 Fabiano Pinheiro da Silva*
Fabiano Pinheiro da Silva* Marcel Cerqueira César Machado
Marcel Cerqueira César Machado
- Laboratório de Emergências Clínicas, Faculdade de Medicina FMUSP, Universidade de São Paulo, São Paulo, Brazil
Aging is a continuous process promoted by both intrinsic and extrinsic factors that each trigger a multitude of molecular events. Increasing evidence supports a central role for inflammation in this progression. Here, we discuss how the low-grade chronic inflammation that characterizes aging is tightly interconnected with other important aspects of this process, such as DNA damage, mitochondrial dysfunction, and epigenetic changes. Similarly, inflammation also plays a critical role in many morbid conditions that affect patients who are admitted to Intensive Care. Although the inflammatory response is low grade and persistent in healthy aging while it is acute and severe in critically ill states, we hypothesize that both situations have important interconnections. Here, we performed an extensive review of the literature to investigate this potential link. Because sepsis is the most extensively studied disease and is the leading cause of death in Critical Care, we focus our discussion on comparing the inflammatory profile of healthy older people with that of patients in septic shock to explain why we believe that both situations have synergistic effects, leading to critically ill aged patients having a worse prognosis when compared with critically ill young patients.
Introduction
Over time, improved health conditions have led to a steady growth in the older population, resulting in a substantial increase in the number of critically ill-aged patients. In addition, advanced age is associated with a worse outcome in all of the most frequent critical care conditions (1).
Chronic, low-grade, systemic inflammation, and the deregulation of several innate and acquired immune responses have been reported in seniors (2). The signaling pathways implicated in this scenario, called inflammaging, create a complex network (3–7) that is probably triggered and perpetuated by prolonged exposure to varied exogenous and endogenous factors, such as infection, tissue injury, DNA damage, mitochondrial dysfunction, intestinal barrier failure, and dysbiosis (8–10) and may contribute to the increased risk of acute illnesses, disability, and death in this population (11). Indeed, inappropriate inflammation and metabolic stress lead to the accumulation of senescent cells, which are characterized by transcriptional and epigenetic alterations that determine cell cycle arrest, as well as aberrant mRNA production and maturation, chromatin structure changes, and impaired proteostasis (12, 13). Moreover, emergency myelopoiesis and the persistence of immature myeloid cell progenitors have been shown to be significant contributors to dysfunctional inflammation in sepsis and are emerging in aging research (14, 15).
Pathogen-associated molecular patterns are molecules shared by many microorganisms, but not found in mammals, that are recognized by the immune system and activate cell defense. Several host factors are also able to alarm the immune system, even in sterile conditions. Indeed, persistent or recurrent contact with microbes (16), as well as with non-infectious danger signals, induce the production of damage-associated molecular patterns (DAMPs), which accumulate during aging (17). Examples include adenosine triphosphate, high mobility group box 1 protein, oxidized lipoproteins, heat shock proteins, and urate and cholesterol crystals. These DAMPs are able to activate membrane receptors and cytosolic receptors (including inflammasomes) or act directly at the nuclear level, inducing gene transcription (16, 18). Moreover, accumulating evidence suggests that mitochondrial and genomic DNA and histones also activate danger signals and induce systemic protection (19). For example, levels of circulating cell-free DNA, presumably released from damaged or dying cells, are increased in older adults (20) and are associated with both mortality and the magnitude of the inflammatory response (21–23). Similarly, critical inflammatory conditions, such as sepsis, are also characterized by high levels of circulating host DNA (24, 25).
Many nucleic acid molecular sensors have been found (26). CpG-enriched DNA, such as mitochondrial and bacterial DNA, are mostly recognized by TLR9 (21, 27), but other systems to detect not only mitochondrial and microbial DNA but also the nuclear DNA that migrates to the cytosol under pathologic conditions have been described (28–31). Furthermore, inside the nucleus, a sophisticated system of DNA sensors is able to detect DNA damage and activate immune signaling (31). In parallel, harmful DNA coming from pathogens, apoptotic cells, or DNA replication byproducts can be directly degraded by DNases to avoid the excessive activation of immune cascades (31).
Enduring Permanent Aggression: From Mitochondrial Dysfunction to Genomic Instability
Aging is accompanied by a decline in mitochondrial function in all tissues; however, some tissues, such as the muscles, are particularly affected (32). Beyond their function in bioenergetics, growing research suggests that mitochondria participate in many other mechanisms that are deregulated in senescence (33). Indeed, mitochondria are important organelles in the maintenance of stem cells (12), the activation of the unfolded protein response (34), the regulation of innate and adaptive immune pathways (35, 36), and the modulation of the metabolic profile of the cell (32). As such, mitochondria are deeply integrated into cellular homeostasis (37).
Similarly, mitochondrial dysfunction is a common finding in a wide range of patients in critical care conditions (38–41). Pro-inflammatory mediators and oxidative stress impair the function of the respiratory chain enzyme complexes and damage the mitochondrial structure, including their genes (42, 43). While the genomic DNA can be affected in a similar manner, the mitochondrial DNA is likely more vulnerable to this type of damage due to its close location to the electron transport chain, its lack of protective histones, the limited efficiency of the mtDNA repair mechanisms, and the fact that, like bacterial DNA, it exclusively contains coding regions (44, 45). However, a single-cell contains thousands of mitochondrial genomes, and mutations of all of them in the same gene are unlikely, putting the nuclear genome at a higher risk (46). An excellent publication from Patananan et al. describes the current challenges to therapeutically modifying the mitochondrial genome and the important concept of heteroplasmy (47). Furthermore, emerging evidence suggests that DNA damage activates signaling from the nucleus to the mitochondria, creating complex networks that are crucial for mitochondria maintenance (48).
The genetic lesions arising from DNA damage include point mutations, translocations, gains, losses, strand breaks, and telomere shortening. DNA lesions occur frequently, even under physiological conditions (49). These genetic changes have a well-established impact on the aging process and have been largely investigated as both a cause and consequence of chronic low-grade inflammation. The same process may occur in the critically ill; however, it seems to occur over a short-time period and be massive in this population, while it is low-grade and persistent during aging.
The occurrence of DNA damage induces further immune activation, promoting a vicious circle with disastrous consequences (50). Indeed, there is increasing evidence pointing to reciprocal interactions between DNA damage, DNA repair, and the immune system (51). In the short term, depending on the intensity of damage or the presence of deficient DNA repair responses (52–54), genotoxic stress has the potential to induce aberrant cell responses, apoptosis, organ failure, and immunosuppression (19, 43). The occurrence and magnitude of acute DNA lesions in the setting of severe systemic inflammation, however, remain to be confirmed. In the long term, these plausible DNA mutations and deletions can significantly impact patient quality of life and predispose the survivors of inflammatory catastrophes to several morbid consequences. In this regard, a recent publication from our group showed that sepsis induces telomere shortening (55), confirming that inflammation affects telomere length (56) and that stress-induced premature senescence is a telomere-dependent process (57). Despite that, however, the other evidence in the literature addressing this topic is controversial and/or indirect. The phenotype of sepsis survivors, for example, resembles accelerated aging, and sepsis survivors suffer from a higher risk of additional morbidities, such as cardiovascular disease, cognitive impairment, tumor progression, and possibly death, for years following the sepsis event (58–60).
Since genomic instability is a hallmark of aging, genetic damage secondary to severe infection or other causes of critical illnesses may have a larger impact in seniors than in young patients. We believe that this is a critical factor that partially explains the worse outcome of older people, compared with the young, when affected by overwhelming inflammatory syndromes.
Redox reactions generate oxidatively modified signaling biomolecules that are crucial for the generation of appropriate innate and adaptive cell responses (35) and for many other fundamental biological processes (61–63). Reactive oxygen and nitrogen species have important physiological effects and display various well-established specific signaling functions, but they need to be tightly regulated; otherwise, they can generate significant tissue damage (64). Oxidative damage occurs to a larger extent both in older people and in the presence of acute or chronic inflammatory processes. It can lead to substantial DNA damage and significantly contribute to genomic instability and mitochondrial dysfunction. A DNA microarray study performed by our group found that the oxidative phosphorylation and the mitochondrial dysfunction pathways were the most-enriched pathways in septic patients of advanced age when compared with the young septic group (65).
The Epigenetic Code: An Additional Layer of Complexity
Epigenetic changes involve various histone marks, DNA methylation, nucleosome positioning, and mechanisms governed by non-coding RNAs that are able to repress or activate transcription (66). Epigenetic alterations are important aspects in the regulation of aging, linking environmental factors with the genetic profile (67). We believe epigenetic modifications may be implicated in the global modifications to the cell response that manifest during the evolution of catastrophic inflammatory processes, especially those caused by overwhelming infection. In support of this hypothesis, a recent publication reported that sepsis in humans induces selective and precise chromatin modifications in distinct promoter regions of immunologically relevant genes (68). Cellular dysfunction secondary to genetic and epigenetic changes in the course of major inflammatory syndromes may be stochastic and partially reversible, even though some organs and tissues appear to be more strongly affected. Moreover, DNA regions that are robustly activated are likely more influenced since they are less protected than the heterochromatin. Unfortunately, this topic remains obscure in the critically ill. However, it could potentially explain, for example, the long-term cognitive impairment that is frequently detected in survivors of septic shock and other intriguing findings, such as the phenomenon of endotoxin tolerance (69).
There is a significant interconnection between the DNA damage response and epigenetic changes. DNA damage is a serious threat to cell viability, compromising the integrity of both the genome and the epigenome. The DNA damage response can lead to significant alterations in chromatin structure, affecting chromatin components and epigenetic marks, with major implications for cell metabolism (70). As stated by López-León and Goya, “aging seems to be characterized by a progressive depression of the transcriptional activity of chromatin” (71); this has been partially attributed to a reduction in DNA methylation and an altered chromatin architecture (72). The end result of epigenetic changes is aberrant gene expression, reactivation of transposable elements, and genomic instability (73).
There is increasing evidence that together with the above discussed factors, microRNAs and long non-coding RNAs also play a key role in fundamental epigenetic processes, with important implications for the aging process and various morbid states (74–76).
Aging and Critical Illnesses: From Low-grade to Explosive Inflammation
The treatment of critically ill aged patients is challenging. Older people frequently exhibit atypical symptomatology, due to comorbidities and dysfunctions throughout all body systems that are related to the aging process (77).
Sepsis is a disease of the elderly. The incidence of sepsis increases exponentially with age, and sepsis-associated long-term sequelae particularly affect older patients. Sepsis survivors are at substantial risk for poor quality of life, functional disability, and cognitive impairment. As advances in medicine and quality of life extend the life expectancy worldwide, a growing number of aged patients need critical care (78). A recent study demonstrated a significant rise in survivorship after sepsis in the United States, caused by a rising incidence of sepsis rather than improvements in its case fatality rate, generating a substantial population burden of aged patients with disabilities (79).
The reason for the higher susceptibility to infection and increased mortality in older adults remains in debate (80). The basal inflammatory state found in healthy seniors suggests that aged people possess a limited capacity to control inflammation. Similarly, the critically ill are frequently affected by overwhelming inflammatory syndromes, where the host response is the major cause of damage. Examples include diseases such as septic shock, severe acute pancreatitis, burns, trauma, ischemia reperfusion injuries, and hemorrhages. As discussed earlier, the chronic low-grade inflammation in the elderly and the explosive inflammation in the critically ill share several commonalities. We propose that, together, these processes may have synergistic effects, leading to a worse outcome (Figure 1).
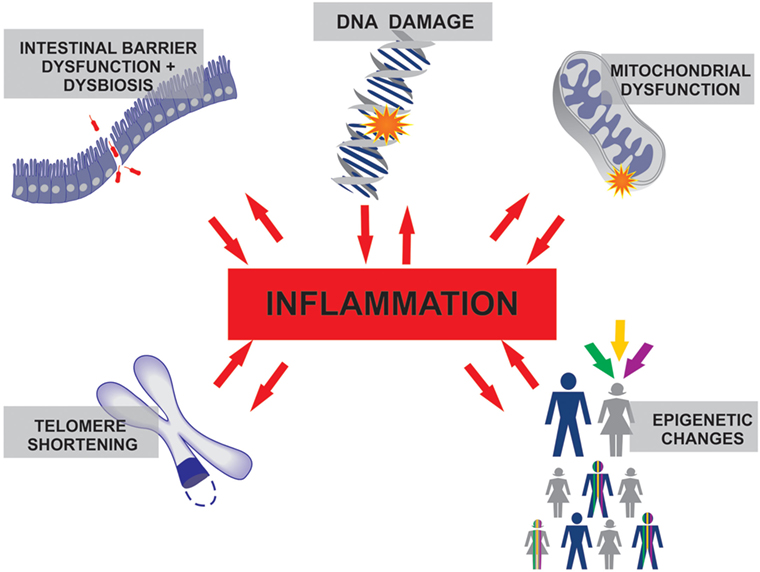
Figure 1. The inflammation that characterizes healthy aging and the inflammatory processes in several critical care conditions differ in duration and intensity, but both involve similar molecular interconnections where inflammation plays a central role.
Notably, these synergistic effects have interesting peculiarities. A study performed by our group found that older people are as immunocompetent as young individuals regarding the cytokines, chemokines, and growth factors produced in response to devastating infections. After our analysis of several inflammatory mediators in the plasma of critically ill individuals, we were unable to find any reason that could serve to better explain why the aged show an increased susceptibility and mortality to septic shock (81). As detailed in the section below, this phenomenon can be partially explained by the fact that aged people probably display a prolonged inflammatory systemic response under acute stress conditions, when compared with the systemic response of the young, even though both groups share the same ability to trigger and sustain the same intensity of inflammatory signaling in the acute phase (81). Moreover, in a study performed in rats, we were able to demonstrate that despite a similar systemic response, aged rats show increased intestinal gene expression levels of TNFα, α-defensin 5, and α-defensin 7, when compared with young rats (82). Similarly, other work from our group demonstrated greater gene expression levels of COX-2 and intercellular junction proteins in the guts of aged rats with acute pancreatitis when compared with young rats in the same conditions, suggesting that, in situations of intestinal damage, the young animals are better able to restore intestinal barrier integrity (83). The results of these studies strongly suggest that the inflammatory response of the elderly is compartmentalized, with significant differences in the inflammatory profile depending on the organ under investigation.
For many years, the catastrophic systemic inflammation associated with many critical care diseases has been attributed to a massive and transient activation of the innate immune system, followed by a period of immunosuppression (84, 85). Seminal high-throughput gene expression studies performed in septic patients by the Wong group, however, challenge this theory. Indeed, instead of the classical biphasic curve, they have consistently detected an elliptical curve, formed by the persistent activation of innate immune genes in conjunction with widespread repression of gene programs corresponding to the adaptive immune system (86–88). Confirming these findings, a similar pattern was found in trauma patients (89), suggesting that infectious and non-infectious systemic inflammation in the critically ill may involve analogous cell responses. Notably, more recent studies are finding subtle differences in the transcriptional program of different acute stress conditions and even in different subsets of the same morbid process (90).
Maintenance of the Intestinal Epithelial Barrier and the Human Microbiome
The intestinal mucosal barrier is a fundamental line of defense against undesirable luminal contents, such as microorganisms, toxins, and antigens, preventing their entrance into the bloodstream. It is mostly composed of epithelial cells, immune components, and mucus. Some researchers also consider the microbiome as part of the intestinal barrier (9, 91), since it helps to maintain the integrity of the intestinal barrier, providing nutrients and protecting against pathogens.
Aged people are in a persistent systemic inflammatory state that may be partially attributed to increased bacterial translocation, secondary to intestinal barrier dysfunction (92). As people age, the intestinal barrier weakens, partially due to decreased levels of tight junctions connecting epithelial cells, and the enteric immune system becomes ineffective (93). Indeed, higher plasma levels of lipopolysaccharide can be detected in the blood of older subjects, when compared with young individuals (94, 95). Furthermore, there is a shift in the intestinal microbiome after the age of 65, with an increased abundance of Bacteroidetes phyla (96) and a reduction in the capacity of the microbiota to carry out metabolic processes, such as short-chain fatty acid production (97). We propose that these previous alterations to the intestinal barrier in the elderly are probably exacerbated during systemic inflammation processes. In support of this idea, Zhang et al. recently demonstrated that neutrophil activation and aging are both affected by the intestinal microbiota and that depletion of the microbiome with broad-spectrum antibiotics significantly reduces the number of circulating aged neutrophils, ameliorating inflammation-related organ damage in a model of endotoxin shock (98). Another recent publication showed that germ-free mice do not display the increase in circulating cytokines that is a hallmark of aging and that co-housing germ-free mice with old, but not young, conventionally raised mice reconstitutes this phenotype; the authors concluded that, in mice, intestinal permeability increases with age due to microbial dysbiosis (99). Taken together, these observations suggest that the increased mortality of aged patients in critical care conditions is probably due to a prolonged systemic inflammatory response, at least partially caused by increased bacterial translocation and defective bacterial clearance (100).
Microbiome studies are challenging because there is extensive inter-individual variability, even among healthy subjects. Genetic, lifestyle, and environmental factors, such as diet, physical activity, geography, and exposure to xenobiotics, all cause substantial modifications to the intestinal microbiome. However, despite this extensive interindividual variability, specialists agree on the existence of a global human core microbiota (101, 102).
Commensal bacteria have much shorter generation times than humans and consequently undergo rapid evolutionary changes, adapting quickly to environmental changes. Unfortunately, external forces sometimes shape a microbiome that is detrimental to the host, a state called dysbiosis. Once established, dysbiosis can exert profound effects on the immune system, creating a feedback loop in which host factors and the microbiome (cell components and metabolites) regulate each other, perpetuating the dysbiotic state (103). It is well established that the intestinal microbiota is severely modified during critical illnesses. However, it remains unclear which change occurs first.
The causal mechanisms of dysbiosis in the critically ill are not completely understood, but they likely result from many intrinsic and extrinsic factors, such as widespread antibiotics use, hypoxic injury, inflammation, intestinal dysmotility, epithelial barrier disruption, vasopressors treatment, and sedation (104). The intestinal microbiome of the critically ill differs substantially from that of healthy individuals and is characterized by lower phylogenetic diversity, commensal microbe loss, and pathobiont overgrowth (105, 106).
Concluding Remarks
Older adults in critical care conditions develop a peculiar inflammatory response, which is associated with poorer outcomes. Current treatments are unspecific and mainly rely on life support techniques. Novel strategies are under investigation (99), and Personalized Medicine has been widely discussed to improve care of the critically ill (107); however, to a large extend, these proposals still remain experimental and hypothetical, without impacting clinical applications. Thus, manipulation of the inflammatory storms that are so frequent in Critical Care remains a challenging task, filled with negative results and nebulous findings. Prolonged hospital stays, recurrent infections, decrepitude, and malnutrition characterize the critically ill population as a whole, but particularly apply to the subset composed of aged adults.
By the other hand, impressive advances in the molecular biology of aging are emerging. Biomarkers of aging have been extensively investigated to guide tailored treatments of the aging process, as well as to detect individuals that age faster (108). Despite the current challenges (109–111), in vivo partial cellular reprogramming (112), direct reprogramming (109), and epigenetic interventions (67) are tentative highways for drug development and captivating platforms to reach this goal. Indeed, the rapid advancement of scientific knowledge in this field provides hope that, in a not-so-distant future, sophisticated medical technologies to delay and even reverse normal aging might be available.
Author Contributions
FPS conceived and wrote the manuscript. MCCM contributed with suggestions and ideas.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
English editing was performed by Edanz.
References
1. Joyce MF, Reich JA. Critical care issues of the geriatric patient. Anesthesiol Clin (2015) 33(3):551–61. doi:10.1016/j.anclin.2015.05.009
2. Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Curr Opin Immunol (2010) 22(4):507–13. doi:10.1016/j.coi.2010.05.003
3. Ruan Q, Qian F, Yu Z. Effects of polymorphisms in immunity-related genes on the immune system and successful aging. Curr Opin Immunol (2014) 29:49–55. doi:10.1016/j.coi.2014.04.003
4. Johnson SC, Dong X, Vijg J, Suh Y. Genetic evidence for common pathways in human age-related diseases. Aging Cell (2015) 14(5):809–17. doi:10.1111/acel.12362
5. Ter Horst R, Jaeger M, Smeekens SP, Oosting M, Swertz MA, Li Y, et al. Host and environmental factors influencing individual human cytokine responses. Cell (2016) 167(4):1111–1124e1113. doi:10.1016/j.cell.2016.10.018
6. Xia S, Zhang X, Zheng S, Khanabdali R, Kalionis B, Wu J, et al. An update on inflamm-aging: mechanisms, prevention, and treatment. J Immunol Res (2016) 2016:8426874. doi:10.1155/2016/8426874
7. Fulop T, Witkowski JM, Le Page A, Fortin C, Pawelec G, Larbi A. Intracellular signalling pathways: targets to reverse immunosenescence. Clin Exp Immunol (2017) 187(1):35–43. doi:10.1111/cei.12836
8. Goldberg EL, Dixit VD. Drivers of age-related inflammation and strategies for healthspan extension. Immunol Rev (2015) 265(1):63–74. doi:10.1111/imr.12295
9. Cesar Machado MC, da Silva FP. Intestinal barrier dysfunction in human pathology and aging. Curr Pharm Des (2016) 22(30):4645–50. doi:10.2174/1381612822666160510125331
10. He S, Sharpless NE. Senescence in health and disease. Cell (2017) 169(6):1000–11. doi:10.1016/j.cell.2017.05.015
11. High KP, Akbar AN, Nikolich-Zugich J. Translational research in immune senescence: assessing the relevance of current models. Semin Immunol (2012) 24(5):373–82. doi:10.1016/j.smim.2012.04.007
12. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi:10.1016/j.cell.2013.05.039
13. Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine (2017) 21:21–8. doi:10.1016/j.ebiom.2017.04.013
14. Flores RR, Clauson CL, Cho J, Lee BC, McGowan SJ, Baker DJ, et al. Expansion of myeloid-derived suppressor cells with aging in the bone marrow of mice through a NF-kappaB-dependent mechanism. Aging Cell (2017) 16(3):480–7. doi:10.1111/acel.12571
15. Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, et al. Human myeloid-derived suppressor cells are associated with chronic immune suppression after severe sepsis/septic shock. Ann Surg (2017) 265(4):827–34. doi:10.1097/SLA.0000000000001783
16. Kang JW, Kim SJ, Cho HI, Lee SM. DAMPs activating innate immune responses in sepsis. Ageing Res Rev (2015) 24(Pt A):54–65. doi:10.1016/j.arr.2015.03.003
17. Kapetanovic R, Bokil NJ, Sweet MJ. Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: a ticking time bomb in ageing. Ageing Res Rev (2015) 24(Pt A):40–53. doi:10.1016/j.arr.2015.02.005
18. Feldman N, Rotter-Maskowitz A, Okun E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res Rev (2015) 24(Pt A):29–39. doi:10.1016/j.arr.2015.01.003
19. Poliezhaieva T, Ermolaeva MA. DNA damage in protective and adverse inflammatory responses: friend of foe? Mech Ageing Dev (2017) 165(Pt A):47–53. doi:10.1016/j.mad.2016.06.004
20. Bandaranayake T, Shaw AC. Host resistance and immune aging. Clin Geriatr Med (2016) 32(3):415–32. doi:10.1016/j.cger.2016.02.007
21. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
22. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi:10.1038/ni.1980
23. Jylhava J, Jylha M, Lehtimaki T, Hervonen A, Hurme M. Circulating cell-free DNA is associated with mortality and inflammatory markers in nonagenarians: the Vitality 90+ Study. Exp Gerontol (2012) 47(5):372–8. doi:10.1016/j.exger.2012.02.011
24. Rhodes A, Cecconi M. Cell-free DNA and outcome in sepsis. Crit Care (2012) 16(6):170. doi:10.1186/cc11508
25. Hamaguchi S, Akeda Y, Yamamoto N, Seki M, Yamamoto K, Oishi K, et al. Origin of circulating free DNA in sepsis: analysis of the CLP mouse model. Mediators Inflamm (2015) 2015:614518. doi:10.1155/2015/614518
26. Gasser S, Zhang WY, Tan NY, Tripathi S, Suter MA, Chew ZH, et al. Sensing of dangerous DNA. Mech Ageing Dev (2016) 165(Pt A):33–46. doi:10.1016/j.mad.2016.09.001
27. Magna M, Pisetsky DS. The alarmin properties of DNA and DNA-associated nuclear proteins. Clin Ther (2016) 38(5):1029–41. doi:10.1016/j.clinthera.2016.02.029
28. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature (2013) 498(7454):380–4. doi:10.1038/nature12306
29. Xiao TS, Fitzgerald KA. The cGAS-STING pathway for DNA sensing. Mol Cell (2013) 51(2):135–9. doi:10.1016/j.molcel.2013.07.004
30. Liu L, Yang M, Kang R, Dai Y, Yu Y, Gao F, et al. HMGB1-DNA complex-induced autophagy limits AIM2 inflammasome activation through RAGE. Biochem Biophys Res Commun (2014) 450(1):851–6. doi:10.1016/j.bbrc.2014.06.074
31. Nakad R, Schumacher B. DNA damage response and immune defense: links and mechanisms. Front Genet (2016) 7:147. doi:10.3389/fgene.2016.00147
32. Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell (2016) 61(5):654–66. doi:10.1016/j.molcel.2016.01.028
33. Hill S, Van Remmen H. Mitochondrial stress signaling in longevity: a new role for mitochondrial function in aging. Redox Biol (2014) 2:936–44. doi:10.1016/j.redox.2014.07.005
34. Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell (2011) 144(1):79–91. doi:10.1016/j.cell.2010.12.016
35. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18(5):488–98. doi:10.1038/ni.3704
36. Sancho D, Enamorado M, Garaude J. Innate immune function of mitochondrial metabolism. Front Immunol (2017) 8:527. doi:10.3389/fimmu.2017.00527
37. Bohovych I, Khalimonchuk O. Sending out an SOS: mitochondria as a signaling hub. Front Cell Dev Biol (2016) 4:109. doi:10.3389/fcell.2016.00109
38. Wendel M, Heller AR. Mitochondrial function and dysfunction in sepsis. Wien Med Wochenschr (2010) 160(5–6):118–23. doi:10.1007/s10354-010-0766-5
39. Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth (2011) 107(1):57–64. doi:10.1093/bja/aer093
40. Chakraborty M, Hickey AJ, Petrov MS, Macdonald JR, Thompson N, Newby L, et al. Mitochondrial dysfunction in peripheral blood mononuclear cells in early experimental and clinical acute pancreatitis. Pancreatology (2016) 16(5):739–47. doi:10.1016/j.pan.2016.06.659
41. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Landi F, et al. Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int J Mol Sci (2017) 18(5):ii:E933. doi:10.3390/ijms18050933
42. Suliman HB, Carraway MS, Piantadosi CA. Postlipopolysaccharide oxidative damage of mitochondrial DNA. Am J Respir Crit Care Med (2003) 167(4):570–9. doi:10.1164/rccm.200206-518OC
43. Kozlov AV, Lancaster JR Jr, Meszaros AT, Weidinger A. Mitochondria-meditated pathways of organ failure upon inflammation. Redox Biol (2017) 13:170–81. doi:10.1016/j.redox.2017.05.017
44. Ozawa T. Mitochondrial genome mutation in cell death and aging. J Bioenerg Biomembr (1999) 31(4):377–90. doi:10.1023/A:1005479920097
45. Van Remmen H, Richardson A. Oxidative damage to mitochondria and aging. Exp Gerontol (2001) 36(7):957–68. doi:10.1016/S0531-5565(01)00093-6
46. Lieber MR, Karanjawala ZE. Ageing, repetitive genomes and DNA damage. Nat Rev Mol Cell Biol (2004) 5(1):69–75. doi:10.1038/nrm1281
47. Patananan AN, Wu TH, Chiou PY, Teitell MA. Modifying the mitochondrial genome. Cell Metab (2016) 23(5):785–96. doi:10.1016/j.cmet.2016.04.004
48. Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol (2016) 17(5):308–21. doi:10.1038/nrm.2016.14
49. Lindahl T. The intrinsic fragility of DNA (Nobel Lecture). Angew Chem Int Ed Engl (2016) 55(30):8528–34. doi:10.1002/anie.201602159
50. Ermolaeva MA, Schumacher B. Systemic DNA damage responses: organismal adaptations to genome instability. Trends Genet (2014) 30(3):95–102. doi:10.1016/j.tig.2013.12.001
51. Ioannidou A, Goulielmaki E, Garinis GA. DNA damage: from chronic inflammation to age-related deterioration. Front Genet (2016) 7:187. doi:10.3389/fgene.2016.00187
52. Bauer NC, Corbett AH, Doetsch PW. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res (2015) 43(21):10083–101. doi:10.1093/nar/gkv1136
53. Gaillard H, Aguilera A. Transcription as a threat to genome integrity. Annu Rev Biochem (2016) 85:291–317. doi:10.1146/annurev-biochem-060815-014908
54. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen (2017) 58(5):235–63. doi:10.1002/em.22087
55. Oliveira NM, Rios EC, de Lima TM, Victorino VJ, Barbeiro H, Pinheiro da Silva F, et al. Sepsis induces telomere shortening: a potential mechanism responsible for delayed pathophysiological events in sepsis survivors? Mol Med (2016) 22:886–91. doi:10.2119/molmed.2016.00225
56. Zhang J, Rane G, Dai X, Shanmugam MK, Arfuso F, Samy RP, et al. Ageing and the telomere connection: an intimate relationship with inflammation. Ageing Res Rev (2016) 25:55–69. doi:10.1016/j.arr.2015.11.006
57. de Magalhães JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev (2017). doi:10.1016/j.mad.2017.07.001
58. Mota JM, Leite CA, Souza LE, Melo PH, Nascimento DC, de-Deus-Wagatsuma VM, et al. Post-sepsis state induces tumor-associated macrophage accumulation through CXCR4/CXCL12 and favors tumor progression in mice. Cancer Immunol Res (2016) 4(4):312–22. doi:10.1158/2326-6066.CIR-15-0170
59. Shankar-Hari M, Ambler M, Mahalingasivam V, Jones A, Rowan K, Rubenfeld GD. Evidence for a causal link between sepsis and long-term mortality: a systematic review of epidemiologic studies. Crit Care (2016) 20:101. doi:10.1186/s13054-016-1276-7
60. Shankar-Hari M, Rubenfeld GD. Understanding long-term outcomes following sepsis: implications and challenges. Curr Infect Dis Rep (2016) 18(11):37. doi:10.1007/s11908-016-0544-7
61. Forman HJ. Redox signaling: an evolution from free radicals to aging. Free Radic Biol Med (2016) 97:398–407. doi:10.1016/j.freeradbiomed.2016.07.003
62. Wang X, Hai C. Novel insights into redox system and the mechanism of redox regulation. Mol Biol Rep (2016) 43(7):607–28. doi:10.1007/s11033-016-4022-y
63. Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem (2017) 86:715. doi:10.1146/annurev-biochem-061516-045037
64. Adams L, Franco MC, Estevez AG. Reactive nitrogen species in cellular signaling. Exp Biol Med (Maywood) (2015) 240(6):711–7. doi:10.1177/1535370215581314
65. Vieira da Silva Pellegrina D, Severino P, Vieira Barbeiro H, Maziero Andreghetto F, Tadeu Velasco I, Possolo, et al. Septic shock in advanced age: transcriptome analysis reveals altered molecular signatures in neutrophil granulocytes. PLoS One (2015) 10(6):e0128341. doi:10.1371/journal.pone.0128341
66. Booth LN, Brunet A. The aging epigenome. Mol Cell (2016) 62(5):728–44. doi:10.1016/j.molcel.2016.05.013
67. Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol (2015) 16(10):593–610. doi:10.1038/nrm4048
68. Weiterer S, Uhle F, Lichtenstern C, Siegler BH, Bhuju S, Jarek M, et al. Sepsis induces specific changes in histone modification patterns in human monocytes. PLoS One (2015) 10(3):e0121748. doi:10.1371/journal.pone.0121748
69. Collins PE, Carmody RJ. The regulation of endotoxin tolerance and its impact on macrophage activation. Crit Rev Immunol (2015) 35(4):293–323. doi:10.1615/CritRevImmunol.2015015495
70. Dabin J, Fortuny A, Polo SE. Epigenome maintenance in response to DNA damage. Mol Cell (2016) 62(5):712–27. doi:10.1016/j.molcel.2016.04.006
71. López-Leon M, Goya RG. The emerging view of aging as a reversible epigenetic process. Gerontology (2017) 63(5):426–31. doi:10.1159/000477209
72. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell (2016) 166(4):822–39. doi:10.1016/j.cell.2016.07.050
73. Pal S, Tyler JK. Epigenetics and aging. Sci Adv (2016) 2(7):e1600584. doi:10.1126/sciadv.1600584
74. Flynn RA, Chang HY. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell (2014) 14(6):752–61. doi:10.1016/j.stem.2014.05.014
75. Kim DH, Marinov GK, Pepke S, Singer ZS, He P, Williams B, et al. Single-cell transcriptome analysis reveals dynamic changes in lncRNA expression during reprogramming. Cell Stem Cell (2015) 16(1):88–101. doi:10.1016/j.stem.2014.11.005
76. Victoria B, Nunez Lopez YO, Masternak MM. MicroRNAs and the metabolic hallmarks of aging. Mol Cell Endocrinol (2017) 455:131–47. doi:10.1016/j.mce.2016.12.021
77. Clifford KM, Dy-Boarman EA, Haase KK, Maxvill K, Pass SE, Alvarez CA. Challenges with diagnosing and managing sepsis in older adults. Expert Rev Anti Infect Ther (2016) 14(2):231–41. doi:10.1586/14787210.2016.1135052
78. Liang SY. Sepsis and other infectious disease emergencies in the elderly. Emerg Med Clin North Am (2016) 34(3):501–22. doi:10.1016/j.emc.2016.04.005
79. Iwashyna TJ, Cooke CR, Wunsch H, Kahn JM. Population burden of long-term survivorship after severe sepsis in older Americans. J Am Geriatr Soc (2012) 60(6):1070–7. doi:10.1111/j.1532-5415.2012.03989.x
80. Kale SS, Yende S. Effects of aging on inflammation and hemostasis through the continuum of critical illness. Aging Dis (2011) 2(6):501–11.
81. Pinheiro da Silva F, Zampieri FG, Barbeiro DF, Barbeiro HV, Goulart AC, Torggler Filho F, et al. Septic shock in older people: a prospective cohort study. Immun Ageing (2013) 10(1):21. doi:10.1186/1742-4933-10-21
82. Cunha DM, Koike MK, Barbeiro DF, Barbeiro HV, Hamasaki MY, Coelho Neto GT, et al. Increased intestinal production of alpha-defensins in aged rats with acute pancreatic injury. Exp Gerontol (2014) 60:215–9. doi:10.1016/j.exger.2014.11.008
83. Barbeiro DF, Koike MK, Coelho AM, da Silva FP, Machado MC. Intestinal barrier dysfunction and increased COX-2 gene expression in the gut of elderly rats with acute pancreatitis. Pancreatology (2016) 16(1):52–6. doi:10.1016/j.pan.2015.10.012
84. Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med (1996) 24(7):1125–8. doi:10.1097/00003246-199607000-00010
85. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med (2003) 348(2):138–50. doi:10.1056/NEJMra021333
86. Tang BM, Huang SJ, McLean AS. Genome-wide transcription profiling of human sepsis: a systematic review. Crit Care (2010) 14(6):R237. doi:10.1186/cc9392
87. Wong HR. Genome-wide expression profiling in pediatric septic shock. Pediatr Res (2013) 73(4 Pt 2):564–9. doi:10.1038/pr.2013.11
88. Maslove DM, Wong HR. Gene expression profiling in sepsis: timing, tissue, and translational considerations. Trends Mol Med (2014) 20(4):204–13. doi:10.1016/j.molmed.2014.01.006
89. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med (2011) 208(13):2581–90. doi:10.1084/jem.20111354
90. Severino P, Silva E, Baggio-Zappia GL, Brunialti MK, Nucci LA, Junior OR, et al. Gene expression profiling of mononuclear cells from patients with sepsis secondary to community-acquired pneumonia. Genom Data (2014) 2:332–4. doi:10.1016/j.gdata.2014.10.004
91. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol (2014) 14(3):141–53. doi:10.1038/nri3608
92. Man AL, Gicheva N, Nicoletti C. The impact of ageing on the intestinal epithelial barrier and immune system. Cell Immunol (2014) 289(1–2):112–8. doi:10.1016/j.cellimm.2014.04.001
93. Lustgarten MS. Classifying aging as a disease: the role of microbes. Front Genet (2016) 7:212. doi:10.3389/fgene.2016.00212
94. Qian F, Wang X, Zhang L, Chen S, Piecychna M, Allore H, et al. Age-associated elevation in TLR5 leads to increased inflammatory responses in the elderly. Aging Cell (2012) 11(1):104–10. doi:10.1111/j.1474-9726.2011.00759.x
95. Ghosh S, Lertwattanarak R, Garduno Jde J, Galeana JJ, Li J, Zamarripa F, et al. Elevated muscle TLR4 expression and metabolic endotoxemia in human aging. J Gerontol A Biol Sci Med Sci (2015) 70(2):232–46. doi:10.1093/gerona/glu067
96. Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4586–91. doi:10.1073/pnas.1000097107
97. Woodmansey EJ, McMurdo ME, Macfarlane GT, Macfarlane S. Comparison of compositions and metabolic activities of fecal microbiotas in young adults and in antibiotic-treated and non-antibiotic-treated elderly subjects. Appl Environ Microbiol (2004) 70(10):6113–22. doi:10.1128/AEM.70.10.6113-6122.2004
98. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nature (2015) 525(7570):528–32. doi:10.1038/nature15367
99. Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe (2017) 21(4):455–466e454. doi:10.1016/j.chom.2017.03.002
100. Nacionales DC, Szpila B, Ungaro R, Lopez MC, Zhang J, Gentile LF, et al. A detailed characterization of the dysfunctional immunity and abnormal myelopoiesis induced by severe shock and trauma in the aged. J Immunol (2015) 195(5):2396–407. doi:10.4049/jimmunol.1500984
101. Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, et al. Population-level analysis of gut microbiome variation. Science (2016) 352(6285):560–4. doi:10.1126/science.aad3503
102. Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science (2016) 352(6285):565–9. doi:10.1126/science.aad3369
103. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol (2017) 17(4):219–32. doi:10.1038/nri.2017.7
104. Haak BW, Wiersinga WJ. The role of the gut microbiota in sepsis. Lancet Gastroenterol Hepatol (2017) 2(2):135–43. doi:10.1016/S2468-1253(16)30119-4
105. Dickson RP. The microbiome and critical illness. Lancet Respir Med (2016) 4(1):59–72. doi:10.1016/S2213-2600(15)00427-0
106. McDonald D, Ackermann G, Khailova L, Baird C, Heyland D, Kozar R, et al. Extreme dysbiosis of the microbiome in critical illness. mSphere (2016) 1(4):e199–196. doi:10.1128/mSphere.00199-16
107. Pinheiro da Silva F, Cesar Machado MC. Personalized medicine for sepsis. Am J Med Sci (2015) 350(5):409–13. doi:10.1097/MAJ.0000000000000558
108. Sebastiani P, Thyagarajan B, Sun F, Schupf N, Newman AB, Montano M, et al. Biomarker signatures of aging. Aging Cell (2017) 16(2):329–38. doi:10.1111/acel.12557
109. Srivastava D, DeWitt N. In vivo cellular reprogramming: the next generation. Cell (2016) 166(6):1386–96. doi:10.1016/j.cell.2016.08.055
110. von Joest M, Bua Aguin S, Li H. Genomic stability during cellular reprogramming: mission impossible? Mutat Res (2016) 788:12–6. doi:10.1016/j.mrfmmm.2016.01.001
111. Marion RM, Blasco MA. Long live partial reprogramming. Circ Res (2017) 120(9):1381–3. doi:10.1161/CIRCRESAHA.117.310594
Keywords: aging, systemic inflammation, immunity, sepsis, critical care
Citation: Pinheiro da Silva F and Machado MCC (2017) Septic Shock and the Aging Process: A Molecular Comparison. Front. Immunol. 8:1389. doi: 10.3389/fimmu.2017.01389
Received: 30 August 2017; Accepted: 09 October 2017;
Published: 25 October 2017
Edited by:
Valquiria Bueno, Federal University of São Paulo, BrazilReviewed by:
Scott Brakenridge, University of Florida, United StatesSinisa Savic, University of Leeds, United Kingdom
Copyright: © 2017 Pinheiro da Silva and Machado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabiano Pinheiro da Silva, cGluaGVpcm9mYWJpYW5vQGhvdG1haWwuY29t