 Yifan Zhan
Yifan Zhan Emma M. Carrington
Emma M. Carrington Yuxia Zhang3
Yuxia Zhang3 Andrew M. Lew
Andrew M. Lew
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 13 December 2017
Sec. T Cell Biology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01809
T cells are pivotal in immunity and immunopathology. After activation, T cells undergo a clonal expansion and differentiation followed by a contraction phase, once the pathogen has been cleared. Cell survival and cell death are critical for controlling the numbers of naïve T cells, effector, and memory T cells. While naïve T cell survival has been studied for a long time, more effort has gone into understanding the survival and death of activated T cells. Despite this effort, there is still much to be learnt about T cell survival, as T cells transition from naïve to effector to memory. One key advance is the development of inhibitors that may allow the temporal study of survival mechanisms operating in these distinct cell states. Naïve T cells were highly reliant on BCL-2 and sensitive to BCL-2 inhibition. Activated T cells are remarkably different in their regulation of apoptosis by pro- and antiapoptotic members of the BCL-2 family, rendering them differentially sensitive to antagonists blocking the function of one or more members of this family. Recent progress in understanding other programmed cell death mechanisms, especially necroptosis, suggests a unique role for alternative pathways in regulating death of activated T cells. Furthermore, we highlight a mechanism of epigenetic regulation of cell survival unique to activated T cells. Together, we present an update of our current understanding of the survival requirement of activated T cells.
Death is fundamental to cellular development and response, with immune cells no exception. It facilitates the selection and retention of desirable clonotypes, while ridding the population of superfluous or often harmful cells. As T cells only become functional upon activation, understanding the control and transition of survival mechanisms in naive, activated and memory T cells is crucial to our ability to harness T cell responses or limit pathology in situations where destructive T cells survive. Although the control of naïve T cell survival has been mostly resolved, how activated T cells regulate their survival is less well understood, despite thorough investigation. In this review, the contribution of several cell death pathways to the life and death of activated T cells will be discussed.
T cells consist of many subtypes including TCRαβ-bearing conventional T cells, Treg cells, TCRγδ T cells, and T cells expressing invariant or semi-invariant TCR chains (such as NKT cells and MAIT cells). Most of our current understanding of T cell survival has largely been focused on two types of TCRαβ-bearing T cells: conventional CD4+ and CD8+ T cells. After development in the thymus, conventional T cells exist in naïve form. Upon activation, they undergo clonal expansion and gain different effector functions. A small fraction of activated cells become long-lived memory cells. It has been appreciated for a long time that naïve T cells and activated T cells differ in their survival program (1, 2). In this section, we will discuss the findings from recent studies investigating the role of pro- and antiapoptotic molecules in activated T cells by analysis of their expression patterns, the use of selective inhibitors, and the genetic deletion of genes in these cell death pathways. The use of selective antagonists offers several advantages. First, they allow quantitative dissection of the contributions of individual antiapoptotic molecules. Second, they allow us to separate the developmental or precursor effects from direct effects in the activated cells. The inhibitor approach is particularly useful for the in vitro dissection of survival requirements of T cells. This approach can also be used for dissection of in vivo survival requirement of T cells. However, the in vivo application can be complicated by effects of antagonists on cells other than T cells, which in turn influence T cell survival. Third, and perhaps most importantly, they may have the therapeutic potential for curtailing unwanted T-cell responses.
The BCL-2 family can be separated into three groups, the pro-survival molecules BCL-2, BCL-XL, BCL-W, MCL-1, and A1/BFL1; the group of BH3-only pro-apoptotic molecules BID, BIM, PUMA/BBC3, BAD, NOXA/PMAIP, BIK/BLK/NBK, BMF, and HRK/DP5; and the pro-apoptotic “effectors” BAX and BAK (3) (Figure 1). The interplay of these molecules is a finely orchestrated system. As antiapoptotic proteins sequester BH3 proteins that initiate apoptosis, BH3 proteins require BAX/BAK for apoptosis induction as multiple BH3 proteins fail to induce apoptosis in BAX−/−/BAK−/− system while reintroduction of BAX restores the ability of BH3 proteins to induce apoptosis (4, 5). When BH3 protein function becomes dominant, the pro-apoptotic “effectors” proteins BAX and BAK will permeabilize the mitochondrial outer membrane, leading to cytochrome c release into the cytosol to assemble with APAF-1 and pro-caspase 9 to form the apoptosome, followed by the activation of effector caspases. Our most recent studies suggest that immune cell survival is controlled by the quantitative participation of multiple antiapoptotic proteins (6). Nevertheless, their contribution to T cell survival is not equal, probably related to their dynamic regulation of expression and lifespan. Below we will discuss the BCL-2 antiapoptotic molecules separately.
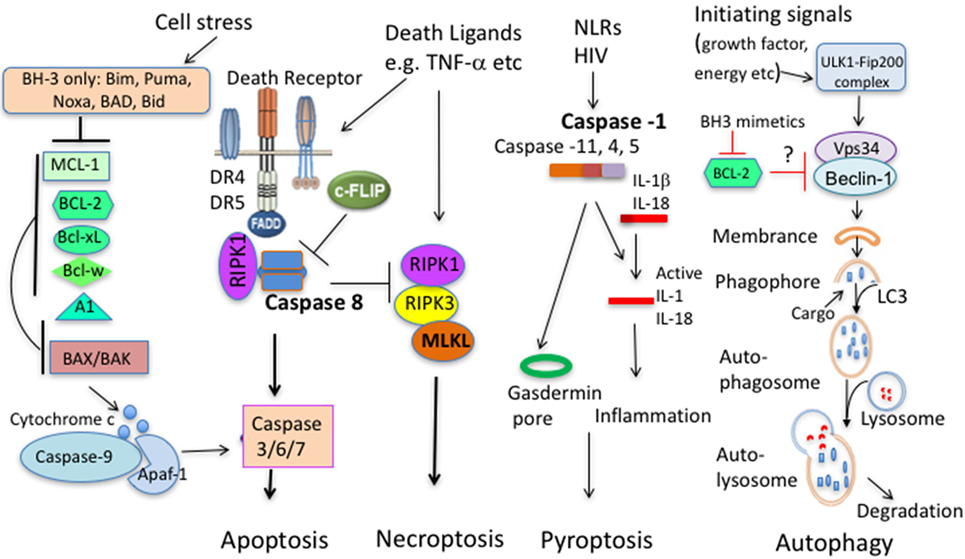
Figure 1. Principal pathways of cell death. Apoptosis comprises of the intrinsic and extrinsic pathway. In the intrinsic pathway, cells sense stress signals, leading to upregulation and activation of BH3 proteins. When antiapoptotic molecules that normally bind and keep BH3 proteins and/or BAX/BAK in check are displaced, BH3 proteins will trigger activation of BAX and BAK. BAX/BAK then mediate cytochrome c release from the mitochondrial outer membrane to the cytosol, activating Caspase-9 and downstream caspases leading to cell demise. In the extrinsic pathway, extracellular ligands engage cell death receptors, leading to formation of the death-inducing signaling complex (DISC) with the adaptor protein Fas-associated death domain protein (FADD) and pro-caspase 8, leading to activation of caspase 8 and subsequent activation of effector caspases and apoptosis. In this pathway, c-FLIP acts as a negative regulator. c-FLIP is structurally highly similar to procaspase-8 but lacks catalytic activity, thus outcompetes caspase 8 binding blunting the death-inducing signal. When extrinsic apoptosis in inhibited (Caspase 8 deficiency, caspase inhibition, and high c-FLIP expression), engagement of death ligand can initiate necroptosis that involves activation of the necroptosome comprising RIPK1, RIPK3, and mixed lineage kinase domain-like (MLKL). Pytoptosis is a type of cell death initiated from activation of several Caspases that cleave IL-1β and IL-18. A downstream molecule Gasdermin is critical for cell death by pyroptosis. Autophagy promotes proteolytic degradation of mitochondria and other cytosolic components at the lysosome. It can promote survival or diminish survival depending on degraded molecules. BCL-2 family members with antiapoptotic and proapoptotic molecules can interact with upstream autophagy signaling molecules.
BCL-2 is the prototype of BCL-2 family members and has been the most extensively studied. Overexpression of BCL-2 delays T-cell death (7, 8) while BCL-2 deficiency reduced T-cell survival (9, 10). Survival of naïve T cells mediated by BCL-2 was largely dependent on IL-7 as BCL-2 rescued the severe defect in T cells in IL-7R-deficient mice (11, 12). Naive T cells almost exclusively express BCL-2 and are heavily dependent on BCL-2 for survival since they show high sensitivity to BCL-2 antagonist ABT-199 (6). Similar findings have also been derived from earlier studies with ABT-737, an inhibitor with a broader binding activity to BCL-2, BCL-xL, and BCL-w (13–17). It is evident from these studies that activated T cells including effector and memory T cells are less sensitive to the inhibitor when compared to naïve T cells. However, there are some variations in sensitivity between activated CD4+ and CD8+ T cells. Compared to CD4+ T cells, CD8+ memory T cells are relatively sensitive to BCL-2 inhibition with ABT-737 (14–16). However, ABT-737 killed only a relatively minor fraction of CD8+ OT-1 T cells under optimal stimulation in vitro (18). As for CD4+ T cells, it is also revealed that Th1 memory cells are long-lived while Th17 cells are short-lived, probably related to their lower expression of BCL-2 (19). It remains to be determined whether functionally different T cells show differential survival requirements for BCL-2. A simple interpretation regarding different sensitivity to BCL-2 antagonist by naïve and activated T cells is that T cells alter their survival program when T cells get activated. They downregulate their BCL-2 sensitivity while at the same time upregulate A1, BCL-xL and MCL-1 (18, 20, 21). This in turn may lead to a higher resistance to BCL-2 inhibitors in vitro and in vivo.
It had been shown that genetic elimination of BCL-xL in the mouse reduces the survival of double-positive thymocytes but not the survival of single-positive thymocytes in culture or peripheral T cells in vivo (22). Early studies demonstrated that BCL-xL was an activation-upregulated antiapoptotic molecule promoting survival of activated T cells and memory T cells (23, 24); however, more recent studies have shown that BCL-xL is dispensable for the generation of effector and memory T cells (25). Recently, highly selective BCL-xL inhibitors have been developed (26), but have shown minimal T-cell killing ability (unpublished). BCL-w is less studied, although it can be expressed by some subsets of T cells (27). The similarity of action on T cells between ABT-737 (antagonizing BCL-2, BCL-xL, and BCL-w) and the BCL-2 specific ABT-199 suggests that BCL-w has little impact on T cell survival (6, 15). No T cell defects have been documented in BCL-w-deficient mice (28).
As global deletion of MCL-1 in mice led to embryonic lethality (29), conditional deletion of MCL-1 has been adopted to examine its contribution to T-cell development and survival (30). It has been shown that the development and maintenance of T cells requires MCL-1, implying that MCL-1 is also important for the survival of naïve T cells (30). These findings are also supported by the characterization of mice with MCL-1 haplodeficiency (6). Conversely, overexpression of MCL-1 can promote T cell development and survival (6, 31). Even for developing T cells, MCL-1 seems to have a unique role in supporting T cell development, as the developmental defects arising from deletion of MCL-1 can be partially rescued by BAK deficiency but not by BAX deficiency or overexpression of BCL-2 (32). During activation, MCL-1, along with A1 and BCL-xL show elevated expression in T cells (18, 21). In a system of inducible deletion of MCL-1, Mx1Cre-induced deletion of Mcl-1 led to massive loss of antigen-specific T cells in LCMV-infected mice (21). Notably, loss of activated T cells with MCL-1 deletion could be rescued with concomitant loss of BAX and BAK but not BIM (21). In contrast, BIM deficiency could rescue T cell defect caused by BCL-2 deficiency (13). Considering that activated T cells expressed not only BIM but also PUMA and NOXA (21), perhaps multiple BH3 proteins participate to regulate cell death. At least in vitro, stimulated human T cells seem to depend on the MCL-1/NOXA axis for survival (33). The recent development of the selective MCL-1 inhibitor (34) also allows timely dissection of MCL-1 contribution to T cell death in vitro (6). It could induce significant death of T cells (6). Together with revelation of the importance of MCL-1 in regulating Treg cell survival (35), MCL-1 is a key antiapoptotic molecule for T cell survival.
A1 was initially identified as a GM-CSF regulated pro-survival gene and its expression restricted to the hematopoietic system (36). In T cells, A1 is largely only induced upon TCR stimulation (18, 37). Several studies have demonstrated an association of A1 upregulation with enhanced T cell survival (38, 39). Due to the quadruplication of A1 genes in mice, mice with a full deletion of A1 genes only recently became available, allowing for a direct assessment of A1’s contribution to T cell survival. Somewhat surprisingly, A1 deficiency has a relatively minor impact on T-cell survival and the induction of T-cell response (40, 41). Nevertheless, when other antiapoptotic molecules were suppressed by inhibitors, A1 deficiency could result in significantly poorer survival of T cells (6).
BIM has been shown to have a dominant role in regulation of T cell survival among the BH3 proteins (13, 42). The deletion of immature autoreactive thymocytes was defective in BIM-deficient mice, leading accumulation of T cells with self-reactivity (43, 44). For mature T cells reaching the periphery, BIM is also important for survival of naïve T cells (45), which was found to be partly regulated via IL-7 signaling (46, 47). Upon activation, BIM is also crucial for the termination of T-cell immune response against acute infection with herpes simplex virus (48). Similarly, peripheral deletion of activated T cells is also mediated by BIM (49, 50). Interestingly, during a chronic infection, activated T cells with different antigen specificity showed differential requirements from BIM (51). It is proposed that infection duration and antigen loads may switch apoptosis pathways for activated T cells (51). Apart from BIM, other BH3 proteins also have a non-redundant role in regulating survival of activated T cells. Deficiency in NOXA could lead to accumulation of activated T cells and immunopathology during chronic LCMV infection (52, 53). Interestingly, PUMA but not NOXA, BID, or BAD was shown to have a non-redundant role in protection from cell death of antigen-specific T cells in HSV-1 infection (54). The basis of the varied requirement for different BH3 proteins remains unclear. Furthermore, BH3 proteins can collaborate to regulate T-cell survival. It has been reported that the combined loss of PUMA and BIM protected mitogen-induced T cell blasts from IL-2 deprivation-induced death more potently than the loss of BIM in vitro (55). BIM also collaborates with NOXA or PUMA to control effector CD8+ T-cell responses during CMV infection, probably by targeting different antiapoptotic molecules (56). Even without infection, simultaneous defects in both BIM and PUMA could lead to severe forms of autoimmunity and organ damage (57). BIM also cooperated with BID for contraction of the anti-viral T cell response (58). Furthermore, three concurrent studies also demonstrated that BIM and Fas, a key molecule of extrinsic apoptosis pathway, also cooperate to regulate different types of T-cell responses (59–61). Thus, BH3 proteins, particularly BIM, are key molecules to limit T-cell survival.
Multidomain pro-apoptotic molecules BAK/BAX are often referred as “effectors” of the intrinsic mitochondrial cell death pathway and are essential, yet each individually redundant, for T-cell apoptosis (62). As individual BH3, only proteins may be redundant and could not rescue the T cell defect caused by MCL-1 deficiency, concomitant loss of BAX and BAK rescued the loss of activated T cells with MCL-1 deletion (21). Thus, BAX and BAK are critical for apoptosis of activated T cells and naïve T cells. However, careful examination has revealed subtle differences between BAK and BAX in apoptosis induction. BAK binds preferentially to MCL-1 and BCL-xL (63). Fittingly, loss of BAK was able to partially rescue T-cell defects caused by conditional deletion of MCL-1, whereas overexpression of BCL-2 or loss of BAX was unable to rescue the cell (32). Notably, chimeric mice reconstituted with BAK−/− bone marrow cells, but not BAX−/− bone marrow cells, developed immunopathology and died prematurely (64). In humans, mutations in BAK have been associated with some forms of autoimmune disease (65). Thus, this group of pro-apoptotic molecules is also dynamically involved in regulation of T-cell survival.
There is great interest to repurpose BCL-2 antagonists that have been approved for cancer treatment to dampen inflammation (66–68). Notably, for T cell-mediated inflammation (collagen-induced arthritis), ABT-737 (antagonizing BCL-2, BCL-xL, and BCL-w) was only effective before but not after induction of disease (67). Differential sensitivity to ABT-737 by naïve T cells and activated T cells may offer an explanation to above discrepancy. Given that a large fraction of activated T cells can still survive even when all antiapoptotic molecules were impaired (6), other mechanisms must contribute to the survival of activated T cells. Here, we provide a brief summary what impact other death pathways have on T-cell survival. Many of these pathways are inter-connected, resulting in a complicated regulatory network balancing T-cell-mediated immunity and tolerance.
Soon after BCL-2 was discovered as a key player of the intrinsic pathway of apoptosis (69), Fas/FasL were discovered as the prototype receptor/ligand pair of the extrinsic (death receptor) pathway of apoptosis (70, 71). Fas, upon engagement to FasL, forms the death-inducing signaling complex with the adaptor protein Fas-associated death domain protein and pro-caspase 8, leading to activation of caspase 8 and subsequent activation of effector caspases and apoptosis (72). Deletion of Fas and FasL in mice resulted in lymphadenopathy and an increase in the unusual TCRαβ+B220+ CD4−CD8− (DN) T cells (73, 74). Human mutations in CD95 also resulted in increased TCRαβ+B220+ CD4−CD8− DN T cells and were associated with the development of autoimmune lymphoproliferative syndrome (75). Thus, the death receptor apoptotic pathway is important for T cell homeostasis.
However, the importance of this pathway in clearance of activated T cells seems to be dependent on the experimental conditions [reviewed in Ref. (42)]. Early on, in vitro induction of activation-induced cell death (AICD) in T cells has been found to be critically dependent on Fas and FasL interaction (76–78). In vivo, deletion of SEB-activated T cells in mice was impaired with defective FasL–Fas pathway in some (79, 80) but not in other studies (49). Similarly, deletion of antigen-activated CD8+ T cells during acute a viral (HSV-1) infection was not affected by Fas deficiency (48, 81) but was impaired during a persistent chronic infection (82). Similarly, work on c-FLIPL in T cells, a classical negative regulator of death receptor/extrinsic pathway signaling, also generated controversial results. c-FLIPL-deficient T cells were shown to display enhanced cell death upon TCR stimulation (83), while an earlier study found that activation-induced death of T cells in c-FLIPL transgenic mice was unaffected (84). The nature of an immune response—acute vs chronic infection, transient vs repeat stimulation or signal strength of T cell activation has been offered as potential explanations for the reported varied dependency (42, 61).
There are two other complex aspects regarding the pathway in regulation of T cell survival. First, AICD could occur via the interaction of death receptor and their ligands other than Fas/FasL. TNF-α/TNF receptor 1 and TRAIL/DR4/DR5 also contribute to AICD (85, 86). For TNF-mediated AICD, it has been reported that soluble TNF-α but not transmembrane TNF-α (tmTNF-α) induced AICD in vitro and in vivo (87). A more recent study showed that tmTNF-α could promote AICD via reverse signaling in which tmTNF-α behaves as a receptor to interact TNFR (88). Furthermore, the ligand binding to death receptor results in not only downstream activation of initiator caspases 8 and 10 (89) but also of prosurvival signaling pathways, including nuclear factor-κB and mitogen-activated protein kinase (89). It remains to be fully appreciated how a final outcome (death vs life) is determined when a ligand binds death receptor.
Second, apart from cooperating with the intrinsic apoptosis pathway to regulate T-cell death (59–61), extrinsic apoptosis pathway is also heavily entangled with programed necroptosis. Despite caspase activity is critical for death receptor-mediated apoptosis, Fas can trigger Caspase-8-independent death of activated human and murine T cells (90, 91). The death pathway (necroptosis) involves the receptor-interacting serine-threonine kinases RIPK 1 and RIPK3 (90, 91) (Figure 1). The involvement of necroptosis in T cell death is discussed in the following section.
Necroptosis requires activation of signaling complex consisting of RIPK1, RIPK3, and mixed lineage kinase domain-like (MLKL). When activity of Caspase 8 is absent or suppressed, three key components will assemble the necroptosome (Figure 1). Both RIPK1 and RIPK3 are key upstream components of TNF signaling and can mediate apoptosis, necroptosis, and inflammation while MLKL, as a downstream signaling molecules is primarily involved in necroptosis (92). At least in vitro, TNF can induce necroptosis in the absence of RIPK1 (93). As for activated T cells, two aforementioned studies demonstrated that necroptosis can occur at certain conditions (90, 91). For death ligand-mediated necrotptosis, RIPK1 is essential for cell death (90). For TCR-mediated necrotptosis (at least without exogenous death ligands), necroptosis occurred in the absence of Caspase 8 is rescued by RIPK3 deficiency and partially rescued by RIPK1 inhibition with necrostatin-1 (91). Notably, necroptosis occurred in vitro in actively proliferating cells (91). In vivo, RIP3 deficiency only prevent the loss of Caspase 8−/− T cells in expansion phase but not in contraction phase during a viral infection (91), implying a stage-specific role. MLKL has been identified as a key player in necroptosis of fibroblasts and macrophages triggered by TNF in conjunction with caspase inhibitors and IAP inhibitors (94, 95). Notably, T cells develop normally in MLKL knockout mice (95, 96). As most myeloid cells constitutively express high levels of MLKL, naïve T cells express low levels of MLKL, but display an increase in MLKL expression upon activation (97). Currently, the importance of MLKL in regulating the death of activated T cells is unknown. Overall, necroptosis of activated T cells is mostly prominent when caspase activity is suppressed. Significance of the pathway in regulation of T cell survival remains to be established.
Pyroptosis describes pro-inflammatory programmed cell death (98). Differing from classical apoptosis, pyroptosis employs inflammatory pyroptotic caspases (caspase-1, -4, -5, -11). Caspase-1-dependent and inflammation-induced pyroptosis is critical for CD4 T-cell death in HIV-infected host (99). It is unclear whether pyroptosis is involved in the death of TCR-triggered T cells.
Autophagy promotes proteolytic degradation of mitochondria and other cytosolic components at the lysosome. It can promote or diminish cell survival depending on degraded molecules. We mainly discuss here the role of autophagy in regulation of T cell survival, although autophagy can influence the induction and maintenance of an immune response independent of cell survival mechanisms (100). Several reports showed that autophagy promotes T-cell survival (101–103). In vitro dissection of contribution of autophagy to AICD had demonstrated that both the activity of upstream kinase AMPK and key downstream molecule LC3 in autophagy signaling was reduced upon AICD induction, leading to the accumulation of damaged mitochondrial and apoptosis progression. T cells from mouse models defective in autophagy had higher sensitivity to AICD (104). Apart from inhibition of autophagy leading to accumulation of damaged mitochondrial, TCR signaling during AICD induction can also lead to mitochondrial fragmentation in a Drp1-dependent fashion, resulting in AICD involving reactive oxygen species and CD95 induction (105). Beyond the potential contribution of autophagy to regulate cell survival at early activation stage, both CD4+ and CD8+ memory T cells have been shown to be preferentially affected by the autophagy process (106, 107). Nevertheless, how this pathway impacts on survival of activated T cells remains to be fully explored. Particularly, how does this pathway interplay with BCL-2-regulated apoptosis? It has been reported that autophagy enhances degradation of pro-apoptotic proteins such as BIM and various caspases (103). Upstream of autophagy signaling, interaction of antiapoptotic molecules and pro-apoptotic molecules with key signaling molecules of autophagy remains controversial. Beclin-1 is a BCL-2-binding protein that is essential to autophagy (108). BCL-2 can inhibit Bectin-1-dependent autophagy to maintain autophagy at levels that are compatible with cell survival (109). However, a more recent study showed that BCL-2 or other antiapoptotic molecules do not directly inhibit components of the autophagic pathway but affect autophagy indirectly by inhibition of Bax/Bak (110). The findings are in contrast to a subsequent study showing that the longer exposure of BH3 mimetic ABT-737 induces autophagy through a BAX and BAK-independent mechanism (111). In addition, it has been shown that BIM directly interacts with Beclin-1 to inhibit autophagy (112). It remains to be investigated how these players in BCL-2-regulated pathways affect autophagy of T cells and the consequences of these effects.
Epigenetic regulation through DNA methylation and histone modification is essential to fine-tune gene expression. Depletion of the methyltransferase SUV39H1, which mediates H3K9 trimethylation in Th2 cells, can lead to the transcription of Th1 cytokine IFN-γ (113). EZH2 (Enhancer of zeste homolog 2) is another histone methyltransferase that catalyzes H3K27me3 and acts primarily as a gene silencer. EZH2 is a key component of the polycomb repressive complex (PRC) 2, which also contains SUZ12 and EED (Figure 2). It has emerged that EZH2 is critically involved in regulation of cell survival and differentiation of activated T cells. T-cell lineage specific deletion of EZH2 (CD4-Cre/EZH2fl/fl) did not alter normal development of CD4 and CD8 T cells. However, it led to enhanced Th1 and Th2 differentiation (114). Counterintuitively, suppression or loss of EZH2 also accelerated the death of activated T cells (97, 115, 116). The precise mechanisms of cell death that cause loss of activated T cells are currently unknown. Expression of both antiapoptotic and pro-apoptotic molecules were higher in activated EZH2−/− T cells, compared to activated WT T cells (116). Notably, deletion of Bim did not prevent the loss of activated EZH2−/− T cells (116). Somewhat surprisingly, despite massive loss of GVDH causing T cells, antileukemia T cells of EZH2−/− donor origin were preserved. Similar to the example of differential requirements for BIM for T cells with different antigen specificity (51), stimulation duration and strength may cause differential dependence on EZH2 for survival. In addition, we found that the induction of MLKL in activated T cells was enhanced by EZH2 deletion (97). This raises the possibility that MLKL-mediated necroptosis contributes to loss of activated T cells in EZH2-deficient mice. It has also been shown that loss of EZH2 in donor T cells has been shown to inhibit GVHD in mice after allogeneic bone marrow transplantation (115, 116), indicating that manipulation of PRC2 signaling may offer an avenue to specifically target activated cells.
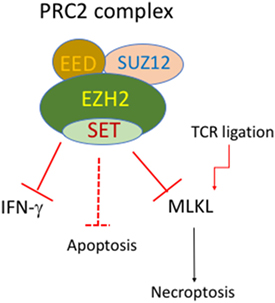
Figure 2. Putative roles of EZH2 and PRC2 complex in regulation of cell death of activated T cells. The polycomb repressive complex (PRC) contains EZH1/2, SUZ12, and EED. EZH2 deficiency in T-cell lineage does not affect normal development of CD4 and CD8 T cells. However, EZH2-deficient T cells display enhanced Th1 differentiation and enhanced cell death upon activation. EZH2-deficient T cells also have enhanced mixed lineage kinase domain-like (MLKL) expression upon TCR stimulation.
T cells are a key component of the immune system and play a critical role in orchestrating the immune responses to self and foreign antigens. The magnitude of the T-cell response is critically regulated by cell survival/death. Conceivably, targeting the survival mechanisms may provide an avenue for immune intervention. Enhancement of T cell survival can be beneficial in situations of immune deficiency, immunization, and cancer immunotherapy. On the other hand, an inappropriate immune response (e.g., autoimmunity and transplant rejection) can be curtailed by inducing T-cell death. We now appreciate that survival control of naïve and activated T cells is different and that multiple pathways contribute to survival control of activated T cells (Table 1). Currently, there are still many unknowns regarding how life and death of activated T cells is regulated. A better understanding of how the survival of T cells, particularly activated T cells, is regulated should increase the potential to harnessing T-cell immune responses.
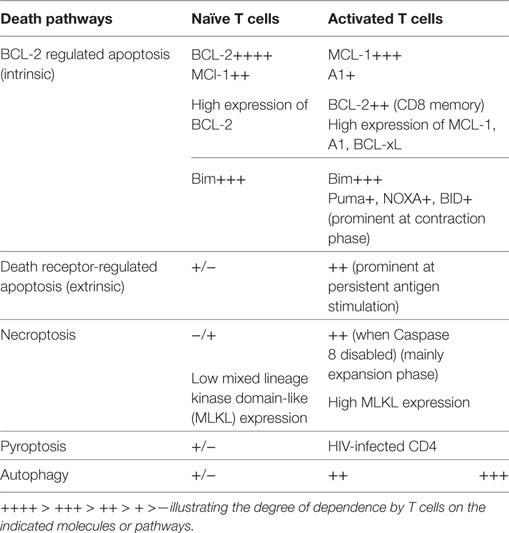
Table 1. Survival requirement of T cells at a glance.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Mr. Michael Zhan for proofreading the manuscript. This work was supported by Rebecca L. Cooper Foundation, National Health and Medical Research Council of Australia (NHMRC) grants (1037321, 1043414, 1080321, 1105209, 1054925), NHMRC Independent Research Institutes Infrastructure Support Scheme grant (361646), and Victorian State Government Operational Infrastructure Support grant. We acknowledge the Wurundjeri people of the Kulin nation as the traditional owners and custodians of the land on which most of the work was performed.
1. Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol (2004) 22:765–87. doi:10.1146/annurev.immunol.22.012703.104554
2. Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol (2007) 7(7):532–42. doi:10.1038/nri2115
3. Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol (2003) 21:71–105. doi:10.1146/annurev.immunol.21.120601.141029
4. Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev (2001) 15(12):1481–6. doi:10.1101/gad.897601
5. Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell (2001) 8(3):705–11. doi:10.1016/S1097-2765(01)00320-3
6. Carrington EM, Zhan Y, Brady JL, Zhang JG, Sutherland RM, Anstee NS, et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ (2017) 24(5):878–88. doi:10.1038/cdd.2017.30
7. Sentman CL, Shutter JR, Hockenbery D, Kanagawa O, Korsmeyer SJ. BCL-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell (1991) 67(5):879–88. doi:10.1016/0092-8674(91)90361-2
8. Strasser A, Harris AW, Cory S. BCL-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell (1991) 67(5):889–99. doi:10.1016/0092-8674(91)90362-3
9. Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell (1993) 75(2):229–40. doi:10.1016/0092-8674(93)80065-M
10. Nakayama K, Nakayama K, Negishi I, Kuida K, Shinkai Y, Louie MC, et al. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science (1993) 261(5128):1584–8.
11. Akashi K, Kondo M, von Freeden-Jeffry U, Murray R, Weissman IL. Bcl-2 rescues T lymphopoiesis in interleukin-7 receptor-deficient mice. Cell (1997) 89(7):1033–41. doi:10.1016/S0092-8674(00)80291-3
12. Maraskovsky E, O’Reilly LA, Teepe M, Corcoran LM, Peschon JJ, Strasser A. Bcl-2 can rescue T lymphocyte development in interleukin-7 receptor-deficient mice but not in mutant rag-1-/- mice. Cell (1997) 89(7):1011–9. doi:10.1016/S0092-8674(00)80289-5
13. Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD, et al. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J Exp Med (2007) 204(7):1665–75. doi:10.1084/jem.20070618
14. Tripathi P, Mitchell TC, Finkelman F, Hildeman DA. Cutting edge: limiting amounts of IL-7 do not control contraction of CD4+ T cell responses. J Immunol (2007) 178(7):4027–31. doi:10.4049/jimmunol.178.7.4027
15. Carrington EM, Vikstrom IB, Light A, Sutherland RM, Londrigan SL, Mason KD, et al. BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc Natl Acad Sci U S A (2010) 107(24):10967–71. doi:10.1073/pnas.1005256107
16. Kurtulus S, Tripathi P, Moreno-Fernandez ME, Sholl A, Katz JD, Grimes HL, et al. Bcl-2 allows effector and memory CD8+ T cells to tolerate higher expression of Bim. J Immunol (2011) 186(10):5729–37. doi:10.4049/jimmunol.1100102
17. Cippa PE, Kraus AK, Lindenmeyer MT, Chen J, Guimezanes A, Bardwell PD, et al. Resistance to ABT-737 in activated T lymphocytes: molecular mechanisms and reversibility by inhibition of the calcineurin-NFAT pathway. Cell Death Dis (2012) 3:e299. doi:10.1038/cddis.2012.38
18. Koenen P, Heinzel S, Carrington EM, Happo L, Alexander WS, Zhang JG, et al. Mutually exclusive regulation of T cell survival by IL-7R and antigen receptor-induced signals. Nat Commun (2013) 4:1735. doi:10.1038/ncomms2719
19. Pepper M, Linehan JL, Pagan AJ, Zell T, Dileepan T, Cleary PP, et al. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat Immunol (2010) 11(1):83–9. doi:10.1038/ni.1826
20. Dzhagalov I, Dunkle A, He YW. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J Immunol (2008) 181(1):521–8. doi:10.4049/jimmunol.181.1.521
21. Tripathi P, Koss B, Opferman JT, Hildeman DA. Mcl-1 antagonizes Bax/Bak to promote effector CD4(+) and CD8(+) T-cell responses. Cell Death Differ (2013) 20(8):998–1007. doi:10.1038/cdd.2013.25
22. Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW, et al. Bclx regulates the survival of double-positive thymocytes. Proc Natl Acad Sci U S A (1995) 92(11):4763–7. doi:10.1073/pnas.92.11.4763
23. Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, et al. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity (1995) 3(1):87–98. doi:10.1016/1074-7613(95)90161-2
24. Mueller DL, Seiffert S, Fang W, Behrens TW. Differential regulation of bcl-2 and bcl-x by CD3, CD28, and the IL-2 receptor in cloned CD4+ helper T cells. A model for the long-term survival of memory cells. J Immunol (1996) 156(5):1764–71.
25. Zhang N, He YW. The antiapoptotic protein Bcl-xL is dispensable for the development of effector and memory T lymphocytes. J Immunol (2005) 174(11):6967–73. doi:10.4049/jimmunol.174.11.6967
26. Brady RM, Vom A, Roy MJ, Toovey N, Smith BJ, Moss RM, et al. De-novo designed library of benzoylureas as inhibitors of BCL-XL: synthesis, structural and biochemical characterization. J Med Chem (2014) 57(4):1323–43. doi:10.1021/jm401948b
27. O’Reilly LA, Print C, Hausmann G, Moriishi K, Cory S, Huang DC, et al. Tissue expression and subcellular localization of the pro-survival molecule Bcl-w. Cell Death Differ (2001) 8(5):486–94. doi:10.1038/sj.cdd.4400835
28. Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N, et al. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci U S A (1998) 95(21):12424–31. doi:10.1073/pnas.95.21.12424
29. Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev (2000) 14(1):23–7. doi:10.1101/gad.14.1.23
30. Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature (2003) 426(6967):671–6. doi:10.1038/nature02067
31. Campbell KJ, Gray DH, Anstee N, Strasser A, Cory S. Elevated Mcl-1 inhibits thymocyte apoptosis and alters thymic selection. Cell Death Differ (2012) 19(12):1962–71. doi:10.1038/cdd.2012.84
32. Dunkle A, Dzhagalov I, He YW. Mcl-1 promotes survival of thymocytes by inhibition of Bak in a pathway separate from Bcl-2. Cell Death Differ (2010) 17(6):994–1002. doi:10.1038/cdd.2009.201
33. Alves NL, Derks IA, Berk E, Spijker R, van Lier RA, Eldering E. The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity (2006) 24(6):703–16. doi:10.1016/j.immuni.2006.03.018
34. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature (2016) 538(7626):477–82. doi:10.1038/nature19830
35. Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3(+) regulatory T cells. Nat Immunol (2013) 14(9):959–65. doi:10.1038/ni.2649
36. Lin EY, Orlofsky A, Berger MS, Prystowsky MB. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J Immunol (1993) 151(4):1979–88.
37. Verschelde C, Walzer T, Galia P, Biemont MC, Quemeneur L, Revillard JP, et al. A1/Bfl-1 expression is restricted to TCR engagement in T lymphocytes. Cell Death Differ (2003) 10(9):1059–67. doi:10.1038/sj.cdd.4401265
38. Gonzalez J, Orlofsky A, Prystowsky MB. A1 is a growth-permissive antiapoptotic factor mediating postactivation survival in T cells. Blood (2003) 101(7):2679–85. doi:10.1182/blood-2002-04-1229
39. Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J Immunol (2002) 169(9):4882–8. doi:10.4049/jimmunol.169.9.4882
40. Schenk RL, Tuzlak S, Carrington EM, Zhan Y, Heinzel S, Teh CE, et al. Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ (2017) 24(3):534–45. doi:10.1038/cdd.2016.156
41. Tuzlak S, Schenk RL, Vasanthakumar A, Preston SP, Haschka MD, Zotos D, et al. The BCL-2 pro-survival protein A1 is dispensable for T cell homeostasis on viral infection. Cell Death Differ (2017) 24(3):523–33. doi:10.1038/cdd.2016.155
42. Bouillet P, O’Reilly LA. CD95, BIM and T cell homeostasis. Nat Rev Immunol (2009) 9(7):514–9. doi:10.1038/nri2570
43. Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science (1999) 286(5445):1735–8. doi:10.1126/science.286.5445.1735
44. Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, et al. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature (2002) 415(6874):922–6. doi:10.1038/415922a
45. Tsukamoto H, Huston GE, Dibble J, Duso DK, Swain SL. Bim dictates naive CD4 T cell lifespan and the development of age-associated functional defects. J Immunol (2010) 185(8):4535–44. doi:10.4049/jimmunol.1001668
46. Pellegrini M, Bouillet P, Robati M, Belz GT, Davey GM, Strasser A. Loss of Bim increases T cell production and function in interleukin 7 receptor-deficient mice. J Exp Med (2004) 200(9):1189–95. doi:10.1084/jem.20041328
47. Li WQ, Guszczynski T, Hixon JA, Durum SK. Interleukin-7 regulates Bim proapoptotic activity in peripheral T-cell survival. Mol Cell Biol (2010) 30(3):590–600. doi:10.1128/MCB.01006-09
48. Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci U S A (2003) 100(24):14175–80. doi:10.1073/pnas.2336198100
49. Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, et al. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity (2002) 16(6):759–67. doi:10.1016/S1074-7613(02)00322-9
50. Davey GM, Kurts C, Miller JF, Bouillet P, Strasser A, Brooks AG, et al. Peripheral deletion of autoreactive CD8 T cells by cross presentation of self-antigen occurs by a Bcl-2-inhibitable pathway mediated by Bim. J Exp Med (2002) 196(7):947–55. doi:10.1084/jem.20020827
51. Grayson JM, Weant AE, Holbrook BC, Hildeman D. Role of Bim in regulating CD8+ T-cell responses during chronic viral infection. J Virol (2006) 80(17):8627–38. doi:10.1128/JVI.00855-06
52. Wensveen FM, van Gisbergen KP, Derks IA, Gerlach C, Schumacher TN, van Lier RA, et al. Apoptosis threshold set by Noxa and Mcl-1 after T cell activation regulates competitive selection of high-affinity clones. Immunity (2010) 32(6):754–65. doi:10.1016/j.immuni.2010.06.005
53. Wensveen FM, Klarenbeek PL, van Gisbergen KP, Pascutti MF, Derks IA, van Schaik BD, et al. Pro-apoptotic protein Noxa regulates memory T cell population size and protects against lethal immunopathology. J Immunol (2013) 190(3):1180–91. doi:10.4049/jimmunol.1202304
54. Fischer SF, Belz GT, Strasser A. BH3-only protein Puma contributes to death of antigen-specific T cells during shutdown of an immune response to acute viral infection. Proc Natl Acad Sci U S A (2008) 105(8):3035–40. doi:10.1073/pnas.0706913105
55. Erlacher M, Labi V, Manzl C, Bock G, Tzankov A, Hacker G, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med (2006) 203(13):2939–51. doi:10.1084/jem.20061552
56. Kurtulus S, Sholl A, Toe J, Tripathi P, Raynor J, Li KP, et al. Bim controls IL-15 availability and limits engagement of multiple BH3-only proteins. Cell Death Differ (2015) 22(1):174–84. doi:10.1038/cdd.2014.118
57. Gray DH, Kupresanin F, Berzins SP, Herold MJ, O’Reilly LA, Bouillet P, et al. The BH3-only proteins Bim and Puma cooperate to impose deletional tolerance of organ-specific antigens. Immunity (2012) 37(3):451–62. doi:10.1016/j.immuni.2012.05.030
58. Masson F, Kupresanin F, Mount A, Strasser A, Belz GT. Bid and Bim collaborate during induction of T cell death in persistent infection. J Immunol (2011) 186(7):4059–66. doi:10.4049/jimmunol.1001918
59. Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity (2008) 28(2):218–30. doi:10.1016/j.immuni.2007.12.014
60. Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK III, Wu T, Li QZ, et al. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity (2008) 28(2):206–17. doi:10.1016/j.immuni.2007.12.015
61. Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity (2008) 28(2):197–205. doi:10.1016/j.immuni.2007.12.017
62. Rathmell JC, Lindsten T, Zong WX, Cinalli RM, Thompson CB. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat Immunol (2002) 3(10):932–9. doi:10.1038/ni834
63. Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev (2005) 19(11):1294–305. doi:10.1101/gad.1304105
64. Mason KD, Lin A, Robb L, Josefsson EC, Henley KJ, Gray DH, et al. Proapoptotic Bak and Bax guard against fatal systemic and organ-specific autoimmune disease. Proc Natl Acad Sci U S A (2013) 110(7):2599–604. doi:10.1073/pnas.1215097110
65. Delgado-Vega AM, Castiblanco J, Gomez LM, Diaz-Gallo LM, Rojas-Villarraga A, Anaya JM. Bcl-2 antagonist killer 1 (BAK1) polymorphisms influence the risk of developing autoimmune rheumatic diseases in women. Ann Rheum Dis (2010) 69(2):462–5. doi:10.1136/ard.2008.100818
66. Bardwell PD, Gu J, McCarthy D, Wallace C, Bryant S, Goess C, et al. The Bcl-2 family antagonist ABT-737 significantly inhibits multiple animal models of autoimmunity. J Immunol (2009) 182(12):7482–9. doi:10.4049/jimmunol.0802813
67. Lawlor KE, Smith SD, van Nieuwenhuijze A, Huang DC, Wicks IP. Evaluation of the Bcl-2 family antagonist ABT-737 in collagen-induced arthritis. J Leukoc Biol (2011) 90(4):819–29. doi:10.1189/jlb.0311174
68. Zhan Y, Carrington EM, Ko HJ, Vikstrom IB, Oon S, Zhang JG, et al. Bcl-2 antagonists kill plasmacytoid dendritic cells from lupus-prone mice and dampen interferon-alpha production. Arthritis Rheumatol (2015) 67(3):797–808. doi:10.1002/art.38966
69. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature (1988) 335(6189):440–2. doi:10.1038/335440a0
70. Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, et al. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell (1991) 66(2):233–43. doi:10.1016/0092-8674(91)90614-5
71. Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell (1993) 75(6):1169–78. doi:10.1016/0092-8674(93)90326-L
72. Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell (1995) 81(4):505–12. doi:10.1016/0092-8674(95)90071-3
73. Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell (1994) 76(6):969–76. doi:10.1016/0092-8674(94)90375-1
74. Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature (1992) 356(6367):314–7. doi:10.1038/356314a0
75. Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science (1995) 268(5215):1347–9. doi:10.1126/science.7539157
76. Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature (1995) 373(6513):441–4. doi:10.1038/373441a0
77. Dhein J, Walczak H, Bäumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature (1995) 373(6513):438–41. doi:10.1038/373438a0
78. Ju ST, Panka DJ, Cui H, Ettinger R, el-Khatib M, Sherr DH, et al. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature (1995) 373(6513):444–8. doi:10.1038/373444a0
79. Mogil RJ, Radvanyi L, Gonzalez-Quintial R, Miller R, Mills G, Theofilopoulos AN, et al. Fas (CD95) participates in peripheral T cell deletion and associated apoptosis in vivo. Int Immunol (1995) 7(9):1451–8. doi:10.1093/intimm/7.9.1451
80. Strasser A, Harris AW, Huang DC, Krammer PH, Cory S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J (1995) 14(24):6136–47.
81. Lohman BL, Welsh RM. Apoptotic regulation of T cells and absence of immune deficiency in virus-infected gamma interferon receptor knockout mice. J Virol (1998) 72(10):7815–21.
82. Balazs M, Martin F, Zhou T, Kearney J. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity (2002) 17(3):341–52. doi:10.1016/S1074-7613(02)00389-8
83. He MX, He YW. A role for c-FLIP(L) in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ (2013) 20(2):188–97. doi:10.1038/cdd.2012.148
84. Lens SM, Kataoka T, Fortner KA, Tinel A, Ferrero I, MacDonald RH, et al. The caspase 8 inhibitor c-FLIP(L) modulates T-cell receptor-induced proliferation but not activation-induced cell death of lymphocytes. Mol Cell Biol (2002) 22(15):5419–33. doi:10.1128/MCB.22.15.5419-5433.2002
85. Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature (1995) 377(6547):348–51. doi:10.1038/377348a0
86. Roberts AI, Devadas S, Zhang X, Zhang L, Keegan A, Greeneltch K, et al. The role of activation-induced cell death in the differentiation of T-helper-cell subsets. Immunol Res (2003) 28(3):285–93. doi:10.1385/IR:28:3:285
87. Muller S, Rihs S, Schneider JM, Paredes BE, Seibold I, Brunner T, et al. Soluble TNF-alpha but not transmembrane TNF-alpha sensitizes T cells for enhanced activation-induced cell death. Eur J Immunol (2009) 39(11):3171–80. doi:10.1002/eji.200939554
88. Zhang H, Wang J, Jia L, Huang J, He C, Hu F, et al. Transmembrane TNF-α promotes activation-induced cell death by forward and reverse signalling. Ongotarget (2017) 8(38):63799–812. doi:10.18632/oncotarget.19124
89. Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J (2009) 23(6):1625–37. doi:10.1096/fj.08-111005
90. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol (2000) 1(6):489–95. doi:10.1038/82732
91. Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med (2011) 208(4):633–41. doi:10.1084/jem.20110251
92. Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science (2014) 343(6177):1357–60. doi:10.1126/science.1249361
93. Moujalled DM, Cook WD, Okamoto T, Murphy J, Lawlor KE, Vince JE, et al. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis (2013) 4:e465. doi:10.1038/cddis.2012.201
94. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signalling downstream of RIP3 kinase. Cell (2012) 148(1–2):213–27. doi:10.1016/j.cell.2011.11.031
95. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity (2013) 39(3):443–53. doi:10.1016/j.immuni.2013.06.018
96. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res (2013) 23(8):994–1006. doi:10.1038/cr.2013.91
97. Zhang Y, Kinkel S, Maksimovic J, Bandala-Sanchez E, Tanzer MC, Naselli G, et al. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood (2014) 124(5):737–49. doi:10.1182/blood-2013-12-544106
98. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 9(3):113–4. doi:10.1016/S0966-842X(00)01936-3
99. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature (2014) 505(7484):509–14. doi:10.1038/nature12940
100. Botbol Y, Guerrero-Ros I, Macian F. Key roles of autophagy in regulating T-cell function. Eur J Immunol (2016) 46(6):1326–34. doi:10.1002/eji.201545955
101. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med (2007) 204(1):25–31. doi:10.1084/jem.20061303
102. McLeod IX, Zhou X, Li QJ, Wang F, He YW. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J Immunol (2011) 187(10):5051–61. doi:10.4049/jimmunol.1100710
103. Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ (2012) 19(1):144–52. doi:10.1038/cdd.2011.78
104. Corrado M, Mariotti FR, Trapani L, Taraborrelli L, Nazio F, Cianfanelli V, et al. Macroautophagy inhibition maintains fragmented mitochondria to foster T cell receptor-dependent apoptosis. EMBO J (2016) 35(16):1793–809. doi:10.15252/embj.201593727
105. Roth D, Krammer PH, Gulow K. Dynamin related protein 1-dependent mitochondrial fission regulates oxidative signalling in T cells. FEBS Lett (2014) 588(9):1749–54. doi:10.1016/j.febslet.2014.03.029
106. Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol (2006) 177(8):5163–8. doi:10.4049/jimmunol.177.8.5163
107. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol (2014) 15(12):1152–61. doi:10.1038/ni.3025
108. Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol (1998) 72(11):8586–96.
109. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell (2005) 122(6):927–39. doi:10.1016/j.cell.2005.07.002
110. Lindqvist LM, Heinlein M, Huang DC, Vaux DL. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc Natl Acad Sci U S A (2014) 111(23):8512–7. doi:10.1073/pnas.1406425111
111. Pedro JM, Wei Y, Sica V, Maiuri MC, Zou Z, Kroemer G, et al. BAX and BAK1 are dispensable for ABT-737-induced dissociation of the BCL2-BECN1 complex and autophagy. Autophagy (2015) 11(3):452–9. doi:10.1080/15548627.2015.1017191
112. Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O, et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell (2012) 47(3):359–70. doi:10.1016/j.molcel.2012.05.040
113. Allan RS, Zueva E, Cammas F, Schreiber HA, Masson V, Belz GT, et al. An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature (2012) 487(7406):249–53. doi:10.1038/nature11173
114. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity (2013) 39(5):819–32. doi:10.1016/j.immuni.2013.09.012
115. He S, Wang J, Kato K, Xie F, Varambally S, Mineishi S, et al. Inhibition of histone methylation arrests ongoing graft-versus-host disease in mice by selectively inducing apoptosis of alloreactive effector T cells. Blood (2012) 119(5):1274–82. doi:10.1182/blood-2011-06-364422
Keywords: activated T cells, apoptosis, necroptosis, epigenetics, autoimmunity, treatment
Citation: Zhan Y, Carrington EM, Zhang Y, Heinzel S and Lew AM (2017) Life and Death of Activated T Cells: How Are They Different from Naïve T Cells? Front. Immunol. 8:1809. doi: 10.3389/fimmu.2017.01809
Received: 20 September 2017; Accepted: 30 November 2017;
Published: 13 December 2017
Edited by:
Loretta Tuosto, Sapienza Università di Roma, ItalyReviewed by:
Silvia Campello, Università di Roma Tor Vergata, ItalyCopyright: © 2017 Zhan, Carrington, Zhang, Heinzel and Lew. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yifan Zhan, emhhbkB3ZWhpLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.