 Ooiean Teng
Ooiean Teng Candice Ke En Ang
Candice Ke En Ang Xue Li Guan
Xue Li Guan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 December 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01836
This article is part of the Research Topic The Mononuclear Phagocyte System in Infectious Disease View all 61 articles
Macrophages are professional phagocytes at the front line of immune defenses against foreign bodies and microbial pathogens. Various bacteria, which are responsible for deadly diseases including tuberculosis and salmonellosis, are capable of hijacking this important immune cell type and thrive intracellularly, either in the cytoplasm or in specialized vacuoles. Tight regulation of cellular metabolism is critical in shaping the macrophage polarization states and immune functions. Lipids, besides being the bulk component of biological membranes, serve as energy sources as well as signaling molecules during infection and inflammation. With the advent of systems-scale analyses of genes, transcripts, proteins, and metabolites, in combination with classical biology, it is increasingly evident that macrophages undergo extensive lipid remodeling during activation and infection. Each bacterium species has evolved its own tactics to manipulate host metabolism toward its own advantage. Furthermore, modulation of host lipid metabolism affects disease susceptibility and outcome of infections, highlighting the critical roles of lipids in infectious diseases. Here, we will review the emerging roles of lipids in the complex host–pathogen relationship and discuss recent methodologies employed to probe these versatile metabolites during the infection process. An improved understanding of the lipid-centric nature of infections can lead to the identification of the Achilles’ heel of the pathogens and host-directed targets for therapeutic interventions. Currently, lipid-moderating drugs are clinically available for a range of non-communicable diseases, which we anticipate can potentially be tapped into for various infections.
Macrophages play a key role as the front line of host defenses against foreign bodies. Complex scavenger receptors (SR), pattern recognition receptors, and other signaling receptors expressed by macrophages make them professional phagocytes and antigen-presenting cells. They are highly specialized in engulfment and digestion of the invading pathogen, followed by presentation of antigens to T cells. Under normal circumstances, cytokines and chemokines are secreted by macrophages once pathogens are detected, to recruit more immune cells to the area of infection for restriction of pathogen invasion. Traditionally, macrophages have been classified as inflammatory macrophages (also known as classically activated or M1 macrophages) or anti-inflammatory macrophages (also known as alternatively activated or M2 macrophages), based on their polarization states and physiological features (1, 2). However, it is increasingly appreciated that these cells display remarkable plasticity and their identities may be far more complex, as described in a comprehensive review by Mosser and Edwards (3). The inflammatory and anti-inflammatory responses of macrophages are tightly regulated at different infection stages and are pathogen-specific. Disturbance in this equilibrium will lead to excessive inflammation, or failure to activate the immune response, and this is often exploited by pathogens through the hijack of host signaling mechanisms to evade clearance by professional phagocytes. In fact, various intracellular pathogens have evolved strategies to reside and thrive in macrophages, despite the bactericidal capacity of these host cells. For instance, Mycobacterium tuberculosis, Legionella pneumophila, and Salmonella enterica serovar Typhimurium enter macrophages and persist in vacuolar compartments by modifying the vacuolar maturation processes, while others, including Listeria monocytogenes, Shigella flexneri, Rickettsia rickettsii, and Mycobacterium marinum escape the phagosome and replicate in the cytosol. M. tuberculosis and Mycobacterium leprae can also escape the phagolysosomal compartment into the cytosol, and this process is mediated by secreted proteins, including culture filtrate protein 10 and early secreted antigenic target 6 kDa (ESAT-6) (4). From the host perspective, it is well established that macrophages make use of soluble proteins for communication with other immune cells for initiation of complex signaling cascades during the infection process. In addition, it is increasingly evident that lipids play an equally important role in macrophage functions, influencing the outcome of infections (5–9).
Lipids are fundamental building blocks of cells and play pivotal roles in diverse biological processes. They are key structural components for cellular membranes. With their hydrophobic and amphipathic properties, lipids are able to form a barrier between cells and their environments, as well as within the cells to produce distinct organelles. These cellular membranes have the ability to mediate cell–cell and intracellular communication by budding, fission, and fusion. Lipids also serve as energy stores in eukaryotic cells, in the form of triglyceride esters and steryl esters in lipid droplets. In addition, they play a key role as first and second messengers in signaling cascades. The functions of membrane lipids as well as the lipid composition of an average mammalian plasma membrane have been thoroughly examined and reviewed previously (7, 10, 11).
In the context of macrophage–intracellular bacteria interactions, the plasma membrane of immune cells will be the first barrier the invading pathogen encounters. To successfully infect the host, pathogens often evolve various strategies to target the lipid-enriched plasma membrane for entry and exit, or to hijack host lipid metabolism to promote their survival (12–14). By contrast, lipids can also provide protection to the host during microbial infections (15), demonstrating that lipids are functionally double-edged swords. Here, we review the confounding nature of lipids in the intimate macrophage–pathogen relationship (Figure 1), with a focus on intracellular bacteria, as well as recent techniques employed to probe the dynamics of lipid metabolism during infections. It should be noted that this is not exhaustive as many other pathogens, including eukaryotic parasites, viruses, and fungal pathogens are capable of manipulating host lipid metabolism as part of their survival strategies (8, 9, 16, 17).
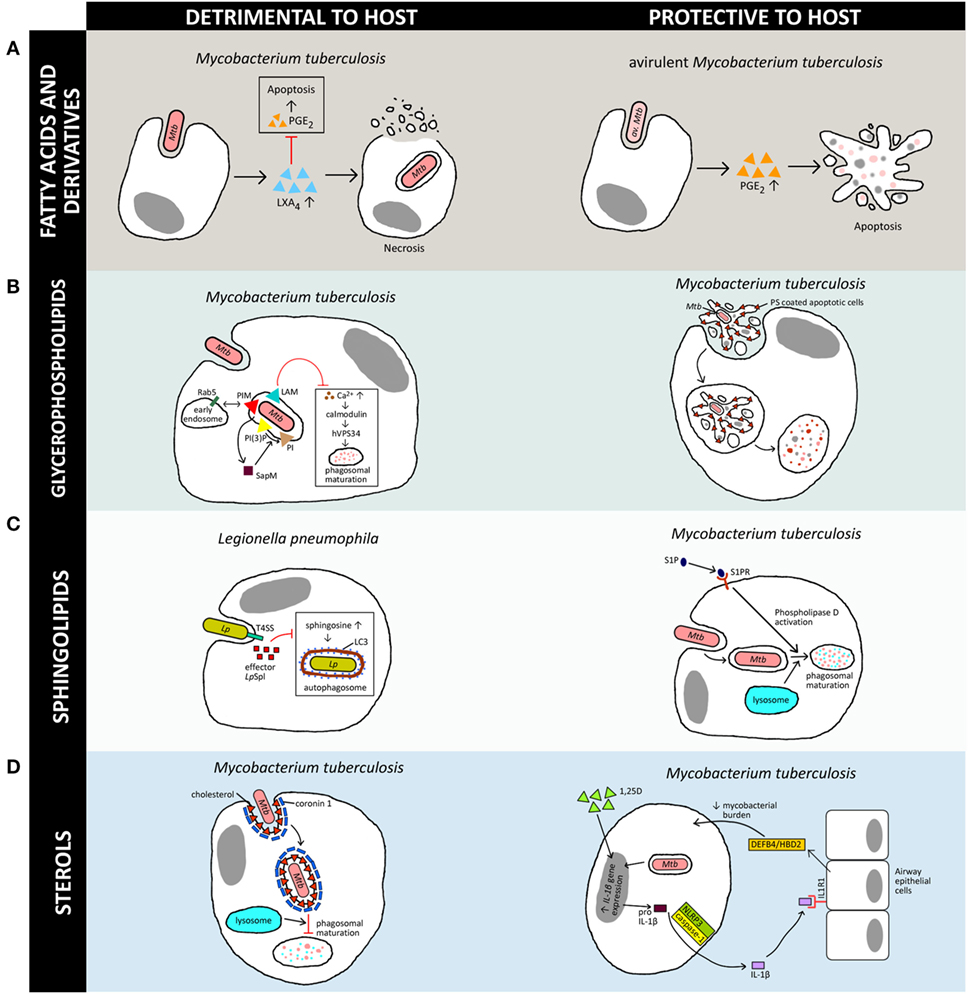
Figure 1. Versatility of lipids in generation of host immune responses against various intracellular pathogens. The schematic diagram illustrates a simplified overview of how the four host-derived lipid classes discussed in this review can be a double-edged sword, either being exploited by the pathogen for its own survival or aiding the host in clearance of the bacteria. Note that there are multiple lipids involved in host–pathogen interactions but only a few representative examples are shown here, and further details can be found in the original works. [(A), left] Induction of LXA4 by virulent M. tuberculosis (Mtb) inhibits PGE2 signaling and promotes necrosis in macrophages (18), whereas [(A), right] induction of PGE2 secretion by avirulent M. tuberculosis (av. Mtb)-infected macrophages leads to apoptosis and protects against mitochondrial inner membrane damage (19). [(B), left] Mtb manipulates host phosphoinositides metabolism to promote their survival in macrophages via inhibition of phagosomal maturation. Mycobacterial phosphatidylinositol mannoside (PIM) stimulates early endosomal fusion by recruiting Rab5. Inhibition of Ca2+ increase by Mycobacterium lipoarabinomannan (LAM) further blocks phagosomal maturation as Ca2+ is required for calmodulin phosphatidylinositol 3-kinase hVPS34 signaling cascade activation. SapM secreted by Mycobacterium inhibits phagosomal-late endosome fusion by hydrolyzing phosphatidylinositol 3-phosphate (20–23). However, [(B), right] redistribution of phosphatidylserine (PS) during apoptosis leads to efferocytosis and restricts the growth of Mtb (24). [(C), left] Effector LpSpl from Legionella pneumophila (Lp) mimics host sphingosine-1-phosphate (S1P) lyase and prevents an increase in sphingosine levels in infected macrophages, inhibiting autophagy (25). On the other hand, [(C), right] S1P is essential for bacterial clearance as it promotes acidification of Mycobacterium-containing phagosomes via phospholipase D activation, which leads to phagosomal maturation and killing of Mtb (26). [(D), left] Accumulation of cholesterol at the Mtb uptake site recruits coronin 1 protein, which inhibits phagosomal maturation (27). [(D), right] The active metabolite of vitamin D (1,25D) controls Mtb infection via macrophage–epithelial paracrine signaling. IL-1β secreted through NLRP3/caspase-1 inflammasome signaling cascade stimulates epithelial cells to produce antimicrobial peptide DEFB4/HBD2, which reduces mycobacterial burden in macrophages (28). In this schematic diagram, triangles represent lipids whereas squares represent proteins.
Macrophages undergo polarization when they encounter foreign bodies, and numerous lines of evidence point toward dynamic lipid remodeling during this defense process. Transcriptional profiling of human macrophage polarization in an in vitro experimental model revealed striking enrichment of genes involved in lipid metabolism as one of the most overrepresented categories of differentially modulated transcripts (29). For M1 macrophages, besides exhibiting preference for aerobic glycolysis, upregulation of cyclooxygenase (COX)-2 and downregulation of COX-1, leukotriene A4 hydrolase, thromboxane A synthase 1, and arachidonate 5-lipoxygenase (5-LO) were observed. On the other hand, M2 macrophages are skewed toward fatty acid oxidation and have upregulated expression of COX-1 and arachidonate 15-lipoxygenase (15-LO) (29–31). Interestingly, opposing regulation of sphingosine and ceramide kinases was observed in both M1 and M2 macrophages (29), highlighting the contrasting nature of lipid metabolism in these distinct macrophage populations.
Receptors involved in binding diverse lipid metabolism products, such as peroxisome proliferator-activated receptor α, β/δ and γ, liver X receptor α and β, CD36, and SR class A-I/II, have also been linked to the polarization of M2 macrophages (32–34). These receptors internalize oxidized and modified lipoproteins and are critical orchestrators of macrophage cholesterol and fatty acid homeostasis (33, 34). M2 macrophages increase oxidized low-density-lipoproteins cholesterol uptake but have similar cholesterol efflux as compared with M1 macrophages. This difference in cholesterol handling leads to increased cholesterol deposition in M2 macrophages (32). It has also been shown that the expression of SR such as CD36 or SR-A1 on M2 macrophages induces endocytosis of triglyceride-rich lipoproteins and generates fatty acids for β-oxidation (32, 35). This aspect of macrophage lipid metabolism is in fact, closely associated with the pathogenesis of atherosclerosis, and the burgeoning field of immunometabolism highlights the critical roles of the cross talk between metabolism and immunity (36).
The dynamics of lipid metabolism during polarization have been further demonstrated at the metabolite level in various studies (37, 38). A shift in the degree of saturation of fatty acids towards monounsaturated fatty acids was observed during human monocyte differentiation to M2 macrophages using macrophage colony stimulating factor (M-CSF), which is in agreement with the induction of sterol regulatory element-binding transcription factor 1c regulated genes, fatty acid synthase, elongation of very long-chain fatty acid-like family member 6 (ELOVL6), and stearoyl-CoA desaturase (37). These observations on the fatty acid composition of glycerophospholipids were further corroborated in a recent study by Zhang and coworkers (38). In addition, an increase in phosphatidylcholine and phosphatidylethanolamine, and a decrease in cholesterol, were observed during macrophage differentiation. Employing a combination of lipidomics, transcriptomics, and pharmacological and genetic manipulations, the direct connection between fatty acid and glycerophospholipid synthesis during the differentiation process was addressed. Furthermore, perturbation of macrophage lipid metabolism was also found to affect the ultrastructural and phagocytic properties of these immune cells (37). These results corroborate earlier findings on the effects of fatty acid unsaturation on macrophage phagocytic activities (39).
Overall, these studies suggest that the polarization of macrophages in vitro is intimately linked to lipid metabolism, and perturbations of host lipid metabolism will affect immune and cellular functions. However, it should be noted that the spectrum of macrophage populations in vivo is more complex (3). This warrants more detailed analyses to determine the exact functions of lipids in the distinct populations and is dependent on environmental cues. We will next examine how macrophage lipids play critical functional roles in the intimate host–microbe relationship.
In macrophages, fatty acids can be synthesized de novo or be taken up by cells through lipolysis of triglyceride-rich lipoproteins particles such as low-density lipoproteins and very-low-density lipoproteins. This is achieved by the expression of SR such as CD36 (40) or SR-B1 (41). The function of fatty acids in various cellular components has a long history and has been extensively reviewed (42). Being the major component of triglycerides, glycerophospholipids, and other complex lipids, fatty acids play pivotal roles as membrane constituents and energy sources, and partake in regulation of signaling pathways as well as cell and tissue metabolism (43). Fatty acids are also substantially found in bacterial cell membranes (44) and serve as precursors for membrane biogenesis. Persistence of M. tuberculosis, an intracellular bacterium and the causative agent of tuberculosis, in mice, is facilitated by isocitrate lyase, an enzyme essential for fatty acid metabolism (45). This highlights the role of fatty acids for maintenance of mycobacterial survival during chronic infections. In fact, besides de novo synthesis, pathogens including M. tuberculosis have evolved multiple strategies to utilize fatty acids derived from their hosts. M. tuberculosis imports fatty acids from host triglycerides for synthesis of its own lipid inclusions and acquisition of dormancy traits in macrophages (46, 47). The mycobacterial protein, LucA, was found to form a complex with Mce1 and Mce4 fatty acid transporters to facilitate cholesterol and fatty acid uptake in infected macrophages (48). In dormant M. tuberculosis, the accumulation of triglycerides is modulated by fatty acid CoA ligase 6 (49), and during nutrient starvation, the bacterium is capable of hydrolyzing these stored triglycerides, owing to its lipase activity (50).
Interestingly, many intracellular pathogens also induce the formation of lipid droplets in the hosts during infections. This has previously been reviewed by Saka and Valdivia (51) and more recently by Barisch and Soldati (52). Besides serving as an energy reservoir, triglycerides found in lipid droplets are also producers of the mediator lipids, eicosanoids, in mammalian cells. Eicosanoids, which are oxidation products of arachidonic acid and other polyunsaturated fatty acids, have assumed a key role as mediators of the immune response during infections (53, 54), and as outlined above, have been shown to be differentially regulated during macrophage polarization. Broadly, it is noted that the effects of eicosanoids in host–pathogen interactions can be classified as pro-inflammatory (54) or anti-inflammatory (55), and they are closely interlinked with their protein counterparts, the cytokines, for orchestration of immune responses. Eicosanoids such as prostaglandin E2 (PGE2) and lipoxin A4 (LXA4) are highly active lipid mediators commonly described in bacterial infections. PGE2 was found to be upregulated by various invasive bacteria, including Mycobacterium bovis bacillus Calmette–Guérin (BCG) (19), L. monocytogenes, Yersinia enterocolitica, Shigella dysenteriae, and the enteroinvasive Escherichia coli (56). These mediator lipids can benefit the host through various mechanisms. In avirulent M. tuberculosis infections, PGE2 has been found to modulate host cell death pathways through the EP2 receptor, which promotes protection of the host against mitochondrial inner membrane perturbation and restricting the spread of bacterium caused by necrosis (57) (Figure 1A, right panel). Interestingly, PGE2 is found to be involved in the repair of host plasma membranes by regulating synaptotagmin 7, the calcium sensing protein involved in lysosome-mediated membrane repair (58). Resealing of membrane lesions is crucial for preventing necrosis and promoting apoptosis, which benefits the host by increasing bacterial clearance.
By contrast, eicosanoids may also bring about detrimental effects to the host, which is to the pathogen’s advantage. PGE2, earlier discussed to be beneficial in mycobacterial infections, can compromise host immunity through inhibition of macrophage maturation (59) and reduction in the release of NAPDH oxidase (60). Upregulation of PGE2 during infections is indeed beneficial for the survival of various pathogens, including Salmonella enterica (61) and Burkholderia pseudomallei (62). During S. enterica serovar Typhimurium infections, the Salmonella pathogenicity island (SPI)-2-encoded SpiC protein activates the ERK1/2 signaling pathway, leading to induction of COX-2 and increased production of PGE2 levels (61). PGE2 impairs killing of Salmonella species in macrophages by inducing IL-10 expression via the protein kinase A pathway (63). During B. pseudomallei infection in macrophages, PGE2 mRNA was rapidly upregulated by over 400-fold at as early as 2h postinfection and promoted B. pseudomallei intracellular survival (62). The suppression of the bactericidal activity of macrophages was associated with decreased nitric oxide production through enhancing the expression of enzyme arginase 2 (62).
LXA4 is another prominent mediator with anti-inflammatory effects (64) generated by 5-LO and 15-LO activities (65). Upregulation of LXA4 levels during mycobacterial infections can lead to inhibition of PGE2, and consequently mitochondrial inner membrane perturbation and macrophage necrosis (57). Unlike PGE2 that protects the host during mycobacterial infections, 5-LO promotes the growth of the bacterium in vivo (66). Virulent M. tuberculosis induces LXA4 and inhibits PGE2 production (18). By inhibiting the production of PGE2, LXA4 impairs apoptotic signaling (67) and promotes necrosis of infected macrophages (18) (Figure 1A, left panel). Furthermore, 5-LO-deficient mice exhibited enhanced expression of IL-12, IFN-γ, and nitrogen oxide synthase 2 in the lungs as compared with wild-type mice, indicating negative regulation of Th1 response by 5-LO during M. tuberculosis infections (66). Similar findings on the negative regulation of Th1 response by 5-LO were observed in Brucella abortus infections in mice (68). Activation of the 5-LO pathway was further demonstrated to impair host T cell immunity by preventing cross-presentation of M. tuberculosis antigen by dendritic cells (18). Interestingly, at the human population level, single nucleotide polymorphisms in eicosanoid receptor gene, EP2, and heterozygosity for leukotriene A4 hydrolase polymorphisms have been reported to modulate host susceptibility to tuberculosis, highlighting the critical roles of eicosanoids in mediating infections (69, 70).
Clearly, as exemplified by the multifaceted functions of eicosanoids, each lipid entity can be a man’s meat and another man’s poison. The type of eicosanoids and the fine balance between the pro-inflammatory and anti-inflammatory mediators are critical determinants of infection outcomes. Should this balance be disrupted, the host will be detrimentally affected either by high pathogen loads due to decreased clearance, or uncontrolled, non-specific inflammation (cytokine storms) leading to poor host outcomes (71). A more refined understanding of the functions of eicosanoids during interactions between specific pathogens and their hosts may potentially pave the future of host-directed therapy against these infections. In fact, a range of drugs (for instance, aspirin) that target eicosanoid biosynthesis are clinically available and can be further evaluated for their effects on bacterial infections.
As one of the main constituents of the mammalian membrane bilayer, glycerophospholipids have been demonstrated to be used by pathogens to evade host defenses. Choline-containing glycerophospholipids such as phosphatidylcholine are predominantly localized to the outer membrane leaflet of eukaryotic cells, while amino-containing glycerophospholipids such as PS and phosphatidylethanolamine are predominantly maintained in the inner membrane leaflet (72). Redistribution of PS to the external surface of the plasma membrane is a key event during apoptosis and has been known as one of the emblematic signals leading to tagging of cells for efferocytosis, a phagocytic process for removal of dead cells (24, 73). This signaling pathway has been shown to be manipulated by L. monocytogenes to ensure their own survival in the infected host (74). The pore-forming toxin listeriolysin O from L. monocytogenes promotes the release of bacteria-containing protrusions from host cell membranes, generating PS-coated vesicles that mimic apoptotic cells. This subsequently promotes efferocytosis by binding to the PS-binding receptor TIM-4 expressed on uninfected macrophages, and eventually facilitates cell-to-cell spread in macrophages in vitro (74). While efferocytosis promotes the spread of infection by L. monocytogenes, this mechanism of cellular clearance appears to be protective to the host for other intracellular bacteria. Notably, efferocytosis has been shown to effectively restrict the growth of M. tuberculosis (75), as the bacteria-infected macrophages are engulfed and killed by uninfected macrophages following apoptosis (75) (Figure 1B, right panel). This process is also effective in limiting the growth of M. marinum in infected macrophages by neutrophils (76).
Phosphoinositides, which are phosphorylated forms of the membrane glycerophospholipid, phosphatidylinositol, are extensively characterized mediators of intracellular signaling and play critical roles during infection. Phosphoinositides can be phosphorylated at the hydroxyl residues at positions 3, 4, or 5 of the inositol ring to produce different phosphoinositide species. These include phosphatidylinositol 3-phosphate [PI(3)P], phosphatidylinositol 4,5-biphosphate, and phosphatidylinositol 3,4,5-triphosphate, which are involved in endocytosis and phagocytosis (77, 78). In fact, different phosphoinositide species exhibit distinct characteristic subcellular distribution patterns due to the organelle-specific phosphoinositide kinases and phosphatases (78). These phosphoinositide species interact with actin-binding proteins through recognition of its head group by PH, PX, ENTH, ANTH, or FYVE domains (20). Many pathogens are able to manipulate host phosphoinositides metabolism to trigger their uptake into macrophages or non-phagocytic cells (21). M. tuberculosis escape phagocytic killing by macrophages through blocking phagosomal maturation via interference of phosphatidylinositol 3-kinase [PI(3)K] signaling (22). M. tuberculosis lipoarabinomannan acts as a phosphatidylinositol analog and inhibits cytosolic calcium increase, leading to the blocking of the Ca2+/calmodulin PI(3)K hVPS34 cascade. This signaling pathway is needed to produce PI(3)P on liposomes or phagosomes and trigger downstream calmodulin kinase II-mediated EEA1 recruitment to phagosomal membranes, which are ultimately required for phagosome maturation (23). In addition, the lipid phosphatase, SapM, which is secreted by the bacterium, is responsible for the inhibition of phagosome-late endosome fusion by hydrolyzing PI(3)P and thus blocking phagosomal maturation (79). Concurrently, mycobacterial phosphatidylinositol mannoside stimulates early endosomal fusion by early recruitment of Rab5, which blocks the acquisition of late endosomal/lysosomal constituents (80) (Figure 1B, left panel). The orchestration of these intracellular signaling events mediated by lipids favors the intracellular persistence of M. tuberculosis, leading to chronic infections.
Similar to M. tuberculosis, Salmonella can infect macrophages and escape killing by these phagocytes to survive and replicate within host cells (81). More extensive research on salmonellosis has been carried out in non-immune epithelial cells since Salmonella initiates its infection via invasion of the intestinal epithelium. Invasion of host cells is dependent on two type III secretion systems (T3SSs) encoded on the SPI-1 and SPI-2 (82–84). One of the effector proteins of the T3SS of Salmonella, SopB (also known as SigD), is a phosphoinositide phosphatase which shows sequence homology to mammalian inositol polyphosphate 4-phosphatases (85) and type II inositol 5-phosphatase synaptojanin (86). The activity of SopB is required for invasion, formation, and maintenance of Salmonella-containing vacuoles (SCVs) in epithelial cells and macrophages (87, 88).
Manipulation of host glycerophospholipid metabolism by bacterial effector proteins is not unique to Salmonella. The Legionella Dot/Icm type IVB secretion system effector protein, VipD, is a phospholipase A1 which binds to and is activated by the endosomal regulator Rab5. The resultant removal of PI(3)P mediated by the bacterial phospholipase blocks endosomal fusion with Legionella-containing vacuoles, shielding the intracellular pathogen from the microbicidal endosomal compartment (89). Besides VipD, two other Legionella effectors, LecE and LpdA, which are localized to Legionella-containing vacuoles, are also capable of manipulating biosynthesis of host glycerophospholipids. Indeed, bacterial phospholipases are well-characterized virulence factors, notably the alpha-toxin from Clostridium perfringens (90). The function of phospholipase C from M. tuberculosis, on the other hand, remains less clear. Originally proposed to promote M. tuberculosis growth in the late stage of infection in mice (91), the role of phospholipase C in mycobacterial virulence has been recently challenged by the works of Le Chevalier and coworkers. In contrast to the works by Raynaud et al. which demonstrated a 1.5-log growth reduction in the phospholipase C mutant (91), the latter study did not detect significant differences between the wild-type and mutant bacteria (92). Clinical M. tuberculosis strains with interruptions of all four mycobacterial phospholipase genes have been isolated in patients with active tuberculosis, supporting a less crucial role of this enzyme in the infectious cycle in vivo (93).
While bacterial phospholipases can be detrimental to the host, conversely, host phospholipase activities can antagonize the survival of intracellular pathogens. In the context of mycobacterial infections, host phospholipase D and lysosomal phospholipase A2 have been implicated in the cells’ ability to control intracellular mycobacterial growth (94, 95). It should be noted that these studies were focused on the enzymes, and the exact lipid mediators remain to be identified. Nonetheless, the cumulative evidence of the involvement of both host and microbial glycerophospholipids metabolizing enzymes in the regulation of the infection process highlights the complexity of interplay of the metabolic networks of two organisms. This intricacy is not restricted to glycerophospholipids and will be reflected in the following subsections on two other major eukaryotic lipid classes, sphingolipids and sterols.
Sphingolipids are another key component of eukaryotic cell membranes. They comprise of a long-chain amino alcohol (also known as a sphingoid base or long-chain base) to which a fatty acid can be covalently linked to form ceramide. Structural variants arise from head group substitutions, as well as chain length differences and hydroxylation of the sphingoid bases and fatty acyl chains, giving rise to tens of thousands of different molecular species with diverse functions (96–98). Sphingolipids are partners with cholesterol in eukaryotic membranes, forming specialized domains (commonly termed as lipid rafts) and serve as signaling platforms and/or entry sites during pathogen invasion. The functions of lipid rafts in host–pathogen interactions have been extensively reviewed (99–102).
Beyond their structural functions, sphingolipids also serve as signaling molecules which mediate the infection process (103, 104). Sphingosine-1-phosphate (S1P) is an active metabolite formed by sphingosine kinases 1 and 2, and are involved in immune cell trafficking, through engagement with G-protein-coupled receptors (S1PR 1–5). S1PRs are expressed on different immune cells, and macrophages mostly express S1PR1 and S1PR2 (105). Recently, it has also been shown that human alveolar macrophages express high levels of S1PR3 and S1PR4, and lower levels of S1RP5 (26). Expression of S1PRs determines the function of an immune cell, as they play critical roles in lymphocyte trafficking, differentiation and triggering of inflammatory responses (106). During mycobacterial infections, phagocytosis is uncoupled from cytosolic calcium level elevation, mediated by the inhibition of macrophage sphingosine kinase activity and consequently, the downregulation of S1P levels. The blockade of phagosome–lysosome fusion allows the intracellular bacterium to avoid the bactericidal phagolysosomes to continue to persist within host cells (107). Stimulation of macrophages with S1P leads to increased killing of internalized Mycobacterium species through the acidification of phagosomes via host phospholipase D (108) (Figure 1C, right panel). Additionally, S1P modulates mycobacterial infections by promoting antigen processing and presentation (109). These studies, taken together, highlight the antimycobacterial properties of the mediator lipid S1P.
The phagolysosomal compartment is clearly crucial for defense against infection with intracellular pathogens. Besides S1P, phagosomal maturation is also regulated by other sphingolipids. Host acid sphingomyelinase (ASMase), which generates the signaling molecule ceramide, is required for the proper fusion of late phagosomes with lysosomes (110). Delivery of ASMase to mycobacteria-containing phagosomes is regulated by sortilin, which requires interactions with adaptor protein AP-1 and monomeric gamma-ear-containing ADP ribosylation factor-binding proteins (GGAs) (111). Sortilin knockout mice are more susceptible to M. tuberculosis infections. Moreover, treatment of mouse macrophages with desipramine, an ASMase inhibitor, resulted in increased mycobacterial survival, indicating that ASMase is required for restricting the growth of M. tuberculosis. The crucial role of this host enzyme in controlling intracellular bacteria can be further illustrated by the increased susceptibility of ASMase knockout mice to L. monocytogenes infections (112). Functional ASMase is also involved in the bactericidal activity of macrophages against S. enterica serovar Typhimurium (25). In contrast to the protective effects of ASMase, neutral sphingomyelinase is associated with superoxide production during M. bovis BCG infections in vitro and in vivo. The superoxide produced during infection inhibits autophagy and therefore reduces bacterial clearance (113).
Various intracellular pathogens have also evolved metabolic strategies to hijack host sphingolipids to promote their pathogenicity. This is evident from the presence of sphingolipid metabolizing genes in bacterial genomes (114), although sphingolipids are synthesized only in eukaryotes (with few exceptions in prokaryotes, such as Sphingomonas species). M. tuberculosis encodes a novel outer membrane protein, Rv0888, which possesses potent sphingomyelinase activity (115). The bacterial protein has been shown to promote intracellular infection in macrophages in vitro, but is not required for virulence of M. tuberculosis in mice (115). While the functions of Rv0888 in mycobacterial infections remain to be clarified, it is interesting to note that a study conducted in as early as 1948 had already demonstrated that sphingomyelin supports the growth of the tubercle bacilli in vitro (116). Besides M. tuberculosis, a phospholipase C with sphingomyelinase activity has been characterized in L. monocytogenes (117). However, its function in Listeria infections has yet to be elucidated. By contrast, L. pneumophila produces an effector protein, LpSpl, which shares structural and sequence homology to the eukaryotic counterpart sphingosine-1 phosphate lyase (118). Similar to the glycerophospholipid metabolizing enzymes, VipD, LecE, and LpdA, outlined above, LpSpl is an effector protein of the Dot/Icm type IVB secretion system. It has been shown that LpSpl activity prevents an increase of sphingosine levels in infected macrophages (Figure 1C, left panel). In addition, the bacterial sphingolipid metabolizing enzyme inhibits autophagy during macrophage infection and is required for efficient infection of mice. This represents a novel mechanism of inhibition of autophagy by an intracellular bacterium through perturbation of host sphingolipid biosynthesis. Strikingly, a similar strategy was observed in the facultative intracellular bacteria B. pseudomallei (119).
Evidently, the balance of host sphingolipids is another determinant of successful infections by intracellular bacteria. S1P is one of the most obvious sphingolipid metabolites which exhibit bactericidal activity. Various drugs which target the S1P axis are available and in fact used in clinical trials for various indications (120). With a deepened understanding of S1P and sphingolipids in inflammation and infection, the appropriate control of the S1P levels and potentially other sphingolipids may be targets for generation of novel drugs in treatment of various infections.
Sterols are major components of many animals (zoosterols) and plants (phytosterols), but only a few bacteria are able to synthesize sterols. In bacterial membranes, hopanoids, a class of pentacyclic triterpenoids, execute the functions of sterols (121). The most well-known zoosterol, cholesterol, plays critical roles in modulating cellular functions. These include regulation of membrane fluidity, phagocytosis, cell signaling, and formation of lipid rafts with glycosphingolipids (27, 122–124). In addition, cholesterol also serves as precursors for bile salts, steroids, and vitamins (125). Cholesterol in mammalian cells can be obtained from exogenous sources such as low-density lipoproteins or endogenously synthesized. Cholesterol accumulation is commonly observed in the form of lipid droplets in infected cells as well as in biopsies, such as in lepromatous leprosy tissues and tuberculosis granulomas (126, 127). At the cellular level, it has been demonstrated that some pathogens utilize cholesterol on lipid rafts for invasion and intracellular replication (99–102). Plasma membrane cholesterol plays an essential role for host cell uptake of M. tuberculosis. In addition, the association of cholesterol with the coronin 1 protein in phagosomal membranes ensures the intracellular survival of Mycobacterium in coronin 1-coated phagosomes by preventing degradation of the tubercle bacilli in lysosomes (128) (Figure 1D, left panel). The need for cholesterol in mycobacterial infections and survival is best emphasized by the presence of multiple genes involved in cholesterol transport and catabolism, although the bacterium does not synthesize cholesterol de novo. These genes include the cholesterol transporter, Mce4 and the transcriptional regulator KstR, which control the expression of bacterial genes involved in sterol catabolism (129–132). Furthermore, mutants lacking genes in cholesterol utilization fail to establish infection in macrophages (133).
Besides Mycobacterium, Listeria species and Salmonella species are also capable of utilizing host cholesterol to promote their survival in macrophages. The cholesterol-binding cytolysin, listeriolysin O, secreted by L. monocytogenes binds to cholesterol embedded in the lipid bilayer of eukaryotic cell cytoplasmic membranes to facilitate bacterial escape from the phagosomal compartment into host cell cytosol (134–136). Listeriolysin O also performs other functions, including modulation of inflammatory responses through activation of caspase-1, leading to IL-18 secretion from the infected macrophages (137). In fact, cholesterol-binding cytolysins are also produced by other pathogens, including perfringolysin O from C. perfringens, and facilitate bacterial escape from phagosomes and survival in macrophages (90). Salmonella, which reside in vacuoles, take a distinct approach. During Salmonella infections, cholesterol accumulates in the SCV (138). SseJ, another effector protein of the T3SS, has been reported to be involved in cholesterol esterification and stabilization of the SCV (139). Interestingly, the modulation of macrophage cholesterol levels with statins has been shown to augment host protection against various intracellular pathogens including M. tuberculosis, M. leprae, S. enterica serovar Typhimurium, and L. monocytogenes (28, 126, 140, 141).
While it has been extensively proven that cholesterol supports the survival of intracellular pathogens within infected macrophages, cholesterol can also exert protective effects on the host during infection by specific intracellular bacteria. The protective role of cholesterol for the host has been shown recently during infection by Coxiella burnetii, the causative agent for Q fever. Mulye et al. (142) reported that increased cholesterol levels in the parasitophorous vacuole by cholesterol supplementation or treatment with U18666A inhibited the growth of intracellular bacteria. Cholesterol induces acidification of the parasitophorous vacuole and eventually, killing of the intracellular bacteria (142). Although this study was performed on mouse embryonic fibroblasts, earlier works on C. burnetii-infected THP-1 macrophage-like cells had also demonstrated inhibition of intracellular C. burnetii growth by U18666A (143), suggesting that alterations in cholesterol levels can affect the survival of this pathogen in immune cells. The biosynthetic intermediates as well as metabolites of cholesterol can also improve resistance of the host cells to infection. During Listeria infection of macrophages, lanosterol, an intermediate of cholesterol biosynthesis, accumulates due to type I IFN-dependent histone deacetylase 1 transcriptional repression of lanosterol-14α-demethylase, the gene product of Cyp51A1. Besides the modulation of IFN-β-stimulated gene expression and the effects on cytokine production, accumulation of lanosterol also leads to an increase of membrane fluidity and ROS production, thus potentiating phagocytosis and the destruction of bacteria (144).
Cholesterol also serves as a precursor for vitamins, including the fat-soluble vitamin D. Interestingly, vitamin D can have protective effects against bacterial infections (145). In the context of mycobacterial infections, 1,25-dihydroxy-vitamin D3 (1,25D), the active metabolite of vitamin D, promotes maturation and activation of human monocytes and macrophages and reduces bacillary replication. In a macrophage–epithelial cell coculture system, 1,25D enhanced IL-1β expression and induced secretion of the cytokine from M. tuberculosis-infected macrophages via the NLRP3/caspase-1 inflammasome pathway. IL-1β secreted from macrophages reduced mycobacterial burden by stimulating the epithelial production of the antimicrobial peptide DEFB4/HBD2. This suggests that the control of M. tuberculosis infection by vitamin D is modulated by macrophage–epithelial paracrine signaling (146) (Figure 1D, right panel). In fact, vitamin D deficiency and vitamin D receptor polymorphism (147) have shown to affect human susceptibility to tuberculosis, and vitamin D-based oxysterols can promote clinical improvements in tuberculosis patients (148). Interestingly, the vitamin D metabolite 25(OH)D3 has also been found to act synergistically with the bacteriostatic antituberculosis drug phenylbutyrate, providing a potential indication of adjunct therapy for tuberculosis through resolution of inflammation and enhancement of bacterial clearance (149).
The possibility of using vitamin D, and potentially sunlight, to fortify antituberculosis resistance is an exciting and straightforward approach for tuberculosis interventions. Definitely, it will be of interest to extend such studies of vitamin D to other microbial infections. However, specifically for cholesterol, with its mixed roles of being both protective and harmful to the host during infection, as well as its central roles in diverse cellular processes, targeting cholesterol metabolism alone may cause undesired complications. Combination treatment or targeting of specific downstream products of cholesterol-induced signaling may be explored as an alternative.
Besides protein-based bacterial virulence factors which are capable of manipulating host lipid metabolism during infection, foreign lipid structures expressed on bacteria surfaces can also act as pathogen-associated molecular patterns which induce immune responses as well as remodeling of lipid metabolism in macrophages. LPS, a major component of the outer membrane of Gram-negative bacteria, is made up of a conserved lipid A, core oligosaccharide regions and a long polysaccharide chain with variable carbohydrate subunits (150). LPS is well known as a ligand for toll-like receptor 4 (TLR4) (151–153) and toll-like receptor 2 (TLR2) (154, 155). Signals triggered by TLR4 upon activation by LPS have been extensively studied and reviewed (151, 156–158). Briefly, this signaling cascade is initiated by LPS binding protein which binds directly with LPS and is transferred to CD14. This then dissociates LPS aggregates into monomeric forms and presents them to the TLR4–MD-2 complex. Binding of LPS to TLR4–MD-2 complex eventually activates NF-κB and IRF3, leading to the production of pro-inflammatory cytokines (via MyD88-dependent pathway) or type-1 interferon (MyD88-independent pathway) (159). In 2013, two independent studies demonstrated that LPS activates inflammasomes via murine caspase-11, independent of TLR4 (160, 161). Following that, Shi and coworkers showed that human caspase-4 and -5 [orthologs of murine caspase-11 (162)] served as intracellular sensors for LPS and lipid A. The binding of LPS to caspase-4 leads to pyroptosis in human monocytes and non-immune cells (163). Due to its ability to trigger strong immune responses, LPS has always been linked to a variety of pathologies such as septic shock and death, while inhibition of LPS-induced signaling pathways often favors the hosts (161, 164–166).
The impact of LPS on host lipid metabolism has been fairly well characterized. Very early studies demonstrated that LPS administration produced hypertriglyceridemia, with increased lipoprotein production and decreased lipoprotein clearance (167, 168). The effect of lipogenesis and lipolysis seemed to be affected by the dosage of LPS, as Feingold and coworkers showed that low doses (10 ng/100 g body weight) induced hepatic secretion of triglyceride, while high doses (50 μg/100 g body weight) decreased the clearance of triglyceride-rich lipoprotein (169). LPS also increased lipid body formation in human macrophages, through increasing the fatty acid uptake and reducing lipolysis, hence increasing triglyceride retention in the cells (170). Furthermore, turnover of glycerophospholipids during LPS stimulation has been observed, which is mediated by induction of acyl-CoA synthetase 1 (171). Accumulation of lipid droplets in non-adipocytic cells has also been reported as a pathological feature in infectious diseases, and it has been demonstrated to be an important site for PGE2 synthesis (172, 173). Lipid body formation is largely dependent on TLRs, especially TLR2 and TLR4, as TLR2-deficient and TLR4-mutated mice both failed to form lipid droplets triggered by LPS (172, 173). When induced by LPS, lipid droplets are formed via the p38 α/β and PI(3)K/Akt pathways (174), and drugs blocking these pathways have been proven to inhibit their formation (174, 175).
A combined lipidomics and transcriptomics study on mouse macrophages upon LPS stimulation by Dennis and coworkers has brought together a system-based overview of the temporal and subcellular dynamics of macrophage lipid remodeling elicited by LPS (176). Treating RAW264.7 cells with Kdo2-lipid A, a chemically defined substructure of LPS, increased COX-2 related lipid metabolites immediately after the stimulation, followed by sterols, sphingolipids, glycerophospholipids, and glycerolipids (176). Based on the reported findings on LPS-induced changes in sphingolipids, KÖberlin and coworkers recently examined the lipid metabolic network of RAW264.7 cells during TLR signaling and showed that genes involving sphingolipids metabolism are tightly modulated upon TLR4 and TLR9 stimulation. Intriguingly, this study revealed that innate immune responses are modulated by a circular network co-regulating sphingolipids and glycerophospholipids (177). These studies have contributed to our current understanding on the complexity of the macrophage lipid metabolic networks during inflammation, and it will be of interest to identify novel small molecules which can target lipid metabolism for immune protection.
Besides the Gram-negative bacteria-specific LPS, lipids produced by other bacteria also possess host immune- and metabolism-modulating functions. M. tuberculosis and M. leprae are notorious for their fat-loving nature as well as their cell walls which are dominated by lipids (178, 179). TDM, also known as cord factor, is a glycolipid abundantly expressed on the cell wall of mycolic acid-containing bacteria such as Mycobacterium, Nocardia, Tsukamurella, Gordona, Rhodococcus, and Corynebacterium species and has previously been identified as a virulence factor (180). The TDM molecule consists of sugar trehalose esterified to two mycolic acid residues, and the length range of residues varies from 20 to 80 carbons depending on the bacterial species. TDM is responsible for the characteristic colony morphology observed in virulent M. tuberculosis due to its hydrophobic nature (181). In addition, at the gene expression level, TDM from M. tuberculosis H37Rv upregulates multiple cytokines (TNF-α, IL-1β, IL-1m, IL-10, and MCSF-1) and chemokines (CCL3, CCL4, CCL7, CCL12, CCR12, CXCL1, CXCL2, and CXCL10) (182), proving the ability of TDM to initiate the macrophage immune responses in vitro (183). Strikingly, TDM alone is sufficient to induce activated foreign body- and hypersensitivity-type granulomas in mice, as well as extensive foam cell formation, illustrating its importance in pathogenesis (127, 184–186). TDM also promotes Mycobacterium survival in vivo by inhibiting phagosome–lysosome fusion and protects the bacteria from destruction within macrophages (187, 188).
Besides its effects on the immune functions of macrophages, TDM has been shown to induce formation of caseating granulomas and foamy macrophages in the absence of Mycobacterium itself (127). The toxicity of TDM is greatly dependent on its surface crystalline structure. This can be enhanced by the presence of oil, as administration of TDM in oil-in-water emulsion has shown to induce higher mortality rates in mice, whereas administration of TDM without oil has no effect (189–193). Interestingly, M. tuberculosis preferentially associates with the lipid droplets in pulmonary lesions to increase the toxicity of TDM and initiation of necrosis (192). Separately, free mycolic acids have also been shown to induce macrophage lipid droplet formation (194). Keto-mycolic acid, the oxygenated form of mycolic acid, was found to transactivate the host orphan lipid-sensing nuclear receptor, testicular receptor 4 (TR4), and induce foamy macrophage formation in vitro and in vivo (195). TR4 is also involved in the polarization of macrophages toward a less microbicidal and an immunomodulatory M2 phenotype, which aids in the survival of M. tuberculosis. With more in-depth understanding of microbial lipid–host receptor interactions, guided designs of structural analogs of lipids to interfere with ligand–receptor binding holds promise as a combinatorial adjunct therapy.
Another mycobacterial lipid virulence factor that is capable of moderating macrophage functions, but differs in function from TDM, is phthiocerol dimycocerosates (PDIM) (196–199). PDIM has been proven to mediate phagosomal escape of M. tuberculosis by enhancing the membrane permeabilizing activity of ESAT-6, increasing phagosomal membrane destabilization and subsequent induction of apoptosis (200, 201). While PDIM and ESAT-6 are known to cause phagosomal membrane rupture, whether this process involves alterations in host lipid metabolism requires further investigation. Probing changes in host lipid metabolism is increasingly possible with the availability of a range of techniques, which we will cover in the next section.
Despite the appreciation of the important roles lipids play in infectious diseases, it is not as well studied as compared with genes and proteins, due to chemical complexity of lipids and the limited availability of tools required. Most of our knowledge on the roles of lipids in the intimate host–pathogen relationship has been gained through genetic and cellular investigations, and only in recent years have sensitive and high resolution technologies for lipid analyses become available for revelation of novel insights of lipid functions during infections.
Traditional methods of lipid analysis include enzyme immunoassays (EIAs) and thin layer chromatography (TLC). EIAs, an established method particularly for cytokine and chemokine measurements in both infection and immunity research, have been commonly used to monitor the levels of various eicosanoids (202, 203). The underlying mechanism of EIA lies in the immunocomplexes formed between the lipids of interest and their antibodies. These assays serve as a fairly rapid method for lipid analyses, provided that the lipids of interest are well defined. Another restriction of EIAs is its limited availability, because lipids generally have poor immunogenicity, and the generation of high affinity anti-lipid antibodies is challenging using traditional hybridoma techniques (204). Furthermore, these assays have limited resolution (i.e., may not be able to separate between lipid molecular species) and sensitivity, and hence are not well suited for very low abundant signaling molecules. The use of TLC for the separation and detection of lipids have been described as early as the 1960s (205) and continues to be commonly used (127, 206, 207). Despite its low sensitivity and resolution, TLC offers the possibility to study the turnover of lipids and capture the temporal dynamics of lipid remodeling when used with radioisotopes labeling (46, 47).
Recently, more advanced methods including gas chromatography–mass spectrometry, liquid chromatography–mass spectrometry (LC–MS), nuclear magnetic resonance (NMR) spectroscopy, and mass spectrometry (MS)-based imaging techniques have revolutionized the fields of metabolomics and lipidomics. These technologies allow for more efficient analysis of the diverse metabolites and lipids in biological samples, with deeper and more sensitive coverage at the lipid molecular species level. Notably, the LC–MS analyses of the monocyte/macrophage lipidome spanning membrane lipids (sterols, glycerophospholipids, and sphingolipids), and mediator and signaling lipids (phosphoinositides, eicosanoids, and phosphorylated sphingoid bases), have contributed to the revelation of the complex metabolic networks during macrophage differentiation (37, 38), TLR activation and signaling (176, 177, 208) as well as the complex host–pathogen interactions (8, 118, 209, 210). In the last 10–15 years, the lipidomes of various other host cell types including epithelial cells (211), platelets (212), and pathogens, including M. tuberculosis (213–215), Clostridium novyi (216), E. coli (217), Enterococcus faecalis (218), and Pseudomonas aeruginosa (219) have been established. The possibility to analyze both host and microbial lipids with high resolution structural information (Figure 2) has opened up new avenues to study the dynamics of lipid metabolism during the complex host–pathogen relationship.
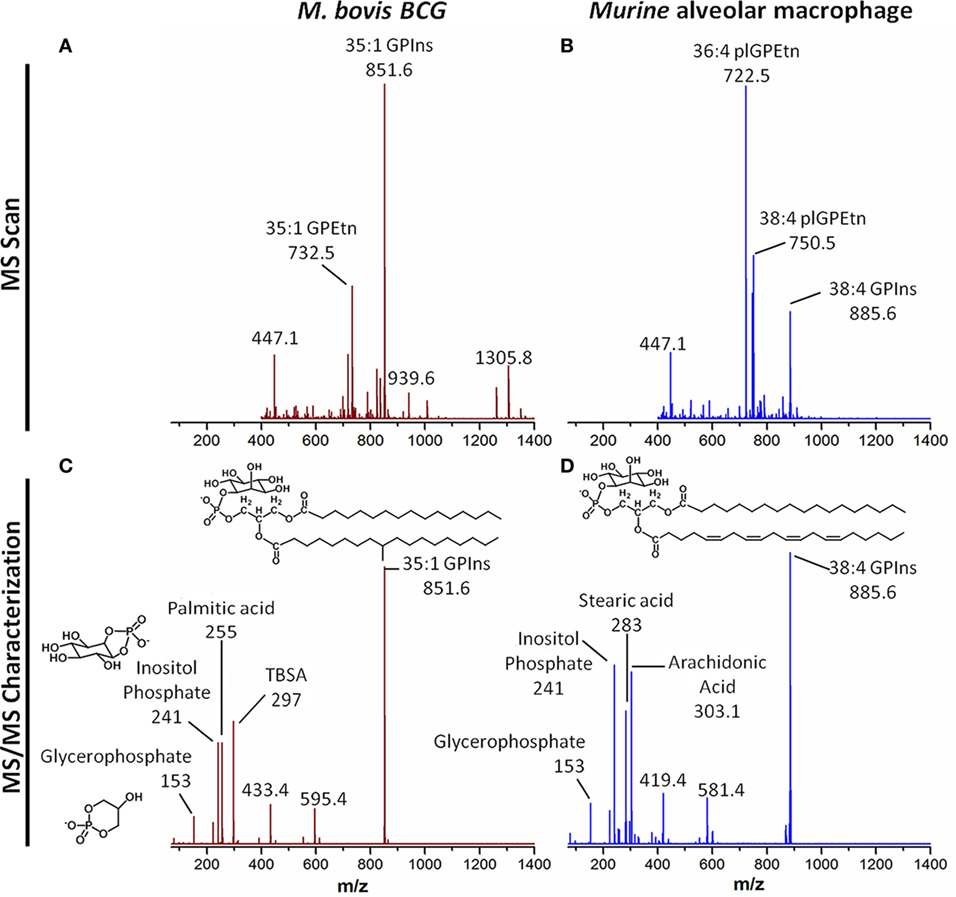
Figure 2. Resolving host and microbial (polar) lipid molecular species by mass spectrometry (MS), a tool for probing host–pathogen interactions. With advances in technologies particularly MS, it is now possible to profile, characterize, and quantify lipids from both microbes and macrophages as well as other host cells and tissues. (A) Single stage MS scan to generate the “lipid profiles” of M. bovis BCG, an experimental surrogate for M. tuberculosis. (B) Single stage MS scan to generate the “lipid profiles” of murine alveolar macrophages. (C) MS/MS of the major phosphatidylinositol from M. bovis BCG, with mass-to-charge ratio (m/z) of 851.6. Collision-induced dissociation reveals the ion contains palmitic acid (m/z 255) and TBSA (m/z 297), as well as the phosphoinositol (m/z 241) and glycerophosphate (m/z 153) headgroups. This confirms the sum composition of fatty acyls to be 35:1 (35 carbons, 1 double bond). (D) MS/MS of the major phosphatidylinositol from murine alveolar macrophages, with m/z of 885.6. Collision-induced dissociation reveals the ion contains stearic acid (m/z 283) and arachidonic acid (m/z 303.1), as well as the phosphoinositol (m/z 241) and glycerophosphate (m/z 153) headgroups. This confirms the sum composite of fatty acyls to be 38:4 (38 carbons, 4 double bonds). The bioanalytical tool can be applied to probe the dynamic changes in levels of thousands of species of lipids from both the host and the pathogens, leading to the identification of novel pathways involved in infection and inflammation. Abbreviations: GPEtn, phosphatidylethanolamine; GPIns, phosphatidylinositol; MS, mass spectrometry; MS/MS, tandem mass spectrometry; m/z: mass-to-charge ratio; plGPEtn, plasmalogen phosphatidylethanolamine; TBSA, tuberculostearic acid.
While LC–MS- and NMR-based lipidomics approaches are extremely powerful, they lack the spatial resolution needed for determining the localization of lipids during the infection process (220), and, importantly, the source of the metabolites in the host–pathogen relationship. MS-based imaging has now offered a new dimension by providing spatial resolution of the metabolites during the infection process by various pathogens (221–223). Alternative novel imaging techniques involve the use of immunofluorescent lipids, isotopic labeling, or lipid-binding probes and microscopy for the spatial resolution and real-time analysis of the host–pathogen interaction, providing extensive information regarding the roles of lipids in pathogenesis and the immune responses. In the study by Barisch and Soldati, pulse-chase experiments using fluorophore-labeled compounds coupled with electron microscopy has provided novel insights about the translocation of host-derived fatty acids in M. marinum to serve as an energy source for the bacteria (224, 225). Metabolic labeling of lipid droplets has also been used for the exploration of the carbon flux within pathogen metabolic networks (47). The lipid-binding properties of bacterial toxins, including perfringolysin O and aerolysin, have also been harnessed as tools to probe lipid localization and signaling (226, 227). In recent years, the specificities of lipid probes have vastly improved, with the recent possibility to study single lipid species in living cells (228).
The current availability and upcoming developments in technologies which provide temporal and/or spatial information of lipids at the single lipid species level are extremely powerful, especially when used in combination with cell and chemical biology, genetics and other systems biology approaches, including proteomics and genomics. Together, these will provide new insights into the complexity of host–pathogen interactions, disease pathogenesis as well as identification of novel markers of inflammation and infection.
Pathogenic infection of macrophages is an intricate process involving numerous sequences of events, during which lipids are clearly instrumental players. Each pathogen has evolved its own strategy to thrive in or kill the host cells, and it is not surprising to find that a single lipid class or species can display contrasting functions during bacterial infections, depending on the bacterial species and even cell types. A single lipid, such as cholesterol, can be utilized by a specific pathogen to promote its own survival in the macrophage and on the other hand, it can also be used by the host to assist in clearance of another bacterial species, making lipids a double-edge sword in the complex host–pathogen relationship. Adding on to the complexity is alterations in the fine chemistry of lipids. For instance, the oxidation of fatty acids can switch its function from being benign to bioactive, or from acting as the protagonist to the antagonist (and vice versa) in this complex interplay between host and pathogen. While we have described in this review how different lipid classes are involved in the infection process, it should be stressed that the system acts as a whole, and multiple components, including different lipid classes and proteins, act in concert to achieve the outcome of an infection. Hence, it is critical to undertake systems-level approaches to dissect the host–pathogen networks. Such in-depth analyses of the complex and interconnected lipid metabolic networks in infection and immunity will contribute to identification of potential lipids or metabolic pathways which can be potentially developed into therapeutics for treatment of infectious diseases or for boosting of immune functions.
OT, CKEA, and XLG contributed to the writing and review of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The research performed in XLG’s group was funded by the Ministry of Education, Singapore (MOE) Tier 1 grant (Reference number: 2015-T1-001-092) and the Nanyang Assistant Professorship (awarded to XLG).
1. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11(10):889–96. doi:10.1038/ni.1937
2. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
3. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi:10.1038/nri2448
4. van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell (2007) 129(7):1287–98. doi:10.1016/j.cell.2007.05.059
5. Boyce JA. Mast cells and eicosanoid mediators: a system of reciprocal paracrine and autocrine regulation. Immunol Rev (2007) 217:168–85. doi:10.1111/j.1600-065X.2007.00512.x
6. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet (2006) 7(8):606–19. doi:10.1038/nrg1879
7. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol (2008) 9(2):162–76. doi:10.1038/nrm2335
8. Wenk MR. Lipidomics of host-pathogen interactions. FEBS Lett (2006) 580(23):5541–51. doi:10.1016/j.febslet.2006.07.007
9. Helms B. Host-pathogen interactions: lipids grease the way. Eur J Lipid Sci Technol (2006) 108(11):895–7. doi:10.1002/ejlt.200600194
10. van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol (2008) 9(2):112–24. doi:10.1038/nrm2330
11. Ingolfsson HI, Melo MN, van Eerden FJ, Arnarez C, Lopez CA, Wassenaar TA, et al. Lipid organization of the plasma membrane. J Am Chem Soc (2014) 136(41):14554–9. doi:10.1021/ja507832e
12. Queiroz A, Riley LW. Bacterial immunostat: Mycobacterium tuberculosis lipids and their role in the host immune response. Rev Soc Bras Med Trop (2017) 50(1):9–18. doi:10.1590/0037-8682-0230-2016
13. Zaas DW, Duncan M, Rae Wright J, Abraham SN. The role of lipid rafts in the pathogenesis of bacterial infections. Biochim Biophys Acta (2005) 1746(3):305–13. doi:10.1016/j.bbamcr.2005.10.003
14. Feingold KR, Grunfeld C. Lipids: a key player in the battle between the host and microorganisms. J Lipid Res (2012) 53(12):2487–9. doi:10.1194/jlr.E033407
15. Ravnskov U. High cholesterol may protect against infections and atherosclerosis. QJM (2003) 96(12):927–34. doi:10.1093/qjmed/hcg150
16. van der Meer-Janssen YP, van Galen J, Batenburg JJ, Helms JB. Lipids in host-pathogen interactions: pathogens exploit the complexity of the host cell lipidome. Prog Lipid Res (2010) 49(1):1–26. doi:10.1016/j.plipres.2009.07.003
17. Herker E, Ott M. Emerging role of lipid droplets in host/pathogen interactions. J Biol Chem (2012) 287(4):2280–7. doi:10.1074/jbc.R111.300202
18. Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol (2010) 11(8):751–8. doi:10.1038/ni.1904
19. Bansal K, Narayana Y, Patil SA, Balaji KN. M. bovis BCG induced expression of COX-2 involves nitric oxide-dependent and -independent signaling pathways. J Leukoc Biol (2009) 85(5):804–16. doi:10.1189/jlb.0908561
20. Lemmon MA. Phosphoinositide recognition domains. Traffic (2003) 4(4):201–13. doi:10.1034/j.1600-0854.2004.00071.x
21. Ireton K, Payrastre B, Chap H, Ogawa W, Sakaue H, Kasuga M, et al. A role for phosphoinositide 3-kinase in bacterial invasion. Science (1996) 274(5288):780–2. doi:10.1126/science.274.5288.780
22. Fratti RA, Backer JM, Gruenberg J, Corvera S, Deretic V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol (2001) 154(3):631–44. doi:10.1083/jcb.200106049
23. Vergne I, Chua J, Deretic V. Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J Exp Med (2003) 198(4):653–9. doi:10.1084/jem.20030527
24. Callahan MK, Williamson P, Schlegel RA. Surface expression of phosphatidylserine on macrophages is required for phagocytosis of apoptotic thymocytes. Cell Death Differ (2000) 7(7):645–53. doi:10.1038/sj.cdd.4400690
25. McCollister BD, Myers JT, Jones-Carson J, Voelker DR, Vazquez-Torres A. Constitutive acid sphingomyelinase enhances early and late macrophage killing of Salmonella enterica serovar Typhimurium. Infect Immun (2007) 75(11):5346–52. doi:10.1128/IAI.00689-07
26. Barnawi J, Tran H, Jersmann H, Pitson S, Roscioli E, Hodge G, et al. Potential link between the sphingosine-1-phosphate (S1P) system and defective alveolar macrophage phagocytic function in chronic obstructive pulmonary disease (COPD). PLoS One (2015) 10(10):e0122771. doi:10.1371/journal.pone.0122771
27. Simons K, Ikonen E. Functional rafts in cell membranes. Nature (1997) 387(6633):569–72. doi:10.1038/42408
28. Parihar SP, Guler R, Lang DM, Suzuki H, Marais AD, Brombacher F. Simvastatin enhances protection against Listeria monocytogenes infection in mice by counteracting Listeria-induced phagosomal escape. PLoS One (2013) 8(9):e75490. doi:10.1371/journal.pone.0075490
29. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol (2006) 177(10):7303–11. doi:10.4049/jimmunol.177.10.7303
30. Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol (2010) 185(1):605–14. doi:10.4049/jimmunol.0901698
31. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab (2006) 4(1):13–24. doi:10.1016/j.cmet.2006.08.006
32. Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, et al. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem (2012) 287(15):11629–41. doi:10.1074/jbc.M111.338673
33. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem (2002) 277(51):49982–8. doi:10.1074/jbc.M209649200
34. Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res (2011) 108(8):985–95. doi:10.1161/CIRCRESAHA.110.233775
35. Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol (2014) 15(9):846–55. doi:10.1038/ni.2956
36. Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nat Rev Immunol (2011) 11(2):81. doi:10.1038/nri2922
37. Ecker J, Liebisch G, Englmaier M, Grandl M, Robenek H, Schmitz G. Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes. Proc Natl Acad Sci U S A (2010) 107(17):7817–22. doi:10.1073/pnas.0912059107
38. Zhang C, Wang Y, Wang F, Wang Z, Lu Y, Xu Y, et al. Quantitative profiling of glycerophospholipids during mouse and human macrophage differentiation using targeted mass spectrometry. Sci Rep (2017) 7(1):412. doi:10.1038/s41598-017-00341-2
39. Schroit AJ, Gallily R. Macrophage fatty acid composition and phagocytosis: effect of unsaturation on cellular phagocytic activity. Immunology (1979) 36(2):199–205.
40. Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem (1993) 268(16):11811–6.
41. Spady DK, Kearney DM, Hobbs HH. Polyunsaturated fatty acids up-regulate hepatic scavenger receptor B1 (SR-BI) expression and HDL cholesteryl ester uptake in the hamster. J Lipid Res (1999) 40(8):1384–94.
42. Alvarez-Curto E, Milligan G. Metabolism meets immunity: the role of free fatty acid receptors in the immune system. Biochem Pharmacol (2016) 114:3–13. doi:10.1016/j.bcp.2016.03.017
43. Calder PC. Functional roles of fatty acids and their effects on human health. JPEN J Parenter Enteral Nutr (2015) 39(1 Suppl):18S–32S. doi:10.1177/0148607115595980
44. Cho KY, Salton MR. Fatty acid composition of bacterial membrane and wall lipids. Biochim Biophys Acta (1966) 116(1):73–9. doi:10.1016/0005-2760(66)90093-2
45. McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature (2000) 406(6797):735–8. doi:10.1038/35021074
46. Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog (2011) 7(6):e1002093. doi:10.1371/journal.ppat.1002093
47. Lee W, VanderVen BC, Fahey RJ, Russell DG. Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J Biol Chem (2013) 288(10):6788–800. doi:10.1074/jbc.M112.445056
48. Nazarova EV, Montague CR, La T, Wilburn KM, Sukumar N, Lee W, et al. Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. Elife (2017) 6:e26969. doi:10.7554/eLife.26969
49. Daniel J, Sirakova T, Kolattukudy P. An acyl-CoA synthetase in Mycobacterium tuberculosis involved in triacylglycerol accumulation during dormancy. PLoS One (2014) 9(12):e114877. doi:10.1371/journal.pone.0114877
50. Deb C, Daniel J, Sirakova TD, Abomoelak B, Dubey VS, Kolattukudy PE. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J Biol Chem (2006) 281(7):3866–75. doi:10.1074/jbc.M505556200
51. Saka HA, Valdivia R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu Rev Cell Dev Biol (2012) 28:411–37. doi:10.1146/annurev-cellbio-092910-153958
52. Barisch C, Soldati T. Breaking fat! How mycobacteria and other intracellular pathogens manipulate host lipid droplets. Biochimie (2017) 141:54–61. doi:10.1016/j.biochi.2017.06.001
53. Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med (2008) 14(10):461–9. doi:10.1016/j.molmed.2008.08.005
54. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol (2015) 15(8):511–23. doi:10.1038/nri3859
55. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol (2008) 8(5):349–61. doi:10.1038/nri2294
56. Eckmann L, Stenson WF, Savidge TC, Lowe DC, Barrett KE, Fierer J, et al. Role of intestinal epithelial cells in the host secretory response to infection by invasive bacteria. Bacterial entry induces epithelial prostaglandin H synthase-2 expression and prostaglandin E2 and F2alpha production. J Clin Invest (1997) 100(2):296–309. doi:10.1172/JCI119535
57. Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med (2008) 205(12):2791–801. doi:10.1084/jem.20080767
58. Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol (2009) 10(8):899–906. doi:10.1038/ni.1758
59. Zaslona Z, Serezani CH, Okunishi K, Aronoff DM, Peters-Golden M. Prostaglandin E2 restrains macrophage maturation via E prostanoid receptor 2/protein kinase A signaling. Blood (2012) 119(10):2358–67. doi:10.1182/blood-2011-08-374207
60. Serezani CH, Chung J, Ballinger MN, Moore BB, Aronoff DM, Peters-Golden M. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am J Respir Cell Mol Biol (2007) 37(5):562–70. doi:10.1165/rcmb.2007-0153OC
61. Uchiya K, Nikai T. Salmonella enterica serovar Typhimurium infection induces cyclooxygenase 2 expression in macrophages: involvement of Salmonella pathogenicity island 2. Infect Immun (2004) 72(12):6860–9. doi:10.1128/IAI.72.12.6860-6869.2004
62. Asakrah S, Nieves W, Mahdi Z, Agard M, Zea AH, Roy CJ, et al. Post-exposure therapeutic efficacy of COX-2 inhibition against Burkholderia pseudomallei. PLoS Negl Trop Dis (2013) 7(5):e2212. doi:10.1371/journal.pntd.0002212
63. Uchiya K, Groisman EA, Nikai T. Involvement of Salmonella pathogenicity island 2 in the up-regulation of interleukin-10 expression in macrophages: role of protein kinase A signal pathway. Infect Immun (2004) 72(4):1964–73. doi:10.1128/IAI.72.4.1964-1973.2004
64. Kieran NE, Maderna P, Godson C. Lipoxins: potential anti-inflammatory, proresolution, and antifibrotic mediators in renal disease. Kidney Int (2004) 65(4):1145–54. doi:10.1111/j.1523-1755.2004.00487.x
65. Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci U S A (1984) 81(17):5335–9. doi:10.1073/pnas.81.17.5335
66. Bafica A, Scanga CA, Serhan C, Machado F, White S, Sher A, et al. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J Clin Invest (2005) 115(6):1601–6. doi:10.1172/JCI23949
67. Prieto P, Cuenca J, Traves PG, Fernandez-Velasco M, Martin-Sanz P, Bosca L. Lipoxin A4 impairment of apoptotic signaling in macrophages: implication of the PI3K/Akt and the ERK/Nrf-2 defense pathways. Cell Death Differ (2010) 17(7):1179–88. doi:10.1038/cdd.2009.220
68. Fahel JS, de Souza MB, Gomes MT, Corsetti PP, Carvalho NB, Marinho FA, et al. 5-Lipoxygenase negatively regulates Th1 response during Brucella abortus infection in mice. Infect Immun (2015) 83(3):1210–6. doi:10.1128/IAI.02592-14
69. Liang L, Zhang Q, Luo LL, Yue J, Zhao YL, Han M, et al. Polymorphisms in the prostaglandin receptor EP2 gene confers susceptibility to tuberculosis. Infect Genet Evol (2016) 46:23–7. doi:10.1016/j.meegid.2016.10.016
70. Tobin DM, Vary JC Jr, Ray JP, Walsh GS, Dunstan SJ, Bang ND, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell (2010) 140(5):717–30. doi:10.1016/j.cell.2010.02.013
71. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell (2012) 148(3):434–46. doi:10.1016/j.cell.2011.12.023
72. Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys (2010) 39:407–27. doi:10.1146/annurev.biophys.093008.131234
73. Schroit AJ, Madsen JW, Tanaka Y. In vivo recognition and clearance of red blood cells containing phosphatidylserine in their plasma membranes. J Biol Chem (1985) 260(8):5131–8.
74. Czuczman MA, Fattouh R, van Rijn JM, Canadien V, Osborne S, Muise AM, et al. Listeria monocytogenes exploits efferocytosis to promote cell-to-cell spread. Nature (2014) 509(7499):230–4. doi:10.1038/nature13168
75. Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe (2012) 12(3):289–300. doi:10.1016/j.chom.2012.06.010
76. Yang CT, Cambier CJ, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L. Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe (2012) 12(3):301–12. doi:10.1016/j.chom.2012.07.009
77. Defacque H, Bos E, Garvalov B, Barret C, Roy C, Mangeat P, et al. Phosphoinositides regulate membrane-dependent actin assembly by latex bead phagosomes. Mol Biol Cell (2002) 13(4):1190–202. doi:10.1091/mbc.01-06-0314
78. De Matteis MA, Godi A. PI-loting membrane traffic. Nat Cell Biol (2004) 6(6):487–92. doi:10.1038/ncb0604-487
79. Vergne I, Chua J, Lee HH, Lucas M, Belisle J, Deretic V. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2005) 102(11):4033–8. doi:10.1073/pnas.0409716102
80. Vergne I, Fratti RA, Hill PJ, Chua J, Belisle J, Deretic V. Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol Biol Cell (2004) 15(2):751–60. doi:10.1091/mbc.E03-05-0307
81. Alpuche-Aranda CM, Racoosin EL, Swanson JA, Miller SI. Salmonella stimulate macrophage macropinocytosis and persist within spacious phagosomes. J Exp Med (1994) 179(2):601–8. doi:10.1084/jem.179.2.601
82. Drecktrah D, Knodler LA, Galbraith K, Steele-Mortimer O. The Salmonella SPI1 effector SopB stimulates nitric oxide production long after invasion. Cell Microbiol (2005) 7(1):105–13. doi:10.1111/j.1462-5822.2004.00436.x
83. Ochman H, Soncini FC, Solomon F, Groisman EA. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc Natl Acad Sci U S A (1996) 93(15):7800–4. doi:10.1073/pnas.93.15.7800
84. Shea JE, Hensel M, Gleeson C, Holden DW. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc Natl Acad Sci U S A (1996) 93(6):2593–7. doi:10.1073/pnas.93.6.2593
85. Norris FA, Wilson MP, Wallis TS, Galyov EE, Majerus PW. SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc Natl Acad Sci U S A (1998) 95(24):14057–9. doi:10.1073/pnas.95.24.14057
86. Marcus SL, Wenk MR, Steele-Mortimer O, Finlay BB. A synaptojanin-homologous region of Salmonella typhimurium SigD is essential for inositol phosphatase activity and Akt activation. FEBS Lett (2001) 494(3):201–7. doi:10.1016/S0014-5793(01)02356-0
87. Terebiznik MR, Vieira OV, Marcus SL, Slade A, Yip CM, Trimble WS, et al. Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat Cell Biol (2002) 4(10):766–73. doi:10.1038/ncb854
88. Hernandez LD, Hueffer K, Wenk MR, Galan JE. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science (2004) 304(5678):1805–7. doi:10.1126/science.1098188
89. Gaspar AH, Machner MP. VipD is a Rab5-activated phospholipase A1 that protects Legionella pneumophila from endosomal fusion. Proc Natl Acad Sci U S A (2014) 111(12):4560–5. doi:10.1073/pnas.1316376111
90. O’Brien DK, Melville SB. Effects of Clostridium perfringens alpha-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect Immun (2004) 72(9):5204–15. doi:10.1128/IAI.72.9.5204-5215.2004
91. Raynaud C, Guilhot C, Rauzier J, Bordat Y, Pelicic V, Manganelli R, et al. Phospholipases C are involved in the virulence of Mycobacterium tuberculosis. Mol Microbiol (2002) 45(1):203–17. doi:10.1046/j.1365-2958.2002.03009.x
92. Le Chevalier F, Cascioferro A, Frigui W, Pawlik A, Boritsch EC, Bottai D, et al. Revisiting the role of phospholipases C in virulence and the lifecycle of Mycobacterium tuberculosis. Sci Rep (2015) 5:16918. doi:10.1038/srep16918
93. Viana-Niero C, de Haas PE, van Soolingen D, Leao SC. Analysis of genetic polymorphisms affecting the four phospholipase C (plc) genes in Mycobacterium tuberculosis complex clinical isolates. Microbiology (2004) 150(Pt 4):967–78. doi:10.1099/mic.0.26778-0
94. Auricchio G, Garg SK, Martino A, Volpe E, Ciaramella A, De Vito P, et al. Role of macrophage phospholipase D in natural and CpG-induced antimycobacterial activity. Cell Microbiol (2003) 5(12):913–20. doi:10.1046/j.1462-5822.2003.00330.x
95. Schneider BE, Behrends J, Hagens K, Harmel N, Shayman JA, Schaible UE. Lysosomal phospholipase A2: a novel player in host immunity to Mycobacterium tuberculosis. Eur J Immunol (2014) 44(8):2394–404. doi:10.1002/eji.201344383
96. Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol (2003) 4(5):397–407. doi:10.1038/nrm1103
97. Luberto C, Hannun YA. Sphingolipid metabolism in the regulation of bioactive molecules. Lipids (1999) 34(Suppl):S5–11. doi:10.1007/BF02562221
98. Merrill AH Jr, Schmelz EM, Dillehay DL, Spiegel S, Shayman JA, Schroeder JJ, et al. Sphingolipids – the enigmatic lipid class: biochemistry, physiology, and pathophysiology. Toxicol Appl Pharmacol (1997) 142(1):208–25. doi:10.1006/taap.1996.8029
99. van der Goot FG, Harder T. Raft membrane domains: from a liquid-ordered membrane phase to a site of pathogen attack. Semin Immunol (2001) 13(2):89–97. doi:10.1006/smim.2000.0300
100. Manes S, del Real G, Martinez AC. Pathogens: raft hijackers. Nat Rev Immunol (2003) 3(7):557–68. doi:10.1038/nri1129
101. Rosenberger CM, Brumell JH, Finlay BB. Microbial pathogenesis: lipid rafts as pathogen portals. Curr Biol (2000) 10(22):R823–5. doi:10.1016/S0960-9822(00)00788-0
102. Riethmuller J, Riehle A, Grassme H, Gulbins E. Membrane rafts in host-pathogen interactions. Biochim Biophys Acta (2006) 1758(12):2139–47. doi:10.1016/j.bbamem.2006.07.017
104. Heung LJ, Luberto C, Del Poeta M. Role of sphingolipids in microbial pathogenesis. Infect Immun (2006) 74(1):28–39. doi:10.1128/IAI.74.1.28-39.2006
105. Fueller M, Wang DA, Tigyi G, Siess W. Activation of human monocytic cells by lysophosphatidic acid and sphingosine-1-phosphate. Cell Signal (2003) 15(4):367–75. doi:10.1016/S0898-6568(02)00117-1
106. Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol (2008) 8(10):753–63. doi:10.1038/nri2400
107. Malik ZA, Thompson CR, Hashimi S, Porter B, Iyer SS, Kusner DJ. Cutting edge: Mycobacterium tuberculosis blocks Ca2+ signaling and phagosome maturation in human macrophages via specific inhibition of sphingosine kinase. J Immunol (2003) 170(6):2811–5. doi:10.4049/jimmunol.170.6.2811
108. Garg SK, Volpe E, Palmieri G, Mattei M, Galati D, Martino A, et al. Sphingosine 1-phosphate induces antimicrobial activity both in vitro and in vivo. J Infect Dis (2004) 189(11):2129–38. doi:10.1086/386286
109. Santucci MB, Greco E, De Spirito M, Arcovito G, De Angelis G, Cauda R, et al. Sphingosine 1-phosphate promotes antigen processing and presentation to CD4+ T cells in Mycobacterium tuberculosis-infected monocytes. Biochem Biophys Res Commun (2007) 361(3):687–93. doi:10.1016/j.bbrc.2007.07.087
110. Utermohlen O, Herz J, Schramm M, Kronke M. Fusogenicity of membranes: the impact of acid sphingomyelinase on innate immune responses. Immunobiology (2008) 213(3–4):307–14. doi:10.1016/j.imbio.2007.10.016
111. Vazquez CL, Rodgers A, Herbst S, Coade S, Gronow A, Guzman CA, et al. The proneurotrophin receptor sortilin is required for Mycobacterium tuberculosis control by macrophages. Sci Rep (2016) 6:29332. doi:10.1038/srep29332
112. Utermohlen O, Karow U, Lohler J, Kronke M. Severe impairment in early host defense against Listeria monocytogenes in mice deficient in acid sphingomyelinase. J Immunol (2003) 170(5):2621–8. doi:10.4049/jimmunol.170.5.2621
113. Li C, Peng H, Japtok L, Seitz A, Riehle A, Wilker B, et al. Inhibition of neutral sphingomyelinase protects mice against systemic tuberculosis. Front Biosci (Elite Ed) (2016) 8:311–25. doi:10.2741/e769
114. Flores-Diaz M, Monturiol-Gross L, Naylor C, Alape-Giron A, Flieger A. Bacterial sphingomyelinases and phospholipases as virulence factors. Microbiol Mol Biol Rev (2016) 80(3):597–628. doi:10.1128/MMBR.00082-15
115. Speer A, Sun J, Danilchanka O, Meikle V, Rowland JL, Walter K, et al. Surface hydrolysis of sphingomyelin by the outer membrane protein Rv0888 supports replication of Mycobacterium tuberculosis in macrophages. Mol Microbiol (2015) 97(5):881–97. doi:10.1111/mmi.13073
116. Dubos RJ. The effect of sphingomyelin on the growth of tubercle bacilli. J Exp Med (1948) 88(1):73–9. doi:10.1084/jem.88.1.73
117. Montes LR, Goni FM, Johnston NC, Goldfine H, Alonso A. Membrane fusion induced by the catalytic activity of a phospholipase C/sphingomyelinase from Listeria monocytogenes. Biochemistry (2004) 43(12):3688–95. doi:10.1021/bi0352522
118. Rolando M, Escoll P, Nora T, Botti J, Boitez V, Bedia C, et al. Legionella pneumophila S1P-lyase targets host sphingolipid metabolism and restrains autophagy. Proc Natl Acad Sci U S A (2016) 113(7):1901–6. doi:10.1073/pnas.1522067113
119. McLean CJ, Marles-Wright J, Custodio R, Lowther J, Kennedy AJ, Pollock J, et al. Characterization of homologous sphingosine-1-phosphate lyase isoforms in the bacterial pathogen Burkholderia pseudomallei. J Lipid Res (2017) 58(1):137–50. doi:10.1194/jlr.M071258
120. Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov (2013) 12(9):688–702. doi:10.1038/nrd4099
121. Poralla K, Kannenberg E. Hopanoids: sterol equivalents in bacteria. In: Fuller G, Nes WD, editors. Ecology and Metabolism of Plant Lipids. ACS Symposium Series. (Vol. 325). Washington, DC: American Chemical Society (1987). p. 239–51.
122. Riobo NA. Cholesterol and its derivatives in Sonic Hedgehog signaling and cancer. Curr Opin Pharmacol (2012) 12(6):736–41. doi:10.1016/j.coph.2012.07.002
123. Haines TH. Do sterols reduce proton and sodium leaks through lipid bilayers? Prog Lipid Res (2001) 40(4):299–324. doi:10.1016/S0163-7827(01)00009-1
124. Yeagle PL. Cholesterol and the cell membrane. Biochim Biophys Acta (1985) 822(3–4):267–87. doi:10.1016/0304-4157(85)90011-5
125. Berg J, Tymoczko J, Stryer L. Important derivatives of cholesterol include bile salts and steroid hormones. 5th ed. Biochemistry. New York: W. H. Freeman (2002).
126. Mattos KA, Oliveira VC, Berredo-Pinho M, Amaral JJ, Antunes LC, Melo RC, et al. Mycobacterium leprae intracellular survival relies on cholesterol accumulation in infected macrophages: a potential target for new drugs for leprosy treatment. Cell Microbiol (2014) 16(6):797–815. doi:10.1111/cmi.12279
127. Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med (2010) 2(7):258–74. doi:10.1002/emmm.201000079
128. Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science (2000) 288(5471):1647–50. doi:10.1126/science.288.5471.1647
129. Kendall SL, Withers M, Soffair CN, Moreland NJ, Gurcha S, Sidders B, et al. A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol Microbiol (2007) 65(3):684–99. doi:10.1111/j.1365-2958.2007.05827.x
130. Kendall SL, Burgess P, Balhana R, Withers M, Ten Bokum A, Lott JS, et al. Cholesterol utilization in mycobacteria is controlled by two TetR-type transcriptional regulators: kstR and kstR2. Microbiology (2010) 156(Pt 5):1362–71. doi:10.1099/mic.0.034538-0
131. Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A (2008) 105(11):4376–80. doi:10.1073/pnas.0711159105
132. Griffin JE, Pandey AK, Gilmore SA, Mizrahi V, McKinney JD, Bertozzi CR, et al. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem Biol (2012) 19(2):218–27. doi:10.1016/j.chembiol.2012.08.012
133. Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, et al. igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol (2009) 191(16):5232–9. doi:10.1128/JB.00452-09
134. Portnoy DA, Jacks PS, Hinrichs DJ. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med (1988) 167(4):1459–71. doi:10.1084/jem.167.4.1459
135. Birmingham CL, Canadien V, Kaniuk NA, Steinberg BE, Higgins DE, Brumell JH. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature (2008) 451(7176):350–4. doi:10.1038/nature06479
136. Bhakdi S, Tranum-Jensen J, Sziegoleit A. Mechanism of membrane damage by streptolysin-O. Infect Immun (1985) 47(1):52–60.
137. Hara H, Tsuchiya K, Nomura T, Kawamura I, Shoma S, Mitsuyama M. Dependency of caspase-1 activation induced in macrophages by Listeria monocytogenes on cytolysin, listeriolysin O, after evasion from phagosome into the cytoplasm. J Immunol (2008) 180(12):7859–68. doi:10.4049/jimmunol.180.12.7859
138. Catron DM, Sylvester MD, Lange Y, Kadekoppala M, Jones BD, Monack DM, et al. The Salmonella-containing vacuole is a major site of intracellular cholesterol accumulation and recruits the GPI-anchored protein CD55. Cell Microbiol (2002) 4(6):315–28. doi:10.1046/j.1462-5822.2002.00198.x
139. Nawabi P, Catron DM, Haldar K. Esterification of cholesterol by a type III secretion effector during intracellular Salmonella infection. Mol Microbiol (2008) 68(1):173–85. doi:10.1111/j.1365-2958.2008.06142.x
140. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis (2014) 209(5):754–63. doi:10.1093/infdis/jit550
141. Catron DM, Lange Y, Borensztajn J, Sylvester MD, Jones BD, Haldar K. Salmonella enterica serovar Typhimurium requires nonsterol precursors of the cholesterol biosynthetic pathway for intracellular proliferation. Infect Immun (2004) 72(2):1036–42. doi:10.1128/IAI.72.2.1036-1042.2004
142. Mulye M, Samanta D, Winfree S, Heinzen RA, Gilk SD. Elevated cholesterol in the Coxiella burnetii intracellular niche is bacteriolytic. mBio (2017) 8(1):e02313–16. doi:10.1128/mBio.02313-16
143. Czyz DM, Potluri LP, Jain-Gupta N, Riley SP, Martinez JJ, Steck TL, et al. Host-directed antimicrobial drugs with broad-spectrum efficacy against intracellular bacterial pathogens. mBio (2014) 5(4):e01534–14. doi:10.1128/mBio.01534-14
144. Araldi E, Fernandez-Fuertes M, Canfran-Duque A, Tang W, Cline GW, Madrigal-Matute J, et al. Lanosterol modulates TLR4-mediated innate immune responses in macrophages. Cell Rep (2017) 19(13):2743–55. doi:10.1016/j.celrep.2017.05.093
145. Bikle DD. Vitamin D and the immune system: role in protection against bacterial infection. Curr Opin Nephrol Hypertens (2008) 17(4):348–52. doi:10.1097/MNH.0b013e3282ff64a3
146. Verway M, Bouttier M, Wang TT, Carrier M, Calderon M, An BS, et al. Vitamin D induces interleukin-1beta expression: paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLoS Pathog (2013) 9(6):e1003407. doi:10.1371/journal.ppat.1003407
147. Wilkinson RJ, Llewelyn M, Toossi Z, Patel P, Pasvol G, Lalvani A, et al. Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: a case-control study. Lancet (2000) 355(9204):618–21. doi:10.1016/S0140-6736(99)02301-6
148. Salahuddin N, Ali F, Hasan Z, Rao N, Aqeel M, Mahmood F. Vitamin D accelerates clinical recovery from tuberculosis: results of the SUCCINCT study [supplementary cholecalciferol in recovery from tuberculosis]. A randomized, placebo-controlled, clinical trial of vitamin D supplementation in patients with pulmonary tuberculosis’. BMC Infect Dis (2013) 13:22. doi:10.1186/1471-2334-13-22
149. Coussens AK, Wilkinson RJ, Martineau AR. Phenylbutyrate is bacteriostatic against Mycobacterium tuberculosis and regulates the macrophage response to infection, synergistically with 25-hydroxy-vitamin D3. PLoS Pathog (2015) 11(7):e1005007. doi:10.1371/journal.ppat.1005007
150. Raetz CR, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in Gram-negative bacteria. Annu Rev Biochem (2007) 76:295–329. doi:10.1146/annurev.biochem.76.010307.145803
151. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature (1997) 388(6640):394–7. doi:10.1038/41131
152. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282(5396):2085–8. doi:10.1126/science.282.5396.2085
153. Akashi S, Saitoh S, Wakabayashi Y, Kikuchi T, Takamura N, Nagai Y, et al. Lipopolysaccharide interaction with cell surface toll-like receptor 4-MD-2: higher affinity than that with MD-2 or CD14. J Exp Med (2003) 198(7):1035–42. doi:10.1084/jem.20031076
154. Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, et al. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature (1998) 395(6699):284–8. doi:10.1038/26239
155. Kirschning CJ, Wesche H, Merrill Ayres T, Rothe M. Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J Exp Med (1998) 188(11):2091–7. doi:10.1084/jem.188.11.2091
156. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4(7):499–511. doi:10.1038/nri1391
157. Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem (2002) 71:635–700. doi:10.1146/annurev.biochem.71.110601.135414
158. Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med (2013) 45:e66. doi:10.1038/emm.2013.97
159. Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol (2001) 167(10):5887–94. doi:10.4049/jimmunol.167.10.5887
160. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science (2013) 341(6151):1250–3. doi:10.1126/science.1240988
161. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341(6151):1246–9. doi:10.1126/science.1240248
162. Ng TM, Monack DM. Revisiting caspase-11 function in host defense. Cell Host Microbe (2013) 14(1):9–14. doi:10.1016/j.chom.2013.06.009
163. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514(7521):187–92. doi:10.1038/nature13683
164. Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, et al. Severe impairment of interleukin-1 and toll-like receptor signalling in mice lacking IRAK-4. Nature (2002) 416(6882):750–6. doi:10.1038/nature736
165. Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol (1999) 162(7):3749–52.
166. Knuefermann P, Nemoto S, Misra A, Nozaki N, Defreitas G, Goyert SM, et al. CD14-deficient mice are protected against lipopolysaccharide-induced cardiac inflammation and left ventricular dysfunction. Circulation (2002) 106(20):2608–15. doi:10.1161/01.CIR.0000038110.69369.4C
167. Lequire VS, Hutcherson JD, Hamilton RL, Gray ME. The effects of bacterial endotoxin on lipide metabolism. I. The responses of the serum lipides of rabbits to single and repeated injections of Shear’s polysaccharide. J Exp Med (1959) 110(2):293–309. doi:10.1084/jem.110.2.293
168. Kaufmann RL, Matson CF, Beisel WR. Hypertriglyceridemia produced by endotoxin: role of impaired triglyceride disposal mechanisms. J Infect Dis (1976) 133(5):548–55. doi:10.1093/infdis/133.5.548
169. Feingold KR, Staprans I, Memon RA, Moser AH, Shigenaga JK, Doerrler W, et al. Endotoxin rapidly induces changes in lipid metabolism that produce hypertriglyceridemia: low doses stimulate hepatic triglyceride production while high doses inhibit clearance. J Lipid Res (1992) 33(12):1765–76.
170. Huang YL, Morales-Rosado J, Ray J, Myers TG, Kho T, Lu M, et al. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J Biol Chem (2014) 289(5):3001–12. doi:10.1074/jbc.M113.524587
171. Rubinow KB, Wall VZ, Nelson J, Mar D, Bomsztyk K, Askari B, et al. Acyl-CoA synthetase 1 is induced by Gram-negative bacteria and lipopolysaccharide and is required for phospholipid turnover in stimulated macrophages. J Biol Chem (2013) 288(14):9957–70. doi:10.1074/jbc.M113.458372
172. D’Avila H, Melo RC, Parreira GG, Werneck-Barroso E, Castro-Faria-Neto HC, Bozza PT. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol (2006) 176(5):3087–97. doi:10.4049/jimmunol.176.5.3087
173. Pacheco P, Bozza FA, Gomes RN, Bozza M, Weller PF, Castro-Faria-Neto HC, et al. Lipopolysaccharide-induced leukocyte lipid body formation in vivo: innate immunity elicited intracellular loci involved in eicosanoid metabolism. J Immunol (2002) 169(11):6498–506. doi:10.4049/jimmunol.169.11.6498
174. Khatchadourian A, Bourque SD, Richard VR, Titorenko VI, Maysinger D. Dynamics and regulation of lipid droplet formation in lipopolysaccharide (LPS)-stimulated microglia. Biochim Biophys Acta (2012) 1821(4):607–17. doi:10.1016/j.bbalip.2012.01.007
175. Maya-Monteiro CM, Almeida PE, D’Avila H, Martins AS, Rezende AP, Castro-Faria-Neto H, et al. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J Biol Chem (2008) 283(4):2203–10. doi:10.1074/jbc.M706706200
176. Dennis EA, Deems RA, Harkewicz R, Quehenberger O, Brown HA, Milne SB, et al. A mouse macrophage lipidome. J Biol Chem (2010) 285(51):39976–85. doi:10.1074/jbc.M110.182915
177. Koberlin MS, Snijder B, Heinz LX, Baumann CL, Fauster A, Vladimer GI, et al. A conserved circular network of coregulated lipids modulates innate immune responses. Cell (2015) 162(1):170–83. doi:10.1016/j.cell.2015.05.051
178. Jankute M, Cox JA, Harrison J, Besra GS. Assembly of the mycobacterial cell wall. Annu Rev Microbiol (2015) 69:405–23. doi:10.1146/annurev-micro-091014-104121
179. Kaur G, Kaur J. Multifaceted role of lipids in Mycobacterium leprae. Future Microbiol (2017) 12:315–35. doi:10.2217/fmb-2016-0173
180. Bloch H, Sorkin E, Erlenmeyer H. A toxic lipid component of the tubercle bacillus (cord factor). I. Isolation from petroleum ether extracts of young bacterial cultures. Am Rev Tuberc (1953) 67(5):629–43.
181. Hunter RL, Venkataprasad N, Olsen MR. The role of trehalose dimycolate (cord factor) on morphology of virulent M. tuberculosis in vitro. Tuberculosis (2006) 86(5):349–56. doi:10.1016/j.tube.2005.08.017
182. Sakamoto K, Kim MJ, Rhoades ER, Allavena RE, Ehrt S, Wainwright HC, et al. Mycobacterial trehalose dimycolate reprograms macrophage global gene expression and activates matrix metalloproteinases. Infect Immun (2013) 81(3):764–76. doi:10.1128/IAI.00906-12
183. Geisel RE, Sakamoto K, Russell DG, Rhoades ER. In vivo activity of released cell wall lipids of Mycobacterium bovis bacillus Calmette-Guerin is due principally to trehalose mycolates. J Immunol (2005) 174(8):5007–15. doi:10.4049/jimmunol.174.8.5007
184. Yamagami H, Matsumoto T, Fujiwara N, Arakawa T, Kaneda K, Yano I, et al. Trehalose 6,6’-dimycolate (cord factor) of Mycobacterium tuberculosis induces foreign-body- and hypersensitivity-type granulomas in mice. Infect Immun (2001) 69(2):810–5. doi:10.1128/IAI.69.2.810-815.2001
185. Perez RL, Roman J, Roser S, Little C, Olsen M, Indrigo J, et al. Cytokine message and protein expression during lung granuloma formation and resolution induced by the mycobacterial cord factor trehalose-6,6’-dimycolate. J Interferon Cytokine Res (2000) 20(9):795–804. doi:10.1089/10799900050151067
186. Bekierkunst A, Yarkoni E. Granulomatous hypersensitivity to trehalose-6,6’-dimycolate (cord factor) in mice infected with BCG. Infect Immun (1973) 7(4):631–8.
187. Silva CL, Ekizlerian SM, Fazioli RA. Role of cord factor in the modulation of infection caused by mycobacteria. Am J Pathol (1985) 118(2):238–47.
188. Indrigo J, Hunter RL Jr, Actor JK. Cord factor trehalose 6,6’-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology (2003) 149(Pt 8):2049–59. doi:10.1099/mic.0.26226-0
189. Bloch H, Noll H. Studies on the virulence of tubercle bacilli; the effect of cord factor on murine tuberculosis. Br J Exp Pathol (1955) 36(1):8–17.
190. Retzinger GS, Meredith SC, Hunter RL, Takayama K, Kezdy FJ. Identification of the physiologically active state of the mycobacterial glycolipid trehalose 6,6’-dimycolate and the role of fibrinogen in the biologic activities of trehalose 6,6’-dimycolate monolayers. J Immunol (1982) 129(2):735–44.
191. Yarkoni E, Rapp HJ. Toxicity of emulsified trehalose-6,6’-dimycolate (cord factor) in mice depends on size distribution of mineral oil droplets. Infect Immun (1978) 20(3):856–60.
192. Hunter RL, Olsen M, Jagannath C, Actor JK. Trehalose 6,6’-dimycolate and lipid in the pathogenesis of caseating granulomas of tuberculosis in mice. Am J Pathol (2006) 168(4):1249–61. doi:10.2353/ajpath.2006.050848
193. Yarkoni E, Rapp HJ. Influence of oil and Tween concentrations on enhanced endotoxin lethality in mice pretreated with emulsified trehalose-6,6’-dimycolate (cord factor). Infect Immun (1979) 24(2):571–2.
194. Korf J, Stoltz A, Verschoor J, De Baetselier P, Grooten J. The Mycobacterium tuberculosis cell wall component mycolic acid elicits pathogen-associated host innate immune responses. Eur J Immunol (2005) 35(3):890–900. doi:10.1002/eji.200425332
195. Dkhar HK, Nanduri R, Mahajan S, Dave S, Saini A, Somavarapu AK, et al. Mycobacterium tuberculosis keto-mycolic acid and macrophage nuclear receptor TR4 modulate foamy biogenesis in granulomas: a case of a heterologous and noncanonical ligand-receptor pair. J Immunol (2014) 193(1):295–305. doi:10.4049/jimmunol.1400092
196. Astarie-Dequeker C, Le Guyader L, Malaga W, Seaphanh FK, Chalut C, Lopez A, et al. Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog (2009) 5(2):e1000289. doi:10.1371/journal.ppat.1000289
197. Rousseau C, Winter N, Pivert E, Bordat Y, Neyrolles O, Ave P, et al. Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell Microbiol (2004) 6(3):277–87. doi:10.1046/j.1462-5822.2004.00368.x
198. Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature (2014) 505(7482):218–22. doi:10.1038/nature12799
199. Barry CE III. Interpreting cell wall ‘virulence factors’ of Mycobacterium tuberculosis. Trends Microbiol (2001) 9(5):237–41. doi:10.1016/S0966-842X(01)02018-2
200. Berg JM, Tymoczko JL, Stryer L. Section 26.4, important derivatives of cholesterol include bile salts and steroid hormones. Biochemistry. 5th ed. New York: W. H. Freeman (2002). Available from: https://www.ncbi.nlm.nih.gov/books/NBK22339//
201. Quigley J, Hughitt VK, Velikovsky CA, Mariuzza RA, El-Sayed NM, Briken V. The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of Mycobacterium tuberculosis. mBio (2017) 8(2):e00148–17. doi:10.1128/mBio.00148-17
202. Wenk MR. Lipidomics: new tools and applications. Cell (2010) 143(6):888–95. doi:10.1016/j.cell.2010.11.033
203. Peres CM, de Paula L, Medeiros AI, Sorgi CA, Soares EG, Carlos D, et al. Inhibition of leukotriene biosynthesis abrogates the host control of Mycobacterium tuberculosis. Microbes Infect (2007) 9(4):483–9. doi:10.1016/j.micinf.2007.01.006
204. Singh KV, Kaur J, Varshney GC, Raje M, Suri CR. Synthesis and characterization of hapten-protein conjugates for antibody production against small molecules. Bioconjug Chem (2004) 15(1):168–73. doi:10.1021/bc034158v
205. Brieskorn CH, Wenger E. [Analysis of ethereal sage oils by means of gas and thin layer chromatography. Part II. On the constituents of Salvia off. L]. Arch Pharm Ber Dtsch Pharm Ges (1960) 29(3/65):21–6. doi:10.1002/ardp.19602930105
206. Tashiro Y, Inagaki A, Shimizu M, Ichikawa S, Takaya N, Nakajima-Kambe T, et al. Characterization of phospholipids in membrane vesicles derived from Pseudomonas aeruginosa. Biosci Biotechnol Biochem (2011) 75(3):605–7. doi:10.1271/bbb.100754
207. Henderson JC, O’Brien JP, Brodbelt JS, Trent MS. Isolation and chemical characterization of lipid A from Gram-negative bacteria. J Vis Exp (2013) 79:e50623. doi:10.3791/50623
208. Buczynski MW, Stephens DL, Bowers-Gentry RC, Grkovich A, Deems RA, Dennis EA. TLR-4 and sustained calcium agonists synergistically produce eicosanoids independent of protein synthesis in RAW264.7 cells. J Biol Chem (2007) 282(31):22834–47. doi:10.1074/jbc.M701831200
209. Blaho VA, Buczynski MW, Brown CR, Dennis EA. Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of Lyme arthritis. J Biol Chem (2009) 284(32):21599–612. doi:10.1074/jbc.M109.003822
210. Tam VC. Lipidomic profiling of bioactive lipids by mass spectrometry during microbial infections. Semin Immunol (2013) 25(3):240–8. doi:10.1016/j.smim.2013.08.006
211. Sampaio JL, Gerl MJ, Klose C, Ejsing CS, Beug H, Simons K, et al. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci U S A (2011) 108(5):1903–7. doi:10.1073/pnas.1019267108
212. Slatter DA, Aldrovandi M, O’Connor A, Allen SM, Brasher CJ, Murphy RC, et al. Mapping the human platelet lipidome reveals cytosolic phospholipase A2 as a regulator of mitochondrial bioenergetics during activation. Cell Metab (2016) 23(5):930–44. doi:10.1016/j.cmet.2016.04.001
213. Sartain MJ, Dick DL, Rithner CD, Crick DC, Belisle JT. Lipidomic analyses of Mycobacterium tuberculosis based on accurate mass measurements and the novel “Mtb LipidDB”. J Lipid Res (2011) 52(5):861–72. doi:10.1194/jlr.M010363
214. Crick PJ, Guan XL. Lipid metabolism in mycobacteria – insights using mass spectrometry-based lipidomics. Biochim Biophys Acta (2016) 1861(1):60–7. doi:10.1016/j.bbalip.2015.10.007
215. Mahrous EA, Lee RB, Lee RE. A rapid approach to lipid profiling of mycobacteria using 2D HSQC NMR maps. J Lipid Res (2008) 49(2):455–63. doi:10.1194/jlr.M700440-JLR200
216. Guan Z, Johnston NC, Aygun-Sunar S, Daldal F, Raetz CR, Goldfine H. Structural characterization of the polar lipids of Clostridium novyi NT. Further evidence for a novel anaerobic biosynthetic pathway to plasmalogens. Biochim Biophys Acta (2011) 1811(3):186–93. doi:10.1016/j.bbalip.2010.12.010
217. Oursel D, Loutelier-Bourhis C, Orange N, Chevalier S, Norris V, Lange CM. Lipid composition of membranes of Escherichia coli by liquid chromatography/tandem mass spectrometry using negative electrospray ionization. Rapid Commun Mass Spectrom (2007) 21(11):1721–8. doi:10.1002/rcm.3013
218. Rashid R, Cazenave-Gassiot A, Gao IH, Nair ZJ, Kumar JK, Gao L, et al. Comprehensive analysis of phospholipids and glycolipids in the opportunistic pathogen Enterococcus faecalis. PLoS One (2017) 12(4):e0175886. doi:10.1371/journal.pone.0175886
219. Benamara H, Rihouey C, Abbes I, Ben Mlouka MA, Hardouin J, Jouenne T, et al. Characterization of membrane lipidome changes in Pseudomonas aeruginosa during biofilm growth on glass wool. PLoS One (2014) 9(9):e108478. doi:10.1371/journal.pone.0108478
220. Barcelo-Coblijn G, Fernandez JA. Mass spectrometry coupled to imaging techniques: the better the view the greater the challenge. Front Physiol (2015) 6:3. doi:10.3389/fphys.2015.00003
221. Golf O, Strittmatter N, Karancsi T, Pringle SD, Speller AV, Mroz A, et al. Rapid evaporative ionization mass spectrometry imaging platform for direct mapping from bulk tissue and bacterial growth media. Anal Chem (2015) 87(5):2527–34. doi:10.1021/ac5046752
222. Hulme HE, Meikle LM, Wessel H, Strittmatter N, Swales J, Thomson C, et al. Mass spectrometry imaging identifies palmitoylcarnitine as an immunological mediator during Salmonella Typhimurium infection. Sci Rep (2017) 7(1):2786. doi:10.1038/s41598-017-03100-5
223. Desbenoit N, Saussereau E, Bich C, Bourderioux M, Fritsch J, Edelman A, et al. Localized lipidomics in cystic fibrosis: TOF-SIMS imaging of lungs from Pseudomonas aeruginosa-infected mice. Int J Biochem Cell Biol (2014) 52:77–82. doi:10.1016/j.biocel.2014.01.026
224. Barisch C, Paschke P, Hagedorn M, Maniak M, Soldati T. Lipid droplet dynamics at early stages of Mycobacterium marinum infection in Dictyostelium. Cell Microbiol (2015) 17(9):1332–49. doi:10.1111/cmi.12437
225. Barisch C, Soldati T. Mycobacterium marinum degrades both triacylglycerols and phospholipids from its Dictyostelium host to synthesise its own triacylglycerols and generate lipid inclusions. PLoS Pathog (2017) 13(1):e1006095. doi:10.1371/journal.ppat.1006095
226. Maekawa M, Yang Y, Fairn GD. Perfringolysin O theta toxin as a tool to monitor the distribution and inhomogeneity of cholesterol in cellular membranes. Toxins (Basel) (2016) 8(3):67. doi:10.3390/toxins8030067
227. Diep DB, Nelson KL, Raja SM, Pleshak EN, Buckley JT. Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin aerolysin. J Biol Chem (1998) 273(4):2355–60. doi:10.1074/jbc.273.4.2355
Keywords: lipids, metabolism, macrophage, intracellular bacteria, infection, immunity, tuberculosis, salmonellosis
Citation: Teng O, Ang CKE and Guan XL (2017) Macrophage–Bacteria Interactions—A Lipid-Centric Relationship. Front. Immunol. 8:1836. doi: 10.3389/fimmu.2017.01836
Received: 20 October 2017; Accepted: 05 December 2017;
Published: 20 December 2017
Edited by:
Céline Cougoule, Centre national de la recherche scientifique (CNRS), FranceReviewed by:
Thierry Soldati, Université de Genève, SwitzerlandCopyright: © 2017 Teng, Ang and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue Li Guan, eHVlbGkuZ3VhbkBudHUuZWR1LnNn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.