 Jia Liu
Jia Liu Fei Wang
Fei Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 August 2017
Sec. Multiple Sclerosis and Neuroimmunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01005
This article is part of the Research Topic Humoral and Cellular Immunity in Neurodegenerative Diseases: Pathogenesis, Diagnostics and Treatment View all 13 articles
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects upper motor neurons (MNs) comprising the corticospinal tract and lower MNs arising from the brain stem nuclei and ventral roots of the spinal cord, leading to fatal paralysis. Currently, there are no effective therapies for ALS. Increasing evidence indicates that neuroinflammation plays an important role in ALS pathogenesis. The neuroinflammation in ALS is characterized by infiltration of lymphocytes and macrophages, activation of microglia and reactive astrocytes, as well as the involvement of complement. In this review, we focus on the key cellular players of neuroinflammation during the pathogenesis of ALS by discussing not only their detrimental roles but also their immunomodulatory actions. We will summarize the pharmacological therapies for ALS that target neuroinflammation, as well as recent advances in the field of stem cell therapy aimed at modulating the inflammatory environment to preserve the remaining MNs in ALS patients and animal models of the disease.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive degeneration of upper and lower motor neurons (MNs) in the brain and spinal cord. ALS usually begins in limb or bulbar muscles, spreads to other body regions, and eventually ends with respiratory muscle dysfunction. The majority (more than 90%) of ALS is sporadic, whereas a minor fraction (about 5–10%) is familial. Mutations in Cu/Zn superoxide dismutase 1 (SOD1), the first identified gene in ALS, characterize more than 20% of familial and 1–4% of sporadic ALS cases (1, 2). To date, more than 20 gene mutations have been identified in familial ALS, and hexarepeat expansion on chromosome 9 open reading frame 72 (C9orf72) is reported to be the most frequent genetic cause of familial ALS (40%) (2, 3). Scientific advances in genetic studies have enabled the identification of genes contributing to ALS pathogenesis. However, no effective therapy is currently available for ALS patients. Riluzole, licensed by the Food and Drug Administration (FDA) in 1996, is the only drug that could extend the survival of ALS patients by about 3 months (4, 5). Therefore, it is urgent to identify new potential therapeutic target(s) for the development of more effective and beneficial treatments for ALS patients. In this review, we discuss not only the detrimental roles but also the immunomodulatory actions of key inflammatory cellular players in ALS. In addition, we summarize current pharmacological strategies targeting neuroinflammation and finally explore advances in stem cell therapy aimed at modulating the inflammatory environment to preserve the remaining MNs in ALS patients and animal models of the disease.
Despite numerous preclinical and clinical studies that have been conducted to evaluate the underlying cause of MN degeneration, the exact pathogenic mechanism of ALS is still far from being understood. Several pathways, including oxidative stress, mitochondrial dysfunction, axonal damage, excitotoxicity, neuroinflammation, and protein aggregation, have been suggested to be involved in ALS pathogenesis (6). It seems that multiple factors, rather than a single mechanism, contribute to the development and progression of ALS.
Increased levels of free radicals were found in cerebrospinal fluid (CSF), serum, and urine samples of both familial and sporadic ALS patients (7–9). In fact, SOD1 plays a crucial role in the clearance of reactive oxygen species (ROS) and the aberrant activity of mutant human SOD1 (mSOD1) in ALS leads to oxidative damage (10). However, other ALS-related proteins, such as mutant TAR DNA-binding protein-43 (TDP-43) and other still unknown factors in sporadic ALS, may promote oxidative stress in MNs (11).
Accumulation of misfolded mSOD1 in mitochondria leads to morphological alternations such as vacuolated and dilated organelles with disorganized cristae and membranes in spinal MNs and skeletal muscles of both ALS patients and SOD1G93A mice (12, 13), as well as physiological changes including abnormal release of adenosine triphosphate (ATP) and ROS, impaired energy homeostasis, and unusual activation of apoptosis (12–14). The presence of mSOD1 also contributes to altered mitochondrial calcium buffering capacity and reduced calcium uptake from the cytoplasm in SOD1G93A mice (15, 16). In addition, axonal transport of mitochondria along microtubules is disrupted in ALS, leading to metabolic changes in neurons (12, 17).
Increased glutamate levels in CSF (18) and therapeutic benefits achieved by riluzole, an anti-excitotoxic drug (19), implicate excitotoxicity as a mechanism contributing to MN injury in ALS. Low endogenous calcium buffering capacity in ALS-vulnerable spinal and brainstem MNs makes them more susceptible to excitotoxic insults (20). It was reported that sera from ALS patients could induce abnormal N-methylaspartate receptor activation (21). Glutamate transport deficits have been identified in motor cortex and spinal cord of ALS patients and transgenic mSOD1 mouse models. Especially affected is astroglial-specific excitatory amino-acid transporter 2 (EAAT2) (22–24), leading to increased synaptic glutamate concentration and overstimulation of postsynaptic glutamate receptors, which contributes to excitotoxic neuronal degeneration (8, 25, 26). Guo et al. observed a delay in MN degeneration and disease progression in transgenic mSOD1 mice overexpressing EAAT2 (27), suggesting the loss of EAAT2 contributes to MN degeneration in ALS.
Aberrant mutant or misfolded proteins (e.g., SOD1, TDP-43, and FUS/TLS RNA-/DNA-binding protein) aggregate in the cytoplasm, nucleus, or extracellular matrix leading to cellular organellar damage and neuronal dysfunction in ALS (8, 28–32). Misfolded mSOD1 aggregates formed pore-like structures in lipid membrane that allowed the influx of calcium (33) and were even able to activate microglia in vitro (34). The ubiquitin proteasome system (UPS) and autophagy play a central role in degrading misfolded proteins and thus preventing their aggregation. Impairment of autophagy in MNs may result in the accumulation of misfolded proteins and cell death (35). Alternation of UPS and activation of autophagy have been observed in spinal MNs of mSOD1 mice (36) and in postmortem samples of ALS cases (37). The enhancement of autophagy could improve the clearance of misfolded protein aggregates and neuronal survival in ALS models (38, 39).
Analysis of CSF and postmortem spinal cord samples from ALS cases revealed increased microglial activation and lymphocyte permeation (40, 41), indicating that neuroinflammation may play a role in MN degeneration. Further investigation revealed that microglia were activated in the early stages of ALS and played an either deleterious or beneficial role (42, 43). Moreover, astrocytes acquired toxic properties and subsequently contributed to MN death (44), while infiltrated T lymphocytes controlled microglial response by limiting their detrimental effects and enhancing their neuroprotective capacity (45). In addition, the breakdown of blood–brain barrier and blood–spinal cord barrier also contributed to early MN degeneration in ALS patients and mice (46, 47), while restoration of the barrier integrity delayed the onset of neurodegeneration and disease progression (48).
Neuroinflammation, characterized by microglial and astrocyte activation, T lymphocyte infiltration, and overproduction of inflammatory cytokines, has been demonstrated in association with neuronal loss in both animal and human tissues, even during the presymptomatic phase of ALS (49). Accumulating evidence from preclinical work has implicated immune cells in either exerting deleterious or protective effects on MN survival depending on the stage of disease progression; however, the mechanism is far from being fully elucidated.
Microglia are the first line of immune defense in the brain and spinal cord. They survey the surrounding environment and respond to “danger signals” from damaged tissues. It has been reported that injured MNs and astrocytes release misfolded proteins (such as mSOD1) in ALS, which activate microglia through CD14, toll-like receptor (TLR) 2, TLR4, and scavenger receptor dependent pathways (34, 50, 51). Direct evidence was provided using positron emission tomography (PET) that widespread microglial activation was present in the brain of living ALS patients and SOD1G93A mice (52–54), with a significant correlation between the intensity of microglial activation in the motor cortex and the severity of clinical MN deficits (54). Studies in mSOD1 transgenic mice further revealed that the replacement of mSOD1 microglia with wild-type microglia, as well as reduced mSOD1 expression in microglia, postponed MN degeneration and extended the lifespan of the animals (55, 56). A recent work by O’Rourke et al. (57) demonstrated that C9orf72 expression was highest in myeloid cells, and the loss of C9orf72 function in mice led to defects in lysosomal trafficking, decreased ability of microglia to clear aggregated proteins, as well as altered microglial responses, and neuroinflammation. Similar results were observed in macrophages. In particular, even haploinsufficiency of C9orf72 appears to contribute to altered inflammatory responses in macrophages. These findings suggest that C9orf72 may have a dual effect on both neurons and myeloid cells.
In addition, extracellular ATP released by dying and abnormally functioning neurons may activate microglia through the ionotropic P2X and metabotropic P2Y purinergic receptors, followed by inflammatory reactions (58). The expression level of P2X7 was increased in activated microglia from postmortem spinal cord of ALS patients (59), as well as in SOD1G93A mice (58). Furthermore, it was observed that the upregulation of P2X4, P2X7, and P2Y6 receptors in mSOD1 microglia, in particular P2X7, was associated with reduced ATP hydrolysis in the same ALS microglia, which led to increased production of tumor necrosis factor (TNF)-α and cyclooxygenase-2 (COX-2) with consequent toxicity to neuronal cells (60). This toxicity through activation of P2X7 was also confirmed in mSOD1 astrocytes (61). Studies in advance have shown that microglia-mediated deleterious effects in ALS could be prevented by genetic ablation of P2X7 receptor or by using specific antagonists to the receptor (58, 59, 62). However, further work displayed the complex role of P2X7 in ALS pathogenesis. Apolloni et al. found that constitutive deletion of P2X7 receptor aggravated disease progression, exacerbated astrogliosis, microgliosis, and motoneuron loss, activated MAPKs pathway, as well as increased the release of proinflammatory markers such as nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) and inducible nitric oxide synthase in the lumbar spinal cord of end-stage (23 weeks of age) SOD1G93A mice (63). Furthermore, these studies demonstrated that only the administration of P2X7 antagonist, Brilliant Blue G, starting at late presymptomatic stage (100 days/14 weeks of age) significantly enhanced MN survival in lumbar spinal cord through reducing microgliosis and modulating the expression of inflammatory markers, accompanied by delayed onset and improved motor functions (64). The key feature that emerged from these results might be the dual action of P2X7 and the existence of a time window concerning its beneficial role in ALS. The dual action of P2X7 during ALS progression might correspond to the switch of microglia from protective M2 to deleterious M1 phenotype. Therefore, precocious ablation of the receptor is detrimental, and its pharmacological blockade at early presymptomatic phase or after disease onset might be too early or too late. Thus, only interventions within an effective therapeutic window may provide positive outcomes in ALS patients.
Once activated, microglia display distinct and plastic phenotypes, with either neurotoxic or neuroprotective function, depending on the state of activation and disease stage. During the early slow progressive stage of ALS, microglia display an M2 phenotype with upregulated expression of M2 markers such as CD206 and Ym1, promote tissue repair and regeneration, and interact with protective signals such as CD200 and fractalkine (51, 65). As the disease progresses, injured MNs release “danger signals” (e.g., mSOD1) that induce microglia to acquire an M1 phenotype with enhanced secretion of NOX2, ROS, and proinflammatory cytokines (e.g., TNF-α, IL-1, and IL-6) (66, 67). In vitro co-culture studies further demonstrate that early stage M2 microglia enhanced MN survival, while end-stage M1 microglia were toxic to MNs (66).
Genes linked to ALS are not only expressed in MNs but also in astrocytes (68–71). Astrocytes expressing mSOD1 have been shown in vitro and in vivo to be toxic to both normal MNs and MNs derived from embryonic stem cell (ESC) carrying mSOD1 gene (71–73). Interestingly, mSOD1-expressing astrocytes selectively caused death of spinal MNs in ALS, but not spinal GABAergic or dorsal root ganglion neurons or ESC-derived interneurons (71). Selective silencing or blockage of mSOD1 gene in astrocytes, or transplantation of healthy astrocytes, could attenuate astrocyte-mediated toxicity and MN loss, delayed disease progression, and prolonged the lifespan of mSOD1 mice (74–77). Conversely, transplantation of mSOD1-expressing astrocytes induced focal MN degeneration and death in the spinal cord of wild-type rats (78). In addition, astrocytes reprogrammed from ALS patient fibroblasts impaired the survival of MNs (79, 80). Thus, the expression of ALS-associated mutant proteins in astrocytes contributes to non-cell autonomous toxicity. Qian et al. (79) recently showed that both MNs and non-MNs degenerated in the spinal cord transplanted with ALS astrocytes. Importantly, they observed that non-MNs were lost earlier than MNs, suggesting that non-cell autonomous toxicity by ALS astrocytes on neural degeneration is not specific to MNs, and non-MNs might mediate the degeneration.
As discussed above, astrocytes play an important role in ALS; however, it is still unclear how ALS-associated mutant proteins contribute to the dysfunction of astrocytes and how dysregulated astrocytes exert non-cell autonomous toxicity to MNs. In normal conditions, astrocytes clear excess glutamate from synaptic clefts through glutamate transporters. In sporadic and familial ALS patients as well as mSOD1 mice, the loss of glutamate transporter, EAAT2/GLT-1, led to less efficient uptake of glutamate by astrocytes and therefore exacerbated MN degeneration (22, 78, 81, 82). Mitochondrial defects in mSOD1 but not wild-type astrocytes were reported to be toxic for MNs (83), and this could be prevented by antioxidants and nitric oxide synthase inhibitors (83, 84). Ferraiuolo et al. revealed the metabolic dysfunction in ALS astrocytes, particularly in the astrocyte lactate efflux transporter, with resultant decrease in spinal cord lactate levels (85). In addition, astrocytes from postmortem tissues from ALS cases and SOD1G93A mice were reported to exert toxic effects on MNs by secreting inflammatory mediators such as prostaglandin E2, leukotriene B4, nitric oxide, and NOX2 (76, 84, 86). Recently, astrocytes have been demonstrated to trigger MN death by activating a caspase-independent form of programmed cell death called necroptosis, which involves the loss of plasma membrane integrity through receptor interacting serine/threonine-protein kinase 1 (RIPK1) and mixed lineage kinase domain-like (MLKL) (87). Inhibition of the key necroptosis effectors, RIPK1 or MLKL, could protect MNs against sporadic ALS astroglial toxicity and delay the onset of motor dysfunction (87, 88), therefore suggesting these as potential new therapeutic targets.
In SOD1G93A mice, CD4+ T cells were observed in lumbar spinal cords at early stages of the disease, while at the end stage, both CD4+ and CD8+ T cells were present (45, 89). Genetic removal of CD4+ T cells or functional total T cells accelerated disease progression with upregulated expression of NOX2 and proinflammatory cytokines in mSOD1 mice (90, 91), while reconstitution of T cells could prolong the survival of mSOD1 mice and inhibited the activation of M1 microglia (91). The neuroprotection associated with CD4+ T cells in ALS is probably due to their interactions with microglia and astrocytes. Beers et al. (92) reported that endogenous regulatory T cells (Tregs) were increased in spinal cords of mSOD1 mice at early slow progressive disease stage, accompanied by increased expression of IL-4 and levels of protective M2 microglia, while these decreased when disease rapidly accelerated with loss of forkhead box P3 (FoxP3) expression in Tregs. Passive transfer of Tregs obtained from donor mSOD1 mice during the stable stage without ex vivo activation extended the stable disease progression phage and lengthened survival of recipient mSOD1 mice (92). Accordingly, in ALS patients, the number of Tregs decreased in blood and spinal cord tissues accompanied by reduced expression of FoxP3, transforming growth factor-β, IL-4 and GATA binding protein 3 (Gata-3) at the rapidly progressive stage, and the number of Tregs was inversely correlated with the progression rates and severity (92, 93). A further analysis revealed that ALS patients with low FoxP3 levels showed more rapid progression, indicating that low FoxP3 expression might be predictive of future rapid disease progression (93). However, it is still unknown whether Tregs from ALS patients still keep their suppressive capabilities. Recently, Beers et al. (94) demonstrated that Tregs from both slowly and rapidly progressing ALS patients were dysfunctional and exhibited reduced suppressive capabilities on the proliferation of responder T lymphocytes compared to Tregs from healthy volunteers. Moreover, Treg suppressive deficiency correlated with disease burden and rates of disease progression. This study further illustrated that the loss of suppressive capabilities of Tregs from ALS patients was not permanent. Tregs regained their suppressive capabilities when removed from their environment and expanded in vitro, thus suggesting a potential novel therapeutic strategy for ALS patients.
Evidence has been provided that the expression of Th17 related cytokines (IL-17 and IL-23) was elevated in blood, CSF, and spinal cord tissues from ALS patients, suggesting Th17 might also play a role in ALS pathogenesis (95–97). However, little is known about Th17 in mSOD1 transgenic mice. Another T-cell subpopulation participating in ALS is natural killer T (NKT) cells. Rentzos et al. reported that NKT cells were increased in peripheral blood of ALS patients (98). Moreover, NKT cell levels and activation state increased in the spinal cord, spleen, and liver of mSOD1 mice, and immunomodulation of NKT cells led to the reduction of MN loss, delayed disease onset, and prolonged the life span of mSOD1 mice (99).
Ample previous evidence has uncovered the role of activated microglia in the CNS and altered T cells in the peripheral nervous system of ALS patients and mSOD1 mice as discussed above. However, much less is known about the role of peripheral macrophages and monocytes in MN degeneration. Several studies have reported the activation of monocytes in the peripheral blood of ALS patients (100), as well as the increased invasion of peripheral monocytes into the spinal cord of ALS patients and mice (101, 102), which contributed to MN loss. Activated monocytes from ALS patients exhibited decelerated phagocytosis, altered adhesion behavior, and impaired proinflammatory cytokine secretion (102, 103). A recent study revealed 233 differentially expressed genes in ALS monocytes. Among these was a unique inflammation-related gene expression profile, including IL-1β, IL-8, FOSB, CXCL1, and CXCL2, suggesting ALS monocytes were skewed toward a proinflammatory state in the peripheral circulation, which might play a role in rapidly progressing ALS (104). Murdock et al. further suggested that an increased ratio of neutrophils to monocytes displayed a better correlation with disease progression (105). Another recent study has observed macrophage-mediated inflammation in the skeletal muscle of familial ALS rats (106), which might be another therapeutic target for novel treatment of ALS.
The complement system plays a crucial role in the recruitment of mononuclear cells and macrophages (mediated by C3a and C5a) and in the deposition of cytotoxic pore-forming membrane attack complex (formed by C5–C9) on the cell surface. Multiple complement components including C1q, C5a, and C5b-9 are elevated in the plasma, CSF, and spinal cord of ALS patients and transgenic mSOD1 mice (107–110). However, the role of the complement pathway in ALS pathogenesis is still controversial. Complement factor C1q was observed to colocalize with neurofilament and deposited on motor end plates in intercostal muscle of ALS patients, suggesting that complement activation may precede end-plate denervation in human ALS (111). Lobsiger et al. showed that genetic deletion of C1q and C3 from ALS mice did not affect the overall onset and progression of the disease, thus suggesting that C1q induction and classical or alternative complement pathway activation do not contribute significantly to mSOD1-mediated ALS pathogenesis in mice (112). On the contrary, others have reported that terminal complement activation and C5a production occurred in skeletal muscle tissue of SOD1G93A mice (113). Local activation and increased expression of C5a–C5aR1 signaling contributed to the recruitment of macrophages that might have accelerated muscle denervation and MN death in SOD1G93A mice (113, 114). Selective inhibition of C5a–C5aR1 signaling ameliorated disease pathology, reduced motor symptoms, and extended the survival of SOD1G93A mice (110, 115).
Multiple preclinical studies and clinical trials have been conducted to search for the underlying cause of MN degeneration; however, the exact mechanism remains largely unclear. Therefore, the development of effective and targeted therapies is still underway worldwide. In recent years, increased interest has been focused on finding appropriate tools to effectively target neuroinflammation in ALS. Multiple compounds with anti-inflammatory properties have been reported to enhance MN survival in transgenic mice, even though none has been shown to be effective for ALS patients (116). A number of neuroprotective agents targeting neuroinflammatory pathways in ALS are summarized in Table 1.
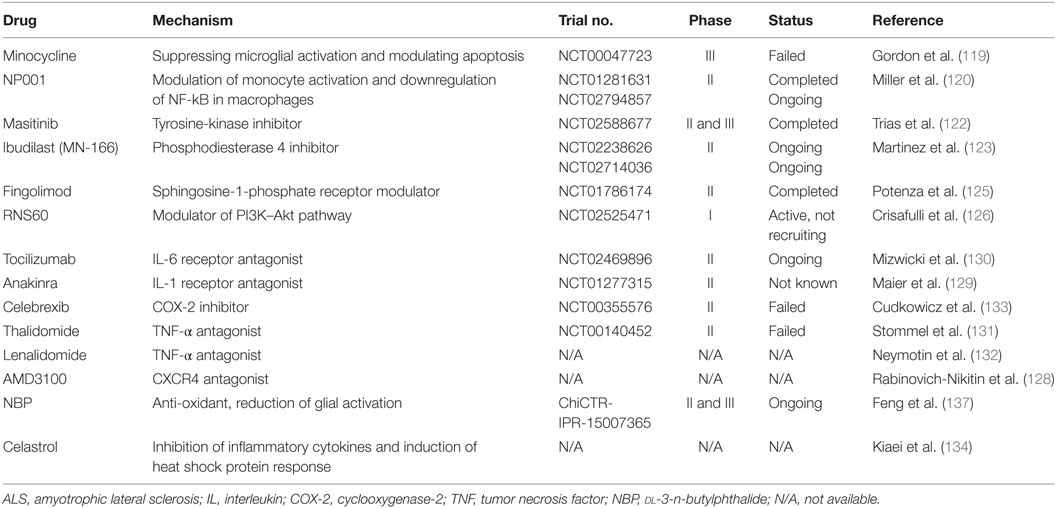
Table 1. Summary of drugs and compounds targeting neuroinflammation in ALS.
Minocycline, a broad-spectrum tetracycline antibiotic, was shown to reduce the loss of MNs, delay disease onset, and extend survival of SOD1G93A mice (117), probably by suppressing microglial activation and modulating apoptosis (118). Unfortunately, a phase III trial (ClinicalTrials.gov Identifier: NCT00047723) revealed harmful effects after its continuous administration to ALS patients (119), limiting its further application. NP001 is a small molecule that regulates monocytes and macrophages by switching them from an inflammatory phenotype to a basal non-inflammatory phenotype. In the completed phase I and II trials in ALS patients (ClinicalTrials.gov Identifier: NCT01091142, NCT01281631), NP001 was found to be safe, well tolerated, and presented a positive trend in slowing disease progression (120, 121). Currently, a second phase II trial is ongoing to confirm these data (ClinicalTrials.gov Identifier: NCT02794857). Masitinib is a tyrosine-kinase inhibitor that was observed to decrease aberrant glial cells, microgliosis, and MN degeneration in the spinal cord of mSOD1 mice, and therefore prolonged survival of the animals (122). A phase II and III clinical trial aimed at assessing the efficacy and safety of masitinib in combination with riluzole in the treatment of ALS patients (ClinicalTrials.gov Identifier: NCT02588677) has recently completed with a total of 394 patients enrolled, and the final results are expected to be announced soon. Ibudilast (MN-166), a non-selective phosphodiesterase 4 inhibitor, is reported to modulate the production of proinflammatory agents from resident immune cells, and also influences their survival and activation (123). Two clinical trials with ibudilast are ongoing in ALS patients. One is administration of ibudilast in combination with riluzole to evaluate its general safety and tolerability (ClinicalTrials.gov Identifier: NCT02238626). Another trial is to investigate the impact of ibudilast on neuroinflammation measured by PET imaging and blood biomarkers (ClinicalTrials.gov Identifier: NCT02714036). Fingolimod, a modulator of sphingosine-1-phosphate receptor, is the first oral drug approved by FDA for the treatment of relapsing remitting multiple sclerosis, because it reduces the number of circulating lymphocytes in peripheral blood by sequestering them in secondary lymphoid organs through its modulation of the sphingosine-1-phosphate receptor (124). In a recent preclinical study, fingolimod improved the outcome and survival rate of mSOD1 mice, and the beneficial effect was associated with modulation of microglial activation and innate immunity (125). A phase II trial evaluating the safety and tolerability of fingolimod in ALS patients (ClinicalTrials.gov Identifier: NCT01786174) has recently been completed with no major safety issue reported.
Blockage of proinflammatory mediators (e.g., IL-6, IL-1, TNF-α, COX-2, and CXCR4) in ALS to modulate neuroinflammation and decrease MN death is another strategy that resulted in delayed symptom onset and prolonged survival of mSOD1 mice, and clinical trials for these compounds are ongoing (4, 123, 126–134) (Table 1). dl-3-n-Butylphthalide (NBP) was initially isolated from the seeds of Apium graveolens (celery) and has been approved by the State FDA of China for clinical use since 2002. NBP has been demonstrated to exert neuroprotective roles in cerebral ischemia and vascular dementia (135, 136). In preclinical work, NBP reduced glial cell activation, attenuated MN death, and thus prolonged the survival interval of SOD1G93A mice (137–139), suggesting this compound might be a novel treatment for ALS. A phase II and III multicenter, randomized, double-blind, placebo-controlled clinical trial with oral administration of NBP in ALS patients is ongoing in China to evaluate its efficacy and safety (Chictr.org.cn Identifier: ChiCTR-IPR-15007365).
Stem cell therapy offers a promising alternative for ALS, and preclinical studies have demonstrated that cell-based treatment could increase MN survival and delay disease progression. Different sources and types of cells have been and/or are being tested in preclinical stages and clinical trials for ALS (140, 141).
Mesenchymal stem cells (MSCs) derived from adipose or bone marrow tissue are the most studied cells in ALS animals and clinical trials. Studies with MSCs grafted in mSOD1 mice have reported beneficial effects on disease progression including attenuated neuroinflammation, improved motor function, reduced loss of MNs, and prolonged survival (142, 143). MSCs may also serve as a carrier to deliver neurotrophic factors (144). Due to the positive results obtained from in vivo models of the disease, numerous cell-based clinical trials have used MSCs and reported the feasibility, safety, and immunomodulatory effects of administration of MSCs to ALS patients (142, 145, 146). However, adverse effects of MSCs, such as enhancing tumor growth and metastases (147, 148), and triggering ROS generation and inflammation (149), have also been reported. Thus, more careful and detailed studies concerning the clinical safety and efficacy of MSCs should be further performed.
Embryonic stem cells have the potential to differentiate into any cell types of the three germ layers, including specific cells of neuronal or glial fates. However, whether MNs derived from ESCs could exert beneficial effects on ALS animals is still controversial (150, 151). The application of ESCs is also limited due to the difficulty of generating high-purity lineage-specific cell lines without risk of tumorigenesis and ethical issues. The generation of induced pluripotent stem cells (iPSCs) provides an alternative therapeutic approach to cell-based treatment for ALS. Autologous iPSCs might represent an ideal cell source that can be derived from patients’ own somatic cells with similar features as ESCs, but with no ethical issues, reduced risk of immune rejection, and less need for immunosuppressive drugs. Transplantation of iPSC-derived neural cells into SOD1G93A mice showed survival of donor cells, axonal sprouting, and reduction of macro- and microgliosis (152, 153). However, the use of iPSCs in the clinic is still under debate due to its capacity of tumor formation after transplantation, inefficient in vitro differentiation methods, and inherent genetic deficits from donors. Thus, it is still a big challenge to produce safe iPSC-derived neural cells for clinical cell therapy.
Neural stem/progenitor cells (NPCs) are multipotent cells committed to the neural cell lineage that can self-renew and be readily expanded in vitro. NPCs derived from an already formed nervous system have not been reported to cause tumor formation with metastasis after transplantation, which is an important criterion for a donor cell population in clinical cell transplantation. However, the limited replication and decreased differentiation potential with time, as well as the ethical concerns imposed by NPC origins and derivation should not be ignored. NPCs, derived from adult and fetal CNS tissues, as well as from ESCs and iPSCs as mentioned above, have been reported to survive, differentiate into mature neural cells, and may delay disease progression in association with extended lifespan after transplantation in ALS animals (154). The potential mechanism of grafted NPCs to repair injured MNs has been widely investigated. It has been demonstrated that NPCs may provide an effective treatment by directly replacing the lost and damaged neural cells, enhancing functional synaptic formation, providing neuroprotection by producing neurotrophic factors, or even stimulating endogenous neurogenesis (153, 155–158). As discussed previously, neuroinflammation is a key player in ALS pathogenesis that contributes to MN degeneration. However, it is still unclear whether grafted NPCs are able to modulate the inflammatory environment around damaged MNs in ALS and how donor NPCs interact with host immune cells. NPCs were reported to inhibit T-cell proliferation and promote apoptosis of encephalitogenic CNS-infiltrating T cells through apoptotic receptor ligands (e.g., FasL and Apo3L) and release of soluble mediators (e.g., nitric oxide synthase, leukemia inhibitor factor, heme oxygenase-1, and prostaglandin E2) in the chronic inflammatory environment of experimental autoimmune encephalitis (159–164). In spinal cord injury, grafted mouse NPCs could lead to a reprogramming of the local inflammatory microenvironment from a “hostile” to an “instructive” state by increasing the proportion of Tregs and reducing that of M1 macrophages (165). In our previous work with in vitro allogeneic co-culture settings, we demonstrated that human fetal-derived NPCs reduced proliferation of peripheral blood mononuclear cells via the upregulation of Tregs (166) and influenced allogeneic microglial activation and functional activity through enhanced CD200-CD200R interaction between NPCs and microglia (167). A recent work by Gao et al. indicated that induced NPCs directly reprogrammed from mouse embryonic fibroblasts were able to influence microglia activation and the acquisition of neuroprotective phenotypes via CXCL12/CXCR4 signaling (168). Further studies concerning the immunomodulatory effects of grafted NPCs in mSOD1 rodents and even ALS patients are urgently needed. Due to the encouraging results of preclinical studies with neural cell therapy, several clinical trials with human NPCs or human glial restricted progenitor cells are either completed or ongoing (Table 2). Till now, the donor cells have been well tolerated and no safety issue has been reported (169–175).
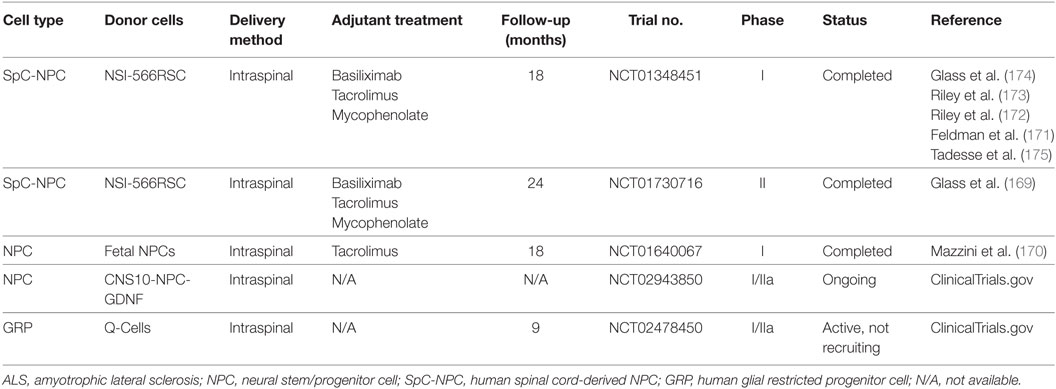
Table 2. Clinical trials of human NPCs in ALS.
Effective therapy for ALS is still in its infancy, even after years of intense investigation with chemical compounds and cell-based treatment. The pathogenesis involved in MN death in ALS is complex, and neuroinflammation has been accepted as a key contributor to MN degeneration and disease progression. However, a big challenge regarding the development of new therapies for ALS patients is the failure to translate positive preclinical results into successful clinical practice. Several issues should be taken into consideration when designing therapeutic strategies targeting neuroinflammation in ALS. First, most preclinical ALS studies invariably employ the mSOD1 transgenic rodents (generally SOD1G93A mice), which is a limited disease condition related to a small subgroup of ALS patients with SOD1 gene mutation. Therefore, transgenic mouse models with different gene mutations (e.g., TDP-43 and C9orf72) should be employed in preclinical studies to find out whether they share common mechanisms involved in neuroinflammation. Second, therapeutic strategies in transgenic animals are usually applied during presymptomatic or slowly progressive disease stage. Despite promising treatment outcomes in these preclinical studies, they cannot be translated into patients because most ALS patients are identified and diagnosed during the late and rapidly progressive phase. Third, the inflammatory environment of ALS varies with disease progression, and involves both neurotoxic and neuroprotective aspects. Thus, specific therapeutic timing may influence the pathogenic target and choice of drugs. Fourth, the heterogeneity of patients may also contribute to the failed translation of therapeutic effects from homogeneous transgenic animals to ALS patients. In addition, one should consider promoting anti-inflammatory and neuroprotective properties of immune cells, instead of simply and completely suppressing inflammatory and immune responses to achieve precise and personalized treatment for ALS patients.
JL and FW wrote the manuscript and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from the National Natural Science Foundation of China for Young Scholars (Grant No. 81501010) and from the Science and Technology Foundation for Selected Overseas Scholars of Ministry of Human Resources and Social Security of China.
1. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature (1993) 362:59–62. doi:10.1038/362059a0
2. Corcia P, Couratier P, Blasco H, Andres CR, Beltran S, Meininger V, et al. Genetics of amyotrophic lateral sclerosis. Rev Neurol (Paris) (2017) 173:254–62. doi:10.1016/j.neurol.2017.03.030
3. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron (2011) 72:245–56. doi:10.1016/j.neuron.2011.09.011
4. Kumar V, Islam A, Hassan MI, Ahmad F. Therapeutic progress in amyotrophic lateral sclerosis-beginning to learning. Eur J Med Chem (2016) 121:903–17. doi:10.1016/j.ejmech.2016.06.017
5. Gordon P, Corcia P, Meininger V. New therapy options for amyotrophic lateral sclerosis. Expert Opin Pharmacother (2013) 14:1907–17. doi:10.1517/14656566.2013.819344
6. Geevasinga N, Menon P, Özdinler PH, Kiernan MC, Vucic S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat Rev Neurol (2016) 12:651–61. doi:10.1038/nrneurol.2016.140
7. Simpson EP, Henry YK, Henkel JS, Smith RG, Appel SH. Increased lipid peroxidation in sera of ALS patients: a potential biomarker of disease burden. Neurology (2004) 62:1758–65. doi:10.1212/WNL.62.10.1758
8. Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int (2015) 6:171. doi:10.4103/2152-7806.169561
9. Chang Y, Kong Q, Shan X, Tian G, Ilieva H, Cleveland DW, et al. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One (2008) 3:e2849. doi:10.1371/journal.pone.0002849
10. Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee MK, et al. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science (1996) 271:515–8. doi:10.1126/science.271.5248.515
11. Duan W, Li X, Shi J, Guo Y, Li Z, Li C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience (2010) 169:1621–9. doi:10.1016/j.neuroscience.2010.06.018
12. Magrane J, Manfredi G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal (2009) 11:1615–26. doi:10.1089/ARS.2009.2604
13. Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol (2007) 66:10–6. doi:10.1097/nen.0b013e31802c396b
14. Pasinelli P, Houseweart MK, Brown RH Jr, Cleveland DW. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A (2000) 97:13901–6. doi:10.1073/pnas.240305897
15. Fuchs A, Kutterer S, Mühling T, Duda J, Schütz B, Liss B, et al. Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Physiol (2013) 591:2723–45. doi:10.1113/jphysiol.2012.247981
16. Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, et al. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem (2006) 96:1349–61. doi:10.1111/j.1471-4159.2006.03619.x
17. Mórotz GM, De Vos KJ, Vagnoni A, Ackerley S, Shaw CE, Miller CC. Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum Mol Genet (2012) 21:1979–88. doi:10.1093/hmg/dds011
18. Shaw PJ, Forrest V, Ince PG, Richardson JP, Wastell HJ. CSF and plasma amino acid levels in motor neuron disease: elevation of CSF glutamate in a subset of patients. Neurodegeneration (1995) 4:209–16. doi:10.1006/neur.1995.0026
19. Zhu Y, Fotinos A, Mao LL, Atassi N, Zhou EW, Ahmad S, et al. Neuroprotective agents target molecular mechanisms of disease in ALS. Drug Discov Today (2015) 20:65–75. doi:10.1016/j.drudis.2014.08.016
20. Alexianu ME, Ho BK, Mohamed AH, La Bella V, Smith RG, Appel SH. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol (1994) 36:846–58. doi:10.1002/ana.410360608
21. Texido L, Hernandez S, Martin-Satue M, Povedano M, Casanovas A, Esquerda J, et al. Sera from amyotrophic lateral sclerosis patients induce the non-canonical activation of NMDA receptors “in vitro”. Neurochem Int (2011) 59:954–64. doi:10.1016/j.neuint.2011.07.006
22. Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A (2002) 99:1604–9. doi:10.1073/pnas.032539299
23. Trotti D, Rolfs A, Danbolt NC, Brown RH Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci (1999) 2:848. doi:10.1038/12227
24. Bristol LA, Rothstein JD. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol (1996) 39:676–9. doi:10.1002/ana.410390519
25. Sunico CR, Domínguez G, García-Verdugo JM, Osta R, Montero F, Moreno-López B. Reduction in the motoneuron inhibitory/excitatory synaptic ratio in an early-symptomatic mouse model of amyotrophic lateral sclerosis. Brain Pathol (2011) 21:1–15. doi:10.1111/j.1750-3639.2010.00417.x
26. Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, et al. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron (1998) 20:589–602. doi:10.1016/S0896-6273(00)80997-6
27. Guo H, Lai L, Butchbach ME, Stockinger MP, Shan X, Bishop GA, et al. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet (2003) 12:2519–32. doi:10.1093/hmg/ddg267
28. Walker LC, LeVine H. The cerebral proteopathies: neurodegenerative disorders of protein conformation and assembly. Mol Neurobiol (2000) 21:83–95. doi:10.1385/MN:21:1-2:083
29. van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet (2017). doi:10.1016/S0140-6736(17)31287-4
30. Coan G, Mitchell CS. An assessment of possible neuropathology and clinical relationships in 46 sporadic amyotrophic lateral sclerosis patient autopsies. Neurodegener Dis (2015) 15:301–12. doi:10.1159/000433581
31. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (2006) 314:130–3. doi:10.1126/science.1134108
32. Dormann D, Haass C. TDP-43 and FUS: a nuclear affair. Trends Neurosci (2011) 34:339–48. doi:10.1016/j.tins.2011.05.002
33. Allen MJ, Lacroix JJ, Ramachandran S, Capone R, Whitlock JL, Ghadge GD, et al. Mutant SOD1 forms ion channel: implications for ALS pathophysiology. Neurobiol Dis (2012) 45:831–8. doi:10.1016/j.nbd.2011.08.031
34. Roberts K, Zeineddine R, Corcoran L, Li W, Campbell IL, Yerbury JJ. Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia (2013) 61:409–19. doi:10.1002/glia.22444
35. Cipolat Mis MS, Brajkovic S, Frattini E, Di Fonzo A, Corti S. Autophagy in motor neuron disease: key pathogenetic mechanisms and therapeutic targets. Mol Cell Neurosci (2016) 72:84–90. doi:10.1016/j.mcn.2016.01.012
36. Li L, Zhang X, Le W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy (2008) 4:290–3. doi:10.4161/auto.5524
37. Sasaki S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol (2011) 70:349–59. doi:10.1097/NEN.0b013e3182160690
38. Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol (2014) 10:677–85. doi:10.1038/nchembio.1563
39. Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev (2009) 23:2294–306. doi:10.1101/gad.1830709
40. Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol (2011) 10:253–63. doi:10.1016/S1474-4422(11)70015-1
41. Süssmuth SD, Brettschneider J, Ludolph AC, Tumani H. Biochemical markers in CSF of ALS patients. Curr Med Chem (2008) 15:1788–801. doi:10.2174/092986708785133031
42. Lee J, Hyeon SJ, Im H, Ryu H, Kim Y, Ryu H. Astrocytes and microglia as non-cell autonomous players in the pathogenesis of ALS. Exp Neurobiol (2016) 25:233–40. doi:10.5607/en.2016.25.5.233
43. Volpe CM, Nogueira-Machado JA. Is innate immunity and inflammasomes involved in pathogenesis of amyotrophic lateral sclerosis (ALS)? Recent Pat Endocr Metab Immune Drug Discov (2015) 9:40–5. doi:10.2174/1872214809666150407111420
44. Peric M, Mitrecic D, Andjus PR. Targeting astrocytes for treatment in amyotrophic lateral sclerosis. Curr Pharm Des (2017). doi:10.2174/1381612823666170615110446
45. Hooten KG, Beers DR, Zhao W, Appel SH. Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics (2015) 12:364–75. doi:10.1007/s13311-014-0329-3
46. Garbuzova-Davis S, Hernandez-Ontiveros DG, Rodrigues MC, Haller E, Frisina-Deyo A, Mirtyl S, et al. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res (2012) 1469:114–28. doi:10.1016/j.brainres.2012.05.056
47. Garbuzova-Davis S, Haller E, Saporta S, Kolomey I, Nicosia SV, Sanberg PR. Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res (2007) 1157:126–37. doi:10.1016/j.brainres.2007.04.044
48. Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A (2014) 111:E1035–42. doi:10.1073/pnas.1401595111
49. Komine O, Yamanaka K. Neuroinflammation in motor neuron disease. Nagoya J Med Sci (2015) 77:537–49.
50. Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol (2013) 8:888–99. doi:10.1007/s11481-013-9489-x
51. Appel SH, Zhao W, Beers DR, Henkel JS. The microglial-motoneuron dialogue in ALS. Acta Myol (2011) 30:4–8.
52. Gargiulo S, Anzilotti S, Coda AR, Gramanzini M, Greco A, Panico M, et al. Imaging of brain TSPO expression in a mouse model of amyotrophic lateral sclerosis with (18)F-DPA-714 and micro-PET/CT. Eur J Nucl Med Mol Imaging (2016) 43:1348–59. doi:10.1007/s00259-016-3311-y
53. Corcia P, Tauber C, Vercoullie J, Arlicot N, Prunier C, Praline J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One (2012) 7:e52941. doi:10.1371/journal.pone.0052941
54. Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis (2004) 15:601–9. doi:10.1016/j.nbd.2003.12.012
55. Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science (2006) 312:1389–92. doi:10.1126/science.1123511
56. Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A (2006) 103:16021–6. doi:10.1073/pnas.0607423103
57. O’Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AK, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science (2016) 351:1324–9. doi:10.1126/science.aaf1064
58. Volonté C, Apolloni S, Parisi C, Amadio S. Purinergic contribution to amyotrophic lateral sclerosis. Neuropharmacology (2016) 104:180–93. doi:10.1016/j.neuropharm.2015.10.026
59. Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, et al. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol (2006) 6:12. doi:10.1186/1471-2377-6-12
60. D’Ambrosi N, Finocchi P, Apolloni S, Cozzolino M, Ferri A, Padovano V, et al. The proinflammatory action of microglial P2 receptors is enhanced in SOD1 models for amyotrophic lateral sclerosis. J Immunol (2009) 183:4648–56. doi:10.4049/jimmunol.0901212
61. Gandelman M, Peluffo H, Beckman JS, Cassina P, Barbeito L. Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: implications for amyotrophic lateral sclerosis. J Neuroinflammation (2010) 7:33. doi:10.1186/1742-2094-7-33
62. Apolloni S, Parisi C, Pesaresi MG, Rossi S, Carrì MT, Cozzolino M, et al. The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J Immunol (2013) 190:5187–95. doi:10.4049/jimmunol.1203262
63. Apolloni S, Amadio S, Montilli C, Volonté C, D’Ambrosi N. Ablation of P2X7 receptor exacerbates gliosis and motoneuron death in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet (2013) 22:4102–16. doi:10.1093/hmg/ddt259
64. Apolloni S, Amadio S, Parisi C, Matteucci A, Potenza RL, Armida M, et al. Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis Model Mech (2014) 7:1101–9. doi:10.1242/dmm.017038
65. Blasco H, Corcia P, Pradat PF, Bocca C, Gordon PH, Veyrat-Durebex C, et al. Metabolomics in cerebrospinal fluid of patients with amyotrophic lateral sclerosis: an untargeted approach via high-resolution mass spectrometry. J Proteome Res (2013) 12:3746–54. doi:10.1021/pr400376e
66. Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol (2012) 237:147–52. doi:10.1016/j.expneurol.2012.06.011
67. Beers DR, Zhao W, Liao B, Kano O, Wang J, Huang A, et al. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav Immun (2011) 25:1025–35. doi:10.1016/j.bbi.2010.12.008
68. Forsberg K, Andersen PM, Marklund SL, Brännström T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol (2011) 121:623–34. doi:10.1007/s00401-011-0805-3
69. Zhang H, Tan CF, Mori F, Tanji K, Kakita A, Takahashi H, et al. TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol (2008) 115:115–22. doi:10.1007/s00401-007-0285-7
70. Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron (1997) 18:327–38. doi:10.1016/S0896-6273(00)80272-X
71. Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci (2007) 10:615–22. doi:10.1038/nn1876
72. Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell (2008) 3:637–48. doi:10.1016/j.stem.2008.09.017
73. Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci (2007) 10:608–14. doi:10.1038/nn1885
74. Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci (2008) 11:251–3. doi:10.1038/nn2047
75. Lepore AC, Rauck B, Dejea C, Pardo AC, Rao MS, Rothstein JD, et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci (2008) 11:1294–301. doi:10.1038/nn.2210
76. Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol (2011) 29:824–8. doi:10.1038/nbt.1957
77. Wang L, Gutmann DH, Roos RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet (2011) 20:286–93. doi:10.1093/hmg/ddq463
78. Papadeas ST, Kraig SE, O’Banion C, Lepore AC, Maragakis NJ. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc Natl Acad Sci U S A (2011) 108:17803–8. doi:10.1073/pnas.1103141108
79. Qian K, Huang H, Peterson A, Hu B, Maragakis NJ, Ming G-L, et al. Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Reports (2017) 8:843–55. doi:10.1016/j.stemcr.2017.03.003
80. Meyer K, Ferraiuolo L, Miranda CJ, Likhite S, McElroy S, Renusch S, et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci U S A (2014) 111:829–32. doi:10.1073/pnas.1314085111
81. Pardo AC, Wong V, Benson LM, Dykes M, Tanaka K, Rothstein JD, et al. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp Neurol (2006) 201:120–30. doi:10.1016/j.expneurol.2006.03.028
82. Dunlop J, Beal McIlvain H, She Y, Howland DS. Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis. J Neurosci (2003) 23:1688–96.
83. Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de León A, et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci (2008) 28:4115–22. doi:10.1523/JNEUROSCI.5308-07.2008
84. Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell (2008) 3:649–57. doi:10.1016/j.stem.2008.10.001
85. Ferraiuolo L, Higginbottom A, Heath PR, Barber S, Greenald D, Kirby J, et al. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain (2011) 134:2627–41. doi:10.1093/brain/awr193
86. Hensley K, Abdel-Moaty H, Hunter J, Mhatre M, Mou S, Nguyen K, et al. Primary glia expressing the G93A-SOD1 mutation present a neuroinflammatory phenotype and provide a cellular system for studies of glial inflammation. J Neuroinflammation (2006) 3:2. doi:10.1186/1742-2094-3-2
87. Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron (2014) 81:1001–8. doi:10.1016/j.neuron.2014.01.011
88. Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science (2016) 353:603–8. doi:10.1126/science.aaf6803
89. Bowerman M, Vincent T, Scamps F, Perrin FE, Camu W, Raoul C. Neuroimmunity dynamics and the development of therapeutic strategies for amyotrophic lateral sclerosis. Front Cell Neurosci (2013) 7:214. doi:10.3389/fncel.2013.00214
90. Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, et al. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A (2008) 105:17913–8. doi:10.1073/pnas.0804610105
91. Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A (2008) 105:15558–63. doi:10.1073/pnas.0807419105
92. Beers DR, Henkel JS, Zhao W, Wang J, Huang A, Wen S, et al. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain (2011) 134:1293–314. doi:10.1093/brain/awr074
93. Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med (2013) 5:64–79. doi:10.1002/emmm.201201544
94. Beers DR, Zhao W, Wang J, Zhang X, Wen S, Neal D, et al. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight (2017) 2:e89530. doi:10.1172/jci.insight.89530
95. Saresella M, Piancone F, Tortorella P, Marventano I, Gatti A, Caputo D, et al. T helper-17 activation dominates the immunologic milieu of both amyotrophic lateral sclerosis and progressive multiple sclerosis. Clin Immunol (2013) 148:79–88. doi:10.1016/j.clim.2013.04.010
96. Rentzos M, Rombos A, Nikolaou C, Zoga M, Zouvelou V, Dimitrakopoulos A, et al. Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation? Acta Neurol Scand (2010) 122:425–9. doi:10.1111/j.1600-0404.2010.01333.x
97. Fiala M, Chattopadhay M, La Cava A, Tse E, Liu G, Lourenco E, et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J Neuroinflammation (2010) 7:76. doi:10.1186/1742-2094-7-76
98. Rentzos M, Evangelopoulos E, Sereti E, Zouvelou V, Marmara S, Alexakis T, et al. Alterations of T cell subsets in ALS: a systemic immune activation? Acta Neurol Scand (2012) 125:260–4. doi:10.1111/j.1600-0404.2011.01528.x
99. Finkelstein A, Kunis G, Seksenyan A, Ronen A, Berkutzki T, Azoulay D, et al. Abnormal changes in NKT cells, the IGF-1 axis, and liver pathology in an animal model of ALS. PLoS One (2011) 6:e22374. doi:10.1371/journal.pone.0022374
100. Mantovani S, Garbelli S, Pasini A, Alimonti D, Perotti C, Melazzini M, et al. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J Neuroimmunol (2009) 210:73–9. doi:10.1016/j.jneuroim.2009.02.012
101. Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest (2012) 122:3063–87. doi:10.1172/JCI62636
102. Zondler L, Müller K, Khalaji S, Bliederhäuser C, Ruf WP, Grozdanov V, et al. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol (2016) 132:391–411. doi:10.1007/s00401-016-1548-y
103. Zondler L, Feiler MS, Freischmidt A, Ruf WP, Ludolph AC, Danzer KM, et al. Impaired activation of ALS monocytes by exosomes. Immunol Cell Biol (2017) 95:207–14. doi:10.1038/icb.2016.89
104. Zhao W, Beers DR, Hooten KG, Sieglaff DH, Zhang A, Kalyana-Sundaram S, et al. Characterization of gene expression phenotype in amyotrophic lateral sclerosis monocytes. JAMA Neurol (2017) 74:677–85. doi:10.1001/jamaneurol.2017.0357
105. Murdock BJ, Bender DE, Kashlan SR, Figueroa-Romero C, Backus C, Callaghan BC, et al. Increased ratio of circulating neutrophils to monocytes in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm (2016) 3:e242. doi:10.1212/NXI.0000000000000242
106. Van Dyke JM, Smit-Oistad IM, Macrander C, Krakora D, Meyer MG, Suzuki M. Macrophage-mediated inflammation and glial response in the skeletal muscle of a rat model of familial amyotrophic lateral sclerosis (ALS). Exp Neurol (2016) 277:275–82. doi:10.1016/j.expneurol.2016.01.008
107. Mantovani S, Gordon R, Macmaw JK, Pfluger CM, Henderson RD, Noakes PG, et al. Elevation of the terminal complement activation products C5a and C5b-9 in ALS patient blood. J Neuroimmunol (2014) 276:213–8. doi:10.1016/j.jneuroim.2014.09.005
108. Heurich B, El Idrissi NB, Donev RM, Petri S, Claus P, Neal J, et al. Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J Neuroimmunol (2011) 235:104–9. doi:10.1016/j.jneuroim.2011.03.011
109. Woodruff TM, Costantini KJ, Taylor SM, Noakes PG. Role of complement in motor neuron disease: animal models and therapeutic potential of complement inhibitors. Adv Exp Med Biol (2008) 632:143–58. doi:10.1007/978-0-387-78952-1_11
110. Woodruff TM, Costantini KJ, Crane JW, Atkin JD, Monk PN, Taylor SM, et al. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol (2008) 181:8727–34. doi:10.4049/jimmunol.181.12.8727
111. Bahia El Idrissi N, Bosch S, Ramaglia V, Aronica E, Baas F, Troost D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J Neuroinflammation (2016) 13:72. doi:10.1186/s12974-016-0538-2
112. Lobsiger CS, Boillée S, Pozniak C, Khan AM, McAlonis-Downes M, Lewcock JW, et al. C1q induction and global complement pathway activation do not contribute to ALS toxicity in mutant SOD1 mice. Proc Natl Acad Sci U S A (2013) 110:E4385–92. doi:10.1073/pnas.1318309110
113. Wang HA, Lee JD, Lee KM, Woodruff TM, Noakes PG. Complement C5a-C5aR1 signalling drives skeletal muscle macrophage recruitment in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Skelet Muscle (2017) 7:10. doi:10.1186/s13395-017-0128-8
114. Lee JD, Kamaruzaman NA, Fung JN, Taylor SM, Turner BJ, Atkin JD, et al. Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation (2013) 10:119. doi:10.1186/1742-2094-10-119
115. Lee JD, Kumar V, Fung JN, Ruitenberg MJ, Noakes PG, Woodruff TM. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Br J Pharmacol (2017) 174:689–99. doi:10.1111/bph.13730
116. Petrov D, Mansfield C, Moussy A, Hermine O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front Aging Neurosci (2017) 9:68. doi:10.3389/fnagi.2017.00068
117. Van Den Bosch L, Tilkin P, Lemmens G, Robberecht W. Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport (2002) 13:1067–70. doi:10.1097/00001756-200206120-00018
118. Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol (2004) 3:744–51. doi:10.1016/S1474-4422(04)00937-8
119. Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol (2007) 6:1045–53. doi:10.1016/S1474-4422(07)70270-3
120. Miller RG, Block G, Katz JS, Barohn RJ, Gopalakrishnan V, Cudkowicz M, et al. Randomized phase 2 trial of NP001-a novel immune regulator: safety and early efficacy in ALS. Neurol Neuroimmunol Neuroinflamm (2015) 2:e100. doi:10.1212/NXI.0000000000000100
121. Miller RG, Zhang R, Block G, Katz J, Barohn R, Kasarskis E, et al. NP001 regulation of macrophage activation markers in ALS: a phase I clinical and biomarker study. Amyotroph Lateral Scler Frontotemporal Degener (2014) 15:601–9. doi:10.3109/21678421.2014.951940
122. Trias E, Ibarburu S, Barreto-Núñez R, Babdor J, Maciel TT, Guillo M, et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation (2016) 13:177. doi:10.1186/s12974-016-0620-9
123. Martinez A, Palomo Ruiz MD, Perez DI, Gil C. Drugs in clinical development for the treatment of amyotrophic lateral sclerosis. Expert Opin Investig Drugs (2017) 26:403–14. doi:10.1080/13543784.2017.1302426
124. Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med (2010) 362:402–15. doi:10.1056/NEJMoa0907839
125. Potenza RL, De Simone R, Armida M, Mazziotti V, Pèzzola A, Popoli P, et al. Fingolimod: a disease-modifier drug in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics (2016) 13:918–27. doi:10.1007/s13311-016-0462-2
126. Crisafulli SG, Brajkovic S, Cipolat Mis MS, et al. Therapeutic strategies under development targeting inflammatory mechanisms in amyotrophic lateral sclerosis. Mol Neurobiol (2017). doi:10.1007/s12035-017-0532-4
127. Goyal NA, Mozaffar T. Experimental trials in amyotrophic lateral sclerosis: a review of recently completed, ongoing and planned trials using existing and novel drugs. Expert Opin Investig Drugs (2014) 23:1541–51. doi:10.1517/13543784.2014.933807
128. Rabinovich-Nikitin I, Ezra A, Barbiro B, Rabinovich-Toidman P, Solomon B. Chronic administration of AMD3100 increases survival and alleviates pathology in SOD1(G93A) mice model of ALS. J Neuroinflammation (2016) 13:123. doi:10.1186/s12974-016-0587-6
129. Maier A, Deigendesch N, Muller K, Weishaupt JH, Krannich A, Röhle R, et al. Interleukin-1 antagonist anakinra in amyotrophic lateral sclerosis – a pilot study. PLoS One (2015) 10:e0139684. doi:10.1371/journal.pone.0139684
130. Mizwicki MT, Fiala M, Magpantay L, Aziz N, Sayre J, Liu G, et al. Tocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am J Neurodegener Dis (2012) 1:305–15.
131. Stommel EW, Cohen JA, Fadul CE, Cogbill CH, Graber DJ, Kingman L, et al. Efficacy of thalidomide for the treatment of amyotrophic lateral sclerosis: a phase II open label clinical trial. Amyotroph Lateral Scler (2009) 10:393–404. doi:10.3109/17482960802709416
132. Neymotin A, Petri S, Calingasan NY, Wille E, Schafer P, Stewart C, et al. Lenalidomide (Revlimid) administration at symptom onset is neuroprotective in a mouse model of amyotrophic lateral sclerosis. Exp Neurol (2009) 220:191–7. doi:10.1016/j.expneurol.2009.08.028
133. Cudkowicz ME, Shefner JM, Schoenfeld DA, Zhang H, Andreasson KI, Rothstein JD, et al. Trial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol (2006) 60:22–31. doi:10.1002/ana.20903
134. Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener Dis (2005) 2:246–54. doi:10.1159/000090364
135. Jia J, Wei C, Liang J, Zhou A, Zuo X, Song H, et al. The effects of dl-3-n-butylphthalide in patients with vascular cognitive impairment without dementia caused by subcortical ischemic small vessel disease: a multicentre, randomized, double-blind, placebo-controlled trial. Alzheimers Dement (2016) 12:89–99. doi:10.1016/j.jalz.2015.04.010
136. Cui LY, Zhu YC, Gao S, Wang JM, Peng B, Ni J, et al. Ninety-day administration of dl-3-n-butylphthalide for acute ischemic stroke: a randomized, double-blind trial. Chin Med J (Engl) (2013) 126:3405–10. doi:10.3760/cma.j.issn.0366-6999.20123240
137. Feng XH, Yuan W, Peng Y, Liu MS, Cui LY. Therapeutic effects of dl-3-n-butylphthalide in a transgenic mouse model of amyotrophic lateral sclerosis. Chin Med J (Engl) (2012) 125:1760–6. doi:10.3760/cma.j.issn.0366-6999.2012.10.014
138. Feng X, Peng Y, Liu M, Cui L. dl-3-n-Butylphthalide extends survival by attenuating glial activation in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology (2012) 62:1004–10. doi:10.1016/j.neuropharm.2011.10.009
139. Zhou QM, Zhang JJ, Li S, Chen S, Le WD. n-Butylidenephthalide treatment prolongs life span and attenuates motor neuron loss in SOD1G93A mouse model of amyotrophic lateral sclerosis. CNS Neurosci Ther (2017) 23:375–85. doi:10.1111/cns.12681
140. Czarzasta J, Habich A, Siwek T, Czapliński A, Maksymowicz W, Wojtkiewicz J. Stem cells for ALS: an overview of possible therapeutic approaches. Int J Dev Neurosci (2017) 57:46–55. doi:10.1016/j.ijdevneu.2017.01.003
141. Goutman SA, Chen KS, Feldman EL. Recent advances and the future of stem cell therapies in amyotrophic lateral sclerosis. Neurotherapeutics (2015) 12:428–48. doi:10.1007/s13311-015-0339-9
142. Hajivalili M, Pourgholi F, Kafil HS, Jadidi-Niaragh F, Yousefi M. Mesenchymal stem cells in the treatment of amyotrophic lateral sclerosis. Curr Stem Cell Res Ther (2016) 11:41–50. doi:10.2174/1574888X10666150902095031
143. Boido M, Piras A, Valsecchi V, Spigolon G, Mareschi K, Ferrero I, et al. Human mesenchymal stromal cell transplantation modulates neuroinflammatory milieu in a mouse model of amyotrophic lateral sclerosis. Cytotherapy (2014) 16:1059–72. doi:10.1016/j.jcyt.2014.02.003
144. Bonafede R, Mariotti R. ALS pathogenesis and therapeutic approaches: the role of mesenchymal stem cells and extracellular vesicles. Front Cell Neurosci (2017) 11:80. doi:10.3389/fncel.2017.00080
145. Syková E, Rychmach P, Drahorádová I, Konrádová Š, Růžičková K, Voříšek I, et al. Transplantation of mesenchymal stromal cells in patients with amyotrophic lateral sclerosis: results of phase I/IIa clinical trial. Cell Transplant (2017) 26:647–58. doi:10.3727/096368916X693716
146. Mazzini L, Ferrero I, Luparello V, Rustichelli D, Gunetti M, Mareschi K, et al. Mesenchymal stem cell transplantation in amyotrophic lateral sclerosis: a phase I clinical trial. Exp Neurol (2010) 223:229–37. doi:10.1016/j.expneurol.2009.08.007
147. Zhang T, Lee YW, Rui YF, Cheng TY, Jiang XH, Li G. Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res Ther (2013) 4:70. doi:10.1186/scrt221
148. Ramasamy R, Lam EW, Soeiro I, Tisato V, Bonnet D, Dazzi F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia (2007) 21:304–10. doi:10.1038/sj.leu.2404489
149. Horn AP, Bernardi A, Luiz Frozza R, Grudzinski PB, Hoppe JB, de Souza LF, et al. Mesenchymal stem cell-conditioned medium triggers neuroinflammation and reactive species generation in organotypic cultures of rat hippocampus. Stem Cells Dev (2011) 20:1171–81. doi:10.1089/scd.2010.0157
150. Lopez-Gonzalez R, Kunckles P, Velasco I. Transient recovery in a rat model of familial amyotrophic lateral sclerosis after transplantation of motor neurons derived from mouse embryonic stem cells. Cell Transplant (2009) 18:1171–81. doi:10.3727/096368909X12483162197123
151. Deshpande DM, Kim YS, Martinez T, Carmen J, Dike S, Shats I, et al. Recovery from paralysis in adult rats using embryonic stem cells. Ann Neurol (2006) 60:32–44. doi:10.1002/ana.20901
152. Nizzardo M, Bucchia M, Ramirez A, Trombetta E, Bresolin N, Comi GP, et al. iPSC-derived LewisX+CXCR4+beta1-integrin+ neural stem cells improve the amyotrophic lateral sclerosis phenotype by preserving motor neurons and muscle innervation in human and rodent models. Hum Mol Genet (2016) 25:3152–63. doi:10.1093/hmg/ddw163
153. Popescu IR, Nicaise C, Liu S, Bisch G, Knippenberg S, Daubie V, et al. Neural progenitors derived from human induced pluripotent stem cells survive and differentiate upon transplantation into a rat model of amyotrophic lateral sclerosis. Stem Cells Transl Med (2013) 2:167–74. doi:10.5966/sctm.2012-0042
154. Haidet-Phillips AM, Maragakis NJ. Neural and glial progenitor transplantation as a neuroprotective strategy for amyotrophic lateral sclerosis (ALS). Brain Res (2015) 1628:343–50. doi:10.1016/j.brainres.2015.06.035
155. Knippenberg S, Rath KJ, Böselt S, Thau-Habermann N, Schwarz SC, Dengler R, et al. Intraspinal administration of human spinal cord-derived neural progenitor cells in the G93A-SOD1 mouse model of ALS delays symptom progression, prolongs survival and increases expression of endogenous neurotrophic factors. J Tissue Eng Regen Med (2017) 11:751–64. doi:10.1002/term.1972
156. Lee HJ, Kim KS, Ahn J, Bae HM, Lim I, Kim SU. Human motor neurons generated from neural stem cells delay clinical onset and prolong life in ALS mouse model. PLoS One (2014) 9:e97518. doi:10.1371/journal.pone.0097518
157. Kondo T, Funayama M, Tsukita K, Hotta A, Yasuda A, Nori S, et al. Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of ALS mice. Stem Cell Reports (2014) 3:242–9. doi:10.1016/j.stemcr.2014.05.017
158. Hefferan MP, Galik J, Kakinohana O, Sekerkova G, Santucci C, Marsala S, et al. Human neural stem cell replacement therapy for amyotrophic lateral sclerosis by spinal transplantation. PLoS One (2012) 7:e42614. doi:10.1371/journal.pone.0042614
159. Einstein O, Fainstein N, Vaknin I, Mizrachi-Kol R, Reihartz E, Grigoriadis N, et al. Neural precursors attenuate autoimmune encephalomyelitis by peripheral immunosuppression. Ann Neurol (2007) 61:209–18. doi:10.1002/ana.21033
160. Einstein O, Grigoriadis N, Mizrachi-Kol R, Reinhartz E, Polyzoidou E, Lavon I, et al. Transplanted neural precursor cells reduce brain inflammation to attenuate chronic experimental autoimmune encephalomyelitis. Exp Neurol (2006) 198:275–84. doi:10.1016/j.expneurol.2005.11.007
161. Pluchino S, Zanotti L, Rossi B, Brambilla E, Ottoboni L, Salani G, et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature (2005) 436:266–71. doi:10.1038/nature03889
162. Bonnamain V, Mathieux E, Thinard R, Thébault P, Nerrière-Daguin V, Lévêque X, et al. Expression of heme oxygenase-1 in neural stem/progenitor cells as a potential mechanism to evade host immune response. Stem Cells (2012) 30:2342–53. doi:10.1002/stem.1199
163. Wang L, Shi J, van Ginkel FW, Lan L, Niemeyer G, Martin DR, et al. Neural stem/progenitor cells modulate immune responses by suppressing T lymphocytes with nitric oxide and prostaglandin E2. Exp Neurol (2009) 216:177–83. doi:10.1016/j.expneurol.2008.11.017
164. Cao W, Yang Y, Wang Z, Liu A, Fang L, Wu F, et al. Leukemia inhibitory factor inhibits T helper 17 cell differentiation and confers treatment effects of neural progenitor cell therapy in autoimmune disease. Immunity (2011) 35:273–84. doi:10.1016/j.immuni.2011.06.011
165. Cusimano M, Biziato D, Brambilla E, Donegà M, Alfaro-Cervello C, Snider S, et al. Transplanted neural stem/precursor cells instruct phagocytes and reduce secondary tissue damage in the injured spinal cord. Brain (2012) 135:447–60. doi:10.1093/brain/awr339
166. Liu J, Götherström C, Forsberg M, Samuelsson EB, Wu J, Calzarossa C, et al. Human neural stem/progenitor cells derived from embryonic stem cells and fetal nervous system present differences in immunogenicity and immunomodulatory potentials in vitro. Stem Cell Res (2013) 10:325–37. doi:10.1016/j.scr.2013.01.001
167. Liu J, Hjorth E, Zhu M, Calzarossa C, Samuelsson EB, Schultzberg M, et al. Interplay between human microglia and neural stem/progenitor cells in an allogeneic co-culture model. J Cell Mol Med (2013) 17:1434–43. doi:10.1111/jcmm.12123
168. Gao M, Dong Q, Yao H, Zhang Y, Yang Y, Dang Y, et al. Induced neural stem cells modulate microglia activation states via CXCL12/CXCR4 signaling. Brain Behav Immun (2017) 59:288–99. doi:10.1016/j.bbi.2016.09.020
169. Glass JD, Hertzberg VS, Boulis NM, Riley J, Federici T, Polak M, et al. Transplantation of spinal cord-derived neural stem cells for ALS: analysis of phase 1 and 2 trials. Neurology (2016) 87:392–400. doi:10.1212/WNL.0000000000002889
170. Mazzini L, Gelati M, Profico DC, Sgaravizzi G, Projetti Pensi M, Muzi G, et al. Human neural stem cell transplantation in ALS: initial results from a phase I trial. J Transl Med (2015) 13:17. doi:10.1186/s12967-014-0371-2
171. Feldman EL, Boulis NM, Hur J, Johe K, Rutkove SB, Federici T, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann Neurol (2014) 75:363–73. doi:10.1002/ana.24113
172. Riley J, Glass J, Feldman EL, Polak M, Bordeau J, Federici T, et al. Intraspinal stem cell transplantation in ALS: a phase I trial, cervical microinjection and final surgical safety outcomes. Neurosurgery (2014) 74:77–87. doi:10.1227/NEU.0000000000000156
173. Riley J, Federici T, Polak M, Kelly C, Glass J, Raore B, et al. Intraspinal stem cell transplantation in amyotrophic lateral sclerosis: a phase I safety trial, technical note, and lumbar safety outcomes. Neurosurgery (2012) 71:405–416; discussion 416. doi:10.1227/NEU.0b013e31825ca05f
174. Glass JD, Boulis NM, Johe K, Rutkove SB, Federici T, Polak M, et al. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients. Stem Cells (2012) 30:1144–51. doi:10.1002/stem.1079
Keywords: amyotrophic lateral sclerosis, neuroinflammation, microglia, astrocytes, neural stem cells, regulatory T cells, dl-3-n-butylphthalide
Citation: Liu J and Wang F (2017) Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 8:1005. doi: 10.3389/fimmu.2017.01005
Received: 01 July 2017; Accepted: 07 August 2017;
Published: 21 August 2017
Edited by:
Andras Palotas, Asklepios-Med (Private Medical Practice and Research Center), HungaryReviewed by:
Marinos Kallikourdis, Humanitas Università, ItalyCopyright: © 2017 Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Liu, amlhbGl1MjAxM0B5ZWFoLm5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.