 Deepti Pilli1†
Deepti Pilli1† Alicia Zou
Alicia Zou Fabienne Brilot
Fabienne Brilot- 1Brain Autoimmunity Group, Institute for Neuroscience and Muscle Research, The Kids Research Institute at The Children’s Hospital at Westmead, University of Sydney, Sydney, NSW, Australia
- 2Brain and Mind Centre, University of Sydney, Sydney, NSW, Australia
It is being increasingly recognized that a dysregulation of the immune system plays a vital role in neurological disorders and shapes the treatment of the disease. Aberrant T cell responses, in particular, are key in driving autoimmunity and have been traditionally associated with multiple sclerosis. Yet, it is evident that there are other neurological diseases in which autoreactive T cells have an active role in pathogenesis. In this review, we report on the recent progress in profiling and assessing the functionality of autoreactive T cells in central nervous system (CNS) autoimmune disorders that are currently postulated to be primarily T cell driven. We also explore the autoreactive T cell response in a recently emerging group of syndromes characterized by autoantibodies against neuronal cell-surface proteins. Common methodology implemented in T cell biology is further considered as it is an important determinant in their detection and characterization. An improved understanding of the contribution of autoreactive T cells expands our knowledge of the autoimmune response in CNS disorders and can offer novel methods of therapeutic intervention.
Introduction
Autoimmunity is believed to be the underlying cause in a growing number of neurological disorders. Although the precise mechanisms that trigger autoimmunity have not been fully elucidated, it is known that a dysregulation in T cells is a key component, given their constitutive role in immunosurveillance (1). The archetypal neurological disease mediated primarily by T cells is multiple sclerosis (MS) (2, 3). It has been studied extensively for many years in both humans and animal models, and an informed understanding of MS has laid the groundwork for further studies in other suspected autoimmune neurological disorders. In particular, Rasmussen’s encephalitis (RE) (4) and a spectrum of paraneoplastic syndromes (5, 6) are hypothesized to be T cell driven. In other disorders, like amytrophic lateral sclerosis (ALS), T cells may conversely play a neuroprotective role (7). In addition to a dysfunctional cellular immunity, effector molecules of humoral immunity, such as autoantibodies, may concomitantly participate in autoimmunity. Although paraneoplastic syndromes have been associated with specific autoantibodies, the search for autoantibodies in autoimmune central nervous system (CNS) diseases such as MS, RE, and ALS is still ongoing.
Indeed, in recent years, a growing number of autoantibodies targeting neuronal receptors or synaptic proteins of the CNS are proving to be useful biomarkers of various neurological diseases treatable with immunotherapy (8–15). This has spurred intensive investigations to understand the mechanisms behind autoantibody responses, with emerging evidence suggesting a pathogenic role. However, it is well established that the production and sustenance of immunoglobulin-G (IgG) autoantibodies and autoantibody-producing B cells necessitates the involvement of T cells reactive against a shared protein antigen (16–19). Although this aspect of adaptive immunity has been explored less thoroughly in autoantibody-associated neuroimmune disorders, this premise has broadened studies to focus on cellular responses in the following autoantibody-associated diseases: neuromyelitis optica (NMO), acute disseminated encephalomyelitis (ADEM), stiff person syndrome (SPS), and anti-N-methyl-d-aspartate receptor (anti-NMDAR) encephalitis.
In this review, we explore the accumulating evidence of cellular immune responses in various disorders of the CNS that are predominantly T cell-driven, as well as the more newly classified group of autoantibody-associated syndromes. In particular, we focus on findings in humans, as many studies conducted in animal models are reviewed elsewhere or have been recently reviewed (1, 20–23). Common methods implemented in the study of T cell biology are also evaluated.
Immunosurveillance of T Cells in the CNS
The CNS has been traditionally viewed as an immune privilege site that is inaccessible to T cells and other immune cells. However, it is now well recognized that T cells actively survey the CNS in the healthy state to ensure host defense against infections. Central and effector memory T cells constantly patrol the brain and spinal cord for pathogens via the cerebrospinal fluid (CSF) that bathes these structures (24–26). In fact, around 80% of immune cells in the CSF are T cells (27). As they travel through the subarachnoid space between the meninges, T cells interact with resident antigen-presenting cells (APCs) to sample antigens, including parenchyma-derived antigens in the interstitial fluid that drains into the CSF (28). Memory T cells can then be restimulated upon recognition of a pathogen as part of the host response.
Moreover, recent evidence confirms the presence of lymphatic vessels within the meninges of healthy mice that resemble traditional lymphatic vessels found in the periphery, both structurally and functionally (29, 30). These meningeal vessels line the dural sinuses and drain cells and fluid of the subarachnoid space directly into the deep cervical lymph node. Notably, T cells were identified in these meningeal lymphatics (30), indicating a travel route between the CNS and lymph nodes in the steady state. Together, this refutes previous notions that immune cell entry into the CNS was restricted by the apparent absence of lymphatic drainage, and further supports the concept of immunosurveillance by T cells in the CNS.
Although the blood–brain barrier (BBB) and blood–CSF barrier shielding the CNS were seen to be another mechanism exempting the CNS from immune monitoring, various adhesion molecules on their surface enable T cell migration. Egress from the blood to the CSF is dependent on the expression of P-selectin in choroid plexus stroma vessels and meningeal vessels (24, 31). In addition, the interaction of α4β1 integrin with vascular cell adhesion molecule 1 (VCAM1) on endothelial cells of the BBB is important in facilitating T cell movement into the perivascular space, as evidenced by the efficacy of natalizumab in reducing inflammation in MS (32–34).
As will be discussed later, the importance of immunosurveillance in maintaining homeostasis in the CNS is particularly evident when it is disrupted by immunosuppression. Under immunosuppression, the mobilization of immune cells into the CNS is hindered, making the body more susceptible to opportunistic infections by agents such as, JC polyoma virus (JCV), herpes simplex virus, toxoplasmosis, and Cryptococcus (35–37). With the host immune response dampened and the CNS unguarded, the pathogenic response goes unchecked, leading to potentially fatal diseases.
T Cell-Mediated CNS Diseases
Multiple Sclerosis
Multiple sclerosis is a common chronic inflammatory disease of the CNS resulting in the demyelination of neurons. Damage to the myelin sheath surrounding neuronal axons leads to the progressive loss of neurological function and affects over two million people globally (38, 39). The majority of MS patients (85%) experience a relapsing-remitting disease course (40), who can transition into a secondary progressive disease form after approximately 10 years of primary disease (41, 42). The remaining 15% of patients follow a primary progressive disease course characterized by a steady decline in neurological function from the initial attack (38). Lesions, or plaques, are traditionally thought to present in the white matter of the brain and spinal cord. However, recent studies have shown gray matter lesions to accrue through the MS disease course and dominate in progressive disease (43–49).
The current consensus argues in favor of MS as an autoimmune disease mediated by self-reactive, myelin-specific T cells (1, 38, 46), with additional components of genetic susceptibility and environmental factors (41, 50). In terms of genetic susceptibility, MS has been strongly associated with different HLA class II haplotypes, including HLA-DR15 and HLA-DQ6, although their contributions to clinical disease have yet to be uncovered (51–53). It is hypothesized that these MHC molecules are able to present target autoantigens to autoreactive components of the adaptive immune system (54, 55). Several immune system genes have also been implicated in MS disease susceptibility, including those that code for IL-17 and IL-2 receptor (51, 52).
However, genetics only partially contribute to the risk of MS disease development. A significant proportion of disease risk can be directly correlated with various lifestyle and environmental factors including vitamin D deficiency, Epstein-Barr virus (EBV) infection, and smoking (56, 57). Epidemiological studies show that there is a strong association between MS prevalence and the angle of latitude. This trend may be attributed to exposure to solar radiation and vitamin D, with vitamin D-deficient individuals more prone to developing disease (58, 59). Several studies have also shown that MS patients with lower serum levels of vitamin D to be more susceptible to relapses (60–63). Additionally, EBV infection has been strongly linked to MS disease initiation (64). While up to 95% of the general population is seropositive for the virus by early adulthood, the risk of MS will be 15 fold greater in the seropositive population than the seronegative cohort (65). Certain sequences of EBV have been hypothesized to share homology with components of the CNS, suggesting that MS autoimmunity may be initiated by molecular mimicry (66, 67).
The MS autoimmune hypothesis is supported by the predominant presence of activated T cells in active plaques (41, 50, 65, 68–70). Myelin-specific T cells are first activated in the peripheral compartment, after which they cross the BBB into the CNS as they gain the expression of the appropriate adhesion molecules and homing receptors (55, 71). Once inside the CNS, T cells are then reactivated by CNS autoantigens presented by CNS-resident APCs, contributing to the clinical disease and demyelination (72, 73).
CD4+ T cells have been the focus of MS autoimmunity for decades, as MHC class II-restricted T cells are preferentially activated by EAE disease induction (38). Self-reactive CD4+ T cells have been shown to recognize proteins of the myelin sheath, including myelin basic protein (MBP) (74, 75), myelin-associated glycoprotein (MAG) (76, 77), and myelin oligodendrocyte glycoprotein (MOG) (78) in both MS patients and healthy donors (69). However, CD4+ T cells in MS patients display an activated or memory phenotype with increased avidity to myelin proteins, compared to naive myelin-specific CD4+ T cells isolated from controls (79–82). Previously, myelin-specific CD4+ T cells in MS were thought to contribute to Th1-mediated inflammation, in contrast to the Th2-mediated response of myelin-reactive T cells isolated from healthy donors (83, 84). However, recent studies have demonstrated the importance of IL-23 in MS. IL-23 is necessary for the regulation of the proinflammatory IL-17-secreting Th17 cell lineage, which have been described as the pathogenic mediators of several autoimmune diseases (85–87). This is supported by the upregulation of IL-17 gene expression in the brain lesions of MS patients as measured by microarray analysis (88, 89). The levels of Th17 cells in the CSF of relapsing MS patients were elevated in comparison to non-inflammatory neurological disease controls, whereas there were no differences in the percentages of IFN-γ-secreting Th1 cells (90). Th17 cells were also raised in the peripheral compartments of MS patients during relapses, implicating their possible relevance to disease activity (91, 92). Th17 cells were additionally demonstrated to home to active regions of lesions and areas of inflammatory demyelination, and are a major constituent of perivascular cuffs (93). The high expression of granzyme B by myelin-specific Th17 cells also promotes the death of human neurons (94). These works combined strongly insinuates Th17 cells as a potential mediator of MS pathogenesis.
The focus of research in MS has recently shifted from a predominantly CD4+ T cell field to include CD8+ cytotoxic T cells as a novel effector cell type in MS pathology (95, 96). CD8+ T cells have been shown to outnumber CD4+ T cells in MS plaques up to 10-fold at all stages of disease progression (47, 97–100). Oligoclonal expansion within the CD8+ T cell compartment is elevated compared to CD4+ T cells in lesions, CSF, and peripheral blood of MS patients (95, 96, 101). In contrast to its constitutive low expression in the CNS, MHC class I is highly upregulated on neurons and glial cells within MS lesions, which proposes that CD8+ T cells may be interacting with these cells (100, 102, 103). A significant number of activated or memory CD8+ T cells are capable of secreting the proinflammatory cytokine, IL-17, similar to the Th17 cells mentioned earlier (93). Histological analysis of MS lesions has also revealed that granzyme B-positive CD8+ T cells are often located adjacent to regions of demyelination (47, 104, 105). Expectedly, the levels of CD8+ T cells within lesions have been positively correlated with the magnitude of axonal injury (106). These findings encourage the hypothesis that CD8+ T cells play a role in the demyelination of axons in MS lesions.
The monoclonal antibody natalizumab is successfully used as an immunosuppressant in diseases such as MS. Natalizumab therapy is administered to MS patients who are unresponsive to first-line immunotherapies, as well as those with severe clinical disease (107). It has shown a 68% reduction in the annualized relapse rate of MS patients and has decreased the probability of sustained disability progression by 42% over the course of 2 years (108). Natalizumab targets the α4β1 integrin on T cells, thereby preventing T cells binding to VCAM1 on endothelial cells of the BBB and subsequent egress into the CNS. This is evidenced by the significant decline of several populations of T cells in the CSF of natalizumab-treated MS patients compared to controls (34). As such, immune surveillance within the CNS is compromised, which can lead to various inherited or acquired immune deficiencies (37). In particular, the use of natalizumab has been associated with opportunistic infections, most significantly JCV infection or reactivation leading to potentially lethal progressive multifocal leukoencephalopathy (inflammation of white matter in the brain) in 4 out of 1,000 treated patients (37).
Based on a review of the literature, it seems apparent that MS is a multifaceted autoimmune disease with potential contributions from Th17 cells and CD8+ T cells in demyelination (Table 1). Although T cell dependency is well established, the quest for potential autoantibodies in MS is still going strong (109). Popularly studied autoantigens in this field include MOG and aquaporin 4 (AQP4), although extensive research into these targets reveal that they are not, in fact, associated with MS (11, 110).
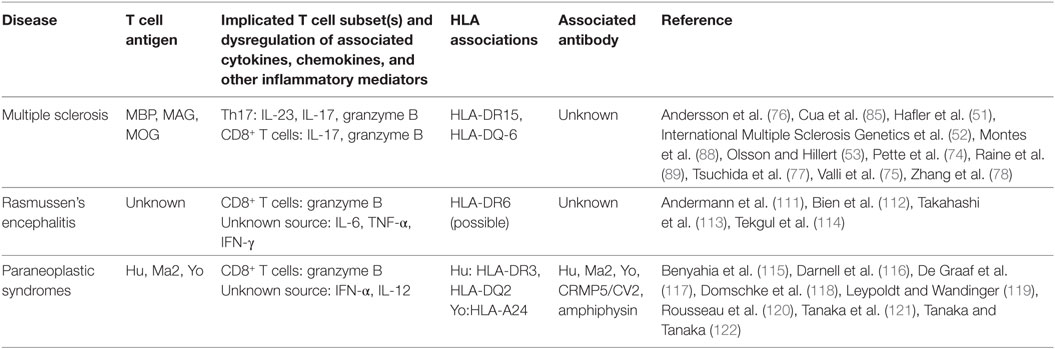
Table 1. Summary of findings of T cell activity in T cell-associated central nervous system diseases.
Rasmussen’s Encephalitis
Rasmussen’s encephalitis is a chronic pediatric inflammatory neurological disorder characterized by drug-resistant focal seizures, unihemispheric inflammation and atrophy, and unilateral movement disorders accompanied by progressive neurological decline (112, 123, 124). Lymphocytic and microglial nodules are commonly observed upon histopathological analysis of RE brain specimens, along with perivascular cuffing of infiltrating T cells, neuron and astrocyte death, and gliosis of the diseased hemisphere (4, 124, 125). RE has not yet been associated with any disease-specific autoantibodies, and the presence of autoantibodies to glutamate receptor 3 is secondary to and not causative of disease (124, 126–129). In fact, RE has been hypothesized to be a T cell-mediated disease based on the dominant influx of CD8+ T cells into active brain lesions at the initiation of disease (4) (Table 1). 7% of these infiltrating CD8+ cells are granzyme B-positive, and vesicles were often found positioned adjacent to MHC class I-expressing neurons and astrocytes with their granules polarized toward their target(s), suggesting a cytotoxic T cell-mediated disease course of RE (112). In addition to increased levels of granzyme B at initial stages of disease (113), Tekgul et al. revealed raised concentrations of the cytokine IL-6 in the CSF of RE patients compared to controls (114). A correlation was then established between the magnitude of neuronal death and inflammation with the level of IL-6 in the CNS of these patients, based on magnetic resonance spectroscopy (114). The overproduction of IL-6 has been attributed to overstimulation of TNF-α early in disease (113). Takahashi et al. have also shown that excessive IFN-γ production during the early stages of disease induces the secretion of IL-12 from macrophages (113).
Analysis of the T cell receptor (TCR) repertoire in the CNS and periphery of RE patients revealed clonal expansions of CD8+ T cells in both compartments, suggesting the presence of an antigen-specific T cell response (130). This is in contrast to normal TCR distribution in stroke patient controls (130). The number of peripheral CD8+ T cell clones has also been shown to correlate with the magnitude of unihemispheric atrophy (131). Although the disease epitope for RE has not yet been elucidated, the identification of a CD8+ T cell-mediated response in this disease expands potential treatment options, as seizures may be refractory and poorly responsive to anti-epileptic drugs (124). As a result, some patients may require invasive procedures such as hemispherectomy to regulate seizure frequency (124). Novel T cell-specific immunotherapies, like T cell blockade from the CNS with natalizumab, are therefore a promising alternative (124, 132, 133).
Paraneoplastic Syndromes
Diseases in which the body’s immune system is altered in response to cancer are termed paraneoplastic syndromes. When paraneoplastic syndromes disturb the CNS, the effects can be far more severe than the initiating tumor, with significant disability taking hold over short periods of time (134). In CNS paraneoplastic syndromes, paraneoplastic antibodies are present at higher titers in CSF versus serum, insinuating that they are synthesized intrathecally (135). These onconeuronal IgG antibodies target intracellular neuronal antigens expressed ectopically by the tumor (136). Paraneoplastic antibodies are important biomarkers of disease, but appear unrelated to pathogenesis (137). Instead, pathogenesis may be mediated by T cells targeting the same autoantigens as the onconeural antibodies present (5) (Table 1). This hypothesis is supported by the presence of disease-specific T cells in the peripheral blood and CSF of patients with anti-Yo (cdr2) (116) and anti-Hu antibodies (115, 120). Extensive T cell infiltration into the CNS in patients with anti-Ma2 (138) and anti-Hu antibody-associated paraneoplastic encephalitis (139) has also been observed, and along with poor responses to humoral immunotherapies (140–142), supports a T cell-mediated pathogenesis of CNS paraneoplastic syndromes.
CD8+ cytotoxic T cells have been implicated in paraneoplastic limbic encephalitis and are associated with autoantibodies against intracellular antigens, mainly Hu (139) and Ma2, as well as CRMP5/CV2 and amphiphysin (5). In comparison to encephalitides with neuronal cell-surface antigen-directed autoantibodies, T cells in anti-Ma2 and anti-Hu paraneoplastic encephalitis are preferentially skewed toward a CD8+ phenotype, with a significantly higher number of activated cytotoxic granzyme B-positive cells found in close proximity to injured neurons (143, 144).
There is a limited number of studies that detail the cytokine profile in paraneoplastic patients. Autoreactive T cells in paraneoplastic breast cancer patients were found in association with elevated intratumoural levels of IFN-α and IL-12, a correlation unseen in antibody-negative breast cancer patients (118). As IL-12 is concomitant with T cell activation and function, the increase of this cytokine likely promotes the expansion of autoreactive T cells in paraneoplastic syndromes (118, 145). These results collectively suggest that paraneoplastic encephalitides are mediated by cytotoxic, antigen-specific CD8+ T cells, in which onconeuronal antibodies may exist as an epiphenomenon.
Amyotrophic Lateral Sclerosis
Amytrophic lateral sclerosis is a neurodegenerative disease of the motor neurons resulting in progressive muscle paralysis. The underlying mechanisms of ALS have not yet been elucidated, and therapies to modify or delay the advancement of the disease are still being trialed. Intriguingly, recent studies have shown that CD4+ T cells infiltrating the spinal cord in ALS patients and mice lie adjacent to degenerating motor neurons and activated microglia (7, 146–148). However, global immunosuppression does not appear to be effective in ALS treatment, suggesting that these T cells may, in fact, rescue motor neuron death (7). There have been several studies investigating the neuroprotective mechanisms of CD4+ T cells following injury (7, 149–152). The data collected thus far suggests that CD4+ T cells in ALS mediate motor neuron survival in a highly regulated process (7).
T Cells in Antibody-Associated CNS Diseases
Neuromyelitis Optica
The most compelling evidence for autoreactive T cell involvement in an autoantibody-associated disease comes from studies in neuromyelitis optica (Table 2). NMO is an aggressive demyelinating disease that is distinguished from MS by the presence of specific IgG1 antibodies against AQP4 (153, 154), a water channel abundantly expressed by astrocytes in the CNS. Anti-AQP4 antibodies are detected in a significant proportion (up to 75%) of NMO patients (110, 155) and have become an important diagnostic tool. However, involvement of other immune mechanisms has been theorized as several lines of evidence, while inconclusive, indicate that anti-AQP4 antibodies alone do not induce complete pathogenesis. For example, there are incongruences in NMO induction in animal models by passive transfer of anti-AQP4 IgG alone (156–158), and high titers of anti-AQP4 antibodies were detected in humans during remission (159, 160). Furthermore, B cell-targeted immunotherapies do not always ameliorate the disease (161, 162).
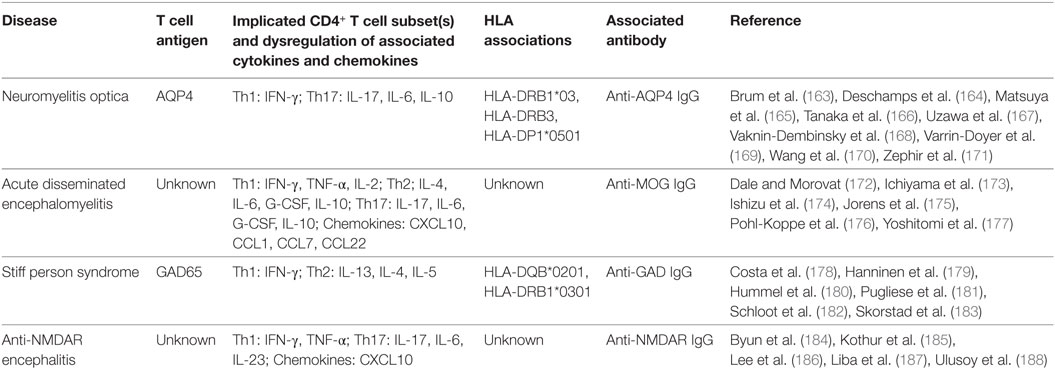
Table 2. Summary of findings of T cell activity in antibody-associated central nervous system diseases.
Since the early description of CD3+ T cells in active NMO lesions (189), there is mounting evidence of cellular involvement in NMO. In fact, activated T cells infiltrate NMO patient-derived lesions (190) and clonal expansion of T cells was reported (191). Efforts have been made to define the immunodominant epitope by identifying which peptide from a human AQP4 (hAQP4) peptide library induced the greatest T cell proliferation when cultured with peripheral blood mononuclear cells (PBMCs) from anti-AQP4 antibody-positive NMO patients compared to MS subjects and healthy controls (165, 168, 169). However, further studies are required to precisely define the dominant target region as these epitopes differed greatly between studies. This discrepancy could be due to different populations with varied HLA associations, or different stages of the disease in which subjects were sampled. Indeed, a longitudinal analysis of NMO patients has demonstrated a change in reactivity and specificity of T cells toward the hAQP4 peptides over time (168). Relapses in the disease were associated with elevated CD69+ activated T cells compared to remission (165), highlighting the possible intermittent role of T cells during an NMO attack, and thus emphasizing the importance of understanding the T cell response to monitor the disease course.
Cytokine profiling helps elucidate the functional properties of AQP4-specific T cells and has revealed that these cells exhibit predominantly a Th17 bias but also a Th1 response. Compared to MS patients or healthy controls, increased secretion of IL-17, IL-10, IL-6, and IFN-γ have been reported in the CSF (166, 167), peripheral blood (168, 169, 192), and epitope-specific T cell lines derived from NMO patients (168). Secretion of IL-17 from Th17-biased AQP4-specific T cells promoted neutrophil infiltration, which was consistent with pathological findings (169, 189). In particular, elevated IL-6, a cytokine important for Th17 differentiation, may promote survival of AQP4-specific Th17 cells while suppressing FOXP3+ Treg function (193–195). Furthermore, tocilizumab, a monoclonal antibody against IL-6 receptor, ameliorated the disease in NMO patients unresponsive to standard immunotherapy (196, 197).
Genetics may be a determinant of autoimmunity and indeed, there appears to be a HLA haplotype association in NMO. Depending on the ethnicity of the cohort, there is an over-representation of HLA-DRB1*03, HLA-DRB3, or HLA-DPB1*0501 in anti-AQP4 antibody-positive NMO patients (163, 164, 169–171, 198). Interestingly, Varrin-Doyer et al. demonstrated that the hAQP4 epitope they identified induced the highest T cell reactivity in NMO patients that were HLA-DR carriers (169). However, there needs to be more definitive analysis as a distinct HLA allele could not be determined based on the T cell response to a different set of AQP4 epitopes (165).
While the triggers of autoimmunity remain elusive, like in many other autoimmune diseases, molecular mimicry has been implicated in the generation of AQP4-specific T cells. In addition to proposing AQP4-specific T cell epitopes, Varrin-Doyer et al. revealed a 90% homology between the immunodominant AQP4 epitope and Clostridium perfringens adenosine triphosphate-binding cassette transporter permease, and a 60–70% homology to other commensal and pathogenic Clostridium species (169). Not only could these microbes serve to display cross-reactive determinants, the Clostridium species may also augment a Th17-biased response as demonstrated in mice (169, 199). Nevertheless, further investigations into molecular mimicry are required to ascertain the extent of its contribution to the development of AQP4-specific T cells.
Acute Disseminated Encephalomyelitis
Acute disseminated encephalomyelitis is a monophasic inflammatory demyelinating disease predominantly affecting children. It can have postinfectious origins but in a subset of patients (27–47%) (200), extensive evidence implicates pathogenic autoantibodies against MOG, a protein on the outer surface of the myelin sheath (201–206). Interestingly, findings predating the discovery of anti-MOG antibodies in ADEM (205) provide support for an autoimmune T cell response.
The majority of literature supporting T cell involvement in ADEM stems indirectly from analyses of chemokines and cytokines (Table 2). Concurrent recruitment of Th1 and Th2 cells has been proposed as there was an increase in their signature chemokines, CXCL10, CCL1, CCL7, and CCL22 in the CSF of adults with ADEM compared to MS and healthy controls and was correlated with an increase in pleocytosis (207). Dysregulation in cytokine production was not distinguished in adults, but IFN-γ, TNF-α, IL-2, IL-10, IL-6, and G-CSF were upregulated in separate pediatric ADEM cohorts (172–176), further supporting the contribution of Th1 and Th2 cells. Pohl-Koppe et al. hypothesized that Th1 cells contribute to the deleterious effects of the disease, while Th2 cells predominate in the recovery of ADEM as they reported an absence of IFN-γ but an increase in IL-4 in patients during the recovery phase (176). Consistent with this, there was increased IFN-γ+CD3+ T cells in the peripheral blood during the acute stage of ADEM (177).
Conversely, as IL-6, G-CSF, and IL-10 are pleiotropic, their elevation along with IL-17A, but little Th1 and no Th2 cytokines, in the CSF of anti-MOG antibody-positive children favors a Th17 phenotype (208). Interestingly, this increase in Th17 cytokines correlated with an increase in B cell-associated cytokines and chemokines, suggesting possible interactions between multiple cell types in mediating demyelination (208). Likewise, CSF IL-6 levels correlated with the presence of plasma anti-MOG antibodies in acquired demyelinating syndromes like ADEM (209). It can then be proposed that, like in NMO, IL-6 signaling is a suitable target for treatment in anti-MOG antibody-positive patients resistant to conventional immunotherapy (196, 197). These preliminary, albeit conflicting, reports of functional helper T cells warrant investigations into autoreactive T cells themselves, but also in combination with the recent developments in anti-MOG antibodies to assess the interplay between the humoral and cellular components of the autoimmune response in ADEM (201, 204, 210).
Stiff Person Syndrome and Other Anti-Glutamic Acid Decarboxylase Glutamic Acid Decarboxylase (GAD) Antibody-Associated Neurological Disorders
Markedly high titers of autoantibodies against glutamic acid decarboxylase (GAD) are a hallmark of non-paraneoplastic SPS and variants of cerebellar ataxia, limbic encephalitis, and epilepsy (211–214). As GAD is an enzyme involved in the synthesis of the inhibitory neurotransmitter γ–aminobutyric (GABA), the current hypothesis is that anti-GAD antibodies disrupt GABAergic signaling. Indeed, in vitro and in vivo studies demonstrate the potential pathogenicity of anti-GAD antibodies (215–217). The salient question remains, however, of the mechanism underlying autoantibody recognition of a cytoplasmic antigen like GAD, which is unlike other known extracellular antigens targeted by pathogenic autoantibodies (218).
Given the variety of anti-GAD antibody-associated neurological disorders, it is plausible that antigen-specific T cells play an additional role in pathogenesis that differentiates the diseases (Table 2). This is an important aspect for investigation but there is a paucity of studies examining cellular mechanisms despite reports of CNS infiltration of lymphocytes in these patients (219). In a study comparing SPS with cerebellar ataxia associated with polyendocrine autoimmunity (CAPA), both cohorts presented with high titers of anti-GAD antibodies (178). Yet, cell proliferation and the percentage of HLADR+CD3+ activated T cells in response to GAD65 protein was significantly greater in SPS but not CAPA. Monitoring the course of SPS revealed a constant reactivity of CD4+ T cells against GAD65, which notably correlated with high anti-GAD antibody titers (179). A few other groups have identified GAD65-specific T cells in the blood but these were weakly responsive to GAD65 (180, 182, 220, 221). To this end, Skorstad et al. argue that GAD65-specific T cells largely reside in the CNS along with B cells to collaborate in the intrathecal production of anti-GAD antibodies as they were more successful in identifying and cloning GAD65-specific T cells from CSF than from blood (183). Furthermore, using overlapping GAD65 peptides, putative T cell epitopes have been identified but differ between studies and depending on whether T cell lines were generated from the blood or CSF (179, 182, 183, 220). As has been demonstrated with anti-GAD65 antibody epitopes (222) [recently reviewed in Ref. (223)], differences in T cell epitopes have been shown to be a distinguishing factor between SPS and type 1 diabetes, another anti-GAD antibody-associated disease (220, 221).
Exploration of the cytokine environment to determine the phenotype of T cells has indicated largely a Th2 bias. Secretion of IL-13, IL-4, and IL-5 reported in SPS (179, 182, 183) supports a non-inflammatory environment wherein disease is driven by autoantibodies. IFN-γ production, indicative of a Th1 response, was also recorded (182, 183) and subsequently reduced upon treatment with immunotherapy, and coincided with clinical improvement (180). It was proposed that high levels of IFN-γ production occurs in the early phase of SPS but is later exceeded by significant production of IL-13, alluding to a shift from Th1 to Th2 (179). While low or undetectable in SPS, there was a notable production of IFN-γ, and hence a dominant Th1 response is observed in CAPA (178) and type 1 diabetes (220).
T cell involvement is further supported by preliminary findings on HLA allele correlations in SPS. Pugliese et al. described a strong association between SPS and carriers of HLA-DQB1*0201 haplotype (181). HLA-DRB1*0301 has additionally been proposed as a correlate of SPS but the validity of this finding is hampered by small sample size (178, 179).
Anti-NMDAR Encephalitis
Anti-NMDAR encephalitis is the prototypic autoimmune encephalitis associated with autoantibodies against cell-surface antigens. Discovery of the specific anti-NMDAR antibody (224) has sparked considerable interest in humoral mechanisms of this disease [recently reviewed in Ref. (225)], leading to the current hypothesis that anti-NMDAR antibodies exhibit pathogenic effects via internalization of the surface receptor, thereby resulting in reversible NMDAR hypofunction (226–228).
To date, there are limited and small studies investigating cellular responses in anti-NMDAR encephalitis but nevertheless, prompt further exploration (Table 2). Evidence of T cell involvement derives from cytokine and chemokine profiling and mainly favors a Th17 response. Based on significantly elevated serum levels of IL-17 and IL-6 in anti-NMDAR antibody-positive patients compared to controls (184), Byun et al. hypothesized that undetected Th17 cells secrete IL-17, which promotes a positive feedback loop of IL-6 signaling that facilitates intrathecal antibody production observed in most patients (224, 229). In line with this finding, targeting the IL-6 receptor with tocilizumab in rituximab-resistant patients with suspected autoimmune encephalitis demonstrated marked improvements (186), as was also seen in NMO. Upregulation of serum IL-23 strengthens the case for Th17 activity (188). There appears to be some heterogeneity in T cell lineage as higher levels IFN-γ and TNF-α were observed in the CSF, indicative of a Th1 cytokine dysregulation (185, 187). Consistent with T cell involvement in anti-NMDAR encephalitis was the increased level of T cell-related chemokine CXCL10 in patient CSF, which correlated with CSF pleocytosis (187).
On the other hand, there is contentious evidence of T cell involvement. Immunopathological analysis of brain sections from anti-NMDAR encephalitis patients show some to no evidence of T cell infiltration in the parenchyma and perivascular space, which disqualifies CD8+ cytotoxic T cells as drivers of the disease (143, 230–232). However, this does not preclude the possibility of NMDAR-specific T cell involvement in B cell activation in the periphery, prior to anti-NMDAR antibodies trafficking to the CNS.
Evaluating T Cell Detection Methods
Choice of methods is a key determinant in discovering and studying T cell biology. An important consideration is the rarity of antigen-specific T cells, especially the proportion reactive against auto-antigens which is typically less than 0.01% of the total T cell repertoire (233). It is therefore imperative that sensitive yet specific techniques are implemented when analyzing antigen-specific T cells. Widely used T cell detection methods can be broadly distinguished into two categories: techniques that identify and assess specificity and assays that examine the functionality (Table 3).
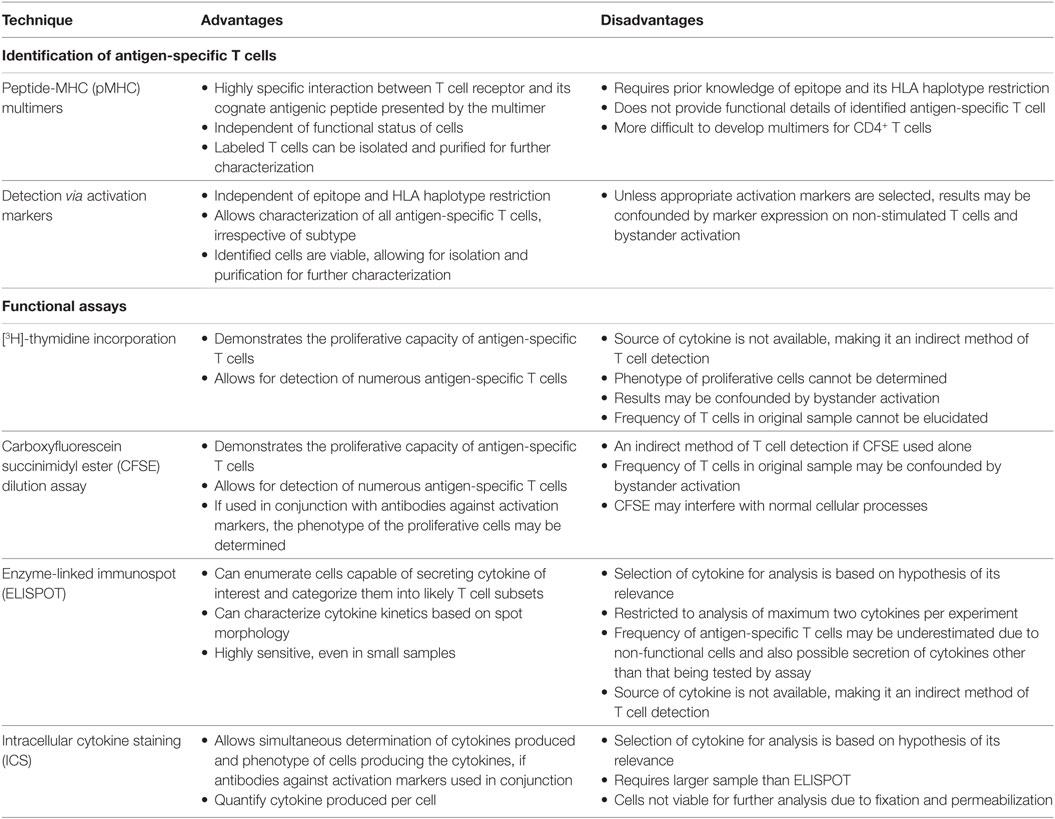
Table 3. Evaluation of major techniques used in the analysis of human antigen-specific T cells.
An important tool for identification of antigen-specific T cells is peptide-MHC (pMHC) multimers. This rapidly evolving technology involves the formation of a complex of peptide-loaded MHC monomers via biotinylation with a fluorescently labeled streptavidin (Figure 1), which increases binding avidity (234) and overcomes the issue of low affinity binding and fast dissociation rate between TCRs and pMHC monomers (235). The value of this method lies in the direct and specific recognition and isolation of T cells via flow cytometry that is independent of their biological activity, such as anergic cells that are incapable of proliferation and cytokine production (236–239). However, this is a double-edged sword, as knowledge of the functional characteristics of the identified T cells allows for a deeper understanding of their response. In addition, a major drawback of this approach is that it necessitates knowledge of the T cell epitope and its MHC haplotype association (236). While pMHC multimers are extensively used for study of CD8+ T cells, their use in the study of MHC class II-restricted CD4+ T cells is challenged by the lower frequency of antigen-specific CD4+ T cells in the peripheral blood (240) but also largely by the difficulty in creating them because of variations in MHC structure and TCR affinity (236, 238).
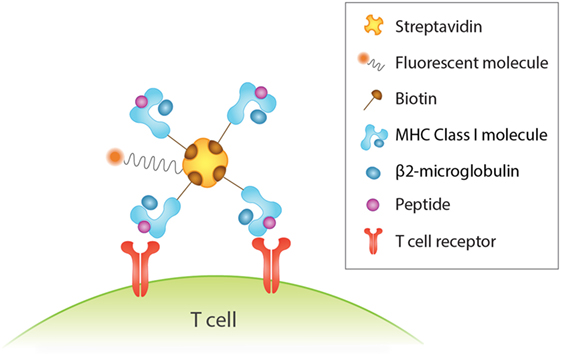
Figure 1. Detection of human antigen-specific T cells with peptide-MHC (pMHC) multimer. Binding four pMHC monomers, for instance, via biotin–streptavidin interactions increases binding avidity between antigen and T cell receptor. This in turn enhances the sensitivity and specificity of antigen-specific T cells detected by flow cytometry analysis via the fluorescent streptavidin.
Alternatively, antigen-specific T cells can be directly identified and isolated for downstream functional characterization by probing for activation markers expressed on the surface of T cells upon antigenic stimulation. A major advantage of this technique is that unlike pMHC multimers, it does not require knowledge of the antigenic epitope and associated MHC haplotypes (236) and is effective for studying CD4+ T cells. A multitude of activation markers have been proposed, including CD25, CD69, CD40L, CD134, CD137, and HLA-DR (241–245). Such markers are favorable indicators of an antigenic-specific response as their surface expression is contingent on activation, but absent or minimally expressed in the resting phase, and occurs for a transient period of time. Moreover, like pMHC multimer staining, expression of some activation markers is irrespective of T cell function and its differentiation state (236, 244), allowing for an unbiased characterization of antigen-specific T cells.
Detection of antigen-specific T cells is a vital step, but determining the functional capacity of the identified cells in producing a robust immune response is equally important and relies on functional assays (Figure 2). Methods measuring [3H]-thymidine incorporation into lymphocyte DNA (246) and dilution of carboxyfluorescein succinimidyl ester (CFSE) dye bound to amine groups of intracellular molecules (247) during cell division have been a mainstay for evaluating the proliferation of lymphocytes in response to antigens. These procedures circumvent the issue of low frequency of target cells. However, as it is often PBMCs that are cultured, and not sorted T cells, there is the possibility of bystander activation, which decreases the specificity of the response observed and the frequency of the antigen-specific T cells cannot be accurately extrapolated. In the case of [3H]-thymidine incorporation assay, the proliferative cell subpopulation cannot be phenotyped, whereas with CFSE dilution assay, cell subpopulations may be delineated with surface markers and flow cytometry analysis. However, the dye can interfere with expression of activation markers (248).
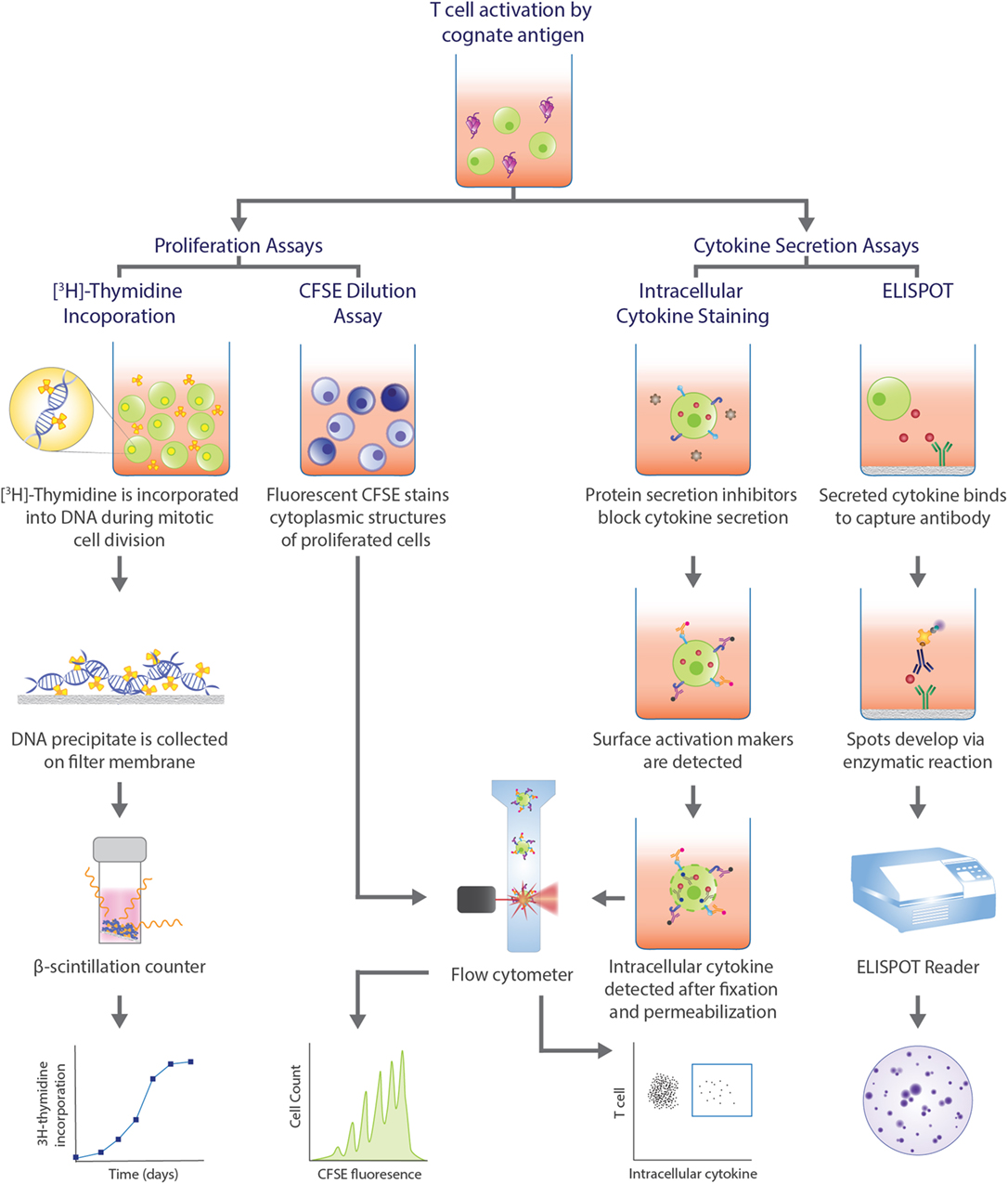
Figure 2. Functional assays commonly used in human T cell studies. Functional assays can be categorized into those that assess proliferative capacity of antigen-specific T cells and assays that analyze cytokine profiles upon T cell recognition of cognate antigen and subsequent activation. Proliferation assays can measure the amount of radioactive [3H]-thymidine incorporated into the DNA during cell division, with greater radioactivity indicating greater cell division. Alternatively, the level of fluorescence emitted by cells stained with carboxyfluorescein succinimidyl ester (CFSE) can be detected by flow cytometry, with greater number divisions correlating with lower fluorescence. In intracellular cytokine staining (ICS), protein secretion inhibitors, such as brefeldin A or monensin, allows for examination of cytokine production within a cell. Staining surface activation markers allows for phenotyping. Following fixation and permeabilization, the trapped intracellular cytokines are stained with fluorescent antibodies which can be detected via flow cytometry. Enzyme-linked immunospot (ELISPOT) is a popular method to assess cytokine secretion. The cytokine of interest secreted from an activated T cell is bound to a capture antibody on a PVDF bottom well. A biotinylated detection antibody also binds to the cytokine and facilitates the interaction between streptavidin-conjugated enzyme and its substrate to produce a color spot. Spots are quantified with a ELISPOT plate reader. Each spot represents one reactive cell.
Functional assays that examine cytokine production allows for classification of the cells into different subsets that are distinguished by different effector functions. This is particularly valuable for CD4+ T cells that can be categorized as Th1, Th2, and Th17 cells, for example. Two classic procedures that assess cytokine production are enzyme-linked immunospot (ELISPOT) and intracellular cytokine staining (ICS) (Figure 2). While an ELISPOT detects secreted cytokines induced by antigens, ICS reveals cytokine production within the golgi/ER bodies upon permeabilization of the cell and treatment with protein secretion inhibitors, such as brefeldin A or monensin. Both techniques are sensitive for detection of antigen-specific T cells and allow for enumeration and characterization at a single-cell level. However, as both techniques also depend on postulating cytokines relevant for the disease, it is possible that the frequency of cytokine-producing cells is underestimated if cells secrete cytokines other than the one of interest. Additionally, the breadth of cytokine analysis is limited in an ELISPOT to only two cytokines for a given experiment (249). The source of the cytokine cannot be determined with an ELISPOT, making it an indirect method of T cell identification. Conversely, ICS allows for the simultaneous detection of cytokines and the phenotype of the cytokine-producing cells by the addition of activation marker fluorescent antibodies. Hence, pairing with pMHC multimer or activation marker staining enhances the specificity of the reaction observed and provides a complete assessment of the antigen-specific response.
Pre-enrichment is a modification often made to the above methods to overcome the issue of low target cells and further improve the sensitivity offered by flow cytometry. A common approach is in vitro expansion, wherein PBMCs are cultured in the presence of the antigen over one to two weeks to preferentially grow antigen-specific T cells. However, similar to proliferation assays, a drawback of this procedure is the risk of activating non-T cells present in the sample, thereby increasing background and reducing the accuracy of the quoted frequency of antigen-specific T cells (236). Alternatively, enriching the target cell population via magnetic separation greatly increases sensitivity and provides insight into previously unidentified T cell subpopulations, provided that highly specific markers were utilized, whether that be pMHC multimers or activation markers (236, 238).
Employing the right technique may lead to discovery of T cells in disease. Each method explores different aspects of T cell biology. Taking this into consideration and the rarity of antigen-specific T cells, it is advantageous to integrate the various approaches for a more reliable and holistic understanding of the cellular mechanisms at play.
Future Directions
Cellular immunity is a key player in the autoimmune response, as evidenced by the growing number of studies in both T cell-mediated and antibody-associated CNS disorders. Yet, there is much to be learnt of T cell contribution to the complexities of CNS autoimmunity. Investigating the underlying cellular mechanisms can deepen our understanding of disease pathogenesis, especially in the expanding range of neurological diseases recently associated with antibodies, in patients seronegative for antibodies but suspected to have immune dysregulation, in differentiating clinically similar diseases with heterogeneous pathology, and in conditions currently classified as idiopathic. Importantly, this new found knowledge can lead to the development of improved diagnostic tools and also translate into novel immunotherapeutics that are more targeted against T cells or their cytokines, like tocilizumab and IL-17-directed secukinumab (250), which can be more effective than the current treatment regime given the unique environment of the CNS.
Author Contributions
DP and AZ performed the literature review, wrote the manuscript, and contributed equally to the work. DP and FT designed the figures. FB conceived the concept, critically revised, and oversaw the process of manuscript preparation. All authors read, edited, and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank remaining members of the Brain Autoimmunity Group for their valuable comments on the manuscript.
Funding
The authors received funding from the Australian National Health and Medical Research Council (NHMRC), Multiple Sclerosis Research Australia, Star Scientific Foundation, Tourette Syndrome Association (USA), and the Petre Foundation (AU).
References
1. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol (2009) 9(6):393–407. doi: 10.1038/nri2550
2. Compston A, Coles A. Multiple sclerosis. Lancet (2008) 372(9648):1502–17. doi:10.1016/S0140-6736(08)61620-7
3. Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol (2015) 14(4):406–19. doi:10.1016/S1474-4422(14)70305-9
4. Farrell MA, Droogan O, Secor DL, Poukens V, Quinn B, Vinters HV. Chronic encephalitis associated with epilepsy: immunohistochemical and ultrastructural studies. Acta Neuropathol (1995) 89(4):313–21. doi:10.1007/BF00309624
5. Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol (2008) 7(4):327–40. doi:10.1016/S1474-4422(08)70060-7
6. Melzer N, Meuth SG, Wiendl H. Paraneoplastic and non-paraneoplastic autoimmunity to neurons in the central nervous system. J Neurol (2013) 260(5):1215–33. doi:10.1007/s00415-012-6657-5
7. Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci (2004) 24(18):4333–9. doi:10.1523/JNEUROSCI.5276-03.2004
8. Dalmau J, Rosenfeld MR. Autoimmune encephalitis update. Neuro Oncol (2014) 16(6):771–8. doi:10.1093/neuonc/nou030
9. Hoftberger R. Neuroimmunology: an expanding frontier in autoimmunity. Front Immunol (2015) 6:206. doi:10.3389/fimmu.2015.00206
10. Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol (2014) 76(2):168–84. doi:10.1002/ana.24200
11. Ramanathan S, Dale RC, Brilot F. Anti-MOG antibody: the history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun Rev (2016) 15(4):307–24. doi:10.1016/j.autrev.2015.12.004
12. Ramanathan S, Mohammad SS, Brilot F, Dale RC. Autoimmune encephalitis: recent updates and emerging challenges. J Clin Neurosci (2014) 21(5):722–30. doi:10.1016/j.jocn.2013.07.017
13. Sinmaz N, Amatoury M, Merheb V, Ramanathan S, Dale RC, Brilot F. Autoantibodies in movement and psychiatric disorders: updated concepts in detection methods, pathogenicity, and CNS entry. Ann N Y Acad Sci (2015) 1351:22–38. doi:10.1111/nyas.12764
14. van Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PA. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev (2014) 13(3):299–312. doi:10.1016/j.autrev.2013.10.016
15. Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry (2012) 83(6):638–45. doi:10.1136/jnnp-2011-301237
16. Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol (2015) 15(3):185–9. doi:10.1038/nri3803
17. Mitchison NA. T-cell–B-cell cooperation. Nat Rev Immunol (2004) 4(4):308–12. doi:10.1038/nri1334
18. Toellner KM, Luther SA, Sze DM, Choy RK, Taylor DR, MacLennan IC, et al. T helper 1 (Th1) and Th2 characteristics start to develop during T cell priming and are associated with an immediate ability to induce immunoglobulin class switching. J Exp Med (1998) 187(8):1193–204. doi:10.1084/jem.187.8.1193
19. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol (2016) 34:335–68. doi:10.1146/annurev-immunol-041015-055605
20. McPherson RC, Anderton SM. Adaptive immune responses in CNS autoimmune disease: mechanisms and therapeutic opportunities. J Neuroimmune Pharmacol (2013) 8(4):774–90. doi:10.1007/s11481-013-9453-9
21. Petermann F, Korn T. Cytokines and effector T cell subsets causing autoimmune CNS disease. FEBS Lett (2011) 585(23):3747–57. doi:10.1016/j.febslet.2011.03.064
22. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine (2015) 74(1):5–17. doi:10.1016/j.cyto.2014.09.011
23. Sie C, Korn T, Mitsdoerffer M. Th17 cells in central nervous system autoimmunity. Exp Neurol (2014) 262(Pt A):18–27. doi:10.1016/j.expneurol.2014.03.009
24. Kivisakk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T, et al. Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci U S A (2003) 100(14):8389–94. doi:10.1073/pnas.1433000100
25. Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol (2003) 3(7):569–81. doi:10.1038/nri1130
26. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401(6754):708–12. doi:10.1038/44385
27. Svenningsson A, Andersen O, Edsbagge M, Stemme S. Lymphocyte phenotype and subset distribution in normal cerebrospinal fluid. J Neuroimmunol (1995) 63(1):39–46. doi:10.1016/0165-5728(95)00126-3
28. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12(9):623–35. doi:10.1038/nri3265
29. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med (2015) 212(7):991–9. doi:10.1084/jem.20142290
30. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523(7560):337–41. doi:10.1038/nature14432
31. Carrithers MD, Visintin I, Kang SJ, Janeway CA Jr. Differential adhesion molecule requirements for immune surveillance and inflammatory recruitment. Brain (2000) 123(Pt 6):1092–101. doi:10.1093/brain/123.6.1092
32. Khatri BO, Man S, Giovannoni G, Koo AP, Lee JC, Tucky B, et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology (2009) 72(5):402–9. doi:10.1212/01.wnl.0000341766.59028.9d
33. Man S, Tucky B, Bagheri N, Li X, Kochar R, Ransohoff RM. Alpha4 integrin/FN-CS1 mediated leukocyte adhesion to brain microvascular endothelial cells under flow conditions. J Neuroimmunol (2009) 210(1–2):92–9. doi:10.1016/j.jneuroim.2009.03.008
34. Stuve O, Marra CM, Jerome KR, Cook L, Cravens PD, Cepok S, et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol (2006) 59(5):743–7. doi:10.1002/ana.20858
35. Ellwardt E, Walsh JT, Kipnis J, Zipp F. Understanding the role of T cells in CNS homeostasis. Trends Immunol (2016) 37(2):154–65. doi:10.1016/j.it.2015.12.008
36. Kappos L, Bates D, Edan G, Eraksoy M, Garcia-Merino A, Grigoriadis N, et al. Natalizumab treatment for multiple sclerosis: updated recommendations for patient selection and monitoring. Lancet Neurol (2011) 10(8):745–58. doi:10.1016/S1474-4422(11)70149-1
37. Warnke C, Olsson T, Hartung HP. PML: the dark side of immunotherapy in multiple sclerosis. Trends Pharmacol Sci (2015) 36(12):799–801. doi:10.1016/j.tips.2015.09.006
38. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol (2015) 15(9):545–58. doi:10.1038/nri3871
39. Polman CH, Reingold SC, Edan G, Filippi M, Hartung H-P, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol (2005) 58(6):840–6. doi:10.1002/ana.20703
40. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology (1996) 46(4):907. doi:10.1212/WNL.46.4.907
41. Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat Rev Immunol (2009) 9(6):440–7. doi:10.1038/nri2548
42. Weinshenker BG, Bass B, Rice GP, Noseworthy J, Carriere W, Baskerville J, et al. The natural history of multiple sclerosis: a geographically based study. 2. Predictive value of the early clinical course. Brain (1989) 112(Pt 6):1419–28. doi:10.1093/brain/112.6.1419
43. BØ L, Vedeler CA, Nyland HI, Trapp BD, MØRk SJ. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol (2003) 62(7):723–32. doi:10.1093/jnen/62.7.723
44. Calabrese M, De Stefano N, Atzori M, Bernardi V, Mattisi I, Barachino L, et al. Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch Neurol (2007) 64(10):1416–22. doi:10.1001/archneur.64.10.1416
45. Fisher E, Lee JC, Nakamura K, Rudick RA. Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann Neurol (2008) 64(3):255–65. doi:10.1002/ana.21436
46. Huseby ES, Kamimura D, Arima Y, Parello CS, Sasaki K, Murakami M. Role of T cell-glial cell interactions in creating and amplifying central nervous system inflammation and multiple sclerosis disease symptoms. Front Cell Neurosci (2015) 9:295. doi:10.3389/fncel.2015.00295
47. Lassmann H, Bruck W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol (2007) 17(2):210–8. doi:10.1111/j.1750-3639.2007.00064.x
48. Ontaneda D, Hyland M, Cohen JA. Multiple sclerosis: new insights in pathogenesis and novel therapeutics. Annu Rev Med (2012) 63(1):389–404. doi:10.1146/annurev-med-042910-135833
49. Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol (2001) 50(3):389. doi:10.1002/ana.1123
50. Martin R, McFarland HF. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol (2007) 8(9):913–9. doi:10.1038/ni1507
51. Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med (2007) 357(9):851–62. doi:10.1056/NEJMoa073493
52. International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature (2011) 476(7359):214–9. doi:10.1038/nature10251
53. Olsson T, Hillert J. The genetics of multiple sclerosis and its experimental models. Curr Opin Neurol (2008) 21(3):255–60. doi:10.1097/WCO.0b013e3282fd10cc
54. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell (2014) 157(5):1073–87. doi:10.1016/j.cell.2014.03.047
55. Huseby ES, Huseby PG, Shah S, Smith R, Stadinski BD. Pathogenic CD8 T cells in multiple sclerosis and its experimental models. Front Immunol (2012) 3:64. doi:10.3389/fimmu.2012.00064
56. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol (2017) 13(1):25–36. doi:10.1038/nrneurol.2016.187
57. van der Mei I, Lucas RM, Taylor BV, Valery PC, Dwyer T, Kilpatrick TJ, et al. Population attributable fractions and joint effects of key risk factors for multiple sclerosis. Mult Scler (2016) 22(4):461–9. doi:10.1177/1352458515594040
58. Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA (2006) 296(23):2832–8. doi:10.1001/jama.296.23.2832
59. Munger KL, Zhang SM, O’Reilly E, Hernan MA, Olek MJ, Willett WC, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology (2004) 62(1):60–5. doi:10.1212/01.WNL.0000101723.79681.38
60. Mowry EM, Krupp LB, Milazzo M, Chabas D, Strober JB, Belman AL, et al. Vitamin D status is associated with relapse rate in pediatric-onset multiple sclerosis. Ann Neurol (2010) 67(5):618–24. doi:10.1002/ana.21972
61. Mowry EM, Waubant E, McCulloch CE, Okuda DT, Evangelista AA, Lincoln RR, et al. Vitamin D status predicts new brain magnetic resonance imaging activity in multiple sclerosis. Ann Neurol (2012) 72(2):234–40. doi:10.1002/ana.23591
62. Runia TF, Hop WC, de Rijke YB, Buljevac D, Hintzen RQ. Lower serum vitamin D levels are associated with a higher relapse risk in multiple sclerosis. Neurology (2012) 79(3):261–6. doi:10.1212/WNL.0b013e31825fdec7
63. Simpson S Jr, Taylor B, Blizzard L, Ponsonby AL, Pittas F, Tremlett H, et al. Higher 25-hydroxyvitamin D is associated with lower relapse risk in multiple sclerosis. Ann Neurol (2010) 68(2):193–203. doi:10.1002/ana.22043
64. Ascherio A. Environmental factors in multiple sclerosis. Expert Rev Neurother (2013) 13(12 Suppl):3–9. doi:10.1586/14737175.2013.865866
65. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol (2007) 61(4):288–99. doi:10.1002/ana.21117
66. Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol (2012) 42(1):102–11. doi:10.1007/s12016-011-8294-7
67. Wandinger K, Jabs W, Siekhaus A, Bubel S, Trillenberg P, Wagner H, et al. Association between clinical disease activity and Epstein-Barr virus reactivation in MS. Neurology (2000) 55(2):178–84. doi:10.1212/WNL.55.2.178
68. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med (2007) 204(12):2899–912. doi:10.1084/jem.20071030
69. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol (2005) 23(1):683–747. doi:10.1146/annurev.immunol.23.021704.115707
70. Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, et al. Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain (2009) 132(12):3318–28. doi:10.1093/brain/awp200
71. Kidd G, Kivisäkk P, Ransohoff RM. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol (2003) 3(7):569–81. doi:10.1038/nri1130
72. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev (2006) 213(1):48–65. doi:10.1111/j.1600-065X.2006.00441.x
73. González H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflammation (2014) 11(1):201. doi:10.1186/s12974-014-0201-8
74. Pette M, Fujita K, Wilkinson D, Altmann DM, Trowsdale J, Giegerich G, et al. Myelin autoreactivity in multiple sclerosis: recognition of myelin basic protein in the context of HLA-DR2 products by T lymphocytes of multiple-sclerosis patients and healthy donors. Proc Natl Acad Sci U S A (1990) 87(20):7968–72. doi:10.1073/pnas.87.20.7968
75. Valli A, Sette A, Kappos L, Oseroff C, Sidney J, Miescher G, et al. Binding of myelin basic protein peptides to human histocompatibility leukocyte antigen class II molecules and their recognition by T cells from multiple sclerosis patients. J Clin Invest (1993) 91(2):616–28. doi:10.1172/JCI116242
76. Andersson M, Yu M, Söderström M, Weerth S, Baig S, Linington C, et al. Multiple MAG peptides are recognized by circulating T and B lymphocytes in polyneuropathy and multiple sclerosis. Eur J Neurol (2002) 9(3):243–51. doi:10.1046/j.1468-1331.2002.00391.x
77. Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci U S A (1994) 91(23):10859–63. doi:10.1073/pnas.91.23.10859
78. Zhang J, Markovic-Plese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med (1994) 179(3):973–84. doi:10.1084/jem.179.3.973
79. Bielekova B, Sung M-H, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol (2004) 172(6):3893. doi:10.4049/jimmunol.172.6.3893
80. Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. A marker of activated/memory T cells. J Clin Invest (1998) 101(4):725–30. doi:10.1172/JCI1528
81. Scholz C, Patton KT, Anderson DE, Freeman GJ, Hafler DA. Expansion of autoreactive T cells in multiple sclerosis is independent of exogenous B7 costimulation. J Immunol (1998) 160(3):1532.
82. Zang YCQ, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, et al. Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol (2004) 172(8):5120. doi:10.4049/jimmunol.172.8.5120
83. Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, et al. High prevalence of autoreactive, neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood (2004) 103(11):4222–31. doi:10.1182/blood-2003-11-4025
84. Frohman EM, Racke MK, Raine CS. Multiple sclerosis – the plaque and its pathogenesis. N Engl J Med (2006) 354(9):942–55. doi:10.1056/NEJMra052130
85. Cua DJ, Wiekowski M, Sedgwick JD, Chen Y, Murphy CA, Kastelein RA, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421(6924):744–8. doi:10.1038/nature01355
86. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi:10.1084/jem.20041257
87. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441(7090):231–4. doi:10.1038/nature04754
88. Montes M, Zhang X, Berthelot L, Laplaud D-A, Brouard S, Jin J, et al. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin Immunol (2009) 130(2):133–44. doi:10.1016/j.clim.2008.08.030
89. Raine CS, Strober S, Oksenberg JR, Klonowski P, Langer-Gould A, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med (2002) 8(5):500–8. doi:10.1038/nm0502-500
90. Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain (2009) 132(12):3329–41. doi:10.1093/brain/awp289
91. Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol (2009) 65(5):499–509. doi:10.1002/ana.21652
92. Matusevicius D, Kivisäkk P, He B, Kostulas N, Özenci V, Fredrikson S, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler (1999) 5(2):101–4. doi:10.1177/135245859900500206
93. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol (2008) 172(1):146–55. doi:10.2353/ajpath.2008.070690
94. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med (2007) 13(10):1173–5. doi:10.1038/nm1651
95. Friese MA, Fugger L. Autoreactive CD8+ T cells in multiple sclerosis: a new target for therapy? Brain (2005) 128(Pt 8):1747–63. doi:10.1093/brain/awh578
96. Goverman J, Perchellet A, Huseby ES. The role of CD8(+) T cells in multiple sclerosis and its animal models. Curr Drug Targets Inflamm Allergy (2005) 4(2):239. doi:10.2174/1568010053586264
97. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med (2000) 192(3):393–404. doi:10.1084/jem.192.3.393
98. Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci (1983) 62(1):219–32. doi:10.1016/0022-510X(83)90201-0
99. Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol (1986) 19(6):578–87. doi:10.1002/ana.410190610
100. Traugott U, Reinherz EL, Raine CS. Multiple sclerosis: distribution of T cell subsets within active chronic lesions. Science (1983) 219(4582):308–10. doi:10.1126/science.6217550
101. Junker A, Ivanidze J, Malotka J, Eiglmeier I, Lassmann H, Wekerle H, et al. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain (2007) 130(11):2789–99. doi:10.1093/brain/awm214
102. Bartlett PF, Wong GHW, Schrader JW, Battye F, Clark-Lewis I. Inducible expression of H-2 and Ia antigens on brain cells. Nature (1984) 310(5979):688–91. doi:10.1038/310688a0
103. Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science (1995) 269(5223):549–52. doi:10.1126/science.7624779
104. Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T Lymphocytes in Autoimmune and Degenerative CNS Diseases. England: Elsevier Ltd (2002). p. 313–9.
105. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, et al. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol (2006) 65(2):124–41. doi:10.1097/01.jnen.0000199572.96472.1c
106. Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Brück W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain (2000) 123(Pt 6):1174–83. doi:10.1093/brain/123.6.1174
107. Warnke C, Menge T, Hartung HP, Racke MK, Cravens PD, Bennett JL, et al. Natalizumab and progressive multifocal leukoencephalopathy: what are the causal factors and can it be avoided? Arch Neurol (2010) 67(8):923–30. doi:10.1001/archneurol.2010.161
108. Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med (2006) 354(9):899–910. doi:10.1056/NEJMoa044397
109. Probstel AK, Sanderson NS, Derfuss T. B cells and autoantibodies in multiple sclerosis. Int J Mol Sci (2015) 16(7):16576–92. doi:10.3390/ijms160716576
110. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med (2005) 202(4):473–7. doi:10.1084/jem.20050304
111. Andermann E, Oguni H, Guttmann RD, Osterland CK, Antel JP, Eeg-Olofisson O, et al. Genetic aspects of chronic encephalitis. In: Andermann F, editor. Chronic Encephalitis and Epilepsy. Boston: Butterworth-Heinemann (1991). p. 167–75.
112. Bien CG, Urbach H, Deckert M, Schramm J, Wiestler OD, Lassmann H, et al. Diagnosis and staging of Rasmussen’s encephalitis by serial MRI and histopathology. Neurology (2002) 58(2):250. doi:10.1212/WNL.58.2.250
113. Takahashi Y, Mine J, Kubota Y, Yamazaki E, Fujiwara T. A substantial number of Rasmussen syndrome patients have increased IgG, CD4+ T cells, TNFalpha, and granzyme B in CSF. Epilepsia (2009) 50(6):1419–31. doi:10.1111/j.1528-1167.2008.01977.x
114. Tekgul H, Polat M, Kitis O, Serdaroglu G, Tosun A, Terlemez S, et al. T-cell subsets and interleukin-6 response in Rasmussen’s encephalitis. Pediatr Neurol (2005) 33(1):39–45. doi:10.1016/j.pediatrneurol.2005.01.007
115. Benyahia B, Liblau R, Merle-Béral H, Tourani JM, Dalmau J, Delattre JY. Cell-mediated autoimmunity in paraneoplastic neurological syndromes with anti-Hu antibodies. Ann Neurol (1999) 45(2):162–7. doi:10.1002/1531-8249(199902)45:23.0.CO;2-R
116. Darnell JC, Albert ML, Darnell RB. Cdr2, a target antigen of naturally occuring human tumor immunity, is widely expressed in gynecological tumors. Cancer Res (2000) 60(8):2136.
117. de Graaf MT, de Beukelaar JW, Haasnoot GW, Levering WH, Rogemond V, Didelot A, et al. HLA-DQ2+ individuals are susceptible to Hu-Ab associated paraneoplastic neurological syndromes. J Neuroimmunol (2010) 226(1–2):147–9. doi:10.1016/j.jneuroim.2010.05.035
118. Domschke C, Schuetz F, Ge Y, Rom J, Zorn M, Sinn HP, et al. Determination of paraneoplastic autoimmune responses by tumor cell biology and intratumoral IFN-alpha/IL-12 in breast cancer patients. Cancer Immunol Immunother (2011) 60(3):401–11. doi:10.1007/s00262-010-0956-4
119. Leypoldt F, Wandinger KP. Paraneoplastic neurological syndromes. Clin Exp Immunol (2014) 175(3):336–48. doi:10.1111/cei.12185
120. Rousseau A, Benyahia B, Dalmau J, Connan F, Guillet JG, Delattre JY, et al. T cell response to Hu-D peptides in patients with anti-Hu syndrome. J Neurooncol (2005) 71(3):231–6. doi:10.1007/s11060-004-1723-1
121. Tanaka Y, Suzuki N, Takao M, Ichikawa A, Susumu N, Aoki D. Paraneoplastic cerebellar degeneration with fallopian tube adenocarcinoma. Gynecol Oncol (2005) 99(2):500–3. doi:10.1016/j.ygyno.2005.06.064
122. Tanaka M, Tanaka K. HLA A24 in paraneoplastic cerebellar degeneration with anti-Yo antibody. Neurology (1996) 47(2):606–7. doi:10.1212/WNL.47.2.606
123. Rasmussen T, Olszewski J, Lloydsmith D. Focal seizures due to chronic localized encephalitis. Neurology (1958) 8(6):435–45. doi:10.1212/WNL.8.6.435
124. Varadkar S, Bien CG, Kruse CA, Jensen FE, Bauer J, Pardo CA, et al. Rasmussen’s encephalitis: clinical features, pathobiology, and treatment advances. Lancet Neurol (2014) 13(2):195. doi:10.1016/S1474-4422(13)70260-6
125. Bien CG, Granata T, Antozzi C, Cross JH, Dulac O, Kurthen M, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain (2005) 128(Pt 3):454–71. doi:10.1093/brain/awh415
126. Mantegazza R, Bernasconi P, Baggi F, Spreafico R, Ragona F, Antozzi C, et al. Antibodies against GluR3 peptides are not specific for Rasmussen’s encephalitis but are also present in epilepsy patients with severe, early onset disease and intractable seizures. J Neuroimmunol (2002) 131(1):179–85. doi:10.1016/S0165-5728(02)00261-8
127. Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science (1994) 265(5172):648–51. doi:10.1126/science.8036512
128. Watson R, Jiang Y, Bermudez I, Houlihan L, Clover L, McKnight K, et al. Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology (2004) 63(1):43–50. doi:10.1212/01.WNL.0000132651.66689.0F
129. Wiendl H, Bien CG, Bernasconi P, Fleckenstein B, Elger CE, Dichgans J, et al. GluR3 antibodies: prevalence in focal epilepsy but no specificity for Rasmussen’s encephalitis. Neurology (2001) 57(8):1511. doi:10.1212/WNL.57.8.1511
130. Schwab N, Bien CG, Waschbisch A, Becker A, Vince GH, Dornmair K, et al. CD8+ T-cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain (2009) 132(5):1236–46. doi:10.1093/brain/awp003
131. Schneider-Hohendorf T, Mohan H, Bien CG, Breuer J, Becker A, Gorlich D, et al. CD8(+) T-cell pathogenicity in Rasmussen encephalitis elucidated by large-scale T-cell receptor sequencing. Nat Commun (2016) 7:11153. doi:10.1038/ncomms11153
132. Bien CG, Schramm J. Treatment of Rasmussen encephalitis half a century after its initial description: promising prospects and a dilemma. Epilepsy Res (2009) 86(2):101–12. doi:10.1016/j.eplepsyres.2009.06.001
133. Bittner S, Simon OJ, Göbel K, Bien CG, Meuth SG, Wiendl H. Rasmussen encephalitis treated with natalizumab. Neurology (2013) 81(4):395–7. doi:10.1212/WNL.0b013e31829c5ceb
134. Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain (2000) 123(Pt 7):1481–94. doi:10.1093/brain/123.7.1481
135. Furneaux HM, Rosenblum MK, Dalmau J, Wong E, Woodruff P, Graus F, et al. Selective expression of Purkinje-cell antigens in tumor tissue from patients with paraneoplastic cerebellar degeneration. N Engl J Med (1990) 322(26):1844–51. doi:10.1056/NEJM199006283222604
136. McKeon A, Pittock SJ. Paraneoplastic encephalomyelopathies: pathology and mechanisms. Acta Neuropathol (2011) 122(4):381–400. doi:10.1007/s00401-011-0876-1
137. Albert ML, Austin LM, Darnell RB. Detection and treatment of activated T cells in the cerebrospinal fluid of patients with paraneoplastic cerebellar degeneration. Ann Neurol (2000) 47(1):9. doi:10.1002/1531-8249(200001)47:13.3.CO;2-9
138. Blumenthal DT, Salzman KL, Digre KB, Jensen RL, Dunson WA, Dalmau J. Early pathologic findings and long-term improvement in anti-Ma2-associated encephalitis. Neurology (2006) 67(1):146–9. doi:10.1212/01.wnl.0000223647.83708.20
139. Bernal F, Graus F, Pifarré À, Saiz A, Benyahia B, Ribalta T. Immunohistochemical analysis of anti-Hu-associated paraneoplastic encephalomyelitis. Acta Neuropathol (2002) 103(5):509–15. doi:10.1007/s00401-001-0498-0
140. Graus F, Vega F, Delattre JY, Bonaventura I, Reñé R, Arbaiza D, et al. Plasmapheresis and antineoplastic treatment in CNS paraneoplastic syndromes with antineuronal autoantibodies. Neurology (1992) 42(3 Pt 1):536–40. doi:10.1212/WNL.42.3.536
141. Shams’ili S, Grefkens J, Leeuw B, Bent M, Hooijkaas H, Holt B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain (2003) 126(Pt 6):1409–18. doi:10.1093/brain/awg133
142. Uchuya M, Graus F, Vega F, Reñé R, Delattre JY. Intravenous immunoglobulin treatment in paraneoplastic neurological syndromes with antineuronal autoantibodies. J Neurol Neurosurg Psychiatry (1996) 60(4):388–92. doi:10.1136/jnnp.60.4.388
143. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain (2012) 135(Pt 5):1622–38. doi:10.1093/brain/aws082
144. Melzer N, Meuth SG, Wiendl H. CD8+ T cells and neuronal damage: direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. FASEB J (2009) 23(11):3659–73. doi:10.1096/fj.09-136200
145. Sommerfeldt N, Schutz F, Sohn C, Forster J, Schirrmacher V, Beckhove P. The shaping of a polyvalent and highly individual T-cell repertoire in the bone marrow of breast cancer patients. Cancer Res (2006) 66(16):8258–65. doi:10.1158/0008-5472.CAN-05-4201
146. Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A (2008) 105(40):15558–63. doi:10.1073/pnas.0807419105
147. Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, et al. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A (2008) 105(46):17913–8. doi:10.1073/pnas.0804610105
148. Mantovani S, Garbelli S, Pasini A, Alimonti D, Perotti C, Melazzini M, et al. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J Neuroimmunol (2009) 210(1–2):73–9. doi:10.1016/j.jneuroim.2009.02.012
149. Carson MJ. Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia (2002) 40(2):218–31. doi:10.1002/glia.10145
150. Hammarberg H, Lidman O, Lundberg C, Eltayeb SY, Gielen AW, Muhallab S, et al. Neuroprotection by encephalomyelitis: rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J Neurosci (2000) 20(14):5283–91.
151. Moalem G, Monsonego A, Shani Y, Cohen IR, Schwartz M. Differential T cell response in central and peripheral nerve injury: connection with immune privilege. FASEB J (1999) 13(10):1207–17.
152. Serpe CJ, Coers S, Sanders VM, Jones KJ. CD4+ T, but not CD8+ or B, lymphocytes mediate facial motoneuron survival after facial nerve transection. Brain Behav Immun (2003) 17(5):393–402. doi:10.1016/S0889-1591(03)00028-X
153. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet (2004) 364(9451):2106–12. doi:10.1016/S0140-6736(04)17551-X
154. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology (2006) 66(10):1485–9. doi:10.1212/01.wnl.0000216139.44259.74
155. Waters P, Jarius S, Littleton E, Leite MI, Jacob S, Gray B, et al. Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol (2008) 65(7):913–9. doi:10.1001/archneur.65.7.913
156. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol (2009) 66(5):617–29. doi:10.1002/ana.21802
157. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol (2009) 66(5):630–43. doi:10.1002/ana.21837
158. Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, et al. Anti-aquaporin-4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen-specific T cells. Biochem Biophys Res Commun (2010) 394(1):205–10. doi:10.1016/j.bbrc.2010.02.157
159. Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain (2008) 131(Pt 11):3072–80. doi:10.1093/brain/awn240
160. Nishiyama S, Ito T, Misu T, Takahashi T, Kikuchi A, Suzuki N, et al. A case of NMO seropositive for aquaporin-4 antibody more than 10 years before onset. Neurology (2009) 72(22):1960–1. doi:10.1212/WNL.0b013e3181a82621
161. Jacob A, Weinshenker BG, Violich I, McLinskey N, Krupp L, Fox RJ, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol (2008) 65(11):1443–8. doi:10.1001/archneur.65.11.noc80069
162. Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol (2011) 68(11):1412–20. doi:10.1001/archneurol.2011.154
163. Brum DG, Barreira AA, dos Santos AC, Kaimen-Maciel DR, Matiello M, Costa RM, et al. HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult Scler (2010) 16(1):21–9. doi:10.1177/1352458509350741
164. Deschamps R, Paturel L, Jeannin S, Chausson N, Olindo S, Bera O, et al. Different HLA class II (DRB1 and DQB1) alleles determine either susceptibility or resistance to NMO and multiple sclerosis among the French Afro-Caribbean population. Mult Scler (2011) 17(1):24–31. doi:10.1177/1352458510382810
165. Matsuya N, Komori M, Nomura K, Nakane S, Fukudome T, Goto H, et al. Increased T-cell immunity against aquaporin-4 and proteolipid protein in neuromyelitis optica. Int Immunol (2011) 23(9):565–73. doi:10.1093/intimm/dxr056
166. Tanaka M, Matsushita T, Tateishi T, Ochi H, Kawano Y, Mei FJ, et al. Distinct CSF cytokine/chemokine profiles in atopic myelitis and other causes of myelitis. Neurology (2008) 71(13):974–81. doi:10.1212/01.wnl.0000326589.57128.c3
167. Uzawa A, Mori M, Ito M, Uchida T, Hayakawa S, Masuda S, et al. Markedly increased CSF interleukin-6 levels in neuromyelitis optica, but not in multiple sclerosis. J Neurol (2009) 256(12):2082–4. doi:10.1007/s00415-009-5274-4
168. Vaknin-Dembinsky A, Brill L, Kassis I, Petrou P, Ovadia H, Ben-Hur T, et al. T-cell responses to distinct AQP4 peptides in patients with neuromyelitis optica (NMO). Mult Scler Relat Disord (2016) 6:28–36. doi:10.1016/j.msard.2015.12.004
169. Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, Cree BA, et al. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol (2012) 72(1):53–64. doi:10.1002/ana.23651
170. Wang H, Dai Y, Qiu W, Zhong X, Wu A, Wang Y, et al. HLA-DPB1 0501 is associated with susceptibility to anti-aquaporin-4 antibodies positive neuromyelitis optica in southern Han Chinese. J Neuroimmunol (2011) 233(1–2):181–4. doi:10.1016/j.jneuroim.2010.11.004
171. Zephir H, Fajardy I, Outteryck O, Blanc F, Roger N, Fleury M, et al. Is neuromyelitis optica associated with human leukocyte antigen? Mult Scler (2009) 15(5):571–9. doi:10.1177/1352458508102085
172. Dale RC, Morovat A. Interleukin-6 and oligoclonal IgG synthesis in children with acute disseminated encephalomyelitis. Neuropediatrics (2003) 34(3):141–5. doi:10.1055/s-2003-41281
173. Ichiyama T, Shoji H, Kato M, Sawaishi Y, Ozawa H, Matsubara T, et al. Cerebrospinal fluid levels of cytokines and soluble tumour necrosis factor receptor in acute disseminated encephalomyelitis. Eur J Pediatr (2002) 161(3):133–7. doi:10.1007/s00431-001-0888-2
174. Ishizu T, Minohara M, Ichiyama T, Kira R, Tanaka M, Osoegawa M, et al. CSF cytokine and chemokine profiles in acute disseminated encephalomyelitis. J Neuroimmunol (2006) 175(1–2):52–8. doi:10.1016/j.jneuroim.2006.03.020
175. Jorens PG, VanderBorght A, Ceulemans B, Van Bever HP, Bossaert LL, Ieven M, et al. Encephalomyelitis-associated antimyelin autoreactivity induced by streptococcal exotoxins. Neurology (2000) 54(7):1433–41. doi:10.1212/WNL.54.7.1433
176. Pohl-Koppe A, Burchett SK, Thiele EA, Hafler DA. Myelin basic protein reactive Th2 T cells are found in acute disseminated encephalomyelitis. J Neuroimmunol (1998) 91(1–2):19–27. doi:10.1016/S0165-5728(98)00125-8
177. Yoshitomi T, Matsubara T, Nishikawa M, Katayama K, Ichiyama T, Hayashi T, et al. Increased peripheral blood interferon gamma-producing T cells in acute disseminated encephalomyelitis. J Neuroimmunol (2000) 111(1–2):224–8. doi:10.1016/S0165-5728(00)00358-1
178. Costa M, Saiz A, Casamitjana R, Castaner MF, Sanmarti A, Graus F, et al. T-cell reactivity to glutamic acid decarboxylase in stiff-man syndrome and cerebellar ataxia associated with polyendocrine autoimmunity. Clin Exp Immunol (2002) 129(3):471–8. doi:10.1046/j.1365-2249.2002.01931.x
179. Hanninen A, Soilu-Hanninen M, Hampe CS, Deptula A, Geubtner K, Ilonen J, et al. Characterization of CD4+ T cells specific for glutamic acid decarboxylase (GAD65) and proinsulin in a patient with stiff-person syndrome but without type 1 diabetes. Diabetes Metab Res Rev (2010) 26(4):271–9. doi:10.1002/dmrr.1083
180. Hummel M, Durinovic-Bello I, Bonifacio E, Lampasona V, Endl J, Fessele S, et al. Humoral and cellular immune parameters before and during immunosuppressive therapy of a patient with stiff-man syndrome and insulin dependent diabetes mellitus. J Neurol Neurosurg Psychiatry (1998) 65(2):204–8. doi:10.1136/jnnp.65.2.204
181. Pugliese A, Solimena M, Awdeh ZL, Alper CA, Bugawan T, Erlich HA, et al. Association of HLA-DQB1*0201 with stiff-man syndrome. J Clin Endocrinol Metab (1993) 77(6):1550–3. doi:10.1210/jcem.77.6.8263140
182. Schloot NC, Batstra MC, Duinkerken G, De Vries RR, Dyrberg T, Chaudhuri A, et al. GAD65-reactive T cells in a non-diabetic stiff-man syndrome patient. J Autoimmun (1999) 12(4):289–96. doi:10.1006/jaut.1999.0280
183. Skorstad G, Hestvik AL, Vartdal F, Holmoy T. Cerebrospinal fluid T cell responses against glutamic acid decarboxylase 65 in patients with stiff person syndrome. J Autoimmun (2009) 32(1):24–32. doi:10.1016/j.jaut.2008.10.002
184. Byun JI, Lee ST, Moon J, Jung KH, Sunwoo JS, Lim JA, et al. Distinct intrathecal interleukin-17/interleukin-6 activation in anti-N-methyl-d-aspartate receptor encephalitis. J Neuroimmunol (2016) 297:141–7. doi:10.1016/j.jneuroim.2016.05.023
185. Kothur K, Wienholt L, Mohammad SS, Tantsis EM, Pillai S, Britton PN, et al. Utility of CSF cytokine/chemokines as markers of active intrathecal inflammation: comparison of demyelinating, anti-NMDAR and enteroviral encephalitis. PLoS One (2016) 11(8):e0161656. doi:10.1371/journal.pone.0161656
186. Lee WJ, Lee ST, Moon J, Sunwoo JS, Byun JI, Lim JA, et al. Tocilizumab in autoimmune encephalitis refractory to rituximab: an institutional cohort study. Neurotherapeutics (2016) 13(4):824–32. doi:10.1007/s13311-016-0442-6
187. Liba Z, Kayserova J, Elisak M, Marusic P, Nohejlova H, Hanzalova J, et al. Anti-N-methyl-d-aspartate receptor encephalitis: the clinical course in light of the chemokine and cytokine levels in cerebrospinal fluid. J Neuroinflammation (2016) 13(1):55. doi:10.1186/s12974-016-0507-9
188. Ulusoy C, Tuzun E, Kurtuncu M, Turkoglu R, Akman-Demir G, Eraksoy M. Comparison of the cytokine profiles of patients with neuronal-antibody-associated central nervous system disorders. Int J Neurosci (2012) 122(6):284–9. doi:10.3109/00207454.2011.648762
189. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain (2002) 125(Pt 7):1450–61. doi:10.1093/brain/awf151
190. Pohl M, Kawakami N, Kitic M, Bauer J, Martins R, Fischer MT, et al. T cell-activation in neuromyelitis optica lesions plays a role in their formation. Acta Neuropathol Commun (2013) 1:85. doi:10.1186/2051-5960-1-85
191. Warabi Y, Yagi K, Hayashi H, Matsumoto Y. Characterization of the T cell receptor repertoire in the Japanese neuromyelitis optica: T cell activity is up-regulated compared to multiple sclerosis. J Neurol Sci (2006) 249(2):145–52. doi:10.1016/j.jns.2006.06.011
192. Wang HH, Dai YQ, Qiu W, Lu ZQ, Peng FH, Wang YG, et al. Interleukin-17-secreting T cells in neuromyelitis optica and multiple sclerosis during relapse. J Clin Neurosci (2011) 18(10):1313–7. doi:10.1016/j.jocn.2011.01.031
193. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol (2015) 16(5):448–57. doi:10.1038/ni.3153
194. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A (2008) 105(47):18460–5. doi:10.1073/pnas.0809850105
195. Uzawa A, Mori M, Arai K, Sato Y, Hayakawa S, Masuda S, et al. Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler (2010) 16(12):1443–52. doi:10.1177/1352458510379247
196. Ayzenberg I, Kleiter I, Schroder A, Hellwig K, Chan A, Yamamura T, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol (2013) 70(3):394–7. doi:10.1001/jamaneurol.2013.1246
197. Kieseier BC, Stuve O, Dehmel T, Goebels N, Leussink VI, Mausberg AK, et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol (2013) 70(3):390–3. doi:10.1001/jamaneurol.2013.668
198. Matsushita T, Matsuoka T, Isobe N, Kawano Y, Minohara M, Shi N, et al. Association of the HLA-DPB1*0501 allele with anti-aquaporin-4 antibody positivity in Japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens (2009) 73(2):171–6. doi:10.1111/j.1399-0039.2008.01172.x
199. Farkas AM, Panea C, Goto Y, Nakato G, Galan-Diez M, Narushima S, et al. Induction of Th17 cells by segmented filamentous bacteria in the murine intestine. J Immunol Methods (2015) 421:104–11. doi:10.1016/j.jim.2015.03.020
200. Reindl M, Di Pauli F, Rostasy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol (2013) 9(8):455–61. doi:10.1038/nrneurol.2013.118
201. Brilot F, Dale RC, Selter RC, Grummel V, Kalluri SR, Aslam M, et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol (2009) 66(6):833–42. doi:10.1002/ana.21916
202. Dale RC, Tantsis EM, Merheb V, Kumaran RY, Sinmaz N, Pathmanandavel K, et al. Antibodies to MOG have a demyelination phenotype and affect oligodendrocyte cytoskeleton. Neurol Neuroimmunol Neuroinflamm (2014) 1(1):e12. doi:10.1212/NXI.0000000000000012
203. Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation (2011) 8:184. doi:10.1186/1742-2094-8-184
204. McLaughlin KA, Chitnis T, Newcombe J, Franz B, Kennedy J, McArdel S, et al. Age-dependent B cell autoimmunity to a myelin surface antigen in pediatric multiple sclerosis. J Immunol (2009) 183(6):4067–76. doi:10.4049/jimmunol.0801888
205. O’Connor KC, McLaughlin KA, De Jager PL, Chitnis T, Bettelli E, Xu C, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med (2007) 13(2):211–7. doi:10.1038/nm1488
206. Probstel AK, Dornmair K, Bittner R, Sperl P, Jenne D, Magalhaes S, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology (2011) 77(6):580–8. doi:10.1212/WNL.0b013e318228c0b1
207. Franciotta D, Zardini E, Ravaglia S, Piccolo G, Andreoni L, Bergamaschi R, et al. Cytokines and chemokines in cerebrospinal fluid and serum of adult patients with acute disseminated encephalomyelitis. J Neurol Sci (2006) 247(2):202–7. doi:10.1016/j.jns.2006.05.049
208. Kothur K, Wienholt L, Tantsis EM, Earl J, Bandodkar S, Prelog K, et al. B Cell, Th17, and neutrophil related cerebrospinal fluid cytokine/chemokines are elevated in MOG antibody associated demyelination. PLoS One (2016) 11(2):e0149411. doi:10.1371/journal.pone.0149411
209. Horellou P, Wang M, Keo V, Chretien P, Serguera C, Waters P, et al. Increased interleukin-6 correlates with myelin oligodendrocyte glycoprotein antibodies in pediatric monophasic demyelinating diseases and multiple sclerosis. J Neuroimmunol (2015) 289:1–7. doi:10.1016/j.jneuroim.2015.10.002
210. Lalive PH, Menge T, Delarasse C, Della Gaspera B, Pham-Dinh D, Villoslada P, et al. Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc Natl Acad Sci U S A (2006) 103(7):2280–5. doi:10.1073/pnas.0510672103
211. Honnorat J, Saiz A, Giometto B, Vincent A, Brieva L, de Andres C, et al. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies: study of 14 patients. Arch Neurol (2001) 58(2):225–30. doi:10.1001/archneur.58.2.225
212. Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol (2010) 67(4):470–8. doi:10.1002/ana.21917
213. Peltola J, Kulmala P, Isojarvi J, Saiz A, Latvala K, Palmio J, et al. Autoantibodies to glutamic acid decarboxylase in patients with therapy-resistant epilepsy. Neurology (2000) 55(1):46–50. doi:10.1212/WNL.55.1.46
214. Solimena M, Folli F, Denis-Donini S, Comi GC, Pozza G, De Camilli P, et al. Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med (1988) 318(16):1012–20. doi:10.1056/NEJM198804213181602
215. Dinkel K, Meinck HM, Jury KM, Karges W, Richter W. Inhibition of gamma-aminobutyric acid synthesis by glutamic acid decarboxylase autoantibodies in stiff-man syndrome. Ann Neurol (1998) 44(2):194–201. doi:10.1002/ana.410440209
216. Jarius S, Stich O, Speck J, Rasiah C, Wildemann B, Meinck HM, et al. Qualitative and quantitative evidence of anti-glutamic acid decarboxylase-specific intrathecal antibody synthesis in patients with stiff person syndrome. J Neuroimmunol (2010) 229(1–2):219–24. doi:10.1016/j.jneuroim.2010.07.019
217. Levy LM, Levy-Reis I, Fujii M, Dalakas MC. Brain gamma-aminobutyric acid changes in stiff-person syndrome. Arch Neurol (2005) 62(6):970–4. doi:10.1001/archneur.62.6.970
218. Vincent A. Stiff, twitchy or wobbly: are GAD antibodies pathogenic? Brain (2008) 131(Pt 10):2536–7. doi:10.1093/brain/awn221
219. Warren JD, Scott G, Blumbergs PC, Thompson PD. Pathological evidence of encephalomyelitis in the stiff man syndrome with anti-GAD antibodies. J Clin Neurosci (2002) 9(3):328–9. doi:10.1054/jocn.2001.1014
220. Lohmann T, Hawa M, Leslie RD, Lane R, Picard J, Londei M. Immune reactivity to glutamic acid decarboxylase 65 in stiffman syndrome and type 1 diabetes mellitus. Lancet (2000) 356(9223):31–5. doi:10.1016/S0140-6736(00)02431-4
221. Lohmann T, Londei M, Hawa M, Leslie RD. Humoral and cellular autoimmune responses in stiff person syndrome. Ann N Y Acad Sci (2003) 998:215–22. doi:10.1196/annals.1254.024
222. Raju R, Foote J, Banga JP, Hall TR, Padoa CJ, Dalakas MC, et al. Analysis of GAD65 autoantibodies in stiff-person syndrome patients. J Immunol (2005) 175(11):7755–62. doi:10.4049/jimmunol.175.11.7755
223. Sinmaz N, Nguyen T, Tea F, Dale RC, Brilot F. Mapping autoantigen epitopes: molecular insights into autoantibody-associated disorders of the nervous system. J Neuroinflammation (2016) 13(1):219. doi:10.1186/s12974-016-0678-4
224. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol (2008) 7(12):1091–8. doi:10.1016/S1474-4422(08)70224-2
225. Dalmau J. NMDA receptor encephalitis and other antibody-mediated disorders of the synapse: the 2016 Cotzias Lecture. Neurology (2016) 87(23):2471–82. doi:10.1212/WNL.0000000000003414
226. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci (2010) 30(17):5866–75. doi:10.1523/JNEUROSCI.0167-10.2010
227. Mikasova L, De Rossi P, Bouchet D, Georges F, Rogemond V, Didelot A, et al. Disrupted surface cross-talk between NMDA and ephrin-B2 receptors in anti-NMDA encephalitis. Brain (2012) 135(Pt 5):1606–21. doi:10.1093/brain/aws092
228. Planaguma J, Leypoldt F, Mannara F, Gutierrez-Cuesta J, Martin-Garcia E, Aguilar E, et al. Human N-methyl-d-aspartate receptor antibodies alter memory and behaviour in mice. Brain (2015) 138(Pt 1):94–109. doi:10.1093/brain/awu310
229. Irani SR, Bera K, Waters P, Zuliani L, Maxwell S, Zandi MS, et al. N-methyl-d-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain (2010) 133(Pt 6):1655–67. doi:10.1093/brain/awq113
230. Camdessanche JP, Streichenberger N, Cavillon G, Rogemond V, Jousserand G, Honnorat J, et al. Brain immunohistopathological study in a patient with anti-NMDAR encephalitis. Eur J Neurol (2011) 18(6):929–31. doi:10.1111/j.1468-1331.2010.03180.x
231. Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology (2011) 77(6):589–93. doi:10.1212/WNL.0b013e318228c136
232. Tuzun E, Zhou L, Baehring JM, Bannykh S, Rosenfeld MR, Dalmau J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol (2009) 118(6):737–43. doi:10.1007/s00401-009-0582-4
233. Bacher P, Schink C, Teutschbein J, Kniemeyer O, Assenmacher M, Brakhage AA, et al. Antigen-reactive T cell enrichment for direct, high-resolution analysis of the human naive and memory Th cell repertoire. J Immunol (2013) 190(8):3967–76. doi:10.4049/jimmunol.1202221
234. Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science (1996) 274(5284):94–6. doi:10.1126/science.274.5284.94
235. Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, et al. Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol (1998) 16:523–44. doi:10.1146/annurev.immunol.16.1.523
236. Bacher P, Scheffold A. Flow-cytometric analysis of rare antigen-specific T cells. Cytometry A (2013) 83(8):692–701. doi:10.1002/cyto.a.22317
237. Bill JR, Kotzin BL. Use of soluble MHC class II/peptide multimers to detect antigen-specific T cells in human disease. Arthritis Res (2002) 4(4):261–5. doi:10.1186/ar417
238. Davis MM, Altman JD, Newell EW. Interrogating the repertoire: broadening the scope of peptide-MHC multimer analysis. Nat Rev Immunol (2011) 11(8):551–8. doi:10.1038/nri3020
239. Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med (1999) 5(6):677–85. doi:10.1038/9525
240. Laughlin EM, Miller JD, James E, Fillos D, Ibegbu CC, Mittler RS, et al. Antigen-specific CD4+ T cells recognize epitopes of protective antigen following vaccination with an anthrax vaccine. Infect Immun (2007) 75(4):1852–60. doi:10.1128/IAI.01814-06
241. Caruso A, Licenziati S, Corulli M, Canaris AD, De Francesco MA, Fiorentini S, et al. Flow cytometric analysis of activation markers on stimulated T cells and their correlation with cell proliferation. Cytometry (1997) 27(1):71–6. doi:10.1002/(SICI)1097-0320(19970101)27:13.0.CO;2-O
242. Frentsch M, Arbach O, Kirchhoff D, Moewes B, Worm M, Rothe M, et al. Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nat Med (2005) 11(10):1118–24. doi:10.1038/nm1292
243. Testi R, D’Ambrosio D, De Maria R, Santoni A. The CD69 receptor: a multipurpose cell-surface trigger for hematopoietic cells. Immunol Today (1994) 15(10):479–83. doi:10.1016/0167-5699(94)90193-7
244. Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood (2007) 110(1):201–10. doi:10.1182/blood-2006-11-056168
245. Zaunders JJ, Munier ML, Seddiki N, Pett S, Ip S, Bailey M, et al. High levels of human antigen-specific CD4+ T cells in peripheral blood revealed by stimulated coexpression of CD25 and CD134 (OX40). J Immunol (2009) 183(4):2827–36. doi:10.4049/jimmunol.0803548
246. Madhavan H. Simple laboratory methods to measure cell proliferation using DNA synthesis property. J Stem Cells Regen Med (2007) 3(1):12–4.
247. Mannering SI, Morris JS, Jensen KP, Purcell AW, Honeyman MC, van Endert PM, et al. A sensitive method for detecting proliferation of rare autoantigen-specific human T cells. J Immunol Methods (2003) 283(1–2):173–83. doi:10.1016/j.jim.2003.09.004
248. De Clerck LS, Bridts CH, Mertens AM, Moens MM, Stevens WJ. Use of fluorescent dyes in the determination of adherence of human leucocytes to endothelial cells and the effect of fluorochromes on cellular function. J Immunol Methods (1994) 172(1):115–24. doi:10.1016/0022-1759(94)90384-0
249. Mobs C, Schmidt T. Research techniques made simple: monitoring of T-cell subsets using the ELISPOT assay. J Invest Dermatol (2016) 136(6):e55–9. doi:10.1016/j.jid.2016.04.009
Keywords: autoreactive T cells, central nervous system autoimmune diseases, neuroimmunology, autoantibodies, multiple sclerosis, T cell detection
Citation: Pilli D, Zou A, Tea F, Dale RC and Brilot F (2017) Expanding Role of T Cells in Human Autoimmune Diseases of the Central Nervous System. Front. Immunol. 8:652. doi: 10.3389/fimmu.2017.00652
Received: 31 January 2017; Accepted: 17 May 2017;
Published: 07 June 2017
Edited by:
Jens Geginat, Istituto Nazionale Genetica Molecolare (INGM), ItalyReviewed by:
Richa Hanamsagar, Massachusetts General Hospital, United StatesAnne Kathrin Mausberg, Essen University Hospital, Germany
Copyright: © 2017 Pilli, Zou, Tea, Dale and Brilot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabienne Brilot, ZmFiaWVubmUuYnJpbG90QHN5ZG5leS5lZHUuYXU=
†These authors have contributed equally to this work.