 Vasco Rodrigues1
Vasco Rodrigues1 Nicolas Ruffin
Nicolas Ruffin Philippe Benaroch
Philippe Benaroch- 1Institut Curie, PSL Research University, INSERM U932, Paris, France
- 2Institut Curie, PSL Research University, UMR3216, Paris, France
Cells of the myeloid lineage, particularly macrophages, serve as primary hosts for HIV in vivo, along with CD4 T lymphocytes. Macrophages are present in virtually every tissue of the organism, including locations with negligible T cell colonization, such as the brain, where HIV-mediated inflammation may lead to pathological sequelae. Moreover, infected macrophages are present in multiple other tissues. Recent evidence obtained in humanized mice and macaque models highlighted the capacity of macrophages to sustain HIV replication in vivo in the absence of T cells. Combined with the known resistance of the macrophage to the cytopathic effects of HIV infection, such data bring a renewed interest in this cell type both as a vehicle for viral spread as well as a viral reservoir. While our understanding of key processes of HIV infection of macrophages is far from complete, recent years have nevertheless brought important insight into the uniqueness of the macrophage infection. Productive infection of macrophages by HIV can occur by different routes including from phagocytosis of infected T cells. In macrophages, HIV assembles and buds into a peculiar plasma membrane-connected compartment that preexists to the infection. While the function of such compartment remains elusive, it supposedly allows for the persistence of infectious viral particles over extended periods of time and may play a role on viral transmission. As cells of the innate immune system, macrophages have the capacity to detect and respond to viral components. Recent data suggest that such sensing may occur at multiple steps of the viral cycle and impact subsequent viral spread. We aim to provide an overview of the HIV–macrophage interaction along the multiple stages of the viral life cycle, extending when pertinent such observations to additional myeloid cell types such as dendritic cells or blood monocytes.
Introduction
The introduction of antiretroviral therapy (ART) to treat HIV infection in the mid 1990s was met with extraordinary success and dramatically improved the lives of patients, by turning a deadly infection into a manageable chronic disease. However, while able to prevent progression to AIDS, ART cannot eradicate HIV from the body, and a viral reservoir quickly rebounds after interruption of the therapy. In addition, HIV patients under suppressive therapy are at elevated risk of developing several non-AIDS related diseases, including cognitive impairment and cardiovascular problems.
HIV mainly replicates in CD4 T cells and macrophages in the body. Loss of CD4 T cells has long been known as the major pathological event leading to AIDS. In macrophages, HIV infection does not induce immediate cell death and viral replication proceeds for extended periods of time.
Macrophages maintain tissue homeostasis by performing crucial housekeeping tasks. Their ubiquitous distribution in the body allows HIV to disseminate into organs and tissues and establish compartmentalized infection. Macrophages are also an important effector arm of the innate immune system. These cells detect HIV infection and express cellular factors that severely restrain the capacity of the virus to replicate.
Here, we discuss the interplay between HIV and macrophages. We review recent work highlighting the unique interaction between HIV and macrophages, at the cellular level. We further discuss evidence pointing to a role for macrophages as cellular reservoirs of HIV during ART and how they participate in the pathological morbidities that prevail in patients under therapy.
Macrophage Ontogeny and Function
Macrophages populate virtually all tissues of the body, where they perform a multitude of functions that are essential for tissue homeostasis, architecture, and protection (1). This wide range of macrophage action was described more than a century ago by Elie Metchnikoff. In his pioneering work, Metchnikoff observed the swarming and subsequent clearance of foreign objects by phagocytic cells in starfish larvae and water fleas (1). He correctly foresaw the importance of macrophages in the removal of obsolete cells, pathogen elimination, or sterile inflammation (2, 3).
Tissue macrophages have classically been considered as originating exclusively and in a continuous manner from bone marrow-derived monocytes, as part of the mononuclear–phagocyte system, a concept put forward by Van Furth in the 1970s (4). However, fate-mapping studies over the past decade have drastically changed our views on macrophage ontogeny. It is now widely accepted that many tissues are seeded with macrophages derived from the yolk sac or the fetal liver, during embryonic development [reviewed in Ref. (5)]. Once at their site of residency, macrophages proliferate locally to maintain a population size able to meet the requirements of the developing tissue or organ (6). The ability to self-renew suggests the existence of a subpopulation of tissue-resident macrophages with stem cell properties and capable of asymmetric cell division, but no such cell has yet been described in the tissues (6), with the possible exception of a subpopulation of epidermal Langerhans cells (7). Alternatively, the whole population of macrophages residing in a given tissue may be endowed with self-renewal potential, as suggested in studies with microglial cells or peritoneal macrophages (8–10). In some tissues, such as the brain or the liver, the resident macrophage population appears to be exclusively derived from embryonic cells throughout all adulthood (6). While monocytes may infiltrate these tissues under inflammatory or pathologic conditions, and differentiate into macrophages, they do not become part of the stable resident population (11). In stark contrast, embryonic macrophages that seed the gut prenatally appear to be completely replaced by monocyte-derived cells after birth (12). The factors that dictate this differential capacity of embryonically or monocyte-derived cells to stably engraft different tissues are not well understood and are an area of active research (6).
In their tissues of residency, macrophages perform a wide range of tasks. Some of these functions, such as apoptotic cell removal or extracellular matrix (MA) remodeling, are required in all tissues to different extents, indicating that macrophages are engaged in cross talks with their local microenvironment. The capacity to perform such general functions appears to be imprinted in the whole macrophage lineage and possibly involves the role of master transcription regulators such as PU (13). Other functions, by contrast, are specific to certain tissues. For instance, alveolar macrophages are specialized in clearing excessive surfactant, while macrophages of the red pulp of the spleen recycle iron from senescent erythrocytes (6). These site-specific functions are presumably imprinted on macrophages by tissue-specific signals and will induce transcriptional programs that define macrophage populations in different tissues (13).
Tissue macrophages are further subjected to environmental cues that occur in non-homeostatic conditions such as inflammation. Evolution has shaped macrophages as primary tissue sentinels (14). These cells are equipped with a broad range of receptors capable of detecting molecular patterns from all classes of microbes and multiple types of tissue damage, as well as receptors for chemokines and cytokines produced by immune cells (1). Integration of these multiple signals leads to what is commonly known as macrophage polarization (15). For instance, in an infected/inflamed tissue, macrophages may encounter microbial products such as LPS or be exposed to T cell-derived IFN-γ, leading to a polarized state known as M1, that is highly efficient in killing intracellular or ingested pathogens (15). Importantly, the majority of the knowledge gathered on macrophage polarization derives from well-defined in vitro experiments (16). These studies led to the M1 versus M2 model of macrophage polarization, which is unlikely to capture the complexity and the diversity of signals that macrophages can integrate in vivo. Furthermore, these polarizing stimuli act upon macrophages with previously imprinted tissue-specific programs. As such, similar polarizing signals probably lead to distinct phenotypes in macrophages from different tissues (13).
A wealth of information on the ontogeny, differentiation, and function of macrophages, derived from multiple studies in recent years, has profound implications in how we perceive the role of the macrophage in pathological settings, such as cancer, metabolic disease, or infections like HIV.
Macrophages During Acute and Chronic HIV Infection
The first description that tissue macrophages were permissive to HIV infection and capable of replicating the virus came in 1986, from the lab of Robert Gallo (17), amid the fast-paced period that characterized the early years of HIV/AIDS research. That very same pioneering study further provided the initial evidence that macrophages produce HIV for extended periods of time, hence coping with viral-induced cytopathy (17). In addition, the study revealed that in macrophages, HIV accumulates in apparent intracellular compartments absent from T cells.
HIV can infect macrophages as these cells express both the viral entry receptor, CD4, and co-receptors, CCR5 and CXCR4, that bind the viral envelop protein, gp120. Macrophage infection by HIV requires initial adsorption of the virus to the cell surface, mediated by lectin-like receptors, integrins, and heparan sulfate proteoglycans (18). Entry then probably takes place following virion internalization into macropinosomes (19) or endosomes, where fusion between the viral envelope and the host cell appears to occur (20), as recently proposed by a study following the internalization of fluorescent quantum dots encapsulated by infectious HIV-1 particles in primary macrophages (21).
Classically, macrophage-tropic viruses (M-tropic) were thought to exclusively employ CCR5 for entry (R5 viruses), while CXCR4-using strains (X4 viruses) were viewed as unable to enter macrophages and establish productive infection (22). This simplified categorization of macrophage tropism based on co-receptor usage has been proven imperfect as many R5 viruses are unable to infect macrophages (23), whereas some X4 isolates can (24). While co-receptor usage may frequently predict macrophage tropism, categorizing a virus as M-tropic requires demonstration of its ability to replicate in vitro in macrophages, although, understandably, this may not be a practical approach to test every isolate (18).
Transmitted/founder (T/F) viruses are the viral variants that initiate infection in a new host, at genital or rectal mucosal surfaces. Their sequences can be inferred by the mathematical modeling of virus evolution after single-genome amplification analysis of the plasma viral population (25). These types of analyses, across multiple studies, support the idea that most infections are initiated by a single or a very limited number of founder viruses (26). Biological characterization of T/F viruses demonstrated that they are usually unable to replicate in macrophages (27, 28), possibly due to the lower densities of the CD4 molecule on the macrophage surface, as compared with CD4 T cells (29). This suggests that macrophages are not an important source of viral replication in the initial stages of infection, emerging only later, as the virus adapts to infect cells with a lower CD4 density at the surface. In agreement, studies with mucosal explants from the human reproductive tract (30–32), or in non-human primates (33) support the idea that CD4+ T cells are the crucial targets at very early time points of infection.
Following migration from mucosal entry points into regional lymph nodes, via yet poorly described mechanisms, HIV rapidly disseminates systemically in the host. In SIV-infected rhesus macaques, viral spread to distal tissues such as the gastrointestinal (GI) tract or the spleen can be detected as early as 1 day after intravaginal inoculation and systemic distribution of SIV was observed by day 7 (34). This rapid but clinically silent spread is followed by the acute phase of HIV infection characterized by unrestrained viral replication in multiple tissues (35–37).
Macrophages are likely targets of HIV during the acute phase of infection, as viral nucleic acids have been detected in tissue macrophages from multiple organs in infected patients. These include Kupffer cells in the liver (38), microglial cells in the brain (39), alveolar macrophages in the lung (40), and intestinal macrophages obtained from several segments of the GI tract (41, 42). Importantly, replication-competent virus can be recovered from cultures of macrophages purified from lymphoid tissues of acutely infected rhesus macaques (43), implying that productive infection is taking place. It remains unclear how HIV disseminates to establish infection in these cells and tissues.
Monocytes can seed many tissues and differentiate locally into macrophages, turning this cell type into a potential vehicle for HIV dissemination across the myeloid compartment. Several reports claim indeed that replication of HIV-1 can take place in vivo in monocytes, even in patients under ART (44–46). Infected monocytes have been proposed to play a key role in viral dissemination to the brain due to their capacity to cross the blood–brain barrier (47), see Ref. (48).
At their sites of residency, macrophages constitutively patrol the tissues for danger signals, while also performing several housekeeping tasks. Interestingly, through the action of the viral accessory protein Nef, HIV is capable of reprogramming the migration of macrophages and selectively promotes a mesenchymal type of migration, while inhibiting the amoeboid type (49). The mesenchymal mode of migration is characterized by extensive extracellular MA remodeling, thus allowing the invasion of dense microenvironments, which may further promote viral dissemination and persistence. The relevance of these findings is supported by the increased accumulation of macrophages in the tissues of mice engineered to express the HIV Nef protein (49).
Alternatively, migratory, infected CD4+ T cells may serve as vehicles for HIV systemic dissemination, as suggested by intravital microscopy in infected humanized mice (50), and possibly transmit the virus to tissue-resident macrophages. Interestingly, during the acute phase, SIV-DNA-positive myeloid cells present in lymphoid tissues also contain rearranged T cell receptor DNA (51). This suggests that phagocytosis of infected T cells allows macrophages to acquire viral DNA, which is presumably taken to the macrophage degradative compartments for destruction preventing potential infection of macrophages (51). However, at least in vitro, cultured macrophages become productively infected after ingesting infected T cells (52). This mode of direct T cell-to-macrophage HIV transmission results in more efficient macrophage infection than exposure to cell-free virus (52). While this mechanism has yet to be demonstrated in vivo, it seemingly represents a strategy employed by HIV to maximize its spread, by exploiting the extensive phagocytic capacity of macrophages (53).
Chronic untreated HIV infection leads to extensive depletion of the body’s CD4+ T cell pool and progression to AIDS (54). Concurrently, the viral population evolves to become more M-tropic (55), presumably because extensive CD4+ T cell loss makes the macrophage the most abundant cell target in advanced disease. Rhesus macaques treated with an antibody depleting CD4+ T cells before SIV infection mimic this advanced stage AIDS (56). In these animals, macrophages represent about 80% of the SIV-RNA+ cells in the tissues with evidence of productive infection of macrophages from lymphoid tissues and the brain, frequently associated with activation markers. Remarkably, plasma viral loads were two logs higher in depleted animals as compared with CD4+ T cell-sufficient controls, which led to rapid disease progression (56). Thus, in the context of CD4+ T cell depletion that might reflect the advanced AIDS status, extensive activation and viral replication in macrophages drives a precipitous progression of clinical disease.
Macrophage Sensing of HIV and Intrinsic Restrictions to Viral Replication
The macrophage paradox refers to the fact that macrophages represent both the first line of defense against many pathogens, including viruses, and yet are exploited by many of these pathogens as their favorite cellular niche for replication (57). Such is the case of HIV-1, which efficiently replicates in macrophages. However, as sentinel cells, macrophages are equipped with a range of sensors that detect ongoing infection at many steps of the viral cycle and trigger cellular responses that will activate antiviral immunity (58). A number of these induced antiviral effector genes, known as restrictions factors, will block infection at specific steps of the viral life cycle (59). Thus, from the viral perspective, the extent to which HIV-1 replicates in macrophages must be tightly regulated as to ensure viral transmission/dissemination, while avoiding significant antiviral responses. Such delicate balance is achieved by a combination of precise employment of viral accessory proteins and usurpation of the normal function of cellular factors. Complete reviews devoted to the various restriction factors are available (59), we will focus here on factors that play important roles in infected macrophages.
Studies examining the initial stages of the viral cycle, i.e., upon viral entry and retro-transcription (RT) of the viral RNA into cDNA in the cytosol, proposed that HIV-1 escapes early innate sensing in myeloid cells before viral DNA integration. This would result from a combination of shielding the newly synthesized cDNA by viral and cellular factors (60, 61), and maintenance of very low levels of cytosolic viral cDNA due to the action of the cellular nuclease TREX1 (62, 63). However, induction of a weak, yet detectable, interferon-stimulated gene (ISG) response after HIV-1 infection of macrophages has also been reported (64, 65), with type I IFNs levels remaining undetectable (64, 66).
Examining these apparent discrepancies, we recently confirmed this transient response of monocyte-derived macrophages (MDMs) to HIV-1 infection, detectable as soon as 6 h postexposure and peaking at 24 h (67). Such response induces an ISG signature and depends on the induction of low levels of type I IFN. This sensing step is macrophage specific as it does not occur in monocyte-derived dendritic cells (MDDCs) exposed to HIV-1 (Decalf et al., unpublished results). The signal inducing the early ISG wave preceded reverse transcription but required viral fusion. Virus-like particles devoid of their genome, but capable of fusing, elicited a similar ISG response, indicating that viral nucleic acids were not implicated is this sensing step (67). Importantly, this early and transient ISG induction alone conferred partial protection to macrophages against subsequent HIV-1 infection. Different viruses carrying different envelopes and thus entering MDM although different receptors exhibited similar capacities to induce this response (67). The actual sensor of viral entry involved in this process remains to be identified. Membrane perturbations, such as fusion events, can elicit antiviral responses in macrophages, via the stimulator of IFN genes (STING)/tank-binding kinase-1 (TBK-1)/interferon-responsive factor-3 (IRF-3) pathway (68, 69). Thus, it is tempting to consider that the plasma membrane of the macrophage represents its first line of defense, and that sensing membrane perturbations, like viral entry, as soon as it occurs would be advantageous for the rapidity of the establishment of the antiviral response (Figure 1).
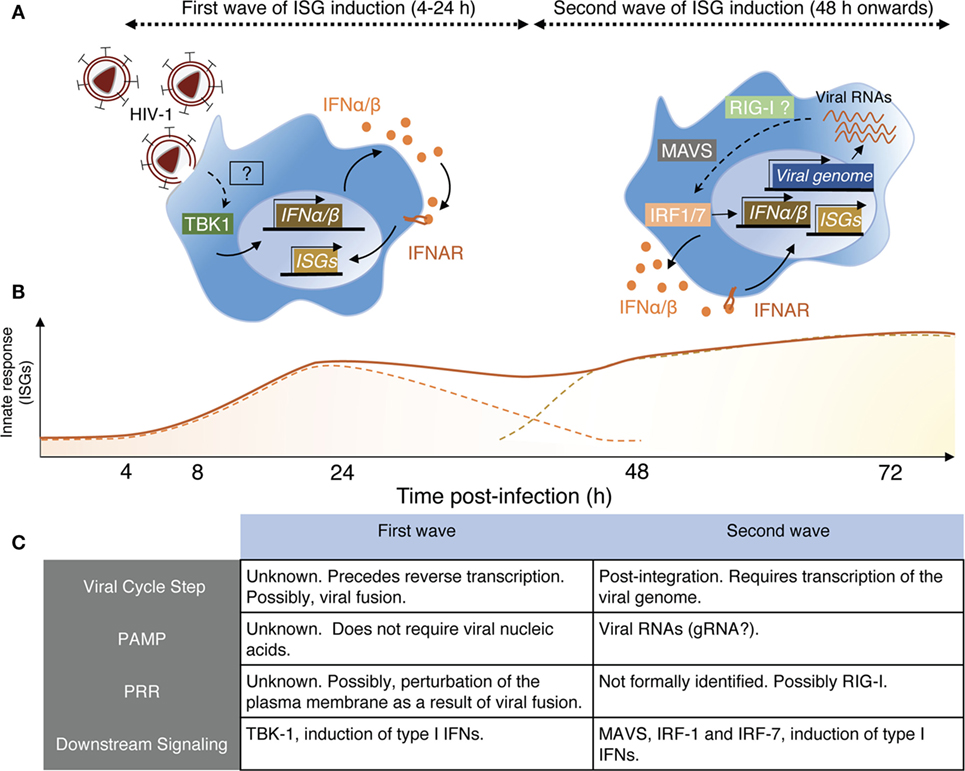
Figure 1. Schematic view of HIV-1 sensing by macrophages. (A) Macrophages sense HIV-1 at two independent steps of the viral cycle. Left panel—Early sensing of HIV-1 by macrophages requires viral fusion with the plasma membrane but precedes retro-transcription (RT). This sensing step is detectable by 4 h after cell exposure to the virus and declines after 24 h when RT is inhibited. While the actual sensor involved remains to be identified, it activates the kinase tank-binding kinase-1 (TBK1), leading to production of type I IFN, signaling via IFNAR, and triggering of interferon-stimulated genes (ISGs). Right panel—The second wave on HIV-1 sensing is measurable only 48 h after cell exposure to the virus. It requires integration of the viral genome in the host DNA and transcription of viral RNAs, which appear to be the viral component triggering the late ISG response. Here also, the actual sensor remains to be identified, but retinoic acid-inducible gene I (RIG-I) is a likely candidate as the signaling cascade involves the adaptor MAVS and IRF-1 and IRF-7, leading to type I IFN production. (B) Schematic representation of the two ISG waves induced by the sensing steps described in panel (A). The full line represents the putative measurable ISG response, whereas the dashed lines indicate the contribution of the individual waves for the measurable response. (C) This table resumes the main characteristics associated with the two sensing mechanisms through which macrophages detect HIV-1 and was established based on Ref. (67, 70).
Retro-transcription represents a very specific and mandatory step for retroviral replication and is the target of SAM domain- and HD domain-containing protein 1 (SAMHD1), a major restriction factor for HIV-1 replication in macrophages and other cells of the myeloid lineage, such as DCs (71, 72). Upon fusion of the HIV envelope with the host cell membrane, the viral capsid (CA) is released into the cytoplasm and the viral reverse transcriptase initiates reverse transcription of the viral RNA genome. SAMHD1 restricts HIV infection by depleting the cytosolic pool of dNTPs available for reverse transcription, via its deoxynucleoside triphosphate triphosphohydrolase activity (73) and possibly also by directly attacking viral RNA via its ribonuclease activity (74). While HIV-1 has no known factor to counteract SAMHD1 restriction, HIV-2 and several SIV strains encode the accessory protein Vpx that targets SAMHD1 for degradation (71, 72). Yet, HIV-1 is capable of replicating in macrophages, suggesting alternative mechanisms to bypass restriction.
Cyclin-dependent kinases (CDKs) phosphorylate SAMHD1 in proliferating cells, halting its activity (75, 76). Interestingly, primary macrophages in culture spontaneously and temporarily enter a G1-like state, without progressing to actual cell division, leading to CDK1 induction that limits the levels of SAMHD1 in its active form (77). This provides HIV-1 with a window of opportunity, as the virus preferentially infects these G1-like phase macrophages (77). Importantly, microglial and peritoneal macrophages recovered from mouse tissues similarly exhibit spontaneous cycling between G0 and the G1 state, suggesting a relevance for this mechanism in vivo (77). Other recent studies reported elevated levels of different members of the cyclin family in macrophages, rendering them permissive to HIV-1 infection, further supporting an important role for cell cycle proteins in the mechanism through which HIV-1 bypasses SAMDH1 restriction (78, 79).
Packaging Vpx into HIV-1 virions leads to a strong increase in infection efficiency of macrophages and DCs (80). However, such increased infectivity comes at the cost of detection of the viral cDNA by the cytoplasmic DNA sensor cGAS and induction of antiviral type I IFN (63). This suggests that HIV-1, unlike HIV-2, adopts a strategy of co-habitation with SAMHD1 in myeloid cells to avoid triggering antiviral immunity (81).
There is, however, a second phase of ISG induction in macrophages, peaking around 96 h postinfection that requires retro-transcription (67) and viral integration (70). This response is induced by detection of newly transcribed viral RNA by the RNA sensor retinoic acid-inducible gene I, and it requires the activity of the trans-activating (tat) HIV-1 accessory protein, responsible for the elongation of HIV-1 transcripts (70). Together, the two sensing steps confer an ISG signature in HIV-1-infected macrophages that may contribute to maintaining a low level of viral replication in this cell type (67, 70) (Figure 1).
Once viral proteins are produced, a key step in the assembly of new viral particles is the incorporation of the viral envelope. Two recently identified restriction factors, active in macrophages, target the viral envelope and thus reduce infectivity. Membrane-associated RING-CH 8 is highly expressed in myeloid cells where it retains the viral envelope glycoproteins intracellularly hence impairing their incorporation into the budding viral particles (82). The guanylate binding protein-5 (GBP5) is highly inducible by type I IFN and interferes with processing, trimming and incorporation of the HIV-1 envelope, rendering the produced virions less infectious (83). Interestingly, the viral genes encoding Env and the HIV-1 accessory protein Vpu are expressed from the same bicistronic RNA (84). Deletion of Vpu from the viral genome enhances Env expression and renders HIV-1 less susceptible to GBP5 restriction (83). These observations may explain the high frequencies of defective Vpu gene observed in M-tropic HIV-1 strains (85).
Not surprisingly, restriction also takes place during the late phase of HIV replication cycle in macrophages. Tetherin (or BST2) is an interferon-inducible transmembrane protein that restricts HIV-1 particle release by inserting its C-terminal end into the viral lipid bilayer (86). As a result, newly formed virions are unable to leave the surface of infected T cells; i.e., they stay tethered (87). Tetherin activity is counteracted by Vpu that mediates its surface downregulation and degradation (88). HIV-1 infection of macrophages upregulates tetherin in an apparently IFN-independent but Nef-dependent manner (89). However, despite the presence of Vpu, HIV-1-infected macrophages still express detectable levels of tetherin that appears to partially restrict viral particle release (89).
The interferon-induced transmembrane (IFITM) proteins belong to a small family of highly related proteins and act has as broad restriction factors able to interfere with the replication of many viruses. These relatively short proteins (around 130 aa) were initially characterized for their capacity to protect IFITM expressing cells from HIV-1 infection (90, 91). In addition, viral particles produced by IFITM expressing cells exhibit a reduced infectivity due to their incorporation of IFITM into the viral envelope (92). Whether present in the membrane of the target cell or the viral particles, IFITM proteins appear to impair viral fusion. Of note, endogenous IFITM3 also inhibits cell-to-cell transmission, and its inhibitory effect is stronger when it is present in viral particles than when part of the target cell membrane (92). The mechanism of action of IFITM is still unclear but probably relies on their capacity to reduce the fluidity of the membranes where they insert, thereby preventing viral fusion (or hemifusion). The precise topology of IFITM proteins is probably key to understand how they work, but this aspect is still debated. Mutagenesis studies combined with secondary structure predictions indicate that IFITM3 is a type 2 transmembrane protein that possesses an amphipathic helix adjacent to two palmitoylated cysteines (93). Silencing of IFITM1, 2, and 3 in HIV-1 producing cells resulted in increased infectivity of the viruses released (94). Importantly, the impact of such silencing was more striking in MDM than in any other cell type (94), raising the possibility that IFITM expression induced early in HIV-1-exposed MDM, as we documented (67), reduces the infectivity of the viral progeny.
In conclusion, low-level sensing of HIV-1 by macrophages results in the induction of a panoply of restriction factors that severely restricts infection. This antiviral state still allows for a certain degree of replication-competent viral production and may explain why HIV-1-infected macrophages are so resistant to the cytopathic effects that the virus exhibits in other cell types. The net effect, however, is that macrophages produce HIV-1 for extended periods of time which may favor long-term viral persistence.
Cell Biology of HIV Assembly in Macrophages: From GAG Synthesis to Particle Release
In CD4+ T cells and model cell lines, HIV assembly and budding take place at the plasma membrane. By contrast, in infected macrophages HIV buds into an apparently intracellular compartment, known as the virus-containing compartment (VCC). The VCC is a unique compartment with topological and biochemical properties distinct from late endosomes or multivesicular bodies, see Ref. (95). Indeed, the VCC (i) lacks classical markers of endosomal and lysosomal compartments (96, 97); (ii) possesses a near neutral pH (97); and (iii) is connected to the extracellular milieu making its lumen accessible to small membrane-impermeable dies (96, 98–100). The compartment consists of a complex membranous system of interconnected tubules and vesicles enclosing immature and mature viral particles in its lumen as well as budding virions in its limiting membrane (99) (Figure 2). The limiting membrane of the VCC possesses specific biophysical properties due in part to the particular membrane topology of the proteins that are inserted and the presence of high levels of cholesterol (101).
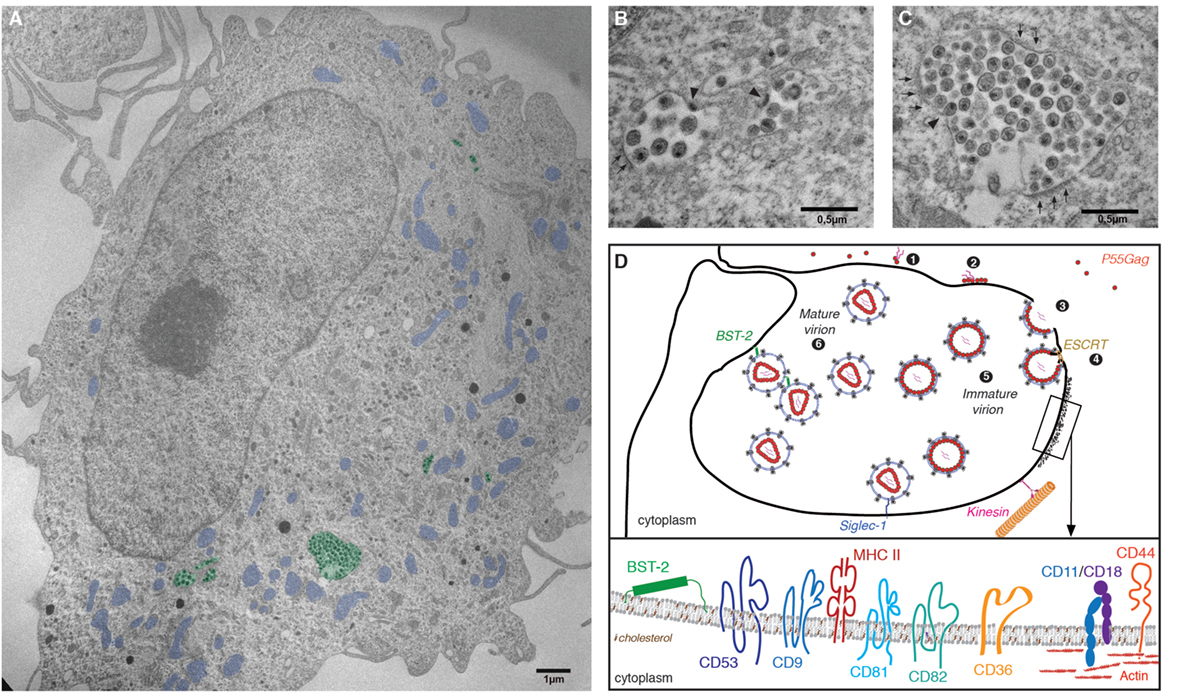
Figure 2. The virus-containing compartment (VCC) in macrophages. (A) Electron micrograph depicting HIV-1-infected monocyte-derived macrophages (MDMs). MDMs were differentiated from monocytes purified from the peripheral blood of healthy human donors, by culture over 7 days in the presence of M-cerebrospinal fluid. Cells were then infected with HIV-1 NL-AD8 and fixed and embedded in epon 5 days postinfection. Ultrathin sections were processed for electron microscopy and imaged using a Philips 120 keV. For clarity, VCC is pseudo-colored in green and mitochondria in blue. (B,C) Magnifications of VCC present in the MDM depicted in panel (A). Arrowheads indicate viral buds. A thick molecular coat, electron dense and often associated with the VCC limiting membrane of the VCC can be seen in panel (C), see arrowheads. (D) Schematic representation of the late phases of the HIV life cycle in macrophages. (1) Gag monomers initiate oligomerization in the cytoplasm, forming dimers with the viral genomic RNA and (2) subsequently bind the plasma membrane via interactions with acidic phospholipids. (3) In macrophages, high-order Gag multimerization and formation of a viral bud only occurs at the limiting membrane of the VCC. (4) Gag subsequently recruits the components of the ESCRT complex that ensure fission of the budding viral particle into the lumen of the VCC. (5) Immature viral particles accumulate inside the VCC and (6) convert into mature viral particles via the activity of the viral protease. The restriction factor BST2/tetherin and Siglec-1/CD169 may contribute to the retention of viral particles within the lumen of the VCC. The limiting membrane of the VCC is tightly associated with the microtubule network on which kinesins may drive the transport of the VCC toward the cell periphery. Inset: Magnification of a region associated with a molecular coat: the VCC limiting membrane constitutes a platform for viral assembly where Gag oligomerization will lead to virus production. Therefore, the viral and VCC membranes share similar composition. They are enriched in particular lipids such as cholesterol and transmembrane proteins. These include the tetraspanins CD9, CD53, CD81, and CD82, the scavenger receptor CD36, the integrins CD18/CD11, or the surface glycoprotein CD44 (see text for details and references). The electron micrograph depicted in this figure panel results from original work performed in our lab and has not been published elsewhere previously.
Evidence supports the notion that the VCC originates from intracellular sequestration of domains of the plasma membrane with a specific protein and lipid composition (102, 103). Cell surface proteins found in the limiting membrane of the VCC include the tetraspanins CD9, CD53, CD81, and CD82 (96, 104) that provide membrane rigidity; the scavenger receptor CD36 (105) that contains two cytoplasmic tails; CD44 (98), a receptor with promiscuous capacity to bind numerous ligands; and MHC II complexes (106) (Figure 2). The restriction factors IFITM, with their peculiar topology of membrane insertion, are incorporated into viral membranes, especially in viruses produced by MDM (94), and may therefore also associate with the limiting membrane of the VCC, potentially contributing to a decreased membrane fluidity. As in T cells, the ESCRT complex is in charge of promoting the fission of nascent particles (107) and ESCRT III proteins that are key players in the late abscission process have been found at the VCC limiting membrane (108). Further supporting a surface origin for the VCC, fluorescence recovery after photobleaching (FRAP) experiments demonstrated that the VCC limiting membrane and the plasma membrane are in rapid equilibrium (102).
Given that newly formed particles bud away from the cytosol toward the lumen of the VCC, they become wrapped with VCC-derived membrane (Figure 2). The proteomic analysis of purified HIV-1 particles released by infected MDM published more than 10 years ago (109) thus reflects the protein composition of the VCC limiting membrane, and still represents a valuable source of information. Studies published thereafter indeed confirmed not only the presence of the given proteins at the VCC limiting membrane but also revealed their role in the viral assembly process [see, for instance, CD36 (105), IFITM (94), and CD18 or Filamin A (110)].
An issue debated since the initial characterization of the VCC is whether some degree of viral budding can also occur at the surface of the macrophage. Indeed, some studies claimed that budding at the surface occurs alongside budding at the VCC (99, 111). A recent study provided an exhaustive examination of the site of virion assembly in individual macrophages by using a viral mutant arrested at the budding stage (103). Although such arrested buds could potentially move toward the plasma membrane, the analysis indicated that only about 5–12% of budding events occur at the cell surface. Moreover, these events were concentrated in the region where the VCC connected with the surface (103). These observations confirm that the VCC is the primary site of HIV assembly in macrophages, while budding at the surface is, at best, rare.
Thus, the specific lipid and protein composition of the limiting membrane of the VCC seem to constitute an assembly platform (101, 112), and presumably possess specific properties required for assembly and budding. However, which cellular and viral determinants direct HIV assembly and budding in macrophages to the VCC, remain poorly elucidated. The Gag polyprotein precursor drives the multistep process of HIV assembly and coordinates the activity of the cellular players involved. Recent studies indicate that viral genomic RNA is an important player/cofactor in the viral assembly process. Using elegant cross-linking immunoprecipitation sequencing coupled to membrane flotations, Kutluay et al. established that assembly starts in the cytoplasm where monomers and dimers of newly synthesized Gag bound to viral genome accumulate (113, 114). Oligomerization of Gag requires interactions with membrane enriched in cholesterol (115), via its MA domain that directly binds to membrane phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) (116) [see Ref. (112)]. The levels of PI(4,5)P2 in the VCC are similar to those at the surface (102) and hence should not be a determining factor that directs viral assembly to the VCC. Assembly of the viral particle at the membrane is coordinated via lateral interactions promoted by the CA and nucleocapsid (NC) domains of Gag, leading to a nascent virion that buds off the membrane (117). Gag possesses two RNA binding domains (NC and MA) with different specificities (115). In addition, the RNA binding specificities for the viral genome versus cellular RNA, i.e., mRNA and tRNA, present in the cytosol change during these processes finally promoting packaging of viral genomic RNA into the nascent viral particles (115).
Aiming to elucidate the role of the viral factors dictating the assembly site of HIV in macrophages, Inlora and colleagues evaluated the impact of targeted deletions or mutations in the different Gag domains, on virion assembly at the VCC (118). Viral mutants unable of high-order multimerization, due to NC substitutions, distributed equally between the VCC and the cell surface (118). This indicates that Gag initially binds the membrane arbitrarily at the surface membrane or the VCC and initiates low-order multimerization. However, in macrophages, high-order multimerization and complete virion assembly occur at the VCC but not at the surface.
Assembly of the polyprotein Gag precursor leads to the formation of a bud head and stalk. Gag further coordinates the recruitment of important cellular factors. These include the ESCRT proteins; key players critically required for proper abscission of the nascent viral particles. Gag recruits components of the ESCRT complex through its late p6 C-ter domain, thus promoting severance of the viral stalk and particle release in the VCC. The precise molecular mechanisms involved in membrane abscission have stimulated numerous studies and corresponding excellent reviews [see, for instance, Ref. (107, 119)], and thus will not be discussed here.
The molecular players implicated in the establishment and maintenance of the VCC’s intricate architecture remain poorly characterized. The cytoskeleton appears to play a key role in maintaining the integrity of the VCC. Indeed, a meshwork of filamentous actin surrounds the VCC and treating MDMs with actin-depolymerizing agents causes dispersion of the VCC throughout the cell (102). Moreover, the VCC limiting membrane is often surrounded by an electron dense and thick molecular coat visible by EM and containing CD18, a β2 integrin and its associated α integrins CD11b and CD11c (Figure 2) (120). Proteins known to interact with integrins are also associated with the molecular coat, including actin and focal adhesion scaffold/linker proteins such as talin, vinculin, and paxillin (120). These adherent complexes appear to be involved in the maintenance of the architecture of the VCC; however, CD18 silencing does not affect the amount of virus released nor its infectivity (120). Links between VCC and the autophagy machinery in HIV-1-infected macrophages have been investigated in a few studies that should be extended in the future (121).
The VCC limiting membrane appears tightly associated with the microtubule network and disruption of this network by nocodazole exposure leads to relocalization of the VCC into the perinuclear area (122). This suggested that kinesins, molecular motors associated with microtubules, could be involved in maintaining the correct positioning of the compartment. The kinesin II, KIF3A, is closely associated with VCCs in infected macrophages (122). Moreover, time-lapse microscopy in primary MDM showed paired movements of Gag and KIF3A. Importantly, KIF3A silencing in HIV-1-infected macrophages reduced viral particle release and increased intracellular Gag and the VCC volume. Overall, KIF3A is likely involved in the transport of the VCC along microtubules and toward the macrophage periphery or provides a force for particle release from the VCC (122). Other molecular motors are likely involved in the transport and positioning of the VCC but also in transport steps of viral components toward the assembly sites, i.e., the VCC (123). Future work will aim to establish at the molecular level how viral release from VCC is regulated.
While the VCC is defined by its viral content, the question of its induction by HIV or its existence in macrophages before infection was of interest. Analysis of primary MDMs by ultrastructural approaches suggested that indeed similar compartments are present in non-infected macrophages (96, 99, 120). Confocal microscopy confirmed the presence in non-infected MDMs of compartments sharing specific features with the VCC; i.e., containing CD36 and CD9, rapid accessibility to small dextran and therefore connected to the external medium (105). The direct proof of HIV capacity to highjack such compartments was obtained when transduced macrophages exhibiting CD36-GFP+ compartments were subjected to time-lapse epifluorescent microscopy after infection with HIV-1 Gag-iCherry allowing the visualization in real time of Gag recruitment and accumulation only to preexisting CD36+ compartments (105). Thus, in macrophages, HIV-1 hijacks these CD36+ compartments for viral assembly (105), and expands them (96).
Viral particles accumulate in the VCC over time (100). Infected MDMs initially contain sparse, barely filled VCCs, but as the time after infection progresses the compartments become crowded with virions and Brownian-like movements of particles within the VCC become highly limited as shown by FRAP experiments (100). In parallel, viral release into the extracellular media decreases over time, suggesting that viral particles remain within the VCC (100). The restriction factor tetherin is concentrated in the VCC where it may connect viral particles to each other or to the VCC limiting membrane, hence apparently tethering virions to the compartment (89, 124). Indeed, silencing tetherin expression increases viral release and reduces the size of the compartment (89).
Whether HIV-1 release from the VCC is inducible remains indeed elusive. The connections between the VCC and the membrane appear too narrow to allow passive viral diffusion to the extracellular media (99). The release of HIV-1 from infected MDMs can be induced by exposure to extracellular ATP, via activation of the P2X7 purinergic receptor that triggers a drastic remodeling of the cytoskeleton and the VCC, accompanied by sudden release of the viral particles packed within the VCC (125). However, we also propose that the highly dynamic nature of the plasma membrane of macrophages, which is subjected to a very active flux of exocytosis and endocytosis/phagocytosis, may promote the temporary widening of the VCC connections to the plasma membrane and allow the release of viral particles to the extracellular media. Supporting this hypothesis, antibodies specific for VCC components, such as tetraspanins (96), gp120 (126), or CD36 (105) had access to the VCC only after several hours of incubation at 37°C and not at 4°C. These observations suggest that the access of extracellular molecules to the VCC depends on the dynamics of the macrophage’s plasma membrane, which possibly promotes a frequent opening of the connections between the lumen of the compartment and the surface. Future insight into the role of the cytoskeleton and its associated proteins in regulating the dynamics of the VCC and its impact on viral release may open new avenues to pharmacologically target the VCC (123, 127).
Whether tissue macrophages in vivo possess VCCs remains poorly investigated. Indeed, our current view of the VCC results almost exclusively from studies in vitro with MDMs. Of note, former ultrastructural analysis of tissue macrophages from patient’s organs confirmed the presence of mature and immature virions in intracellular compartments (128, 129). Accumulation of viral particles within VCC in macrophages in vivo could be advantageous for the virus to be less accessible to (i) soluble immune mediators such as neutralizing antibodies as observed in vitro (126), (ii) to innate sensors that are cytoplasmic or endosomal, and (iii) to anti-pathogen effector mechanisms such as reactive oxygen species or acidic pH.
Direct cell-to-cell transfer is more efficient for spreading HIV infection than the cell-free route (130). This process involves the formation of a stable interface between an infected and an uninfected cell, known as viral synapse. Interaction between the viral envelope present at the membrane of the infected cell and CD4+ in the target cell initiates formation of the viral synapse, which is then maintained via additional interactions between cell adhesion molecules (131). Directed release leads to virion accumulation at the viral synapse and efficient infection of the target cell (132). Macrophages can transfer HIV directly to T cells (133), and the VCC appears to play a role in this process. Real-time imaging suggested a recruitment of the compartment to the proximity of viral synapses leading to subsequent T cell infection (134, 135), and the VCC markers CD9, CD18, and CD81 were found enriched at the macrophage to T cell interface (124). However, the precise mechanisms underlying viral transfer from infected macrophages to target cells (T cells or macrophages) remains incompletely understood as compared with T cell to T cell transfer and thus deserve further studies.
Despite their monocytic origin MDDCs are rather resistant to HIV-1 infection as compared with MDMs. Yet, MDDCs, when activated by LPS, can capture and retain vial particles in surface-connected compartments that bear some resemblance with the VCC. Although activated MDDCs are resistant to the infection and do not produce new viral progeny, they can efficiently transfer captured viruses to activated CD4+ T cells that get then productively infected. This transfer mode known as infection in trans [see accompanying review by Izquierdo-Useros or Izquierdo-Useros et al. (136)] appears to be highly related to the virological synapse established between HIV-1-infected T cells and target cells (137). This process is thought to play an important role in viral dissemination at the early stages of HIV-1 infection at mucosal entry sites (138). Intravital microscopy revealed that subcapsular sinus macrophages from the peripheral lymph nodes can capture and transfer HIV-1 to target cells without getting productively infected in the process (139). Viral capture is mediated by the sialoadhesin CD169/SIGLEC1 that binds gangliosides embedded in the envelope glycoprotein (139). Interestingly, CD169-mediated capture of HIV-1 in macrophages leads to virion retention in the VCC and subsequent transfer to and productive infection of CD4+ T cells (140). Remarkably, viral particles captured via CD169 intermingled with virions endogenously produced by the macrophage in the same VCCs (140), suggesting that the VCC is not only the site of HIV budding and assembly in macrophages but also a compartment of retention of particles captured from the extracellular media. Indeed, compartments with topologies resembling the VCC have been described in the past after exposure of macrophages to various particulate matter, including latex, cholesterol, or low-density lipoprotein (95).
Macrophages and the HIV Reservoir in the Post-Art Era
While ART can efficiently prevent AIDS by restoring CD4+ T cell counts and suppressing viral load to undetectable levels, it fails to provide a sterilizing cure (141). A viral reservoir remains stable in HIV-infected patients under prolonged therapy (142, 143), and is responsible for the quick viral rebound observed within weeks after ART interruption (144). The cumulative toxicity and the cost associated with lifelong ART made it imperative to devise new strategies to eliminate or curb the viral reservoir (145). Unfortunately, such goal has remained elusive.
At the cellular level, the HIV reservoir during ART is mainly composed of resting memory CD4+ T cells that are latently infected; i.e., cells bearing integrated, transcriptionally silent, but replication-competent proviruses (146). The mechanism behind HIV latency is not fully understood but likely results from multiple factors acting together, such as sequestration of cellular transcription factors in the cytoplasm, epigenetic regulation, or the action of transcriptional repressors [reviewed in Ref. (147)]. T cells with central memory (TCM), transitional memory (TTM), and effector memory (TEM) phenotypes contain the highest levels of latent HIV-1 (148–150). A major breakthrough in the characterization of the latent CD4+ reservoir is the recent identification of CD32a as a surface marker highly enriched in circulating cells harboring replication-competent quiescent proviruses (151). The HIV reservoir is seeded very early after infection (within 2–3 days) (152, 153), and its size remains stable even after years of suppressive therapy (154, 155).
Whether residual viral replication under ART, the so-called active reservoir, contributes to HIV persistence is a rather contentious issue in the field (156). Low-level viremia (“Blips”) can be detected in patients under therapy (157, 158). However, HIV shows little sign of genetic evolution during ART (159, 160), and the emergence of drug-resistant virus is remarkably low (158, 161), suggesting that ongoing residual replication does not significantly influence long-term viral persistence. However, a recent high-depth temporal analysis of the phylogeny of viral sequences from the blood and lymph node of patients under ART revealed a constant replenishment of the circulating reservoir as a result of low-level viral replication in sanctuary sites within lymphoid tissues (162). Experimental evidence for the existence of such lymphoid sanctuaries has emerged in the last years. B cell follicles and more specifically germinal centers have long been known as primary sites of HIV replication (163), possibly due to lower antiretroviral drug penetration (164), exclusion of cytotoxic CD8+ T cells (165, 166), and retention of infectious virions within immune complexes on the surface of follicular dendritic cells (167). In patients under treatment and with an aviremic status, CD4+ T cells with a T follicular helper phenotype that reside within germinal centers are the major source of residual infectious virus and contain the highest levels of HIV DNA (168–170).
These recent advances highlight the importance of accurately defining the HIV reservoir, with respect to its cellular and anatomic composition (171). However, studies evaluating the importance of cellular reservoirs other than CD4+ T lymphocytes have been scarce and, for the most part, non-conclusive. Analysis of the rebounding viral sequences in patients after therapy interruption revealed that they differ from proviral sequences integrated in resting CD4 T cells (172). Recovery of M-tropic sequences among the pool of rebounding virus has been recently reported (173). However, there is no readily available method to measure viral rebound from macrophages or other cellular reservoirs, equivalent to the viral outgrowth assay (VOA) typically used to quantify the latent CD4+ reservoir (171). Usually, VOAs require the culture of several million purified cells for accurate quantification of the CD4 reservoir, due to the rarity of latently infected cells (150), making an adaptation of the assay to tissue macrophages unfeasible. Alternatively, quantification of viral nucleic acids provides a more practical manner to evaluate the macrophage reservoir (174). In patients under ART, HIV DNA and/or RNA has been detected in alveolar (175) and duodenal (41) macrophages, microglia in the brain (39), as well as in liver Kupffer cells (176).
Evaluation of the HIV reservoir based on cell-associated DNA or RNA tends to largely overestimate the pool of replication-competent virus, as many defective viral genomes accumulate in patients (177, 178). Also, macrophages may acquire viral DNA via phagocytosis of infected T cells (51), as discussed previously. In the absence of a reliable outgrowth assay to measure the macrophage HIV reservoir from treated patients, animal models become a valuable alternative.
In a recent study with Asian macaques chronically infected with SIV, replication-competent virus could be recovered from macrophages purified from the spleen and mesenteric lymph nodes, using a modified version of the VOA (43). However, in macaques that had been under ART for 5 months, no replication-competent virus could be recovered after macrophage culture, from any of the animals under study and despite the presence of detectable SIV-DNA in macrophages in 40% of the animals (43). A possible limitation of this study is the small number of macrophages that could be purified for the viral outgrowth experiments. However, in parallel experiments, replicating virus could be recovered after culture of purified memory CD4+ T cells from all animals under therapy (43). This suggests that, in the SIV model, the macrophage reservoir, if existent, is clearly smaller than the memory CD4 reservoir.
T cell-deficient humanized mice can be generated by transferring CD34+ human hematopoietic cells into NOD/SCID mice. These mice are reconstituted with human myeloid cells and B cells, but completely devoid of T cells (179). As only macrophages can sustain HIV replication, these myeloid-only mice (MoM) provide a valuable model to test the HIV macrophage reservoir without the confounding effects conferred by the presence of the more abundant CD4+ T cell compartment. When infected with macrophage-tropic HIV-1, MoM present sustained viremia, and viral nucleic acids were detected in macrophages of the liver, bone marrow, spleen, lungs, and brain (179). Initiation of ART in infected MoM leads to undetectable viremia within 2 weeks, and a drastic reduction in the levels of HIV DNA and RNA in the tissues. Importantly, ART interruption led to viral rebound about 7 weeks later, although only in three out of nine animals under study (180). This represents a significantly longer period of viral remission after treatment interruption when compared with T cell-sufficient humanized mice, where rebound occurs within 1–2 weeks after ART interruption (181). Due to the short life span of the MoM mice, the authors could not extend the study and evaluate whether the non-rebounding group of mice would eventually show signs of reactivation of the infection (180).
The absence of T cells in the MoM model of HIV infection, certainly constitute a large deviation from the regular course of HIV infection in humans. Nevertheless, these observations provide the first direct evidence for HIV persistence in macrophages, in the setting of suppressive therapy. Whether such persistence is due to latent infection or ongoing residual replication remains unclear from the data available, as both viral DNA and RNA dropped to undetectable levels in most treated mice (180). Efforts to purge the CD4+ T cell reservoir have mostly employed the “shock and kill” approach (145); a latency-reversal agent (LRA) is initially administered to reactivate viral production in latently infected cells (shock), and followed by an immune-modulatory intervention that renders infected cells susceptible to destruction by the immune system (kill) (182). However, HIV latency in macrophages is not well understood (183), and LRAs that efficiently purge the CD4 reservoir may not have a similar effect in latently infected macrophages. Worse, if the macrophage reservoir is maintained mostly via residual ongoing replication or retention of infectious viral particles within VCCs then LRA therapy will be of negligible effect.
Definitive proof of the macrophage reservoir will require demonstration in human patients under therapy. This will likely require more sensitive methods of measuring the reservoir. A promising alternative is the recent report of an in vivo VOA, wherein cells from patients with undetectable viremia are transplanted into humanized mice (184). This method appears more sensitive than in vitro VOAs, although its widespread application will likely be hampered by the costs associated (185).
Macrophages During Chronic Disease in Treated HIV-1 Infection
Introduction of ART effectively halted the AIDS pandemic, improved health, and prolonged the life of patients. However, a new group of problems, commonly known as “non-AIDS-related conditions,” is emerging in HIV patients with long-term suppressed viremia (186). People living with HIV are at increased risk of developing, among others, cardiovascular and neurocognitive disease, osteoporosis, or cancer (187).
Persistent inflammation appears to lie at the origin of these pathologies, although its causes remain incompletely elucidated and may involve multiple factors. Microbial translocation across the gut mucosa is a well-established cause of systemic inflammation during HIV-1 (188, 189). Long-term suppressive therapy does not completely reconstitute the pool of CD4+ T cells in the gut mucosa, particularly those of the Th17 subset (190). This leads to loss of integrity of the epithelial mucosa and translocation of bacterial products through the lamina propria to mesenteric lymph nodes and extranodal sites (189). These microbial products engage pattern-recognition receptors in cells of the innate immune system, particularly monocytes, macrophages and DCs, leading to widespread production of inflammatory mediators (191). Indeed, myeloid cell-derived biomarkers of microbial translocation, such as IL-6, soluble CD14 (sCD14) or sCD163 are found elevated in ART-treated individuals, as compared with age-matched controls, and are strongly associated with premature mortality of HIV-infected individuals (191, 192). This persistent pro-inflammatory state appears to feedback on intestinal macrophages as they become unable to phagocyte microbial debris in the lamina propria and are thus unable to halt this inflammatory cycle (193). Importantly, persistent systemic inflammation drives the occurrence of non-AIDS comorbidities. For instance, inflammatory monocytes migrate to the heart and contribute to HIV-associated myocarditis (194).
Before the implementation of ART, more than half of HIV-infected patients exhibited HIV-1-associated dementia (HAD); a broad term used to described symptoms of cognitive impairment, including psychiatric disorders, loss of motor coordination, and in severe cases, HIV-1-associated encephalitis (195). The incidence of HAD has dramatically decreased with ART, but a set of milder cognitive problems have emerged and are collectively known as HIV-associated neurological disorders (HAND) that still affect about half of the HIV-1-infected population (196).
Residual viral replication in the CNS and neuronal death has been proposed has an explanation for the occurrence of HAND (197). This effect is probably indirect, as neurons and cells of the macroglia do not support productive HIV infection. Instead, residual viral replication in macrophages leads to the production of inflammatory mediators with neurotoxic action (196). While it is challenging to assess productive infection of brain-resident cells, macrophages are known to contain the highest levels of viral nucleic acids of all CNS cell populations (198). Both perivascular macrophages and microglial cells are targeted by HIV in the CNS (199) and HIV DNA has been detected in brain macrophages from patients under long-term therapy (39). HIV RNA persists in the cerebrospinal fluid (CSF) of ART-treated patients even after suppression of plasma viral RNA to undetectable levels (200), and genetic analysis revealed a significant compartmentalization between the CSF and plasma viral populations (201). Importantly, viruses isolated from the CSF are frequently M-tropic (202). Taken together, these studies suggest that the CNS is a tissue reservoir of HIV-1 during ART, likely maintained through low-level viral replication in resident macrophages (203).
Systemic inflammation is also likely to play a role in the progression of HAND. There is a strong association between circulating levels of sCD14 and the development of neurological disorders in HIV-1 infected individuals (204). It has been proposed that microbial products activate circulating monocytes, particularly those of the CD16+ subset that subsequently cross the blood–brain barrier and differentiate into a pro-inflammatory macrophage population in the brain by producing chemokines, cytokines, and neurotoxic factors such as nitric oxide (48). Supporting this model, CD14+CD16+ cells accumulate in the white matter and perivascular space of brains from non-treated patients (205). Also, these CD14+CD16+ monocytes are capable of transmigrating across in vitro models of the blood–brain barrier in response to the chemokine CCL2 (206).
These selected examples highlight the pathological role played by macrophages and other myeloid cells in non-AIDS conditions that afflict HIV-1-infected patients with suppressed viremia. It is thus crucial to devise new therapies able to complement ART and capable of targeting the persistent inflammation that drives these morbidities.
Conclusion
Because of their localization in many tissues, their long life span and the unique nature of their interaction with HIV-1, macrophages play a key role in HIV-1 pathogenesis. The last decade of research brought new and exciting insight into the ontogeny and functional specialization of tissue-resident macrophages. Whether these recently described macrophage properties, such as their proliferative potential, are explored HIV for dissemination and persistence remains unknown, but this subject should deserve increased interest in future studies. Macrophages are susceptible to HIV-1 infection but also sense the virus and thus participate in the general immune activation observed in infected patients. However, initiation of the antiviral immune response relies on the DC population whose capacity to perform antigen presentation and deal with infection is highly organized in space and time. This dual function appears to be ensured by a division of labor between DC subsets (207). While cDC1 (CD141+) are resistant to productive HIV-1 infection, they can cross-present viral antigens derived from cDC2 (CD1c+) that are susceptible to HIV-1. Thus, this dissociation of the viral infection and the antigen presentation function provides to DC populations the capacity to elicit antiviral immune responses and prime T cell responses (207). How macrophages cross talk with DCs and contribute to the antiviral response remains obscure. Reciprocally, how HIV succeeds to cope with these immune cells specialized in antiviral immunity is similarly incompletely understood. Future work addressing these questions should keep on producing exciting results enlightening our general comprehension of the virus–immune system relationships.
Author Contributions
VR and PB wrote the review. MS-R performed the electron microscopy. NR, PB, and VR designed the cartoon. All the authors edited the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Nicolas Manel at the Institut Curie for critical reading of the manuscript.
Funding
This work was supported by grants from «Agence Nationale de Recherche contre le SIDA et les hépatites virales» (ANRS), «Ensemble contre le SIDA» (Sidaction), ANR-10-IDEX-0001-02 PSL and ANR-11-LABX-0043 to PB. VR and NR were supported by postdoctoral fellowships from ANRS.
References
1. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14:986–95. doi:10.1038/ni.2705
2. Cavaillon JM. The historical milestones in the understanding of leukocyte biology initiated by Elie Metchnikoff. J Leukoc Biol (2011) 90:413–24. doi:10.1189/jlb.0211094
3. Gordon S. Phagocytosis: the legacy of Metchnikoff. Cell (2016) 166:1065–8. doi:10.1016/j.cell.2016.08.017
4. Van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ (1972) 46:845–52.
5. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity (2016) 44:439–49. doi:10.1016/j.immuni.2016.02.024
6. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol (2015) 33:643–75. doi:10.1146/annurev-immunol-032414-112220
7. Ghigo C, Mondor I, Jorquera A, Nowak J, Wienert S, Zahner SP, et al. Multicolor fate mapping of Langerhans cell homeostasis. J Exp Med (2013) 210:1657–64. doi:10.1084/jem.20130403
8. Alliot F, Lecain E, Grima B, Pessac B. Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proc Natl Acad Sci U S A (1991) 88:1541–5. doi:10.1073/pnas.88.4.1541
9. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330:841–5. doi:10.1126/science.1194637
10. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38:792–804. doi:10.1016/j.immuni.2013.04.004
11. Ajami B, Bennett JL, Krieger C, Mcnagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci (2011) 14:1142–9. doi:10.1038/nn.2887
12. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol (2014) 15:929–37. doi:10.1038/ni.2967
13. Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol (2016) 17:9–17. doi:10.1038/ni.3320
14. Franken L, Schiwon M, Kurts C. Macrophages: sentinels and regulators of the immune system. Cell Microbiol (2016) 18:475–87. doi:10.1111/cmi.12580
15. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi:10.1146/annurev-physiol-022516-034339
16. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11:723–37. doi:10.1038/nri3073
17. Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science (1986) 233:215–9. doi:10.1126/science.3014648
18. Duncan CJ, Sattentau QJ. Viral determinants of HIV-1 macrophage tropism. Viruses (2011) 3:2255–79. doi:10.3390/v3112255
19. Marechal V, Prevost MC, Petit C, Perret E, Heard JM, Schwartz O. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J Virol (2001) 75:11166–77. doi:10.1128/JVI.75.22.11166-11177.2001
20. Van Wilgenburg B, Moore MD, James WS, Cowley SA. The productive entry pathway of HIV-1 in macrophages is dependent on endocytosis through lipid rafts containing CD4. PLoS One (2014) 9:e86071. doi:10.1371/journal.pone.0086071
21. Li Q, Li W, Yin W, Guo J, Zhang ZP, Zeng D, et al. Single-particle tracking of human immunodeficiency virus type 1 productive entry into human primary macrophages. ACS Nano (2017) 11:3890–903. doi:10.1021/acsnano.7b00275
22. Carter CA, Ehrlich LS. Cell biology of HIV-1 infection of macrophages. Annu Rev Microbiol (2008) 62:425–43. doi:10.1146/annurev.micro.62.081307.162758
23. Peters PJ, Sullivan WM, Duenas-Decamp MJ, Bhattacharya J, Ankghuambom C, Brown R, et al. Non-macrophage-tropic human immunodeficiency virus type 1 R5 envelopes predominate in blood, lymph nodes, and semen: implications for transmission and pathogenesis. J Virol (2006) 80:6324–32. doi:10.1128/JVI.02328-05
24. Jayakumar P, Berger I, Autschbach F, Weinstein M, Funke B, Verdin E, et al. Tissue-resident macrophages are productively infected ex vivo by primary X4 isolates of human immunodeficiency virus type 1. J Virol (2005) 79:5220–6. doi:10.1128/JVI.79.8.5220-5226.2005
25. Boltz VF, Rausch J, Shao W, Hattori J, Luke B, Maldarelli F, et al. Ultrasensitive single-genome sequencing: accurate, targeted, next generation sequencing of HIV-1 RNA. Retrovirology (2016) 13:87. doi:10.1186/s12977-016-0321-6
26. Joseph SB, Swanstrom R, Kashuba AD, Cohen MS. Bottlenecks in HIV-1 transmission: insights from the study of founder viruses. Nat Rev Microbiol (2015) 13:414–25. doi:10.1038/nrmicro3471
27. Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med (2009) 206:1273–89. doi:10.1084/jem.20090378
28. Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, et al. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol (2012) 86:2715–28. doi:10.1128/JVI.06157-11
29. Fackler OT, Murooka TT, Imle A, Mempel TR. Adding new dimensions: towards an integrative understanding of HIV-1 spread. Nat Rev Microbiol (2014) 12:563–74. doi:10.1038/nrmicro3309
30. Hladik F, Sakchalathorn P, Ballweber L, Lentz G, Fialkow M, Eschenbach D, et al. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity (2007) 26:257–70. doi:10.1016/j.immuni.2007.01.007
31. Saba E, Grivel JC, Vanpouille C, Brichacek B, Fitzgerald W, Margolis L, et al. HIV-1 sexual transmission: early events of HIV-1 infection of human cervico-vaginal tissue in an optimized ex vivo model. Mucosal Immunol (2010) 3:280–90. doi:10.1038/mi.2010.2
32. King DF, Siddiqui AA, Buffa V, Fischetti L, Gao Y, Stieh D, et al. Mucosal tissue tropism and dissemination of HIV-1 subtype B acute envelope-expressing chimeric virus. J Virol (2013) 87:890–9. doi:10.1128/JVI.02216-12
33. Spira AI, Marx PA, Patterson BK, Mahoney J, Koup RA, Wolinsky SM, et al. Cellular targets of infection and route of viral dissemination after an intravaginal inoculation of simian immunodeficiency virus into rhesus macaques. J Exp Med (1996) 183:215–25. doi:10.1084/jem.183.1.215
34. Barouch DH, Ghneim K, Bosche WJ, Li Y, Berkemeier B, Hull M, et al. Rapid inflammasome activation following mucosal SIV infection of rhesus monkeys. Cell (2016) 165:656–67. doi:10.1016/j.cell.2016.03.021
35. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med (2004) 200:749–59. doi:10.1084/jem.20040874
36. Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, et al. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature (2005) 434:1148–52. doi:10.1038/nature03513
37. Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature (2005) 434:1093–7. doi:10.1038/nature03501
38. Hufert FT, Schmitz J, Schreiber M, Schmitz H, Racz P, Von Laer DD. Human Kupffer cells infected with HIV-1 in vivo. J Acquir Immune Defic Syndr (1993) 6:772–7.
39. Churchill MJ, Gorry PR, Cowley D, Lal L, Sonza S, Purcell DF, et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol (2006) 12:146–52. doi:10.1080/13550280600748946
40. Jambo KC, Banda DH, Kankwatira AM, Sukumar N, Allain TJ, Heyderman RS, et al. Small alveolar macrophages are infected preferentially by HIV and exhibit impaired phagocytic function. Mucosal Immunol (2014) 7:1116–26. doi:10.1038/mi.2013.127
41. Zalar A, Figueroa MI, Ruibal-Ares B, Bare P, Cahn P, De Bracco MM, et al. Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Antiviral Res (2010) 87:269–71. doi:10.1016/j.antiviral.2010.05.005
42. Yukl SA, Sinclair E, Somsouk M, Hunt PW, Epling L, Killian M, et al. A comparison of methods for measuring rectal HIV levels suggests that HIV DNA resides in cells other than CD4+ T cells, including myeloid cells. AIDS (2014) 28:439–42. doi:10.1097/QAD.0000000000000166
43. Dinapoli SR, Ortiz AM, Wu F, Matsuda K, Twigg HL III, Hirsch VM, et al. Tissue-resident macrophages can contain replication-competent virus in antiretroviral-naive, SIV-infected Asian macaques. JCI Insight (2017) 2:e91214. doi:10.1172/jci.insight.91214
44. Sonza S, Mutimer HP, Oelrichs R, Jardine D, Harvey K, Dunne A, et al. Monocytes harbour replication-competent, non-latent HIV-1 in patients on highly active antiretroviral therapy. AIDS (2001) 15:17–22. doi:10.1097/00002030-200101050-00005
45. Zhu T, Muthui D, Holte S, Nickle D, Feng F, Brodie S, et al. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J Virol (2002) 76:707–16. doi:10.1128/JVI.76.2.707-716.2002
46. Ellery PJ, Tippett E, Chiu YL, Paukovics G, Cameron PU, Solomon A, et al. The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol (2007) 178:6581–9. doi:10.4049/jimmunol.178.10.6581
47. Pulliam L, Gascon R, Stubblebine M, Mcguire D, Mcgrath MS. Unique monocyte subset in patients with AIDS dementia. Lancet (1997) 349:692–5. doi:10.1016/S0140-6736(96)10178-1
48. Williams DW, Veenstra M, Gaskill PJ, Morgello S, Calderon TM, Berman JW. Monocytes mediate HIV neuropathogenesis: mechanisms that contribute to HIV associated neurocognitive disorders. Curr HIV Res (2014) 12:85–96. doi:10.2174/1570162X12666140526114526
49. Verollet C, Souriant S, Bonnaud E, Jolicoeur P, Raynaud-Messina B, Kinnaer C, et al. HIV-1 reprograms the migration of macrophages. Blood (2015) 125:1611–22. doi:10.1182/blood-2014-08-596775
50. Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, Von Andrian UH, et al. HIV-infected T cells are migratory vehicles for viral dissemination. Nature (2012) 490:283–7. doi:10.1038/nature11398
51. Calantone N, Wu F, Klase Z, Deleage C, Perkins M, Matsuda K, et al. Tissue myeloid cells in SIV-infected primates acquire viral DNA through phagocytosis of infected T cells. Immunity (2014) 41:493–502. doi:10.1016/j.immuni.2014.08.014
52. Baxter AE, Russell RA, Duncan CJ, Moore MD, Willberg CB, Pablos JL, et al. Macrophage infection via selective capture of HIV-1-infected CD4+ T cells. Cell Host Microbe (2014) 16:711–21. doi:10.1016/j.chom.2014.10.010
53. Sattentau QJ, Stevenson M. Macrophages and HIV-1: an unhealthy constellation. Cell Host Microbe (2016) 19:304–10. doi:10.1016/j.chom.2016.02.013
54. Moir S, Chun TW, Fauci AS. Pathogenic mechanisms of HIV disease. Annu Rev Pathol (2011) 6:223–48. doi:10.1146/annurev-pathol-011110-130254
55. Richards KH, Aasa-Chapman MM, Mcknight A, Clapham PR. Modulation of HIV-1 macrophage-tropism among R5 envelopes occurs before detection of neutralizing antibodies. Retrovirology (2010) 7:48. doi:10.1186/1742-4690-7-48
56. Micci L, Alvarez X, Iriele RI, Ortiz AM, Ryan ES, Mcgary CS, et al. CD4 depletion in SIV-infected macaques results in macrophage and microglia infection with rapid turnover of infected cells. PLoS Pathog (2014) 10:e1004467. doi:10.1371/journal.ppat.1004467
57. Price JV, Vance RE. The macrophage paradox. Immunity (2014) 41:685–93. doi:10.1016/j.immuni.2014.10.015
58. Altfeld M, Gale M Jr. Innate immunity against HIV-1 infection. Nat Immunol (2015) 16:554–62. doi:10.1038/ni.3157
59. Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol (2015) 16:546–53. doi:10.1038/ni.3156
60. Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity (2013) 39:1132–42. doi:10.1016/j.immuni.2013.11.002
61. Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature (2013) 503:402–5. doi:10.1038/nature12769
62. Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol (2010) 11:1005–13. doi:10.1038/ni.1941
63. Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science (2013) 341:903–6. doi:10.1126/science.1240933
64. Nasr N, Maddocks S, Turville SG, Harman AN, Woolger N, Helbig KJ, et al. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood (2012) 120:778–88. doi:10.1182/blood-2012-01-407395
65. Diget EA, Zuwala K, Berg RK, Laursen RR, Soby S, Ostergaard L, et al. Characterization of HIV-1 infection and innate sensing in different types of primary human monocyte-derived macrophages. Mediators Inflamm (2013) 2013:208412. doi:10.1155/2013/208412
66. Woelk CH, Ottones F, Plotkin CR, Du P, Royer CD, Rought SE, et al. Interferon gene expression following HIV type 1 infection of monocyte-derived macrophages. AIDS Res Hum Retroviruses (2004) 20:1210–22. doi:10.1089/aid.2004.20.1210
67. Decalf J, Desdouits M, Rodrigues V, Gobert FX, Gentili M, Marques-Ladeira S, et al. Sensing of HIV-1 entry triggers a type I interferon response in human primary macrophages. J Virol (2017) 91:e147–117. doi:10.1128/JVI.00147-17
68. Noyce RS, Taylor K, Ciechonska M, Collins SE, Duncan R, Mossman KL. Membrane perturbation elicits an IRF3-dependent, interferon-independent antiviral response. J Virol (2011) 85:10926–31. doi:10.1128/JVI.00862-11
69. Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol (2012) 13:737–43. doi:10.1038/ni.2350
70. Nasr N, Alshehri AA, Wright TK, Shahid M, Heiner BM, Harman AN, et al. Mechanism of interferon-stimulated gene induction in HIV-1-infected macrophages. J Virol (2017) 91:e744–717. doi:10.1128/JVI.00744-17
71. Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature (2011) 474:658–61. doi:10.1038/nature10195
72. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature (2011) 474:654–7. doi:10.1038/nature10117
73. Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol (2012) 13:223–8. doi:10.1038/ni0612-621c
74. Ryoo J, Choi J, Oh C, Kim S, Seo M, Kim SY, et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat Med (2014) 20:936–41. doi:10.1038/nm.3626
75. Cribier A, Descours B, Valadao AL, Laguette N, Benkirane M. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep (2013) 3:1036–43. doi:10.1016/j.celrep.2013.03.017
76. Pauls E, Ruiz A, Badia R, Permanyer M, Gubern A, Riveira-Munoz E, et al. Cell cycle control and HIV-1 susceptibility are linked by CDK6-dependent CDK2 phosphorylation of SAMHD1 in myeloid and lymphoid cells. J Immunol (2014) 193:1988–97. doi:10.4049/jimmunol.1400873
77. Mlcochova P, Sutherland KA, Watters SA, Bertoli C, De Bruin RA, Rehwinkel J, et al. A G1-like state allows HIV-1 to bypass SAMHD1 restriction in macrophages. EMBO J (2017) 36:604–16. doi:10.15252/embj.201696025
78. Kyei GB, Cheng X, Ramani R, Ratner L. Cyclin L2 is a critical HIV dependency factor in macrophages that controls SAMHD1 abundance. Cell Host Microbe (2015) 17:98–106. doi:10.1016/j.chom.2014.11.009
79. Badia R, Pujantell M, Riveira-Munoz E, Puig T, Torres-Torronteras J, Marti R, et al. The G1/S specific cyclin D2 is a regulator of HIV-1 restriction in non-proliferating cells. PLoS Pathog (2016) 12:e1005829. doi:10.1371/journal.ppat.1005829
80. Sunseri N, O’Brien M, Bhardwaj N, Landau NR. Human immunodeficiency virus type 1 modified to package Simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J Virol (2011) 85:6263–74. doi:10.1128/JVI.00346-11
81. Lahaye X, Manel N. Viral and cellular mechanisms of the innate immune sensing of HIV. Curr Opin Virol (2015) 11:55–62. doi:10.1016/j.coviro.2015.01.013
82. Tada T, Zhang Y, Koyama T, Tobiume M, Tsunetsugu-Yokota Y, Yamaoka S, et al. MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat Med (2015) 21:1502–7. doi:10.1038/nm.3956
83. Krapp C, Hotter D, Gawanbacht A, Mclaren PJ, Kluge SF, Sturzel CM, et al. Guanylate binding protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe (2016) 19:504–14. doi:10.1016/j.chom.2016.02.019
84. Schwartz S, Felber BK, Fenyo EM, Pavlakis GN. Env and Vpu proteins of human immunodeficiency virus type 1 are produced from multiple bicistronic mRNAs. J Virol (1990) 64:5448–56.
85. Thomas ER, Dunfee RL, Stanton J, Bogdan D, Kunstman K, Wolinsky SM, et al. High frequency of defective vpu compared with tat and rev genes in brain from patients with HIV type 1-associated dementia. AIDS Res Hum Retroviruses (2007) 23:575–80. doi:10.1089/aid.2006.0246
86. Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature (2008) 451:425–30. doi:10.1038/nature06553
87. Perez-Caballero D, Zang T, Ebrahimi A, Mcnatt MW, Gregory DA, Johnson MC, et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell (2009) 139:499–511. doi:10.1016/j.cell.2009.08.039
88. Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe (2008) 3:245–52. doi:10.1016/j.chom.2008.03.001
89. Chu H, Wang JJ, Qi M, Yoon JJ, Chen X, Wen X, et al. Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe (2012) 12:360–72. doi:10.1016/j.chom.2012.07.011
90. Lu J, Pan Q, Rong L, He W, Liu SL, Liang C. The IFITM proteins inhibit HIV-1 infection. J Virol (2011) 85:2126–37. doi:10.1128/JVI.01531-10
91. Jia R, Pan Q, Ding S, Rong L, Liu SL, Geng Y, et al. The N-terminal region of IFITM3 modulates its antiviral activity by regulating IFITM3 cellular localization. J Virol (2012) 86:13697–707. doi:10.1128/JVI.01828-12
92. Compton AA, Bruel T, Porrot F, Mallet A, Sachse M, Euvrard M, et al. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe (2014) 16:736–47. doi:10.1016/j.chom.2014.11.001
93. Chesarino NM, Compton AA, Mcmichael TM, Kenney AD, Zhang L, Soewarna V, et al. IFITM3 requires an amphipathic helix for antiviral activity. EMBO Rep (2017) 18:1740–51. doi:10.15252/embr.201744100
94. Tartour K, Appourchaux R, Gaillard J, Nguyen XN, Durand S, Turpin J, et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology (2014) 11:103. doi:10.1186/s12977-014-0103-y
95. Tan J, Sattentau QJ. The HIV-1-containing macrophage compartment: a perfect cellular niche? Trends Microbiol (2013) 21:405–12. doi:10.1016/j.tim.2013.05.001
96. Deneka M, Pelchen-Matthews A, Byland R, Ruiz-Mateos E, Marsh M. In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J Cell Biol (2007) 177:329–41. doi:10.1083/jcb.200609050
97. Jouve M, Sol-Foulon N, Watson S, Schwartz O, Benaroch P. HIV-1 buds and accumulates in “nonacidic” endosomes of macrophages. Cell Host Microbe (2007) 2:85–95. doi:10.1016/j.chom.2007.06.011
98. Welsch S, Keppler OT, Habermann A, Allespach I, Krijnse-Locker J, Krausslich HG. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog (2007) 3:e36. doi:10.1371/journal.ppat.0030036
99. Welsch S, Groot F, Krausslich HG, Keppler OT, Sattentau QJ. Architecture and regulation of the HIV-1 assembly and holding compartment in macrophages. J Virol (2011) 85:7922–7. doi:10.1128/JVI.00834-11
100. Gaudin R, Berre S, Cunha De Alencar B, Decalf J, Schindler M, Gobert FX, et al. Dynamics of HIV-containing compartments in macrophages reveal sequestration of virions and transient surface connections. PLoS One (2013) 8:e69450. doi:10.1371/journal.pone.0069450
101. Waheed AA, Freed EO. Lipids and membrane microdomains in HIV-1 replication. Virus Res (2009) 143:162–76. doi:10.1016/j.virusres.2009.04.007
102. Mlcochova P, Pelchen-Matthews A, Marsh M. Organization and regulation of intracellular plasma membrane-connected HIV-1 assembly compartments in macrophages. BMC Biol (2013) 11:89. doi:10.1186/1741-7007-11-89
103. Nkwe DO, Pelchen-Matthews A, Burden JJ, Collinson LM, Marsh M. The intracellular plasma membrane-connected compartment in the assembly of HIV-1 in human macrophages. BMC Biol (2016) 14:50. doi:10.1186/s12915-016-0272-3
104. Pelchen-Matthews A, Kramer B, Marsh M. Infectious HIV-1 assembles in late endosomes in primary macrophages. J Cell Biol (2003) 162:443–55. doi:10.1083/jcb.200304008
105. Berre S, Gaudin R, Cunha De Alencar B, Desdouits M, Chabaud M, Naffakh N, et al. CD36-specific antibodies block release of HIV-1 from infected primary macrophages and its transmission to T cells. J Exp Med (2013) 210:2523–38. doi:10.1084/jem.20130566
106. Raposo G, Moore M, Innes D, Leijendekker R, Leigh-Brown A, Benaroch P, et al. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic (2002) 3:718–29. doi:10.1034/j.1600-0854.2002.31004.x
107. Lippincott-Schwartz J, Freed EO, Van Engelenburg SB. A consensus view of ESCRT-mediated human immunodeficiency virus type 1 abscission. Annu Rev Virol (2017) 4:309–25. doi:10.1146/annurev-virology-101416-041840
108. Welsch S, Habermann A, Jager S, Muller B, Krijnse-Locker J, Krausslich HG. Ultrastructural analysis of ESCRT proteins suggests a role for endosome-associated tubular-vesicular membranes in ESCRT function. Traffic (2006) 7:1551–66. doi:10.1111/j.1600-0854.2006.00489.x
109. Chertova E, Chertov O, Coren LV, Roser JD, Trubey CM, Bess JW Jr, et al. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J Virol (2006) 80:9039–52. doi:10.1128/JVI.01013-06
110. Dotson D, Woodruff EA, Villalta F, Dong X. Filamin A is involved in HIV-1 Vpu-mediated evasion of host restriction by modulating tetherin expression. J Biol Chem (2016) 291:4236–46. doi:10.1074/jbc.M115.708123
111. Bennett AE, Narayan K, Shi D, Hartnell LM, Gousset K, He H, et al. Ion-abrasion scanning electron microscopy reveals surface-connected tubular conduits in HIV-infected macrophages. PLoS Pathog (2009) 5:e1000591. doi:10.1371/journal.ppat.1000591
112. Mariani C, Desdouits M, Favard C, Benaroch P, Muriaux DM. Role of Gag and lipids during HIV-1 assembly in CD4(+) T cells and macrophages. Front Microbiol (2014) 5:312. doi:10.3389/fmicb.2014.00312
113. Kutluay SB, Bieniasz PD. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog (2010) 6:e1001200. doi:10.1371/journal.ppat.1001200
114. Kutluay SB, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, et al. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell (2014) 159:1096–109. doi:10.1016/j.cell.2014.09.057
115. Ono A, Waheed AA, Freed EO. Depletion of cellular cholesterol inhibits membrane binding and higher-order multimerization of human immunodeficiency virus type 1 Gag. Virology (2007) 360:27–35. doi:10.1016/j.virol.2006.10.011
116. Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J Virol (2008) 82:2405–17. doi:10.1128/JVI.01614-07
117. Balasubramaniam M, Freed EO. New insights into HIV assembly and trafficking. Physiology (Bethesda) (2011) 26:236–51. doi:10.1152/physiol.00051.2010
118. Inlora J, Chukkapalli V, Bedi S, Ono A. Molecular determinants directing HIV-1 Gag assembly to virus-containing compartments in primary macrophages. J Virol (2016) 90:8509–19. doi:10.1128/JVI.01004-16
119. Schoneberg J, Lee IH, Iwasa JH, Hurley JH. Reverse-topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol (2017) 18:5–17. doi:10.1038/nrm.2016.121
120. Pelchen-Matthews A, Giese S, Mlcochova P, Turner J, Marsh M. beta2 Integrin adhesion complexes maintain the integrity of HIV-1 assembly compartments in primary macrophages. Traffic (2012) 13:273–91. doi:10.1111/j.1600-0854.2011.01306.x
121. Dinkins C, Pilli M, Kehrl JH. Roles of autophagy in HIV infection. Immunol Cell Biol (2015) 93:11–7. doi:10.1038/icb.2014.88
122. Gaudin R, De Alencar BC, Jouve M, Berre S, Le Bouder E, Schindler M, et al. Critical role for the kinesin KIF3A in the HIV life cycle in primary human macrophages. J Cell Biol (2012) 199:467–79. doi:10.1083/jcb.201201144
123. Gaudin R, De Alencar BC, Arhel N, Benaroch P. HIV trafficking in host cells: motors wanted! Trends Cell Biol (2013) 23:652–62. doi:10.1016/j.tcb.2013.09.004
124. Giese S, Marsh M. Tetherin can restrict cell-free and cell-cell transmission of HIV from primary macrophages to T cells. PLoS Pathog (2014) 10:e1004189. doi:10.1371/journal.ppat.1004189
125. Graziano F, Desdouits M, Garzetti L, Podini P, Alfano M, Rubartelli A, et al. Extracellular ATP induces the rapid release of HIV-1 from virus containing compartments of human macrophages. Proc Natl Acad Sci U S A (2015) 112:E3265–73. doi:10.1073/pnas.1500656112
126. Koppensteiner H, Banning C, Schneider C, Hohenberg H, Schindler M. Macrophage internal HIV-1 is protected from neutralizing antibodies. J Virol (2012) 86:2826–36. doi:10.1128/JVI.05915-11
127. Graziano F, Vicenzi E, Poli G. Immuno-pharmacological targeting of virus-containing compartments in HIV-1-infected macrophages. Trends Microbiol (2016) 24:558–67. doi:10.1016/j.tim.2016.02.018
128. Orenstein JM, Meltzer MS, Phipps T, Gendelman HE. Cytoplasmic assembly and accumulation of human immunodeficiency virus types 1 and 2 in recombinant human colony-stimulating factor-1-treated human monocytes: an ultrastructural study. J Virol (1988) 62:2578–86.
129. Orenstein JM. Replication of HIV-1 in vivo and in vitro. Ultrastruct Pathol (2007) 31:151–67. doi:10.1080/01913120701344343
130. Sattentau Q. Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol (2008) 6:815–26. doi:10.1038/nrmicro1972
131. Law KM, Satija N, Esposito AM, Chen BK. Cell-to-cell spread of HIV and viral pathogenesis. Adv Virus Res (2016) 95:43–85. doi:10.1016/bs.aivir.2016.03.001
132. Agosto LM, Uchil PD, Mothes W. HIV cell-to-cell transmission: effects on pathogenesis and antiretroviral therapy. Trends Microbiol (2015) 23:289–95. doi:10.1016/j.tim.2015.02.003
133. Groot F, Welsch S, Sattentau QJ. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood (2008) 111:4660–3. doi:10.1182/blood-2007-12-130070
134. Gousset K, Ablan SD, Coren LV, Ono A, Soheilian F, Nagashima K, et al. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog (2008) 4:e1000015. doi:10.1371/journal.ppat.1000015
135. Duncan CJ, Williams JP, Schiffner T, Gartner K, Ochsenbauer C, Kappes J, et al. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J Virol (2014) 88:2025–34. doi:10.1128/JVI.03245-13
136. Izquierdo-Useros N, Lorizate M, Mclaren PJ, Telenti A, Krausslich HG, Martinez-Picado J. HIV-1 capture and transmission by dendritic cells: the role of viral glycolipids and the cellular receptor Siglec-1. PLoS Pathog (2014) 10:e1004146. doi:10.1371/journal.ppat.1004146
137. Piguet V, Sattentau Q. Dangerous liaisons at the virological synapse. J Clin Invest (2004) 114:605–10. doi:10.1172/JCI22812
138. Coleman CM, Gelais CS, Wu L. Cellular and viral mechanisms of HIV-1 transmission mediated by dendritic cells. Adv Exp Med Biol (2013) 762:109–30. doi:10.1007/978-1-4614-4433-6_4
139. Sewald X, Ladinsky MS, Uchil PD, Beloor J, Pi R, Herrmann C, et al. Retroviruses use CD169-mediated trans-infection of permissive lymphocytes to establish infection. Science (2015) 350:563–7. doi:10.1126/science.aab2749
140. Hammonds JE, Beeman N, Ding L, Takushi S, Francis AC, Wang JJ, et al. Siglec-1 initiates formation of the virus-containing compartment and enhances macrophage-to-T cell transmission of HIV-1. PLoS Pathog (2017) 13:e1006181. doi:10.1371/journal.ppat.1006181
141. Chun TW, Moir S, Fauci AS. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat Immunol (2015) 16:584–9. doi:10.1038/ni.3152
142. Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science (1997) 278:1295–300. doi:10.1126/science.278.5341.1295
143. Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science (1997) 278:1291–5. doi:10.1126/science.278.5341.1291
144. Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A (1999) 96:15109–14. doi:10.1073/pnas.96.26.15109
145. Margolis DM, Garcia JV, Hazuda DJ, Haynes BF. Latency reversal and viral clearance to cure HIV-1. Science (2016) 353:aaf6517. doi:10.1126/science.aaf6517
146. Barton K, Winckelmann A, Palmer S. HIV-1 reservoirs during suppressive therapy. Trends Microbiol (2016) 24:345–55. doi:10.1016/j.tim.2016.01.006
147. Dahabieh MS, Battivelli E, Verdin E. Understanding HIV latency: the road to an HIV cure. Annu Rev Med (2015) 66:407–21. doi:10.1146/annurev-med-092112-152941
148. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med (2009) 15:893–900. doi:10.1038/nm.1972
149. Buzon MJ, Sun H, Li C, Shaw A, Seiss K, Ouyang Z, et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med (2014) 20:139–42. doi:10.1038/nm.3445
150. Soriano-Sarabia N, Bateson RE, Dahl NP, Crooks AM, Kuruc JD, Margolis DM, et al. Quantitation of replication-competent HIV-1 in populations of resting CD4+ T cells. J Virol (2014) 88:14070–7. doi:10.1128/JVI.01900-14
151. Descours B, Petitjean G, Lopez-Zaragoza JL, Bruel T, Raffel R, Psomas C, et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature (2017) 543:564–7. doi:10.1038/nature21710
152. Chun TW, Engel D, Berrey MM, Shea T, Corey L, Fauci AS. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci U S A (1998) 95:8869–73. doi:10.1073/pnas.95.15.8869
153. Whitney JB, Hill AL, Sanisetty S, Penaloza-Macmaster P, Liu J, Shetty M, et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature (2014) 512:74–7. doi:10.1038/nature13594
154. Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med (2003) 9:727–8. doi:10.1038/nm880
155. Crooks AM, Bateson R, Cope AB, Dahl NP, Griggs MK, Kuruc JD, et al. Precise quantitation of the latent HIV-1 reservoir: implications for eradication strategies. J Infect Dis (2015) 212:1361–5. doi:10.1093/infdis/jiv218
156. Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, Swanstrom R. HIV reservoirs: what, where and how to target them. Nat Rev Microbiol (2016) 14:55–60. doi:10.1038/nrmicro.2015.5
157. Dornadula G, Zhang H, Vanuitert B, Stern J, Livornese L Jr, Ingerman MJ, et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA (1999) 282:1627–32. doi:10.1001/jama.282.17.1627
158. Nettles RE, Kieffer TL, Kwon P, Monie D, Han Y, Parsons T, et al. Intermittent HIV-1 viremia (Blips) and drug resistance in patients receiving HAART. JAMA (2005) 293:817–29. doi:10.1001/jama.293.7.817
159. Josefsson L, Von Stockenstrom S, Faria NR, Sinclair E, Bacchetti P, Killian M, et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc Natl Acad Sci U S A (2013) 110:E4987–96. doi:10.1073/pnas.1308313110
160. Kearney MF, Spindler J, Shao W, Yu S, Anderson EM, O’Shea A, et al. Lack of detectable HIV-1 molecular evolution during suppressive antiretroviral therapy. PLoS Pathog (2014) 10:e1004010. doi:10.1371/journal.ppat.1004010
161. Siliciano JD, Siliciano RF. Recent trends in HIV-1 drug resistance. Curr Opin Virol (2013) 3:487–94. doi:10.1016/j.coviro.2013.08.007
162. Lorenzo-Redondo R, Fryer HR, Bedford T, Kim EY, Archer J, Pond SLK, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature (2016) 530:51–6. doi:10.1038/nature16933
163. Fox CH, Tenner-Racz K, Racz P, Firpo A, Pizzo PA, Fauci AS. Lymphoid germinal centers are reservoirs of human immunodeficiency virus type 1 RNA. J Infect Dis (1991) 164:1051–7. doi:10.1093/infdis/164.6.1051
164. Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci U S A (2014) 111:2307–12. doi:10.1073/pnas.1318249111
165. Connick E, Mattila T, Folkvord JM, Schlichtemeier R, Meditz AL, Ray MG, et al. CTL fail to accumulate at sites of HIV-1 replication in lymphoid tissue. J Immunol (2007) 178:6975–83. doi:10.4049/jimmunol.178.11.6975
166. Mylvaganam GH, Rios D, Abdelaal HM, Iyer S, Tharp G, Mavinger M, et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV infection. Proc Natl Acad Sci U S A (2017) 114:1976–81. doi:10.1073/pnas.1621418114
167. Keele BF, Tazi L, Gartner S, Liu Y, Burgon TB, Estes JD, et al. Characterization of the follicular dendritic cell reservoir of human immunodeficiency virus type 1. J Virol (2008) 82:5548–61. doi:10.1128/JVI.00124-08
168. Perreau M, Savoye AL, De Crignis E, Corpataux JM, Cubas R, Haddad EK, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med (2013) 210:143–56. doi:10.1084/jem.20121932
169. Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, et al. PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat Med (2016) 22:754–61. doi:10.1038/nm.4113
170. Moukambi F, Rodrigues V, Fortier Y, Rabezanahary H, Borde C, Krust B, et al. CD4 T follicular helper cells and HIV infection: friends or enemies? Front Immunol (2017) 8:135. doi:10.3389/fimmu.2017.00135
171. Kandathil AJ, Sugawara S, Balagopal A. Are T cells the only HIV-1 reservoir? Retrovirology (2016) 13:86. doi:10.1186/s12977-016-0323-4
172. Chun TW, Davey RT Jr, Ostrowski M, Shawn Justement J, Engel D, Mullins JI, et al. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat Med (2000) 6:757–61. doi:10.1038/77481
173. Clayton KL, Garcia JV, Clements JE, Walker BD. HIV infection of macrophages: implications for pathogenesis and cure. Pathog Immun (2017) 2:179–92. doi:10.20411/pai.v2i2.204
174. Eriksson S, Graf EH, Dahl V, Strain MC, Yukl SA, Lysenko ES, et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog (2013) 9:e1003174. doi:10.1371/journal.ppat.1003174
175. Cribbs SK, Lennox J, Caliendo AM, Brown LA, Guidot DM. Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res Hum Retroviruses (2015) 31:64–70. doi:10.1089/AID.2014.0133
176. Kandathil AJ, Durand CM, Quinn J, Cameron AM, Thomas DL, Balagopal A. Liver macrophages and HIV persistence. Conference on Retroviruses and Opportunistic Infections. Seattle, WA (2015).
177. Sanchez G, Xu X, Chermann JC, Hirsch I. Accumulation of defective viral genomes in peripheral blood mononuclear cells of human immunodeficiency virus type 1-infected individuals. J Virol (1997) 71:2233–40.
178. Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med (2016) 22:1043–9. doi:10.1038/nm.4156
179. Honeycutt JB, Wahl A, Baker C, Spagnuolo RA, Foster J, Zakharova O, et al. Macrophages sustain HIV replication in vivo independently of T cells. J Clin Invest (2016) 126:1353–66. doi:10.1172/JCI84456
180. Honeycutt JB, Thayer WO, Baker CE, Ribeiro RM, Lada SM, Cao Y, et al. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat Med (2017) 23:638–43. doi:10.1038/nm.4319
181. Honeycutt JB, Wahl A, Archin N, Choudhary S, Margolis D, Garcia JV. HIV-1 infection, response to treatment and establishment of viral latency in a novel humanized T cell-only mouse (TOM) model. Retrovirology (2013) 10:121. doi:10.1186/1742-4690-10-121
182. Archin NM, Sung JM, Garrido C, Soriano-Sarabia N, Margolis DM. Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat Rev Microbiol (2014) 12:750–64. doi:10.1038/nrmicro3352
183. Kumar A, Abbas W, Herbein G. HIV-1 latency in monocytes/macrophages. Viruses (2014) 6:1837–60. doi:10.3390/v6041837
184. Metcalf Pate KA, Pohlmeyer CW, Walker-Sperling VE, Foote JB, Najarro KM, Cryer CG, et al. A murine viral outgrowth assay to detect residual HIV type 1 in patients with undetectable viral loads. J Infect Dis (2015) 212:1387–96. doi:10.1093/infdis/jiv230
185. Henrich TJ, Deeks SG, Pillai SK. Measuring the size of the latent human immunodeficiency virus reservoir: the present and future of evaluating eradication strategies. J Infect Dis (2017) 215:S134–41. doi:10.1093/infdis/jiw648
186. Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet (2013) 382:1525–33. doi:10.1016/S0140-6736(13)61809-7
187. Guaraldi G, Orlando G, Zona S, Menozzi M, Carli F, Garlassi E, et al. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin Infect Dis (2011) 53:1120–6. doi:10.1093/cid/cir627
188. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med (2006) 12:1365–71. doi:10.1038/nm1511
189. Marchetti G, Tincati C, Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev (2013) 26:2–18. doi:10.1128/CMR.00050-12
190. Hunt PW. Th17, gut, and HIV: therapeutic implications. Curr Opin HIV AIDS (2010) 5:189–93. doi:10.1097/COH.0b013e32833647d9
191. Marchetti G, Bellistri GM, Borghi E, Tincati C, Ferramosca S, La Francesca M, et al. Microbial translocation is associated with sustained failure in CD4+ T-cell reconstitution in HIV-infected patients on long-term highly active antiretroviral therapy. AIDS (2008) 22:2035–8. doi:10.1097/QAD.0b013e3283112d29
192. Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis (2011) 203:780–90. doi:10.1093/infdis/jiq118
193. Estes JD, Gordon SN, Zeng M, Chahroudi AM, Dunham RM, Staprans SI, et al. Early resolution of acute immune activation and induction of PD-1 in SIV-infected sooty mangabeys distinguishes nonpathogenic from pathogenic infection in rhesus macaques. J Immunol (2008) 180:6798–807. doi:10.4049/jimmunol.180.10.6798
194. Walker JA, Beck GA, Campbell JH, Miller AD, Burdo TH, Williams KC. Anti-alpha4 integrin antibody blocks monocyte/macrophage traffic to the heart and decreases cardiac pathology in a SIV infection model of AIDS. J Am Heart Assoc (2015) 4:e001932. doi:10.1161/JAHA.115.001932
195. Dinapoli SR, Hirsch VM, Brenchley JM. Macrophages in progressive human immunodeficiency virus/simian immunodeficiency virus infections. J Virol (2016) 90:7596–606. doi:10.1128/JVI.00672-16
196. Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder – pathogenesis and prospects for treatment. Nat Rev Neurol (2016) 12:309. doi:10.1038/nrneurol.2016.53
197. Heaton RK, Franklin DR Jr, Deutsch R, Letendre S, Ellis RJ, Casaletto K, et al. Neurocognitive change in the era of HIV combination antiretroviral therapy: the longitudinal CHARTER study. Clin Infect Dis (2015) 60:473–80. doi:10.1093/cid/ciu862
198. Cosenza MA, Zhao ML, Si Q, Lee SC. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol (2002) 12:442–55. doi:10.1111/j.1750-3639.2002.tb00461.x
199. Thompson KA, Cherry CL, Bell JE, Mclean CA. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol (2011) 179:1623–9. doi:10.1016/j.ajpath.2011.06.039
200. Eden A, Fuchs D, Hagberg L, Nilsson S, Spudich S, Svennerholm B, et al. HIV-1 viral escape in cerebrospinal fluid of subjects on suppressive antiretroviral treatment. J Infect Dis (2010) 202:1819–25. doi:10.1086/657342
201. Dahl V, Gisslen M, Hagberg L, Peterson J, Shao W, Spudich S, et al. An example of genetically distinct HIV type 1 variants in cerebrospinal fluid and plasma during suppressive therapy. J Infect Dis (2014) 209:1618–22. doi:10.1093/infdis/jit805
202. Sturdevant CB, Joseph SB, Schnell G, Price RW, Swanstrom R, Spudich S. Compartmentalized replication of R5 T cell-tropic HIV-1 in the central nervous system early in the course of infection. PLoS Pathog (2015) 11:e1004720. doi:10.1371/journal.ppat.1004720
203. Fois AF, Brew BJ. The potential of the CNS as a reservoir for HIV-1 infection: implications for HIV eradication. Curr HIV/AIDS Rep (2015) 12:299–303. doi:10.1007/s11904-015-0257-9
204. Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One (2008) 3:e2516. doi:10.1371/journal.pone.0002516
205. Fischer-Smith T, Croul S, Sverstiuk AE, Capini C, L’Heureux D, Regulier EG, et al. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol (2001) 7:528–41. doi:10.1080/135502801753248114
206. Buckner CM, Calderon TM, Willams DW, Belbin TJ, Berman JW. Characterization of monocyte maturation/differentiation that facilitates their transmigration across the blood-brain barrier and infection by HIV: implications for NeuroAIDS. Cell Immunol (2011) 267:109–23. doi:10.1016/j.cellimm.2010.12.004
Keywords: macrophages, sensing, viral assembly, antiretroviral therapy, reservoir, virus-containing compartment, restriction factors
Citation: Rodrigues V, Ruffin N, San-Roman M and Benaroch P (2017) Myeloid Cell Interaction with HIV: A Complex Relationship. Front. Immunol. 8:1698. doi: 10.3389/fimmu.2017.01698
Received: 05 October 2017; Accepted: 17 November 2017;
Published: 30 November 2017
Edited by:
Christel Vérollet, Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Florent Ginhoux, Singapore Immunology Network (A*STAR), SingaporeQuentin James Sattentau, University of Oxford, United Kingdom
Copyright: © 2017 Rodrigues, Ruffin, San-Roman and Benaroch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippe Benaroch, cGhpbGlwcGUuYmVuYXJvY2hAY3VyaWUuZnI=