 Dong Sun Oh
Dong Sun Oh Ji Eun Oh
Ji Eun Oh Hi Eun Jung
Hi Eun Jung Heung Kyu Lee
Heung Kyu Lee- 1Biomedical Science and Engineering Interdisciplinary Program, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, South Korea
- 2Laboratory of Host Defenses, Graduate School of Medical Science and Engineering, KAIST, Daejeon, South Korea
- 3KAIST Institute for Health Science and Technology, KAIST, Daejeon, South Korea
Respiratory syncytial virus (RSV) is a major cause of respiratory viral infections in infants and children. Alveolar macrophages (AMs) play a crucial role in combatting airborne pathogens, strongly express CD169, and are localized in the lung alveoli. Therefore, we used CD169-diphtheria toxin receptor (DTR) transgenic mice to explore the roles of CD169+ cells in immune responses to mucosal RSV infection. The administration of diphtheria toxin to CD169-DTR mice induced specific AM depletion and reduced the recruitment of Ly6Chi monocytes. Notably, CD169+ cell depletion reduced levels of innate cytokines, such as interferon-β, IL-6, and TNF-α, in bronchoalveolar lavage fluid during RSV infection without affecting the production of proinflammatory chemokines. Moreover, the depletion of CD169+ cells increased the recruitment of inflammatory cells to the lung during the early stage of RSV infection, although not during the later stages of RSV infection. Furthermore, the depletion of CD169+ cells reduced the recruitment of effector CD8+ T cells to the lungs after RSV mucosal infection. Our findings suggest that modulating the number of CD169+ cells to enhance immune responses to RSV infection may be useful as a new therapeutic strategy.
Introduction
Respiratory syncytial virus (RSV) is a major respiratory pathogen and the leading cause of bronchiolitis and infant hospital admissions throughout the developed world (1–3). In addition, RSV is a primary cause of opportunistic respiratory infections in elderly and immunocompromised patients (4). Despite the many attempts to develop an RSV vaccine, none are currently licensed, likely due to the lack of sufficient understanding of the host antiviral response to RSV (5, 6). When infecting the lung mucosa, RSV directly or indirectly targets various cell types, including primary epithelial cells, alveolar macrophages (AMs), and dendritic cells (DCs) (7, 8). Thus, the identification of candidate cells to target for vaccine delivery may result in an effective strategy to generate anti-RSV immunity.
Alveolar macrophages localize in the alveoli where they play crucial roles in the maintenance of lung homeostasis and the clearance of airway dust, constituting a front line of defense against airborne pathogens such as RSV (9, 10). Several studies using clodronate liposomes to induce irreversible damage and apoptosis in AMs have shown that AMs play a role in RSV infection (11–13). However, although clodronate liposomes can be used to deplete AMs, they also deplete other lung phagocytic cells, such as DCs, neutrophils, and monocytes (14–16). Therefore, more specific models of AM depletion are needed to study the roles of AMs in RSV mucosal infection.
Type I interferons (IFNs) are important cytokines in the innate immune response to viral infection (17) that modulate the expression of numerous genes involved in host defense and initiate the adaptive immune response to RSV infection (18–21). Recently, type I IFNs and their downstream signaling pathways were shown to be critical for the induction of lung inflammation in response to RSV infection in mice (22). Furthermore, the mitochondrial antiviral-signaling protein (MAVS)-dependent RIG-I-like receptors pathway in AMs was found to be required for type I IFN production in the lungs of RSV-infected mice (23). CD169, also known as Siglec-1 or sialoadhesin, is a sialic acid- and glycoprotein-binding adhesion molecule expressed by AMs (24). The administration of diphtheria toxin (DT) to CD169-diphtheria toxin receptor (DTR) transgenic mice causes the effective and specific ablation of AMs (25), making this a useful model in which to study the roles of these cells.
Respiratory syncytial virus-specific effector CD8+ T cells have been reported to play an essential role in recovery from RSV infection (26); however, RSV possesses a mechanism that suppresses lung CD8+ T cell effector and memory activity (27). CXCL9/10, which are IFN-inducible chemokines, are required for effector CD8+ T cell recruitment into sites of infection and for the amplification of immune responses (28, 29). Thus, to overcome RSV-mediated CD8+ T cell suppression, it is important to identify the cell type responsible for CXCL9/10 secretion. Previously, CXCL9/10 was found to be secreted mainly by lung epithelial cells (30, 31); however, little is known regarding the role of infiltrating immune cells in CXCL9/10 production and effector CD8+ T cell recruitment to the lungs during mucosal RSV infection.
Here, we elucidate the roles of CD169+ cells in antiviral immune responses against mucosal RSV infection. DT administration to CD169-DTR mice was found to induce specific AM depletion and to reduce the recruitment of Ly6Chi monocytes. Notably, the depletion of CD169+ cells diminished the secretion of innate cytokines such as IFN-β, IL-6, and TNF-α in bronchoalveolar lavage (BAL) fluid during RSV infection without affecting proinflammatory chemokine production. The depletion of CD169+ cells increased the recruitment of inflammatory cells to the lungs during the early stage of RSV infection, but not during later stages of RSV infection. Finally, we found that, although depletion of CD169+ cells reduced CXCL9/10 production and recruitment of effector CD8+ T cells to the lungs after RSV mucosal infection, CD169+ cells were dispensable for CD8+ T cell priming against RSV infection.
Materials and Methods
Ethics Statement
All animal procedures met the relevant legal and ethical requirements and were in accordance with the guidelines and protocols (KA2013-55) for rodent research approved by the Institutional Animal Care and Use Committee of KAIST.
Mice
CD169-DTR (Siglec1<tm1 (HBEGF) Mtka>) mice were kindly provided by Dr. Masato Tanaka of the Tokyo University of Pharmacy and Life Sciences (Tokyo, Japan). We used heterozygous CD169-DTR mice and wild-type (WT) littermates as controls in our experiments. The mice were bred and housed in an SPF facility at the KAIST.
RSV Infection In Vivo
As previously reported (32), we depleted CD169+ cells in 8- to 15-week-old male WT and CD169-DTR mice by treating the mice intraperitoneally with 40 ng/g DT (Sigma Aldrich, MO, USA) 1 day before infection with RSV. We grew the A2 strain of RSV, kindly provided by Dr. Jun Chang (Ewha Womans University, Seoul, Korea), in HEp-2 cells. We titrated the virus for infectivity as previously described (33). We anesthetized the mice with a mixture of 80 mg/kg ketamine (Youhanyanghaeng, Seoul, Korea) and 16 mg/kg xylazine (BAYER Korea, Ansan, Korea) in PBS (Genedepot, TX, USA) and then inoculated them intranasally with 1.0 × 107 plaque-forming units of RSV.
Single-Cell Isolation
We analyzed immune cells in lungs of mice according to previously described methods (34). Briefly, lungs were removed from mice, minced into small pieces, and then digested with a mixture of 2 mg/ml collagenase IV (Worthington Biochemical Corp., NJ, USA) and 30 µg/ml DNase I (Roche, Basel, Switzerland) in Dulbecco’s modified Eagle’s medium (Welgene, Daegu, Korea) for 30 min at 37°C. Digested lung tissues were then passed through 70-µm cell strainers prior to Percoll® (GE Healthcare, Uppsala, Sweden) density-gradient (30/70%) centrifugation to isolate the leukocytes. Red blood cells were lysed using an in-house ACK lysis buffer.
Flow Cytometry
Single-cell suspensions were pretreated with anti-CD16/32 (clone 2.4G2; TONBO Biosciences, CA, USA) antibody to block Fc receptors prior to staining with anti-mouse CD3ε (clone 145-2C11), CD11b (clone M1/70), CD11c (clone N418), CD44 (clone IM7), CD62L (clone MEL-14), CD64 (clone X54-5/7.1.1), Ly6C (clone AL-21), Ly6G (clone 1A8), Siglec-F (clone E50-2440), and Siglec H (clone 551) (BD Biosciences, CA, USA), CD103 (clone 2E7) and MHC Class II (clone M5/114.15.2) (eBioscience, CA, USA), CD4 (clone GK1.5) and CD8α (clone 53-6.7) (TONBO Biosciences, CA, USA), CD45.2 (clone 104) (Biolegend, CA, USA), and CD169 (clone MOMA-1; AbD Serotec, NC, USA). DAPI (Invitrogen, CA, USA) or propidium iodide (eBioscience) was used to exclude dead cells.
To analyze RSV-specific CD8+ T cells, we prepared H-2Db tetramers specific for the RSV M187–195 peptide (NAITNAKII) using the protocol of the NIH Tetramer Core Facility. We stained the cells with APC-labeled tetramers prior to surface staining. Intracellular cytokine staining was performed based on a previously described method (35). Briefly, we incubated the cells with 50 ng/ml phorbol myristate acetate (Sigma Aldrich), 1 µg/ml ionomycin (Sigma Aldrich), and 2 µM GolgiStop (BD Biosciences) for 5 h at 37°C. For the RSV-specific restimulation of CD8+ cells, we cultured the cells in the presence of 5 µg/ml M187–195 peptide (NAITNAKII) and 2 µM GolgiStop for 5 h. We then stained the cells with anti-mouse CD4, CD8α, CD11b, CD44, and CD45.2 prior to fixation and permeabilization using the Cytofix/Cytoperm Kit (BD Biosciences) according to the manufacturer’s protocol. We used IFN-γ antibody (clone XMG1.2; BD Biosciences) to detect intracellular cytokines. All samples were analyzed on an LSR Fortessa cell analyzer (BD Biosciences), and data were analyzed using the FlowJo software (Tree Star, OR, USA).
Cytometric Bead Array and ELISA
To perform BAL on the mice, RSV-infected mice were euthanized at indicated time points after infection. The thorax of each mouse was then opened, and a 20 GA × 1.16 IN (1.1 mm × 30 mm) I.V. catheter (Sewon Medical, Seoul, Korea) was introduced into the trachea at the cricothyroid membrane. The trachea was then flushed with 500 µl PBS, and the resulting fluid was aspirated. Collected BAL fluids were stored at −80°C. IFN-β and TNF-α (Biolegend, CA, USA), IL-6 (BD Biosciences), and CXCL9 and CXCL10 (R&D systems, MN, USA) were then measured in the BAL fluid samples using ELISA kits according to the manufacturer’s protocols. To measure the proinflammatory chemokines in the BAL fluids, the LEGENDplex™ bead-based immunoassay (Biolegend) was utilized according to the manufacturer’s instructions.
Quantitative RT-PCR
To isolate the total RNA from mock-infected or RSV-infected, DT-treated WT and CD169-DTR mice, the lungs were removed from the mice and homogenized in a bullet blender (Next Advance, New York, NY, USA) and RNA was isolated using an RNeasy Mini kit (Qiagen, CA, USA) according to the manufacturer’s protocol. cDNA was synthesized using NobleZyme™ M-MLV reverse transcriptase (Noble Bio, Suwon, Korea) according to the manufacturer’s instructions. Quantitative PCR was performed using the CFX96 real-time PCR system (Bio-Rad, CA, USA) with SYBR Green-based quantification (Toyobo, Osaka, Japan). The following gene-specific forward (F) and reverse (R) primers were used (36): Oas1 (F) GCCTGATCCCAGAATCTATGC and (R) GAGCAACTCTAGGGCGTACTG; Isg15 (F) GGTGTCCGTGACTAACTCCAT and (R) TGGAAAGGGTAAGACCGTCCT; and Hprt (F) GTTGGATACAGGCCAGACTTTGTTG and (R) GAGGGTAGGCTGGCCTATTGGCT. PCR results were normalized against Hprt and are displayed as the fold difference relative to the mock-infected WT control mice.
RSV Titers in the Lungs
Previously published procedures (33) were used to quantitate RSV titers in lungs. Briefly, RSV-infected mice were euthanized and lungs were collected and stored in PBS prior to processing through 70-µm cell strainers and collection of the supernatants. The RSV titers in the supernatants were measured using a plaque assay on HEp-2 cell monolayers.
Statistical Analysis
Data are expressed as the mean ± SE. Differences between groups were analyzed using Student’s t-tests. All statistical analyses were performed using the GraphPad Prism 7 software (GraphPad). Differences were considered statistically significant when P < 0.05.
Results
Depletion of AMs and Reduction of Monocyte Recruitment following DT Administration to CD169-DTR Mice
CD169 is a known marker of AMs (24), and a previous study showed that DT administration to CD169-DTR mice results in significant AM depletion and induces slight neutrophil recruitment to the lungs but does not contribute to disease in infection experiments (25). We first verified that DT treatment depletes AMs in CD169-DTR mice and determined whether it also affects other myeloid cells in the lungs. Flow cytometric analysis of the lung immune cells (see Figure S1 in Supplementary Material for the gating strategy) from DT-treated WT and CD169-DTR mice revealed significant AM depletion and slightly decreased levels of Ly6Chi monocytes in the CD169-DTR mice. Furthermore, DT treatment increased the neutrophil levels in the lungs of the mice but did not affect the eosinophils, pDCs, CD11b+ DCs, or CD103+ DCs (Figures 1A,B). Next, we confirmed that T cells, NK cells, monocytes, neutrophils, and other myeloid cells in mouse lungs do not express CD169 (Figure S2 in Supplementary Material). These results suggest that AMs may be involved in the recruitment of Ly6Chi monocytes to the lungs in steady-state conditions. Together, our results suggest that the DT treatment results in an at least 95% depletion of AMs and partially affects the recruitment of monocytes to the lungs of CD169-DTR mice.
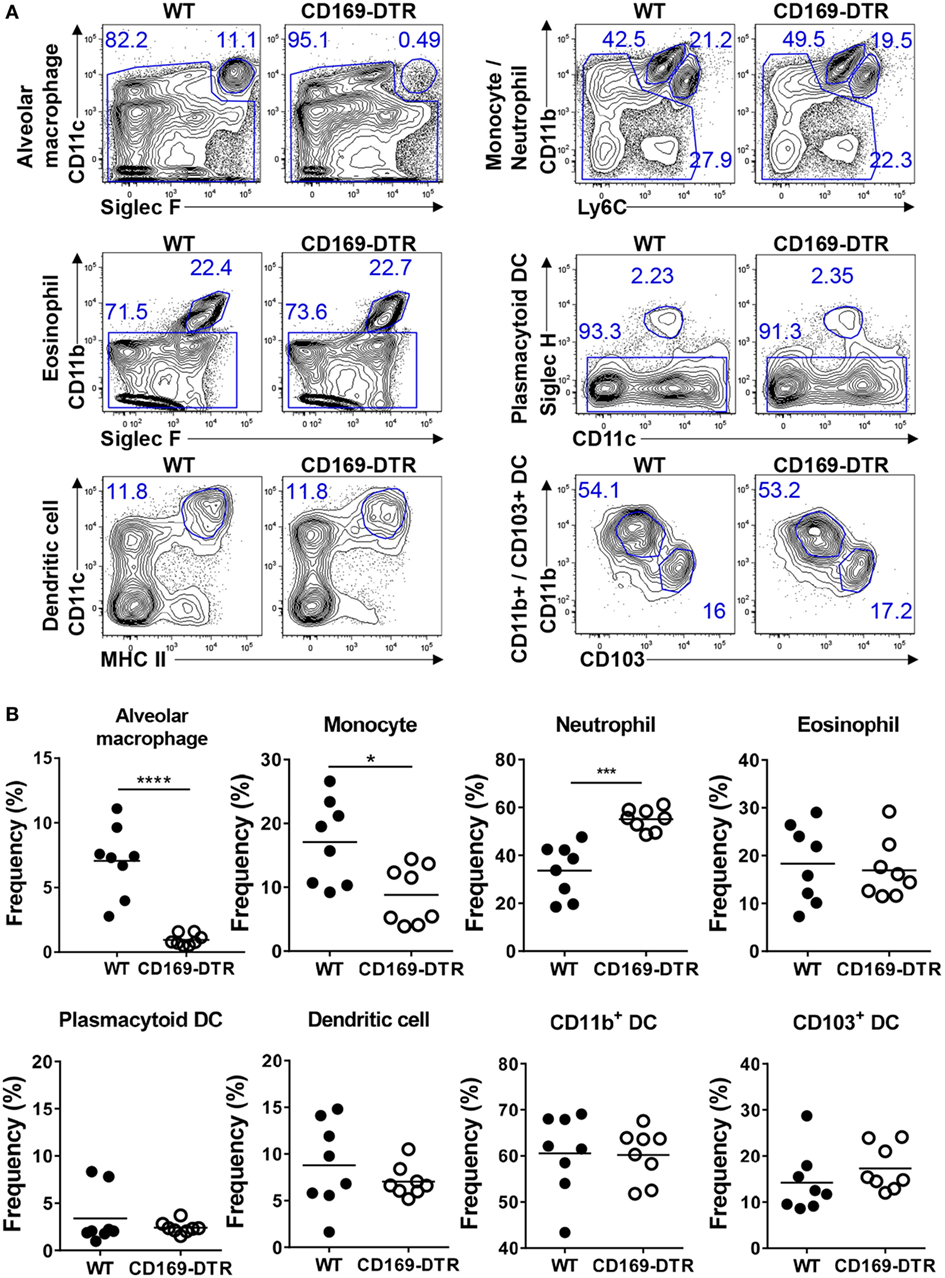
Figure 1. Diphtheria toxin (DT) administration to CD169-diphtheria toxin receptor (DTR) mice induces alveolar macrophage (AM) depletion and reduces monocyte recruitment. (A) Lung cells isolated from wild-type (WT) and CD169-DTR mice 1 day after DT treatment were stained with the indicated antibodies and analyzed by flow cytometry. AMs, monocytes, neutrophils, eosinophils, plasmacytoid dendritic cells, dendritic cells (DCs), CD11b+ DCs, and CD103+ DCs were defined by their surface marker expression. The gating strategy is described in Figure S1 in Supplementary Material. (B) Results are shown as dot graphs. Each dot represents an individual mouse (n = 8 in each group). Statistically significant differences are indicated: *P < 0.05, ***P < 0.001, ****P < 0.0001. Data are representative of three independent experiments.
CD169+ Cell Depletion Diminishes Innate Cytokine Levels in BAL Fluids during RSV Infection
During RSV mucosal infection in mice, IFN-α/β receptor signaling plays a critical role in inducing lung inflammation and limiting viral infection (22), and AMs are major producers of type I IFNs (23). To confirm this, we measured IFN-β, a type I IFN, and the proinflammatory cytokines IL-6 and TNF-α by ELISA in BAL fluids from RSV-infected WT and CD169-DTR mice. Similar to previous results, levels of IFN-β, IL-6, and TNF-α were significantly reduced in BAL fluid from CD169-DTR mice (Figures 2A,B). Furthermore, the mRNA levels of Oas1 and Isg15, two IFN-stimulated genes, were decreased in the RSV-infected lung tissues of the CD169-DTR mice (Figure 2C). Next, we evaluated the levels of proinflammatory chemokines, which are important in inflammatory cell recruitment to the lungs and extravasation into the airways. We found no significant differences in the levels of CXCL1, CCL2, CCL3, CCL4, CCL11, or CXCL13 in the BAL fluid from WT and CD169-DTR mice following RSV infection (Figure 2D). Our results suggest that CD169+ cells are important for both early type I IFN secretion and proinflammatory cytokine secretion but are dispensable for proinflammatory chemokine production.
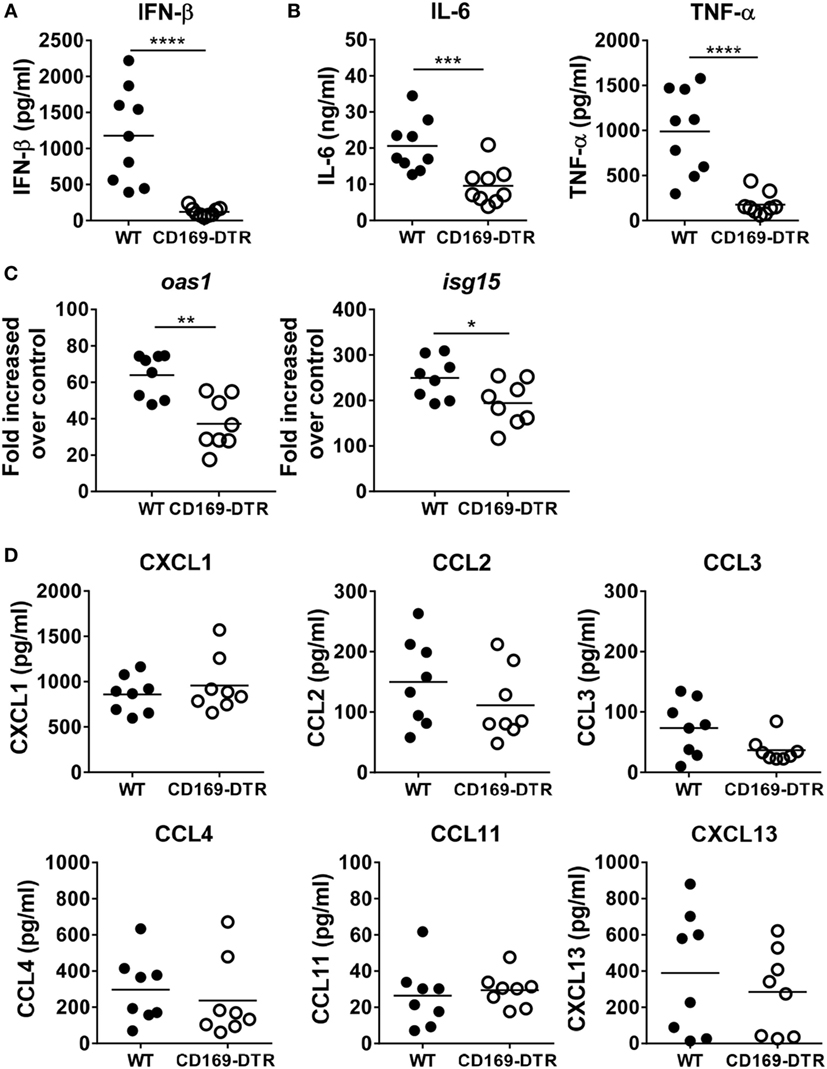
Figure 2. CD169+ cell depletion diminishes innate cytokine secretion into bronchoalveolar lavage (BAL) fluid during respiratory syncytial virus (RSV) infection. At the 9 h later of RSV infection, BAL fluid was collected from diphtheria toxin (DT)-treated wild-type (WT) and CD169-diphtheria toxin receptor (DTR) mice. The levels of interferon (IFN)-β (A) and IL-6 and TNF-α (B) were measured by ELISA. Each dot represents an individual mouse (n = 9 in each group). (C) At the 1 day later of RSV infection, the levels of Oas1 and Isg15 mRNA expression in lung tissues from DT-treated WT and CD169-DTR mice were measured using real-time quantitative PCR. Hprt was used as an internal control. (D) At the 9 h later of RSV infection, the mouse chemokines CXCL1, CCL2, CCL3, CCL4, CCL11, and CXCL13 were monitored using a cytometric bead array. Each dot represents an individual mouse (n = 8 in each group). Statistically significant differences between groups are indicated: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are representative of three independent experiments.
CD169+ Cell Depletion Increases Lung Recruitment of Inflammatory Cells Exclusively during the Early Stage of RSV Infection
CD169+ AMs undergo apoptosis during RSV infection, an event that may contribute to host defense against intracellular pathogens or viruses (37). In the absence of AMs, virus-infected mice show more severe airway inflammation and enhanced lung pathology (25), suggesting that AMs play an important role in the regulation of the inflammatory response to virus infection. To understand the role of CD169+ cells in the trafficking of innate inflammatory cells to the site of infection, we infected DT-treated WT and CD169-DTR mice with RSV, and after the indicated number of days, mice were euthanized and lungs were harvested for analysis by flow cytometry. As the RSV infection progressed, the frequency and number of AMs in the lungs of the mice decreased (Figure 3B). The frequency and number of infiltrating Ly6Chi monocytes and neutrophils (Figure 3A) and eosinophils (Figure 3C) increased in the CD169-DTR mice after RSV infection, whereas the frequency and number of DCs (Figure 3D) were unchanged.
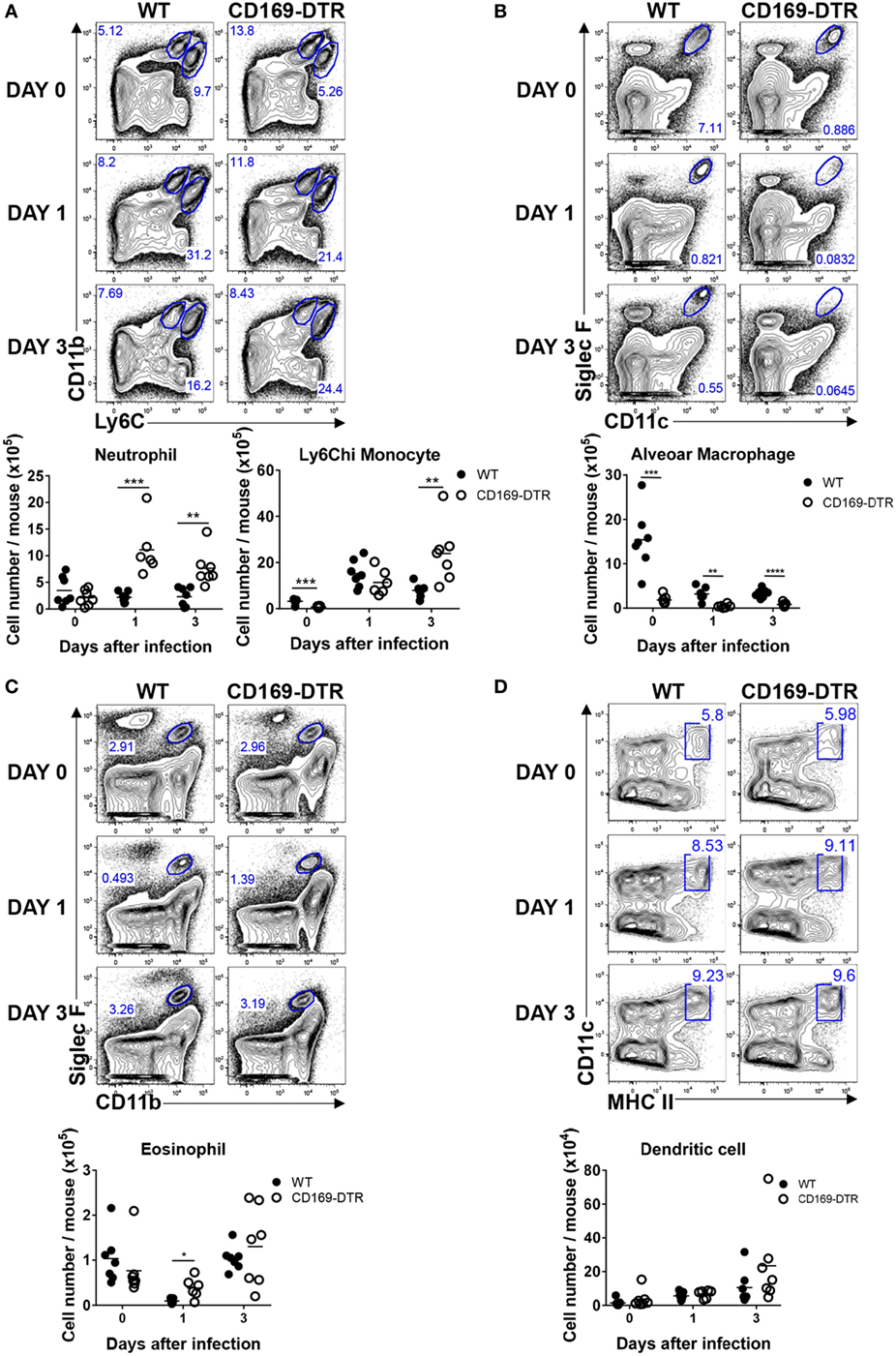
Figure 3. CD169+ cell depletion increases the lung recruitment of inflammatory cells during early respiratory syncytial virus (RSV) infection. At the indicated time points during RSV infection, the lungs of diphtheria toxin-treated wild-type (WT) and CD169-diphtheria toxin receptor (DTR) mice were collected, processed, and analyzed by flow cytometry. The numbers of monocytes and neutrophils (A), alveolar macrophages (B), eosinophils (C), and dendritic cells (D) indicate the frequencies in gated cells. Cell number results are shown in the graphs; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are representative of two independent experiments. Each dot represents an individual mouse (n = 7 in each group). Statistically significant differences between groups are indicated: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are representative of two independent experiments.
Next, we examined the recruitment of innate inflammatory cells to the site of infection during the later stages of RSV infection. AMs in CD169-DTR mice were still depleted by DT treatment after 5 days of RSV infection (Figure 4A). No substantial differences in the frequency or number of Ly6Chi monocytes and neutrophils (Figure 4B), CD64hi inflammatory monocytes (Figure 4C), DCs (Figure 4D), or eosinophils (Figure 4E) were found between the WT and CD169-DTR mice. Collectively, our results demonstrate that the removal of CD169+ cells increases the recruitment of inflammatory cells specifically during the early stage of RSV infection.
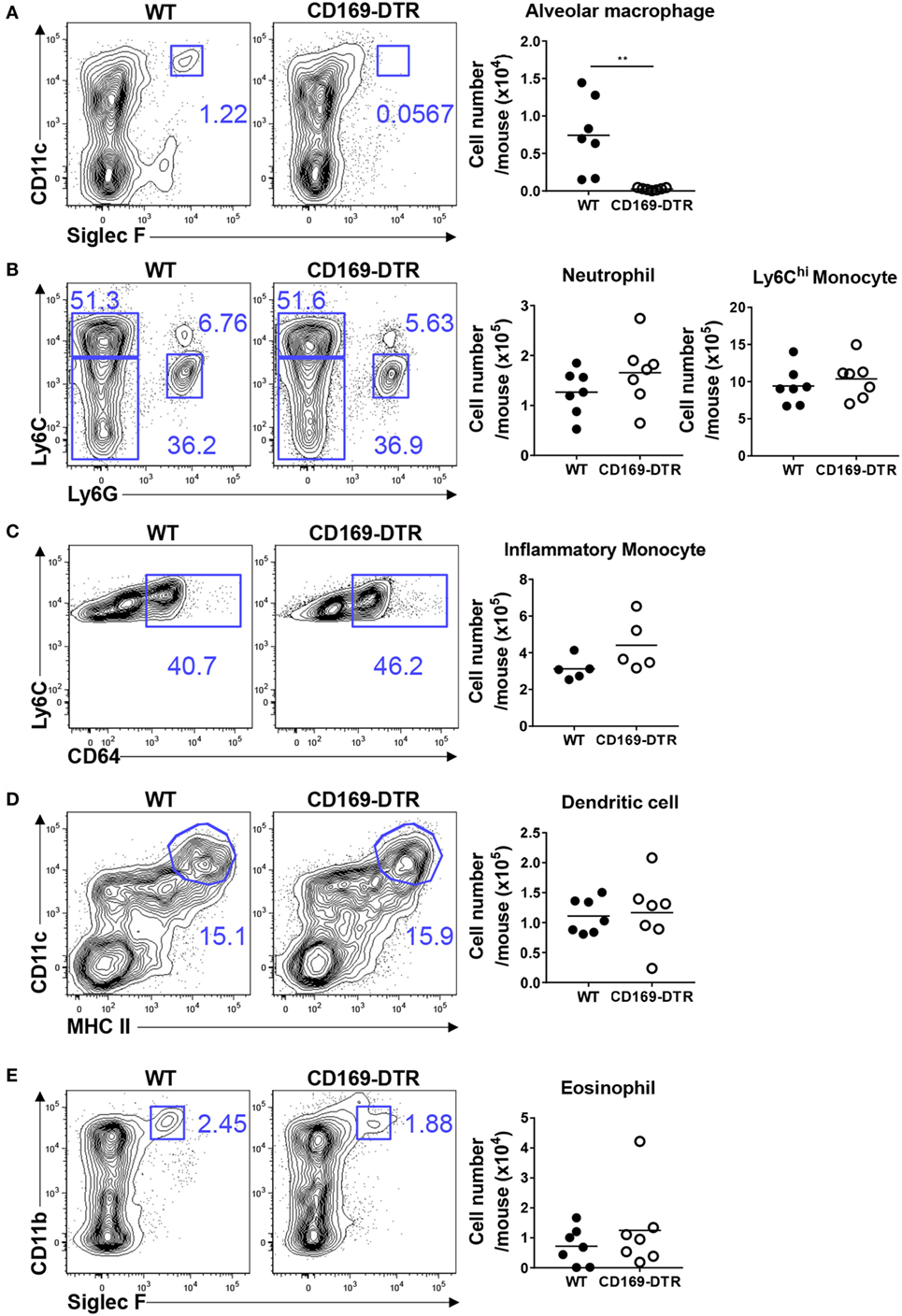
Figure 4. CD169+ cell depletion does not affect the lung recruitment of inflammatory cells 5 days after respiratory syncytial virus (RSV) infection. Five days after RSV infection, the lungs of diphtheria toxin-treated wild-type (WT) and CD169-diphtheria toxin receptor (DTR) mice were collected, processed, and analyzed by flow cytometry. The numbers of alveolar macrophages (A), monocytes and neutrophils (B), inflammatory monocytes (C), dendritic cells (D), and eosinophils (E) indicate the frequencies in gated cells. Cell number results are shown in the graphs. Each dot represents an individual mouse (n = 7 in each group). Statistically significant differences between groups are indicated: **P < 0.01. Data are representative of two independent experiments.
CD169+ Cells Are Dispensable for the Adaptive Immune Response against RSV
Next, we examined the role of CD169+ cells in adaptive immunity to RSV. While adaptive immune responses to RSV infection are required for efficient viral clearance, they also contribute to RSV-induced disease (38). We infected DT-treated WT and CD169-DTR mice with RSV and used intracellular cytokine staining to monitor IFN-γ production from activated lung CD44hi CD4+ and CD44hi CD8+ T cells after stimulation with PMA/ionomycin (for CD4+ T cells) or H-2Db-restricted M187–195 peptides (for CD8+ T cells). There was no substantial difference in the Th1 response (Figures 5A,B) or the CTL response (Figures 5C,D) between the WT and CD169-DTR mice. Therefore, we concluded that CD169+ cells are dispensable for the adaptive immune response against RSV.
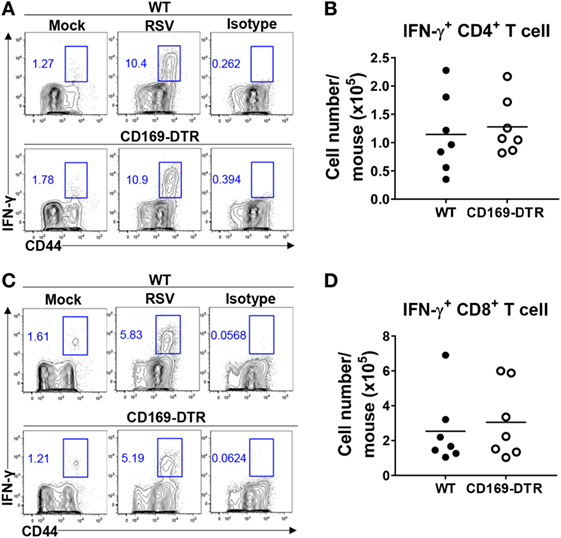
Figure 5. CD169+ cell depletion is dispensable for the adaptive immune response against respiratory syncytial virus (RSV). Seven days after RSV infection, interferon (IFN)-γ production by activated CD4+ or CD8+ T cells from lungs was measured by intracellular cytokine staining after stimulation with PMA and ionomycin (for CD4+ T cells) (A) or H-2Db-restricted M187–195 peptides (for CD8+ T cells) (C). The frequencies and absolute cell numbers of CD44+ IFN-γ+ CD4+ (A,B) and CD8+ (C,D) T cells were assessed. Cell number results from RSV-infected mice are shown in the graphs. Each dot represents an individual mouse (n = 7 in each group). Data are representative of two independent experiments.
Depletion of CD169+ Cells Reduces the Recruitment of Effector CD8+ T Cells to the Lungs after RSV Infection
An efficient antiviral response induces the recruitment of RSV-specific CD4+ and CD8+ T cells to the infection site by the CXCL9 and CXCL10 chemokines, which are induced by IFNs and the chemokine receptor CXCR3. CXCL9 and CXCL10 protect the host by reducing the viral load and pathogenesis (30, 39). To understand the role of CD169+ cells in the recruitment of effector T cells to the infection site, we measured the CXCL9 and CXCL10 levels in BAL fluid from DT-treated WT and CD169-DTR mice after RSV infection. As shown in Figure 6A, both CXCL9 and CXCL10 were decreased in BAL fluid from CD169-DTR mice following RSV infection.
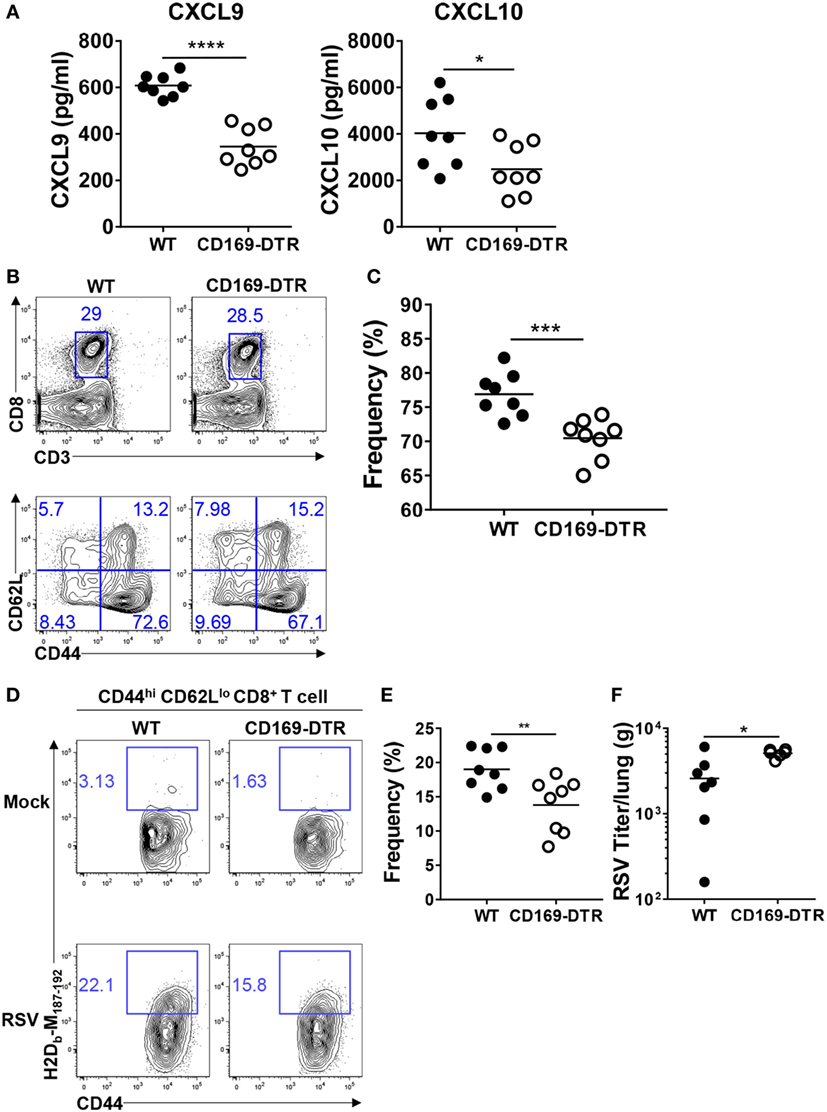
Figure 6. CD169+ cell depletion reduces the recruitment of effector CD8+ T cells in lungs after respiratory syncytial virus (RSV) infection. At the indicated time points after RSV infection, bronchoalveolar lavage fluid was collected from diphtheria toxin-treated wild-type (WT) and CD169-diphtheria toxin receptor (DTR) mice. The levels of CXCL9 and CXCL10 in the lavage fluids were measured by ELISA (A). Seven days after RSV infection, the frequencies of CD8+ T cells (B) and CD44hi CD62Llo CD8+ T cells (C) were assessed by flow cytometry. (D) RSV M peptide-specific CD44hi CD62Llo CD8+ T cells were detected by flow cytometry. (E) Frequencies of CD44hi CD62Llo tetramer+ CD8+ T cells were quantified from flow cytometry data of RSV-infected mice. (F) Five days after infection, RSV viral titers from lung homogenates were measured on HEp-2 cells. Each dot represents an individual mouse (n = 8 in each group). Statistically significant differences between groups are indicated: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are representative of two independent experiments.
Next, we confirmed that the reduction of CXCL9 and CXCL10 reduced the entry of effector T cells into the infection site. DT-treated WT and CD169-DTR mice were infected with RSV, and CD44hi CD62Llo CD4+ and CD44hi CD62Llo CD8+ T cells were assessed by flow cytometry on day 7 after infection. There was no significant difference in the recruitment of CD44hi CD62Llo CD4+ T cells to the lungs (Figure S3 in Supplementary Material); however, the recruitment of CD44hi CD62Llo CD8+ T cells was decreased in the CD169-DTR mice (Figures 6B,C). Staining with RSV-specific tetramers showed that the frequency of RSV-specific CD44hi CD62Llo CD8+ T cells was decreased in the lungs of the RSV-infected CD169-DTR mice (Figures 6D,E).
Finally, we determined whether the effects of the CD169+ cell depletion on innate and adaptive immunity influence RSV replication. Our results show that viral clearance was impaired 5 days after RSV infection in CD169-DTR mice (Figure 6F). Taken together, these results show that CD169+ cells contribute to the recruitment of effector CD8+ T cells to the site of RSV infection and also to the clearance of the virus from the lung mucosa.
Discussion
In this study, we investigated the roles of CD169+ cells in immune responses against mucosal RSV infection. DT administration to CD169-DTR mice was found to specifically induce the depletion of AMs and reduce the recruitment of Ly6Chi monocytes. Notably, CD169+ cell depletion diminished the levels of innate cytokines, such as IFN-β, IL-6, and TNF-α, in BAL fluid from RSV-infected mice without affecting the production of proinflammatory chemokines. Moreover, the depletion of CD169+ cells increased the recruitment of inflammatory cells to the lungs exclusively during the early stage of RSV infection. Furthermore, the depletion of CD169+ cells reduced the recruitment of effector CD8+ T cells to the lungs after RSV mucosal infection. However, the CD169+ cells were dispensable for the adaptive immune response against RSV.
The lung macrophages comprise AMs in the luminal area of alveoli and interstitial macrophages (IMs) within the lung parenchymal interstitium. IMs do not express Siglec-F or CD169 (40) but show characteristics of pro-fibrotic cells in a bleomycin-mediated lung fibrosis model (41). Moreover, in contrast to AMs, IMs play a role in the production of IL-10 (42). The results from this study demonstrate that lung IMs have a unique role in the lung and alter DC functions to prevent ovalbumin/lipopolysaccharide-induced airway allergies in mice. Further studies are required to elucidate the role of IMs in RSV mucosal infection. However, we established the role of AMs in RSV infection by specifically depleting them by DT administration in a CD169-DTR mouse model. Whereas very long-term administration of DT to these mice promotes reduced erythroblast numbers (43), it does not induce any systemic toxic effect (32, 44). Importantly, our studies were performed after short-term administration with a lower dose of DT compared with previous studies.
Our results suggest that AMs contribute to the recruitment of Ly6Chi monocytes to the lungs in steady-state conditions. Although macrophages can be a source of monocyte chemoattractants (45), DT treatment did not alter the levels of inflammatory chemoattractants in BAL fluid from CD169-DTR mice. Recent research using a murine colitis model showed that CD169+ macrophages contribute to the accumulation of colitis-associated CD169− Ly6Chi CD64lo monocytes in the colon mucosa in a CCL8-dependent manner (46). In agreement with this, we found that the levels of CCL8 mRNA in the lungs were decreased in CD169-DTR mice after RSV infection (data not shown). Further research is needed to determine whether AMs are involved in CCL8-dependent monocyte recruitment to the lungs.
Various types of lung cells, such as epithelial cells, macrophages, fibroblasts, DCs, and pDCs, are known to secrete type I IFNs during RSV infection. Alveolar-located DCs send their dendrites trans-epithelially into airspace and assess particulate antigens (47) or RSV virions (48) and have functions to produce various inflammatory mediators, including type I IFNs (49). In addition, alveolar DCs can be infected with RSV in vitro or in vivo and produce type I IFNs and proinflammatory cytokines (48, 50). Similarly, AMs are known to produce type I IFNs after virus infection (51–53) and are known to be resistant to RSV replication, which may help to sustain their activity (54). Our results support previous claims that AMs are major IFN producers in the lungs after RSV infection, while some studies suggested that pDCs are the source of type I IFNs during RSV infection (55, 56). Moreover, our results show that AMs contribute to the production of proinflammatory cytokines such as IL-6 and TNF-α during RSV infection. Although primary-airway epithelial cells and AMs have been associated with the production of proinflammatory chemokines (57, 58), our results show that AMs are dispensable for proinflammatory chemokine production during RSV infection, suggesting that airway epithelial cells or other cells are sufficient for their production.
Alveolar macrophages undergo apoptosis in the early phase of RSV infection, resulting in a reduction of the disease severity and the eventual resolution of inflammation (37). Consistent with previous results, RSV infection in our mouse model reduced AM levels, an effect that was sustained even 5 days after infection. Not only do AMs play a crucial role in the maintenance of lung homeostasis and the clearance of airway dust, but also there is increasing evidence indicating that severe pulmonary disease caused by RSV infection in infancy is associated with recurrent wheezing and the development of asthma later in childhood (59, 60). Recent studies indicated that AMs regulate inflammatory immune responses in the airways and that these cells have a critical role in asthma (61). In a mouse model of house dust mite-induced asthma in which AMs are depleted using clodronate liposomes, Th2 cytokines, such as IL-4, IL-5, and IL-13, and inflammatory cytokines and eosinophil recruitment were increased in BAL fluid, suggesting an immunosuppressive role of AMs (62). Our results suggest a possible mechanism of asthma development following RSV infection in which the failure of homeostasis is due to the induction of AM apoptosis.
During the early phase of RSV infection, neutrophil and monocyte recruitment, which is characteristic of virus-mediated inflammation, was slightly increased in the lungs of CD169-DTR mice. However, the recruitment of these cells was comparable between the WT and CD169-DTR mice during the later stages of RSV infection. This suggests that AMs are required for protection against RSV-induced tissue injury or excessive inflammatory signaling in the early phase of the infection, but their importance decreases at later time points due to RSV-induced apoptosis. The relationship between AMs and RSV-induced eosinophilia, which have been proposed to play roles in the pathogenesis of lower respiratory tract disease (63, 64), is unclear. Our data suggest that AMs are required to resolve eosinophilia during the early stage of RSV infection but not during the later stages. Further studies are needed to determine whether AMs are associated with chemokine production important for eosinophil recruitment.
A previous study noted that MAVS is important for sensing RSV (23). In MAVS-deficient mice, AMs did not produce type I IFNs and there was a defect in the recruitment of inflammatory monocytes to the lungs downstream of type I IFN signaling (23). Our results show that AM depletion reduces the secretion of type I IFN but does not affect the recruitment of CD64hi inflammatory monocytes. This suggests that other factors are involved in the recruitment of CD64hi inflammatory monocytes to the lungs.
While the adaptive T cell immune response to RSV is important for the promotion of viral clearance, a dysregulated T cell response to RSV can induce immunopathology. Macrophage depletion by clodronate liposomes did not affect the recruitment of activated CD4+ or CD8+ T cells, weight loss, or virus-induced changes in mouse lung function (12). Although AMs suppress the migration (65) and antigen-presentation capacity (66) of DCs and increase the trafficking of antigen to draining lymph nodes (65), our results show that AM depletion in CD169-DTR mice does not affect DCs or their subset frequency and has no effect on the adaptive CD4+ and CD8+ T cell immune responses to RSV infection. CXCL9 and CXCL10 play important roles in RSV-infected mice by recruiting virus-specific T cells to the lungs and promoting viral clearance (39, 67). Multiple cell populations, including epithelial cells and fibroblasts, may induce the production of CXCL9 and CXCL10 in the airways (68). Moreover, type I IFN production provides additional increases in CXCL9 and CXCL10 production, thereby enhancing the antiviral response (69). Our results newly demonstrate roles of AMs in CXCL9 and CXCL10 production, and as a result, in the recruitment of antigen-specific effector CD8+ T cells to the lungs. Furthermore, our results demonstrate that AMs are required for RSV clearance.
Recently, a “prime-and-pull” vaccine strategy was developed to induce local resident memory T cells at an infection site using a model of genital mucosal HSV-2 infection (70). This strategy comprises a conventional vaccination to provoke a systemic T cell responses (i.e., the prime) and administration CXCL9/10 chemokines to the genital tract to recruit activated T cells (i.e., the pull), thereby establishing long-term protective immunity. This strategy provoked more efficient protective T cell responses in the lungs after flu infection (71). The results from our study suggest that AMs are the major producer of CXCL9/10 in mucosal RSV infection. Thus, AMs can be considered novel targets for the development of vaccines designed to provoke more efficient local memory T cell immunity via secretion of CXCL9/10.
In conclusion, our study reveals that AMs are responsible for the production of type I IFNs and proinflammatory cytokines and for the recruitment of effector CD8+ T cells via CXCL9 and CXCL10 production. Furthermore, AMs are required for viral clearance in RSV mucosal infection. Therefore, AMs might provide a novel target for the development of an RSV vaccine using the prime-and-pull strategy to stimulate AMs to induce type I IFN and CXCL9 and CXCL10 secretions (70).
Ethics Statement
All animal procedures met the relevant legal and ethical requirements and were in accordance with the guidelines and protocols (KA2013-55) for rodent research approved by the Institutional Animal Care and Use Committee of KAIST.
Author Contributions
DO and HL designed the study. DO, JO, and HJ performed the research. DO and HL conceived the study, analyzed the data, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the members of the Laboratory of Host Defenses for helpful advice on experiments and data discussions. The authors also thank Hye Jee Yoo and Hye Sun Jang for technical help.
Funding
This study was supported by the National Research Foundation (NRF-2016R1A2B2015028, NRF-2015R1A4A1042416, and NRF-2013R1A1A2063347) and KAIST Future Systems Healthcare Project funded by the Ministry of Science, ICT, and Future Planning of Korea.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00819/full#supplementary-material.
References
1. Collins PL, Graham BS. Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol (2008) 82(5):2040–55. doi:10.1128/JVI.01625-07
2. Borchers AT, Chang C, Gershwin ME, Gershwin LJ. Respiratory syncytial virus – a comprehensive review. Clin Rev Allergy Immunol (2013) 45(3):331–79. doi:10.1007/s12016-013-8368-9
3. Haynes LM. Progress and challenges in RSV prophylaxis and vaccine development. J Infect Dis (2013) 208(Suppl 3):S177–83. doi:10.1093/infdis/jit512
4. Whimbey E, Englund JA, Couch RB. Community respiratory virus infections in immunocompromised patients with cancer. Am J Med (1997) 102(3A):10–8; discussion 25–6. doi:10.1016/S0002-9343(97)80004-6
5. Storey S. Respiratory syncytial virus market. Nat Rev Drug Discov (2010) 9(1):15–6. doi:10.1038/nrd3075
6. Openshaw PJ, Culley FJ, Olszewska W. Immunopathogenesis of vaccine-enhanced RSV disease. Vaccine (2001) 20(Suppl 1):S27–31. doi:10.1016/S0264-410X(01)00301-2
7. Kim TH, Lee HK. Innate immune recognition of respiratory syncytial virus infection. BMB Rep (2014) 47(4):184–91. doi:10.5483/BMBRep.2014.47.4.050
8. Kim TH, Lee HK. Differential roles of lung dendritic cell subsets against respiratory virus infection. Immune Netw (2014) 14(3):128–37. doi:10.4110/in.2014.14.3.128
9. Thepen T, Van Rooijen N, Kraal G. Alveolar macrophage elimination in vivo is associated with an increase in pulmonary immune response in mice. J Exp Med (1989) 170(2):499–509. doi:10.1084/jem.170.2.499
10. Harmsen AG, Muggenburg BA, Snipes MB, Bice DE. The role of macrophages in particle translocation from lungs to lymph nodes. Science (1985) 230(4731):1277–80. doi:10.1126/science.4071052
11. Haeberle HA, Takizawa R, Casola A, Brasier AR, Dieterich HJ, Van Rooijen N, et al. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J Infect Dis (2002) 186(9):1199–206. doi:10.1086/344644
12. Pribul PK, Harker J, Wang B, Wang H, Tregoning JS, Schwarze J, et al. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol (2008) 82(9):4441–8. doi:10.1128/JVI.02541-07
13. Reed JL, Brewah YA, Delaney T, Welliver T, Burwell T, Benjamin E, et al. Macrophage impairment underlies airway occlusion in primary respiratory syncytial virus bronchiolitis. J Infect Dis (2008) 198(12):1783–93. doi:10.1086/593173
14. Van Lent PL, Holthuysen AE, Van Rooijen N, Van De Putte LB, Van Den Berg WB. Local removal of phagocytic synovial lining cells by clodronate-liposomes decreases cartilage destruction during collagen type II arthritis. Ann Rheum Dis (1998) 57(7):408–13. doi:10.1136/ard.57.7.408
15. Leenen PJ, Radosevic K, Voerman JS, Salomon B, van Rooijen N, Klatzmann D, et al. Heterogeneity of mouse spleen dendritic cells: in vivo phagocytic activity, expression of macrophage markers, and subpopulation turnover. J Immunol (1998) 160(5):2166–73.
16. McGill J, Van Rooijen N, Legge KL. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J Exp Med (2008) 205(7):1635–46. doi:10.1084/jem.20080314
17. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi:10.1038/nri3787
18. Liu P, Jamaluddin M, Li K, Garofalo RP, Casola A, Brasier AR. Retinoic acid-inducible gene I mediates early antiviral response and toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J Virol (2007) 81(3):1401–11. doi:10.1128/JVI.01740-06
19. Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol (2008) 82(1):335–45. doi:10.1128/JVI.01080-07
20. Yoboua F, Martel A, Duval A, Mukawera E, Grandvaux N. Respiratory syncytial virus-mediated NF-kappa B p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKK beta. J Virol (2010) 84(14):7267–77. doi:10.1128/JVI.00142-10
21. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity (2013) 38(5):855–69. doi:10.1016/j.immuni.2013.05.007
22. Goritzka M, Durant LR, Pereira C, Salek-Ardakani S, Openshaw PJ, Johansson C. Alpha/beta interferon receptor signaling amplifies early proinflammatory cytokine production in the lung during respiratory syncytial virus infection. J Virol (2014) 88(11):6128–36. doi:10.1128/JVI.00333-14
23. Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med (2015) 212(5):699–714. doi:10.1084/jem.20140825
24. Ducreux J, Crocker PR, Vanbever R. Analysis of sialoadhesin expression on mouse alveolar macrophages. Immunol Lett (2009) 124(2):77–80. doi:10.1016/j.imlet.2009.04.006
25. Purnama C, Ng SL, Tetlak P, Setiagani YA, Kandasamy M, Baalasubramanian S, et al. Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur J Immunol (2014) 44(7):2003–12. doi:10.1002/eji.201344359
26. Lee S, Stokes KL, Currier MG, Sakamoto K, Lukacs NW, Celis E, et al. Vaccine-elicited CD8+ T cells protect against respiratory syncytial virus strain A2-line19F-induced pathogenesis in BALB/c mice. J Virol (2012) 86(23):13016–24. doi:10.1128/JVI.01770-12
27. Chang J, Braciale TJ. Respiratory syncytial virus infection suppresses lung CD8+ T-cell effector activity and peripheral CD8+ T-cell memory in the respiratory tract. Nat Med (2002) 8(1):54–60. doi:10.1038/nm0102-54
28. Haeberle HA, Kuziel WA, Dieterich HJ, Casola A, Gatalica Z, Garofalo RP. Inducible expression of inflammatory chemokines in respiratory syncytial virus-infected mice: role of MIP-1alpha in lung pathology. J Virol (2001) 75(2):878–90. doi:10.1128/JVI.75.2.878-890.2001
29. Tripp RA, Jones L, Anderson LJ. Respiratory syncytial virus G and/or SH glycoproteins modify CC and CXC chemokine mRNA expression in the BALB/c mouse. J Virol (2000) 74(13):6227–9. doi:10.1128/JVI.74.13.6227-6229.2000
30. Jafri HS, Chavez-Bueno S, Mejias A, Gomez AM, Rios AM, Nassi SS, et al. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J Infect Dis (2004) 189(10):1856–65. doi:10.1086/386372
31. Villenave R, Thavagnanam S, Sarlang S, Parker J, Douglas I, Skibinski G, et al. In vitro modeling of respiratory syncytial virus infection of pediatric bronchial epithelium, the primary target of infection in vivo. Proc Natl Acad Sci U S A (2012) 109(13):5040–5. doi:10.1073/pnas.1110203109
32. Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, et al. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity (2011) 34(1):85–95. doi:10.1016/j.immuni.2010.12.011
33. Kim S, Joo DH, Lee JB, Shim BS, Cheon IS, Jang JE, et al. Dual role of respiratory syncytial virus glycoprotein fragment as a mucosal immunogen and chemotactic adjuvant. PLoS One (2012) 7(2):e32226. doi:10.1371/journal.pone.0032226
34. Shim YR, Lee HK. Caspase-1 independent viral clearance and adaptive immunity against mucosal respiratory syncytial virus infection. Immune Netw (2015) 15(2):73–82. doi:10.4110/in.2015.15.2.73
35. Oh JE, Kim BC, Chang DH, Kwon M, Lee SY, Kang D, et al. Dysbiosis-induced IL-33 contributes to impaired antiviral immunity in the genital mucosa. Proc Natl Acad Sci U S A (2016) 113(6):E762–71. doi:10.1073/pnas.1518589113
36. Oh JE, Lee MS, Kim YJ, Lee HK. OASL1 deficiency promotes antiviral protection against genital herpes simplex virus type 2 infection by enhancing type I interferon production. Sci Rep (2016) 6:19089. doi:10.1038/srep19089
37. Aberdein JD, Cole J, Bewley MA, Marriott HM, Dockrell DH. Alveolar macrophages in pulmonary host defence the unrecognized role of apoptosis as a mechanism of intracellular bacterial killing. Clin Exp Immunol (2013) 174(2):193–202. doi:10.1111/cei.12170
38. Varga SM, Braciale TJ. The adaptive immune response to respiratory syncytial virus. Curr Top Microbiol Immunol (2013) 372:155–71. doi:10.1007/978-3-642-38919-1_8
39. Lindell DM, Lane TE, Lukacs NW. CXCL10/CXCR3-mediated responses promote immunity to respiratory syncytial virus infection by augmenting dendritic cell and CD8(+) T cell efficacy. Eur J Immunol (2008) 38(8):2168–79. doi:10.1002/eji.200838155
40. Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol Med (2016) 22(4):303–16. doi:10.1016/j.molmed.2016.02.004
41. Ji WJ, Ma YQ, Zhou X, Zhang YD, Lu RY, Sun HY, et al. Temporal and spatial characterization of mononuclear phagocytes in circulating, lung alveolar and interstitial compartments in a mouse model of bleomycin-induced pulmonary injury. J Immunol Methods (2014) 403(1–2):7–16. doi:10.1016/j.jim.2013.11.012
42. Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Invest (2009) 119(12):3723–38. doi:10.1172/JCI39717
43. Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med (2013) 19(4):429–36. doi:10.1038/nm.3057
44. Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest (2007) 117(8):2268–78. doi:10.1172/JCI31990
45. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11(11):762–74. doi:10.1038/nri3070
46. Asano K, Takahashi N, Ushiki M, Monya M, Aihara F, Kuboki E, et al. Intestinal CD169(+) macrophages initiate mucosal inflammation by secreting CCL8 that recruits inflammatory monocytes. Nat Commun (2015) 6:7802. doi:10.1038/ncomms8802
47. Thornton EE, Looney MR, Bose O, Sen D, Sheppard D, Locksley R, et al. Spatiotemporally separated antigen uptake by alveolar dendritic cells and airway presentation to T cells in the lung. J Exp Med (2012) 209(6):1183–99. doi:10.1084/jem.20112667
48. Hillyer P, Mane VP, Chen A, Dos Santos MB, Schramm LM, Shepard RE, et al. Respiratory syncytial virus infection induces a subset of types I and III interferons in human dendritic cells. Virology (2017) 504:63–72. doi:10.1016/j.virol.2017.01.017
49. Guilliams M, Lambrecht BN, Hammad H. Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol (2013) 6(3):464–73. doi:10.1038/mi.2013.14
50. Rudd BD, Luker GD, Luker KE, Peebles RS, Lukacs NW. Type I interferon regulates respiratory virus infected dendritic cell maturation and cytokine production. Viral Immunol (2007) 20(4):531–40. doi:10.1089/vim.2007.0057
51. Jewell NA, Vaghefi N, Mertz SE, Akter P, Peebles RS Jr, Bakaletz LO, et al. Differential type I interferon induction by respiratory syncytial virus and influenza A virus in vivo. J Virol (2007) 81(18):9790–800. doi:10.1128/JVI.00530-07
52. Demoor T, Petersen BC, Morris S, Mukherjee S, Ptaschinski C, Nagata DED, et al. IPS-1 signaling has a nonredundant role in mediating antiviral responses and the clearance of respiratory syncytial virus. J Immunol (2012) 189(12):5942–53. doi:10.4049/jimmunol.1201763
53. Smit JJ, Rudd BD, Lukacs NW. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J Exp Med (2006) 203(5):1153–9. doi:10.1084/jem.20052359
54. Ravi LI, Li L, Sutejo R, Chen H, Wong PS, Tan BH, et al. A systems-based approach to analyse the host response in murine lung macrophages challenged with respiratory syncytial virus. BMC Genomics (2013) 14:190. doi:10.1186/1471-2164-14-190
55. Schijf MA, Lukens MV, Kruijsen D, van Uden NO, Garssen J, Coenjaerts FE, et al. Respiratory syncytial virus induced type I IFN production by pDC is regulated by RSV-infected airway epithelial cells, RSV-exposed monocytes and virus specific antibodies. PLoS One (2013) 8(11):e81695. doi:10.1371/journal.pone.0081695
56. Wang H, Peters N, Schwarze J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J Immunol (2006) 177(9):6263–70. doi:10.4049/jimmunol.177.9.6263
57. Noah TL, Becker S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am J Physiol (1993) 265(5 Pt 1):L472–8.
58. Meusel TR, Imani F. Viral induction of inflammatory cytokines in human epithelial cells follows a p38 mitogen-activated protein kinase-dependent but NF-kappa B-independent pathway. J Immunol (2003) 171(7):3768–74. doi:10.4049/jimmunol.171.7.3768
59. Henderson J, Hilliard TN, Sherriff A, Stalker D, Al Shammari N, Thomas HM. Hospitalization for RSV bronchiolitis before 12 months of age and subsequent asthma, atopy and wheeze: a longitudinal birth cohort study. Pediatr Allergy Immunol (2005) 16(5):386–92. doi:10.1111/j.1399-3038.2005.00298.x
60. Sigurs N, Gustafsson PM, Bjarnason R, Lundberg F, Schmidt S, Sigurbergsson F, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med (2005) 171(2):137–41. doi:10.1164/rccm.200406-730OC
61. Balhara J, Gounni AS. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol (2012) 5(6):605–9. doi:10.1038/mi.2012.74
62. Zaslona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, et al. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol (2014) 193(8):4245–53. doi:10.4049/jimmunol.1400580
63. Lindemans CA, Kimpen JL, Luijk B, Heidema J, Kanters D, van der Ent CK, et al. Systemic eosinophil response induced by respiratory syncytial virus. Clin Exp Immunol (2006) 144(3):409–17. doi:10.1111/j.1365-2249.2006.03084.x
64. Matthews SP, Tregoning JS, Coyle AJ, Hussell T, Openshaw PJ. Role of CCL11 in eosinophilic lung disease during respiratory syncytial virus infection. J Virol (2005) 79(4):2050–7. doi:10.1128/JVI.79.4.2050-2057.2005
65. Jakubzick C, Tacke F, Llodra J, van Rooijen N, Randolph GJ. Modulation of dendritic cell trafficking to and from the airways. J Immunol (2006) 176(6):3578–84. doi:10.4049/jimmunol.176.6.3578
66. Holt PG. Antigen presentation in the lung. Am J Respir Crit Care Med (2000) 162(4 Pt 2):S151–6. doi:10.1164/ajrccm.162.supplement_3.15tac2
67. Vissers M, Schreurs I, Jans J, Heldens J, de Groot R, de Jonge MI, et al. Antibodies enhance CXCL10 production during RSV infection of infant and adult immune cells. Cytokine (2015) 76(2):458–64. doi:10.1016/j.cyto.2015.07.024
68. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol (2011) 89(2):207–15. doi:10.1038/icb.2010.158
69. Yarilina A, Ivashkiv LB. Type I interferon: a new player in TNF signaling. Curr Dir Autoimmun (2010) 11:94–104. doi:10.1159/000289199
70. Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature (2012) 491(7424):463–7. doi:10.1038/nature11522
Keywords: alveolar macrophage, respiratory syncytial virus, type I interferon, CD169, CXCL9/10
Citation: Oh DS, Oh JE, Jung HE and Lee HK (2017) Transient Depletion of CD169+ Cells Contributes to Impaired Early Protection and Effector CD8+ T Cell Recruitment against Mucosal Respiratory Syncytial Virus Infection. Front. Immunol. 8:819. doi: 10.3389/fimmu.2017.00819
Received: 21 April 2017; Accepted: 28 June 2017;
Published: 13 July 2017
Edited by:
Marcello Chieppa, IRCCS “de Bellis”, ItalyReviewed by:
Laurent Pierre Nicod, University of Lausanne, SwitzerlandRosalinda Sorrentino, University of Salerno, Italy
Copyright: © 2017 Oh, Oh, Jung and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heung Kyu Lee, aGV1bmdreXUubGVlJiN4MDAwNDA7a2Fpc3QuYWMua3I=