 David R. Withers
David R. Withers Matthew R. Hepworth
Matthew R. Hepworth
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 October 2017
Sec. NK and Innate Lymphoid Cell Biology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01298
This article is part of the Research Topic Differentiation and Mechanisms of Activation of Innate Lymphoid Cells View all 11 articles
The maintenance of mammalian health requires the generation of appropriate immune responses against a broad range of environmental and microbial challenges, which are continually encountered at barrier tissue sites including the skin, lung, and gastrointestinal tract. Dysregulated barrier immune responses result in inflammation, both locally and systemically in peripheral organs. Group 3 innate lymphoid cells (ILC3) are constitutively present at barrier sites and appear to be highly specialized in their ability to sense a range of environmental and host-derived signals. Under homeostatic conditions, ILC3 respond to local cues to maintain tissue homeostasis and restrict inflammatory responses. In contrast, perturbations in the tissue microenvironment resulting from disease, infection, or tissue damage can drive dysregulated pro-inflammatory ILC3 responses and contribute to immunopathology. The tone of the ILC3 response is dictated by a balance of “exogenous” signals, such as dietary metabolites and commensal microbes, and “endogenous” host-derived signals from stromal cells, immune cells, and the nervous system. ILC3 must therefore have the capacity to simultaneously integrate a wide array of complex and dynamic inputs in order to regulate barrier function and tissue health. In this review, we discuss the concept of ILC3 as a “communications hub” in the intestinal tract and associated lymphoid tissues and address the variety of signals, derived from multiple biological systems, which are interpreted by ILC3 to modulate the release of downstream effector molecules and regulate cell–cell crosstalk. Successful integration of environmental cues by ILC3 and downstream propagation to the broader immune system is required to maintain a tolerogenic and anti-inflammatory tone and reinforce barrier function, whereas dysregulation of ILC3 responses can contribute to the onset or progression of clinically relevant chronic inflammatory diseases.
Innate lymphoid cells (ILCs) constitute a family of tissue-resident innate lymphocytes with increasingly appreciated roles in tissue homeostasis, immunity, and inflammation (1–5). A rapidly developing body of evidence derived from mouse and human studies has begun to demonstrate how ILCs play critical, non-redundant roles in maintaining tissue health or in driving disease pathology. ILCs possess several characteristics that make them particularly suited to rapidly respond to perturbations in tissue homeostasis, infection, or tissue damage including (i) constitutive presence in barrier tissues and lymphoid organs, (ii) a “poised” transcriptional and epigenetic landscape, and (iii) the ability to respond rapidly and robustly to signals in the tissue microenvironment.
As reviewed extensively elsewhere (1–5), ILCs can be subdivided into groups on the basis of transcription factor expression and cytokine secretion profile. In this review, we will focus on group 3 innate lymphoid cells (ILC3), which are characterized by the expression of the transcription factor retinoic acid (RA)-related orphan receptor γ isoform t (RORγt) and the capacity to produce the cytokines interleukin (IL)-17A, IL-17F, IL-22, and GM-CSF. ILC3 differ from other ILC groups in that they constitute at least two bona fide subsets that are transcriptionally, developmentally, and functionally distinct and inhabit distinct tissue microenvironments [reviewed in Ref. (6)]. In mice, these subsets are distinguished by surface expression of natural cytotoxity receptors (NCR), particularly NKp46 (termed NCR+ ILC3), and molecules and receptors historically associated with fetal lymphoid tissue inducer (LTi) cells, such as lymphotoxin and the chemokine receptor CCR6 (termed LTi-like ILC3) (5, 6). ILC3 are found in a range of tissues and organs, most notably the gastrointestinal tract and associated lymphoid tissues, although the relative distribution of ILC3 subsets differs dependent upon tissue location. Indeed, while NCR+ ILC3 are the most prevalent ILC3 subset in the small intestine, LTi-like ILC3 appear to dominate in the colon and lymphoid tissues. Importantly, emerging evidence suggests that these two subsets also play distinct functional roles that relate to tissue-specific biological challenges.
Tissue-resident ILC3 sense and respond to a wide range of environmental and host-derived signals within the local microenvironment and integrate these cues to modulate cell-intrinsic transcription and to relay information to other cells, either through the production of cytokines or through cell–cell interactions. Indeed, as discussed below, ILC3 concurrently sense a multitude of soluble signals and environmental cues, which may change dynamically following infection or tissue damage, thus posing the question as to how ILC3 integrate and interpret these signals to respond appropriately. The balance of ILC3 effector functions may set the immunological tone of the tissue and help orchestrate the wider immune response. Although other ILC subsets also share the capacity to respond to a wide range of cues within the tissue microenvironment [reviewed in detail elsewhere—(4, 5, 7)], here, we will discuss the concept of ILC3 as tissue-resident sentinels and key “communications hubs” of the intestinal immune response.
ILC3 are derived from fetal liver progenitors during embryogenesis and are among the first lymphocytes to seed barrier tissues, in particular the intestinal tract, prior to birth (8, 9). In this context, ILC3 are among the first-responders to colonization by commensal microbes, as well as diet-derived antigens introduced following weaning. Furthermore, these cells are central organizers of secondary lymphoid tissue organogenesis (10). ILC3 are therefore in prime position to shape the emerging mucosal immune system. Developmentally both ILC3 subsets derive from a common lymphoid progenitor ancestor; however, recent evidence has highlighted a divergent developmental relationship between ILC3 subsets and other ILC family members. The development of the wider ILC family is mediated through a series of transcriptional decisions that generate distinct progenitor populations with increasing commitment toward the ILC lineage, as reviewed extensively elsewhere (3, 11, 12). A key stage in this development is the bifurcation of the “cytotoxic” ILC lineage (i.e., classical NK cells), from the remaining “helper” ILC lineage (ILC1, ILC2, and ILC3). This is characterized by the development of a common helper ILC progenitor (CHILP) that expresses high levels of the transcription factor ID2 and IL-7R and which is able to generate ILC1, ILC2, and both ILC3 subsets but has lost the potential to generate progeny from other lymphocyte lineages (13). Additionally, a subset of ID2+ progenitors develops downstream of the CHILP, characterized by the expression of the transcription factor PLZF and surface PD-1—termed the common ILC precursor (ILCp) (14, 15). However, while ILCp can give rise to ILC1, ILC2, and NCR+ ILC3, they are unable to generate LTi-like ILC3 progeny (14), suggesting that a bifurcation of ILC3 subset development occurs during the progression from CHILP to ILCp. In line with these findings, LTi-like ILC3 have been demonstrated to derive from an ID2+ α4β7+ CXCR5+ progenitor cell that diverges upstream of ILCp, prior to the acquisition of PLZF (16–18). As such LTi-like ILC3 develop via a distinct route from the remaining ILC family members, including NCR+ ILC3, which may underlie key transcriptional and functional differences recently highlighted between ILC3 subsets (6, 19–21). Moreover, recent studies have uncovered plasticity among ILC3 and other ILC populations that is dictated by changes in the cytokine milieu within the tissue microenvironment (22–24). Thus, in addition to transcriptional decisions made during development, plasticity of mature ILC3 may shape the composition of these cells in tissues.
Seeding of intestinal tissues and associated lymphoid structures by ILC3 occurs during embryogenesis and is further regulated by environmental signals encountered following birth. Seminal studies demonstrated LTi-like ILC3 are present in the fetal gut, whereas NKp46+ ILC3 are largely absent but proliferate rapidly following birth to become the dominant ILC3 subset in the small intestine (8). ILC3 subset maturation and migration to the gut is in part dictated by maternal, microbial and dietary signals (Figure 1; Inputs). The extent to which ILC3 can directly sense microbial-derived cues remains poorly understood. Indeed, unlike many myeloid cell populations, ILC3 do not appear to express toll-like receptors or other canonical pattern recognition receptors. Rather ILC3 responses to microbial cues are dependent upon other key intermediaries, such as resident mononuclear phagocyte (MNP) populations that convey information to ILC3 (detailed below). ILC3 are particularly sensitive to alterations in the microbiota and their numbers are modulated following neonatal colonization by commensal microbes, in part through an IL-25-dependent negative feedback mechanism that restricts expansion of ILC3 in a microbiota-dependent manner (9). Recent studies suggest that ILC3 are directly regulated by bacterial metabolites, such as short-chain fatty acids (SCFAs) produced through microbial metabolism of dietary fiber. Consistent with this, NCR+ ILC3 present in the Peyer’s patches (PP) of the ileum were found to express Gpr109a—the receptor for butyrate—and SCFA signaling acted to restrict NCR+ ILC3 numbers and cytokine production (25). Additionally, NCR+ ILC3 are dependent upon the aryl hydrocarbon receptor (Ahr) for their development and persistence in the small intestine (26–29). Ahr ligands can be derived from intestinal bacteria, as well as dietary and endogenous sources and further regulate cytokine production of mature adult ILC3, while immunoglobulin-bound Ahr ligands are also transferred from the mother to promote the migration of NCR+ ILC3 to the neonatal small intestine (30).
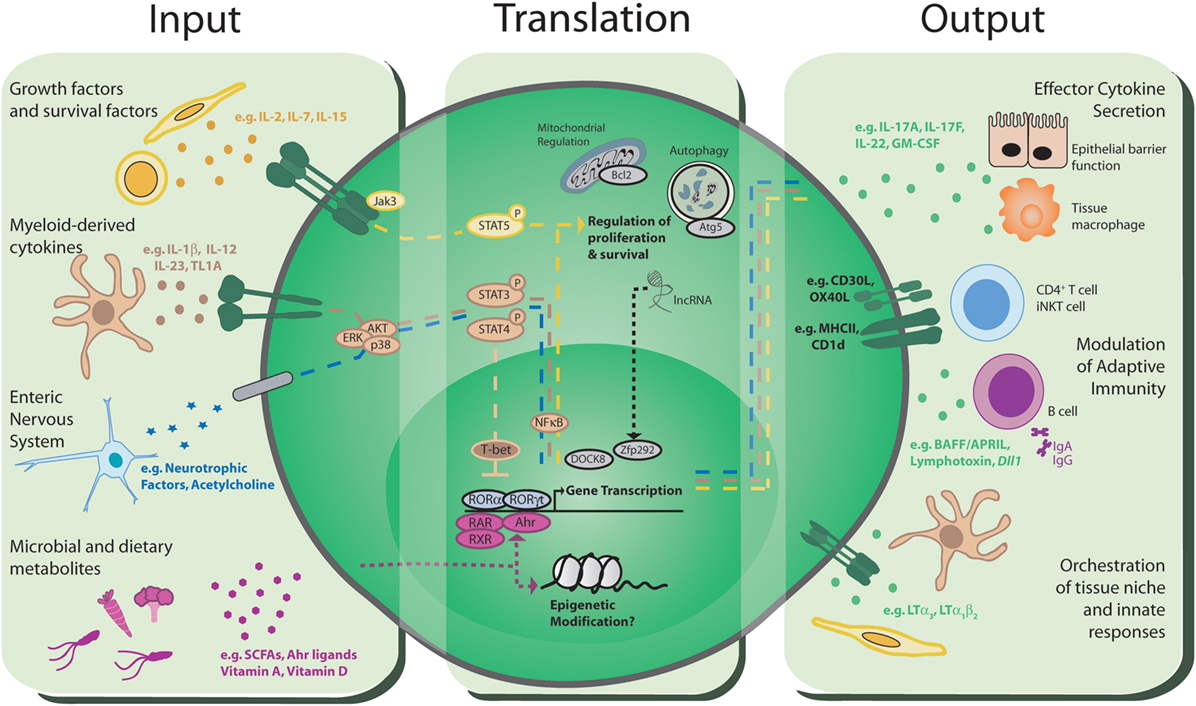
Figure 1. Group 3 innate lymphoid cells (ILC3): communications hubs of the intestinal immune system. Input: ILC3 have the capacity to receive environmental cues and host-derived signals from a range of sources including the immune system, stroma, the enteric nervous system, the microbiota, and the diet. Translation: these signals are integrated through a range of intracellular signaling mechanisms, including phosphorylation of STAT proteins, ligand-binding transcription factors, epigenetic modification, and multiple intracellular processes with the potential to support long-term survival of ILC3. Integration of multiple signals likely happens simultaneously, indicating a complicated and dynamic process that determines the threshold, magnitude, and type of ILC3 responses that are propagated to communicate with other cells in the intestinal environment. Output: translated signals are conveyed to other cells within the local environment through a variety of distinct mechanisms, most notably via the production of effector cytokines and growth factors that modulate and orchestrate the wider immune response and intestinal barrier function. The nature and balance of the signals perceived by ILC3 ultimately determine the downstream effector response, and dysregulation of homeostatic signals in disease may dramatically alter ILC3 responses and drive pro-inflammatory phenotypes. Finally, direct cell–cell communication through antigen presentation and co-stimulatory molecules can directly regulate the adaptive immune response.
In addition to microbial metabolites, ILC3 are subjected to further direct modulation by dietary cues. Maternal retinoids transferred during embryogenesis favor the development and maturation of LTi-like ILC3 by promoting and stabilizing RORγt expression (31), while dietary vitamin A-derived RA is required for the maintenance of adult small intestinal ILC3 subsets (32–34). In support of a role for dietary vitamins in the regulation of ILC3 responses, human ILC3 were found to up-regulate vitamin D receptor expression following cytokine stimulation and vitamin D treatment subsequently resulted in suppression of the IL-23R, implicating vitamin D as a regulator of ILC3 function (35). In line with this vitamin D receptor-deficient mice exhibit elevated ILC3 numbers and IL-22 production (36). Taken together, these studies implicate dietary vitamins as key modulators of ILC3 numbers and function and demonstrate the capacity of ILC3 to directly sense dietary signals.
In addition to environmental cues derived from the microbiota and diet, the numbers and relative balance of ILC3 subsets at steady state are additionally regulated by host-derived inputs (Figure 1; Inputs). In contrast to the dynamic changes observed within the neonatal intestine, adult ILC3 populations appear to be stable and long-lived with little to no replenishment from the bone marrow under homeostatic conditions (37). Current understanding suggests that ILC3 are maintained through self-renewal and/or replenished from tissue-resident precursors and several groups have reported the presence of tissue-resident RORγt+ progenitors that can give rise to mature ILC3 subsets in both mice and humans (38, 39). In particular, intestinal resident NKp46−CCR6−RORγt+ ILC3 precursors give rise to NCR+ ILC3, and to a lesser extent the LTi-like ILC3 compartment, a process that is regulated by local tissue cytokines and Notch signaling (40–44). Adult ILC3 subsets differ in their tissue localization; CCR6+ ILC3 are largely restricted to lymphoid structures such as the mesenteric lymph node and cryptopatches and ILFs within the small intestine and colon (45, 46), while NCR+ ILC3 are largely excluded from lymph nodes and colon but reside within both the lamina propria and ILFs of the small intestine, with localization of NCR+ ILC3 dictated in part through CXCR6 expression (47–51). Thus, local signals perceived by ILC3 subsets likely dictate their maintenance and survival within specific tissue microenvironments.
While the cell-extrinsic and cell-intrinsic signals that support ILC3 under homeostatic conditions remain incompletely defined, recent evidence suggests that ILC3 subsets are differentially dependent on constitutive signals received via pro-survival cytokines for their maintenance. ILC3 constitutively express high levels of common γ chain cytokine family receptors including IL-2R, IL-7R, and IL-15R. Of these, the IL-7:IL-7R interaction has been most extensively studied and IL-7 deficient mice have reduced numbers of lymph nodes, consistent with a loss of LTi cells in the embryo, and decreased ILC3 numbers in the adult (40, 52–54). Furthermore, enhanced signaling through IL-7Rα increases LTi-like cell numbers in the adult (53), arguing that IL-7 has the capacity to directly regulate ILC3 numbers. Notably overexpression of TSLP, which signals through a heterodimer of TSLPR and IL-7R, could overcome the effect of IL-7 deficiency, further arguing that other signals can compensate (55). A more recent study demonstrated that residual numbers of all ILC groups persist in the absence of IL-7, with IL-15 necessary to support the survival of the remaining NCR+ ILC3, but not LTi-like ILC3, in the intestinal tract (56). Provision of survival signals such as IL-7, IL-15, and TSLP is mediated largely through critical interactions with non-hematopoietic, stromal cell populations (Figure 1; Inputs). Studies of IL-7 reporter mice identified marginal reticular cells as a key source of this cytokine in lymph nodes (57), although analysis of IL-7 mRNA indicates that fibroblastic reticular cells (FRCs) also produce IL-7 (58). Strikingly, production of IL-7 by stromal cells requires signaling through the lymphotoxin β-receptor (59), indicating that continued interactions between stroma and lymphotoxin expressing immune cells support normal FRC function, which in turn maintains ILC3. Similarly, FRCs in lymph nodes and PP are a key source of IL-15, which supports NCR+ ILC3 as well as NK cells and ILC1 (56, 60). Thus, the stromal infrastructure of tissue microenvironments provides much of the survival requirements of ILC3. However, there is evidence that some LTi-like ILC3 persist in the absence of both of IL-7 and IL-15 (56), arguing for a role for other cytokine signals or indicating that alternative mechanisms of survival exist. Given that LTi-like ILC3 also highly express CD25 (IL-2R) and are enriched in lymphoid structures in close proximity to proliferating T cells (61, 62), it is possible that IL-2 may play an additional role in the long-term maintenance of LTi-like ILC3. Together these findings suggest that stromal cells form critical niches within secondary and tertiary lymphoid tissues that provide signals required to support ILC3 survival.
As addressed earlier, ILC3 have the capacity to receive input from cells and soluble molecules within their local tissue microenvironment (Figure 1; Inputs). In addition to stromal cells, other innate immune cells, such as MNPs, play key roles in both homeostatic and effector ILC3 responses (63, 64). Within the intestine it is clear that IL-23, produced by CX3CR1+ MNPs, is a key regulator of ILC3 function (65–67). CX3CR1+ MNPs were observed to cluster with ILC3 in distinct intestinal lymphoid tissues, such as cryptopatches. Following weaning sensing of the microbiota by intestinal lymphoid tissue, MNPs result in local production of IL-23 and the induction of IL-22 from lymphoid tissue-resident LTi-like ILC3 (46). Moreover, depletion of CX3CR1+ MNPs results in a failure to control Citrobacter rodentium infection due to impaired IL-22 production by ILC3. In addition to IL-23, IL-1β drives IL-22 production by ILC3 and this is further augmented by TL1A, again produced by CX3CR1+ MNPs (67). The balance of cytokine signals perceived by ILC3 is also critical in determining phenotype and function. Indeed, intestinal ILC3 exhibit significant plasticity and signals including IL-12, IL-15, and IL-18 released in the context of infection and inflammation promote the progressive up-regulation of T-bet and production of pro-inflammatory cytokines such as IFN-γ and TNF-α by NCR+ ILC3 and a subsequent loss of RORγt expression by this subset, resulting in these cells being labeled “ex-ILC3” (40, 41). Conversely, upon resolution of infection or inflammation, restoration of homeostatic levels of MNP-derived signals including IL-23 and IL-1β favors the reconversion of inflammatory “ex-ILC3” back to an RORγt+ NCR+ ILC3 phenotype (43, 68).
ILC3 may also respond to other fundamental innate immune factors, such as the complement system. A subset of ILC3 appear to be sensitive to the complement cascade via expression of the C3aR (69), while Complement Factor P—a positive regulator of the alternative complement pathway—was found to directly bind NKp46 (70), suggesting NCR+ ILC3 in particular may sense pathogen infection via interactions with the complement cascade. Beyond interactions with the immune system, an increasing body of evidence indicates intestinal resident ILC3 can directly sense cues from the enteric nervous system. In a series of studies, LTi-like ILC3 were demonstrated to express RET—a receptor for neurotrophic factors (71, 72). RET expression is required for PP formation and accumulation of lymphotoxin-producing LTi cells, although this occurs indirectly via ligation of RET on lymphoid tissue-initiator cells and induction of chemokine production to sequester immune cells, including LTi cells, to drive PP formation. Nonetheless, adult CCR6+ LTi-like ILC3 express RET and can directly respond to glial-derived neurotrophic factor (GDNF) family ligands. Production of GDNF by intestinal glial cells in response to stimulation by microbial ligands acts to reinforce intestinal barrier function via regulation of IL-22 transcription and secretion by LTi-like ILC3 (72). Interestingly, enteric neurons also have the capacity to provide RA signals, which are critical for the maturation of LTi cells (73), thus suggesting that alternative, non-dietary sources of RA may also play roles in ILC function and immune homeostasis. In addition to local neuronal signals, systemic nervous signals may modulate the tone and magnitude of ILC3 responses. Vagal nerve innervation of the colon is required for the formation of tertiary lymphoid structures, via regulation of local chemokine production (74). Although a direct effect of vagal denervation on ILC3 was not demonstrated in this study, more recent evidence suggests that the vagal nerve acts to regulate ILC3 responses to bacterial pathogens in the peritoneal cavity (75). This effect was in part dependent upon the ability of NCR+ ILC3 to enzymatically generate lipid precursors that were in turn metabolized by resident macrophages to promote resolution of inflammation (75). Together these findings suggest signals from both tissue-resident and systemic neurons may directly regulate ILC3 numbers and function during homeostasis or following infection. Interestingly, ILC3 may also play important roles in the central nervous system and a recent study demonstrated that NCR+ ILC3 are present in the meninges and promote neuroinflammation in a model of multple sclerosis by licensing entry of inflammatory Th17 cells into the brain (76). It is highly likely that other signals from the nervous system and beyond may impact upon ILC3 function. For example, host-derived lipid mediators—such as prostaglandin E2—have been demonstrated to directly promote homeostatic ILC3 cytokine production (77). Similarly, endocrine signals, particularly sex hormones, are long appreciated to regulate innate immunity. In this regard testosterone directly inhibits ILC2 responses (78); however, the capacity of androgens, estrogens, or other hormones to modulate ILC3 is yet to be investigated.
The ability of tissue ILC3 to sense a breadth of exogenous and endogenous cues of significantly varying natures provokes the question as to how these various inputs are simultaneously integrated, and prioritized, in order to control ILC3 function. While significant advances have been made in understanding the effector functions of ILC3, as well as the transcriptional decisions that determine ILC development, relatively little is known about the signaling pathways and transcriptional dynamics that act to integrate external cues and determine the nature and magnitude of downstream ILC3 responses. Advances in RNA sequencing and epigenetic profiling have begun to redress this balance and allowed for enhanced resolution of individual cellular states among ILC populations (18–21, 79, 80). Furthermore, comparison of the transcriptional networks of ILC3 subsets and their Th17 counterparts will likely prove useful in identifying common and distinct signaling pathways utilized by RORγt-expressing cells (81). While significant overlap may be expected in this case, it is critical to note that ILC3 and Th17 cells also demonstrate key difference in their transcriptional regulation. For example, while Th17 is acutely dependent on RORγt for the maintenance of a mature functional phenotype, ILC3 were able to maintain core effector functions and phenotype following deletion of RORγt (82). Surprisingly, human IL-22-producing ILC3 can be generated from circulating ILCps even when derived from Rorc-deficient donors (83). Thus, it is likely that ILC3 also exhibit unique and differing signaling transduction pathways and transcriptional regulation that underlie their innate functions (Figure 1; Translation). As detailed earlier, ILC3 transcription dynamics are acutely modified by dietary and bacterial metabolites via Ahr and it is likely epigenetic changes may be imprinted via histone deacetylases downstream of SCFA-sensing, as in other lymphocytes (84).
ILC3 additionally integrate multiple soluble signals including common gamma chain cytokines and growth factors—many of which induce signal transduction via phosphorylation of STAT5 (85). Thus, pSTAT5 and downstream signal activation (ERK/AKT) are likely to play key roles in orchestrating intracellular responses in ILC3. Similarly, both IL-23 and RET signals are transduced in part by pSTAT3 to regulate IL-22 production (72, 86). Thus, it is likely that a threshold of intracellular signaling downstream of multiple receptors, sensing cues from multiple biological systems, acts to establish the tone and magnitude of the ILC3 response (Figure 1; Translation). The signaling pathway engaged following stimulation by cytokines or other cues is likely to determine the biological processes that are modulated. For example, ILC3 from mice with mutations in Jak signaling exhibited an impaired ability to phosphorylate STAT5 in response to IL-7, while inhibiting Jak3 signaling in mature human ILC3 suppressed proliferation of these cells but not cytokine production (87).
Maintenance of ILC3 subsets is likely to be regulated transcriptionally via multiple mechanisms (Figure 1; Translation). Mice lacking the scaffolding protein dedicator of cytokinesis 8 (DOCK8) demonstrate reduced ILC3 numbers and defective immunity to C. rodentium, in part due to a reduced ability of DOCK8-deficient ILC3 to respond to IL-7 and IL-23 and an increased rate of apoptosis (88). Similarly, survival of ILC3 is modulated via long non-coding RNAs that orchestrate downstream gene accessibility (89). In particular, lncKdm2b expression by ILC3 controls activation of transcription factors, including Zfp292, and recruitment of chromatin organizational machinery that promote ILC3 maintenance in vivo (89). ILC3 survival may also be dependent upon expression of anti-apoptotic machinery. Indeed, LTi-like ILC3 highly express the anti-apoptotic molecule Bcl-2, expression of which fluctuates following perturbation of local cytokine signals such as IL-7 (56). In addition, regulation of intracellular organelle degradation through autophagy has been implicated in ILC survival (90). The autophagy related factor Atg5 was found to be required for ILC family development, suggesting that autophagy may play a role in ILC3 persistence and maintenance (90). Nonetheless, and despite these advances, many of the precise mechanisms through which ILC3 maintenance is instructed via changes in gene expression or epigenetics and the environmental signals that are required to promote ILC3 survival are yet to be defined.
ILC3 possess multiple mechanisms through which they relay host and environmental signals to orchestrate the intestinal immune response and to maintain tissue function (Figure 1; Outputs). ILC3 were initially identified as potent sources of effector cytokines, most notably IL-22. ILC3-derived IL-22 plays critical roles in regulating host-commensal bacteria interactions in the healthy intestine in addition to mediating responses to enteric pathogen infections (27, 86, 91–98). IL-22 mediates its effects on IL-22R-expressing non-hematopoietic cells, including intestinal epithelial cells, epithelia-associated stem cells, and Paneth cells (99–101), and promotes barrier function and segregation of commensal bacteria from the underlying immune system via induction of epithelial tight junction proteins (97, 102), fucosylation of epithelial cell-associated glycans (103–105), secretion of mucin, and production of anti-microbial peptides (e.g., S100A8/A9 and RegIIIβ/γ), which together induce bacterial killing and prevent translocation of commensal organisms into the circulation and peripheral organs (102).
Although the importance of ILC3 in regulating intestinal health and inflammation has largely been ascribed to the production of IL-22, ILC3 produce several other cytokines that contribute to intestinal immune responses. In particular, ILC3 subsets have the capacity to produce IL-17A and IL-17F (2). Although both embryonic LTi and adult ILC3 isolated from the small intestine can be induced to secrete IL-17A upon ex vivo stimulation (9), fate mapping of IL-17A producing cells revealed that only a small proportion of ILC3 in the intestine exhibit a history of IL-17A expression (106). Nonetheless, ILC3-derived IL-17A may contribute to the formation of pulmonary tertiary lymphoid structures following infection and inflammation (107), can contribute to host immunity to fungal and bacterial pathogens (63, 108, 109), and has been implicated in the pathogenesis of obesity-associated airway hyper reactivity (110). Additionally, dysregulated IL-17A production by ILC3 may act to exacerbate inflammation and disease pathology in a range of inflammatory diseases, including psoriasis and IBD (65, 111, 112). IL-17F has high homology to IL-17A and can be secreted as either a homodimer or heterodimer with IL-17A (113). ILC3 are a dominant source of IL-17F after induction of colitis in Rag1−/− mice (65, 114), upon oral infection with Candida albicans and following skin wounding (108, 115), suggesting that IL-17F production by ILC3 may play critical roles in inflammation and immunity or resolving immune responses. Although IL-17F has largely been ascribed pro-inflammatory roles and may play pathogenic roles in colitis models (116, 117), it can also synergize with IL-22 to enhance production of anti-microbial peptides (118). Despite this evidence, the exact role of ILC3-derived IL-17F and its mechanisms of action in infection and disease, and how its effects differ from those of IL-17A, remain incompletely understood.
Recent studies have additionally highlighted ILC3 as a potent source of the cytokine and growth factor GM-CSF (67, 119–121). GM-CSF modulates myelopoiesis in the bone marrow, as well as extramedullary hematopoiesis in tissues, and acts on mature peripheral myeloid cells including monocytes, macrophages and neutrophils by regulating their activation, maturation, and migration into tissues (122). ILC3 are the predominant source of GM-CSF at steady state in the intestinal tract, with both NCR+ and LTi-like ILC3 capable of GM-CSF secretion. Under homeostatic conditions ILC3-derived GM-CSF acts to maintain immune tolerance by regulating DC subsets that further promote regulatory T-cell populations (120). Thus, constitutive homeostatic ILC3-derived GM-CSF secretion acts to maintain a tolerogenic environment. The interplay between MNPs and ILC3 in the intestine is bidirectional. Indeed, microbiota-dependent signals, including IL-1β, IL-23, and TL1A, derived from intestinal MNPs act to potentiate GM-CSF production by ILC3 (67, 120), suggesting the potential for a regulatory feedback loop in the intestine regulated by ILC3-derived GM-CSF-dependent crosstalk with myeloid cells. This crosstalk may also be important in the context of intestinal inflammation and following perturbation of intestinal barrier function as MNP-derived cytokines were found to regulate ILC3 production of IL-22, in addition to GM-CSF, in mouse models of colitis and human IBD patients (67). In contrast to a tissue-protective role for ILC3-derived GM-CSF two independent studies demonstrated that ILC3-derived GM-CSF acted to exacerbate intestinal pathology in an innate cell driven model of colitis (anti-CD40 treatment of Rag1−/− mice), in part through recruitment of inflammatory monocytes (121, 123). Furthermore, onset of colitis results in migration of ILC3 out of intestinal cryptopatches and into the lamina propria in a GM-CSF-dependent manner (121), further demonstrating that GM-CSF-dependent ILC3-MNP crosstalk may dictate the migration and localization of immune populations within the intestinal tissue.
Embryonic LTi cells are required to generate secondary lymphoid tissues, and this role has been expertly reviewed before (10, 124) Further to this role in establishing the microenvironments that foster B- and T-cell responses, more recent studies have revealed that CCR6+ LTi-like ILC3 contribute to the regulation of adaptive immune responses via both indirect and direct interactions with the adaptive immune system (Figure 1; Outputs). CCR6+ ILC3 reside within the spleen, mucosal-associated lymphoid tissues, and lymph nodes—particularly those draining mucosal sites such as the mesenteric and mediastinal lymph nodes (62). Phenotypically, adult LTi-like CCR6+ ILC3 are very similar to the embryonic LTi population but additionally express molecules such as OX40L and CD30L that may foster interactions with lymphocytes (125–128). Interestingly, expression of OX40L can be induced in embryonic LTi cells through ex vivo culture with inflammatory cytokines, such as TL1A (129). Whether embryonic-derived LTi persist in the neonate and adult, and for how long, is unclear—but given the presence of long-lived LTi-like cells in the adult and the functional heterogeneity of ILC3 subsets, the potential persistence of bona fide embryonic LTi cells after birth needs to be addressed. It is striking that CCR6+ ILC3 in adult PP, LNs, and spleen associate with stromal populations that closely resemble the embryonic “organizer” cells through which fetal LTi orchestrate lymphorganogenesis (57, 130). Thus, tissue microenvironments fostered early in the life in secondary lymphoid tissues by ILC3 may be maintained in the adult. In support of this, restoration of the splenic white pulp architecture after viral infection was delayed in the absence of ILC3 (131).
Direct regulation of CD4+ T cells by ILC3 can be mediated through MHCII-dependent antigen presentation. ILC3-conditional deletion of MHCII resulted in moderate colitis due to a failure to control T-cell responses to commensal bacteria (61, 132). Thus, within the gastrointestinal tract, ILC3 appear to play a crucial suppressive role in regulating CD4 T-cell responses to commensal organisms to maintain tissue homeostasis. Mechanistically, ILC3 were found to control commensal bacteria-specific CD4+ T-cell responses in part by outcompeting T cells for IL-2, thus starving that T cells of growth factors needed for proliferation and resulting in apoptosis (61). Similarly, ILC3 may regulate the T-cell pool by controlling the availability of other critical growth cytokines in lymphoid tissues, including IL-7 (133). Further investigations have indicated that the outcome of ILC3:T-cell interactions may be determined by the specific tissue environment, with splenic ILC3 found to support, rather than suppress, CD4 T-cell responses in an MHCII-dependent manner (134). This discrepancy could be partially explained by the ability of cytokines to modulate ILC3—for example, IL-1β can induce ILC3 expression of co-stimulatory molecules (e.g., CD80 and CD86) and alter the nature of ILC3 antigen presentation (134). In addition to the presentation of protein-derived antigenic peptides via MHCII, LTi-like ILC3 have the capacity to present lipid antigens to iNKT cells via surface CD1d (135). Furthermore, ILC3 are required to suppress homeostatic CD8+ T-cell expansion in neonatal mice (136).
ILC3 also have the capacity to modulate humoral immunity (Figure 1; Outputs). ILC3 present in the spleen and PP support innate T-cell-independent IgA production through production of secreted and membrane bound lymphotoxin, which supports local DC populations and aids IgA class switching (137–139). Similarly, splenic ILC3 provide B-cell growth factors, including BAFF/APRIL and Dll1, to enhance local Ab production by marginal zone innate B cells (119). Despite these advances, the full extent and nature of the crosstalk between ILC3 and other lymphocyte populations, and how these signals are integrated alongside those provided by traditional antigen-presenting cell populations such as DC and B cells, remain to be fully elucidated.
Here, we have highlighted the roles of ILC3 in integrating signals from the environment and relaying information to surrounding immune and non-immune cells, thus functioning as a critical communications hub within intestinal tissue. Through being able to respond to both epithelial and myeloid-derived cytokines, vitamins, metabolites, and also neuropeptides, ILC3 integrate a wealth of regional cues to maintain the appropriate balance of key effector molecules and ensure local tissue homeostasis. Thus, while ILC3 have a clear protective role in the tissue, dysregulation or dramatic changes in environmental cues can result in disrupted ILC3 communication and may contribute to disease pathology, in part via altered ILC3 effector functions. For example, dysregulated cytokine production in the context of mouse models of colitis can promote ILC3 production of disease driving pro-inflammatory cytokines such as IFN-γ and IL-17A (40, 68, 111). Similarly, while ILC3-derived IL-22 is critical for supporting homeostatic intestinal barrier function, epithelial cell repair and regeneration, chronic overproduction of IL-22 by ILC3 may promote colorectal cancers (140, 141). Interestingly, genetic polymorphisms associated with chronic inflammatory disease or cancer may also alter the inflammatory milieu and have the potential to drive dysregulated ILC3 communication. For example, Card 9 deficiency results in disrupted IL-1β production and impacts upon epithelial cell proliferation and colitis-associated cancer due to perturbed ILC3-associated IL-22 production (142). It is likely that many other polymorphisms seen in patients with intestinal inflammatory disorders, including IL-23R, IL-10/IL-10R, Atg16l1, and Nod2, also impact upon ILC3 function either directly or by altering the integration of tissue-specific signals that are sensed, interpreted or propagated by ILC3. Dysregulation of protective ILC3 functions in intestinal disease is also not limited to effector cytokine production. Indeed, ILC3-intrinsic expression of MHCII has been observed to be reduced in two separate cohorts of Crohn’s patients and found to correlate with enhanced Th17 responses in disease (61, 143), suggesting that the altered intestinal tissue environment may impact upon antigen presentation by these cells.
Infections may additionally disrupt ILC3 populations in the gut, resulting in a loss of their normal sentinel activity, the breakdown of gut barrier integrity, and intestinal pathology. In particular, infection with human or simian immunodeficiency viruses (HIV/SIV) results in depletion of ILC3 from the intestinal mucosa and lymph nodes (144–147). Thus, further investigation is needed to understand how alterations in ILC-related signals result in loss of these cells and how this balance can be redressed to restore homeostatic numbers and functions of ILC3. Finally, as the next generation of anti-inflammatory therapeutics enter the clinics, their relative impact on beneficial intestinal ILC3 need to be thoroughly addressed. In particular, monoclonal antibodies and small molecule inhibitors targeting common pathways shared by Th17 and ILC3 (e.g., anti-IL-23, anti-IL-12, anti-IL-17A, and small molecule antagonists of RORγt) have the potential to suppress inflammation but may have long-term consequences for patients by disrupting protective ILC3 pathways. In this regard, an increased understanding of the differences and similarities between Th17 and ILC3 regulation is required to guide therapeutic interventions and treatment regimen. Promisingly, recent studies suggest that acute targeting of RORγt effectively reduces Th17-driven inflammation while leaving protective ILC3 responses intact (82), although chronic inhibition of this master transcription factor may eventually negatively impact upon ILC3 responses. Furthermore, ILC3-derived cytokines may have beneficial roles in maintaining epithelial barrier function and healthy host–microbe interactions, thus targeting that these cytokines and their receptors in the context of Th17-driven inflammation may result in undesirable consequences. Indeed, treatment with neutralizing monoclonal antibodies against IL-17A or its receptor has been reported to worsen disease and increase incidence of adverse effects in several IBD patient cohorts (148, 149), further highlighting the need to understand the potential repercussions of emerging therapeutics on ILC3. Future studies will lead to a further understanding of how these critical innate immune sentinels are regulated in order to harness their protective functions to maintain tissue health, while suppressing dysregulated responses that exacerbate disease.
This review was jointly written by both authors, who contributed equally.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Research in the Hepworth lab is supported by a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (Grant Number 105644/Z/14/Z). Research in the Withers lab is supported by a Wellcome Trust Senior Research Fellowship (Grant Number 110199/Z/15/Z). The authors thank members of the Withers and Hepworth lab for critical feedback and Juliane Poschinski for the figure illustration.
1. Sonnenberg GF, Artis D. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity (2012) 37(4):601–10. doi:10.1016/j.immuni.2012.10.003
2. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi:10.1038/nri3365
3. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity (2014) 41(3):354–65. doi:10.1016/j.immuni.2014.09.005
4. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science (2015) 348(6237):aaa6566. doi:10.1126/science.aaa6566
5. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17(7):765–74. doi:10.1038/ni.3489
6. Melo-Gonzalez F, Hepworth MR. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology (2017) 150(3):265–75. doi:10.1111/imm.12697
7. Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells – how did we miss them? Nat Rev Immunol (2013) 13(2):75–87. doi:10.1038/nri3349
8. Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, et al. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science (2010) 330(6004):665–9. doi:10.1126/science.1194597
9. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol (2011) 12(4):320–6. doi:10.1038/ni.2002
10. van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol (2010) 10(9):664–74. doi:10.1038/nri2832
11. Serafini N, Vosshenrich CA, Di Santo JP. Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol (2015) 15(7):415–28. doi:10.1038/nri3855
12. Zook EC, Kee BL. Development of innate lymphoid cells. Nat Immunol (2016) 17(7):775–82. doi:10.4038/ni.3481
13. Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157(2):340–56. doi:10.1016/j.cell.2014.03.030
14. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature (2014) 508(7496):397–401. doi:10.1038/nature13047
15. Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, et al. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature (2016) 539(7627):102–6. doi:10.1038/nature20105
16. Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol (2004) 5(1):64–73. doi:10.1038/ni1022
17. Cherrier M, Sawa S, Eberl G. Notch, Id2, and RORgammat sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J Exp Med (2012) 209(4):729–40. doi:10.1084/jem.20111594
18. Ishizuka IE, Chea S, Gudjonson H, Constantinides MG, Dinner AR, Bendelac A, et al. Single-cell analysis defines the divergence between the innate lymphoid cell lineage and lymphoid tissue-inducer cell lineage. Nat Immunol (2016) 17(3):269–76. doi:10.1038/ni.3344
19. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol (2015) 16(3):306–17. doi:10.1038/ni.3094
20. Bjorklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, et al. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol (2016) 17(4):451–60. doi:10.1038/ni.3368
21. Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell (2016) 166(5):1231–46.e13. doi:10.1016/j.cell.2016.07.043
22. Montaldo E, Juelke K, Romagnani C. Group 3 innate lymphoid cells (ILC3s): origin, differentiation, and plasticity in humans and mice. Eur J Immunol (2015) 45(8):2171–82. doi:10.1002/eji.201545598
23. Almeida FF, Belz GT. Innate lymphoid cells: models of plasticity for immune homeostasis and rapid responsiveness in protection. Mucosal Immunol (2016) 9(5):1103–12. doi:10.1038/mi.2016.64
24. Lim AI, Verrier T, Vosshenrich CA, Di Santo JP. Developmental options and functional plasticity of innate lymphoid cells. Curr Opin Immunol (2017) 44:61–8. doi:10.1016/j.coi.2017.03.010
25. Kim SH, Cho BH, Kiyono H, Jang YS. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Sci Rep (2017) 7(1):3980. doi:10.1038/s41598-017-02729-6
26. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (2011) 334(6062):1561–5. doi:10.1126/science.1214914
27. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol (2012) 13(2):144–51. doi:10.1038/ni.2187
28. Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity (2012) 36(1):92–104. doi:10.1016/j.immuni.2011.11.011
29. Qiu J, Guo X, Chen ZM, He L, Sonnenberg GF, Artis D, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity (2013) 39(2):386–99. doi:10.1016/j.immuni.2013.08.002
30. Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The maternal microbiota drives early postnatal innate immune development. Science (2016) 351(6279):1296–302. doi:10.1126/science.aad2571
31. van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature (2014) 508(7494):123–7. doi:10.1038/nature13158
32. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science (2014) 343(6169):432–7. doi:10.1126/science.1247606
33. Goverse G, Labao-Almeida C, Ferreira M, Molenaar R, Wahlen S, Konijn T, et al. Vitamin A controls the presence of RORgamma+ innate lymphoid cells and lymphoid tissue in the small intestine. J Immunol (2016) 196(12):5148–55. doi:10.4049/jimmunol.1501106
34. Wilhelm C, Harrison OJ, Schmitt V, Pelletier M, Spencer SP, Urban JF Jr, et al. Critical role of fatty acid metabolism in ILC2-mediated barrier protection during malnutrition and helminth infection. J Exp Med (2016) 213(8):1409–18. doi:10.1084/jem.20151448
35. Konya V, Czarnewski P, Forkel M, Rao A, Kokkinou E, Villablanca EJ, et al. Vitamin D downregulates the IL-23 receptor pathway in human mucosal group 3 innate lymphoid cells. J Allergy Clin Immunol (2017). doi:10.1016/j.jaci.2017.01.045
36. Chen J, Waddell A, Lin YD, Cantorna MT. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol (2015) 8(3):618–26. doi:10.1038/mi.2014.94
37. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350(6263):981–5. doi:10.1126/science.aac9593
38. Montaldo E, Teixeira-Alves LG, Glatzer T, Durek P, Stervbo U, Hamann W, et al. Human RORgammat(+)CD34(+) cells are lineage-specified progenitors of group 3 RORgammat(+) innate lymphoid cells. Immunity (2014) 41(6):988–1000. doi:10.1016/j.immuni.2014.11.010
39. Scoville SD, Mundy-Bosse BL, Zhang MH, Chen L, Zhang X, Keller KA, et al. A progenitor cell expressing transcription factor RORgammat generates all human innate lymphoid cell subsets. Immunity (2016) 44(5):1140–50. doi:10.1016/j.immuni.2016.04.007
40. Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity (2010) 33(5):736–51. doi:10.1016/j.immuni.2010.10.017
41. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature (2013) 494(7436):261–5. doi:10.1038/nature11813
42. Chea S, Perchet T, Petit M, Verrier T, Guy-Grand D, Banchi EG, et al. Notch signaling in group 3 innate lymphoid cells modulates their plasticity. Sci Signal (2016) 9(426):ra45. doi:10.1126/scisignal.aaf2223
43. Verrier T, Satoh-Takayama N, Serafini N, Marie S, Di Santo JP, Vosshenrich CA. Phenotypic and functional plasticity of murine intestinal NKp46+ group 3 innate lymphoid cells. J Immunol (2016) 196(11):4731–8. doi:10.4049/jimmunol.1502673
44. Viant C, Rankin LC, Girard-Madoux MJ, Seillet C, Shi W, Smyth MJ, et al. Transforming growth factor-beta and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci Signal (2016) 9(426):ra46. doi:10.1126/scisignal.aaf2176
45. Eberl G. Inducible lymphoid tissues in the adult gut: recapitulation of a fetal developmental pathway? Nat Rev Immunol (2005) 5(5):413–20. doi:10.1038/nri1600
46. Savage AK, Liang HE, Locksley RM. The development of steady-state activation hubs between adult LTi ILC3s and primed macrophages in small intestine. J Immunol (2017) 199:1912–22. doi:10.4049/jimmunol.1700155
47. Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol (2009) 10(1):75–82. doi:10.1038/ni.1681
48. Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol (2009) 10(1):83–91. doi:10.1038/ni.1684
49. Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci U S A (2011) 108(45):18324–9. doi:10.1073/pnas.1112064108
50. Reynders A, Yessaad N, Vu Manh TP, Dalod M, Fenis A, Aubry C, et al. Identity, regulation and in vivo function of gut NKp46+RORgammat+ and NKp46+RORgammat- lymphoid cells. EMBO J (2011) 30(14):2934–47. doi:10.1038/emboj.2011.201
51. Satoh-Takayama N, Serafini N, Verrier T, Rekiki A, Renauld JC, Frankel G, et al. The chemokine receptor CXCR6 controls the functional topography of interleukin-22 producing intestinal innate lymphoid cells. Immunity (2014) 41(5):776–88. doi:10.1016/j.immuni.2014.10.007
52. Meier D, Bornmann C, Chappaz S, Schmutz S, Otten LA, Ceredig R, et al. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity (2007) 26(5):643–54. doi:10.1016/j.immuni.2007.04.009
53. Schmutz S, Bosco N, Chappaz S, Boyman O, Acha-Orbea H, Ceredig R, et al. Cutting edge: IL-7 regulates the peripheral pool of adult ROR gamma+ lymphoid tissue inducer cells. J Immunol (2009) 183(4):2217–21. doi:10.4049/jimmunol.0802911
54. Chappaz S, Gartner C, Rodewald HR, Finke D. Kit ligand and Il7 differentially regulate Peyer’s patch and lymph node development. J Immunol (2010) 185(6):3514–9. doi:10.4049/jimmunol.1000665
55. Chappaz S, Finke D. The IL-7 signaling pathway regulates lymph node development independent of peripheral lymphocytes. J Immunol (2010) 184(7):3562–9. doi:10.4049/jimmunol.0901647
56. Robinette ML, Bando JK, Song W, Ulland TK, Gilfillan S, Colonna M. IL-15 sustains IL-7R-independent ILC2 and ILC3 development. Nat Commun (2017) 8:14601. doi:10.1038/ncomms14601
57. Hoorweg K, Narang P, Li Z, Thuery A, Papazian N, Withers DR, et al. A stromal cell niche for human and mouse type 3 innate lymphoid cells. J Immunol (2015) 195(9):4257–63. doi:10.4049/jimmunol.1402584
58. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol (2007) 8(11):1255–65. doi:10.1038/ni1513
59. Chai Q, Onder L, Scandella E, Gil-Cruz C, Perez-Shibayama C, Cupovic J, et al. Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity (2013) 38(5):1013–24. doi:10.1016/j.immuni.2013.03.012
60. Gil-Cruz C, Perez-Shibayama C, Onder L, Chai Q, Cupovic J, Cheng HW, et al. Fibroblastic reticular cells regulate intestinal inflammation via IL-15-mediated control of group 1 ILCs. Nat Immunol (2016) 17(12):1388–96. doi:10.1038/ni.3566
61. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science (2015) 348(6238):1031–5. doi:10.1126/science.aaa4812
62. Mackley EC, Houston S, Marriott CL, Halford EE, Lucas B, Cerovic V, et al. CCR7-dependent trafficking of RORgamma(+) ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat Commun (2015) 6:5862. doi:10.1038/ncomms6862
63. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med (2009) 206(1):35–41. doi:10.1084/jem.20072713
64. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity (2012) 36(2):276–87. doi:10.1016/j.immuni.2011.12.011
65. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208(6):1127–33. doi:10.1084/jem.20101712
66. Manta C, Heupel E, Radulovic K, Rossini V, Garbi N, Riedel CU, et al. CX(3)CR1(+) macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium. Mucosal Immunol (2013) 6(1):177–88. doi:10.1038/mi.2012.61
67. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med (2014) 211(8):1571–83. doi:10.1084/jem.20140678
68. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity (2015) 43(1):146–60. doi:10.1016/j.immuni.2015.06.019
69. Quell KM, Karsten CM, Kordowski A, Almeida LN, Briukhovetska D, Wiese AV, et al. Monitoring C3aR expression using a floxed tdtomato-C3aR reporter knock-in mouse. J Immunol (2017) 199(2):688–706. doi:10.4049/jimmunol.1700318
70. Narni-Mancinelli E, Gauthier L, Baratin M, Guia S, Fenis A, Deghmane AE, et al. Complement factor P is a ligand for the natural killer cell-activating receptor NKp46. Sci Immunol (2017) 2(10). doi:10.1126/sciimmunol.aam9628
71. Veiga-Fernandes H, Coles MC, Foster KE, Patel A, Williams A, Natarajan D, et al. Tyrosine kinase receptor RET is a key regulator of Peyer’s patch organogenesis. Nature (2007) 446(7135):547–51. doi:10.1038/nature05597
72. Ibiza S, Garcia-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature (2016) 535(7612):440–3. doi:10.1038/nature18644
73. van de Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, et al. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol (2009) 10(11):1193–9. doi:10.1038/ni.1789
74. Olivier BJ, Cailotto C, van der Vliet J, Knippenberg M, Greuter MJ, Hilbers FW, et al. Vagal innervation is required for the formation of tertiary lymphoid tissue in colitis. Eur J Immunol (2016) 46(10):2467–80. doi:10.1002/eji.201646370
75. Dalli J, Colas RA, Arnardottir H, Serhan CN. Vagal regulation of group 3 innate lymphoid cells and the immunoresolvent PCTR1 controls infection resolution. Immunity (2017) 46(1):92–105. doi:10.1016/j.immuni.2016.12.009
76. Kwong B, Rua R, Gao Y, Flickinger J Jr, Wang Y, Kruhlak MJ, et al. T-bet-dependent NKp46+ innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat Immunol (2017) 18:1117–27. doi:10.1038/ni.3816
77. Duffin R, O’Connor RA, Crittenden S, Forster T, Yu C, Zheng X, et al. Prostaglandin E(2) constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science (2016) 351(6279):1333–8. doi:10.1126/science.aad9903
78. Laffont S, Blanquart E, Savignac M, Cenac C, Laverny G, Metzger D, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med (2017) 214(6):1581–92. doi:10.1084/jem.20161807
79. Chea S, Schmutz S, Berthault C, Perchet T, Petit M, Burlen-Defranoux O, et al. Single-cell gene expression analyses reveal heterogeneous responsiveness of fetal innate lymphoid progenitors to notch signaling. Cell Rep (2016) 14(6):1500–16. doi:10.1016/j.celrep.2016.01.015
80. Shih HY, Sciume G, Mikami Y, Guo L, Sun HW, Brooks SR, et al. Developmental acquisition of regulomes underlies innate lymphoid cell functionality. Cell (2016) 165(5):1120–33. doi:10.1016/j.cell.2016.04.029
81. Koues OI, Collins PL, Cella M, Robinette ML, Porter SI, Pyfrom SC, et al. Distinct gene regulatory pathways for human innate versus adaptive lymphoid cells. Cell (2016) 165(5):1134–46. doi:10.1016/j.cell.2016.04.014
82. Withers DR, Hepworth MR, Wang X, Mackley EC, Halford EE, Dutton EE, et al. Transient inhibition of ROR-gammat therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat Med (2016) 22(3):319–23. doi:10.1038/nm.4046
83. Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell (2017) 168(6):1086–100.e10. doi:10.1016/j.cell.2017.02.021
84. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504(7480):451–5. doi:10.1038/nature12726
85. Wei L, Laurence A, O’Shea JJ. New insights into the roles of Stat5a/b and Stat3 in T cell development and differentiation. Semin Cell Dev Biol (2008) 19(4):394–400. doi:10.1016/j.semcdb.2008.07.011
86. Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity (2014) 40(1):25–39. doi:10.1016/j.immuni.2013.10.021
87. Robinette ML, Cella M, Telliez JB, Ulland TK, Barrow AD, Capuder K, et al. Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol (2017). doi:10.1038/mi.2017.38
88. Singh AK, Eken A, Fry M, Bettelli E, Oukka M. DOCK8 regulates protective immunity by controlling the function and survival of RORgammat+ ILCs. Nat Commun (2014) 5:4603. doi:10.1038/ncomms5603
89. Liu B, Ye B, Yang L, Zhu X, Huang G, Zhu P, et al. Long noncoding RNA lncKdm2b is required for ILC3 maintenance by initiation of Zfp292 expression. Nat Immunol (2017) 18(5):499–508. doi:10.1038/ni.3712
90. O’Sullivan TE, Geary CD, Weizman OE, Geiger TL, Rapp M, Dorn GW II, et al. Atg5 is essential for the development and survival of innate lymphocytes. Cell Rep (2016) 15(9):1910–9. doi:10.1016/j.celrep.2016.04.082
91. Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity (2008) 29(6):958–70. doi:10.1016/j.immuni.2008.11.001
92. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med (2008) 14(3):282–9. doi:10.1038/nm1720
93. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature (2009) 457(7230):722–5. doi:10.1038/nature07537
94. Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci U S A (2010) 107(24):10961–6. doi:10.1073/pnas.1005641107
95. Satoh-Takayama N, Lesjean-Pottier S, Sawa S, Vosshenrich CA, Eberl G, Di Santo JP. Lymphotoxin-beta receptor-independent development of intestinal IL-22-producing NKp46+ innate lymphoid cells. Eur J Immunol (2011) 41(3):780–6. doi:10.1002/eji.201040851
96. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity (2011) 34(1):122–34. doi:10.1016/j.immuni.2010.12.009
97. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science (2012) 336(6086):1321–5. doi:10.1126/science.1222551
98. Rankin LC, Girard-Madoux MJ, Seillet C, Mielke LA, Kerdiles Y, Fenis A, et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat Immunol (2016) 17(2):179–86. doi:10.1038/ni.3332
99. Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity (2012) 37(2):339–50. doi:10.1016/j.immuni.2012.05.028
100. Aparicio-Domingo P, Romera-Hernandez M, Karrich JJ, Cornelissen F, Papazian N, Lindenbergh-Kortleve DJ, et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J Exp Med (2015) 212(11):1783–91. doi:10.1084/jem.20150318
101. Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature (2015) 528(7583):560–4. doi:10.1038/nature16460
102. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol (2011) 12(5):383–90. doi:10.1038/ni.2025
103. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science (2014) 345(6202):1254009. doi:10.1126/science.1254009
104. Pham TA, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP, et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe (2014) 16(4):504–16. doi:10.1016/j.chom.2014.08.017
105. Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature (2014) 514(7524):638–41. doi:10.1038/nature13823
106. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol (2011) 12(3):255–63. doi:10.1038/ni.1993
107. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol (2011) 12(7):639–46. doi:10.1038/ni.2053
108. Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol (2013) 190(2):521–5. doi:10.4049/jimmunol.1202924
109. Xiong H, Keith JW, Samilo DW, Carter RA, Leiner IM, Pamer EG. Innate lymphocyte/Ly6C(hi) monocyte crosstalk promotes Klebsiella pneumoniae clearance. Cell (2016) 165(3):679–89. doi:10.1016/j.cell.2016.03.017
110. Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med (2014) 20(1):54–61. doi:10.1038/nm.3423
111. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464(7293):1371–5. doi:10.1038/nature08949
112. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol (2014) 134(4):984–91. doi:10.1038/jid.2013.477
113. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol (2017) 18(6):612–21. doi:10.1038/ni.3742
114. Eken A, Singh AK, Treuting PM, Oukka M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol (2014) 7(1):143–54. doi:10.1038/mi.2013.33
115. Li Z, Hodgkinson T, Gothard EJ, Boroumand S, Lamb R, Cummins I, et al. Epidermal Notch1 recruits RORgamma(+) group 3 innate lymphoid cells to orchestrate normal skin repair. Nat Commun (2016) 7:11394. doi:10.1038/ncomms11394
116. Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, et al. Regulation of inflammatory responses by IL-17F. J Exp Med (2008) 205(5):1063–75. doi:10.1084/jem.20071978
117. O’Connor W Jr, Zenewicz LA, Flavell RA. The dual nature of T(H)17 cells: shifting the focus to function. Nat Immunol (2010) 11(6):471–6. doi:10.1038/ni.1882
118. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203(10):2271–9. doi:10.1084/jem.20061308
119. Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol (2014) 15(4):354–64. doi:10.1038/ni.2830
120. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science (2014) 343(6178):1249288. doi:10.1126/science.1249288
121. Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife (2016) 5:e10066. doi:10.7554/eLife.10066
122. Becher B, Tugues S, Greter M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity (2016) 45(5):963–73. doi:10.1016/j.immuni.2016.10.026
123. Song C, Lee JS, Gilfillan S, Robinette ML, Newberry RD, Stappenbeck TS, et al. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J Exp Med (2015) 212(11):1869–82. doi:10.1084/jem.20151403
124. Cupedo T, Kraal G, Mebius RE. The role of CD45+CD4+CD3- cells in lymphoid organ development. Immunol Rev (2002) 189:41–50. doi:10.1034/j.1600-065X.2002.18905.x
125. Withers DR, Jaensson E, Gaspal F, McConnell FM, Eksteen B, Anderson G, et al. The survival of memory CD4+ T cells within the gut lamina propria requires OX40 and CD30 signals. J Immunol (2009) 183(8):5079–84. doi:10.4049/jimmunol.0901514
126. Kim S, Han S, Withers DR, Gaspal F, Bae J, Baik S, et al. CD117(+) CD3(-) CD56(-) OX40Lhigh cells express IL-22 and display an LTi phenotype in human secondary lymphoid tissues. Eur J Immunol (2011) 41(6):1563–72. doi:10.1002/eji.201040915
127. Withers DR, Gaspal FM, Bekiaris V, McConnell FM, Kim M, Anderson G, et al. OX40 and CD30 signals in CD4(+) T-cell effector and memory function: a distinct role for lymphoid tissue inducer cells in maintaining CD4(+) T-cell memory but not effector function. Immunol Rev (2011) 244(1):134–48. doi:10.1111/j.1600-065X.2011.01057.x
128. Withers DR, Gaspal FM, Mackley EC, Marriott CL, Ross EA, Desanti GE, et al. Cutting edge: lymphoid tissue inducer cells maintain memory CD4 T cells within secondary lymphoid tissue. J Immunol (2012) 189(5):2094–8. doi:10.4049/jimmunol.1201639
129. Kim MY, Toellner KM, White A, McConnell FM, Gaspal FM, Parnell SM, et al. Neonatal and adult CD4+ CD3- cells share similar gene expression profile, and neonatal cells up-regulate OX40 ligand in response to TL1A (TNFSF15). J Immunol (2006) 177(5):3074–81. doi:10.4049/jimmunol.177.5.3074
130. Katakai T, Suto H, Sugai M, Gonda H, Togawa A, Suematsu S, et al. Organizer-like reticular stromal cell layer common to adult secondary lymphoid organs. J Immunol (2008) 181(9):6189–200. doi:10.4049/jimmunol.181.9.6189
131. Scandella E, Bolinger B, Lattmann E, Miller S, Favre S, Littman DR, et al. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat Immunol (2008) 9(6):667–75. doi:10.1038/ni.1605
132. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature (2013) 498(7452):113–7. doi:10.1038/nature12240
133. Martin CE, Spasova DS, Frimpong-Boateng K, Kim HO, Lee M, Kim KS, et al. Interleukin-7 availability is maintained by a hematopoietic cytokine sink comprising innate lymphoid cells and T cells. Immunity (2017) 47(1):171–82.e4. doi:10.1016/j.immuni.2017.07.005
134. von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc Natl Acad Sci U S A (2014) 111(35):12835–40. doi:10.1073/pnas.1406908111
135. Saez de Guinoa J, Jimeno R, Farhadi N, Jervis PJ, Cox LR, Besra GS, et al. CD1d-mediated activation of group 3 innate lymphoid cells drives IL-22 production. EMBO Rep (2016) 18:39–47. doi:10.15252/embr.201642412
136. Bank U, Deiser K, Finke D, Hammerling GJ, Arnold B, Schuler T. Cutting edge: innate lymphoid cells suppress homeostatic T cell expansion in neonatal mice. J Immunol (2016) 196(9):3532–6. doi:10.4049/jimmunol.1501643
137. Tsuji M, Suzuki K, Kitamura H, Maruya M, Kinoshita K, Ivanov II, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity (2008) 29(2):261–71. doi:10.1016/j.immuni.2008.05.014
138. Kruglov AA, Grivennikov SI, Kuprash DV, Winsauer C, Prepens S, Seleznik GM, et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science (2013) 342(6163):1243–6. doi:10.1126/science.1243364
139. Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, Cyster JG. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science (2016) 352(6287):aaf4822. doi:10.1126/science.aaf4822
140. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491(7423):259–63. doi:10.1038/nature11535
141. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med (2013) 210(5):917–31. doi:10.1084/jem.20122308
142. Bergmann H, Roth S, Pechloff K, Kiss EA, Kuhn S, Heikenwalder M, et al. Card9-dependent IL-1beta regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. Eur J Immunol (2017) 47(8):1342–53. doi:10.1002/eji.201646765
143. Li J, Doty AL, Tang Y, Berrie D, Iqbal A, Tan SA, et al. Enrichment of IL-17A+ IFN-gamma+ and IL-22+ IFN-gamma+ T cell subsets is associated with reduction of NKp44+ ILC3s in the terminal ileum of Crohn’s disease patients. Clin Exp Immunol (2017) 190:143–53. doi:10.1111/cei.12996
144. Li H, Richert-Spuhler LE, Evans TI, Gillis J, Connole M, Estes JD, et al. Hypercytotoxicity and rapid loss of NKp44+ innate lymphoid cells during acute SIV infection. PLoS Pathog (2014) 10(12):e1004551. doi:10.1371/journal.ppat.1004551
145. Zhang Z, Cheng L, Zhao J, Li G, Zhang L, Chen W, et al. Plasmacytoid dendritic cells promote HIV-1-induced group 3 innate lymphoid cell depletion. J Clin Invest (2015) 125(9):3692–703. doi:10.1172/JCI82124
146. Kloverpris HN, Kazer SW, Mjosberg J, Mabuka JM, Wellmann A, Ndhlovu Z, et al. Innate lymphoid cells are depleted irreversibly during acute HIV-1 infection in the absence of viral suppression. Immunity (2016) 44(2):391–405. doi:10.1016/j.immuni.2016.01.006
147. Kramer B, Goeser F, Lutz P, Glassner A, Boesecke C, Schwarze-Zander C, et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog (2017) 13(5):e1006373. doi:10.1371/journal.ppat.1006373
148. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61(12):1693–700. doi:10.1136/gutjnl-2011-301668
Keywords: innate lymphoid cells, group 3 innate lymphoid cell, intestinal inflammation, interleukin-22, inflammatory bowel disease
Citation: Withers DR and Hepworth MR (2017) Group 3 Innate Lymphoid Cells: Communications Hubs of the Intestinal Immune System. Front. Immunol. 8:1298. doi: 10.3389/fimmu.2017.01298
Received: 04 August 2017; Accepted: 27 September 2017;
Published: 16 October 2017
Edited by:
Chiara Romagnani, Deutsches Rheuma Forschungszentrum, GermanyReviewed by:
Burkhard Ludewig, Kantonsspital St. Gallen, SwitzerlandCopyright: © 2017 Withers and Hepworth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David R. Withers, ZC53aXRoZXJzQGJoYW0uYWMudWs=;
Matthew R. Hepworth, bWF0dGhldy5oZXB3b3J0aEBtYW5jaGVzdGVyLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.