 Wenzhe Song
Wenzhe Song Roberta Mazzieri1
Roberta Mazzieri1 Glenda C. Gobe
Glenda C. Gobe
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 13 September 2017
Sec. Cancer Immunity and Immunotherapy
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01106
The tumor microenvironment determines development and progression of many cancers. Epithelial–mesenchymal transition (EMT) is fundamental to tumor progression and metastasis not only by increasing invasiveness but also by increasing resistance to cell death, senescence, and various cancer therapies; determining inflammation and immune surveillance; and conferring stem cell properties. It does this by enabling polarized epithelial cells to transform into cells with a mesenchymal, and therefore motile, phenotype. Tumor-associated macrophages (TAMs) are key cells of the tumor microenvironment that orchestrate the connection between inflammation and cancer. Activation of EMT often requires crosstalk between cancer cells and components of the local tumor microenvironment, including TAMs. In this review, clinical and experimental evidence is presented for control of TAMs in promoting cancer cell invasion and migration and their interaction with the EMT process in the metastatic cascade. The translational significance of these findings is that the signaling pathways that interconnect TAMs and EMT-modified cancer cells may represent promising therapeutic targets for the treatment of tumor metastasis.
Tumor metastasis is responsible for 90% of cancer-related mortality but is still one of the most poorly understood components of cancer pathogenesis (1). The majority of solid tumors are carcinomas, cancers that originate in the epithelial cell population. Successful invasion-metastasis cascades require several steps that include epithelial–mesenchymal transition (EMT) of cancer cells, invasion through the extracellular matrix (ECM) and stromal cell layers, intravasation into the vasculature lumina, transport through the circulatory system, extravasation into parenchyma of distant tissues and organs, seeding at the premetastatic niche, and finally survival and growth at the metastatic site (1, 2). Chaffer, Weinberg and colleagues have described the complicated and multistep metastatic process in two stages. In the first stage, cancer cells translocate physically from the primary tumor to the site of dissemination; and in the second stage, colonization occurs at the secondary site (3–5). As simple as these processes may sound, the clinical impact of these changes is immense. Many of the events involved in these stages are the result of reciprocal and evolving crosstalk between the tumor microenvironment and carcinoma cells. The EMT program plays an important role in tumor metastasis by disassembling adherens and tight junctions, transforming polarized epithelial cancer cells into a mesenchymal cell phenotype, and facilitating the detachment of mesenchymal cells from initial sites to allow passage through dismantled basement membranes (BMs) (Chaffer and Weinberg’s stage 1) (4). Once they have reached the distant organs (Chaffer and Weinberg’s stage 2), these mesenchymal cells may return to an epithelial phenotype through a mesenchymal–epithelial transition (MET) and thereby regain the ability of cancer cell proliferation and differentiation in metastatic sites (4).
Activation of the EMT program for metastasis typically requires crosstalk between cancer cells and the local microenvironment. Different populations of chronic inflammatory cells, some of which mature to be tumor-associated macrophages (TAMs), influence the local tumor microenvironment. These inflammatory cells play an important role in tumor progression by promoting tumor cell proliferation, matrix remodeling, angiogenesis, suppressed adaptive immunity, and EMT (2, 6). Classically, they do this by producing various cytokines such as the interleukins (ILs), interferon (IFN), and other tumor-promoting factors (7, 8). Research that is more recent has now demonstrated that EMT-mediated premetastatic tumor cells are also involved in the recruitment, activation, and differentiation of TAMs (5). The fundamental concept of EMT stimulating cancer development and progression is well accepted, but the links between EMT and TAMs in these processes present a gap in research. For example, although the tumor inflammatory cell populations appear to promote growth and progression of tumors, the exact interactive pathways are not well defined.
By defining the roles of EMT and TAMs in cancer development, therapeutic strategies that target these processes may be developed to benefit cancer patients (9). The purpose of this review is to provide an overview of experimental and clinical evidence that demonstrates the crosstalk between TAMs and cancer cells undergoing EMT during metastasis and develop an understanding of the translational significance of this information, for development of new diagnostic and therapeutic strategies targeting TAMs and/or EMT.
Epithelial–mesenchymal transition refers to a series of biologic processes that allow polarized epithelial cells, which normally adhere to the BM, to undergo multiple biochemical and molecular changes to transform into mesenchymal-like cells (5, 10, 11). The hallmarks of EMT include loss of cell-to-cell adhesion; loss of apical–basal polarity; and acquisition of increased migratory and invasive properties. There are three different classifications of EMT: type 1 EMT is involved in embryo implantation, embryogenesis, and organ development; type 2 EMT is a process involved in wound healing, tissue regeneration, and organ fibrosis, initiated by the injury; and type 3 EMT is associated with cancer progression and metastasis (12). In a comprehensive review of EMT in cancer development, Micalizzi et al. (13) suggest that cancer cell motility may also use a process of non-EMT epithelial cell plasticity, termed collective migration. The similarities between EMT and collective migration suggest that the epithelial and mesenchymal cell phenotype transition may not always involve two absolute and independent cell states. Instead, a continuous spectrum of epithelial and mesenchymal properties may contribute to cancer progression, which typically lacks the coordinated and orderly induction of complete EMT. In the current review, however, the EMT theory of cancer cell motility forms the basis of discussion.
At the molecular level, the characteristics of EMT include downregulation of epithelial markers, such as E-cadherin, desmoplakin, and cytokeratins, and upregulation of mesenchymal markers, such as N-cadherin, fibronectin, vimentin, and α-smooth muscle actin (α-SMA) (8, 14–17). Activation of transcription factors that include Snail, Twist, ZEB1 and ZEB2, and others, triggers EMT. These transcription factors repress the epithelial cell phenotype and promote mesenchymal characteristics of motility and BM and ECM degradation (14). For example, ZEB1 and ZEB2 directly bind to the promoter of target genes through conserved zinc finger proteins to downregulate expression of E-cadherin (epithelial marker) and induce expression of vimentin (mesenchymal marker) (15, 16). ZEB1 also enhanced the migration of PC-3 human prostate cancer cells through the extracellular barrier and increased metastatic colonization (16). Snail1 and Snail2, which belong to the Snail zinc finger family, are capable of repressing the expression of E-cadherin by directly binding to its promoter and activating the expression of proinvasive vimentin, fibronectin, and the matrix degradation enzymes termed matrix metalloproteinases (MMPs) (17, 18). Overexpression of Snail1 induced prostate cancer cells to degrade and break through BM barriers, thereby invading into the blood system, through directly activating membrane-anchored MMPs, MT1-MMP, or MT2-MMP (19). Snail1 also promoted angiogenesis, another essential process for tumor invasion and metastasis (20). Activated Twist is a member of the basic helix-loop-helix transcription factor family, and it plays a role in tumor invasion and metastasis via downregulating E-cadherin and upregulating N-cadherin (21). Twist also upregulates the expression of MMPs and downregulates the expression of tissue inhibitor of metalloproteinase, a naturally occurring specific inhibitor of MMPs (22).
The movement of tumor cells to distant organ sites requires a risky journey with intravasion into the vasculature, immune attack, and extravasion to a new site for cancer development. Tumor cells that intravasate into the blood vessel lumens and disseminate to new sites of colony formation are termed circulating tumor cells (CTCs) (23). There is limited research on the association between CTCs, platelets, and macrophages; controls on transendothelial migration; and the molecular mechanisms active in these processes. The following section presents some information on the role of EMT in CTC production and motility. Perhaps the pre-eminent molecular pathway involves Notch signaling. The Notch pathway not only acts as a regulator of cell survival and cell proliferation but is also involved in cancer cell intravasation by stimulating transendothelial migration (23). How Notch signaling and EMT control the activity of CTCs is largely unknown. Nonetheless, the number of CTCs in blood is a significant prognostic factor for lung (23), colorectal (24, 25), breast (26), and prostate (27) cancers, and it is likely that CTCs feature in development of most carcinomas. They are found only rarely in normal healthy people and those with non-malignant tumors (28). EMT may help CTCs with cancer dissemination by overcoming detachment-induced cell apoptosis or “anoikis” and promoting survival in the circulation, but how EMT helps the CTCs is still not clear (29). In breast cancer, EMT may prevent CTCs from anoikis by enhancing cancer cell reattachment to leukocytes, platelets, and endothelium. EMT does this via formation of microtubule-based membrane protrusions on the CTCs, called microtentacles, which are formed on the surface of detached cells through expression of Twist1 and Snail1 (30, 31). In addition, neurotrophin receptor-interacting melanoma antigen (NRAGE) interacts with ankyrin-G, a part of the E-cadherin complex, rendering cancer cells sensitive to apoptosis. During oncogenic EMT, loss of E-cadherin downregulates ankyrin-G, enhancing NRAGE translocation to the nucleus, where the NRAGE-TBX2 complex can inhibit p14ARF gene expression to protect cancer cells against anoikis (32).
Some of the surviving CTCs that move through the vascular lumen and lodge on the vascular endothelium may break through the endothelial and pericyte layers and then enter the tissue parenchyma at the secondary organ site. During this process, the adherent cancer cells must communicate with endothelial cells to open their cell junctions, thus allowing the passage of the cancer cells to extravasate across the endothelium to the connective tissue space of the host organ (33). Cancer cells can migrate through the endothelial layer of blood vessels in two basic ways: paracellular migration, where cancer cells cross the endothelial layer by disrupting the cell junctions in the endothelial layer; and transcellular migration by which cancer cells cross the endothelial barriers by traversing through the endothelial cell body (10, 34). An extravasation assay in zebrafish was used to model the roles of EMT in the process of metastasis. Zebrafish are transparent and allow real-time imaging of cell movement in the live animal. The results demonstrated that Twist1, a central protein in EMT, affects intravascular migration of arrested cancer cells, remodels the vasculature, and promotes cancer cell exit from the blood circulation through a β1 integrin-independent mechanism (35). On extravasation into a foreign microenvironment, tumor cells follow one of three alternative courses: cell death, dormancy, or senescence.
Occasionally, EMT-derived mesenchymal cells with CSC-like properties (less than 1% of disseminated cancer cells) undergo MET to be able to initiate cancer cell proliferation and differentiation during colonization (34, 36). Downregulation of Twist1 at the metastatic site was essential for increased proliferation and reversal of EMT-induced growth arrest, which clearly proves the indispensable role of MET for colonization and macrometastasis (37). Figure 1 summarizes the roles of EMT in the process of metastasis.
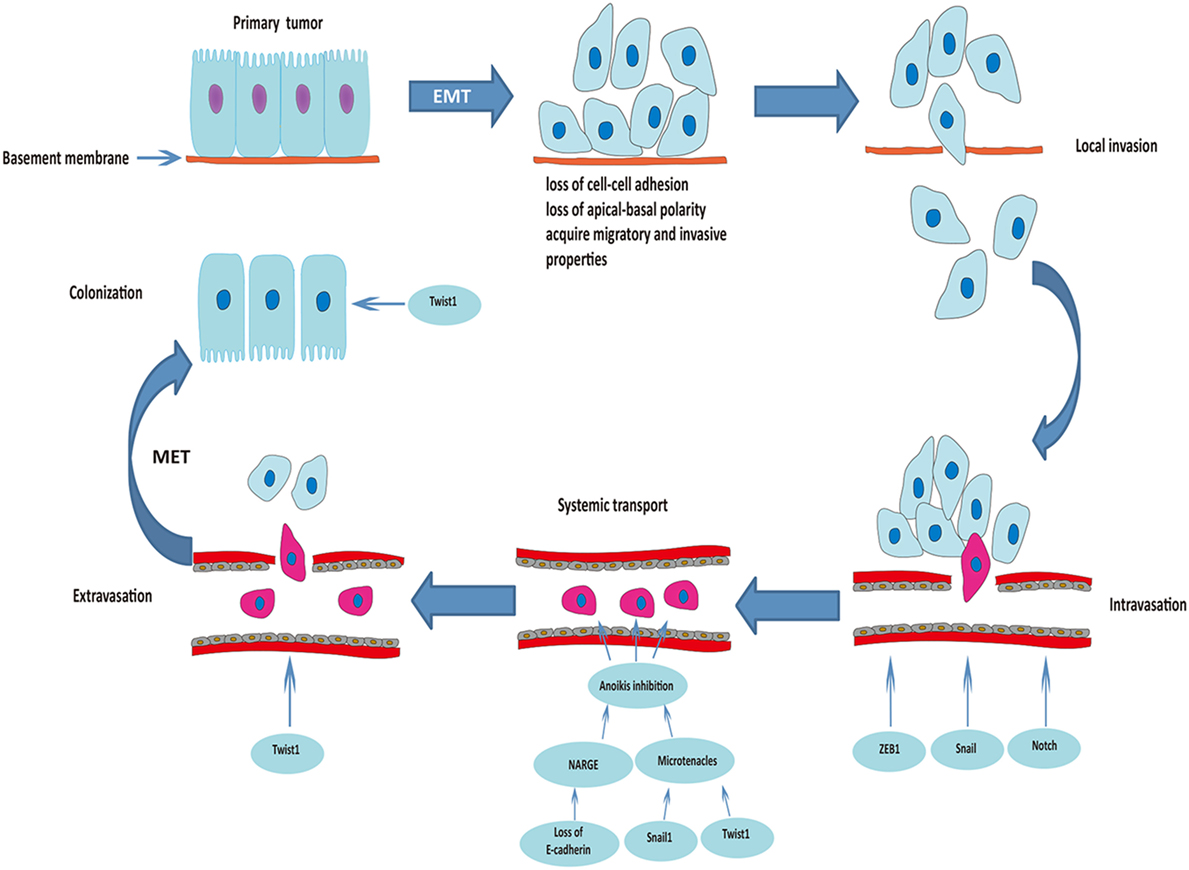
Figure 1. Roles of epithelial–mesenchymal transition (EMT) in the process of cancer metastasis. Triggered by the activation of transcriptional factors, tumor cells of epithelial origin that undergo EMT are transformed into a mesenchymal phenotype, lose cell-to-cell adhesion and apical-basal polarity, and acquire migratory and invasive properties. After degradation of the underlying basement membrane and extracellular matrix, tumor cells invade the neighboring tissue parenchyma (local invasion). The mesenchymal–phenotype tumor cells then invade into the blood or lymphatic vessels (intravasation). ZEB1, Snail, and Notch are involved in this process. Circulating tumor cells (within vessel lumens) overcome the harsh conditions in the blood stream and attach to the vessel wall at a distant site to prepare for escape from the blood circulation. Transcriptional factors Snail1 and Twist1 play an important role in preventing anoikis and maintaining survival of circulating tumor cells. Some of the surviving circulating tumor cells break through the vascular wall and enter the tissue parenchyma at the secondary organ site (extravasation). Finally, some of the EMT-derived mesenchymal cells with cancer stem cell-like properties undergo mesenchymal–epithelial transition (MET) to initiate proliferation of the metastatic clone.
The role of macrophages in cancer is controversial, and many aspects remain unresolved. Macrophage surveillance is essential for preventing the cancer growth, and there is evidence that activated macrophages can identify and kill transformed cells. However, there is also evidence that macrophage depletion has little effect on the host’s susceptibility to cancer (38) and in some cases may even be beneficial to the host. Different subpopulations of macrophages may be responsible for distinct tumor-promoting activities and tumor relapse on different types of therapies. Here, the proposal that TAMs have contrasting roles in cancer depending on their phenotype is discussed. Figure 2 summarizes the role of TAMs in cancer development.
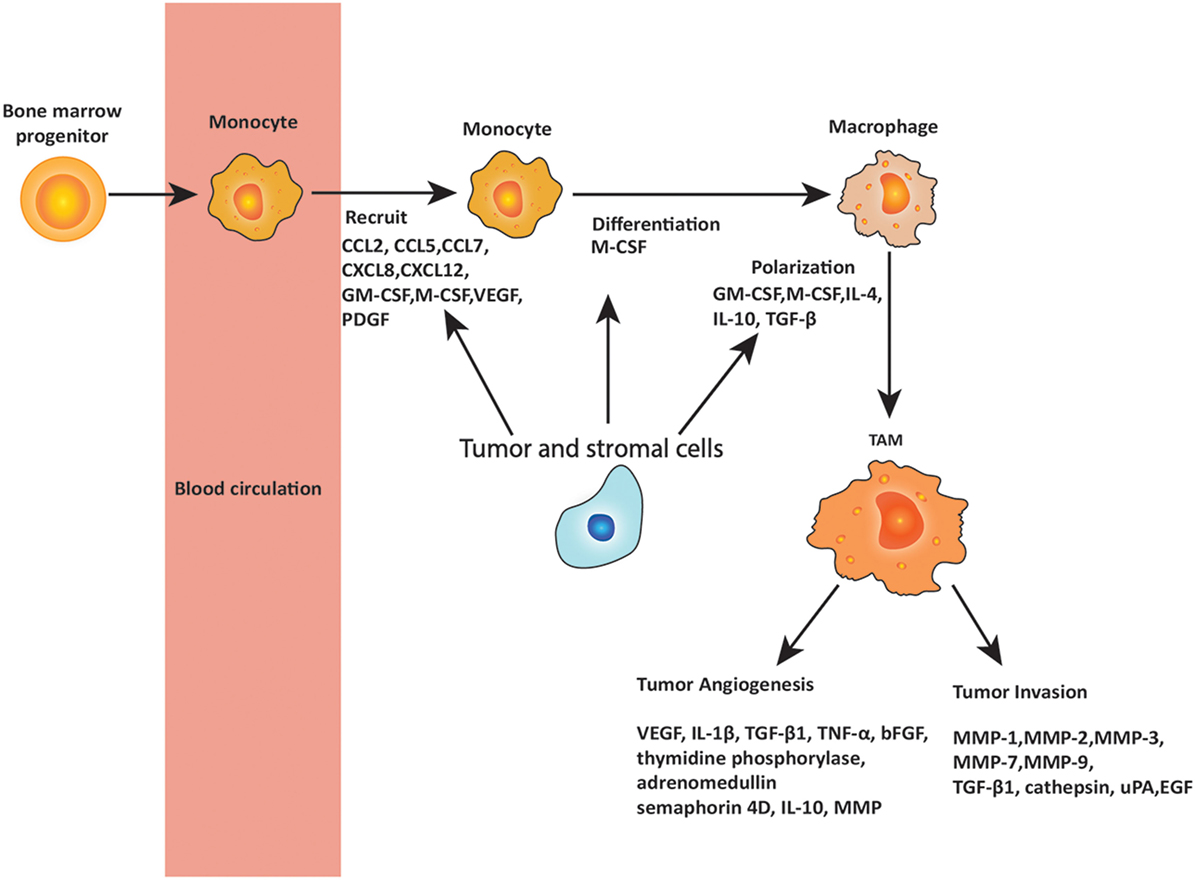
Figure 2. Roles of tumor-associated macrophages (TAMs) in metastasis. Circulating monocytes that are derived from bone marrow progenitors are recruited to the tumor site by chemokines and cytokines secreted by tumor and stromal cells. These chemokines and cytokines include C-C chemokine motif ligand 2 (CCL2), CCL5, CCL7, C-X-C chemokine motif ligand 8 (CXCL8), and CXCL12, as well as granulocyte–macrophage colony-stimulating factor (GM-CSF), macrophage colony-stimulating factor (M-CSF), vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF), of which CCL2 is the most important. In the tumor microenvironment, monocytes are differentiated and polarized to TAMs by the cytokines produced by tumor and stromal cells, including M-CSF, GM-CSF, interleukin 4 (IL-4), IL-10, and transforming growth factor-β (TGF-β). TAMs play important roles in promoting tumor angiogenesis and invasion through releasing various cytokines including VEGF, IL-1β, TGF-β1, matrix metalloproteinases (MMPs), tumor necrosis factor (TNF)-α, basic fibroblast growth factor (bFGF), IL-10, urokinase plasminogen activator (uPA), epidermal growth factor (EGF), and others as noted in the figure.
Macrophages are differentiated immune cells of the myeloid lineage, found in all tissues (39). Most tissue-resident macrophages originate from the embryonic yolk sac and demonstrate tissue-specific functional properties important for the tissue differentiation and homeostasis. To carry out their tissue-specific functions, macrophages respond to local signals released in their niche. They also act as sentinel cells that constantly monitor their tissues for potential threats, such as infection, injury, and tumors, and they respond rapidly by initiating the inflammatory cascade. During inflammation, macrophages also differentiate from circulating monocytes that recruit to the site of inflammation and extravasate into the tissue (40). Importantly, macrophages also play a fundamental role in promoting the resolution of inflammation by clearing apoptotic cells/debris and supporting wound healing and tissue repair programs (41–43). When resident and recruited macrophages activate in response to potentially harmful agents, they can acquire different polarization states. Historically, according to their activation state, macrophages divide into classically activated M1 and alternatively activated M2 macrophages (44, 45). This classification reflects the Th1/Th2 paradigm of T helper cell activation. M1 macrophages, typically, activate with IFN-γ, lipopolysaccharides, viral products, or granulocyte-macrophage colony-stimulating factor (GM-CSF). In contrast, M2 macrophages, typically, activate with IL-4, IL-10, glucocorticoids, or macrophage colony-stimulating factor (M-CSF) (46, 47). M2-like macrophages have been often further divided into M2a (IL-4 induced), M2b (IgG-induced), and M2c (IL-10 and glucocorticoid-induced). More recently a consensus was reached to, instead, define macrophage phenotypes based on the activator used (48, 49). It is important to note that these polarization states are just extremes of a continuum in vivo. Macrophages display a high degree of plasticity, and the different activation states can often coexist or change during disease progression (50). Moreover, according to Mills, who originally developed the M1/M2 paradigm, what really matters is the function that macrophages play within a specific in vivo context: M1/kill and M2/repair (51). Therefore, M1 macrophages were defined originally in vivo by the production of the functional molecule nitric oxide that inhibits proliferation. M1 responses are linked with IL-12 and IL-8/CCL (C-C chemokine motif ligand) production, and cell surface expression of CD80 or 86 that attract killer cells like neutrophils and/or stimulate Th1 responses such as cytotoxic T lymphocytes (52–54). M2 macrophages were defined originally in vivo by the production of ornithine that promotes proliferation. Their responses are associated with transforming growth factor (TGF)-β and other growth factor production, for example, vascular endothelial growth factor (VEGF) or epidermal growth factor (EGF), cell surface expression of CD163 or 206, and the propensity to stimulate Th2 responses such as antibody production and antibody-dependent cell-mediated cytotoxicity. Many macrophage populations express additional cytokines and growth factors, including tumor necrosis factor (TNF)-α, IL-6, IL-1, IL-10, NADPH oxidases, and MMPs. Changes in the relative ratios between pro-inflammatory and immunosuppressive/remodeling factors characterize a more M1-like or M2-like polarization state (55).
Tumor-associated macrophages represent the major type of immune cells that infiltrate the tumor microenvironment (56–58). Over the last decades, many studies have shown a significant link between TAM number/density and a poor prognosis in most tumor types, illustrating the clinical significance of macrophages in tumor progression (58). For example, there is a significant association between higher risk of distant metastasis and poor overall survival with increased density of TAMs in renal cell (59), breast (60, 61), bladder (62), and hepatocellular (63) cancers, and lymphoma (64, 65). It is now clear that dynamic changes in the phenotypes of macrophages occur during the different steps of tumor progression, including invasion, metastasis, seeding, and growth (66). Different subpopulations of TAMs are responsible for distinct tumor-promoting activities and tumor relapse on different types of therapies (for example, irradiation, antiangiogenic therapies, some forms of chemotherapy) (67–70). Currently, it is not known if TAMs derive from resident or recruited macrophages. Small tumors probably interact with the surrounding resident macrophages and, only later as the tumor mass grows and becomes vascularized, are circulating monocytes recruited and differentiate into TAMs. Circulating monocytes usually derive from bone marrow progenitors; however, under abnormal circumstances, such as during tumor development, extramedullary hematopoiesis can also take place in the spleen (71). Peripheral blood monocytes consist of two main populations: inflammatory or classical monocytes (Ly6ChighCX3CR1low CCR2high in mouse and CD14++CD16neg in humans) and patrolling or non-classical monocytes (Ly6Clow CX3CR1high CCR2low and CD14+CD16++). Many chemokines and cytokines secreted by tumor and stromal cells recruit peripheral blood monocytes to the tumor site. These include CCL2, CCL5, CCL7, CXCL8, and CXCL12, as well as GM-CSF, M-CSF, VEGF, and platelet-derived growth factor (71, 72). Among these chemokines, CCL2 is likely the most important in recruitment of TAMs. However, the monocyte subset that recruits to the primary tumor and the involvement of the CCL2-CCR2 chemokine axis appears to be model and tumor type dependent (73).
Within the tumor microenvironment, monocytes are induced to differentiate and polarize into protumoral macrophages through complex signaling networks induced by multiple soluble mediators, such as M-CSF, GM-CSF, and immunosuppressive cytokines such as IL-4, IL-10, and TGF-β (74). TAMs respond by modulating cancer cell proliferation, immune regulation, ECM remodeling, tumor cell invasion and metastasis, lymphangiogenesis and vascular angiogenesis by secreting IL-10, VEGF, prokineticin (Bv8), prostaglandin E2, and MMP-9 (75–77). As the tumor progresses, TAMs are more and more polarized toward an M2-like phenotype and express a typical M2 signature including high levels of MR (CD206), scavenger receptor-A, arginase-1, and low MHC-class II antigens. The roles played by TAMs in promoting tumor angiogenesis, tumor invasion, as well as intravasation, extravasation, and seeding of metastatic tumor cells are relevant to this review.
There is considerable controversy about whether TAMs promote tumor progression and metastasis via their stimulation of tumor angiogenesis (78). An angiogenic switch is crucial for tumor cell survival, proliferation, invasiveness, and metastasis. High levels of TAM infiltration in the primary tumor are usually associated with angiogenesis via upregulation and release of several pro-angiogenic factors that, typically, promote new vessel growth. For example, VEGF-A is an important pro-angiogenic cytokine secreted by TAMs. Under hypoxic conditions, hypoxia-inducible factors (HIF-1α and HIF-2α) increase the production of VEGF-A from TAMs and, thus, promote TAM-induced angiogenesis and metastasis (79). Under normoxic conditions, IL-1β and TGF-β1 released by TAMs promote the expression of VEGF-A in mouse macrophages via HIF-1α/β- and Smad3/4-dependent signaling pathways (80–83). Other pro-angiogenic factors secreted by TAMs, including TNF-α, basic fibroblast growth factor, thymidine phosphorylase, adrenomedullin, and semaphorin 4D, are also involved in angiogenesis and metastasis (84, 85). A specific subpopulation of pro-angiogenic TAMs was identified, characterized by the expression of the angiopoietin receptor Tie2, and endowed with pro-angiogenic and immunosuppressive activities (86, 87). Tie2-expressing monocytes/macrophages express higher levels of CD163 and CD206, indicating that Tie2-expressing myeloid cells show an M2-like phenotype. They also express VEGF, IL-10, and MMP-9. Tie2-expressing monocytes/macrophages recruit to tumors and are required for tumor growth. This makes them excellent candidates for the development of new tumor targeted therapies. Indeed, targeting the angiopoietin 2 (ANG2)/Tie2 pathway, with an anti-ANG2 antibody, inhibits tumor growth and metastasis by disabling the pro-angiogenic activity of Tie2-expressing monocytes/macrophages and impeding the emergence of evasive resistance to antiangiogenic therapy (88).
The tumor microenvironment is important for tumor progression. Among the components of the microenvironment, TAMs are thought to be the major inflammatory component of the tumor support stroma. However, there remain inconsistent data on whether TAMs promote tumor invasion. Here, we present some evidence for a positive link between TAMs and tumor invasion.
Matrix metalloproteinases are a family of zinc-dependent proteases that function to degrade matrix, which includes collagenase (MMP-1), gelatinase A (MMP-2), stromelysin (MMP-3), matrilysin (MMP-7), gelatinase B (MMP-9). MMPs are involved in tumor invasion and metastasis by degrading the BM, activating growth factors, and promoting angiogenesis (89, 90). Macrophage-derived TGF-β1 can promote MMP-9 expression in glioma stem-like cells, thereby enhancing the invasiveness of tumor cells (81). Moreover, specific ablation of MMP-9-positive TAMs with zoledronic acid resulted in reduced angiogenesis (91, 92). Macrophages are also effective producers of additional proteases, including cathepsins and serine proteases such as urokinase-type plasminogen activator. In many tumors, these proteases function to enhance tumor progression and metastasis by proteolytic destruction of the matrix to allow tumor cells to escape from the confines of the BM and to migrate through the dense stoma (93–95).
An important EGFR/CSF-1R paracrine loop exists between macrophages and tumor cells in which CSF-1, produced by carcinoma cells and bound by macrophages, promotes the proliferation, differentiation, and polarization of macrophages toward an M2-like phenotype (96). CSF-1 also stimulates macrophages to release EGF, which promotes tumor cell proliferation and migration. EGF also stimulates the secretion of CSF-1 by tumor cells, thereby forming a positive feedback loop between tumor cells and macrophages (97, 98). Recently, intravital microscopy was used to investigate tumor cell migration toward blood vessels in mouse models of breast cancer (99). By using live cell imaging, perivascular Tie2-expressing macrophages promoted the transient opening of tumor blood vessels in a VEGF-dependent manner. Diffusion of plasma proteins from leaky vessels attracts cancer cells that can easily enter systemic circulation at the open gates (99). Previous work from this group had identified perivascular structures called “tumor microenvironment of metastasis” in which a macrophage, a specialized cancer cell, and a blood vessel establish tripartite contacts. Importantly, “tumor microenvironment of metastasis” cell density predicts distant metastatic recurrence in breast cancer patients independently of other clinical prognostic indicators (100). TAMs are also major contributors in the formation of the metastases at secondary sites of primary tumor, where they prepare a suitable microenvironment for successful colonization (101).
The interactions of cancer cells with various stromal cells in the tumor microenvironment, as well as the accumulation of intrinsic changes in cancer cells, drive progression and metastasis. The tumor microenvironment comprises endothelial cells, pericytes, fibroblasts, ECM, and various bone marrow-derived cells such as macrophages, neutrophils, and mast cells. As a major component of the tumor microenvironment, TAMs act in cancer migration, invasion, and metastasis through processes such as EMT by producing or activating various factors including nuclear factor-κB (NF-κB) and important inflammatory cytokines and growth factors, such as IL-6, TGF-β1, TNF-α, and IL-8 (102). Information on these co-activating factors is now provided.
Nuclear factor-κB consists of a family of transcription factors that play pivotal roles in both TAMs and tumor cells to promote tumor initiation and growth (103, 104). NF-κB activation is also an important event in the acquisition of metastatic potential in cancer cells, and its activation can result from the crosstalk between TAMs and cancer cells. For example, TAMs prime NF-κB activation in both stromal and cancer cells by secreting TNF-α, which in turn upregulates Snail expression (105). In the process of TNF-α-induced EMT, NF-κB regulates phosphorylation and degradation of Snail. Knockdown of Snail by shRNA was able to inhibit migration and invasion of breast cancer cells induced by TNF-α in vitro as well as inflammation factor-mediated metastasis in vivo (106). TAMs also promoted tumor invasion and migration ability and transformation of oral squamous cell carcinoma cells from an epithelial-to-mesenchymal phenotype through activation of the Gas6/Axl-NF-κB signaling pathway (105).
Interleukin-6 is an inflammatory cytokine upregulated in most common human cancers, with increased levels of IL-6 in serum indicating poor prognosis in most cancers (107). IL-6 decreased the expression of E-cadherin in estrogen receptor α-positive human breast cancer cells, with constitutively ectopic IL-6-expressing MCF-7 breast cancer cells (MCF-7IL-6) exhibiting an EMT phenotype characterized by decreased expression of E-cadherin and upregulation of vimentin, N-cadherin, Snail, and Twist (108). In vitro co-culture of human lung adenocarcinoma A549 or H1299 cells with THP-1-derived macrophages upregulated IL-6 and increased the invasion ability of A549 and H1299 cells through EMT by repressing the expression of E-cadherin and promoting vimentin. In addition, the presence of additional anti-IL-6 antibody was able to neutralize the enhanced invasiveness (109).
Transforming growth factor-β1 is an important inflammatory cytokine that is involved in inducing EMT, facilitating tumor cell evasion of immune surveillance, and promoting cancer cell dissemination and metastasis (110). In the tumor microenvironment, TGF-β1 is produced by infiltrating immune cells such as TAMs, myeloid-derived suppressor cells and regulatory T cells, and cells that are major promoters of EMT (7). TGF-β1 derived from TAMs induced and promoted EMT in various cancer cells (111, 112). TGF-β1 also promoted cancer stem cells-like properties in hepatocellular carcinoma Hepa1-6 cells, which underwent EMT and acquired higher invasive ability (113). Once secreted, TGF-β1 can function in an autocrine manner to sustain the mesenchymal and stem cell traits of cancer cells that are undergoing EMT (114).
Macrophages are major producers of TNF-α and are highly responsive to TNF-α. High levels of TNF-α can lead to anticancer effects through activating T cell-mediated immunity, whereas low-level, chronic TNF-α produced by cancer cells and stromal cells may promote tumor growth and metastasis (115). TNF-α can induce EMT and promote tumorigenicity of renal cell carcinoma and increase invasion and migration activities by inhibiting E-cadherin, upregulating vimentin expression and activating MMP-9. The PI3K/Akt/GSK-3β signaling pathway plays an important role in the TNF-α-induced EMT processes of in renal cell carcinomas (116). This knowledge may be a key to targeting TNF-α receptors to promote antitumor immunity. The design of therapeutic antibodies, which engage and activate TNF-α receptors for cancer therapeutics but avoid serious immune-related adverse events, is a current challenge (117).
Interleukin-8, also known as CXCL8, plays a vital role in cancer progression by initiating angiogenesis; recruiting monocytes to the tumor site; and enhancing the proliferation, survival, and metastasis of cancer cells (118, 119). The mechanism of IL-8 involvement in EMT is that Snail activates the expression of IL-8 by binding to its E3/E4 E-box located in the promoter of IL-8 (120). There is also a link between IL-8 secreted by TAMs and tumor EMT. For example, TAMs induce EMT of hepatocellular carcinoma cells through the IL-8 activated JAK2/STAT3/Snail signaling pathway (120, 121).
Tumor-associated macrophages also promote tumor cell invasion and metastasis via the interactions with cancer stem cells. For example, during EMT, increased expression of CD90 and EphA4 control the cell-to-cell interactions between TAMs and cancer stem cells through directly binding with their respective receptors on the surface of these cells (122). For example, Lewis lung carcinoma cells potently activated macrophages leading to production of IL-6 and TNF-α through activation of the TLR family members TLR2 and TLR6 (123). Endothelial activation markers, including vascular cell adhesion molecule-1 (VCAM-1) and vascular adhesion protein-1 (VAP-1), are induced near metastatic tumor cells after their attachment to the vascular bed. Embolus formation of tumor cells enhances VCAM-1 induction. Blocking endothelial activation, with either an anti-VCAM-1 blocking antibody or a VAP-1 inhibitor, resulted in decreased recruitment of macrophages and reduced metastatic cell survival (124).
Are insights gleaned from basic laboratory research useful to the diagnosis and treatment of clinical metastatic disease? The interaction and crosstalk between TAMs and EMT undoubtedly promotes tumor progression, invasiveness, and metastasis, at least in some cancers. As mentioned previously, secretion of various cytokines and chemokines by TAMs orchestrates the crosstalk and promotes adjacent epithelial tumor cells to undergo EMT through a paracrine manner. In turn, cytokines produced by tumor cells also promote the differentiation process in TAMs, thereby forming a positive feedback loop between TAMs and EMT in metastasis (125). Thus, breaking the pathways of crosstalk between TAMs and EMT is a reasonable option for the treatment of tumor metastasis.
In terms of targeting TAMs; several strategies have been promising: blocking recruitment of TAMs; depleting numbers of TAMs by promoting their removal; reversal of immune suppression by switching the M2 to M1 phenotype; and inhibiting TAM-induced tumor angiogenesis (126). For example, blockade of TAM recruitment by the genetic deletion of CSF-1 delayed tumor growth, angiogenesis, and reduced metastasis in several models of cancer (127). Inhibition of CSF-1 or its receptor with antibody, small molecule inhibitors, or antisense RNA reduced the recruitment of macrophages, thereby inhibiting the proliferation of cancer cells and metastasis (128–130). Specific depletion of macrophages in the tumor microenvironment by clodronate-encapsulated liposomes or zoledronic acid significantly inhibited tumor proliferation, angiogenesis, and distant metastasis in liver, lung, and xenograft models of colon carcinoma (131–134). In a breast cancer model, the combination of CpG with an anti-IL-10 receptor antibody switched TAMs from the M2 to the M1 phenotype and triggered an innate immune response of debulking of tumors (135). Trabectedin, a chemotherapeutic agent approved in Europe for the treatment of sarcomas and ovarian cancer, was selectively toxic for TAMs, resulting in reduction of angiogenesis in mouse tumor models (136).
Since EMT is a crucial step in the process of cancer metastasis, targeting transcriptional factors of EMT or EMT-related pathways by miRNA has proven to be an effective strategy for the treatment of tumor metastasis. For example, members of the miR-200 family have emerged as pivotal repressors of EMT and cancer metastasis through ZEB-dependent and ZEB-independent mechanisms (137, 138). Delivery of miR-200 members to different cancer models, including lung, ovarian, renal, and basal-like breast cancers, led to significant reduction in primary tumor burden and distant metastasis (139). Cortez et al. have reported that transiently transfected lung cancer cell line A549 with miR-200c resulted in significantly more sensitivity to the cytotoxic effects of radiation. Systemic delivery of miR-200c enhanced the effects of radiotherapy in a xenograft model of lung cancer (140). The miR-34 family also plays an important role in reducing the viability of cancer stem cells and inhibiting metastasis through downregulating the expression of Snail in a manner of double-negative feedback loop (141, 142). Chen et al. have reported that systemic delivery of miR-34a into experimental lung metastasis models of B16F10 melanoma resulted in the downregulation of the survivin gene and reduction of tumor load in the lung (143).
Given the increasing evidence supporting active crosstalk between TAMs and EMT in tumor metastasis, targeting the signaling pathways in this process is another option for the treatment of invasive cancer. As mentioned above, the signaling pathway induced by TGF-β plays an important role in this crosstalk. Therefore, TGF-β-induced signaling cascades are potential therapeutic targets. For example, CX-4945, a potent and selective inhibitor of protein kinase CK2, inhibited the EMT-mediated migration and invasion of A549 human lung cancer cells by blocking the TGF-β1 signaling pathway (144). Ginsenoside 20 (R)-Rg3, an active component of ginseng, suppressed A549 cell migration and invasion through inhibiting the EMT process induced by TGF-β1 (145). Sorafenib, a tyrosine kinase inhibitor used for the treatment of primary kidney cancer, advanced primary liver cancer, and radioactive iodine-resistant advanced thyroid carcinoma inhibited TGF-β released in TAMs; reduced TGF-β-induced cancer cell proliferation, metastasis, and EMT process in vitro; and partially blocked macrophage activation in vivo (146).
Because NF-κB is another crucial cytokine involved in the crosstalk between TAMs and cancer cell EMT, targeting the NF-κB pathway has become one of the intensely studied strategies for the treatment of TAM-related tumor metastasis. Zoledronic acid is a third-generation bisphosphonate for the treatment of bone metastasis in breast cancer patients and for osteoporosis. Zoledronic acid reversed EMT in triple-negative breast cancer cells by inactivation of NF-κB signaling (147). A somatostatin derivative (smsDX) can block the paracrine loop between TAMs and prostate cancer cells and reduce the risk of migration and invasion through inhibiting activation of NF-κB (148).
A major complication of cancer progression is the metastatic spread of cancer cells from the primary tumor. Similarities between the process of EMT in embryogenesis and wound healing and that seen in spread of epithelial-derived cancers are now becoming clear. There is increasing evidence that the tumor metastatic cascade relies on a complicated interaction between EMT-modified cancer cells and TAMs. Cytokines released by tumor cells promote the recruit of macrophages to the tumor site and differentiate them into TAMs that then become active in the tumor microenvironment. Reciprocally, as a major component of solid tumors, TAMs promote cancer cell invasion and metastasis by secreting various cytokines, for example, TGF-β, NF-κB, VEGF, and CCL18. Thus, a positive feedback circuit forms between TAMs and EMT-modified cancer cells in the tumor microenvironment (125). Despite some residual gaps in knowledge regarding links between EMT, TAMs, and cancer invasiveness, it appears that targeting transcriptional factors and the signaling pathways between TAMs and EMT can break the cycle of crosstalk to combat metastasis. Although most evidence detailed here was from experimental models, some results involving patient-derived cancers support the preclinical experimental data. With further elucidation about the mechanism of tumor metastasis, it may soon be possible to translate these fundamental research discoveries successfully into clinic practice.
WS, RM, TY, and GG contributed equally to deciding on topics of interest in this manuscript. WS wrote the manuscript, and all authors contributed equally to its editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
WS was supported by a Visiting Academic Scholar Research Project, funded by the Government of Jiangsu Province, China during a 12-month research visit to the Centre for Kidney Disease Research, Translational Research Institute, Brisbane, Australia.
1. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell (2007) 147:275–92. doi:10.1016/j.cell.2011.09.024
2. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer (2009) 9:239–52. doi:10.1038/nrc2618
3. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (2011) 331:1559–64. doi:10.1126/science.1203543
4. Chaffer CL, Thompson EW, Williams ED. Mesenchymal transition in development and disease. Cells Tissues Organs (2007) 185:7–19. doi:10.1159/000101298
5. Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev (2016) 35:645–54. doi:10.1007/s10555-016-9648-7
6. Laoui D, Van Overmeire E, Movahedi K, Van den Bossche J, Schouppe E, Mommer C, et al. Mononuclear phagocyte heterogeneity in cancer: different subsets and activation states reaching out at the tumor site. Immunobiology (2011) 216:1192–202. doi:10.1016/j.imbio.2011.06.007
7. Smith HA, Kang Y. The metastasis-promoting roles of tumor-associated immune cells. J Mol Med (Berl) (2013) 91:411–29. doi:10.1007/s00109-013-1021-5
8. Wang X, Wang H, Li G, Song Y, Wang S, Zhu F, et al. Activated macrophages down-regulate expression of E-cadherin in hepatocellular carcinoma cells via NF-kappaB/Slug pathway. Tumour Biol (2014) 35:8893–901. doi:10.1007/s13277-014-2159-7
9. Jinushi M, Komohara Y. Tumor-associated macrophages as an emerging target against tumors: creating a new path from bench to bedside. Biochim Biophys Acta (2015) 1855:123–30. doi:10.1016/j.bbcan.2015.01.002
10. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119:1420–8. doi:10.1172/JCI39104
11. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science (2013) 342:1234850. doi:10.1126/science.1234850
12. Song W, Gobe GC. Understanding molecular pathways and targets of Brachyury in epithelial-mesenchymal transition (EMT) in human cancers. Curr Cancer Drug Targets (2016) 16:586–93. doi:10.2174/1568009616666160328113338
13. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia (2010) 15:117–34. doi:10.1007/s10911-010-9178-9
14. Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci (2009) 66:773–87. doi:10.1007/s00018-008-8465-8
15. Ran J, Lin DL, Wu RF, Chen QH, Huang HP, Qiu NX, et al. ZEB1 promotes epithelial-mesenchymal transition in cervical cancer metastasis. Fertil Steril (2015) 103:1606.e2–14.e2. doi:10.1016/j.fertnstert.2015.03.016
16. Drake JM, Strohbehn G, Bair TB, Moreland JG, Henry MD. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol Biol Cell (2009) 20:2207–17. doi:10.1091/mbc.E08-10-1076
17. Sanchez-Tillo E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci (2012) 69:3429–56. doi:10.1007/s00018-012-1122-2
18. Ota I, Li XY, Hu Y, Weiss SJ. Induction of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration program in cancer cells by Snail1. Proc Natl Acad Sci U S A (2009) 106:20318–23. doi:10.1073/pnas.0910962106
19. Batlle R, Alba-Castellón L, Loubat-Casanovas J, Armenteros E, Francí C, Stanisavljevic J, et al. Snail1 controls TGF-β responsiveness and differentiation of mesenchymal stem cells. Oncogene (2013) 32:3381–9. doi:10.1038/onc.2012.342
20. Suarez-Carmona M, Bourcy M, Lesage J, Leroi N, Syne L, Blacher S, et al. Soluble factors regulated by epithelial-mesenchymal transition mediate tumour angiogenesis and myeloid cell recruitment. J Pathol (2015) 236:491–504. doi:10.1002/path.4546
21. Khan MA, Chen HC, Zhang D, Fu J. Twist: a molecular target in cancer therapeutics. Tumour Biol (2013) 34:2497–506. doi:10.1007/s13277-013-1002-x
22. Zhao XL, Sun T, Che N, Sun D, Zhao N, Dong XY, et al. Promotion of hepatocellular carcinoma metastasis through matrix metalloproteinase activation by epithelial-mesenchymal transition regulator Twist1. J Cell Mol Med (2011) 15:691–700. doi:10.1111/j.1582-4934.2010.01052.x
23. Fusi A, Metcalf R, Krebs M, Dive C, Blackhall F. Clinical utility of circulating tumour cell detection in non-small-cell lung cancer. Curr Treat Options Oncol (2013) 14:610–22. doi:10.1007/s11864-013-0253-5
24. Sonoshita M, Aoki M, Fuwa H, Aoki K, Hosogi H, Sakai Y, et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell (2011) 19:125–37. doi:10.1016/j.ccr.2010.11.008
25. Lim SM, Becker TM, Chua W, Caixeiro NJ, Ng WL, Kienzl N, et al. Circulating tumour cells and circulating free nucleic acid as prognostic and predictive biomarkers in colorectal cancer. Cancer Lett (2014) 346:24–33. doi:10.1016/j.canlet.2013.12.019
26. Ueo H, Sugimachi K, Gorges TM, Bartkowiak K, Yokobori T, Müller V, et al. Circulating tumour cell-derived plastin3 is a novel marker for predicting long-term prognosis in patients with breast cancer. Br J Cancer (2015) 112:1519–26. doi:10.1038/bjc.2015.132
27. Doyen J, Alix-Panabières C, Hofman P, Parks SK, Chamorey E, Naman H, et al. Circulating tumor cells in prostate cancer: a potential surrogate marker of survival. Crit Rev Oncol Hematol (2012) 81:241–56. doi:10.1016/j.critrevonc.2011.05.004
28. Javaid S, Zhang J, Smolen GA, Yu M, Wittner BS, Singh A, et al. MAPK7 regulates EMT features and modulates the generation of CTCs. Mol Cancer Res (2015) 13:934–43. doi:10.1158/1541-7786.MCR-14-0604
29. Kumar S, Park SH, Cieply B, Schupp J, Killiam E, Zhang F, et al. A pathway for the control of anoikis sensitivity by E-cadherin and epithelial-to-mesenchymal transition. Mol Cell Biol (2011) 31:4036–51. doi:10.1128/MCB.01342-10
30. Matrone MA, Whipple RA, Thompson K, Cho EH, Vitolo MI, Balzer EM, et al. Metastatic breast tumors express increased tau, which promotes microtentacle formation and the reattachment of detached breast tumor cells. Oncogene (2010) 29:3217–27. doi:10.1038/onc.2010.68
31. Charpentier M, Martin S. Interplay of stem cell characteristics, EMT, and microtentacles in circulating breast tumor cells. Cancers (Basel) (2013) 5:1545–65. doi:10.3390/cancers5041545
32. Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci (2013) 126:21–9. doi:10.1242/jcs.120907
33. Gomes FG, Nedel F, Alves AM, Nör JE, Tarquinio SB. Tumor angiogenesis and lymphangiogenesis: tumor/endothelial crosstalk and cellular/microenvironmental signaling mechanisms. Life Sci (2013) 92:101–7. doi:10.1016/j.lfs.2012.10.008
34. Shibue T, Weinberg RA. Metastatic colonization: settlement, adaptation and propagation of tumor cells in a foreign tissue environment. Semin Cancer Biol (2011) 21:99–106. doi:10.1016/j.semcancer.2010.12.003
35. Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, et al. Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci (2010) 123:2332–41. doi:10.1242/jcs.069443
36. Sun Y, Ma L. The emerging molecular machinery and therapeutic targets of metastasis. Trends Pharmacol Sci (2015) 36:349–59. doi:10.1016/j.tips.2015.04.001
37. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell (2012) 22:725–36. doi:10.1016/j.ccr.2012.09.022
38. Mosser DM, Edwards JP. Exploring the spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi:10.1038/nri2448
39. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol (2014) 5:514. doi:10.3389/fimmu
40. Kapellos TS, Iqbal AJ. Epigenetic control of macrophage polarisation and soluble mediator gene expression during inflammation. Mediators Inflamm (2016) 2016:6591703. doi:10.1155/2016/6591703
41. Schultze JL, Schmidt SV. Molecular features of macrophage activation. Semin Immunol (2015) 27:416–23. doi:10.1016/j.smim.2016.03.009
42. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204:3037–47. doi:10.1084/jem.20070885
43. Nucera S, Biziato D, De Palma M. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int J Dev Biol (2011) 55:495–503. doi:10.1387/ijdb.103227sn
44. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3:23–35. doi:10.1038/nri978
45. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol (2009) 27:451–83. doi:10.1146/annurev.immunol.021908.132532
46. Bode JG, Ehlting C, Haussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal (2012) 24:1185–94. doi:10.1016/j.cellsig.2012.01.018
47. Lee M, Park CS, Lee YR, Im SA, Song S, Lee CK. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch Pharm Res (2014) 37:1234–40. doi:10.1007/s12272-014-0379-4
48. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
49. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
50. Malyshev I, Malyshev Y. Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage “switch” phenotype. Biomed Res Int (2015) 2015:341308. doi:10.1155/2015/341308
51. Mills CD. Anatomy of a discovery: M1 and M2 macrophages. Front Immunol (2015) 6:212. doi:10.3389/fimmu.2015.00212
52. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25:677–86. doi:10.1016/j.it.2004.09.015
53. Lazennec G, Richmond A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol Med (2010) 16:133–44. doi:10.1016/j.molmed.2010.01.003
54. Schmidt SV, Krebs W, Ulas T, Xue J, Bassler K, Gunther P, et al. The transcriptional regulator network of human inflammatory macrophages is defined by open chromatin. Cell Res (2016) 26:151–70. doi:10.1038/cr.2016.1
55. Hao NB, Lü MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol (2012) 2012:948098. doi:10.1155/2012/948098
56. Cortez-Retamozo V, Etzrod M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A (2012) 109:2491–6. doi:10.1073/pnas.1113744109
57. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett (2008) 267:204–15. doi:10.1016/j.canlet.2008.03.028
58. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One (2012) 7:e50946. doi:10.1371/journal.pone.0050946
59. Hamada I, Kato M, Yamasaki T, Iwabuchi K, Watanabe T, Yamada T, et al. Clinical effects of tumor-associated macrophages and dendritic cells on renal cell carcinoma. Anticancer Res (2002) 22:4281–4.
60. Yuan ZY, Luo RZ, Peng RJ, Wang SS, Xue C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco Targets Ther (2014) 7:1475–80. doi:10.2147/OTT.S61838
61. Gwak JM, Jang MH, Kim DI, Seo AN, Park SY. Prognostic value of tumor-associated macrophages according to histologic locations and hormone receptor status in breast cancer. PLoS One (2015) 10:e0125728. doi:10.1371/journal.pone.0125728
62. Wang B, Liu H, Dong X, Wu S, Zeng H, Liu Z, et al. High CD204+ tumor-infiltrating macrophage density predicts a poor prognosis in patients with urothelial cell carcinoma of the bladder. Oncotarget (2015) 6:20204–14. doi:10.18632/oncotarget.3887
63. Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol (2008) 26:2707–16. doi:10.1200/JCO.2007.15.6521
64. Wang H, Li P, Wang L, Xia Z, Huang H, Lu Y, et al. High numbers of CD68+ tumor-associated macrophages correlate with poor prognosis in extranodal NK/T-cell lymphoma, nasal type. Ann Hematol (2015) 94:1535–44. doi:10.1007/s00277-015-2401-4
65. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med (2010) 362:875–85. doi:10.1056/NEJMoa0905680
66. Wei IH, Harmon CM, Arcerito M, Cheng DF, Minter RM, Simeone DM. Tumor-associated macrophages are a useful biomarker to predict recurrence after surgical resection of nonfunctional pancreatic neuroendocrine tumors. Ann Surg (2014) 260:1088–94. doi:10.1097/SLA.0000000000000262
67. Sugimura K, Miyata H, Tanaka K, Takahashi T, Kurokawa Y, Yamasaki M, et al. High infiltration of tumor-associated macrophages is associated with a poor response to chemotherapy and poor prognosis of patients undergoing neoadjuvant chemotherapy for esophageal cancer. J Surg Oncol (2015) 111:752–9. doi:10.1002/jso.23881
68. Matsuoka Y, Yoshida R, Nakayama H, Nagata M, Hirosue A, Tanaka T, et al. The tumour stromal features are associated with resistance to 5-FU-based chemoradiotherapy and a poor prognosis in patients with oral squamous cell carcinoma. APMIS (2015) 123:205–14. doi:10.1111/apm.12344
69. Zhang B, Zhang Y, Zhao J, Wang Z, Wu T, Ou W, et al. M2-polarized macrophages contribute to the decreased sensitivity of EGFR-TKIs treatment in patients with advanced lung adenocarcinoma. Med Oncol (2014) 31:127. doi:10.1007/s12032-014-0127-0
70. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res (2013) 73:2782–94. doi:10.1158/0008-5472.CAN-12-3981
71. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475:222–5. doi:10.1038/nature10138
72. Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev (2010) 21:41–8. doi:10.1016/j.cytogfr.2009.11.009
73. Van Overmeire E, Laoui D, Keirsse J, Bonelli S, Lahmar Q, Van Ginderachter J. STAT of the union: dynamics of distinct tumor-associated macrophage subsets governed by STAT1. Eur J Immunol (2014) 44:2238–42. doi:10.1002/eji.201444870
74. Ye YZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol (2012) 189:444–53. doi:10.4049/jimmunol.1103248
75. Schmieder A, Michel J, Schönhaar K, Goerdt S, Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol (2012) 22:289–97. doi:10.1016/j.semcancer.2012.02.002
76. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi:10.1016/j.cell.2010.03.014
77. Ngan ES, Sit FY, Lee K, Miao X, Yuan Z, Wang W, et al. Implications of endocrine gland-derived vascular endothelial growth factor/prokineticin-1 signaling in human neuroblastoma progression. Clin Cancer Res (2007) 13:868–75. doi:10.1158/1078-0432.CCR-06-2176
78. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol (2014) 5:75. doi:10.3389/fphys.2014.00075
79. Joshi S, Singh AR, Zulcic M, Durden DL. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1alpha and HIF2alpha stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res (2014) 12:1520–31. doi:10.1158/1541-7786.MCR-13-0682
80. Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J (2003) 17:2115–7. doi:10.1096/fj.03-0329fje
81. Jeon SH, Chae BC, Kim HA, Seo GY, Seo DW, Chun GT, et al. Mechanisms underlying TGF-beta1-induced expression of VEGF and Flk-1 in mouse macrophages and their implications for angiogenesis. J Leukoc Biol (2007) 81:557–666. doi:10.1189/jlb.0806517
82. Kawahara A, Hattori S, Akiba J, Nakashima K, Taira T, Watari K, et al. Infiltration of thymidine phosphorylase-positive macrophages is closely associated with tumor angiogenesis and survival in intestinal type gastric cancer. Oncol Rep (2010) 24:405–15. doi:10.3892/or_00000873
83. Leek RD, Landers R, Fox SB, Ng F, Harris AL, Lewis CE. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br J Cancer (1998) 77:2246–51. doi:10.1038/bjc.1998.373
84. Chen P, Huang Y, Bong R, Ding Y, Song N, Wang X, et al. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin Cancer Res (2011) 17:7230–9. doi:10.1158/1078-0432.CCR-11-1354
85. Sierra JR, Corso S, Caione L, Cepero V, Conrotto P, Cignetti A, et al. Tumor angiogenesis and progression are enhanced by Sema4D produced by tumor-associated macrophages. J Exp Med (2008) 205:1673–85. doi:10.1084/jem.20072602
86. De Palma M, Mazzieri R, Politi LS, Pucci F, Zonari E, Sitia G, et al. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell (2008) 14:299–311. doi:10.1016/j.ccr.2008.09.004
87. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell (2011) 19:512–26. doi:10.1016/j.ccr.2011.02.005
88. Biel NM, Siemann DW. Targeting the angiopoietin-2/Tie-2 axis in conjunction with VEGF signal interference. Cancer Lett (2016) 380(2):525–33. doi:10.1016/j.canlet.2014.09.035
89. Shuman Moss LA, Jensen-Taubman S, Stetler-Stevenson WG. Matrix metalloproteinases: changing roles in tumor progression and metastasis. Am J Pathol (2012) 181:1895–9. doi:10.1016/j.ajpath.2012.08.044
90. Wang FQ, So J, Reierstad S, Fishman DA. Matrilysin (MMP-7) promotes invasion of ovarian cancer cells by activation of progelatinase. Int J Cancer (2005) 114:19–31. doi:10.1002/ijc.20697
91. Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. J Transl Med (2011) 9:177. doi:10.1186/1479-5876-9-177
92. Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest (2004) 114:623–33. doi:10.1172/JCI200422087
93. Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol (2014) 16:876–88. doi:10.1038/ncb3011
94. Sroka IC, Sandoval CP, Chopra H, Gard JM, Pawar SC, Cress AE. Macrophage-dependent cleavage of the laminin receptor alpha6beta1 in prostate cancer. Mol Cancer Res (2011) 9:1319–28. doi:10.1158/1541-7786.MCR-11-0080
95. Marconi C, Bianchini F, Mannini A, Mugnai G, Ruggieri S, Calorini L. Tumoral and macrophage uPAR and MMP-9 contribute to the invasiveness of B16 murine melanoma cells. Clin Exp Metastasis (2008) 25:225–31. doi:10.1007/s10585-007-9136-0
96. Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol (2006) 18:39–48. doi:10.1016/j.coi.2005.11.006
97. Sweet MJ, Hume DA. CSF-1 as a regulator of macrophage activation and immune responses. Arch Immunol Ther Exp (Warsz) (2003) 51:169–77.
98. Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front Immunol (2014) 5:489. doi:10.3389/fimmu.2014.00489
99. Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov (2015) 5:932–43. doi:10.1158/2159-8290.CD-15-0012
100. Robinson BD, Sica GL, Liu YF, Rohan TE, Gertler FB, Condeelis JS, et al. Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin Cancer Res (2009) 15:2433–41. doi:10.1158/1078-0432.CCR-08-2179
101. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One (2009) 4:e6562. doi:10.1371/journal.pone.0006562
102. Catalano V, Turdo A, Di Franco S, Dieli F, Todaro M, Stassi G. Tumor and its microenvironment: a synergistic interplay. Semin Cancer Biol (2013) 23:522–32. doi:10.1016/j.semcancer.2013.08.007
103. Hagemann T, Biswas SK, Lawrence T, Sica A, Lewis CE. Regulation of macrophage function in tumors: the multifaceted role of NF-kappaB. Blood (2009) 113:3139–46. doi:10.1182/blood-2008-12-172825
104. Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell (2009) 15:416–28. doi:10.1016/j.ccr.2009.03.016
105. Lee CH, Liu SY, Chou KC, Yeh CT, Shiah SG, Huang Y, et al. Tumor-associated macrophages promote oral cancer progression through activation of the Axl signaling pathway. Ann Surg Oncol (2014) 21:1031–7. doi:10.1245/s10434-013-3400-0
106. Martinez FO. Analysis of gene expression and gene silencing in human macrophages. Curr Protoc Immunol (2012) 14:14.28.1–23. doi:10.1002/0471142735.im1428s96
107. Heikkila K, Ebrahim S, Lawlor DA. Systematic review of the association between circulating interleukin-6 (IL-6) and cancer. Eur J Cancer (2008) 44:937–45. doi:10.1016/j.ejca.2008.02.047
108. Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene (2009) 28:2940–7. doi:10.1038/onc.2009.180
109. Dehai C, Bo P, Qiang T, Lihua S, Fang L, Shi J, et al. Enhanced invasion of lung adenocarcinoma cells after co-culture with THP-1-derived macrophages via the induction of EMT by IL-6. Immunol Lett (2014) 160:1–10. doi:10.1016/j.imlet.2014.03.004
110. Kahata K, Dadras MS, Moustakas A. TGF-β family signaling in epithelial differentiation and epithelial-mesenchymal transition. Cold Spring Harb Perspect Biol (2017). doi:10.1101/cshperspect.a022194
111. Techasen A, Loilome W, Namwat N, Dokduang H, Jongthawin J, Yongvanit P. Cytokines released from activated human macrophages induce epithelial mesenchymal transition markers of cholangiocarcinoma cells. Asian Pac J Cancer Prev (2012) 13 Suppl:115–8.
112. Kawata M, Koinuma D, Ogami T, Umezawa K, Iwata C, Watabe T, et al. TGF-beta-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J Biochem (2012) 151:205–16. doi:10.1093/jb/mvr136
113. Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett (2014) 352:160–8. doi:10.1016/j.canlet.2014.05.008
114. Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell (2011) 145:926–40. doi:10.1016/j.cell.2011.04.029
115. Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J (2008) 27:1671–81. doi:10.1038/emboj.2008.105
116. Ho MY, Tang SJ, Chuang MJ, Cha TL, Li JY, Sun GH, et al. TNF-alpha induces epithelial-mesenchymal transition of renal cell carcinoma cells via a GSK3beta-dependent mechanism. Mol Cancer Res (2012) 10:1109–19. doi:10.1158/1541-7786.MCR-12-0160
117. Waight JD, Gombos RB, Wilson NS. Harnessing co-stimulatory TNF receptors for cancer immunotherapy: current approaches and future opportunities. Hum Antibodies (2017) 25(3–4):87–109. doi:10.3233/HAB-160308
118. David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R axis: a double agent in tumour immune resistance. Vaccines (Basel) (2016) 4:22. doi:10.3390/vaccines4030022
119. Palena C, Hamilton DH, Fernando RI. Influence of IL-8 on the epithelial-mesenchymal transition and the tumor microenvironment. Future Oncol (2012) 8:713–22. doi:10.2217/fon.12.59
120. Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology (2011) 141:279–91.e1–5. doi:10.1053/j.gastro.2011.04.008
121. Fu XT, Dai Z, Song K, Zhang ZJ, Zhou ZJ, Zhou SL, et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol (2015) 46:587–96. doi:10.3892/ijo.2014.2761
122. Lu H, Clauser KR, Tam WL, Fröse J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol (2014) 16:1105–17. doi:10.1038/ncb3041
123. Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature (2009) 457:102–6. doi:10.1038/nature07623
124. Ferjančič S, Gil-Bernabé AM, Hill SA, Allen PD, Richardson P, Sparey T, et al. VCAM-1 and VAP-1 recruit myeloid cells that promote pulmonary metastasis in mice. Blood (2013) 121:3289–97. doi:10.1182/blood-2012-08-449819
125. Su S, Liu Q, Chen J, Chen J, Chen F, He C, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell (2014) 25:605–20. doi:10.1016/j.ccr.2014.03.021
126. Williams CB, Yeh ES, Soloff AC. Tumor-associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer (2016) 2:15025. doi:10.1038/npjbcancer.2015.25
127. Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M, et al. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med (2009) 206:1089–102. doi:10.1084/jem.20081605
128. Abraham D, Zins K, Sioud M, Lucas T, Schäfer R, Stanley ER, et al. Stromal cell-derived CSF-1 blockade prolongs xenograft survival of CSF-1-negative neuroblastoma. Int J Cancer (2010) 126:1339–52. doi:10.1002/ijc.24859
129. Aharinejad S, Sioud M, Lucas T, Abraham D. Targeting stromal-cancer cell interactions with siRNAs. Methods Mol Biol (2009) 487:243–66. doi:10.1007/978-1-60327-547-7_12
130. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19:1423–37. doi:10.1038/nm.3394
131. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25:846–59. doi:10.1016/j.ccr.2014.05.016
132. Zhang W, Zhu XD, Sun HC, Xiong XQ, Zhuang PY, Xu HX, et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res (2010) 16:3420–30. doi:10.1158/1078-0432.CCR-09-2904
133. Hiraoka K, Zenmyo M, Watari K, Iguchi H, Fotovati A, Kimura YN, et al. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci (2008) 99:1595–602. doi:10.1111/j.1349-7006.2008.00880.x
134. Kruse J, von Bernstorff W, Evert K, Albers N, Hadlich S, Hagemann S, et al. Macrophages promote tumour growth and liver metastasis in an orthotopic syngeneic mouse model of colon cancer. Int J Colorectal Dis (2013) 28:1337–49. doi:10.1007/s00384-013-1703-z
135. Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res (2005) 65:3437–46. doi:10.1158/0008-5472.CAN-04-4262
136. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell (2013) 23:249–62. doi:10.1016/j.ccr.2013.01.008
137. Mongroo PS, Rustgi AK. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol Ther (2010) 10:219–22. doi:10.4161/cbt.10.3.12548
138. Bracken CP, Khew-Goodall Y, Goodall GJ. Network-based approaches to understand the roles of miR-200 and other microRNAs in cancer. Cancer Res (2015) 75:2594–9. doi:10.1158/0008-5472.CAN-15-0287
139. Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C, et al. Tumour angiogenesis regulation by the miR-200 family. Nat Commun (2013) 4:2427. doi:10.1038/ncomms3427
140. Cortez MA, Valdecanas D, Zhang X, Zhan Y, Bhardwaj V, Calin GA, et al. Therapeutic delivery of miR-200c enhances radiosensitivity in lung cancer. Mol Ther (2014) 22:1494–503. doi:10.1038/mt.2014.79
141. Siemens H, Jackstadt R, Hünten S, Kaller M, Menssen A, Götz U, et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle (2011) 10:4256–71. doi:10.4161/cc.10.24.18552
142. Siemens H, Neumann J, Jackstadt R, Mansmann U, Horst D, Kirchner T, et al. Detection of miR-34a promoter methylation in combination with elevated expression of c-Met and beta-catenin predicts distant metastasis of colon cancer. Clin Cancer Res (2013) 19:710–20. doi:10.1158/1078-0432.CCR-12-1703
143. Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther (2010) 18:1650–6. doi:10.1038/mt.2010.136
144. Kim J, Hwan Kim S. CK2 inhibitor CX-4945 blocks TGF-beta1-induced epithelial-to-mesenchymal transition in A549 human lung adenocarcinoma cells. PLoS One (2013) 8:e74342. doi:10.1371/journal.pone.0074342
145. Kim YJ, Choi WI, Jeon BN, Choi KC, Kim K, Kim TJ, et al. Stereospecific effects of ginsenoside 20-Rg3 inhibits TGF-beta1-induced epithelial-mesenchymal transition and suppresses lung cancer migration, invasion and anoikis resistance. Toxicology (2014) 322:23–33. doi:10.1016/j.tox.2014.04.002
146. Wei X, Tang C, Lu X, Liu R, Zhou M, He D, et al. MiR-101 targets DUSP1 to regulate the TGF-beta secretion in sorafenib inhibits macrophage-induced growth of hepatocarcinoma. Oncotarget (2015) 30:18389–405. doi:10.18632/oncotarget.4089
147. Schech AJ, Kazi AA, Gilani RA, Brodie AH. Zoledronic acid reverses the epithelial-mesenchymal transition and inhibits self-renewal of breast cancer cells through inactivation of NF-kappaB. Mol Cancer Ther (2013) 12:1356–66. doi:10.1158/1535-7163.MCT-12-0304
Keywords: tumor microenvironment, tumor-associated macrophages, epithelial–mesenchymal transition, metastasis, EMT
Citation: Song W, Mazzieri R, Yang T and Gobe GC (2017) Translational Significance for Tumor Metastasis of Tumor-Associated Macrophages and Epithelial–Mesenchymal Transition. Front. Immunol. 8:1106. doi: 10.3389/fimmu.2017.01106
Received: 31 May 2017; Accepted: 23 August 2017;
Published: 13 September 2017
Edited by:
Sherven Sharma, VA Greater Los Angeles Healthcare System (VHA), United StatesCopyright: © 2017 Song, Mazzieri, Yang and Gobe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Glenda C. Gobe, Zy5nb2JlQHVxLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.