 Nataša Kopitar-Jerala
Nataša Kopitar-Jerala- Department of Biochemistry, Molecular and Structural Biology, Jožef Stefan Institute, Ljubljana, Slovenia
Inflammation is an essential physiological process, which enables survival during infection and maintains tissue homeostasis. Interferons (IFNs) and pro- and anti-inflammatory cytokines are crucial for appropriate response to pathogens, damaged cells, or irritants in inflammatory response. The inflammasom is multiprotein complex, which initiates cleavage of pro-inflammatory cytokines IL-1β and IL-18 into active forms. In addition, inflammasomes initiate pyroptotic cell death. In the present review, I summarize and analyze recent findings regarding the cross talk of IFNs and inflammasomes.
Introduction
Inflammation is a complex immune response to response to pathogens, damaged cells, or irritants and enables survival during infection or injury and maintains tissue homeostasis (1). In response to an infection, a cascade of signals leads to the recruitment of inflammatory cells (neutrophils and macrophages), which produce cytokines and chemokines (2). The sustained robust inflammation may lead to serious disorders due to the overproduction of inflammatory cytokines and tissue damage (2). However, cytokine secretion from neutrophils and macrophages is tightly regulated on the transcriptional level, and several pro-inflammatory cytokines also have posttranscriptional level of regulation (3). A typical inflammatory response consists of four components: inflammatory inducers, the sensors that detect them, the inflammatory mediators induced by the sensors, and the target tissues that are affected by the inflammatory mediators (1, 3). The innate immune response is involved in various inflammatory processes and has a particularly important role in bacterial and viral infections. Interferons (IFNs) and inflammatory cytokines are crucial molecules in this process, influencing cellular, tissue, and global physiological functions. Immune cells (macrophages, dendritic cells) recognize pathogen-associated molecular patterns (PAMPs) and endogenous danger-associated molecular patterns (DAMPs) (4, 5). Bacterial and viral PAMPs are detected by pattern recognition receptors (PRRs), which are also able to recognize DAMPs—endogenous molecules, released by dying or damaged cells (5–7). PRRs have distinct subcellular localization: toll-like receptors (TLRs) and C-type lectin receptors are transmembrane proteins found in the plasma membrane and endosomes, where they can survey PAMPs and DAMPs in the extracellular milieu. Intracellular PRRs are the retinoic acid-inducible gene I (RIG-I)-like receptor, the AIM2-like receptor (ALR), and the nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins (8). In addition, PRRs that sense cytosolic DNA and trigger the production of type I interferon were described (9). In this review, we discuss recent advances in understanding the role of IFNs in inflammatory response and inflammasome activation.
Interferons
Interferons were first described as an antiviral factor that interferes with viral replication in mammalian cells (10). They are secreted from infected cells and activate innate immune response that promotes not only cytokine production but also natural killer cell functions and antigen presentation (11, 12). On the basis of the structural homology and the specific receptor they associate with, three classes of IFNs have been described (Type I, II, and III) (12). Type-I IFN family includes numerous IFN-α variants (13 in human and 14 in mouse), a single IFN-β; in addition, several other IFNs were reported (IFN-ε, -k, -ω, and -δ) (11, 13). IFN-γ, is the sole type II interferon, is structurally different from the type I and III IFNs, and signals through a different receptor: the IFN-γ receptor (3). IFN-γ can potentiate pro-inflammatory signaling by priming macrophages for antimicrobial actions, since it induces nitric oxide (NO) production and inhibit NLRP3 inflammasome activation (14, 15).
Type I-IFN expression is induced by activation of PRRs and by cytokines (9, 16). While, several different cell types express IFN-β, IFN-α is secreted only by hematopoietic cells, predominately plasmacytoid dendritic cells (17). Type I-IFNs are protective in acute viral infections; however, in bacterial infections, they could have either protective or deleterious roles (18). Type I-IFNs are induced by ssRNA, dsRNA, and cytosolic DNA from viruses or bacteria (19, 20). Type-I and type-II IFNs were reported to promote the expression of over 2,000 IFN-stimulated genes (ISGs), and the ISGs-induced proteins were demonstrated to act by enhancing pathogen detection and restrict the replication of pathogens (21). Several environmental factors, as well as host and pathogen factors, regulate responses of cells to IFN signaling (11).
Toll-like receptors are a family of 13 receptors known as PRRs and play a key role in the innate and adaptive immune response (22). Viral nucleic acids are recognized by endosomal TLR-3 (double-stranded RNA), TLR-7, -8 (single-stranded RNA), and TLR-9 (unmethylated CpG DNA) (4, 19). While TLR7 and TLR9 are expressed in B cells, macrophages, and DCs, TLR8 is expressed in macrophages and DCs In addition, TLR3 is broadly expressed also in non-hematopoietic cells, in humans. Triggering of PRR results in signaling pathways that activate gene transcription by nuclear factor (NF)-κB, as well as interferon regulatory factors (IRFs) and leads to production of type I IFNs and cytokines and chemokines (4, 23, 24). Endosomal TLR3 signals solely via the adaptor TIR domain-containing adaptor inducing IFN-β (TRIF), while TLR7, 8, and 9 depend on myeloid differentiation factor-88. Both pathways subsequently activate the IκB kinase (IKK) complex leading to nuclear translocation of the transcription factor NF-κB to upregulate the expression of inflammatory cytokines and chemokines (25). IRF transcription factors, crucial for the induction of type I IFNs (IFN-α and IFN-β), are also activated by endosomal TLRs. Signaling of TLR receptors and their adaptors result in transcription factors IRF3 and IRF7 activation, while IRF3 is expressed in many different cell types, plasmacytoid dendritic cells are the only cell type that constitutively express IRF7 (11). PRRs also induce activation of pro inflammatory caspases, leading to production of processed mature cytokines. Recently, also epigenetic mechanism that determines cell type-specific differences in IFN and IFN-stimulated gene (ISG) expression in response to exogenous signals was described (26).
Cytosolic DNA sensor proteins include cyclic GMP-AMP synthase (cGAS) (27) and ALR inflammasomes: Aim-2 and IFN-γ-inducible protein 16 (IFI16). Both Aim2 and IFI16 contain HIN200 domain that bind directly to DNA and a pyrin domain (28–30). Moreover, an endoplasmic reticulum-associated molecule referred to as stimulator of interferon genes (STING) was reported to control a signaling pathway important for the detection of cytosolic DNA and type I IFN expression (31, 32). Microbial RNAs are recognized by melanoma differentiation-associated gene 5 and (RIG-I), both of which are expressed in macrophages and non-hematopoietic cells (4, 19). Downstream signaling pathways are transmitted by mitochondrial antiviral mitochondrial antiviral signaling (MAVS), also known as IFN-β promoter stimulator-1 (IPS-1)/virus-induced signaling adaptor (VISA)/Cardif, a transmembrane protein on mitochondria (33). Recently, several excellent reviews describe the mechanism of nucleic acid sensing and signaling in the cytosol (34–36).
Interferon (IFN)-α and IFN-β bind to IFN-α receptor (IFNAR), a heterodimeric transmembrane receptor, which consist of two subunits: IFNAR1 and IFNAR2. Type I IFN-induced canonical signaling pathway IFNAR engagement activated the receptor-associated protein tyrosine kinases Janus kinase 1 (JAK1) and tyrosine kinase 2, which in turn phosphorylated the signal transducer and activator of transcription 1 (STAT1) and STAT2 (37). The activated STAT1 and STAT2 dimerize and rapidly translocate to the nucleus, where they together with IFN-regulatory factor 9 form a trimolecular complex called IFN-stimulated gene factor 3 (ISGF3) (11). ISGF3 binds to DNA sequences, which are known as IFN-stimulated response elements and directly activating the transcription of ISGs. Within a period of hours, however, the signal decays and the STATs are exported back to the cytoplasm for the next round of signaling (38, 39). Interestingly, the affinity of the IFNAR receptor varies between the different type I IFN ligands, due to the activation of different regulatory elements (40). However, the other cytokines activate STAT homodimers that recognize different gamma-activated sequence. Therefore, canonical type I IFN signaling induces a distinct subset of several hundred ISRE-driven ISGs. Cellular responses to IFNAR ligation vary during the course of an immune response and are cell type-and context-dependent (41).
Several different mechanisms were described that suppress type I IFN-mediated responses: downregulation of IFNAR expression on cell surface, induction of negative regulators like ubiquitin carboxy-terminal hydrolase 18 (USP18), and suppressor of cytokine signaling (SOCS). SOCS proteins compete with STATs for binding to IFNAR, while USP18 displaces JAK1 from IFNAR2 (42, 43). In addition, type I IFN responses are regulated by miRNAs (44, 45). During PRR and inflammatory signaling, miR-155 is highly induced (46, 47). It was reported that miR-155 suppressed the expression of IFNAR–JAK–STAT pathway in CD8+ T cells and the consequence of this suppression was enhanced CD8+ T cell responses to viral and bacterial pathogens (47).
Type-I and type-II IFNs are known to promote the expression of over 2,000 ISGs, and the products of ISGs have been shown to act by enhancing pathogen detection and innate immune signaling or restricting intracellular replication of viruses, bacteria, and parasites (21). Protein modification by the ubiquitin-like modifier interferon (IFN)-stimulated gene 15 (ISG15) is strongly induced by type I IFNs and represents one of the major antiviral IFN effector systems (48, 49). Conjugation of ISG15 to its substrates is counteracted by the activity of ubiquitin-specific protease 18 (USP18/UBP43) (50).
Another important group of proteins is superfamily of IFN-induced GTPases. Based on biochemical and structural studies, IFN-induced GTPases are grouped into four families of IFN-inducible, dynamin-like GTPases: the myxovirus resistance proteins (Mx), the immunity-related GTPases, the guanylate-binding proteins (GBPs), and the very large IFN-inducible GTPases (51). IFN-induced GTPases are transcribed in response to type-I, type-II, and type-III IFNs, while the Mx proteins are expressed only in response to type-I and type-III IFNs. TNF-α signaling was proposed to act as an alternative induction route for the GTPase; therefore, IFNs are not the only factors acting as GTPase inducers (52, 53). However, type-II IFNs and type-I IFNs are the strongest inducers, while TNFα and LPS are relatively weak stimuli.
Inflammasomes
Activation of the inflammasome is a key event in inflammatory immune response. The inflammasomes are cytosolic multiprotein complexes that are composed of an inflammasome-initiating sensor, apoptosis-associated speck-like protein containing a CARD (ASC) acts as an adaptor protein and the protease-caspase-1. Inflammasome-initiating sensors include members of the NLRs the pyrin and HIN domain-containing (also known as PYHIN, Aim 2-like receptors, or ALRs; e.g., Aim2), or the TRIM (e.g., pyrin) family (54). Complex assembly leads to caspase-1-dependent cleavage of cytokines pro-interleukin 1β (pro-IL-1β) and pro-IL-18 into secreted mature forms (55–57). In addition, inflammasomes initiate pyroptotic cell death (52, 57, 58). Pyroptosis involves cell swelling, membrane rupture, and release of the cytoplasmic content into the extracellular space (58–60). Pyroptotic cell death is induced by caspase-1 or mouse caspase-11 (human caspase-4/5) cleavage gasdermin D (GSDMD), a pore-forming protein that normally exists in the auto inhibited state (58, 61, 62). Interestingly, since mature IL-1β lacks target sequences, secretion may require pyroptosis of the macrophages (60). However, other mechanisms of IL-1β secretion might also exist, human monocytes were reported to release IL-1β without pyroptosis (63).
Recently, several excellent reviews described mechanism of inflammasome activation (52, 56, 64, 65). Several NLR family members have been described as components of inflammasomes: Nlrp1b inflammasome (66, 67), Naip-Nlrc4 inflammasome (68, 69), the Nlrp6 inflammasome (70), the Nlrp12 inflammasome, the Aim2 inflammasome (28, 71), the RIG-I inflammasome (72), and the IFI16 inflammasome (73). Particularly, the activation of Nlrp3 inflamamsome is well characterized (55, 64, 74, 75). Since it responds to variety of stimuli, many different mechanisms of its activation have been proposed, including the release of oxidized mitochondrial DNA, production of reactive oxygen species and mitochondrial dysfunction, lysosomal destabilization, changes in intracellular calcium levels, the formation of large non-specific membrane. The Nlrp3 inflammasome activation in macrophages requires 2 steps: the first, priming step is provided by TLR signaling that upregulates NLPR3 and pro-IL-1β gene expression. This process is tightly controlled by signals culminating in the activation of NF-κB (76). Moreover, Nlrp3 activation can be regulated through direct posttranslational modifications, such as ubiquitination (77). Recently, several independent studies reported non-canonical inflammasome activation (78–80). While canonical inflammasome activation results in caspase-1 cleavage and activation, the activation of a non-canonical inflammasome results in activation of procaspase-11 (56). The mouse caspase-11 has high similarities to caspase-1 and is orthologous to human caspases-4 and -5 (81, 82). Both caspase-1 and caspase-4/11 could induce pyroptosis, while only caspase-1 processes proforms of IL-1β and IL-18 into secreted mature forms (78, 83). Only caspase-11-deficient mice, but not caspase-1-deficient mice were partially protected from septic death (78, 84). Recent reports showed that caspase-11 was involved in the response to cytosolic LPS, independently of TLR4 and was integral to the pathology of LPS-mediated endotoxic shock in mice (61). Moreover, it was shown that human caspase-4 and caspase-5 and mouse caspase-11 bound directly to LPS in the cytosol (85). With the difference to canonical inflammasome activation were the receptor (Nlrp3) and ASC form a scaffold on which caspase-1 can oligomerize, in non-canonical infalmmasome activation, caspase-11 oligomerization occurs directly upon binding to LPS (85). Human caspase-4/5/or mouse caspase-11 cleave GSDMD, a pore-forming protein that normally exists in the auto inhibited state (58, 61, 62). Furthermore, GSDMD N-terminal domain was found to associate with membranes, including the plasma membrane (86–89). It was reported that canonical Nlrp3 inflammasome activation downstream of caspase-4 and caspase-11 activation was dependent on potassium efflux (90–92). Yang et al. reported that cytosolic LPS stimulation induced caspase-11-dependent cleavage of the pannexin-1 channel followed up by potassium efflux and ATP release (92).
AIM2-like receptor inflammasomes are another class of inflammasomes that function to induce caspase-1 activation and IL-1β cytokine maturation. However, unlike NLR inflammasomes, ALR inflammasomes directly bind their ligand, dsDNA (28–30). While IFI16 recognizes dsDNA in the cytosol and nucleus, while Alm2 is localized only in the cytosol (93). In addition, IFI16 could induce type I IFN expression (30).
Inhibition of inflammasome activation by decoy proteins uses proteins structurally related to components of inflammasome and competing for the same adaptors. The CARD-only proteins and PYD-only proteins (POPs) function as endogenous dominant negative proteins that modulate the activity of inflammasomes and protect from excessive inflammation (94, 95). The genes encoding these decoy proteins, POPs, are located on the same chromosome, in the proximity of genes that encode their ligands: the gene encoding POP1 is located on human chromosome 16 next to the gene encoding ASC (96). POP3 has significant sequence similarity to the PYRIN domain of AIM2 (its target protein), encoded by a neighboring gene (97). Recently, it was demonstrated that POP2 not only prevented inflammasome assembly by binding to ASC but also impaired macrophage priming by inhibiting the activation of non-canonical IKK ε and IκBα (98).
Cross Talk of IFNs and Inflammasomes
Interferons could contribute to inflammasome activation through several different mechanisms (Figure 1). It was reported that type I IFNs are required for the caspase-11 expression, which contributes to activation of non-canonical inflammasome (79). Several recent studies have shown that IFN-inducible endogenous proteins could act also as negative regulators and thus inhibit inflammasome activation (97, 99). Among others, interferon-inducible GBPs not only mediate host resistance to pathogens but also promote inflammasome activation in bacterial infections (100, 101). Also, small proteins that are composed of either a CARD or a PYD only, emerged as important inflammasome regulators (94, 95). It was demonstrated that POP3, which is induced by type I IFNs, interacted with the PYD domain of AIM2 and competed with ASC to inhibit AIM2 inflammasome activation in response to dsDNA, mouse CMV, and modified vaccinia virus Ankara infection (97). Silencing of POP3 in human macrophages enhanced DNA and DNA virus-induced ALR inflammasome formation and hence the maturation and release of IL-1β and IL-18 (97). Not only POPs but also metabolites like 25-hydroxycholesterol, an oxysterol and is derived from cholesterol, suppress inflammasome activation (99). At least in macrophages, IFN-β strongly induced cholesterol 25-hydroxylase, the enzyme that transforms cholesterol into 25-hydroxycholesterol (102, 103). Work of Reboldi et al. showed that 25-hydroxycholesterol inhibited not only pro-IL-1β gene transcription but also the inflammasome activation (99). The authors proposed that 25-hydroxycholesterol antagonized the sterol response element-binding protein processing (99). Moreover, cholesterol 25-hydroxylase-deficient mice showed increased sensitivity to LPS-induced septic shock (99).
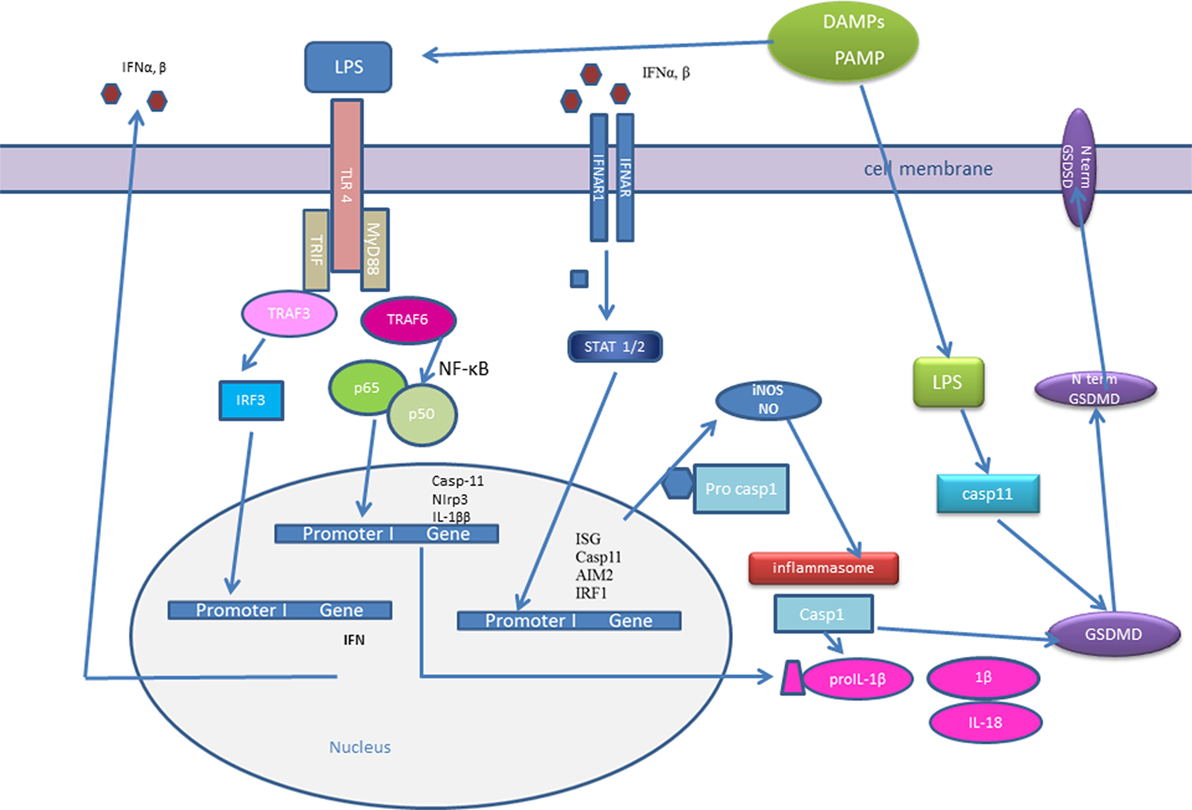
Figure 1. Type I interferons (IFNs) and inflammasome activation. Initial pathogen-associated molecular patterns (PAMPs) recognition by pattern recognition receptors induces IFN-β expression. IFNs could signal in an autocrine or paracrine manner and trigger expression of IFN-stimulated genes (ISGs): interferon regulatory factor (IRF)1, AIM2, caspase-11. Caspase-11 recognizes cytosolic LPS and induces IL-1β processing in an Nlrp3-dependent manner and triggers pyroptosis through gasdermin D (GSDMD) cleavage. Active caspase-1 and caspase-11 cleave GSDMD and the released gasdermin-N domain binds to phosphoinositides in the plasma membrane, oligomerizes to generate membrane pores, and initiates cell death-pyroptosis. IRF induce the expression of guanylate-binding proteins (GBPs), which target vacuolar and cytosolic bacteria, compromise the integrity of bacterial cells, and expose PAMPs like LPS and dsDNA to cytosolic sensors, caspase-11, and AIM2. IFN signaling triggers the expression of inducible nitric oxide synthase (iNOS), which upregulates cellular nitric oxide (NO) levels leading to NLRP3 S-nitrosylation.
Both type-I IFNs and IFN-γ could promote inducible nitric oxide synthase (iNOS), which increases the amount of endogenous NO, expression in macrophages (15, 104). NO plays an important role in a defense against pathogens, it could be oxidized to reactive nitrogen oxide species, that S-nitrosate thiols in proteins (15, 104). Mishra et al. reported that NO inhibited NLRP3 oligomerization by means of direct S-nitrosylation of the NLRP3 protein, preventing full inflammasome assembly (15). Also study by Mao et al. demonstrated that NO prevented the activation of the NLRP3 inflammasome (14). In line with the above results, in iNOS-deficient macrophages, NLRP3 inflammasome activation was enhanced, iNOS-deficient mice had increased mortality from LPS-induced sepsis (14).
In addition, type I IFN signal via STAT1 decreased the activity of Nlrp3 inflammasome that induce caspase-1 to process the IL1-β precursor in response to a large variety of intracellular PAMPs (105). Different mechanisms could contribute to diminished IL1-β processing in IFN-stimulated cells. STAT1 target gene products directly repress NLRP3 inflammasome. Moreover, the IFN-I/STAT1 pathway increases IL-10 synthesis, IL-10-mediated STAT3 activation, and the suppression of IL1-β precursor synthesis by activated STAT3 (106). Guarda et al. showed that IL-1α and IL-1β were downregulated in mice pretreated with poly(I:C), a synthetic RNA analog that strongly induces type-I IFNs (106). In addition, they demonstrated that the recruitment of inflammatory cells (neutrophils and monocytes) into peritoneal cavity was significantly lower in poly(I:C) pretreated mice, than in control animals injected only with LPS. Moreover, they demonstrated that IFN-β suppress not only inflammasome activation and IL-1β secretion but also it rendered the mice more susceptible to Candida albicans infection (106).
Several recent studies reported cross talk between IFNs and inflammasome activation in bacterial infections (79, 100, 101, 107, 108). An early study showed that caspase-11 gene expression in response to LPS and IFN-γ was dependent on NF-κB and STAT-1 signaling (109). Rathinam et al. demonstrated that transcriptional induction of caspase-11 by IFN-β signaling was enough to induce both its expression and auto activation (79). Gurung et al. reported that TLR4–TRIF–IFNβ-induced caspase-11 synthesis is crucial for non-canonical Nlrp3 inflammasome activation in macrophages infected with enteric pathogens Escherichia coli and Citrobacter rodentium (110). IFN-γ could also upregulate caspase-11 expression. Aachoui et al. showed that caspase-1 activity is required upstream of caspase-11 to control infection by cytosolic bacterium Burkholderia thailandensis. Caspase-1-activated IL-18, which further induced IFN-γ to prime caspase-11 and rapidly clear B. thailandensis infection. Whereas IFN-γ was essential, endogenous type I IFNs were insufficient to prime caspase-11 and cleared B. thailandensis (111). Oficjalska et al. reported that IFN-γ-dependent, type I IFN–TRIF-independent signaling pathway was required for in vivo caspase-11 production in intestinal epithelial cells during DSS-induced colitis (112). However, LPS-stimulated macrophages from TRIF-deficient mice had impaired caspase-11 expression, implying a context-dependent role for type I or II IFN in the regulation of caspase-11 activity (79, 112). In addition, IFN-γ induced upregulation of Nlrp3, ASC, and procaspase-1 expression (100, 113, 114). IFN-γ enhanced Aim2-induced IL-1β release or Nlrp3-dependent pro-IL-18 cleavage during HSV-1 and Chlamydia muridarum infections (115, 116).
Upon bacterial infection, IFN-inducible GTPases—GBPs target vacuolar and cytosolic bacteria and compromise the integrity of bacterial cells, thus exposing the microbial ligands LPS and DNA to cytosolic sensors caspase-11 and Aim2 (100, 101). GBPs have also been shown to regulate the entry of LPS into the cytosol by, as yet, poorly defined mechanisms (100). Significant reduction in NLRP3 inflammasome activation was reported in GBP5-deficient macrophages infected with S. typhimurium or treated with potassium efflux agonists (117). However, studies on different mouse strain of GBP5-deficient mice could not confirm the initial results (108, 114). Despite the uncertainty surrounding the role of GBP5 in Nlrp3 inflammasome activation, studies using mice lacking the entire cluster of GBP genes on chromosome 3, have firmly confirmed a functional link between GBPs and the activation of the canonical NLRP3 and AIM2 inflammasomes, as well as the non-canonical caspase-11 inflammasomes. Recently, GBP2 emerged as a critical activator of AIM2 and caspase-11 inflammasomes (100, 101). GBP2 is induced by type I or II IFNs and exposes Gram-negative bacteria-derived LPS to caspase-11 (114). In addition, it was shown that IFN-β boosts canonical AIM2-dependent IL-1β secretion to Francisella tularenis or Listeria monocytogenes (71, 118) and helps to control caspase-11-dependent pyroptosis by Gram-negative bacteria (79, 107).
Type I-IFN signaling is also essential in response to Francisella novicida infection (101, 119). F. novicida DNA is detected by DNA sensor cGAS, which induced STING-dependent production of type I-IFNs (71). Type I-IFNs in act via the transcription factor IRF1, which regulates expression of GBPs and IRG (108, 120) (Figure 2). Interferon response gene B10 together with GBP2, GBP5 work synergistically to rupture F. novicida that have entered the cytoplasm, and their action result in the exposure of F. novicida DNA for sensing by DNA sensor AIM2 (52, 101, 114).
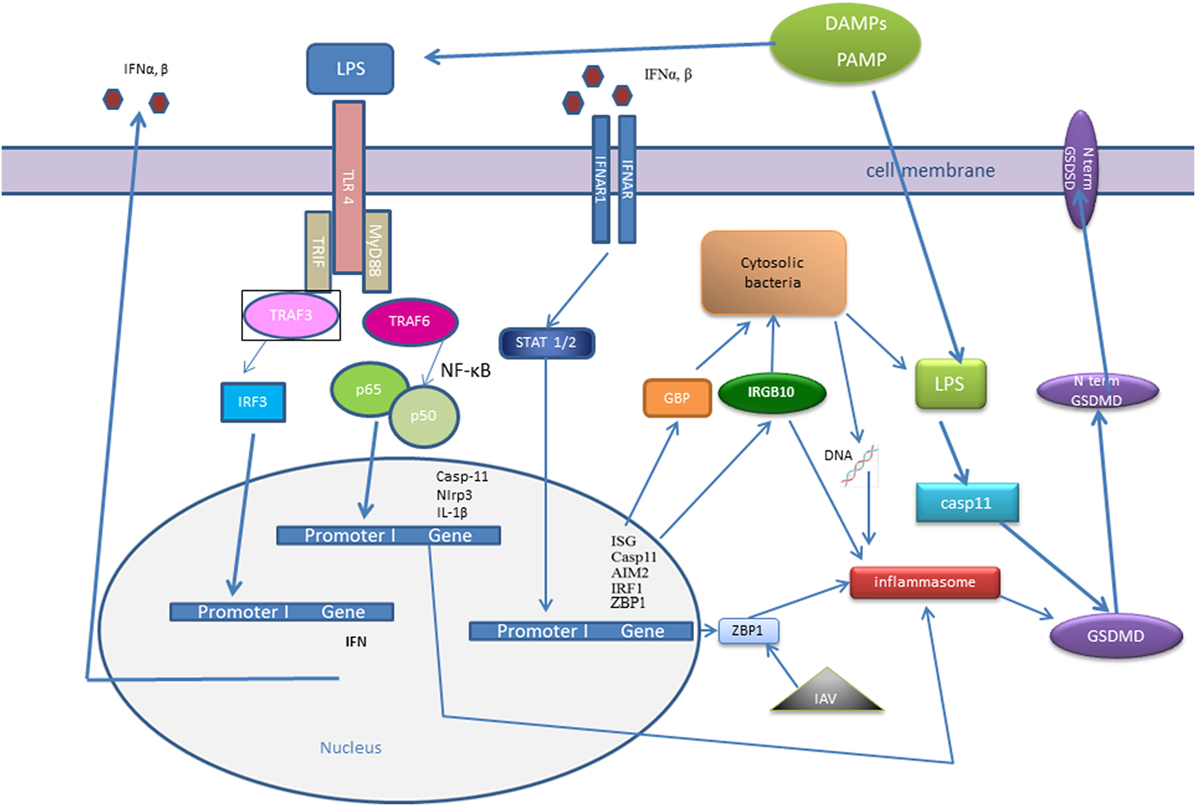
Figure 2. Interferon (IFN) signaling influence recognition of intracellular pathogens—cytosolic bacteria and influenza A virus (IAV). IFNs signaling trigger the transcription factor interferon regulatory factor (IRF)1, which promotes expression of guanylate-binding proteins (GBPs) and interferon response gene B10 (IRGB10). IRGB10, together with GBPs permeabilizes the membrane of Gram-negative bacteria, an action that results in release of bacterial DNA and LPS. Bacterial cytosolic DNA is sensed by Aim2 inflammasome and LPS directly interacts with caspase-11. Type I IFN signaling mediates upregulation of interferon-inducible protein Z-DNA-binding protein 1 (ZBP1), which recognizes the IAV proteins and triggers NLRP3 inflammasome activation, as well as induction of apoptosis, necroptosis, and pyroptosis in IAV-infected cells.
Another IFN-inducible protein, Z-DNA-binding protein 1 (ZBP1), also known as DNA-dependent activator of IFN-regulatory factors (DAI), has been known as a cytosolic DNA sensor for almost a decade (121). However, a recent work demonstrated that ZBP1 could sense the RNA virus, influenza A virus (IAV) proteins: nucleoprotein and polymerase subunit 1. Kuriakose showed that in IAV-infected cells, ZBP1 regulated NLRP3 inflammasome activation, as well as induction of apoptosis, necroptosis, and pyroptosis (122) (Figure 2). ZBP1-deficient mice were protected from mortality during IAV infection, due to reduced inflammatory response (122).
Concluding Remarks
I have summarized considerable, but by no means all evidence documenting the role of IFNs in inflammasome activation and inflammation. Several recent studies reported the essential role of type I IFNs in non-canonical Nlrp3 inflammasome activation and pyroptosis. Different levels of regulation are involved in the cross talk of IFNs in inflammasome. Not only type I-IFNs but also IFN-γ influence caspase-11 expression and consequently pyroptosis. Dysregulated type I-IFN production could lead to a cell death. However, a recent study reported that in the absence of active proapoptotic caspases-3 and -7, mitochondrial outer membrane permeabilization by Bax and Bak resulted in the expression of type I-IFNs. The process was mediated by mitochondrial DNA-dependent activation of the cGAS/STING (123). Particularly, the role of STAT and other protein modification in IFN signaling pathways could give us important insight into the regulatory mechanisms. IFN-induced GBPs were reported to have an important role in caspase-11 activation and pyroptotic cell death. How does the polymorphisms of GBPs influence inflammasome activation and inflammation is yet to be determined. Future research should explore the detailed molecular mechanisms that are responsible for type I IFN-dependent cell death and inflammasome activation in inflammatory response. Moreover, recently, several studies determined the role of cytokines in metabolic reprograming and inflammasome activation (124). The role cross talk of IFNs, inflammasomes, and metabolism could be a future frontier for the cutting edge research. Identification of the factors involved in inflammasome regulation and signaling will lead to the identification of novel targets for therapeutic intervention.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The author acknowledges the financial support from the Slovenian Research Agency (research core funding No. P-0140).
References
1. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell (2010) 140:771–6. doi:10.1016/j.cell.2010.03.006
2. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10:826–37. doi:10.1038/nri2873
3. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi:10.1038/nature07201
4. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi:10.1016/j.cell.2006.02.015
5. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature (2007) 449:819–26. doi:10.1038/nature06246
6. Moresco EM, Lavine D, Beutler B. Toll-like receptors. Curr Biol (2011) 21:039. doi:10.1016/j.cub.2011.05.039
7. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13:325–32. doi:10.1038/ni.2231
8. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
9. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity (2013) 38:870–80. doi:10.1016/j.immuni.2013.05.004
10. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci (1957) 147:258–67. doi:10.1098/rspb.1957.0048
11. Ivashkiv LB, Donlin LT. Regulation of type i interferon responses. Nat Rev Immunol (2014) 14:36–49. doi:10.1038/nri3581
12. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi:10.1038/nri3787
13. de Weerd NA, Nguyen T. The interferons and their receptors – distribution and regulation. Immunol Cell Biol (2012) 90:483–91. doi:10.1038/icb.2012.9
14. Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res (2013) 23:201–12. doi:10.1038/cr.2013.6
15. Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1[beta]. Nat Immunol (2013) 14:52–60. doi:10.1038/ni.2474
16. Goubau D, Deddouche S, Reis ESC. Cytosolic sensing of viruses. Immunity (2013) 38:855–69. doi:10.1016/j.immuni.2013.05.007
17. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science (1999) 284:1835–7. doi:10.1126/science.284.5421.1835
18. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207:2053–63. doi:10.1084/jem.20101664
19. Iwasaki A. A virological view of innate immune recognition. Annu Rev Microbiol (2012) 66:177–96. doi:10.1146/annurev-micro-092611-150203
20. MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol (2012) 12:367–82. doi:10.1038/nri3210
21. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi:10.1146/annurev-immunol-032713-120231
22. Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect (2004) 6:1382–7. doi:10.1016/j.micinf.2004.08.018
23. Apostolou E, Thanos D. Virus infection induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell (2008) 134:85–96. doi:10.1016/j.cell.2008.05.052
24. Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol (2009) 21:317–37. doi:10.1093/intimm/dxp017
25. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11:373–84. doi:10.1038/ni.1863
26. Fang TC, Schaefer U, Mecklenbrauker I, Stienen A, Dewell S, Chen MS, et al. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J Exp Med (2012) 209:661–9. doi:10.1084/jem.20112343
27. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi:10.1126/science.1232458
28. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458:509–13. doi:10.1038/nature07710
29. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi:10.1038/nature07725
30. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11:997–1004. doi:10.1038/ni.1932
31. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455:674–8. doi:10.1038/nature07317
32. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type i interferon-dependent innate immunity. Nature (2009) 461:788–92. doi:10.1038/nature08476
33. Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol (2015) 32:48–53. doi:10.1016/j.coi.2014.12.012
34. Gürtler C, Bowie AG. Innate immune detection of microbial nucleic acids. Trends Microbiol (2013) 21:413–20. doi:10.1016/j.tim.2013.04.004
35. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol (2014) 32:461–88. doi:10.1146/annurev-immunol-032713-120156
36. Dempsey A, Bowie AG. Innate immune recognition of DNA: a recent history. Virology (2015) 480:146–52. doi:10.1016/j.virol.2015.03.013
37. Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity (2012) 36:503–14. doi:10.1016/j.immuni.2012.03.013
38. Levy DE, Darnell JE Jr. STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol (2002) 3:651–62. doi:10.1038/nrm909
39. Rauch I, Müller M, Decker T. The regulation of inflammation by interferons and their STATs. JAKSTAT (2013) 2:e23820. doi:10.4161/jkst.23820
40. Schreiber G, Piehler J. The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol (2015) 36:139–49. doi:10.1016/j.it.2015.01.002
41. van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity (2006) 25:361–72. doi:10.1016/j.immuni.2006.08.014
42. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol (2007) 7:454–65. doi:10.1038/nri2093
43. Sarasin-Filipowicz M. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol Cell Biol (2009) 29:4841–51. doi:10.1128/MCB.00224-09
44. David M. Interferons and microRNAs. J Interferon Cytokine Res (2010) 30:825–8. doi:10.1089/jir.2010.0080
45. Nazarov PV. Interplay of microRNAs, transcription factors and target genes: linking dynamic expression changes to function. Nucleic Acids Res (2013) 41:2817–31. doi:10.1093/nar/gks1471
46. Tili E. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-[alpha] stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol (2007) 179:5082–9. doi:10.4049/jimmunol.179.8.5082
47. Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat Immunol (2013) 14:593–602. doi:10.1038/ni.2576
48. Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol (2008) 8:559–68. doi:10.1038/nri2314
49. Hermann M, Bogunovic D. ISG15: in sickness and in health. Trends Immunol (2017) 38:79–93. doi:10.1016/j.it.2016.11.001
50. Basters A, Geurink PP, Rocker A, Witting KF, Tadayon R, Hess S, et al. Structural basis of the specificity of USP18 toward ISG15. Nat Struct Mol Biol (2017) 6:270–8. doi:10.1038/nsmb.3371
51. Praefcke GJK, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol (2004) 5:133–47. doi:10.1038/nrm1313
52. Meunier E, Broz P. Interferon-inducible GTPases in cell autonomous and innate immunity. Cell Microbiol (2016) 18:168–80. doi:10.1111/cmi.12546
53. Pilla-Moffett D, Barber MF, Taylor GA, Coers J. Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol (2016) 428:3495–513. doi:10.1016/j.jmb.2016.04.032
54. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi:10.1038/nri.2016.58
55. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi:10.1038/nri3452
56. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157:1013–22. doi:10.1016/j.cell.2014.04.007
57. Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol (2015) 25:308–15. doi:10.1016/j.tcb.2014.12.009
58. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42(4):245–54. doi:10.1016/j.tibs.2016.10.004
59. Ashkenazi A, Salvesen G. Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol (2014) 30:337–56. doi:10.1146/annurev-cellbio-100913-013226
60. Broz P. Immunology: caspase target drives pyroptosis. Nature (2015) 526:642–3. doi:10.1038/nature15632
61. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi:10.1038/nature15541
62. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi:10.1038/nature15514
63. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity (2016) 44:833–46. doi:10.1016/j.immuni.2016.01.012
64. Prochnicki T, Mangan MS, Latz E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Res (2016) 5(F1000 Faculty Rev):1469. doi:10.12688/f1000research.8614.1
65. Guo H, Callaway JB, Ting JPY. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi:10.1038/nm.3893
66. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet (2006) 38:240–4. doi:10.1038/ng1724
67. Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity (2012) 37:1009–23. doi:10.1016/j.immuni.2012.08.027
68. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430:213–8. doi:10.1038/nature02664
69. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi:10.1038/nature10510
70. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi:10.1016/j.cell.2011.04.022
71. Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A (2010) 107:9771–6. doi:10.1073/pnas.1003738107
72. Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol (2010) 11:63–9. doi:10.1038/ni.1824
73. Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma associated herpesvirus infection. Cell Host Microbe (2011) 9:363–75. doi:10.1016/j.chom.2011.04.008
74. Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ (2007) 14:10–22. doi:10.1038/sj.cdd.4402038
75. Jabaut J, Ather JL, Taracanova A, Poynter ME, Ckless K. Mitochondria-targeted drugs enhance Nlrp3 inflammasome-dependent IL-1beta secretion in association with alterations in cellular redox and energy status. Free Radic Biol Med (2013) 29:233–45. doi:10.1016/j.freeradbiomed.2013.01.025
76. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, Macdonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183:787–91. doi:10.4049/jimmunol.0901363
77. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell (2013) 49:331–8. doi:10.1016/j.molcel.2012.11.009
78. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi:10.1038/nature10558
79. Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell (2012) 150:606–19. doi:10.1016/j.cell.2012.07.007
80. Broz P, Monack DM. Noncanonical inflammasomes: caspase-11 activation and effector mechanisms. PLoS Pathog (2013) 9:e1003144. doi:10.1371/journal.ppat.1003144
81. Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, et al. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem (1996) 271:20580–7. doi:10.1074/jbc.271.34.20580
82. Kajiwara Y. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol (2014) 193:335–43. doi:10.4049/jimmunol.1303424
83. Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol (2015) 32:78–83. doi:10.1016/j.coi.2015.01.007
84. Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell (1998) 92:501–9. doi:10.1016/S0092-8674(00)80943-5
85. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi:10.1038/nature13683
86. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A (2016) 113:7858–63. doi:10.1073/pnas.1607769113
87. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi:10.1038/nature18590
88. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi:10.1038/nature18629
89. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J (2016) 35:1766–78. doi:10.15252/embj.201694696
90. Rühl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+ efflux. Eur J Immunol (2015) 45:2927–36. doi:10.1002/eji.201545772
91. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung VD. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol (2015) 45:2911–7. doi:10.1002/eji.201545523
92. Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity (2015) 43:923–32. doi:10.1016/j.immuni.2015.10.009
93. Li T, Diner BA, Chen J, Cristea IM. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A (2012) 109:10558–63. doi:10.1073/pnas.1203447109
94. Stehlik C, Dorfleutner A. COPs and POPs: modulators of inflammasome activity. J Immunol (2007) 179:7993–8. doi:10.4049/jimmunol.179.12.7993
95. Le HT, Harton JA. Pyrin- and CARD-only proteins as regulators of NLR functions. Front Immunol (2013) 4:00275. doi:10.3389/fimmu.2013.00275
96. Stehlik C, Krajewska M, Welsh K, Krajewski S, Godzik A, Reed JC. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulation. Biochem J (2003) 373:101–13. doi:10.1042/bj20030304
97. Khare S, Ratsimandresy RA, De Almeida L, Cuda CM, Rellick SL, Misharin AV, et al. The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat Immunol (2014) 15:343–53. doi:10.1038/ni.2829
98. Ratsimandresy RA, Chu LH, Khare S, De Almeida L, Gangopadhyay A, Indramohan M, et al. The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat Commun (2017) 8:15556. doi:10.1038/ncomms15556
99. Reboldi A, Dang EV, Mcdonald JG, Liang G, Russell DW, Cyster JG. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science (2014) 345:679–84. doi:10.1126/science.1254790
100. Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A (2014) 111:6046–51. doi:10.1073/pnas.1321700111
101. Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Rühl S, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol (2015) 16:476–84. doi:10.1038/ni.3119
102. Diczfalusy U, Olofsson KE, Carlsson AM, Gong M, Golenbock DT, Rooyackers O, et al. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J Lipid Res (2009) 50:2258–64. doi:10.1194/jlr.M900107-JLR200
103. Park K, Scott AL. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol (2010) 88:1081–7. doi:10.1189/jlb.0610318
104. Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, et al. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol (2012) 189:5113–7. doi:10.4049/jimmunol.1202479
105. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol (2010) 10:210–5. doi:10.1038/nri2725
106. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity (2011) 34:213–23. doi:10.1016/j.immuni.2011.02.006
107. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature (2012) 490:288–91. doi:10.1038/nature11419
108. Man SM, Karki R, Malireddi RKS, Neale G, Vogel P, Yamamoto M, et al. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol (2015) 16:467–75. doi:10.1038/ni.3118
109. Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem (2002) 277:41624–30. doi:10.1074/jbc.M207852200
110. Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z, et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem (2012) 287:34474–83. doi:10.1074/jbc.M112.401406
111. Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JP, Coers J, et al. Canonical inflammasomes drive IFN-gamma to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe (2015) 18:320–32. doi:10.1016/j.chom.2015.07.016
112. Oficjalska K, Raverdeau M, Aviello G, Wade SC, Hickey A, Sheehan KM, et al. Protective role for caspase-11 during acute experimental murine colitis. J Immunol (2015) 194:1252–60. doi:10.4049/jimmunol.1400501
113. Shenoy AR. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science (2012) 336:481–5. doi:10.1126/science.1217141
114. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature (2014) 509:366–70. doi:10.1038/nature13157
115. Finethy R, Jorgensen I, Haldar AK, De Zoete MR, Strowig T, Flavell RA, et al. Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in chlamydia-infected macrophages. Infect Immun (2015) 83:4740–9. doi:10.1128/IAI.00856-15
116. Strittmatter GE, Sand J, Sauter M, Seyffert M, Steigerwald R, Fraefel C, et al. IFN-gamma primes keratinocytes for HSV-1-induced inflammasome activation. J Invest Dermatol (2016) 136:610–20. doi:10.1016/j.jid.2015.12.022
117. Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science (2012) 336:481–5. doi:10.1126/science.1217141
118. Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med (2007) 204:987–94. doi:10.1084/jem.20062665
119. Fernandes-Alnemri T, Yu J-W, Juliana C, Solorzano L, Kang S, Wu J, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol (2010) 11:385–93. doi:10.1038/ni.1859
120. Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, et al. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell (2016) 167:382–96.e17. doi:10.1016/j.cell.2016.09.012
121. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature (2007) 448:501–5. doi:10.1038/nature06013
122. Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol (2016) 1:12. doi:10.1126/sciimmunol.aag2045
123. Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell (2014) 159:1563–77. doi:10.1016/j.cell.2014.11.037
Keywords: caspase-1, caspase-11, cyclic GMP-AMP synthase, guanylate-binding protein, interferon, inflammasome, macrophages, pyropotosis
Citation: Kopitar-Jerala N (2017) The Role of Interferons in Inflammation and Inflammasome Activation. Front. Immunol. 8:873. doi: 10.3389/fimmu.2017.00873
Received: 26 February 2017; Accepted: 10 July 2017;
Published: 25 July 2017
Edited by:
Claudia U. Duerr, McGill University, CanadaReviewed by:
Ian Gaël Rodrigue-Gervais, Institut national de la recherche scientifique, CanadaEtienne Meunier, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), France
Copyright: © 2017 Kopitar-Jerala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nataša Kopitar-Jerala, bmF0YXNhLmtvcGl0YXJAaWpzLnNp