) and dimerization (not shown) of occludin appear to be key mechanisms that govern occludin function (42).
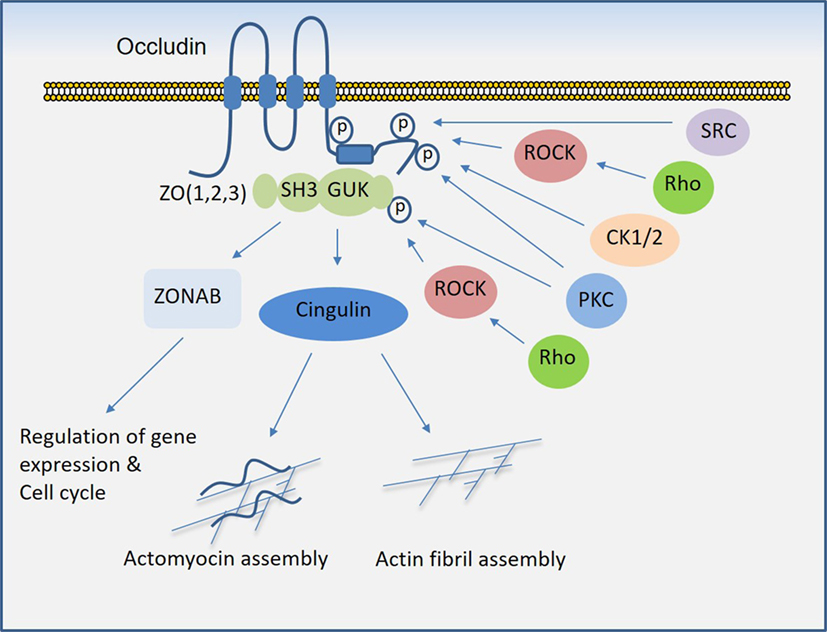
Figure 3. Regulation of occludin mediated cell–cell junction assembly and organization. Occludin via its coiled-coil domain recognizes guanylate kinase (GUK) domain, PDZ, and SH3 domain (43), that further recruits Cingulin and ZONAB that participate in the formation and regulation of the tight junction and paracellular permeability barrier. C-terminal of occludin is subject to phosphorylation at serine/threonine and tyrosine residues by multiple serine/threonine kinases and tyrosine kinases, which in part regulate occludin binding with ZO proteins and its tight junctional function (see the text).
Junctional adhesion molecules are distinct and important cell surface proteins that are involved in the regulation of cell–cell adhesion and barrier. JAMs belong to the Ig superfamily proteins and contain two extracellular Ig-like domains, a single transmembrane domain and a C-terminal cytoplasmic domain (44). The cytoplasmic domain of JAMs contains a PDZ domain, which recruits PDZ-binding proteins such ZO and afadin that connects JAM proteins to actin assembly and regulation of epithelial and endothelial barrier function (44–46). JAM-A regulates the barrier function of tight junctions in both endothelial and epithelial cells (47) and is involved in the migration of endothelial cells (48). JAM-C was proposed to be involved in tumor angiogenesis (49). Furthermore, other JAMs such as CAR and endothelial cell-selective adhesion molecule are also expressed in endothelial cells and are involved in the regulation of permeability, angiogenesis, and cell migration (50, 51).
The control of the endothelial barrier function is largely mediated by cell-to-cell junctions, which include adherens and tight junctions. CAMs are the key mediators of endothelial barrier function. CAMs mediate cell–cell and cell–matrix adhesion and transmit signals across the plasma membrane to process information from the extracellular environment involved in tissue morphogenesis, angiogenesis, and tumor progression (52, 53).
Various proteins and molecules could destabilize endothelial barrier function and stimulate vascular permeability. Proteins and molecules such as Ang2, chemokines, and IL-8 (interleukin-8), bradykinin, histamine, thrombin, fibrinogen, tumor necrosis factor-α (TNF-α), and endotoxins such LPS could destabilize endothelial barrier. However, VEGF, also called vascular permeability factor, is perhaps the most potent factor involved in the disruption of endothelial barrier function and induction of vascular permeability in pathological conditions (16, 54–56). In tumorigenesis, not only does VEGF induce angiogenesis but also mediates disruption of the vascular barrier, resulting in the leaky vessels leading to an increase in tumor cell extravasation and reduced drug delivery to tumor site which is associated with the development of drug resistance and inefficacy (57). Similarly, VEGF causes vascular permeability and edema in various other diseases such as diabetic retinopathy, age-related macular degeneration, and inflammation (58–61). One of VEGF’s receptor, VEGF receptor-2 (VEGFR-2), predominantly mediates VEGF-mediated destabilization of endothelial junctions (16, 62). Upon activation by VEGF, VEGFR-2 undergoes various posttranslational modifications including phosphorylation and methylation, which stimulate its activation and recruitment of signaling proteins to the receptor (63–65). Activation of VEGFR-2 by VEGF stimulates diverse signaling events that affect endothelial cell migration, proliferation, tube formation, and regulation of endothelial junctions. However, the activation of Src family kinases, phosphoinositide 3-kinase (66, 67), and phospholipase Cγ1 (PLCγ1) in particular play major roles in the induction of vascular permeability (16, 67) (Figure 4). In addition, VEGFR-2 can also stimulate permeability by directly targeting endothelial junctional proteins such as VE-cadherin and integrins (16, 62, 68, 69), providing an additional layer of complexity to VEGF-mediated destabilization of endothelial barrier function.
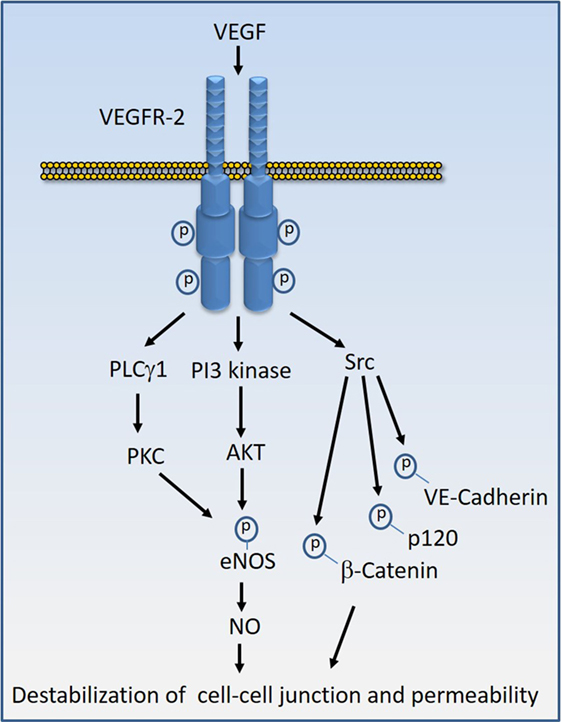
Figure 4. Vascular endothelial growth factor (VEGF)-induced VEGF receptor-2 (VEGFR-2) activation signal transduction that leads to destabilization of cell–cell junctions. Stimulation of VEGFR-2 by VEGF results in the kinase activation of VEGFR-2 and recruitment of diverse signaling proteins to VEGFR-2. The key VEGFR-2 signaling proteins whose activity are linked to vascular permeability include Src family kinases, phosphoinositide 3-kinase (PI3 kinase), and phospholipase Cγ1 (PLCγ1). Src kinases in turn can phosphorylate VE-cadherin and VE-cadherin-associated proteins such as β-catenin and p120 leading destabilization of VE-cadherin mediated endothelial barrier function. Activation of PI3 kinase and PLCγ1 can lead to phosphorylation of eNOS and production of nitric oxide (NO) that leads to interruption of endothelial junctions.
Vascular endothelial cadherin (VE-cadherin also called Cadherin-5 and CD144) is considered a main transmembrane component of endothelial adherens junction (70, 71). Similar to E-cadherin, VE-cadherin binds to members of the armadillo repeat family of proteins, p120-catenin, β-catenin, and plakoglobin through its cytoplasmic C-terminal (72). Inactivation of the VE-cadherin gene in both mouse and zebrafish clearly demonstrated its key role in vascular remodeling (73, 74). VE-cadherin plays an important role in controlling endothelial monolayer permeability and angiogenesis. VEGF-induced tyrosine phosphorylation of VE-cadherin at Y658 and Y731 by Src family kinases appear to play a prominent role in the destabilization of adherens junction and increased permeability of endothelial cells (75, 76). Consistent with the regulatory role of phosphorylation on VE-cadherin, other factors such as TNF-α that stimulate permeability also target VE-cadherin through tyrosine phosphorylation at Y658 and Y731 through proline-rich tyrosine kinase 2 and Rac1/Tiam1 (77). Phosphorylation of Y658 and Y731 disrupt VE-cadherin binding with VE-cadherin-associated proteins such as p120-catenin and β-catenin (78). Underscoring the role of phosphorylation in the regulation of VE-cadherin function, several protein tyrosine phosphatases are known to associate with and dephosphorylate VE-cadherin (79, 80). In view of the fundamental role of VEGF in angiogenesis and its robust action in the destabilization of endothelial barrier function in pathological conditions, VEGF system emerged as a major challenger and provocateur of the endothelial barrier function.
Immunoglobulin (Ig) and proline-rich receptor-1 is expressed in human endothelial and epithelial cells. Unlike the classical cadherins and tight junction proteins such as JAMs, claudins family proteins, and occludin, IGPR-1 expression is restricted to higher mammalians as it is not present in rodents such as mouse or rat (81). However, its closely related protein, transmembrane and immunoglobulin domain1 is expressed in the renal epithelial cells of human and rodents (82). IGPR-1 colocalizes with VE-cadherin in endothelial cells in cell culture and mediates endothelial cell–cell adhesion and its activity is required for angiogenesis in vitro and regulation of cell migration (81). Further studies revealed that it plays an important role in monolayer permeability (83). IGPR-1 is composed of three major domains: extracellular, transmembrane, and intracellular. The extracellular domain of IGPR-1 contains a single Ig domain followed by a single transmembrane domain and a proline-rich intracellular domain (81). The Ig-containing extracellular domain is required for IGPR-1 to mediate endothelial cell–cell interaction and barrier function (83). IGPR-1 is typically present as a disulfide bound cis-dimer, which further forms a trans-dimer complex in a cell density-dependent manner (83) (Figure 5A).
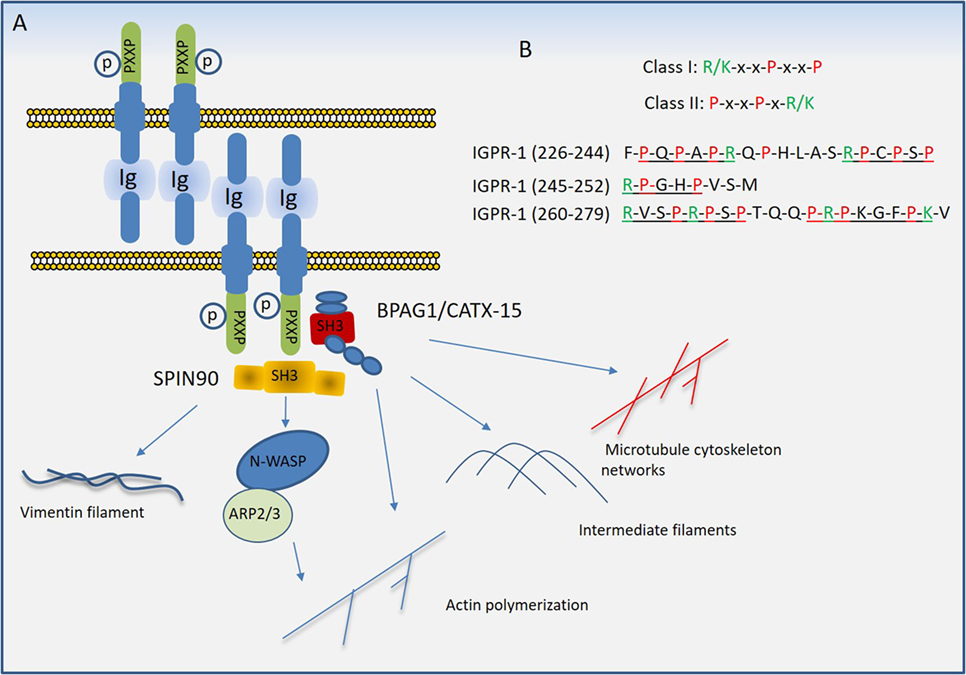
Figure 5. Proposed model of immunoglobulin (Ig) and proline-rich receptor-1 (IGPR-1) mediated regulation of endothelial barrier. (A) Trans-dimeric IGPR-1 undergoes serine phosphorylation at multiple sites. IGPR-1 through its proline-rich motifs recruits SH3 containing proteins, bullous pemphigoid antigen 1 (BPAG1), and SH3 protein interacting with Nck90 (SPIN90)/WISH (SH3 protein interacting with Nck). Interaction of IGPR-1 with BPAG1 and SPIN90 links IGPR-1 to actin fibril assembly, intermediate filament formation, microtubule cytoskeleton networks, and vimentin filament assembly (see text). (B) The conventional proline-rich motifs and the proline-rich motifs on the cytoplasmic domain of IGPR-1 are shown.
Proline rich sequences (PRDs) play a major role in mediating protein–protein interaction in prokaryotes and eukaryotes (84). PRDs are highly versatile and recognize different consensus motifs or canonical sequences in their protein ligands. A conspicuous feature of most binding motifs identified for PDRs is the presence of one or more proline residue that interact with the ligand, while residues that flank the core proline residue determines the selectivity (85). Although various protein domains are known to interact with proline-rich sequences (85), Src homology domain 3 (SH3) and WW domains are the most common domains that interact with PRDs (86). The PRDs of IGPR-1 interact with multiple SH3 domain-containing proteins including SPIN90/WISH (SH3 protein interacting with Nck) and bullous pemphigoid antigen 1 (BPAG1) (81). The cytoplasmic domain of IGPR-1 contains at least five PRDs (Figure 5B), which are variants of canonical class I (R/KxxPxxP) and class II (PxxPxR/K) PRD motifs (85). Furthermore, SH3 domain-containing proteins can interact with PRDs beyond the PXXP motifs, consistent with their versatility in their interaction with other proteins (85, 87).
In addition to being rich in proline residues, the cytoplasmic domain of IGPR-1 also is heavily phosphorylated at serine residues. A recent liquid chromatography–tandem mass spectrometry analysis of IGPR-1 identified seven phosphorylated serine residues on the cytoplasmic domain of IGPR-1, including Ser186, Ser220, Ser238, Ser243, Ser249, Ser262, and Ser266, five of which are located in the proline-rich region (83). Although the functional importance of these phosphorylation sites remains to be determined, phosphorylation of Ser220 is regulated by homophilic trans-dimerization of IGPR-1 and is required for endothelial barrier function and angiogenesis (83).
Although significant work is required to fully understand the signal transduction events orchestrated by IGPR-1, recent studies, however, provide important new insights about signaling of IGPR-1 in endothelial cells (81, 83). Through the screening of a Src-homology3 (SH3) domain array, BPAG1 (or BP230), also called DST and SPIN90/WISH (SH3 protein interacting with Nck), also called NCKIPSD were identified as putative IGPR-1-binding proteins (81). The binding of BPAG1 and SPIN90 with IGPR-1 was further confirmed by recombinant GST-SH3 domain of BPAG1 and SPIN90 in a GST-pull down assay (81).
Bullous pemphigoid antigen 1 is a member of the plakin family proteins, which include desmoplakin, plectin, envoplakin, and periplakin, is involved in cytoskeletal organization (88). BPAG1 is a cytoskeletal linker protein that crosslinks cytoskeletal filaments to membrane-associated complexes at cell junctions in epithelial cells and other cell types (89, 90). BPAG1 is a gigantic protein with 7,570 amino acids and an approximate molecular weight of 834 kDa. However, it is expressed in a various isoforms by mechanism of mRNA alternative splicing, which results in the transcription and translation that generates different isoforms of BPAG1 with varying molecular weights (91). Based on the human genome sequence information, there are 35 different transcripts of BPAG1, many of which are untranslated1 and based on the available human protein sequence data2 there are at least nine isoforms of BPAG1 (Figure 6). BPAG1 is a multidomain protein. It has a conserved N-terminal actin-binding domain, followed by plakin domain which consists of 4–8 spectrin repeats interrupted by a Src-homology3 (SH3) domain. This unique domain is conserved in all plakin family proteins. The C-terminal of BPAG1 composed of additional plakin repeat domains and intermediate filaments binding domain (88, 92–94).
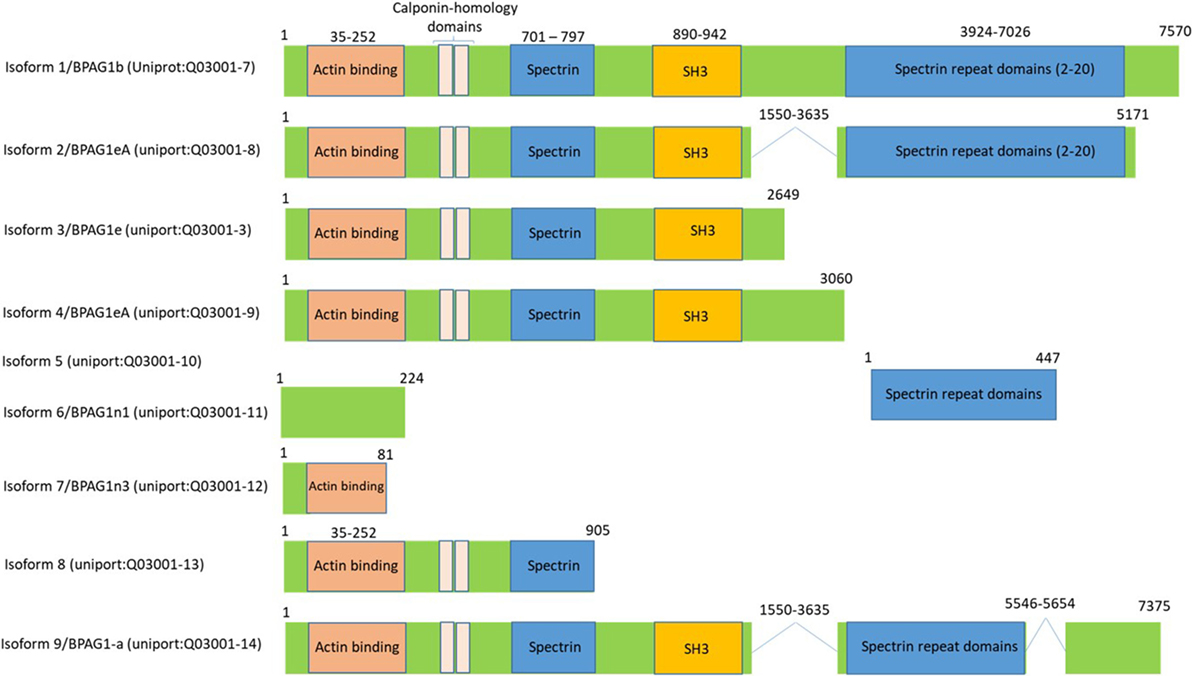
Figure 6. Bullous pemphigoid antigen 1 (BPAG1) is a multidomain protein with various alternatively spliced variants. BPAG1 is a large protein with 7,570 amino acids with multiple domains including N-terminus actin-binding domain, followed by plakin domain which consists of 4–8 spectrin repeats interrupted by a Src-homology3 (SH3) domain. The C-terminal of BPAG1 is composed of additional plakin repeat domains and intermediate filaments binding domain. Various alternatively spliced variants of BPAG1 are also shown.
Despite extensive studies on the functional role of BPAG1 in epithelial cells, expression and importance of BPAG1 in endothelial cells remains virtually unknown. However, analysis of publically available gene array datasets3 indicates that BPAG1 is likely expressed in mouse and human endothelial cells. Based on various recently published gene array analyzes, BPAG1 appears to widely expressed in human vascular endothelial cells derived from lung (95), macrovascular umbilical vein endothelial cells (96), umbilical cord arterial and venous endothelial cells (97), and mouse neonatal retinal endothelial cells (98), suggesting a functional role for BPAG1 in IGPR-1-mediated signal transduction in endothelial cells. Interestingly, one of the major characteristics of epidermolysis bullosa, a neurological condition that causes the skin to blister, is caused by a genetic defect in BPAG1 and it is associated with increased pathological angiogenesis with a leaky vessel (99). Nevertheless, the role of vascular component in epidermolysis bullosa remains unexamined. While the role of BPAG1 in endothelial cells and its possible role in connecting IGPR-1 cytoskeletal filaments remains unresolved, a recent study demonstrated that plectin/epiplakin 1, a closely related protein to BPAG1, is expressed in endothelial cells, which crosslinks vimentin to the actin assembly to regulate vascular integrity (100).
SH3 protein interacting with Nck90/WISH (SH3 protein interacting with Nck 90, also called NCKIPSD and DIP, mDia-interacting protein) is another cytoplasmic signaling protein that interacts with IGPR-1 in endothelial cells (81). SPIN90 is involved in actin polymerization via its interactions with Arp2/3, N-WASP, and actin (101), regulates stress fiber formation (102), Rac-mediated membrane ruffling (103), and binds to Palladin, a cytoskeletal protein that is required for organization of normal actin cytoskeleton, which is important for cell morphology, motility, and cell adhesion (104). SPIN90 is highly expressed in endothelial cells and the siRNA-mediated downregulation of SPIN90 inhibited capillary tube formation, suggesting an important role for SPIN90 in IGPR-1-mediated signaling events in endothelial cells and angiogenesis (81).
Although a significant work remains, the emerging picture of IGPR-1-mediated signal transduction in endothelial cells indicates that IGPR-1 is cross-linked to actin fibril assembly and other cytoskeletal filaments that contributes to endothelial cell adhesion, integrity, and barrier function partly through interaction with SPIN90 and BPAG1. However, deciphering the molecular mechanisms of IGPR-1 in various cell culture systems and animal models other than mouse (IGPR-1 is not expressed mouse or rat) is an important area for future research, which may lead to the discovery of new therapeutic targets for various human diseases associated with endothelial dysfunction.
Altered endothelial barrier function is a hallmark of many human disorders. Understanding the molecular mechanisms of vascular permeability could lead to new therapeutic strategies to prevent vascular leakage and improve drug delivery. Moreover, controlling integrity and function of endothelial cells in organ transplantation could reduce complications associated with transplantation medicine.
The author confirms being the sole contributor of this work and approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part through grants from the NIH (R21CA191970 and R21CA193958 to NR). The author thanks Rachel Ho for reading and commenting on the manuscript.
1. Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol (2006) 291:H985–1002. doi:10.1152/ajpheart.00292.2006
2. Franke WW. Discovering the molecular components of intercellular junctions – a historical view. Cold Spring Harb Perspect Biol (2009) 1:a003061. doi:10.1101/cshperspect.a003061
3. Mateo M, Generous A, Sinn PL, Cattaneo R. Connections matter – how viruses use cell-cell adhesion components. J Cell Sci (2015) 128:431–9. doi:10.1242/jcs.159400
4. Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res (2007) 74:72–84. doi:10.1016/j.mvr.2007.05.003
5. Lee JH, Okada K, Khush K, Kobayashi Y, Sinha S, Luikart H, et al. Coronary endothelial dysfunction and the index of microcirculatory resistance as a marker of subsequent development of cardiac allograft vasculopathy. Circulation (2017) 135:1093–5. doi:10.1161/CIRCULATIONAHA.116.025268
6. Nickel T, Schlichting CL, Weis M. Drugs modulating endothelial function after transplantation. Transplantation (2006) 82:S41–6. doi:10.1097/01.tp.0000231505.91988.26
7. Park-Windhol C, D’Amore PA. Disorders of vascular permeability. Annu Rev Pathol (2016) 11:251–81. doi:10.1146/annurev-pathol-012615-044506
8. Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, et al. The vascular endothelium and human diseases. Int J Biol Sci (2013) 9:1057–69. doi:10.7150/ijbs.7502
9. Daher IN, Yeh ETH. Vascular complications of selected cancer therapies. Nat Clin Pract Cardiovasc Med (2008) 5:797–805. doi:10.1038/ncpcardio1375
10. Suter TM, Ewer MS. Cancer drugs and the heart: importance and management. Eur Heart J (2013) 34:1102–11. doi:10.1093/eurheartj/ehs181
11. Al-Lamki RS, Bradley JR, Pober JS. Endothelial cells in allograft rejection. Transplantation (2008) 86:1340–8. doi:10.1097/TP.0b013e3181891d8b
12. Ramzy D, Rao V, Brahm J, Miriuka S, Delgado D, Ross HJ. Cardiac allograft vasculopathy: a review. Can J Surg (2005) 48:319–27.
13. Matter K, Aijaz S, Tsapara A, Balda MS. Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Curr Opin Cell Biol (2005) 17:453–8. doi:10.1016/j.ceb.2005.08.003
14. Steed E, Balda MS, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol (2010) 20:142–9. doi:10.1016/j.tcb.2009.12.002
15. Forster C. Tight junctions and the modulation of barrier function in disease. Histochem Cell Biol (2008) 130:55–70. doi:10.1007/s00418-008-0424-9
16. Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature (2005) 437:497–504. doi:10.1038/nature03987
17. Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci U S A (1999) 96:511–6. doi:10.1073/pnas.96.2.511
18. Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol (2006) 68:403–29. doi:10.1146/annurev.physiol.68.040104.131404
19. Krause G, Protze J, Piontek J. Assembly and function of claudins: structure-function relationships based on homology models and crystal structures. Semin Cell Dev Biol (2015) 42:3–12. doi:10.1016/j.semcdb.2015.04.010
20. Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta (2008) 1778:631–45. doi:10.1016/j.bbamem.2007.10.018
21. Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol (1999) 147:185–94. doi:10.1083/jcb.147.1.185
22. Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, et al. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol (2003) 105:586–92. doi:10.1007/s00401-003-0688-z
23. Kiuchi-Saishin Y, Gotoh S, Furuse M, Takasuga A, Tano Y, Tsukita S. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J Am Soc Nephrol (2002) 13:875–86.
24. Amasheh S, Meiri N, Gitter AH, Schoneberg T, Mankertz J, Schulzke JD, et al. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci (2002) 115:4969–76. doi:10.1242/jcs.00165
25. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science (1999) 285:103–6. doi:10.1126/science.285.5424.103
26. Yu AS, Enck AH, Lencer WI, Schneeberger EE. Claudin-8 expression in Madin-Darby canine kidney cells augments the paracellular barrier to cation permeation. J Biol Chem (2003) 278:17350–9. doi:10.1074/jbc.M213286200
27. Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature (2007) 446:801–5. doi:10.1038/nature05654
28. Che P, Tang H, Li Q. The interaction between claudin-1 and dengue viral prM/M protein for its entry. Virology (2013) 446:303–13. doi:10.1016/j.virol.2013.08.009
29. Hirase T, Staddon JM, Saitou M, Ando-Akatsuka Y, Itoh M, Furuse M, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci (1997) 110(Pt 14):1603–13.
30. McCarthy KM, Skare IB, Stankewich MC, Furuse M, Tsukita S, Rogers RA, et al. Occludin is a functional component of the tight junction. J Cell Sci (1996) 109(Pt 9):2287–98.
31. Kuwabara H, Kokai Y, Kojima T, Takakuwa R, Mori M, Sawada N. Occludin regulates actin cytoskeleton in endothelial cells. Cell Struct Funct (2001) 26:109–16. doi:10.1247/csf.26.109
32. Harhaj NS, Felinski EA, Wolpert EB, Sundstrom JM, Gardner TW, Antonetti DA. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci (2006) 47:5106–15. doi:10.1167/iovs.06-0322
33. Sundstrom JM, Tash BR, Murakami T, Flanagan JM, Bewley MC, Stanley BA, et al. Identification and analysis of occludin phosphosites: a combined mass spectrometry and bioinformatics approach. J Proteome Res (2009) 8:808–17. doi:10.1021/pr7007913
34. Van Itallie CM, Fanning AS, Holmes J, Anderson JM. Occludin is required for cytokine-induced regulation of tight junction barriers. J Cell Sci (2010) 123:2844–52. doi:10.1242/jcs.065581
35. Chiba H, Osanai M, Murata M, Kojima T, Sawada N. Transmembrane proteins of tight junctions. Biochim Biophys Acta (2008) 1778:588–600. doi:10.1016/j.bbamem.2007.08.017
36. Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem (2009) 284:21036–46. doi:10.1074/jbc.M109.016766
37. Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem (1999) 274:23463–7. doi:10.1074/jbc.274.33.23463
38. DeMaio L, Chang YS, Gardner TW, Tarbell JM, Antonetti DA. Shear stress regulates occludin content and phosphorylation. Am J Physiol Heart Circ Physiol (2001) 281:H105–13.
39. McKenzie JA, Riento K, Ridley AJ. Casein kinase I epsilon associates with and phosphorylates the tight junction protein occludin. FEBS Lett (2006) 580:2388–94. doi:10.1016/j.febslet.2006.03.048
40. Kale G, Naren AP, Sheth P, Rao RK. Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. Biochem Biophys Res Commun (2003) 302:324–9. doi:10.1016/S0006-291X(03)00167-0
41. Tash BR, Bewley MC, Russo M, Keil JM, Griffin KA, Sundstrom JM, et al. The occludin and ZO-1 complex, defined by small angle X-ray scattering and NMR, has implications for modulating tight junction permeability. Proc Natl Acad Sci U S A (2012) 109:10855–60. doi:10.1073/pnas.1121390109
42. Cummins PM. Occludin: one protein, many forms. Mol Cell Biol (2012) 32:242–50. doi:10.1128/MCB.06029-11
43. Pan L, Chen J, Yu J, Yu H, Zhang M. The structure of the PDZ3-SH3-GuK tandem of ZO-1 protein suggests a supramodular organization of the membrane-associated guanylate kinase (MAGUK) family scaffold protein core. J Biol Chem (2011) 286:40069–74. doi:10.1074/jbc.C111.293084
44. Bazzoni G. The JAM family of junctional adhesion molecules. Curr Opin Cell Biol (2003) 15:525–30. doi:10.1016/S0955-0674(03)00104-2
45. Mandai K, Rikitake Y, Shimono Y, Takai Y. Afadin/AF-6 and canoe: roles in cell adhesion and beyond. Prog Mol Biol Transl Sci (2013) 116:433–54. doi:10.1016/B978-0-12-394311-8.00019-4
46. Monteiro AC, Sumagin R, Rankin CR, Leoni G, Mina MJ, Reiter DM, et al. JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol Biol Cell (2013) 24:2849–60. doi:10.1091/mbc.E13-06-0298
47. Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol (2007) 7:467–77. doi:10.1038/nri2096
48. Naik MU, Naik UP. Junctional adhesion molecule-A-induced endothelial cell migration on vitronectin is integrin alpha v beta 3 specific. J Cell Sci (2006) 119:490–9. doi:10.1242/jcs.02771
49. Lamagna C, Hodivala-Dilke KM, Imhof BA, Aurrand-Lions M. Antibody against junctional adhesion molecule-C inhibits angiogenesis and tumor growth. Cancer Res (2005) 65:5703–10. doi:10.1158/0008-5472.CAN-04-4012
50. Inoue M, Ishida T, Yasuda T, Toh R, Hara T, Cangara HM, et al. Endothelial cell-selective adhesion molecule modulates atherosclerosis through plaque angiogenesis and monocyte-endothelial interaction. Microvasc Res (2010) 80:179–87. doi:10.1016/j.mvr.2010.04.005
51. Raschperger E, Thyberg J, Pettersson S, Philipson L, Fuxe J, Pettersson RF. The coxsackie- and adenovirus receptor (CAR) is an in vivo marker for epithelial tight junctions, with a potential role in regulating permeability and tissue homeostasis. Exp Cell Res (2006) 312:1566–80. doi:10.1016/j.yexcr.2006.01.025
52. Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer (2004) 4:118–32. doi:10.1038/nrc1276
53. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev (2006) 86:279–367. doi:10.1152/physrev.00012.2005
54. Bates DO. Vascular endothelial growth factors and vascular permeability. Cardiovasc Res (2010) 87:262–71. doi:10.1093/cvr/cvq105
55. Murohara T, Horowitz JR, Silver M, Tsurumi Y, Chen D, Sullivan A, et al. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation (1998) 97:99–107. doi:10.1161/01.CIR.97.1.99
56. Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science (1983) 219:983–5. doi:10.1126/science.6823562
57. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science (2005) 307:58–62. doi:10.1126/science.1104819
58. Caldwell RB, Bartoli M, Behzadian MA, El-Remessy AE, Al-Shabrawey M, Platt DH, et al. Vascular endothelial growth factor and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev (2003) 19:442–55. doi:10.1002/dmrr.415
59. Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis (2008) 11:109–19. doi:10.1007/s10456-008-9099-z
60. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7:803–15. doi:10.1038/nri2171
61. Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO. Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res (2003) 22:1–29. doi:10.1016/S1350-9462(02)00043-5
62. Suarez S, Ballmer-Hofer K. VEGF transiently disrupts gap junctional communication in endothelial cells. J Cell Sci (2001) 114:1229–35.
63. Hartsough EJ, Meyer RD, Chitalia V, Jiang Y, Marquez VE, Zhdanova IV, et al. Lysine methylation promotes VEGFR-2 activation and angiogenesis. Sci Signal (2013) 6:ra104. doi:10.1126/scisignal.2004289
64. Rahimi N. The ubiquitin-proteasome system meets angiogenesis. Mol Cancer Ther (2012) 11:538–48. doi:10.1158/1535-7163.MCT-11-0555
65. Rahimi N, Costello CE. Emerging roles of post-translational modifications in signal transduction and angiogenesis. Proteomics (2015) 15:300–9. doi:10.1002/pmic.201400183
66. Lal BK, Varma S, Pappas PJ, Hobson RW II, Duran WN. VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc Res (2001) 62:252–62. doi:10.1006/mvre.2001.2338
67. Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol (2002) 39:213–23. doi:10.1016/S1537-1891(03)00010-7
68. Rabiet MJ, Plantier JL, Rival Y, Genoux Y, Lampugnani MG, Dejana E. Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler Thromb Vasc Biol (1996) 16:488–96. doi:10.1161/01.ATV.16.3.488
69. Robinson SD, Reynolds LE, Wyder L, Hicklin DJ, Hodivala-Dilke KM. Beta3-integrin regulates vascular endothelial growth factor-A-dependent permeability. Arterioscler Thromb Vasc Biol (2004) 24:2108–14. doi:10.1161/01.ATV.0000143857.27408.de
70. Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell (2009) 16:209–21. doi:10.1016/j.devcel.2009.01.004
71. Lampugnani MG, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, et al. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol (1992) 118:1511–22. doi:10.1083/jcb.118.6.1511
72. Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell (2013) 26:441–54. doi:10.1016/j.devcel.2013.08.020
73. Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell (1999) 98:147–57. doi:10.1016/S0092-8674(00)81010-7
74. Montero-Balaguer M, Swirsding K, Orsenigo F, Cotelli F, Mione M, Dejana E. Stable vascular connections and remodeling require full expression of VE-cadherin in zebrafish embryos. PLoS One (2009) 4:e5772. doi:10.1371/journal.pone.0005772
75. Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell (1999) 4:915–24. doi:10.1016/S1097-2765(00)80221-X
76. Wallez Y, Cand F, Cruzalegui F, Wernstedt C, Souchelnytskyi S, Vilgrain I, et al. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene (2007) 26:1067–77. doi:10.1038/sj.onc.1209855
77. Cain RJ, Vanhaesebroeck B, Ridley AJ. The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J Cell Biol (2010) 188:863–76. doi:10.1083/jcb.200907135
78. Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem (2005) 280:31906–12. doi:10.1074/jbc.M505568200
79. Fachinger G, Deutsch U, Risau W. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene (1999) 18:5948–53. doi:10.1038/sj.onc.1202992
80. Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J (2002) 21:4885–95. doi:10.1093/emboj/cdf497
81. Rahimi N, Rezazadeh K, Mahoney JE, Hartsough E, Meyer RD. Identification of IGPR-1 as a novel adhesion molecule involved in angiogenesis. Mol Biol Cell (2012) 23:1646–56. doi:10.1091/mbc.E11-11-0934
82. Arafa E, Bondzie PA, Rezazadeh K, Meyer RD, Hartsough E, Henderson JM, et al. TMIGD1 is a novel adhesion molecule that protects epithelial cells from oxidative cell injury. Am J Pathol (2015) 185:2757–67. doi:10.1016/j.ajpath.2015.06.006
83. Wang YH, Meyer RD, Bondzie PA, Jiang Y, Rahimi I, Rezazadeh K, et al. IGPR-1 is required for endothelial cell-cell adhesion and barrier function. J Mol Biol (2016) 428:5019–33. doi:10.1016/j.jmb.2016.11.003
84. Cuadrat RR, da Serra Cruz SM, Tschoeke DA, Silva E, Tosta F, Juca H, et al. An orthology-based analysis of pathogenic protozoa impacting global health: an improved comparative genomics approach with prokaryotes and model eukaryote orthologs. OMICS (2014) 18:524–38. doi:10.1089/omi.2013.0172
85. Li SS. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem J (2005) 390:641–53. doi:10.1042/BJ20050411
86. Zarrinpar A, Bhattacharyya RP, Lim WA. The structure and function of proline recognition domains. Sci STKE (2003) 2003:RE8. doi:10.1126/stke.2003.179.re8
87. Saksela K, Permi P. SH3 domain ligand binding: what’s the consensus and where’s the specificity? FEBS Lett (2012) 586:2609–14. doi:10.1016/j.febslet.2012.04.042
88. Kunzli K, Favre B, Chofflon M, Borradori L. One gene but different proteins and diseases: the complexity of dystonin and bullous pemphigoid antigen 1. Exp Dermatol (2016) 25:10–6. doi:10.1111/exd.12877
89. Jefferson JJ, Leung CL, Liem RK. Plakins: goliaths that link cell junctions and the cytoskeleton. Nat Rev Mol Cell Biol (2004) 5:542–53. doi:10.1038/nrm1425
90. Leung CL, Green KJ, Liem RK. Plakins: a family of versatile cytolinker proteins. Trends Cell Biol (2002) 12:37–45. doi:10.1016/S0962-8924(01)02180-8
91. Fuchs E, Karakesisoglou I. Bridging cytoskeletal intersections. Genes Dev (2001) 15:1–14. doi:10.1101/gad.861501
92. Garcia-Alvarez B, Bobkov A, Sonnenberg A, de Pereda JM. Structural and functional analysis of the actin binding domain of plectin suggests alternative mechanisms for binding to F-actin and integrin beta4. Structure (2003) 11:615–25. doi:10.1016/S0969-2126(03)00090-X
93. Ortega E, Buey RM, Sonnenberg A, de Pereda JM. The structure of the plakin domain of plectin reveals a non-canonical SH3 domain interacting with its fourth spectrin repeat. J Biol Chem (2011) 286:12429–38. doi:10.1074/jbc.M110.197467
94. Sonnenberg A, Liem RK. Plakins in development and disease. Exp Cell Res (2007) 313:2189–203. doi:10.1016/j.yexcr.2007.03.039
95. Cheranova D, Gibson M, Kibiryeva N, Zhang LQ, Ye SQ. Pleiotropic functions of pre-B-cell colony-enhancing factor (PBEF) revealed by transcriptomics of human pulmonary microvascular endothelial cells treated with PBEFsiRNA. Genes Cells (2012) 17:420–30. doi:10.1111/j.1365-2443.2012.01598.x
96. Viemann D, Goebeler M, Schmid S, Nordhues U, Klimmek K, Sorg C, et al. TNF induces distinct gene expression programs in microvascular and macrovascular human endothelial cells. J Leukoc Biol (2006) 80:174–85. doi:10.1189/jlb.0905530
97. Aranguren XL, Agirre X, Beerens M, Coppiello G, Uriz M, Vandersmissen I, et al. Unraveling a novel transcription factor code determining the human arterial-specific endothelial cell signature. Blood (2013) 122:3982–92. doi:10.1182/blood-2013-02-483255
98. Fukushima Y, Okada M, Kataoka H, Hirashima M, Yoshida Y, Mann F, et al. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J Clin Invest (2011) 121:1974–85. doi:10.1172/JCI44900
99. Cianfarani F, Zambruno G, Castiglia D, Odorisio T. Pathomechanisms of altered wound healing in recessive dystrophic epidermolysis bullosa. Am J Pathol (2017) 187:1445–53. doi:10.1016/j.ajpath.2017.03.003
100. Osmanagic-Myers S, Rus S, Wolfram M, Brunner D, Goldmann WH, Bonakdar N, et al. Plectin reinforces vascular integrity by mediating crosstalk between the Vimentin and the actin networks. J Cell Sci (2015) 128:4138–50. doi:10.1242/jcs.172056
101. Kim DJ, Kim SH, Lim CS, Choi KY, Park CS, Sung BH, et al. Interaction of SPIN90 with the Arp2/3 complex mediates lamellipodia and actin comet tail formation. J Biol Chem (2006) 281:617–25. doi:10.1074/jbc.M504450200
102. Satoh S, Tominaga T. mDia-interacting protein acts downstream of Rho-mDia and modifies Src activation and stress fiber formation. J Biol Chem (2001) 276:39290–4. doi:10.1074/jbc.M107026200
103. Lee S, Lee K, Hwang S, Kim SH, Song WK, Park ZY, et al. SPIN90/WISH interacts with PSD-95 and regulates dendritic spinogenesis via an N-WASP-independent mechanism. EMBO J (2006) 25:4983–95. doi:10.1038/sj.emboj.7601349
Keywords: cell adhesion molecules, vascular permeability, endothelial dysfunction, vascular endothelial growth factor A, adherens junctions, gap junctions
Citation: Rahimi N (2017) Defenders and Challengers of Endothelial Barrier Function. Front. Immunol. 8:1847. doi: 10.3389/fimmu.2017.01847
Received: 24 September 2017; Accepted: 06 December 2017;
Published: 18 December 2017
Edited by:
Thomas Luft, University Hospital Heidelberg, GermanyReviewed by:
Luiza Guilherme, University of São Paulo, BrazilCopyright: © 2017 Rahimi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nader Rahimi, bnJhaGltaUBidS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.