
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 September 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01147
This article is part of the Research Topic Reassessing Twenty Years of Vaccine Development Against Tuberculosis View all 9 articles
 Natalie E. Nieuwenhuizen1
Natalie E. Nieuwenhuizen1 Prasad S. Kulkarni2
Prasad S. Kulkarni2 Umesh Shaligram2
Umesh Shaligram2 Mark F. Cotton3
Mark F. Cotton3 Cyrill A. Rentsch4,5
Cyrill A. Rentsch4,5 Bernd Eisele6
Bernd Eisele6 Leander Grode6
Leander Grode6 Stefan H. E. Kaufmann1*
Stefan H. E. Kaufmann1*
The only licensed vaccine against tuberculosis (TB), bacille Calmette–Guérin (BCG), protects against severe extrapulmonary forms of TB but is virtually ineffective against the most prevalent form of the disease, pulmonary TB. BCG was genetically modified at the Max Planck Institute for Infection Biology to improve its immunogenicity by replacing the urease C encoding gene with the listeriolysin encoding gene from Listeria monocytogenes. Listeriolysin perturbates the phagosomal membrane at acidic pH. Urease C is involved in neutralization of the phagosome harboring BCG. Its depletion allows for rapid phagosome acidification and promotes phagolysosome fusion. As a result, BCGΔureC::hly (VPM1002) promotes apoptosis and autophagy and facilitates release of mycobacterial antigens into the cytosol. In preclinical studies, VPM1002 has been far more efficacious and safer than BCG. The vaccine was licensed to Vakzine Projekt Management and later sublicensed to the Serum Institute of India Pvt. Ltd., the largest vaccine producer in the world. The vaccine has passed phase I clinical trials in Germany and South Africa, demonstrating its safety and immunogenicity in young adults. It was also successfully tested in a phase IIa randomized clinical trial in healthy South African newborns and is currently undergoing a phase IIb study in HIV exposed and unexposed newborns. A phase II/III clinical trial will commence in India in 2017 to assess efficacy against recurrence of TB. The target indications for VPM1002 are newborn immunization to prevent TB as well as post-exposure immunization in adults to prevent TB recurrence. In addition, a Phase I trial in non-muscle invasive bladder cancer patients has been completed, and phase II trials are ongoing. This review describes the development of VPM1002 from the drawing board to its clinical assessment.
Infection with Mycobacterium tuberculosis (Mtb) led to 10.4 million recorded cases of tuberculosis (TB) in 2015, with 1.8 million recorded deaths [World Health Organization (WHO) report 2016]. The current therapy involves 6–9 months of antibiotics, with the emergence of multiple drug resistant strains being a continuing obstacle. An attenuated form of the bovine Mycobacterium species, Mycobacterium bovis bacille Calmette–Guerin (BCG) has been in clinical use since 1921 and remains the only licensed vaccine against TB. BCG partially protects against TB meningitis and disseminated TB in infants and has non-specific immunostimulatory effects (1), which reduce general infant mortality by enhancing responses to other infectious diseases (2, 3). However, in all age groups, BCG does not adequately protect against pulmonary TB, the most prevalent form of disease and the route of disease transmission. In addition, BCG can cause severe adverse effects in immunocompromised individuals (4) and hence is contraindicated in HIV-infected individuals, the group that is most vulnerable to TB. However, in the absence of an alternative, BCG continues to be used in the immunization programs of several countries. To overcome these issues, several TB vaccine candidates are under development (5). One of the most advanced among them is BCG ΔureC::hly (VPM1002) (6).
VPM1002 is a recombinant BCG (rBCG) in which the urease C gene has been replaced by the listeriolysin O (LLO) encoding gene (hly) from Listeria monocytogenes (7). Urease C drives neutralization of phagosomes containing mycobacteria by generation of ammonia, thereby inhibiting phagolysosomal maturation and contributing to the survival of mycobacteria inside the macrophage (8, 9). Its depletion allows for rapid phagosome acidification, which promotes phagolysosome fusion and provides the optimal pH for LLO stability (10). LLO is a cholesterol-dependant cytolysin that forms transmembrane β-barrel pores in the phagolysosome membrane, allowing escape of L. monocytogenes into the cytosol (10, 11). Its expression in VPM1002 results in the release of antigens and bacterial DNA into the cytosol, triggering autophagy, inflammasome activation, and apoptosis. VPM1002 has demonstrated substantially increased immunogenicity, efficacy, and safety in preclinical studies, successfully passed Phase I and II clinical trials, and will now enter a Phase II/III clinical trial in India in 2017. This review summarizes the development, preclinical, and clinical testing of VPM1002 (Figure 1).
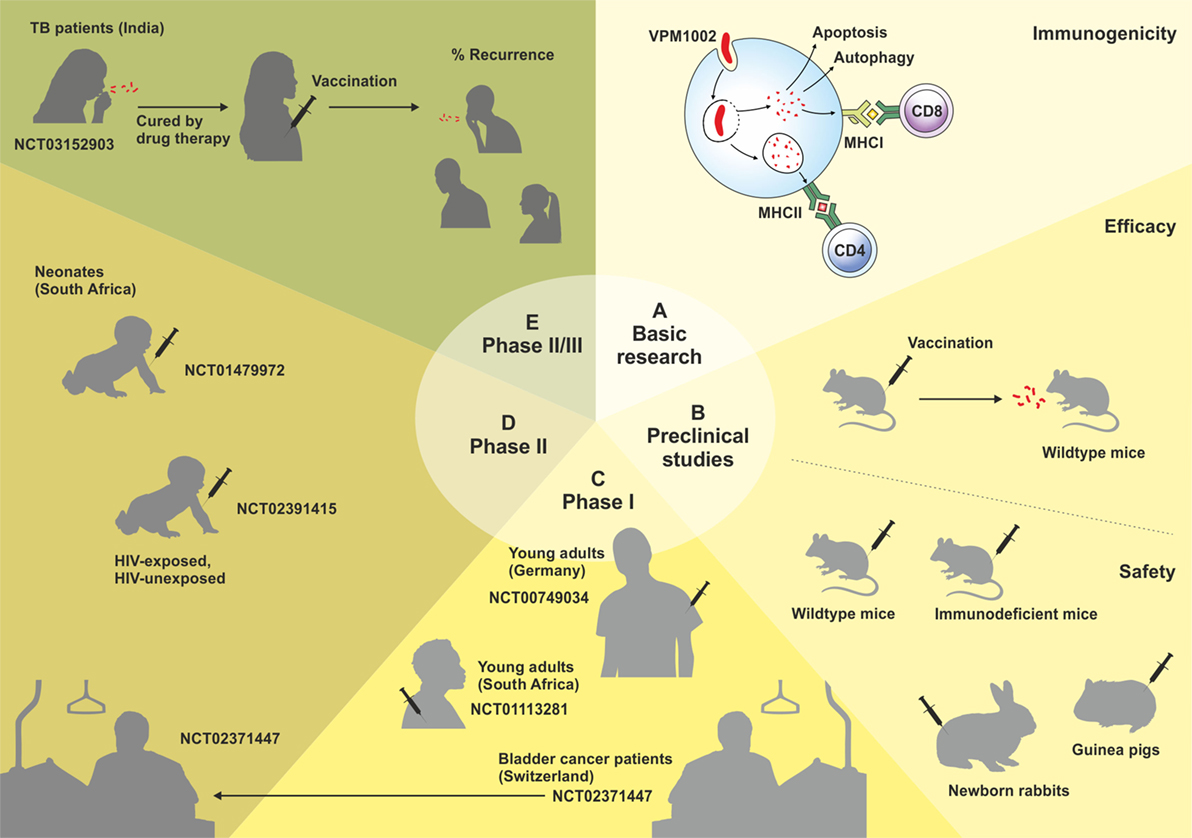
Figure 1. Schematic overview of the development of the VPM1002 vaccine candidate. Clinical trials are labeled by their ClinicalTrials.gov Identifier number.
The attenuation of BCG was achieved by passaging virulent M. bovis in bile-containing medium for 13 years in the laboratory (12), during which time several genome segments were lost, including a segment known as Region of Difference 1 (RD1) which encodes the unique mycobacterial ESX-1 type VII secretion system (13, 14). ESX-1-dependent perturbation of host cell membranes requires direct contact with pathogenic mycobacteria such as Mtb, allowing the bacilli or their antigens to egress the phagosome into the cytosol (15). Mtb antigens are thus accessible to both the endocytic major histocompatibility complex (MHC) class II antigen presentation pathway and the MHC I antigen presentation pathway in the cytosol, and consequently can stimulate CD4+ and CD8+ T-cell subsets, respectively, both of which are required for optimal protection against TB (16–21). In addition, ESX-1 dependent release of Mtb DNA into the cytosol can be detected by host sensors, leading to activation of NLR family pyrin domain-containing 3 (NLRP3) and absent in melanoma 2 inflammasomes, release of interferons, increased autophagy and apoptosis (22–25). Induction of apoptosis in infected host cells generates vesicles carrying mycobacterial antigens that can be phagocytosed by bystander antigen presenting cells, mainly dendritic cells (DCs) and trafficked through MHC I antigen processing pathways to stimulate CD8+ T cells in a process known as cross-priming (26, 27). Mice with deficient cross-presentation due to the absence of annexin 1 show impaired Mtb-specific CD8+ T cells and are highly susceptible to TB (28). Lacking the ESX-1 secretion system, BCG is restricted to the phagosome of host cells, therefore its antigens and bacterial DNA do not enter the cytosol and the antigens are primarily processed by MHC class II pathways, stimulating CD4+ T cell responses (13, 14, 29, 30). BCG induces only weak apoptosis and CD8+ T cell responses (26). Furthermore, both BCG and Mtb inhibit surface MHC II expression, as urease-dependent alkalinization of the phagosome causes intracellular sequestration of MHC II dimers, resulting in suboptimal CD4+ T cell responses (31–33). Phagosomal biology is therefore a clear target for interventions aimed at enhancing T cell responses against mycobacteria.
Originally, VPM1002 was designed to improve accessibility of mycobacterial antigens to the MHC I pathway via cytosolic egression of antigens mediated by LLO perturbation of phagosomal membranes in order to improve induction of CD8+ T cells by the parental BCG strain (34, 35). In addition, leakage of phagolysosomal proteases such as cathepsins into the cytosol could activate caspases, leading to apoptosis and subsequent cross-presentation of mycobacterial antigens, which promotes both MHC I and MHC II restricted T cell stimulation (36). Studies with L. monocytogenes have shown that pore formation by LLO also triggers many downstream effects such as activation of the NLRP3 inflammasome, induction of cytokine expression, activation of kinases, triggering of endocytosis, histone modification and release of calcium from intracellular stores (37). An Hly recombinant strain, hly+ rBCG+, was generated by integrating the hly gene into BCG using the mycobacteria-Escherichia coli shuttle vector pMV306 (34). LLO was detected in the membrane structures, phagosomal space, and cytoplasmic vacuoles of macrophages infected with BCG pMV306::hly, and intracellular persistence of this strain was reduced compared with the parental BCG strain. MHC I presentation of co-phagocytosed soluble protein was improved in macrophages infected with this strain compared to BCG (34) and an in vitro human cytotoxic T lymphocyte (CTL) assay using cultured DCs and T cells from healthy human donors demonstrated that hly+ BCG infection was better at inducing CTL responses than BCG infection (38). In the next generation strain, deletion of ureC was performed to ensure an optimal (acidic) pH for LLO stability; however, absence of ureC also promotes MHCII trafficking to the macrophage surface (31), which would also stimulate CD4+ T cell responses. To generate ΔureC hly+ BCG, the chromosomal integrative shuttle vector pMV306hyg-hly (8) was used to transform M. bovis BCG ΔureC::aph, and hygromycin-resistant clones were selected (35). The vaccine was licensed to Vakzine Projekt Management, and named “VPM1002.” The resistance cassette was subsequently successfully removed, although VPM1002 is equally sensitive to the antimycobacterial agents isoniazid, rifampicin, and ethambutanol in the presence or absence of the hygromycin resistance gene (39).
Increased quantities of mycobacterial antigen were detected in VMP1002 infected macrophages compared to BCG infected macrophages (35), and mycobacterial DNA was detected only in the cytosol of VPM1002 infected but not BCG infected macrophages (29), indicating that expression of LLO in BCG ΔureC::hly allows the escape of bacterial products to the cytosol, presumably by perturbation of the phagosomal membrane. The bacteria themselves do not escape to the cytosol, unlike Mtb bacilli (29, 35). Infection of primary human and mouse macrophages demonstrated increased apoptosis after infection with VPM1002 compared to both BCG and BCG::hly, demonstrating the additional benefit of urease C deletion (35). Membrane disruption can facilitate the release of phagolysosomal proteases such as cathepsins into the cytosol, which are known to induce apoptosis (36, 40). Both the presence of mycobacterial proteins in the cytosol and the induction of apoptosis by perforation of the phagosomal membrane could cause increased trafficking of antigens to MHC I pathways (35). Apoptosis results in an increase in both CD8+ and CD4+ T cell responses in mycobacterial infection, suggesting that DCs may transfer efferocytosed antigens to the endocytic system (27, 36). The priming potential of apoptotic vesicles isolated from BCG and VPM1002 infected mouse macrophages was investigated in a co-culture system with splenic DCs and T cells, and VPM1002-infected apoptotic vesicles induced more profound CD4+ and CD8+ T cell responses compared to those infected with BCG (41). Vesicles from VPM1002 infected macrophages also induced higher production of the T helper type (Th)17-polarizing cytokines interleukin (IL)-6 and IL-23, and the immunoregulatory cytokine IL-10 by bone marrow-derived DCs.
Experiments in THP1 macrophages demonstrated that VPM1002 infection leads to activation of multiple caspases (29). The apoptotic effector caspases 3 and 7 were highly activated by VPM1002 in comparison to BCG, as well as caspase 1, which mediates pyroptosis, an inflammatory form of cell death and is an important regulator of the inflammatory response (42). Inflammasomes are multi-protein complexes composed of intracellular sensors and caspase 1. They control activation of caspase 1, which in turn cleaves the precursors of the cytokines IL-1β and IL-18 into their active forms (43). VPM1002 infection increased production of IL-1β and IL-18, which was dependent on AIM2 inflammasome activation but not on NLRP 1 and 3 inflammasome activation. Furthermore, VPM1002 induced increased levels of the autophagy marker microtubule-associated protein light chain 3 in an AIM2- and stimulator of interferon genes (STING)-dependent manner. The AIM2 inflammasome senses cytosolic DNA and is involved in the induction of caspase 1-dependent pyroptosis (44, 45), while STING acts as an essential adaptor protein in the induction of autophagy by cytosolic DNA (25). Autophagy, a protein degradation process induced by stress conditions such as infection, promotes the delivery of cytosolic antigens to MHC trafficking pathways (46, 47). It has also been shown to contribute to innate immunity against mycobacteria and other intracellular pathogens (48, 49). While autophagy was originally thought to be non-specific, it is now known that it can selectively target intracellular pathogens in a process known as xenophagy that involves ubiquitination of pathogen proteins or pathogen-containing endosomes (50). Intriguingly, gene expression of guanylate-binding proteins (GBPs) was also elevated in VPM1002 infected THP-1 macrophages compared to BCG infected macrophages. Interferon-inducible GBPs have multiple roles in inflammasome activation, autophagy, and lysis of pathogen-containing vacuoles and can even directly target the pathogens themselves (51–54). Whether they play a role in the translocation of mycobacterial components from the phagosome into the cytosol during VMP1002 infection remains to be determined.
Disruption of the VPM1002-containing phagosome membrane by LLO and release of mycobacterial DNA into the cytosol appears to have effects in inducing immune responses that are similar to the effects of ESX-1 activity in Mtb or M. marinum. ESX-1 of M. marinum stimulates autophagosome formation and recruitment to the vacuole; however, unlike LLO it also inhibits autophagic flux, thereby preventing bacterial degradation (49). Testing of vaccine candidates expressing ESX-1 such as Mtb Δppe25-pe19 (55) and BCG expressing ESX-1 of M. marinum (BCG:ESX-1Mmar) (56) demonstrated that ESX-1 was critical for enhancing innate immune responses via phagosome rupture. BCG:ESX-1Mmar induced the cGas/STING/TBK1/IRF-3/type I interferon axis and promoted AIM2 and NLRP3 inflammasome activation, resulting in increased frequencies of antigen-specific CD8+ and CD4+ T cells and increased protection against Mtb compared to BCG (56), while Mtb Δppe25-pe19 also led to enhanced protection. ESX-1 may induce protective immunity by an additional mechanism, as ESAT6 is required for rapid, non-cognate IFN-γ production by CD8+ T cells, mediated by the NLRP3/caspase-1/IL-18 axis (57).
Aerosol challenge of vaccinated BALB/c or C57BL6 mice with 100–200 colony-forming units (CFUs) of Mtb H37Rv or a clinical isolate of the Beijing/W genotype family demonstrated that VPM1002 immunization has significantly greater protective efficacy than the parental BCG strain, with bacterial loads in the lungs typically reduced by one to two logs in late stages of infection (35, 58–61). In a low dose infection (30 CFU), VPM1002 led to an almost 1000-fold reduction of Mtb in the lungs compared to naïve mice at day 200 after infection (35). Homologous boosting with VPM1002 did not improve protection compared to a single immunization (60). However, a post-exposure vaccination model using antibiotics for an extended period and then allowing bacterial regrowth demonstrated that mice with subclinical TB had lower bacterial burdens when vaccinated with VPM1002 compared to BCG, suggesting that VPM1002 could also be considered for use as a post-exposure vaccine (60).
The safety profile of VPM1002 has been evaluated in animal models including mice, guinea pigs, rabbits, and non-human primates (6). In RAG1-/- immunodeficient mice lacking mature T and B cells, bacterial loads were not significantly different in lungs and spleen after vaccination with VPM1002 compared to BCG (35). However, VPM1002 demonstrated substantially lower virulence in severe combined immunodeficiency mice, most likely due to the reduced intracellular persistence of this strain (35, 61). After immunization of wildtype BALB/c or C57BL6 mice, VPM1002 was more rapidly cleared from the draining lymph nodes than BCG and disseminated less to the spleens, where it was also quickly cleared (59, 61). Dissemination to the lungs was observed in BCG vaccinated but not VPM1002 vaccinated mice. Enhanced adaptive immune responses after VPM1002 vaccination are therefore likely to play a role in the reduced dissemination of VPM1002 in immunocompetent mice. Overall, the data demonstrate increased safety and protective efficacy of VPM1002 compared to parental BCG in mice.
In guinea pigs and non-human primates, the safety of VPM1002 was comparable to that of BCG (6, 39). As the primary target population for vaccination against TB is newborns, the safety profiles of VPM1002 and BCG were also compared in newborn rabbits (39). No dissemination to tissues was observed after VPM1002 administration, and the body weight gain was not affected during the 90 days observation period, whereas the body weight was reduced in the BCG vaccinated group compared to the saline control group. No premature mortality was observed in either group. The preclinical safety of VPM1002 is thus supported by a large body of evidence.
Analysis of gene expression in mice early after immunization with VPM1002 demonstrated that, as in THP-1 cells, expression of IL-18 and IL-1β was increased, as well as expression of IFN-inducible genes such as Tmem173 (STING), Gbp’s, and other GTPases (29, 61). Apoptosis was increased in the lymph nodes of VPM1002 immunized mice compared with BCG immunized mice by day 14 (61). Immunization with VMP1002 induced both type 1 and type 17 cytokine responses in mice, whereas BCG induced type 1 responses only (58). After restimulation with PPD, levels of IFN-γ, IL-17, IL-2, IL-6, and GM-CSF were increased in lung cells isolated from VPM1002 immunized mice compared to those from BCG immunized mice, and splenocytes from VPM1002-vaccinated mice also produced more IL-17. Furthermore, percentages of γδ T cells producing IFN-γ and IL-17 were increased after vaccination with VPM1002 (58). Seven days after Mtb challenge, IL-2+TNF+ double cytokine producing cells were increased in the lungs of VPM1002-immunized mice compared with BCG-vaccinated mice, suggesting recall responses, because newly generated T cells take 12–14 days to reach the lungs during Mtb infection (62). IL-2+TNF+ CD4+ T cells typically show a central memory phenotype (TCM) (21), and further studies demonstrated that VPM1002 immunization indeed induces higher frequencies of TCM than immunization with BCG (59, 61). Ag85B-specific CD4+ TCM were significantly increased in the draining lymph nodes of VPM1002-vaccinated compared to BCG-vaccinated mice at day 14 (59).
Bacille Calmette–Guérin induces effector memory CD4+ T (TEM) cells that can control acute infection but appears to induce insufficient numbers of TCM cells for long-term protection (21). Transfer studies demonstrated that TCM cells from VPM1002 infected mice conferred protection against TB infection whereas TEM, T follicular helper (TFH), and naïve T cells did not, at least at the numbers of cells tested (59). These findings concur with other studies in which TCM cells were associated with protection (21). While TEM cells appear early after infection and provide protection by the secretion of effector cytokines such as IFN-γ and TNF-α, TCM cells proliferate in the LN and generate new pools of TEM cells after re-exposure to antigen (59, 63, 64). The TCM cells generated by subcutaneous vaccination with VPM1002 or BCG were found to reside over the long term in the secondary lymphoid organs, rather than in the lung, and to be recruited to the lungs after Mtb challenge (58, 59). Waning of BCG-induced immunity correlates with a decline in T cell functions such as cytokine production and CTL activity and an increase in terminally differentiated, dysfunctional T cells (65). Thus, systemic maintenance of TCM populations over the long term and the rapid recruitment of TCM cells to the lung following Mtb infection remains a key goal in the development of more effective vaccine candidates (59). VPM1002 also induced an increase in mycobacteria-specific immunoglobulin G levels after vaccination compared to BCG, and a concomitant increase in CXCR5-expressing TFH cells (59, 61), which have been associated with decreased lung pathology (66) and stimulate germinal center B cell responses (63). Passive transfer of serum from VPM1002- or BCG-immunized mice on the day of Mtb infection and thrice weekly did not reduce bacterial load at day 14 (59), but growing evidence suggests that antibodies may play a role in protection against Mtb (67–71). Overall, increased protection conferred by VPM1002 immunization in the mouse model was associated with increased numbers of TCM and TFH cells, increased Th17 responses, earlier recruitment of T cells to the lungs following Mtb challenge and increased levels of anti-mycobacterial antibodies (58, 59, 61).
Human data on VPM1002 are available from three clinical trials, all performed with the original hygromycin-resistant strain of VPM1002. Two Phase I studies were performed in healthy adult volunteers, and one Phase IIa study was conducted in healthy newborn infants, one of the intended target populations. In the first Phase I clinical trial (ClinicalTrials.gov Identifier: NCT00749034) conducted in Germany, healthy Caucasian adult males with (W) or without (WO) a history of BCG vaccination received VPM1002 randomized to three escalating doses (N = 30W + 30WO) or BCG at the standard vaccine dose (N = 10W + 10WO) and were followed for 6 months. Single vaccination with VPM1002 up to 5 × 10e5 CFU was safe and well tolerated. The immunogenicity of VPM1002 as measured by IFN-γ release by stimulated T cells was dose dependent. Both VPM1002 and BCG induced multifunctional CD4+ and CD8+ T cell subsets, which are thought to play a role in protection against TB (72–74), with VPM1002 showing an earlier increase in double and triple cytokine producing T cells which remained at heightened levels throughout the study (7). Furthermore, only VPM1002 induced serum antibodies against mycobacterial antigens (7), echoing preclinical studies in which VPM1002 induced higher levels of mycobacteria-specific antibodies than BCG in mice (59, 61). In the second Phase I clinical trial (ClinicalTrials.gov Identifier: NCT01113281), performed in South Africa, 24 healthy male and female adults with a history of BCG immunization, predominantly from the indigenous African population, were vaccinated with VPM1002. The study showed that a single vaccination with VPM1002 is safe, well tolerated and elicits a profound immune response in an African adult population (6).
The Phase IIa clinical trial (ClinicalTrials.gov Identifier: NCT01479972) was the first investigation of VPM1002 in newborns (75). It was conducted in Cape Town, South Africa, a region with a high TB burden. Forty-eight HIV-unexposed, newborn infants were vaccinated with either VPM1002 (n = 36) or BCG (n = 12) through an open label, randomized, controlled design. Polyfunctional CD4+ and CD8+ T cell responses were similar between the groups, and both groups had increased IFN-γ responses after 7 h PPD stimulation at all measured time points post vaccination compared to baseline. Both vaccines induced IL-17 responses; though, unlike BCG, VPM1002 induced increased proportions of CD8+ IL-17+ T cells at day 14 and month 6 time points compared to the baseline. The incidence of abscess formation was lower for VPM1002 compared to BCG. Thus, VPM1002 was safe, well tolerated, and immunogenic in newborn infants.
In addition, a Phase IIb clinical trial is currently ongoing in South Africa (ClinicalTrials.gov Identifier: NCT02391415). This trial is a double-blind, randomized, controlled study to evaluate the safety and immunogenicity of VPM1002 in comparison with BCG in HIV-exposed uninfected (HEU) and HIV-unexposed, BCG-naive newborn infants. The inclusion of HEU infants in the trial is important, as this group comprises 30% of the newborns requiring BCG vaccination in South Africa, and they may be at higher risk of Mtb infection than HIV-unexposed infants. The proportion of HEU may vary in different countries. Previous work from Brazil suggests that HEU infants have poorer T-cell proliferation and lower levels of IFN-γ production compared to HIV-unexposed infants (76). Enrollment of 416 infants has been completed and follow-up is in progress. Follow-up will continue for 12 months, as opposed to 6 months in NCT01479972, enabling collection of preliminary efficacy data.
In addition to its development as a vaccine for newborns, VPM1002 is also being assessed as a post-exposure vaccine for adults, since preclinical studies in mice demonstrated that it reduced bacterial loads in a post exposure model (60). A phase II/III trial has received regulatory approval by the Indian authorities. Once ethics committee approvals are received for all sites, the trial will commence across India (ClinicalTrials.gov Identifier: NCT03152903). The study will be conducted in 2000 adults who were TB patients, but received drug treatment and were cured of disease. In such populations, there is a high risk of recurrence (including re-infection and relapse), especially within 12 months after completing treatment. The multi-centric, placebo-controlled, randomized, controlled study will assess whether VPM1002 can prevent such TB recurrence over a 1-year follow-up period. Currently, no intervention is licensed for this indication, including BCG, which means there is clearly an unmet medical need. The study will also expand the safety database on VPM1002.
Bladder cancer is the ninth most common cancer in the world, and is four times more common in men than in women (77). The main risk factors for developing bladder cancer include smoking, Schistosoma infection (bilharzia), and exposure to industrial chemicals (77, 78). Tumors can be non-muscle invasive, i.e., confined to the mucosa of the bladder wall, or muscle-invasive. More than seventy percent of bladder cancers are detected while they are still non-muscle invasive (79). Due to its immunostimulatory properties, repeated intravesical BCG instillation is the standard adjuvant treatment for intermediate to high-risk non-muscle-invasive bladder cancer (NMIBC) after transurethral resection of the tumors (80–82). BCG therapy reduces the risk of recurrence and the progression to muscle invasive bladder cancer. The repeated instillations require much higher doses and volumes of BCG than vaccination against TB does, and some patients have adverse events that lead to discontinuation of the therapy (83, 84). Adverse events include fever, bladder irritation, decreased bladder capacity, incontinence, hematuria, flu-like symptoms and in approximately 5% of cases, BCG infection (85, 86). Patients undergoing traumatic catheterization are at risk for intraluminal BCG dissemination, resulting in a potentially lethal systemic infection (87).
The precise immune mechanisms by which BCG promotes anti-tumor activity in bladder cancer are not completely resolved, but it is well-established that the ability of BCG to promote Th1 responses is important, as well as the recruitment of neutrophils and innate lymphocytes including natural killer cells (82, 88, 89). Activation of immune cells may lead to elimination of the urothelial cancerous cells that have internalized BCG (82, 90). Increased CD4+ T cell responses have been measured during BCG therapy, and BCG was shown to promote secretion of both Th1- and Th2-type cytokines (88, 91–93). A positive response to BCG therapy (no recurrence or evidence of disease during follow-up examinations) has been associated with an intratumoral Th2 predisposition (increased GATA3) and decreased concentrations of IL-10, combined with a Th1 functional phenotype indicated by increased levels of Th1-related inflammatory metabolites (88). In another study, increased regulatory T cells and tumor-associated macrophages in the tumor microenvironment were also associated with non-responsiveness, while increased GATA3+ and CD4+ T cells were associated with responders (88, 94). BCG Connaught conferred greater 5-year recurrence-free survival than BCG Tice and induced stronger Th1 type responses, BCG-specific CD8+ T cells and T cell recruitment to the bladder (93). Genetic analysis demonstrated several differences between the two strains, including the absence of RD15 in BCG Connaught (93).
Because approximately 30–40% of patients do not respond to BCG therapy and others suffer from adverse events, rBCG technology has been tested for improving the efficacy and tolerability of BCG in bladder cancer therapy (82). rBCGs that have been modified to express immunostimulatory molecules, cytokines, or antigens have been tested in mice for their capacity to induce stronger and more specific immune responses. VPM1002 is currently being evaluated in SAKK 06/14, a Phase I/II trial for immunotherapy in patients with NMIBC (ClinicalTrials.gov Identifier: NCT02371447). The phase I part of the trial has been completed in Switzerland. Intravesical application of VPM1002BC demonstrated that the product is safe and well tolerated in NMIBC patients. The recommended phase II dose has been established as 1–19.2 × 10e8 CFUs of VPM1002BC. The phase II part has been approved by the Swiss and German regulatory authorities and is currently ongoing in both countries.
The available preclinical and clinical data reveal that VPM1002 is immunogenic and may be better than BCG in terms of safety. VPM1002 could be a safe, well-tolerated and efficacious alternative to the BCG vaccine in the future. With an annual capacity of 100 million doses, Serum Institute of India Pvt. Ltd. can meet the global demand for a BCG vaccine and is well poised to supply the new vaccine if efficacy trials are successful. While this vaccine progresses through efficacy trials, next-generation derivatives are being designed and tested in preclinical models aimed at optimizing efficacy and/or safety (61, 95). Furthermore, VPM1002 is currently being tested in goats by the Friedrich Loeffler Institute in Germany for the prevention of M. caprae infection (Menge et al. unpublished data). Infections with M. caprae and M. bovis, closely related species of the same clade that cause TB in goats and cattle, respectively, are of agricultural importance, and can potentially be transmitted to humans (96, 97).
Almost 100 years after the first immunization with BCG, a rBCG vaccine candidate is ready for clinical efficacy testing. This marks a major step forward in the long journey that began when the recombinant vaccine was constructed in the late 1990s and tested in different animal models to determine its safety and protective effect.
NN, LG, and SK wrote and reviewed the manuscript. All other authors (BE, PK, US, CR, and MC) reviewed the manuscript.
SK and LG are co-inventors/patent holders of BCG ΔureC::hly (VPM1002). BE and LG are working for the Vakzine Projekt Management GmbH who is involved with the development of VPM1002. PK and US are employed by Serum Institute of India Pvt. Ltd., which manufactures VPM1002. NN, CR, and MC declare that they have no conflicts of interest.
The authors thank Souraya Sibaei for her help in preparing the manuscript and Diane Schad for excellent graphics work. This work was supported by The European Union’s Seventh Framework Programme (EU FP7) ADITEC (HEALTH-F4-2011-280873); by the EU Horizon 2020 project TBVAC 2020 (grant no. 643381); The Bill & Melinda Gates Foundation (BMGF) GC6-2013, #OPP 1055806 and #OPP 1065330; the Bundesministerium für Bildung und Forschung (BMBF) project “Infect Control 2020” (grant no. 03ZZ0806A).
AIM2, absent in melanoma 2; BCG, bacille Calmette–Guérin; CFUs, colony forming units; CTL, human cytotoxic lymphocyte; DC, dendritic cell; EPI, expanded program of immunization; GBP, guanylate binding protein; HEU, HIV-exposed uninfected; IgG, immunoglobulin G; LC3, microtubule-associated protein light chain 3; LLO, listeriolysin O; MHC, major histocompatibility complex; Mtb, Mycobacterium tuberculosis; NLRP3, NLR family pyrin domain-containing 3; NMIBC, non-muscle-invasive bladder cancer; rBCG, recombinant BCG; RD1, Region of Difference 1; SCID, severe combined immunodeficiency; STING, stimulator of interferon genes; TB, tuberculosis; TCM, central memory T cells; TEM, effector memory T cells; TFH, follicular T cells; Th, T helper cell.
1. Bekkering S, Blok BA, Joosten LA, Riksen NP, van Crevel R, Netea MG.In vitro experimental model of trained innate immunity in human primary monocytes. Clin Vaccine Immunol (2016) 23(12):926–33. doi:10.1128/cvi.00349-16
2. Aaby P, Kollmann TR, Benn CS. Nonspecific effects of neonatal and infant vaccination: public-health, immunological and conceptual challenges. Nat Immunol (2014) 15(10):895–9. doi:10.1038/ni.2961
3. Kandasamy R, Voysey M, McQuaid F, de Nie K, Ryan R, Orr O, et al. Non-specific immunological effects of selected routine childhood immunisations: systematic review. BMJ (2016) 355:i5225. doi:10.1136/bmj.i5225
4. Talbot EA, Perkins MD, Silva SF, Frothingham R. Disseminated bacille Calmette-Guerin disease after vaccination: case report and review. Clin Infect Dis (1997) 24(6):1139–46. doi:10.1086/513642
5. Kaufmann SH, Lange C, Rao M, Balaji KN, Lotze M, Schito M, et al. Progress in tuberculosis vaccine development and host-directed therapies – a state of the art review. Lancet Respir Med (2014) 2(4):301–20. doi:10.1016/S2213-2600(14)70033-5
6. Kaufmann SH, Cotton MF, Eisele B, Gengenbacher M, Grode L, Hesseling AC, et al. The BCG replacement vaccine VPM1002: from drawing board to clinical trial. Expert Rev Vaccines (2014) 13(5):619–30. doi:10.1586/14760584.2014.905746
7. Grode L, Ganoza CA, Brohm C, Weiner J III, Eisele B, Kaufmann SH. Safety and immunogenicity of the recombinant BCG vaccine VPM1002 in a phase 1 open-label randomized clinical trial. Vaccine (2013) 31(9):1340–8. doi:10.1016/j.vaccine.2012.12.053
8. Reyrat JM, Berthet FX, Gicquel B. The urease locus of Mycobacterium tuberculosis and its utilization for the demonstration of allelic exchange in Mycobacterium bovis bacillus Calmette-Guerin. Proc Natl Acad Sci U S A (1995) 92(19):8768–72. doi:10.1073/pnas.92.19.8768
9. Gordon AH, Hart PD, Young MR. Ammonia inhibits phagosome-lysosome fusion in macrophages. Nature (1980) 286(5768):79–80. doi:10.1038/286079a0
10. Hamon MA, Ribet D, Stavru F, Cossart P. Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol (2012) 20(8):360–8. doi:10.1016/j.tim.2012.04.006
11. Shaughnessy LM, Hoppe AD, Christensen KA, Swanson JA. Membrane perforations inhibit lysosome fusion by altering pH and calcium in Listeria monocytogenes vacuoles. Cell Microbiol (2006) 8(5):781–92. doi:10.1111/j.1462-5822.2005.00665.x
13. Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, et al. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect Immun (2006) 74(1):88–98. doi:10.1128/IAI.74.1.88-98.2006
14. Simeone R, Bottai D, Brosch R. ESX/type VII secretion systems and their role in host-pathogen interaction. Curr Opin Microbiol (2009) 12(1):4–10. doi:10.1016/j.mib.2008.11.003
15. Conrad WH, Osman MM, Shanahan JK, Chu F, Takaki KK, Cameron J, et al. Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc Natl Acad Sci U S A (2017) 114(6):1371–6. doi:10.1073/pnas.1620133114
16. Muller I, Cobbold SP, Waldmann H, Kaufmann SH. Impaired resistance to Mycobacterium tuberculosis infection after selective in vivo depletion of L3T4+ and Lyt-2+ T cells. Infect Immun (1987) 55(9):2037–41.
17. Kaufmann SH. Tuberculosis vaccine development: strength lies in tenacity. Trends Immunol (2012) 33(7):373–9. doi:10.1016/j.it.2012.03.004
18. Sharpe S, White A, Sarfas C, Sibley L, Gleeson F, McIntyre A, et al. Alternative BCG delivery strategies improve protection against Mycobacterium tuberculosis in non-human primates: protection associated with mycobacterial antigen-specific CD4 effector memory T-cell populations. Tuberculosis (Edinb) (2016) 101:174–90. doi:10.1016/j.tube.2016.09.004
19. Chen CY, Huang D, Wang RC, Shen L, Zeng G, Yao S, et al. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS Pathog (2009) 5(4):e1000392. doi:10.1371/journal.ppat.1000392
20. Ritz N, Hanekom WA, Robins-Browne R, Britton WJ, Curtis N. Influence of BCG vaccine strain on the immune response and protection against tuberculosis. FEMS Microbiol Rev (2008) 32(5):821–41. doi:10.1111/j.1574-6976.2008.00118.x
21. Lindenstrom T, Knudsen NP, Agger EM, Andersen P. Control of chronic Mycobacterium tuberculosis infection by CD4 KLRG1- IL-2-secreting central memory cells. J Immunol (2013) 190(12):6311–9. doi:10.4049/jimmunol.1300248
22. Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol (2007) 178(5):3143–52. doi:10.4049/jimmunol.178.5.3143
23. Dorhoi A, Nouailles G, Jorg S, Hagens K, Heinemann E, Pradl L, et al. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol (2012) 42(2):374–84. doi:10.1002/eji.201141548
24. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe (2015) 17(6):799–810. doi:10.1016/j.chom.2015.05.003
25. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell (2012) 150(4):803–15. doi:10.1016/j.cell.2012.06.040
26. Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med (2003) 9(8):1039–46. doi:10.1038/nm906
27. Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, et al. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity (2006) 24(1):105–17. doi:10.1016/j.immuni.2005.12.001
28. Tzelepis F, Verway M, Daoud J, Gillard J, Hassani-Ardakani K, Dunn J, et al. Annexin1 regulates DC efferocytosis and cross-presentation during Mycobacterium tuberculosis infection. J Clin Invest (2015) 125(2):752–68. doi:10.1172/JCI77014
29. Saiga H, Nieuwenhuizen N, Gengenbacher M, Koehler AB, Schuerer S, Moura-Alves P, et al. The recombinant BCG ΔureC::hly vaccine targets the AIM2 inflammasome to induce autophagy and inflammation. J Infect Dis (2015) 211(11):1831–41. doi:10.1093/infdis/jiu675
30. Pedrazzini T, Hug K, Louis JA. Importance of L3T4+ and Lyt-2+ cells in the immunologic control of infection with Mycobacterium bovis strain bacillus Calmette-Guerin in mice. Assessment by elimination of T cell subsets in vivo. J Immunol (1987) 139(6):2032–7.
31. Sendide K, Deghmane AE, Reyrat JM, Talal A, Hmama Z. Mycobacterium bovis BCG urease attenuates major histocompatibility complex class II trafficking to the macrophage cell surface. Infect Immun (2004) 72(7):4200–9. doi:10.1128/IAI.72.7.4200-4209.2004
32. Wang Y, Curry HM, Zwilling BS, Lafuse WP. Mycobacteria inhibition of IFN-gamma induced HLA-DR gene expression by up-regulating histone deacetylation at the promoter region in human THP-1 monocytic cells. J Immunol (2005) 174(9):5687–94. doi:10.4049/jimmunol.174.9.5687
33. Fulton SA, Reba SM, Pai RK, Pennini M, Torres M, Harding CV, et al. Inhibition of major histocompatibility complex II expression and antigen processing in murine alveolar macrophages by Mycobacterium bovis BCG and the 19-kilodalton mycobacterial lipoprotein. Infect Immun (2004) 72(4):2101–10. doi:10.1128/IAI.72.4.2101-2110.2004
34. Hess J, Miko D, Catic A, Lehmensiek V, Russell DG, Kaufmann SH. Mycobacterium bovis bacille Calmette-Guerin strains secreting listeriolysin of Listeria monocytogenes. Proc Natl Acad Sci U S A (1998) 95(9):5299–304. doi:10.1073/pnas.95.9.5299
35. Grode L, Seiler P, Baumann S, Hess J, Brinkmann V, Nasser Eddine A, et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guerin mutants that secrete listeriolysin. J Clin Invest (2005) 115(9):2472–9. doi:10.1172/JCI24617
36. Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol (2010) 11(8):751–8. doi:10.1038/ni.1904
37. Podobnik M, Marchioretto M, Zanetti M, Bavdek A, Kisovec M, Cajnko MM, et al. Plasticity of listeriolysin O pores and its regulation by pH and unique histidine [corrected]. Sci Rep (2015) 5:9623. doi:10.1038/srep09623
38. Conradt P, Hess J, Kaufmann SH. Cytolytic T-cell responses to human dendritic cells and macrophages infected with Mycobacterium bovis BCG and recombinant BCG secreting listeriolysin. Microbes Infect (1999) 1(10):753–64. doi:10.1016/S1286-4579(99)80077-X
39. Velmurugan K, Grode L, Chang R, Fitzpatrick M, Laddy D, Hokey D, et al. Nonclinical development of BCG replacement vaccine candidates. Vaccines (Basel) (2013) 1(2):120–38. doi:10.3390/vaccines1020120
40. Leist M, Jaattela M. Triggering of apoptosis by cathepsins. Cell Death Differ (2001) 8(4):324–6. doi:10.1038/sj.cdd.4400859
41. Farinacci M, Weber S, Kaufmann SH. The recombinant tuberculosis vaccine rBCG ΔureC::hly(+) induces apoptotic vesicles for improved priming of CD4(+) and CD8(+) T cells. Vaccine (2012) 30(52):7608–14. doi:10.1016/j.vaccine.2012.10.031
42. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun (2005) 73(4):1907–16. doi:10.1128/IAI.73.4.1907-1916.2005
43. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi:10.1146/annurev.immunol.021908.132715
44. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458(7237):509–13. doi:10.1038/nature07710
45. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458(7237):514–8. doi:10.1038/nature07725
46. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell (2008) 132(1):27–42. doi:10.1016/j.cell.2007.12.018
47. Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med (2009) 15(3):267–76. doi:10.1038/nm.1928
48. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13(10):722–37. doi:10.1038/nri3532
49. Cardenal-Munoz E, Arafah S, Lopez-Jimenez AT, Kicka S, Falaise A, Bach F, et al. Mycobacterium marinum antagonistically induces an autophagic response while repressing the autophagic flux in a TORC1- and ESX-1-dependent manner. PLoS Pathog (2017) 13(4):e1006344. doi:10.1371/journal.ppat.1006344
50. Shibutani ST, Saitoh T, Nowag H, Munz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol (2015) 16(10):1014–24. doi:10.1038/ni.3273
51. Kravets E, Degrandi D, Ma Q, Peulen TO, Klumpers V, Felekyan S, et al. Guanylate binding proteins directly attack Toxoplasma gondii via supramolecular complexes. Elife (2016) 5:e11479. doi:10.7554/eLife.11479
52. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature (2014) 509(7500):366–70. doi:10.1038/nature13157
53. Kim BH, Chee JD, Bradfield CJ, Park ES, Kumar P, MacMicking JD. Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat Immunol (2016) 17(5):481–9. doi:10.1038/ni.3440
54. Man SM, Place DE, Kuriakose T, Kanneganti TD. Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. J Leukoc Biol (2017) 101(1):143–50. doi:10.1189/jlb.4MR0516-223R
55. Sayes F, Pawlik A, Frigui W, Groschel MI, Crommelynck S, Fayolle C, et al. CD4+ T cells recognizing PE/PPE antigens directly or via cross reactivity are protective against pulmonary Mycobacterium tuberculosis infection. PLoS Pathog (2016) 12(7):e1005770. doi:10.1371/journal.ppat.1005770
56. Groschel MI, Sayes F, Shin SJ, Frigui W, Pawlik A, Orgeur M, et al. Recombinant BCG expressing ESX-1 of Mycobacterium marinum combines low virulence with cytosolic immune signaling and improved TB protection. Cell Rep (2017) 18(11):2752–65. doi:10.1016/j.celrep.2017.02.057
57. Kupz A, Zedler U, Staber M, Perdomo C, Dorhoi A, Brosch R, et al. ESAT-6-dependent cytosolic pattern recognition drives noncognate tuberculosis control in vivo. J Clin Invest (2016) 126(6):2109–22. doi:10.1172/JCI84978
58. Desel C, Dorhoi A, Bandermann S, Grode L, Eisele B, Kaufmann SH. Recombinant BCG DeltaureC hly+ induces superior protection over parental BCG by stimulating a balanced combination of type 1 and type 17 cytokine responses. J Infect Dis (2011) 204(10):1573–84. doi:10.1093/infdis/jir592
59. Vogelzang A, Perdomo C, Zedler U, Kuhlmann S, Hurwitz R, Gengenbacher M, et al. Central memory CD4+ T cells are responsible for the recombinant bacillus Calmette-Guerin ΔureC::hly vaccine’s superior protection against tuberculosis. J Infect Dis (2014) 210(12):1928–37. doi:10.1093/infdis/jiu347
60. Gengenbacher M, Kaiser P, Schuerer S, Lazar D, Kaufmann SH. Post-exposure vaccination with the vaccine candidate bacillus Calmette-Guerin ΔureC::hly induces superior protection in a mouse model of subclinical tuberculosis. Microbes Infect (2016) 18(5):364–8. doi:10.1016/j.micinf.2016.03.005
61. Gengenbacher M, Nieuwenhuizen N, Vogelzang A, Liu H, Kaiser P, Schuerer S, et al. Deletion of nuoG from the vaccine candidate Mycobacterium bovis BCG ΔureC::hly improves protection against tuberculosis. MBio (2016) 7(3):e679–616. doi:10.1128/mBio.00679-16
62. Griffiths KL, Ahmed M, Das S, Gopal R, Horne W, Connell TD, et al. Targeting dendritic cells to accelerate T-cell activation overcomes a bottleneck in tuberculosis vaccine efficacy. Nat Commun (2016) 7:13894. doi:10.1038/ncomms13894
63. Chevalier N, Jarrossay D, Ho E, Avery DT, Ma CS, Yu D, et al. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J Immunol (2011) 186(10):5556–68. doi:10.4049/jimmunol.1002828
64. Knudsen NP, Olsen A, Buonsanti C, Follmann F, Zhang Y, Coler RN, et al. Different human vaccine adjuvants promote distinct antigen-independent immunological signatures tailored to different pathogens. Sci Rep (2016) 6:19570. doi:10.1038/srep19570
65. Nandakumar S, Kannanganat S, Posey JE, Amara RR, Sable SB. Attrition of T-cell functions and simultaneous upregulation of inhibitory markers correspond with the waning of BCG-induced protection against tuberculosis in mice. PLoS One (2014) 9(11):e113951. doi:10.1371/journal.pone.0113951
66. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest (2013) 123(2):712–26. doi:10.1172/JCI65728
67. Li H, Wang XX, Wang B, Fu L, Liu G, Lu Y, et al. Latently and uninfected healthcare workers exposed to TB make protective antibodies against Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2017) 114(19):5023–8. doi:10.1073/pnas.1611776114
68. Lu LL, Chung AW, Rosebrock TR, Ghebremichael M, Yu WH, Grace PS, et al. A functional role for antibodies in tuberculosis. Cell (2016) 167(2):433–43.e14. doi:10.1016/j.cell.2016.08.072
69. Zimmermann N, Thormann V, Hu B, Kohler AB, Imai-Matsushima A, Locht C, et al. Human isotype-dependent inhibitory antibody responses against Mycobacterium tuberculosis. EMBO Mol Med (2016) 8(11):1325–39. doi:10.15252/emmm.201606330
70. Prados-Rosales R, Carreno L, Cheng T, Blanc C, Weinrick B, Malek A, et al. Enhanced control of Mycobacterium tuberculosis extrapulmonary dissemination in mice by an arabinomannan-protein conjugate vaccine. PLoS Pathog (2017) 13(3):e1006250. doi:10.1371/journal.ppat.1006250
71. Achkar JM, Chan J, Casadevall A. B cells and antibodies in the defense against Mycobacterium tuberculosis infection. Immunol Rev (2015) 264(1):167–81. doi:10.1111/imr.12276
72. Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol (2008) 8(4):247–58. doi:10.1038/nri2274
73. Wilkinson KA, Wilkinson RJ. Polyfunctional T cells in human tuberculosis. Eur J Immunol (2010) 40(8):2139–42. doi:10.1002/eji.201040731
74. Lindenstrom T, Agger EM, Korsholm KS, Darrah PA, Aagaard C, Seder RA, et al. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J Immunol (2009) 182(12):8047–55. doi:10.4049/jimmunol.0801592
75. Loxton AG, Knaul JK, Grode L, Gutschmidt A, Meller C, Eisele B, et al. Safety and immunogenicity of the recombinant Mycobacterium bovis BCG vaccine VPM1002 in HIV-unexposed newborn infants in South Africa. Clin Vaccine Immunol (2017) 24(2):e439–516. doi:10.1128/CVI.00439-16
76. Mazzola TN, da Silva MT, Abramczuk BM, Moreno YM, Lima SC, Zorzeto TQ, et al. Impaired bacillus Calmette-Guerin cellular immune response in HIV-exposed, uninfected infants. AIDS (2011) 25(17):2079–87. doi:10.1097/QAD.0b013e32834bba0a
77. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol (2017) 71(1):96–108. doi:10.1016/j.eururo.2016.06.010
78. Sanli O, Dobruch J, Knowles MA, Burger M, Alemozaffar M, Nielsen ME, et al. Bladder cancer. Nat Rev Dis Primers (2017) 3:17022. doi:10.1038/nrdp.2017.22
79. Sexton WJ, Wiegand LR, Correa JJ, Politis C, Dickinson SI, Kang LC. Bladder cancer: a review of non-muscle invasive disease. Cancer Control (2010) 17(4):256–68. doi:10.1177/107327481001700406
80. Fuge O, Vasdev N, Allchorne P, Green JS. Immunotherapy for bladder cancer. Res Rep Urol (2015) 7:65–79. doi:10.2147/RRU.S63447
81. Babjuk M, Bohle A, Burger M, Capoun O, Cohen D, Comperat EM, et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: update 2016. Eur Urol (2017) 71(3):447–61. doi:10.1016/j.eururo.2016.05.041
82. Zheng YQ, Naguib YW, Dong Y, Shi YC, Bou S, Cui Z. Applications of bacillus Calmette-Guerin and recombinant bacillus Calmette-Guerin in vaccine development and tumor immunotherapy. Expert Rev Vaccines (2015) 14(9):1255–75.
83. Mostafid AH, Palou Redorta J, Sylvester R, Witjes JA. Therapeutic options in high-risk non-muscle-invasive bladder cancer during the current worldwide shortage of bacille Calmette-Guerin. Eur Urol (2015) 67(3):359–60. doi:10.1016/j.eururo.2014.11.031
84. Chou R, Selph S, Buckley DI, Fu R, Griffin JC, Grusing S, et al. Intravesical therapy for the treatment of nonmuscle invasive bladder cancer: a systematic review and meta-analysis. J Urol (2017) 197(5):1189–99. doi:10.1016/j.juro.2016.12.090
85. Xie J, Codd C, Mo K, He Y. Differential adverse event profiles associated with BCG as a preventive tuberculosis vaccine or therapeutic bladder cancer vaccine identified by comparative ontology-based VAERS and literature meta-analysis. PLoS One (2016) 11(10):e0164792. doi:10.1371/journal.pone.0164792
86. Gonzalez-Del Vecchio M, Ruiz-Serrano MJ, Gijon P, Sanchez-Somolinos M, de Egea V, Garcia de Viedma D, et al. Differences between a probable and proven BCG infection following intravesical instillations: 16 years experience in a tertiary care hospital. Diagn Microbiol Infect Dis (2016) 85(3):338–43. doi:10.1016/j.diagmicrobio.2016.04.006
87. Lukacs S, Tschobotko B, Szabo NA, Symes A. Systemic BCG-osis as a rare side effect of intravesical BCG treatment for superficial bladder cancer. Case Rep Urol (2013) 2013:821526. doi:10.1155/2013/821526
88. Pichler R, Gruenbacher G, Culig Z, Brunner A, Fuchs D, Fritz J, et al. Intratumoral Th2 predisposition combines with an increased Th1 functional phenotype in clinical response to intravesical BCG in bladder cancer. Cancer Immunol Immunother (2017) 66(4):427–40. doi:10.1007/s00262-016-1945-z
89. Suttmann H, Riemensberger J, Bentien G, Schmaltz D, Stockle M, Jocham D, et al. Neutrophil granulocytes are required for effective bacillus Calmette-Guerin immunotherapy of bladder cancer and orchestrate local immune responses. Cancer Res (2006) 66(16):8250–7. doi:10.1158/0008-5472.CAN-06-1416
90. Sapre N, Corcoran NM. Modulating the immune response to bacillus Calmette-Guerin (BCG): a novel way to increase the immunotherapeutic effect of BCG for treatment of bladder cancer? BJU Int (2013) 112(6):852–3. doi:10.1111/bju.12261
91. Ponticiello A, Perna F, Maione S, Stradolini M, Testa G, Terrazzano G, et al. Analysis of local T lymphocyte subsets upon stimulation with intravesical BCG: a model to study tuberculosis immunity. Respir Med (2004) 98(6):509–14. doi:10.1016/j.rmed.2003.12.003
92. Biot C, Rentsch CA, Gsponer JR, Birkhauser FD, Jusforgues-Saklani H, Lemaitre F, et al. Preexisting BCG-specific T cells improve intravesical immunotherapy for bladder cancer. Sci Transl Med (2012) 4(137):137ra72. doi:10.1126/scitranslmed.3003586
93. Rentsch CA, Birkhauser FD, Biot C, Gsponer JR, Bisiaux A, Wetterauer C, et al. Bacillus Calmette-Guerin strain differences have an impact on clinical outcome in bladder cancer immunotherapy. Eur Urol (2014) 66(4):677–88. doi:10.1016/j.eururo.2014.02.061
94. Bahria-Sediki IB, Yousfi N, Paul C, Chebil M, Cherif M, Zermani R, et al. Clinical significance of T-bet, GATA-3, and Bcl-6 transcription factor expression in bladder carcinoma. J Transl Med (2016) 14(1):144. doi:10.1186/s12967-016-0891-z
95. Gengenbacher M, Vogelzang A, Schuerer S, Lazar D, Kaiser P, Kaufmann SH. Dietary pyridoxine controls efficacy of vitamin B6-auxotrophic tuberculosis vaccine bacillus Calmette-Guerin ΔureC::hly Deltapdx1 in mice. MBio (2014) 5(3):e1262–1214. doi:10.1128/mBio.01262-14
96. de la Fuente J, Diez-Delgado I, Contreras M, Vicente J, Cabezas-Cruz A, Tobes R, et al. Comparative genomics of field isolates of Mycobacterium bovis and M. caprae provides evidence for possible correlates with bacterial viability and virulence. PLoS Negl Trop Dis (2015) 9(11):e0004232. doi:10.1371/journal.pntd.0004232
Keywords: tuberculosis, bacille Calmette–Guérin, VPM1002, vaccine, listeriolysin, immune response
Citation: Nieuwenhuizen NE, Kulkarni PS, Shaligram U, Cotton MF, Rentsch CA, Eisele B, Grode L and Kaufmann SHE (2017) The Recombinant Bacille Calmette–Guérin Vaccine VPM1002: Ready for Clinical Efficacy Testing. Front. Immunol. 8:1147. doi: 10.3389/fimmu.2017.01147
Received: 05 July 2017; Accepted: 30 August 2017;
Published: 19 September 2017
Edited by:
Norbert Reiling, Forschungszentrum Borstel (LG), GermanyReviewed by:
Mario M. D’Elios, University of Florence, ItalyCopyright: © 2017 Nieuwenhuizen, Kulkarni, Shaligram, Cotton, Rentsch, Eisele, Grode and Kaufmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan H. E. Kaufmann, a2F1Zm1hbm5AbXBpaWItYmVybGluLm1wZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.