 Nicholas A. J. Dawson
Nicholas A. J. Dawson Jens Vent-Schmidt
Jens Vent-Schmidt Megan K. Levings
Megan K. Levings
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 03 November 2017
Sec. Immunological Tolerance and Regulation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01460
This article is part of the Research Topic Therapeutic Potential of Gene-Modified Regulatory T Cells View all 10 articles
Regulatory T cells (Tregs) are potent suppressors of immune responses and are currently being clinically tested for their potential to stop or control undesired immune responses in autoimmunity, hematopoietic stem cell transplantation, and solid organ transplantation. Current clinical approaches aim to boost Tregs in vivo either by using Treg-promoting small molecules/proteins and/or by adoptive transfer of expanded Tregs. However, the applicability of Treg-based immunotherapies continues to be hindered by technical limitations related to cell isolation and expansion of a pure, well-characterized, and targeted Treg product. Efforts to overcome these limitations and improve Treg-directed therapies are now under intense investigation in animal models and pre-clinical studies. Here, we review cell and protein engineering-based approaches that aim to target different aspects of Treg biology including modulation of IL-2 signaling or FOXP3 expression, and targeted antigen-specificity using transgenic T cell receptors or chimeric antigen receptors. With the world-wide interest in engineered T cell therapy, these exciting new approaches have the potential to be rapidly implemented and developed into therapies that can effectively fine-tune immune tolerance.
Regulatory T cells (Tregs) are essential to maintain self-tolerance and dampen immune responses during infection (1, 2). The best characterized subset of Tregs is defined by high expression of CD25 and FOXP3, the master-regulator of their phenotype and suppressive function (3). The critical role of FOXP3 in controlling Treg development and function is illustrated by the study of Tregs from patients with immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome (4). Depending on the specific mutation, IPEX patients may or may not have circulating FOXP3+ T cells, but even if FOXP3+ T cells are present, they are functionally defective due to inadequate FOXP3 transcriptional function (5–7).
Mechanistically, Tregs suppress the proliferation and function of many immune cells, even at very low Treg:effector cell ratios (2). In terms of suppressive pathways, multiple possibilities have been described, such as immunosuppressive cytokines, contact-dependent cytotoxicity, metabolic disruption, and suppression of antigen presenting cells via co-inhibitory molecule expression. Focusing on human Tregs, there is a dominant role for CTLA-4 and TGF-β. Monogenic mutations affecting CTLA-4 or proteins in its pathway affect Treg function (8, 9) and antibodies that block activation of TGF-β by human Tregs prevent their ability to control xenogeneic graft-versus-host disease (GVHD) (10). An additional aspect of Treg mechanisms is their ability to take on characteristics of other T helper (Th) cells (11, 12) resulting in sub-specialization and enhanced suppression of the Th cell subset they mirror (13). Whether or not these sub-specialized Tregs have unique suppressive mechanisms or are simply better able to traffic to the relevant sites of inflammation remains to be defined.
The immunosuppressive properties of Tregs make them attractive candidates for cellular therapy, particularly for application in conditions such as hematopoietic stem cell transplantation (HSCT), solid organ transplantation, and autoimmunity. However, harnessing Tregs for this purpose has not been trivial due to limitations related to cell isolation and expansion. In this review, we summarize the current state of Treg therapy in the clinic and discuss how engineering strategies can be used to improve upon current approaches.
There are two main approaches to increase Treg numbers and function: in vivo “boosting” using small molecules or proteins and adoptive cellular therapy. To date, the most successful strategy to “boost” Treg in vivo is the use of low-doses of IL-2. When given in limiting concentrations, IL-2 preferentially expands CD25hi Tregs without significantly affecting cells expressing low-levels of CD25, such as resting conventional T (Tconv) cells or NK cells. This concept was first tested for treatment of hepatitis-C-virus-induced vasculitis where low doses of IL-2 induced an increase in circulating Tregs and clinical improvements in 8 of 10 patients (14). Subsequently, the beneficial effect of low-dose IL-2 therapy was also observed in GVHD, alopecia areata, type 1 diabetes (T1D), and systemic lupus erythematosus (15–19). However, a cautionary note is that in one study of T1D where IL-2 therapy was combined with rapamycin, there was an unexpected expansion of NK cells and worsening of disease (20). Thus, this approach may need further refinement to reduce the risk of expanding non-Tregs. Low-dose IL-2 and other strategies for in vivo-boosting of Tregs are discussed extensively in Zhang et al. and Boyman et al. (21, 22).
An alternate to in vivo-boosting is adoptive therapy with ex vivo-enriched, often expanded, Tregs. This method aims to overcome defective or low numbers of Tregs by transfer of a large number of Tregs to re-set the Treg:Tconv cell balance. Adoptive Treg therapy has been applied in the clinic for many years. The first successful study reported that chronic GVHD patients treated with Tregs had a significant reduction in clinical symptoms and immunosuppression (23). Subsequently, Treg therapy has been tested in several other GVHD cohorts, overall showing that infusion of autologous or third party (partially HLA-matched) Tregs is well tolerated, does not inhibit graft-versus-leukemia, and may be protective from GVHD (24, 25).
Adoptive transfer of Tregs has also been applied successfully in autoimmunity and organ transplantation. Children with T1D who received Tregs showed slowed disease progression and long-term preservation of residual beta-cells (26, 27). Adoptive transfer of Tregs in adults with T1D is also well tolerated, with evidence that the cells persist long term (>1 year) (28). A clinical trial of in vitro-expanded naïve Tregs is also underway in Crohn’s Disease, the first application of FOXP3+ Treg immunotherapy for inflammatory bowel disease (IBD) (ISRCTN97547683) (29). In addition, several clinical trials are testing autologous polyclonal or antigen-expanded expanded Tregs in kidney or liver transplantation; these trials are reviewed extensively in Ref. (30–33). To date, all of these studies have shown that adoptive Treg therapy in humans is feasible and safe, and initial data suggest that this approach may also be effective.
With the early success of low-dose IL-2 therapy as an approach to expand Tregs in vivo, there are now several efforts to improve upon this approach by modulating the way IL-2 interacts with its receptors. One strategy to modulate IL-2 is to use IL-2/anti-IL-2 monoclonal antibody (mAb) combination therapy to form “IL-2 complexes” that enhance the half-life of IL-2 after intravenous injection and provide preferential selection of certain immune cell subsets. For example, IL-2 in complex with anti-IL-2 mAbs, JES6-1A12 (mouse), or 5344 (human), preferentially expands Tregs, but not other IL-2-dependent cells such as CD8+ T and NK cells (34). This approach enriches Tregs and treats disease in several different mouse models (22, 34). In 2015, Spangler et al. solved the crystal structure of IL-2/JES6-1A12, showing that this IL-2 complex preferentially binds cells with the trimeric IL-2R (CD25, CD122, and common gamma chain) and not dimeric complexes (CD122 and common gamma chain), thus selecting for Tregs because of their constitutive CD25 expression (35).
Another approach to modulate IL-2 is to directly mutate IL-2 itself to change how it interacts with its receptor complex. Specifically, IL-2 “muteins” have alterations in the CD25-binding domain, thus decrease affinity for CD25, and enabling preferential binding to dimeric IL-2R complexes and activation of NK and CD8+ T cells (36–38). There is also much commercial interest in making IL-2 muteins with the opposite effect: IL-2 muteins that preferentially activate Tregs have led to a $400 million investment from Eli Lilly to Nektar Therapeutics and $300 million from Celgene to Delinia to develop this technology (39).
A final approach to modulate IL-2 signaling is to change IL-2R’s affinity for IL-2. Specifically, it is well established that single nucleotide polymorphisms in the CD25 locus are associated with autoimmunity (40–43). Considering the power of CRISPR/Cas9 technology, in the future it could be possible to edit risk alleles of CD25 into protective alleles or otherwise engineer IL-2 signaling pathways to optimize therapeutic Treg function (44).
A hurdle in Treg therapy is generating sufficient numbers for clinical application (33). Since activated Tconv cells also express CD25 and FOXP3, and downregulate CD127, isolating Tregs on the basis of CD25 and CD127 alone introduces the risk of co-purifying and co-expanding non-Tregs. One way to overcome this limitation is to isolate naive CD45RA+CD25hi cells from blood to enrich for a more homogeneous population (45, 46). However, this also significantly decreases the number of cells with which a culture can be started. Another potential solution to this problem is to isolate Tregs directly from the thymus for application as a third party cell therapy (47).
An additional approach is to find a way to engineer the desired Treg product. Indeed, the possibility of engineering Tregs via over-expression of FOXP3 has been considered since its discovery, with multiple studies showing that viral-mediated overexpression of FOXP3 in mouse or human T cells can induce suppressive function (48). Notably, in order to re-program human T cells into Tregs, FOXP3 has to be expressed at high and stable levels (49, 50); Treg suppressive capacity can be quickly reversed upon removal of FOXP3 (51).
Although FOXP3 is the master Treg transcription factor, evidence that its over-expression alone does not fully recapitulate the Treg gene signature led to the search for other co-factors and the discovery that co-expression of other transcription factors is important for full lineage specification (52). A consideration is whether studies which found that FOXP3 expression alone is not sufficient to induce a complete Treg gene signature considered the time that may be required for epigenetic re-programing to take place. Epigenetic modification and the consequent change in expression of other transcription factors is necessary to stabilize Treg phenotype and function (3). Since these epigenetic changes may require multiple rounds of cell division, re-programing Tconv cells into Tregs may not take place in short-term culture. The first application of FOXP3-engineered Treg therapy will likely happen as gene therapy for IPEX. CD4+ T cells from IPEX patients can be efficiently converted into functional and stable Tregs by FOXP3 gene transfer in vitro (53, 54). Testing these cells in vivo will rigorously determine if they have acquired sufficient Treg function to treat the severe autoimmunity in these patients.
Antigen-specific Tregs have the benefit of being directed toward desired therapeutic antigens, thus increasing their potency up to 100-fold compared to polyclonal Tregs (55). Not only would fewer antigen-specific Tregs need to infused but they would also carry a lower risk of off-target suppression (55, 56). However, antigen-specific Tregs are extremely rare and must undergo significant in vitro expansion to achieve clinical doses. Despite this technical barrier, the testing of antigen-specific Tregs is already underway in the clinic in the context of organ transplantation (31).
Engineering antigen-specific Tregs by genomic modification to confer expression of desired transgenic T cell receptors (TCR) or by chimeric antigen receptors (CARs) represents an exciting approach to solve the challenge of the rarity of antigen-specific Tregs (57). Attempts to re-program the specificity of Tregs have been underway for several years. The first application in human Tregs involved gene transfer of a melanoma-specific, MHC Class I-restricted, TCR (58). These human TCR-transduced Tregs proliferated in response to antigen and suppressed antigen-specific Tconv cells in vitro and in vivo. Similarly, human Tregs transduced with a factor VIII (VIII)-specific TCR suppressed FVIII-specific Tconv cells and anti-FVIII antibody production from primed splenocytes (59). Human Tregs transduced with an islet antigen-specific TCR suppressed antigen-stimulated T cell responses. However, they were less efficient than Tregs expressing a viral antigen-specific TCR (60), possibly due to Treg-specific TCR affinity requirements (61). On the other hand, another study of human Tregs in which multiple class I-restricted TCRs recognizing the same peptide-MHC complex, but with affinities varying up to 3,500-fold, were tested, found TCR affinity had no effect on antigen-specific suppressive function (62). Thus, a consideration for future development of this approach is to find TCRs with an MHC restriction and specificity that would make them applicable in multiple patients, and which possess an optimal affinity for Tregs. TCRs which meet these requirements are most likely to be found in autoimmunity where there are well-known and relatively common MHC-peptide complexes that could be targeted.
Another approach to engineer antigen-specific Tregs is to use a CAR technology, an idea borrowed from cancer immunotherapy. CARs were first described by Eshhar et al. in 1993 (63) and now being applied in humans for cancer immunotherapy (64–66). CARs give T cells the B-cell-like ability to bind to antigen in an MHC-independent manner. Additionally, the modular design of CARs allows for customization of specific regions, such as the signaling domains, to tailor the desired response from the engineered cell (67).
Over the last decade, a number of publications demonstrated the utility of CARs in Tregs (56). All reports used a standard second-generation design and included the CD28 co-stimulatory domain (Table 1) (68). Beginning with mouse models in 2008, Elinav et al. used Tregs from a mouse expressing a transgene for a hapten 2,4,6-trinitrophenol (TNP)-specific CAR (69). They found that transgenic TNP-specific CAR Tregs mediated antigen-specific suppression of effector T cells in vitro as well as in vivo resistance to colitis. The same group then demonstrated that the TNP-CAR could be introduced into mouse Tregs using retroviral-mediated gene transfer, giving these cells the ability to protect from disease in vivo in a dose-dependent manner (70). In a similar system, mouse CAR Tregs specific for a different model antigen, carcinoembryonic antigen (CEA), prevented disease in a model of colitis better than CAR Tregs specific for an irrelevant antigen. Importantly, these CEA-CAR Tregs homed to the location of the antigen (71).
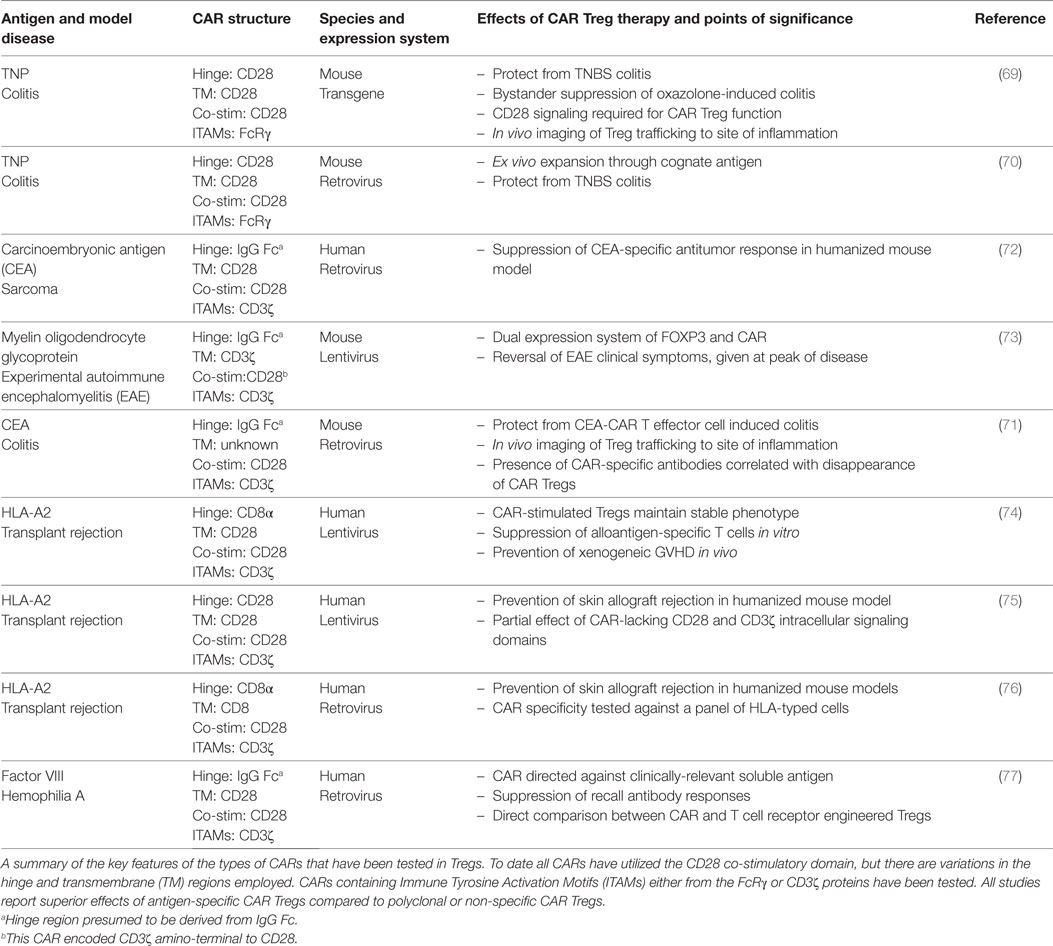
Table 1. Summary of salient details from the current chimeric antigen receptor (CAR) regulatory T cells (Treg) publications.
Apart from these studies in the context of IBD, there is currently only one other report of mouse CAR Tregs. Specifically, in 2012, Fransson et al. developed a CAR specific for myelin oligodendrocyte glycoprotein (MOG), the disease-causing agent for experimental autoimmune encephalomyelitis (EAE) (73). In this study, instead of isolating CD25+FOXP3+ Tregs, lentivirus was used to ectopically express FOXP3 and enforce a Treg phenotype. The resultant MOG-specific CAR Tregs suppressed responder T cell expansion in vitro and reversed symptoms of EAE. Overall, these publications provided important proof-of-concept data supporting the development of CAR Tregs for use in human cells.
Several publications have demonstrated the application of CAR technology to human Tregs. Three reports investigated the utility of expressing a CAR specific for HLA-A*02:01 (A2) to test whether CAR Tregs could be a new approach to control alloreactive T cells that cause rejection in HSCT and solid organ transplantation (74–76). The first publication showed that A2-CAR Tregs are activated and proliferate when stimulated through the CAR via coculture with A2-expressing cells (74). Additionally, A2-CAR Tregs prevented engraftment of A2+ PBMCs and development of xenogeneic GVHD in a humanized mouse model. Two other groups confirmed this approach, showing that A2-CAR Tregs suppress alloimmune responses better than polyclonal Tregs in humanized mouse models of A2+ skin xenografts (75, 76). A2 is an ideal antigen to target with CAR Tregs because it is broadly applicable in the transplant setting due to its high allelic frequency, meaning that a significant proportion of organ transplants could potentially benefit from this therapy (74). Moreover, HLAs in general are likely good targets for CAR Tregs since they are a membrane-bound protein specifically expressed on the transplanted tissues.
Yoon et al. reported the characterization of human CAR Tregs that target FVIII, the protein lacking in hemophilia which is immunogenic in patients receiving FVIII replacement therapy (77). Of specific interest from this study is the finding that a CAR specific for soluble antigens is suitable for use in Tregs, widening the possible antigen-targets that could be considered. This study also demonstrated that both T cell and antibody responses can be controlled by CAR Tregs. Also of note is that this study directly compared the effects of TCR versus CAR-engineered Tregs, finding that antibody recall responses were more effectively controlled by TCR-transgenic Tregs. More research is required to explore similarities and differences between TCR- and CAR-activated Tregs to better understand the affinity requirements and limitations of each approach (Table 2).
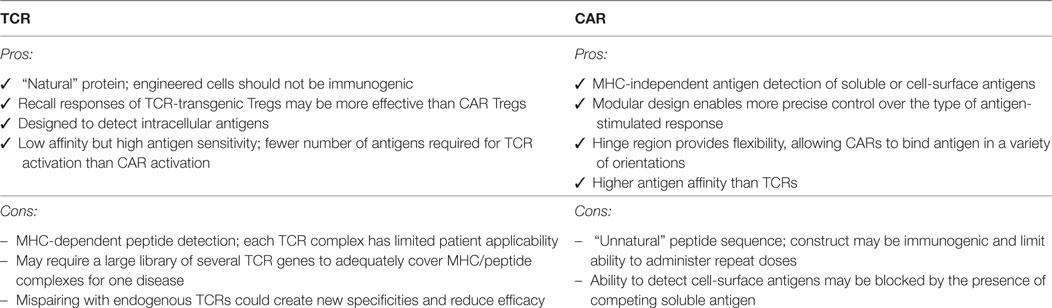
Table 2. Comparison of the benefits and limitations of engineering regulatory T cells (Tregs) to express a defined T cell receptor (TCR) versus chimeric antigen receptor (CAR), see also Harris and Krantz (57).
Many of the fundamental properties of Tregs are similar to Tconv cells so it may be possible to predict some aspects of in vivo Treg behavior on the basis of findings from CAR Tconv cells used in the oncology field. However, Tregs also have many unique properties, such as their strict dependence on other cells for IL-2 and constitutive expression of inhibitory proteins such as CTLA-4 and TGF-β. Thus, there is a need for more detailed studies in animal models to fully appreciate the similarities and differences between the two cell types. For example, will CAR Tregs be able to persist long term even if their antigen is not available? Some research has shown that Tregs have different activation requirements than Tconv cells (62, 78), meaning that optimal proliferation and long-term persistence may require Treg-specific CAR design. Will CAR Tregs traffic to the necessary locations and mediate tolerance? CAR Tconv cells have been found to traffic to the lungs before moving to secondary lymphoid organs and disease sites, delaying their tumor-killing effect (79, 80). If there is similar phenomenon with Tregs then regional cell delivery may be preferred (79). Will CAR Tregs induce tolerance, and if yes, what molecular mechanisms will be necessary? CAR-activated Tregs upregulate CTLA-4, LAP, GARP, and CD39 (74), but it is unknown which pathway(s) are necessary for CAR Treg-mediated suppression. Further, what is the primary target of CAR Treg-mediated suppression? It is unknown whether CAR Tregs suppress immune cells at the site of inflammation, in secondary lymphoid organs, or both. Dissecting the mechanisms important to CAR Treg function may also provide clues as to their primary mode and location of immune suppression. Many of these questions are ideally suited for study in models of transplantation where similar questions with polyclonal or transgenic Tregs have been addressed (55).
Many clinical trials with low-dose IL-2 therapies, expanded polyclonal and antigen-specific Tregs for use in autoimmune diseases, HSCT and solid organ transplantation are underway (18, 31, 33). While initial reports from these trials show that the treatments are well tolerated, the aggregate safety and efficacy data from each approach will greatly inform future studies. Notably, the possible long-term effects, and in particular the potential risk of cancer and infection, of these treatments will not be known for a significant period of time.
We predict that in the next ~5 years there will be a rapid transition from the rather crude current approaches with unmodified IL-2 and/or polyclonal Tregs to engineered approaches that enable precise control over the desired effect (81). It is likely that, as for low-dose IL-2 and polyclonal Treg therapy, transplantation will lead the way in testing these new engineered approaches. HSCT is a setting with a wealth of experience in using engineered T cells for cancer and it would be a natural transition to test engineered Tregs in this context. Moreover, in solid organ transplantation allogeneic HLA antigens represent an ideal target for antigen-specific Tregs because they are only expressed on the transplanted issue, minimizing the risk of off-target suppression (56). Additionally, since solid organ transplant donors and recipients are usually not HLA-matched, there is a large pool of patients that could benefit from this treatment. CAR targets for autoimmunity will be more difficult to identify because there are few truly organ and/or cell-specific antigens that would be suitable CAR targets. This challenge is similar to that faced in oncology, where off-target effects of CAR T cells can have devastating consequences (67, 82). The field of engineered Tregs will benefit greatly from the huge resources being invested into solving this problem in oncology (64–66), creating an ideal landscape to support the rapid development of this next generation of Treg therapies.
ND, JV-S, and ML reviewed the literature and wrote and revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors’ own work in this area is supported by grants from the Canadian Cancer Society Research Institute and the Canadian Institutes of Health Research (CIHR), as well as TxCell. ND holds a CIHR Doctoral Research Award, JV-S holds a Vanier Canada Graduate Scholarship, and ML holds a Salary Award from the BC Children’s Hospital Research Institute.
CEA, carcinoembryonic antigen; EAE, experimental autoimmune encephalomyelitis; IPEX, immunodysregulation polyendocrinopathy enteropathy X-linked; HSCT, hematopoietic stem cell transplantation; GVHD, graft-versus-host disease; MOG, myelin oligodendrocyte glycoprotein; T1D, type 1 diabetes; Th, T helper; TNP, 2,4,6-trinitrophenol; Treg, regulatory T cell; Tconv, conventional T cell.
1. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
2. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol (2017) 5:626. doi:10.1038/nri.2017.75
3. Vent-Schmidt J, Han JM, MacDonald KG, Levings MK. The role of FOXP3 in regulating immune responses. Int Rev Immunol (2014) 33(2):110–28. doi:10.3109/08830185.2013.811657
4. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci (2016). doi:10.1111/nyas.13011
5. Barzaghi F, Passerini L, Gambineri E, Ciullini Mannurita S, Cornu T, Kang ES, et al. Demethylation analysis of the FOXP3 locus shows quantitative defects of regulatory T cells in IPEX-like syndrome. J Autoimmun (2012) 38(1):49–58. doi:10.1016/j.jaut.2011.12.009
6. McMurchy AN, Gillies J, Allan SE, Passerini L, Gambineri E, Roncarolo MG, et al. Point mutants of forkhead box P3 that cause immune dysregulation, polyendocrinopathy, enteropathy, X-linked have diverse abilities to reprogram T cells into regulatory T cells. J Allergy Clin Immunol (2010) 126(6):1242–51. doi:10.1016/j.jaci.2010.09.001
7. Goettel JA, Biswas S, Lexmond WS, Yeste A, Passerini L, Patel B, et al. Fatal autoimmunity in mice reconstituted with human hematopoietic stem cells encoding defective FOXP3. Blood (2015) 125(25):3886–95. doi:10.1182/blood-2014-12-618363
8. Hou TZ, Verma N, Wanders J, Kennedy A, Soskic B, Janman D, et al. Identifying functional defects in patients with immune dysregulation due to LRBA and CTLA-4 mutations. Blood (2017) 129(11):1458–68. doi:10.1182/blood-2016-10-745174
9. Hou TZ, Qureshi OS, Wang CJ, Baker J, Young SP, Walker LS, et al. A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T cells. J Immunol (2015) 194(5):2148–59. doi:10.4049/jimmunol.1401876
10. Cuende J, Lienart S, Dedobbeleer O, van der Woning B, De Boeck G, Stockis J, et al. Monoclonal antibodies against GARP/TGF-beta1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci Transl Med (2015) 7(284):284ra56. doi:10.1126/scitranslmed.aaa1983
11. Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood (2012) 119(19):4430–40. doi:10.1182/blood-2011-11-392324
12. Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N, et al. An Atlas of human regulatory T helper-like cells reveals features of Th2-like Tregs that support a tumorigenic environment. Cell Rep (2017) 20(3):757–70. doi:10.1016/j.celrep.2017.06.079
13. Campbell DJ. Control of regulatory T cell migration, function, and homeostasis. J Immunol (2015) 195(6):2507–13. doi:10.4049/jimmunol.1500801
14. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med (2011) 365(22):2067–77. doi:10.1056/NEJMoa1105143
15. Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol (2015) 15(5):283–94. doi:10.1038/nri3823
16. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med (2013) 5(179):179ra43. doi:10.1126/scitranslmed.3005265
17. Mizui M, Tsokos GC. Low-dose IL-2 in the treatment of lupus. Curr Rheumatol Rep (2016) 18(11):68. doi:10.1007/s11926-016-0617-5
18. Pham MN, von Herrath MG, Vela JL. Antigen-specific regulatory T cells and low dose of IL-2 in treatment of type 1 diabetes. Front Immunol (2015) 6(3):651. doi:10.3389/fimmu.2015.00651
19. Todd JA, Evangelou M, Cutler AJ, Pekalski ML, Walker NM, Stevens HE, et al. Regulatory T cell responses in participants with type 1 diabetes after a single dose of interleukin-2: a non-randomised, open label, adaptive dose-finding trial. PLoS Med (2016) 13(10):e1002139. doi:10.1371/journal.pmed.1002139
20. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes (2012) 61(9):2340–8. doi:10.2337/db12-0049
21. Zhang D, Tu E, Kasagi S, Zanvit P, Chen Q, Chen W. Manipulating regulatory T cells: a promising strategy to treat autoimmunity. Immunotherapy (2015) 7(11):1201–11. doi:10.2217/imt.15.79
22. Boyman O, Kolios AG, Raeber ME. Modulation of T cell responses by IL-2 and IL-2 complexes. Clin Exp Rheumatol (2015) 33(Suppl 92):S54–7.
23. Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol (2009) 133(1):22–6. doi:10.1016/j.clim.2009.06.001
24. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117(3):1061–70. doi:10.1182/blood-2010-07-293795
25. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127(8):1044–51. doi:10.1182/blood-2015-06-653667
26. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care (2012) 35(9):1817–20. doi:10.2337/dc12-0038
27. Marek-Trzonkowska N, Mysliwiec M, Iwaszkiewicz-Grzes D, Gliwinski M, Derkowska I, Zalinska M, et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J Transl Med (2016) 14(1):332. doi:10.1186/s12967-016-1090-7
28. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi:10.1126/scitranslmed.aad4134
29. Canavan JB, Scotta C, Vossenkamper A, Goldberg R, Elder MJ, Shoval I, et al. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut (2016) 65(4):584–94. doi:10.1136/gutjnl-2014-306919
30. Gliwinski M, Iwaszkiewicz-Grzes D, Trzonkowski P. Cell-based therapies with T regulatory cells. BioDrugs (2017) 31:335. doi:10.1007/s40259-017-0228-3
31. Tang Q, Vincenti F. Transplant trials with Tregs: perils and promises. J Clin Invest (2017) 127(7):2505–12. doi:10.1172/JCI90598
32. Romano M, Tung SL, Smyth LA, Lombardi G. Treg therapy in transplantation: a general overview. Transpl Int (2017) 30(8):745–53. doi:10.1111/tri.12909
33. Trzonkowski P, Bacchetta R, Battaglia M, Berglund D, Bohnenkamp HR, ten Brinke A, et al. Hurdles in therapy with regulatory T cells. Sci Transl Med (2015) 7(304):304s18. doi:10.1126/scitranslmed.aaa7721
34. Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: biology, design and application. Trends Immunol (2015) 36(12):763–77. doi:10.1016/j.it.2015.10.003
35. Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity (2015) 42(5):815–25. doi:10.1016/j.immuni.2015.04.015
36. Carmenate T, Pacios A, Enamorado M, Moreno E, Garcia-Martinez K, Fuente D, et al. Human IL-2 mutein with higher antitumor efficacy than wild type IL-2. J Immunol (2013) 190(12):6230–8. doi:10.4049/jimmunol.1201895
37. Rojas G, Carmenate T, Leon K. Molecular dissection of the interactions of an antitumor interleukin-2-derived mutein on a phage display-based platform. J Mol Recognit (2015) 28(4):261–8. doi:10.1002/jmr.2440
38. Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, Ju W, et al. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity (2015) 42(5):826–38. doi:10.1016/j.immuni.2015.04.018
39. Ledford H. Drug companies flock to supercharged T-cells in fight against autoimmune disease. Nat News (2017). doi:10.1038/nature.2017.22393
40. Long A, Buckner JH. Intersection between genetic polymorphisms and immune deviation in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes (2013) 20(4):285–91. doi:10.1097/MED.0b013e32836285b6
41. Alcina A, Fedetz M, Ndagire D, Fernndez O, Leyva L, Guerrero M, et al. IL2RA/CD25 gene polymorphisms: uneven association with multiple sclerosis (MS) and type 1 diabetes (T1D). PLoS One (2009) 4(1):e4137. doi:10.1371/journal.pone.0004137
42. Hinks A, Ke X, Barton A, Eyre S, Bowes J, Worthington J, et al. Association of the IL2RA/CD25 gene with juvenile idiopathic arthritis. Arthritis Rheum (2009) 60(1):251–7. doi:10.1002/art.24187
43. Sebode M, Peiseler M, Franke B, Schwinge D, Schoknecht T, Wortmann F, et al. Reduced FOXP3(+) regulatory T cells in patients with primary sclerosing cholangitis are associated with IL2RA gene polymorphisms. J Hepatol (2014) 60(5):1010–6. doi:10.1016/j.jhep.2013.12.027
44. Simeonov DR, Gowen BG, Boontanrart M, Roth TL, Gagnon JD, Mumbach MR, et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature (2017) 549(7670):111–5. doi:10.1038/nature23875
45. Rossetti M, Spreafico R, Saidin S, Chua C, Moshref M, Leong JY, et al. Ex vivo-expanded but not in vitro-induced human regulatory T cells are candidates for cell therapy in autoimmune diseases thanks to stable demethylation of the FOXP3 regulatory T cell-specific demethylated region. J Immunol (2015) 194(1):113–24. doi:10.4049/jimmunol.1401145
46. Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood (2006) 108(13):4260–7. doi:10.1182/blood-2006-06-027409
47. Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy AN, et al. Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transplant (2016) 16(1):58–71. doi:10.1111/ajt.13456
48. McMurchy AN, Levings MK. In vitro generation of human T regulatory cells: generation, culture, and analysis of FOXP3-transduced T cells. Methods Mol Biol (2013) 946:115–32. doi:10.1007/978-1-62703-128-8_8
49. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther (2008) 16(1):194–202. doi:10.1038/sj.mt.6300341
50. Allan SE, Song-Zhao GX, Abraham T, McMurchy AN, Levings MK. Inducible reprogramming of human T cells into Treg cells by a conditionally active form of FOXP3. Eur J Immunol (2008) 38(12):3282–9. doi:10.1002/eji.200838373
51. Amendola M, Passerini L, Pucci F, Gentner B, Bacchetta R, Naldini L. Regulated and multiple miRNA and siRNA delivery into primary cells by a lentiviral platform. Mol Ther (2009) 17(6):1039–52. doi:10.1038/mt.2009.48
52. Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, et al. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol (2012) 13(10):972–80. doi:10.1038/ni.2420
53. Passerini L, Rossi Mel E, Sartirana C, Fousteri G, Bondanza A, Naldini L, et al. CD4(+) T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med (2013) 5(215):215ra174. doi:10.1126/scitranslmed.3007320
54. Passerini L, Santoni de Sio FR, Porteus MH, Bacchetta R. Gene/cell therapy approaches for immune dysregulation polyendocrinopathy enteropathy X-linked syndrome. Curr Gene Ther (2014) 14(6):422–8. doi:10.2174/1566523214666141001123828
55. Hoeppli RE, MacDonald KG, Levings MK, Cook L. How antigen specificity directs regulatory T-cell function: self, foreign and engineered specificity. HLA (2016) 88(1–2):3–13. doi:10.1111/tan.12822
56. Dawson NAJ, Levings MK. Antigen-specific regulatory T cells: are police CARs the answer? Transl Res (2017) 187:53–8. doi:10.1016/j.trsl.2017.06.009
57. Harris DT, Kranz DM. Adoptive T cell therapies: a comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci (2016) 37(3):220–30. doi:10.1016/j.tips.2015.11.004
58. Brusko TM, Koya RC, Zhu S, Lee MR, Putnam AL, McClymont SA, et al. Human antigen-specific regulatory T cells generated by T cell receptor gene transfer. PLoS One (2010) 5(7):e11726. doi:10.1371/journal.pone.0011726
59. Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood (2015) 125(7):1107–15. doi:10.1182/blood-2014-04-566786
60. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun (2017) 79:63–73. doi:10.1016/j.jaut.2017.01.001
61. Tsang JY, Ratnasothy K, Li D, Chen Y, Bucy RP, Lau KF, et al. The potency of allospecific Tregs cells appears to correlate with T cell receptor functional avidity. Am J Transplant (2011) 11(8):1610–20. doi:10.1111/j.1600-6143.2011.03650.x
62. Plesa G, Zheng L, Medvec A, Wilson CB, Robles-Oteiza C, Liddy N, et al. TCR affinity and specificity requirements for human regulatory T-cell function. Blood (2012) 119(15):3420–30. doi:10.1182/blood-2011-09-377051
63. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A (1993) 90(2):720–4. doi:10.1073/pnas.90.2.720
64. Maus MV, June CH. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res (2016) 22(8):1875–84. doi:10.1158/1078-0432.CCR-15-1433
65. Esensten JH, Bluestone JA, Lim WA. Engineering therapeutic T cells: from synthetic biology to clinical trials. Annu Rev Pathol (2017) 12:305–30. doi:10.1146/annurev-pathol-052016-100304
66. Sadelain M. Chimeric antigen receptors: driving immunology towards synthetic biology. Curr Opin Immunol (2016) 41:68–76. doi:10.1016/j.coi.2016.06.004
67. Chang ZL, Chen YY. CARs: synthetic immunoreceptors for cancer therapy and beyond. Trends Mol Med (2017) 23(5):430–50. doi:10.1016/j.molmed.2017.03.002
68. Oldham RAA, Medin JA. Practical considerations for chimeric antigen receptor design and delivery. Expert Opin Biol Ther (2017) 17(8):961–78. doi:10.1080/14712598.2017.1339687
69. Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology (2008) 134(7):2014–24. doi:10.1053/j.gastro.2008.02.060
70. Elinav E, Adam N, Waks T, Eshhar Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology (2009) 136(5):1721–31. doi:10.1053/j.gastro.2009.01.049
71. Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther (2014) 22(5):1018–28. doi:10.1038/mt.2014.41
72. Hombach AA, Kofler D, Rappl G, Abken H. Redirecting human CD4+CD25+ regulatory T cells from the peripheral blood with pre-defined target specificity. Gene Ther (2009) 16(9):1088–96. doi:10.1038/gt.2009.75
73. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation (2012) 9:112. doi:10.1186/1742-2094-9-112
74. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126(4):1413–24. doi:10.1172/JCI82771
75. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant (2017) 17(4):931–43. doi:10.1111/ajt.14185
76. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant (2017) 17(4):917–30. doi:10.1111/ajt.14175
77. Yoon J, Schmidt A, Zhang AH, Konigs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood (2017) 129(2):238–45. doi:10.1182/blood-2016-07-727834
78. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity (2014) 41(5):722–36. doi:10.1016/j.immuni.2014.10.012
79. Parente-Pereira AC, Burnet J, Ellison D, Foster J, Davies DM, van der Stegen S, et al. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J Clin Immunol (2011) 31(4):710–8. doi:10.1007/s10875-011-9532-8
80. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood (2011) 118(18):4817–28. doi:10.1182/blood-2011-04-348540
81. Lam AJ, Hoeppli RE, Levings MK. Harnessing advances in T regulatory cell biology for cellular therapy in transplantation. Transplantation (2017) 101(10):2277–87. doi:10.1097/TP.0000000000001757
Keywords: regulatory T cells, chimeric antigen receptors, T cell receptor, IL-2, autoimmunity, transplantation, inflammatory bowel disease, immunotherapy
Citation: Dawson NAJ, Vent-Schmidt J and Levings MK (2017) Engineered Tolerance: Tailoring Development, Function, and Antigen-Specificity of Regulatory T Cells. Front. Immunol. 8:1460. doi: 10.3389/fimmu.2017.01460
Received: 17 August 2017; Accepted: 18 October 2017;
Published: 03 November 2017
Edited by:
Herman Waldmann, University of Oxford, United KingdomReviewed by:
Bruce Milne Hall, University of New South Wales, AustraliaCopyright: © 2017 Dawson, Vent-Schmidt and Levings. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Megan K. Levings, bWxldmluZ3NAYmNjaHIuY2E=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.