
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 27 November 2017
Sec. Primary Immunodeficiencies
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01624
This article is part of the Research Topic Primary Immunodeficiencies Worldwide View all 23 articles
 Nikola Schlechter1
Nikola Schlechter1 Brigitte Glanzmann1
Brigitte Glanzmann1 Eileen Garner Hoal1
Eileen Garner Hoal1 Mardelle Schoeman2
Mardelle Schoeman2 Britt-Sabina Petersen3
Britt-Sabina Petersen3 Andre Franke3
Andre Franke3 Yu-Lung Lau4Michael Urban2
Yu-Lung Lau4Michael Urban2 Paul David van Helden1
Paul David van Helden1 Monika Maria Esser5
Monika Maria Esser5 Marlo Möller1
Marlo Möller1 Craig Kinnear1*
Craig Kinnear1*
Primary immunodeficiency disorders (PIDs) render patients vulnerable to infection with a wide range of microorganisms and thus provide good in vivo models for the assessment of immune responses during infectious challenges. Priming of the immune system, especially in infancy, depends on different environmental exposures and medical practices. This may determine the timing and phenotype of clinical appearance of immune deficits as exemplified with early exposure to Bacillus Calmette-Guérin (BCG) vaccination and dissemination in combined immunodeficiencies. Varied phenotype expression poses a challenge to identification of the putative immune deficit. Without the availability of genomic diagnosis and data analysis resources and with limited capacity for functional definition of immune pathways, it is difficult to establish a definitive diagnosis and to decide on appropriate treatment. This study describes the use of exome sequencing to identify a homozygous recessive variant in MAP3K14, NIKVal345Met, in a patient with combined immunodeficiency, disseminated BCG-osis, and paradoxically elevated lymphocytes. Laboratory testing confirmed hypogammaglobulinemia with normal CD19, but failed to confirm a definitive diagnosis for targeted treatment decisions. NIKVal345Met is predicted to be deleterious and pathogenic by two in silico prediction tools and is situated in a gene crucial for effective functioning of the non-canonical nuclear factor-kappa B signaling pathway. Functional analysis of NIKVal345Met- versus NIKWT-transfected human embryonic kidney-293T cells showed that this mutation significantly affects the kinase activity of NIK leading to decreased levels of phosphorylated IkappaB kinase-alpha (IKKα), the target of NIK. BCG-stimulated RAW264.7 cells transfected with NIKVal345Met also presented with reduced levels of phosphorylated IKKα, significantly increased p100 levels and significantly decreased p52 levels compared to cells transfected with NIKWT. Ideally, these experiments would have been conducted in patient-derived immune cells, but we were unable to source these cells from the patient. The functional analysis described in this paper supports previous illustrations of the importance of NIK in human immune responses and demonstrates the involvement of function-altering mutations in MAP3K14 in PIDs. The genomic approach used for this patient demonstrates its value in the diagnosis of an unusual PID and as a tool for detecting rarer mutations to help guide treatment approaches.
Primary immunodeficiency disorders (PIDs) are heritable genetic errors of the immune system that, if left undiagnosed and untreated, may lead to serious, chronic, and in some cases fatal infections and manifestations of autoimmunity (1, 2). PIDs provide in vivo models for identifying factors crucial for human host defense and immune regulation. The nuclear factor-kappa B (NF-κB) family of transcription factors found in mammals is crucial for the expression of developmental, inflammatory, as well as survival genes (3). This pathway consists of a canonical as well as a non-canonical arm, with the former involved in expression of pro-inflammatory genes, and the latter responsible for persistent, slower responses generally not associated with innate immune responses (4, 5).
The non-canonical NF-κB signaling pathway uses NF-κB-inducing kinase (NIK), encoded by mitogen-activated protein kinase (MAP3K14), to integrate signals from various membrane receptors, such as tumor necrosis factor alpha receptor family members (6). Several ligands can activate this pathway, including B cell-activating factor, CD40 ligand (CD40L), lymphotoxin beta, receptor activator of NF-κB ligand (RANKL), and TNF-like weak inducer of apoptosis (TWEAK) (7). Binding of these ligands to their appropriate receptors cause NIK to phosphorylate IkappaB kinase-alpha (IKKα), which activates and targets IKKα to p100, its substrate. p100 is in turn phosphorylated by IKKα, which prompts the ubiquitination and partial degradation of p100 to first produce p52 and second permit the formation of RelB-p52 complexes. These heterodimeric complexes move to the nucleus to activate target genes (3). This non-canonical NF-κB pathway controls lymphoid organogenesis, activation of dendritic cells and B cell maturation and survival, and errors in this pathway are associated with lymphoid disorders (5).
Although mortality rates due to Mycobacterium tuberculosis infections are increased in mice with genetically disrupted NF-κB, the role NF-κB plays in human immune responses to M. tuberculosis is not well understood (8). It has been speculated that some bacterial pathogens misuse specific NF-κB-mediated pathways to promote their survival (9). Activation of autophagy and apoptosis are two processes through which inhibition of NF-κB decreases the amount of intracellular bacilli after M. tuberculosis infection (10, 11). Autophagy is associated with innate and adaptive immune responses, as well as inflammation regulation (12). Ineffective autophagy has been implicated in several human diseases, including infectious diseases and inflammatory disorders (13–16). IkappaB kinase (IKK), the regulator of the NF-κB pathway, is required for autophagy activation in mammals, and inhibition of NF-κB increases autophagosome formation (17). Classic NF-κB is not involved in this response, and the mechanism by which IKK promotes stimulus-induced autophagy is largely unknown (18). NIK as well as IKKα are degraded by autophagy when the function of heat shock protein 90, required for the folding and maturation of certain signaling proteins, is inhibited (19). The processing of p100 and NF-κB activity is thus inhibited (20). However, when heat shock stress activates NF-κB, the autophagy pathway is in turn activated, indicating a close interaction and tight regulation between these two pathways (21).
This study identifies a novel potentially disease-causing variant in NIK in a South African PID patient using whole exome sequencing (WES). In silico analysis predicts the homozygous variant NIKVal345Met to be deleterious and pathogenic. Functional studies with human embryonic kidney (HEK)-293T cells and RAW264.7 cells transfected with either NIKWT or NIKVal345Met showed that this mutation significantly affects the kinase activity of NIK, as well as p100 and p52 levels. The relationship between the non-canonical NF-κB signaling pathway and autophagy was also investigated in an attempt to shed some light on their poorly understood interaction. However, NIKVal345Met did not affect the autophagy pathway.
The proband is a white South African female, from a non-consanguineous marriage, initially diagnosed with humoral immunodeficiency after presenting with a Bacillus Calmette-Guérin (BCG) abscess on the upper leg at the age of 2 years. She received intravenous immunoglobulin (IVIG) replacement therapy. A year later she developed BCG meningitis and received standard treatment in the acute phase with isoniazid (INH), rifampicin (RIF), ethionamide (ETA), and dexamethasone, and thereafter only INH and RIF. At the age of 4 years, diffuse granulomas were identified in her brain. INH, RIF, ETA, and levofloxacin were prescribed for a year, after which the levofloxacin was discontinued. In 2014, at the age of 6 years, she presented with acute loss of consciousness and raised intracranial pressure. Mycobacterium bovis BCG genotypically sensitive to INH and RIF was subsequently cultured from the patient. She was started on a very aggressive 18-month treatment regimen of levofloxacin, terizidone, amikacin IV, linezolid, RIF, INH, para-aminosalisylic acid, as well as continued IVIG replacement therapy. With continued dissemination of BCG in spite of the above treatment and in the absence of a confirmed PID diagnostic category, the patient was not selected for bone marrow transplantation. She proceeded to develop severe neurological, motor, as well as cognitive impairment and is now in total dependency care 6 years after initial presentation.
The study was approved by the Health Research Ethics Committee of Stellenbosch University (approval no. N13/05/075). Written informed consent was granted by the parents of the patient, and this included genetic evaluation of the patient. The parents also consented to the publication of any molecular findings. The study adhered to the ethical guidelines as set out in the “Declaration of Helsinki, 2013” (22). Venous blood for DNA extraction and WES was drawn from the patient and both of her parents. DNA was purified from blood using the Nucleon BACC3 Kit (Amersham Biosciences, Buckinghamshire, UK).
Enrichment and sequencing of the exomes of the proband and both of her healthy parents were performed with Illumina’s TruSeq Exome Enrichment Kit. It targets >20,000 genes with >200,000 exons as well as 9 Mb of predicted microRNA targets with a total size of 62 Mb. Paired-end WES of the three samples was carried out on the Illumina HiSeq 2000, yielding an average of 120 million reads per sample and resulting in an average coverage of the target regions of 60–80× after duplicate removal. The resulting FASTQ file containing the sequencing data underwent quality control in FastQC and was mapped against the human reference genome hg38 using Burrows-Wheeler Aligner (BWA). PCR duplicates were removed using Picard, while SamTools and Genome Analysis Toolkit (GATK) were used in parallel for the detection of single-nucleotide variants (SNVs). The variant calls from both callers were pooled into a single set, after which ANNOVAR was used for SNV annotation and filtering, and to interrogate a number of programs and databases for each called position to generate more evidence of deleterious mutations. The basic filtering options in ANNOVAR used are (1) filtering out common SNVs unlikely to be disease-causing based on a frequency threshold of >1% in the 1000 Genomes Project (1000GP) data (23) and the Exome Sequencing Project 6500 (ESP6500) data (24); (2) restricting the SNVs to those causing amino acid changes in the protein; (3) assessing the impact on protein structure through prediction tools; and (4) the presence of a gene or SNV in Online Mendelian Inheritance in Man (OMIM) or Human Gene Mutation Database (HGMD), which shows known disease associations. All variants with negative Genomic Evolutionary Rate Profiling (GERP)+++ scores as well as all variants with Functional Analysis through Hidden Markov Models (FATHMM) scores greater than 0.1 were also removed. GERP estimates evolutionary constraints at specific positions in an exome and identifies “constrained elements,” where several positions combine to produce a signal indicative of a putative functional element (25). FATHMM is a high-throughput web-server that can predict the functional consequences of coding and non-coding variants (26). Finally, variants were also removed if they were homozygous in either of the healthy parents of the proband.
The 1000GP data, ESP6500, and ExAC Browser were investigated for the presence of all potentially disease-causing variants. Only those present in less than 1% of the population in these databases were considered as candidate variants.
Project HOPE [Have (y)Our Protein Explained] (27), PolyPhen-2 (28), Sorting Tolerant From Intolerant (SIFT) (29), and MutationTaster2 (30) were used to predict the functional and structural causes of the amino acid changes on proteins.
Sanger sequencing was used to validate the WES results and verify whether the potentially disease-causing variants identified were true variants or sequencing artifacts. Only one potentially disease-causing variant was identified, for which the forward and reverse primers F: 5′AGCCCTGGAAACCTCACC and R: 5′TGAGATTGGCGGAATAAGAGA were used to produce a fragment of 455 bp. This was bi-directionally sequenced at the Central Analytical Facility of Stellenbosch University using the BigDye® Terminator v3.1 Cycle Sequencing kit (Perkin-Elmer, Applied Biosystems Inc., Foster City, CA, USA), followed by electrophoresis on an ABI 3130XL Genetic Analyzer (Perkin-Elmer, Applied Biosystems Inc., Foster City, CA, USA).
Two plasmids, pWZL-Neo-Myr-Flag-MAP3K14 and pCR-Flag-IKKalpha, were obtained from the non-profit plasmid repository Addgene (https://www.addgene.org/). pWZL-Neo-Myr-Flag-MAP3K14 was a gift from William Hahn & Jean Zhao (Addgene plasmid # 20640) (31) and pCR-Flag-IKKalpha was a gift from Hiroyasu Nakano (Addgene plasmid # 15467) (32). To generate the pWZL-Neo-Myr-Flag-MAP3K14Val345Met mutant construct, the mutation-specific oligo nucleotide primers F: 5′AAGGCAGCGTGAGCTC and R: 5′CAGAGCATGCACTAGGTAT were used together with the Q5® Site Directed Mutagenesis Kit (New England Biolabs Inc., UK) as per the manufacturer’s instructions. Sanger sequencing was used to confirm successful mutagenesis.
Human embryonic kidney-293T cells and RAW264.7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose with l-glutamine and supplemented with 10% FBS and 1% penicillin/streptomycin. One day before transfection, the cells were trypsinized and seeded into six-well culture plates so that the cells were 50–80% confluent the following day. Plasmid DNA was purified from the IKKα as well as the wild-type and mutant MAP3K14 plasmids using the PureYield™ Plasmid Miniprep System (Promega Corp., USA) according to the manufacturer’s guidelines. The IKKα plasmid DNA combined with either the wild-type or mutant MAP3K14 plasmid DNA were then transfected into the seeded HEK293T, while only the wild-type or mutant MAP3K14 plasmids were transfected into RAW264.7 cells using Lipofectamine™ LTX Reagent and PLUS™ Reagent (Invitrogen, USA) according to the manufacturer’s instructions.
The RAW264.7 cells were divided into stimulated and unstimulated groups, each group consisting of untransfected, NIKVal345Met-transfected, and NIKWT-transfected sub-groups. After transfection of the appropriate plasmids into each sub-group, BCG was added to each well in the six-well plates making up the stimulated group, at a multiplicity of infection of 5. A stock solution BCG with a concentration of 1.46 × 106 CFUs/mL was prepared using BCG Vaccine SSI (Statens Serum Institut, Denmark) and Diluted Sauton SSI (Statens Serum Institut, Denmark). Per well containing 1 × 106 cells growing in 6 mL growing media, 34 µL of this BCG stock solution was added. Cells were stimulated for 16 h at 37°C.
The transfected HEK293T cells used for investigation of autophagy were divided into two groups. One received Bafilomycin A1 (Baf) treatment while the other remained untreated. Baf is a known inhibitor of the late phase of autophagy and prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes (33). A Baf stock solution with a concentration of 1 mM was made using DMSO and Bafilomycin A1, which was further diluted to 1 µM by adding PBS. Each well in one six-well plate seeded with HEK293T cells received 150 µL of the 1 µM Baf stock and 1,350 µL growth media, and constituted the Baf treatment group. Another plate received only 1,500 µL growth media and constituted the control group. The plates were incubated for 16 h at 37°C.
All cells were lysed with lysis buffer [0.05 M Hepes, 0.1 M NaCl, 0.01 M EDTA, 0.17 mM Triton X-100, 4 mM Nappi, 2 mM Na3VO4, and protease inhibitors (Roche)] at 95°C for 5 min and all lysates were stored at −80°C. The lysates were subjected to Sodium dodecyl sulfate polyacrylamide gel electrophoresis SDS/PAGE on Mini-PROTEAN® TGX™ precast polyacrylamide gels [Bio-Rad Laboratories (Pty) Ltd., RSA] containing 1% SDS, after which the proteins were transferred to 0.2-μm-pore polyvinylidine difluoride membranes using the iBlot® Dry Blotting system (Invitrogen, RSA). Membranes were blocked in 5% BSA (w/v) or 5% low fat milk and subsequently probed with the rabbit antibodies phospho-IKKα/β (Ser176/180) (16A6), IKKα, NF-κB p100/p52, NIK (Cell Signaling Technology, Inc., USA; Abcam Inc., UK) and LC3B (Abcam Inc., UK), as well as mouse GAPDH antibody (Santa Cruz Biotechnology Inc., USA). Membranes were then exposed to horseradish peroxidase-conjugated goat anti-rabbit and donkey anti-mouse secondary antibodies (Santa Cruz Biotechnology, Inc., USA) and the proteins were subsequently visualized by enhanced chemiluminescence using Clarity™ Western ECL Substrate (Bio-Rad Laboratories, Inc., USA).
The open platform image analysis tool ImageJ was used for analysis of all western blots. All experiments were done in true biological triplicates and under the same conditions. One-tailed unpaired t-tests were used to determine significant differences between samples. All western blots were normalized using GAPDH.
The patient tested negative for the human immunodeficiency virus as well as Herpes simplex virus and presented with severe hypogammaglobulinemia with decreased immunoglobulin (Ig) levels (IgA < 0.06 G/L, IgM = 0.14 G/L, IgG = 0.42 G/L), increased lymphocyte subset numbers for her age and normal lymphocyte proliferation to mitogens and recall antigens. She was investigated for Mendelian susceptibility to mycobacterial disease (MSMD) by screening STAT1, LRBA, IL12RB1, IL12B, and IFNGR1 for possible disease-causing mutations. No mutations were found in any of these genes. Longitudinal immunological investigation of this patient with persistent, disseminated, treatment-resistant M. bovis BCG infection indicated a dramatic decrease of natural killer (NK) cells, B cells, and CD8 cells over time (Table 1). Her CD27+IgD+ cell population was decreased (2.96%) and class-switched memory B-cells (CD27+IgD−) were also low (0.12%), while CD40 ligand (CD40L) was detected as present. Memory T cells were low in relation to naïve T cells and reduced levels of γ/δ T cells were observed—cells known to be involved in the innate immune reaction against mycobacteria. The patient’s phytohemagglutinin control lymphocyte proliferation was normal on the T Spot TB test with negative proliferation to specific TB antigens. This is what would be expected, since the test is specific for TB antigens, not BCG—the cause of disease dissemination observed in this patient. Upregulation of CD69 on NK cells after interleukin-2 stimulation was slightly decreased. T cell receptor excision circles (TRECs) and kappa-deleting recombination excision circles were clearly visible (AMPATH Laboratories, South Africa). Investigation by WES was thus pursued to assist with establishing a diagnosis for this patient.
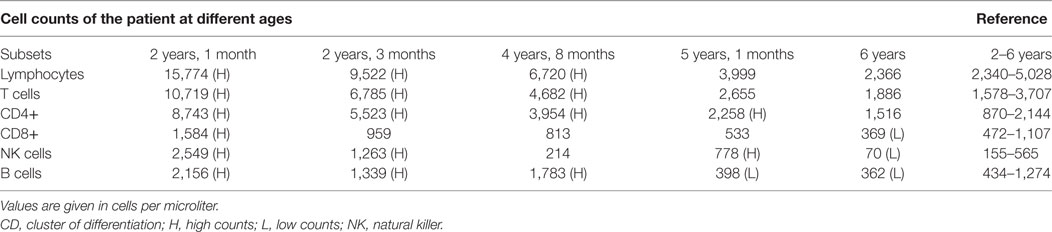
Table 1. Normal cell counts versus patient total cell counts at different ages.
Whole exome sequencing of the proband revealed a total of 23,939 variants, which were reduced to 708 candidate variants after filtering and annotation using ANNOVAR (Table 2). Upon exclusion of all variants present in a homozygous state in either of the healthy parents, 9 homozygous and 23 heterozygous variants remained (Table 3). After interrogating OMIM and HGMD, only one variant was identified as potentially disease-causing according to the function of the gene it is situated in. This homozygous c.G1033A p.Val345Met nucleotide variant is situated in exon 5 of NIK (Table 4). SIFT and PolyPhen-2 predicted this change to be pathogenic and damaging, while MutationTaster2 was not able to generate any results, indicating the uncharacterized state of NIK. The variant is situated at a position that is highly conserved across several different species (Figure 1).
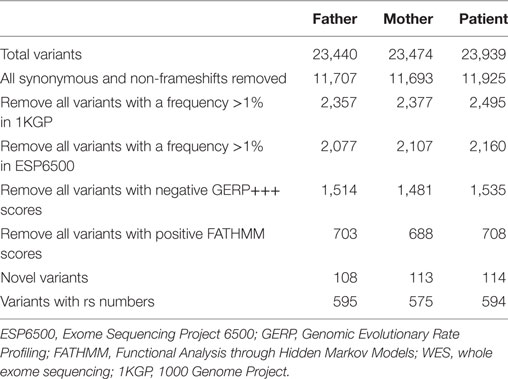
Table 2. Variants identified by WES in the proband and both parents.
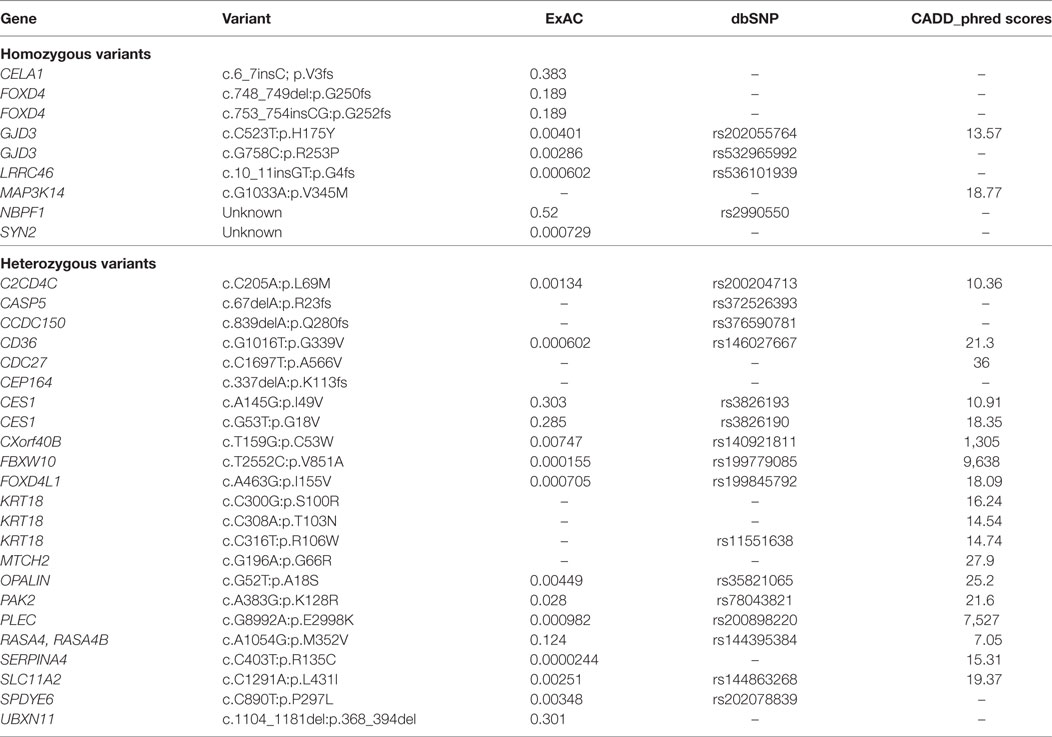
Table 3. Final list of variants identified in the proband after filtering.
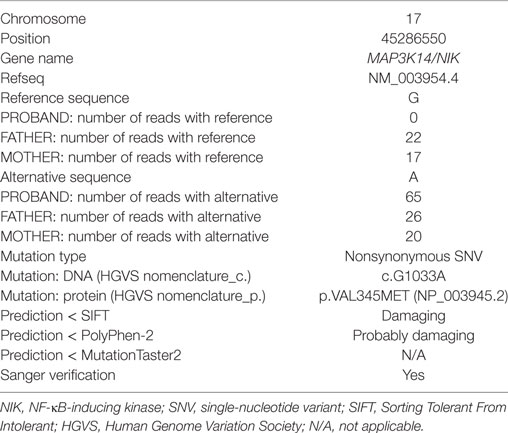
Table 4. Details of the putative disease-causing variant identified in the proband.

Figure 1. ClustalW multiple sequence alignment. The c.G1033A (p.Val345Met) homozygous mutation identified in this patient is situated at a highly conserved position in MAP3K14 (34).
This variant was covered 65× during WES, improving its likelihood of being a true variant rather than a sequencing artifact. The average coverage for all the variants identified in the proband was 105×. Sanger sequencing confirmed this to be a true variant present in a homozygous state in the patient and heterozygous in both unaffected parents (Figure 2). NIKVal345Met was absent from all interrogated databases and therefore previously unidentified in all investigated populations. No missense or loss-of-function mutations in NIK are listed in these databases.
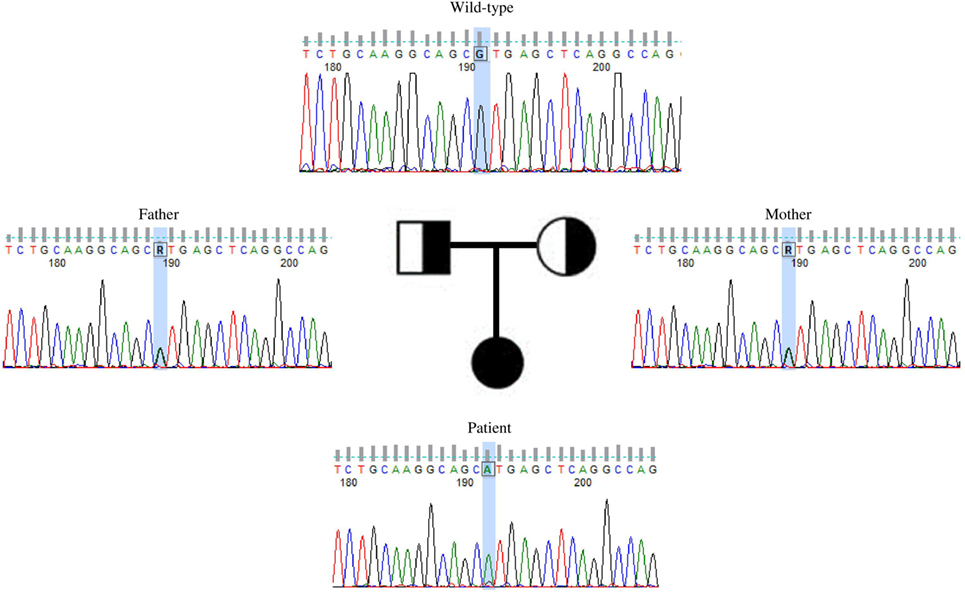
Figure 2. Validation by Sanger sequencing of NIKVal345Met found by whole exome sequencing. Patient’s c.G1033A homozygous mutation, with one mutated allele inherited from each of the healthy, heterozygous parents.
A schematic representation of the amino acid exchange caused by this variant, with wild-type valine (Val) on the left and mutated methionine (Met) on the right, is shown in Figure 3A. Met and Val are both non-polar, hydrophobic, and aliphatic amino acids. Figure 3B shows the difference in size between Val and Met. The main difference is the presence of a C-beta branch in Val: two non-hydrogen substituents attached to its C-beta carbon, instead of only one as for Met. Met also has an additional sulfur atom, which forms very strong amide N–H⋅S hydrogen bonds crucial for controlling the conformational setting of this amino acid (35). Val is bulkier near the protein backbone and more restricted in the conformations the main-chain can adopt. The Val side chain is extremely non-reactive and is rarely directly involved in protein function, although it can play a role in substrate recognition. The site of variation is situated near a conserved site.
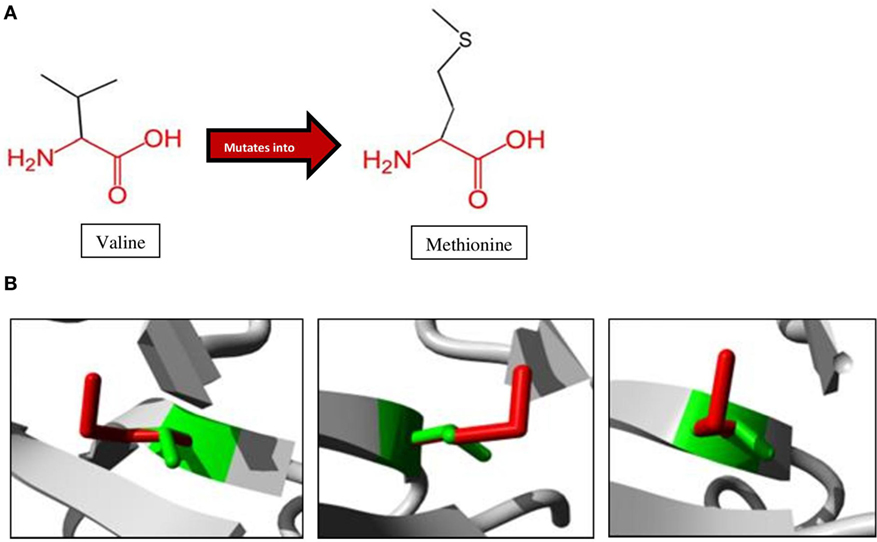
Figure 3. Amino acid changes caused by the mutation identified in the proband. (A) Mutation of Val to Met, as observed at position 345 in NIK of the patient. The conserved backbone is indicated in red, with the unique side-chain in black. (B) Mutant Met (red) is larger than wild-type Val (green), potentially influencing interactions within the protein as well as with other proteins (27).
The HOPE analysis indicated that Val345Met is located 45 amino acid positions before the interpro domain known as mitogen-activated protein kinase kinase kinase 14 (IPR017425), associated with protein kinase and transferase activity. This kinase domain stretches from positions 390 to 660 (Figure 4). Because of its close proximity to the kinase domain, it is tempting to speculate that Val345Met may have an effect on the ability of NIK to phosphorylate IKKα. This domain is in contact with residues from other domains, implying the possibility for the variant to influence correct protein function by inhibiting/altering these interactions.
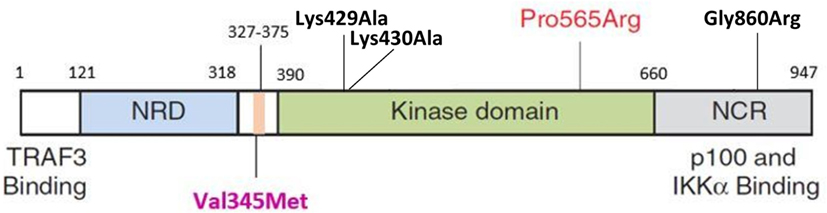
Figure 4. Schematic representation of NIK protein domain structures. The NRD (light blue) contains the BR and the P-RR domains (darker blue). The conserved domain containing V345M (purple label) is indicated in pink and situated before the kinase domain (green). This is followed by the NCR (gray). The P565R amino acid change observed in P1 and P2 by Willmann et al. is situated in the kinase domain. Black labels indicate the mutations previously observed in the murine studies (Lys429Ala; Lys430Ala; Gly860Arg). TRAF3, tumor necrosis factor receptor-associated factor 3; NRD, negative regulatory domain; BR, basic region; P-RR, proline-rich repeat; NCR, non-catalytic region; Lys, lysine; Ala, alanine; Pro, proline; Arg, arginine; Gly, glycine; Val, valine; Met, methionine; IKKα, IkappaB kinase-alpha; NIK, NF-κB inducing kinase (44).
To experimentally assess the effect of the mutation of interest, we analyzed the kinase activity of NIKVal345Met compared to NIKWT by testing NIK-dependent phosphorylation of IKKα. Sanger sequencing first confirmed that the mutagenesis occurred correctly (Figure S1 in Supplementary Material). IKKα in combination with either wild-type or mutant NIK were transfected into HEK293T cells, while only NIK, in either its wild-type or mutant form, was transfected into RAW264.7 cells. The NIK and IKKα antibodies were then used to show that transfection of the plasmids expressing IKKα and mutant and wild-type NIK into HEK293T and RAW264.7 cells was successful. Untransfected HEK293T cells do not contain endogenous NIK, although they do contain IKKα, while RAW264.7 cells contain both NIK and IKKα endogenously. As seen in Figure S2 in Supplementary Material, there was an increase of both the expression of IKKα in both wild-type and mutant NIK cotransfected HEK 293T cells (although this increase did not reach statistical significance; p = 0.0638). Similarly, there was an increase in NIK levels in RAW264.7 cells following transfection with wild-type and mutant NIK constructs (Figure S3 in Supplementary Material). No significant differences are observed between the transfection efficiencies of wild-type- and mutant-transfected cells in either of these cell types.
IkappaB kinase-alpha and phospho-IKKα/β (Ser176/180) antibodies were used to investigate the difference between IKKα and phospho-IKKα levels in NIKWT compared to NIKVal345Met. NIKVal345Met was not thought to affect the production of IKKα and as expected, the level of IKKα was not substantially altered by this mutation (Figure 5A: p = 0.3879; Figures 6A,C,E; unstimulated: p = 0.4757; stimulated: p = 0.1844). However, phosphorylated IKKα levels were significantly reduced by NIKVal345Met in both HEK293T (Figure 5A; p = 0.0353) and RAW264.7 (Figures 6B,D,F; unstimulated: p = 0.0010; stimulated: p < 0.0001) cells, indicating that this mutation alters the kinase activity of NIK. The ratio of phosphorylated IKKα to un-phosphorylated IKKα is shown in Figure 6G, with a significant difference observed between the wild-type-transfected and the mutant-transfected cell groups that have been stimulated with BCG (p = 0.0090).
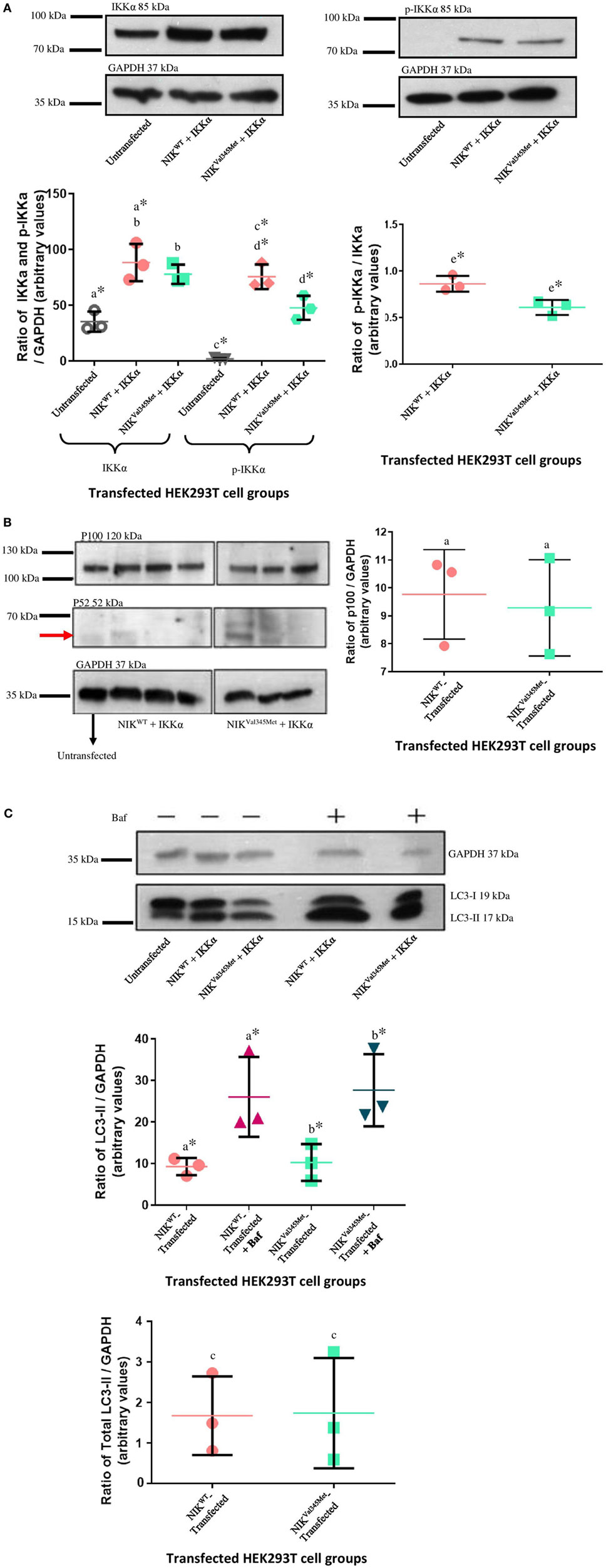
Figure 5. Comparison between NIKVal345Met- and NIKWT-transfected HEK293T cells. (A) No phospho-IKKα was detected in untransfected HEK293T cells, with significant amounts present in transfected cells, proving efficient transfection. A significant decrease in phospho-IKKα is observed in cells transfected with NIKVal345Met compared to NIKWT. The ratio of phospho-IKKα over IKKα is also significantly affected by NIKVal345Met. a: p = 0.0084*; b: p = 0.3879; c: p = 0.0003*; d: p = 0.0353*; e: p = 0.0199*. (B) No quantifiable p52 levels were observed in untransfected, wild-type-transfected, or mutant-transfected samples. p100 was detectable at significant amounts, but no differences between the wild-type and mutant groups were observed. a: p = 0.7409. (C) Significant differences were seen when comparing cells transfected with wild-type NIK and IKKα before and after Baf treatment, as well as mutant NIK and IKKα before and after treatment. However, the difference in autophagy between wild-type-transfected and mutant-transfected groups was not significant. a: p = 0.0418*; b: p = 0.0366*; c: p = 0.9506. Each image is representative of three experiments conducted independently. * indicates significance.
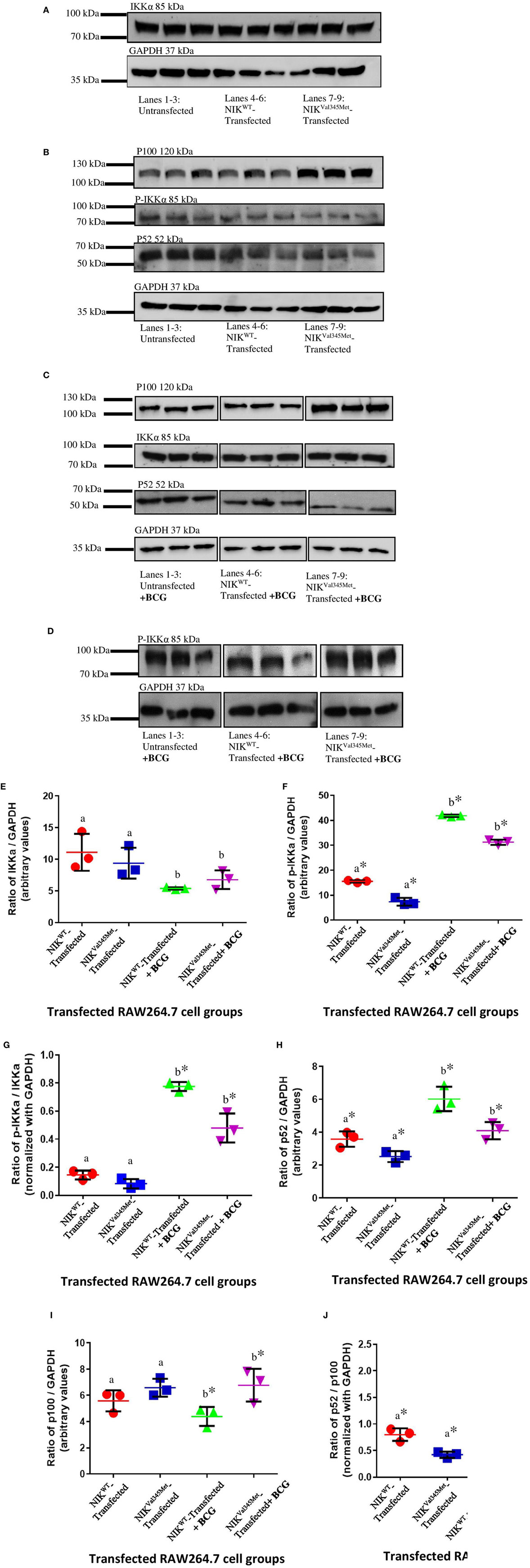
Figure 6. Comparison between NIKVal345Met- and NIKWT-transfected RAW264.7 cells. (A,B) Similar amounts of IKKα was detected in the wild-type and mutant cell groups, indicating that NIKVal345Met had no effect on IKKα production. p100 levels were similar in the wild-type and mutant RAW264.7 cell line groups, with reduced levels of phospho-IKKα and p52 observed in cells transfected with NIKVal345Met compared to NIKWT, without BCG stimulation. (C,D) p100, p52, and phospho-IKKα levels were affected by the Val345Met mutation, with increased p100, and decreased p52 and phospho-IKKα levels observed in BCG-stimulated cells transfected with NIKVal345Met compared to NIKWT. IKKα levels were not affected. (E) Quantification of western blots in (A,C) showed similar amounts of IKKα in wild-type and mutant cell groups, both without and with BCG stimulation. a: p = 0.4757; b: p = 0.1844; (F) quantification of western blots in (B,D) show significant reductions in phospho-IKKα in the mutant-transfected cell groups compared to the wild-type-transfected groups, both without and with BCG stimulation. a: p = 0.0010*; b: p < 0.0001*. (G) The ratio of phosphorylated IKKα over un-phosphorylated IKKα was similar between the wild-type- and mutant-transfected cell groups in the absence of BCG stimulation, but significantly reduced in the mutant-transfected groups compared to the wild-type-transfected groups when stimulated with BCG. a: p = 0.0760; b: p = 0.0090*. (H) Significantly reduced levels of p52 was observed in cells transfected with NIKVal345Met compared to NIKWT, both in the absence and in the presence of BCG stimulation. a: p = 0.0330*; b: p = 0.0208*. (I) p100 levels were similar in cells transfected with NIKVal345Met compared to NIKWT without BCG stimulation. However, p100 levels significantly increased in the NIKVal345Met-transfected groups upon BCG stimulation. a: p = 0.1745; b: p = 0.0458*. (J) The ratio of p52 over p100 differed significantly between cells transfected with NIKVal345Met compared to NIKWT, both without and with BCG stimulation. a: p = 0.0077*; b: p = 0.0053*. The data for each image in this figure is representative of three experiments conducted independently. * indicates significance.
Phosphorylated IKKα results in the ubiquitination and proteosomal degradation of p100 to produce p52. The difference in p100 and p52 levels between cells expressing wild-type and mutant NIK was thus also investigated to determine whether NIKVal345Met has any downstream effects. The ideal would have been to use patient-derived immune cells for these experiments to more accurately measure the downstream effects of the identified mutation. Unfortunately, the patient deteriorated to such an extent that we could not obtain more blood samples from her. HEK293T cells, that are incapable of eliciting an immune response upon infection, were first used. However, no quantifiable levels of p52 were detected (Figure 5B), while no significant differences in the levels of p100 were observed between wild-type- and mutant-transfected groups (Figure 5B; p = 0.7409). p100 and p52 levels were also measured in BCG-stimulated and unstimulated RAW264.7 cells transfected with NIKVal345Met and NIKWT. In BCG-stimulated cells, p100 levels were significantly increased (p = 0.0458) and p52 levels decreased (p = 0.0208) in the mutant compared to the wild-type groups (Figures 6C,H,I). The difference in p100 levels between the mutant and wild-type groups was not significant in BCG-unstimulated RAW264.7 cells (Figures 6B,I; p = 0.1745), although the decrease in p52 levels remained significant in the unstimulated cells (Figures 6B,H; p = 0.0330). The NIKVal345Met variant thus significantly affects the downstream functioning of the non-canonical NF-κB signaling pathway. The ratio of p52 to p100 is shown in Figure 6J, with significant differences observed between the wild-type-transfected and the mutant-transfected cell groups in both BCG-stimulated and unstimulated cell populations (unstimulated: p = 0.0077; stimulated: p = 0.0053).
Autophagy is a dynamic catabolic process during which double-membrane vesicles (autophagosomes) form by engulfing parts of the cytoplasm and subsequently fuse with lysosomes to degrade and recycle their contents (36). LC3 immunoblotting is widely used to measure autophagic activity (37–39). SDS-PAGE and immunoblotting detects two bands of endogenous LC3, namely LC3-I and LC3-II. LC3-I is present in the cytosol, while LC3-II is bound to phosphatidylethanolamine and localized to autophagosomal membranes (40, 41), thus directly correlating with the number of autophagosomes (42). The effect of NIKVal345Met on autophagy was also investigated in this study by measuring LC3-II levels. The western blots and corresponding bar graphs are shown in Figure 5C. Significant differences in LC3-II were observed when comparing cells transfected with NIKWT before and after Baf treatment (p = 0.0418), as well as those infected with NIKVal345Met before and after treatment (p = 0.0366), thus indicating effective functioning of the autophagic pathway. When comparing the difference in LC3-II levels between mutant and wild-type groups (Figure 5C), however, no significant difference was observed (p = 0.9506), indicating that NIKVal345Met has no effect on autophagy.
Phenotype heterogeneity is often observed in patients with mutations in the same gene, and this may in part depend on the patient’s environment (43). We identified a patient with a mutation in NIK where the phenotype was modified by early BCG exposure as part of routine vaccination in the first days of life.
Only one previous study to date has described mutations in NIK in humans and identified a bi-allelic mutation as the cause of a primary immunodeficiency characterized by multifaceted aberrant lymphoid immunity in two patients (44). Patient 1 (P1) was born to consanguineous healthy parents and had a younger brother who died at the age of 2 years from suspected combined immunodeficiency. Decreased IgG and IgA and elevated IgM levels were identified on a single occasion before any treatment was prescribed. Despite treatment (such as regular intravenous Ig substitution and ganciclovir), the patient continued to suffer from multiple episodes of bacterial and viral infections. There was also one documented episode of granulomatous hepatitis and tuberculosis osteomyelitis due to dissemination after BCG vaccination. An allogeneic hematopoietic stem cell transplantation (HSCT) after reduced toxicity conditioning was performed at the age of 9 years, and upon last report in 2014 she remained clinically well. Patient 2 (P2) is a first-degree cousin of P1 also born to consanguineous healthy parents. She presented with severe chronic diarrhea, recurrent lower respiratory tract infections, as well as oral and esophageal candidiasis. She tested positive for Cryptosporidium on one occasion. Her human leukocyte antigen-identical mother was used as a donor for an allogeneic HSCT performed without conditioning at the age of 3 years. As no engraftment was observed after 50 days, the same donor was used and a second transplant was performed. However, the patient died on day 6 following the second HSCT due to rapidly accelerated septic shock and multi-organ failure.
We describe a patient with similar symptoms: initial diagnosis of humoral immunodeficiency with severe hypogammaglobulinemia, decreased memory B cells, B cell lymphopenia, normal T cell proliferation to mitogens and recall antigens, as well as normal IFN-γ production by T cells (Table 1). However, while the patients from the previous study presented with recurrent viral, bacterial, and Cryptosporidium infections, the phenotype of our patient seems to be confined to BCG-osis. Our patient did not present with any other infections. WES and subsequent bioinformatics analysis identified a novel putative disease-causing homozygous variant, c.G1033A, situated in NIK. NIK encodes the 947 residue protein mitogen-activated protein kinase 14, which is a serine/threonine protein kinase involved in NF-κB activity (NM_003954.4). The NIKVal345Met variant identified in this patient has never been described and is predicted to be disease-causing and deleterious by two in silico prediction tools. It is difficult to predict the exact result of this variant on protein function, since NIK is poorly characterized. The variant is situated in a highly conserved region (Figure 4). The domain it is located in is important for NIK’s activity and interacts with residues from other domains, making it possible for this mutation to inhibit the correct functioning of NIK. The involvement of NIK in B cell development and maturation makes it a perfect candidate gene to investigate further for association with primary immunodeficiency and increased susceptibility to TB.
Phenotypic heterogeneity may be due to different effects of a mutation on protein function (43). However, clinical phenotypic variation cannot always be attributed to different functional consequences of a mutation. As an example, the first patient in whom a mutation in TYK2 (member of the JAK family of tyrosine kinases) was identified was diagnosed with hyper-IgE syndrome accompanied by lesions of the skin, BCG disease, as well as fungal and viral infections (45). A second patient, also with small deletions in TYK2 and the absence of protein on western blots like the first patient, presented with BCG disease and brucellosis, but had normal IgE levels and no skin lesions (46). In a further example, three Chinese siblings with intracranial calcifications and epileptic seizures, without severe infectious diseases, were exome sequenced and a mutation in ISG15 was identified (47). Mutations in this gene were previously found to cause MSMD in three unrelated children from two families in Iran and Turkey (48). Subsequently, intracranial calcifications were also identified in other ISG15-deficient patients. The Chinese individuals never received BCG vaccinations, which could explain why they did not have the MSMD phenotype. Even within a single family, mutations in the same gene have been shown to cause very different clinical phenotypes (35). This is due to the range of infectious or environmental exposures, age of exposure and various modifying epigenetic factors that can affect disease presentation. Therefore, clinical outcomes of patients with immunodeficiencies, even with previously described phenotype/genotype associations, cannot always be accurately predicted.
Phenotypic differences observed between the proband in this study and P1 and P2 described by Willmann and colleagues can also be attributed to the location, and thus the effect, of the identified mutations. The previously identified mutation is situate in the kinase domain of NIK, and completely abolishes the functioning of this protein. The mutation described in this study, however, is situated just before the kinase domain (Figure 4) and does not affect the kinase activity of NIK as critically as the mutation previously described (44). This was proven by the decreased, and not abolished, kinase activity of NIK observed in this study.
The genomic approach in unusual presentations like in the presented patient illustrates its value for successful identification of a novel mutation situated in a PID-causing gene. A confirmed molecular diagnosis directs potential treatment approaches such as the indication for stem cell therapy. Moreover, genetic predisposition combined with protein dysfunction studies are already used to tailor-make patient-specific approaches in mycobacterial disease (49). In the era of personalized medical treatment, based on unique genetic features, it is realistic to anticipate that this will also be extended to the specific treatment of variants of genetically susceptible TB.
1000GP, http://www.1000genomes.org
ADMIXTURE, http://www.genetics.ucla.edu/software
WANNOVAR, http://wannovar.wglab.org (accessed 3.10.16)
BWA, https://github.com/lh3/bwa
ClustalW, https://www.ebi.ac.uk/Tools/msa/clustalw2/
ESP6500, http://evs.gs.washington.edu/EVS/
ExAC Browser http://exac.broadinstitute.org/
FastQC, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
GATK suite, www.broadinstitute.org/gatk
HGMD, http://www.hgmd.cf.ac.uk/ac/index.php
MutationTaster-2, http://www.mutationtaster.org/
OMIM, http://www.omim.org/
Picard, http://picard.sourceforge.net
PLINK, http://zzz.bwh.harvard.edu/plink/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Project HOPE, http://www.cmbi.ru.nl/hope/
SamTools, http://www.htslib.org/
SIFT, http://sift.jcvi.org/
Accession numbers: NM_003954.4 and NP_003945.2.
This study was carried out in accordance with the recommendations of the Health Research Ethics Committee of Stellenbosch University (approval no. N13/05/075) with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Health Research Ethics Committee of Stellenbosch University.
CK, EH, PH, and MM conceived the project. Y-LL carried out all testing on MSMD genes. NS carried out all laboratory work and wrote the first draft of the manuscript. B-SP and AF performed the whole exome sequencing and assisted with the bioinformatics analysis. BG and NS performed the bioinformatics analysis and interpreted the data with CK and MM. MS and MU assisted with the genetic counseling. ME was involved in patient recruitment. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to acknowledge the contribution of all study participants, AMPATH Laboratories’ Dr. Sylvia van den Berg and Dr. Cathy van Rooyen, as well as our project funders (The South African National Research Foundation, The South African Medical Research Council, and the Harry Crossley Foundation) for their financial support. This work utilized resources owned and maintained by the Central Analytical Facility of Stellenbosch University for Sanger sequencing. Research reported in this publication was supported by the South African Medical Research Council. The content is solely the responsibility of the authors and does not necessarily represent the official views of the South African Medical Research Council.
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fimmu.2017.01624/full#supplementary-material.
1. Modell F. Immunology today and new discoveries: building upon legacies of Dr. Robert A. Good. Immunol Res (2007) 38(1–3):48–50. doi:10.1007/s12026-007-0049-4
2. Cunningham-Rundles C, Ponda PP. Molecular defects in T- and B-cell primary immunodeficiency diseases. Nat Rev Immunol (2005) 5(11):880–92. doi:10.1038/nri1713
3. Cildir G, Low KC, Tergaonkar V. Noncanonical NF-κB signaling in health and disease. Trends Mol Med (2016) 22(5):414–29. doi:10.1016/j.molmed.2016.03.002
4. Razani B, Reichardt AD, Cheng G. Non-canonical NF-κB signaling activation and regulation: principles and perspectives: non-canonical NF-κB signaling activation and regulation. Immunol Rev (2011) 244(1):44–54. doi:10.1111/j.1600-065X.2011.01059.x
5. Sun S-C. Non-canonical NF-κB signaling pathway. Cell Res (2011) 21(1):71–85. doi:10.1038/cr.2010.177
6. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol (2013) 131(4):959–71. doi:10.1016/j.jaci.2013.01.046
7. Sun S-C. The noncanonical NF-κB pathway. Immunol Rev (2012) 246(1):125–40. doi:10.1111/j.1600-065X.2011.01088.x
8. Yamada H, Mizuno S, Reza-Gholizadeh M, Sugawara I. Relative importance of NF-kappaB p50 in mycobacterial infection. Infect Immun (2001) 69(11):7100–5. doi:10.1128/IAI.69.11.7100-7105.2001
9. Tato CM, Hunter CA. Host-pathogen interactions: subversion and utilization of the NF-kappa B pathway during infection. Infect Immun (2002) 70(7):3311–7. doi:10.1128/IAI.70.7.3311-3317.2002
10. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell (2004) 119(6):753–66. doi:10.1016/j.cell.2004.11.038
11. Yuk JM, Yoshimori T, Jo EK. Autophagy and bacterial infectious diseases. Exp Mol Med (2012) 44(2):99–108. doi:10.3858/emm.2012.44.2.032
12. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469(7330):323–35. doi:10.1038/nature09782
13. Kroemer G, White E. Autophagy for the avoidance of degenerative, inflammatory, infectious, and neoplastic disease. Curr Opin Cell Biol (2010) 22(2):121–3. doi:10.1016/j.ceb.2010.02.003
14. Vece TJ, Watkin LB, Nicholas SK, Canter D, Braun MC, Guillerman RP, et al. Copa syndrome: a novel autosomal dominant immune dysregulatory disease. J Clin Immunol (2016) 36(4):377–87. doi:10.1007/s10875-016-0271-8
15. Hooper KM, Barlow PG, Stevens C, Henderson P. Inflammatory bowel disease drugs: a focus on autophagy. J Crohns Colitis (2016) 11(1):118–27. doi:10.1093/ecco-jcc/jjw127
16. Negroni A, Colantoni E, Vitali R, Palone F, Pierdomenico M, Costanzo M, et al. NOD2 induces autophagy to control AIEC bacteria infectiveness in intestinal epithelial cells. Inflamm Res (2016) 65(10):803–13. doi:10.1007/s00011-016-0964-8
17. Deretic V, Singh S, Master S, Harris J, Roberts E, Kyei G, et al. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol (2006) 8(5):719–27. doi:10.1111/j.1462-5822.2006.00705.x
18. Comb WC, Cogswell P, Sitcheran R, Baldwin AS. IKK-dependent, NF-κB-independent control of autophagic gene expression. Oncogene (2011) 30(14):1727–32. doi:10.1038/onc.2010.553
19. Yan P, Qing G, Qu Z, Wu C-C, Rabson A, Xiao G. Targeting autophagic regulation of NFkappaB in HTLV-I transformed cells by geldanamycin: implications for therapeutic interventions. Autophagy (2007) 3(6):600–3. doi:10.4161/auto.4761
20. Qing G, Yan P, Qu Z, Liu H, Xiao G. Hsp90 regulates processing of NF-kappa B2 p100 involving protection of NF-kappa B-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res (2007) 17(6):520–30. doi:10.1038/cr.2007.47
21. Nivon M, Richet E, Codogno P, Arrigo A-P, Kretz-Remy C. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy (2009) 5(6):766–83. doi:10.4161/auto.8788
22. World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA (2013) 310(20):2191–4. doi:10.1001/jama.2013.281053
23. The 1000 Genomes Project ConsortiumAuton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature (2015) 526(7571):68–74. doi:10.1038/nature15393
24. Exome Variant Server. Seattle, WA: NHLBI GO Exome Sequencing Project (ESP) (2014). Available from: http://evs.gs.washington.edu/EVS/
25. Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol (2010) 6(12):e1001025. doi:10.1371/journal.pcbi.1001025
26. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat (2013) 34(1):57–65. doi:10.1002/humu.22225
27. Venselaar H, Te Beek TAH, Kuipers RKP, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics (2010) 11:548. doi:10.1186/1471-2105-11-548
28. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods (2010) 7(4):248–9. doi:10.1038/nmeth0410-248
29. Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res (2002) 12(3):436–46. doi:10.1101/gr.212802
30. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods (2010) 7(8):575–6. doi:10.1038/nmeth0810-575
31. Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell (2007) 129(6):1065–79. doi:10.1016/j.cell.2007.03.052
32. Nakano H, Shindo M, Sakon S, Nishinaka S, Mihara M, Yagita H, et al. Differential regulation of IkappaB kinase alpha and beta by two upstream kinases, NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc Natl Acad Sci U S A (1998) 95(7):3537–42. doi:10.1073/pnas.95.7.3537
33. Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem (2007) 282(8):5641–52. doi:10.1074/jbc.M609532200
34. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics (2007) 23(21):2947–8. doi:10.1093/bioinformatics/btm404
35. Mundlapati VR, Ghosh S, Bhattacherjee A, Tiwari P, Biswal HS. Critical assessment of the strength of hydrogen bonds between the sulfur atom of methionine/cysteine and backbone amides in proteins. J Phys Chem Lett (2015) 6(8):1385–9. doi:10.1021/acs.jpclett.5b00491
36. Loos B, Hofmeyr J-HS, Müller-Nedebock K, Boonzaaier L, Kinnear C. Autophagic flux, fusion dynamics, and cell death. Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging. Elsevier (2014). p. 39–56. Available from: http://linkinghub.elsevier.com/retrieve/pii/B9780124055292000020
37. Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol (2004) 2(4):301–14. doi:10.1038/nrmicro865
38. Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy (2007) 3(3):181–206. doi:10.4161/auto.3678
39. Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol (2004) 36(12):2491–502. doi:10.1016/j.biocel.2004.02.005
40. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J (2000) 19(21):5720–8. doi:10.1093/emboj/19.21.5720
41. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci (2004) 117(Pt 13):2805–12. doi:10.1242/jcs.01131
42. Loos B, du Toit A, Hofmeyr J-HS. Defining and measuring autophagosome flux – concept and reality. Autophagy (2014) 10(11):2087–96. doi:10.4161/15548627.2014.973338
43. Conley ME, Casanova J-L. Discovery of single-gene inborn errors of immunity by next generation sequencing. Curr Opin Immunol (2014) 30:17–23. doi:10.1016/j.coi.2014.05.004
44. Willmann KL, Klaver S, Doğu F, Santos-Valente E, Garncarz W, Bilic I, et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun (2014) 5:5360. doi:10.1038/ncomms6360
45. Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity (2006) 25(5):745–55. doi:10.1016/j.immuni.2006.09.009
46. Kilic SS, Hacimustafaoglu M, Boisson-Dupuis S, Kreins AY, Grant AV, Abel L, et al. A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr (2012) 160(6):1055–7. doi:10.1016/j.jpeds.2012.01.056
47. Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature (2015) 517(7532):89–93. doi:10.1038/nature13801
48. Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science (2012) 337(6102):1684–8. doi:10.1126/science.1224026
Keywords: nuclear factor-kappa B-inducing kinase, primary immunodeficiency, tuberculosis, whole exome sequencing, BCG dissemination
Citation: Schlechter N, Glanzmann B, Hoal EG, Schoeman M, Petersen B-S, Franke A, Lau Y-L, Urban M, van Helden PD, Esser MM, Möller M and Kinnear C (2017) Exome Sequencing Identifies a Novel MAP3K14 Mutation in Recessive Atypical Combined Immunodeficiency. Front. Immunol. 8:1624. doi: 10.3389/fimmu.2017.01624
Received: 28 February 2017; Accepted: 09 November 2017;
Published: 27 November 2017
Edited by:
Menno C. van Zelm, Monash University, AustraliaReviewed by:
Karin Regine Engelhardt, Newcastle University, United KingdomCopyright: © 2017 Schlechter, Glanzmann, Hoal, Schoeman, Petersen, Franke, Lau, Urban, van Helden, Esser, Möller and Kinnear. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig Kinnear, Z2tpbkBzdW4uYWMuemE=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.