 Benjamin Peschke
Benjamin Peschke Christian W. Keller
Christian W. Keller Patrick Weber1
Patrick Weber1 Isaak Quast
Isaak Quast Jan D. Lünemann
Jan D. Lünemann
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 June 2017
Sec. Molecular Innate Immunity
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00646
This article is part of the Research Topic C1q: A Molecular Bridge to Innate and Adaptive Immunity View all 10 articles
Binding of the complement component C1q to the CH2 domain of antigen-bound immunoglobulin gamma (IgG) activates the classical complement pathway and depends on its close proximity to Fc fragments of neighboring antibodies. IgG subclasses contain a highly conserved asparagine 297 (N)-linked biantennary glycan within their CH2 domains, the core structure of which can be extended with terminal galactose and sialic acid residues. To investigate whether Fc-glycosylation regulates effector functions of human IgG subclasses, we cloned the antigen-binding region of the CD20-specific monoclonal antibody rituximab into IgG isotype expression vectors. We found that Fc-galactosylation enhances the efficacy of CD20-targeting complement-fixing antibodies for C1q binding and complement-mediated tumor cell lysis. Increased efficacies were restricted to IgG1 and IgG3 subclasses indicating that Fc-galactosylation alone is not sufficient for IgG2 and IgG4 to acquire complement-fixing properties. Addition of terminal galactose to the N-glycan specifically improved binding of C1q without changing antigen- and FcγRIIIa-binding affinities of IgG isotypes. These data indicate that Fc galactosylation can be harnessed to enhance the complement-activating properties of IgG1 and IgG3 antibodies.
Immunoglobulin gamma (IgG) antibodies are preeminent effector proteins of the immune system. The bimodal architecture of IgG allows for simultaneous recognition of antigen (by the antigen-binding fragment, Fab) and the initiation of IgG effector functions such as recruitment and activation of leukocytes and antibody-dependent cell-mediated cytotoxicity (ADCC) through interaction with FcγRs as well as complement activation through binding of the constant, crystallizable fragment (Fc) binding of C1q. Human IgG is subdivided in the four isotypes IgG1 to IgG4, numbered based on their abundance in serum (1). Despite being more than 90% identical in amino acid sequence, IgG isotypes differ in key functional regions responsible for flexibility, FcγR binding, and complement fixation (2).
All IgG subclasses contain a highly conserved asparagine-linked (N-)oligosaccharide located in the CH2 domain of the Fc region. Presence of this glycan serves important functions in protein folding and post-translational quality control mechanisms and is essential for antibody-mediated effector functions (3, 4). The biantennary core glycan structure, which is composed of two N-acetylglucosamines (GlcNAc) and three mannose residues, can be further decorated with fucose, bisecting GlcNAc and terminal GlcNAc, galactose, and sialic acid. The highest degree of variability among IgG antibodies stems from the presence or absence of galactose with around 40% containing one galactose, 20–40% two galactoses, and the remainder none (5–7).
Presence or absence of distinct N-glycan residues such as fucose and sialic acid can dramatically alter pro- and anti-inflammatory IgG activities. Removal of the fucose residue from human IgG1 increases antibody-dependent cell-mediated cytotoxicity (ADCC) through improved affinity for Fcγ receptor IIIa (FcγRIIIa) (8, 9). We have previously shown that IgG Fc sialylation of human monoclonal IgG1 molecules impairs their efficacy to induce complement-dependent cytotoxicity (CDC) (10). Here, we determined whether Fc galactosylation of human IgG isotypes (IgG1–4) regulates antibody effector functions.
To test the impact of Fc galactosylation of human IgG isotypes on target cell depletion, we first cloned the sequence encoding the heavy chain (HC) and light chain (LC) antigen-binding regions of the monoclonal CD20-targeting antibody rituximab into human IgG1–4 HC and kappa LC expression vectors. Plasmids were cotransfected in HKB11 cells and antibodies were purified from the supernatant using protein G columns. Next, we either treated the antibodies with recombinant galactosidase to completely remove galactose or with β-1,4-galactosyltransferase in the presence of uridine diphosphate (UDP-) galactose to obtain galactosylated antibodies. After repurification, antibodies were analyzed for purity and integrity by gel electrophoresis and immunoblotting with the galactose-specific Erythrina Cristagalli lectin to confirm the successful generation of glycovariants (Figure 1).
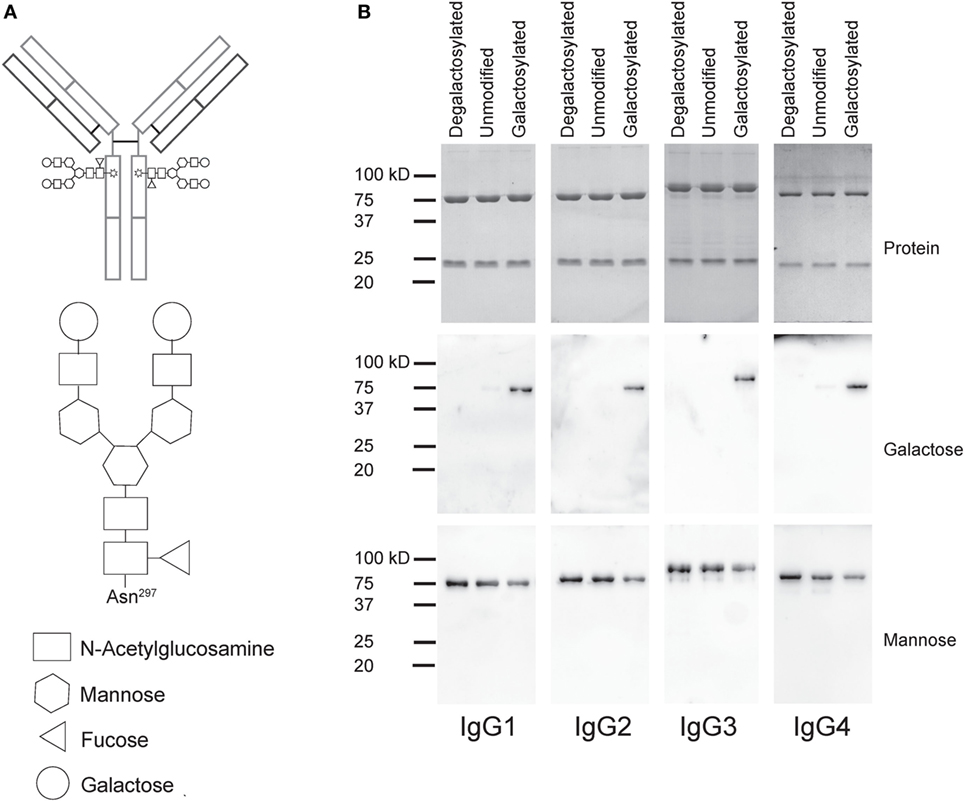
Figure 1. Generation of degalactosylated and galactosylated immunoglobulin gamma (IgG) isotypes derived from rituximab. (A) Schematic depiction of an IgG antibody with its two Fc N-glycans (up) and detailed glycan composition of a fully galactosylated IgG-Fc N-glycan. (B) Gel electrophoresis and coomassie staining to detect total protein showing heavy and light chains of antibodies (up) and immunoblots using galactose- (Erythrina Cristagalli lectin) or mannose- (Lens Culinaris agglutinin) specific lectins.
Modifications in the Fc domain may change the structural properties of the antibody, potentially leading to changes in its antigen-binding region. To determine antigen-binding affinities of IgG isotype glycovariants, we titrated the antibodies on CD20 expressing human Raji-Burkitt’s lymphoma cells and analyzed binding by flow cytometry (Figure 2). For each isotype, galactosylated and degalactosylated glycovariants did not differ in their antigen-binding characteristics.
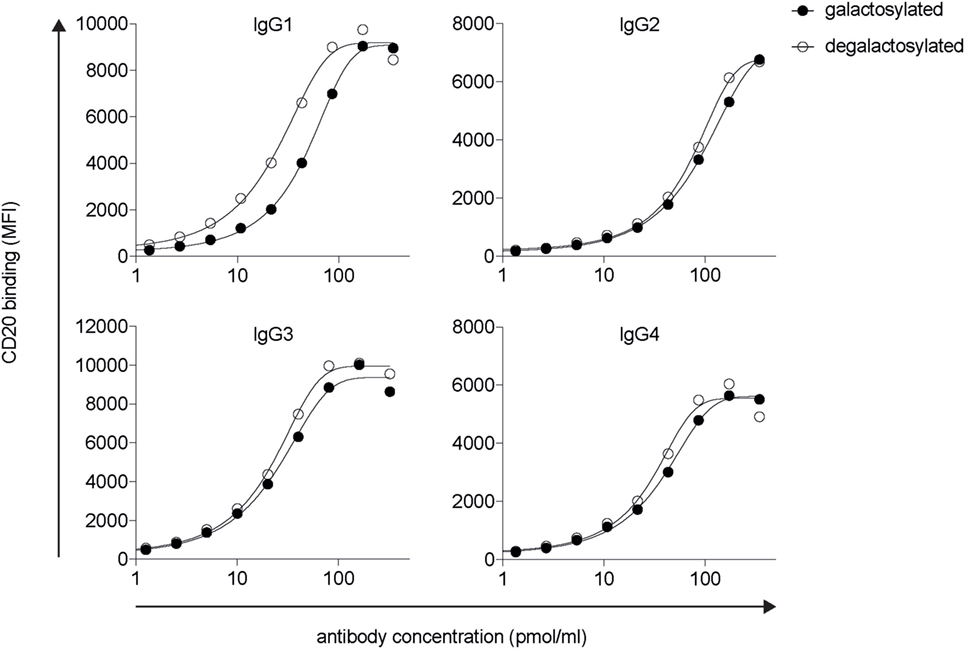
Figure 2. Fc-galactosylation does not increase CD20 binding of rituximab-derived IgG isotypes. Target antigen recognition by galactosylated and degalactosylated human IgG1–4 isotypes specific for CD20 analyzed by flow cytometry via titration on CD20+ Raji cells. MFI, median fluorescence intensity; IgG, immunoglobulin gamma.
The efficacy of IgG isotype-derived glycovariants to induce complement-dependent cytotoxicity (CDC) was determined in Burkitt’s lymphoma-derived Raji cells in the presence of active complement (human serum). Rituximab-derived IgG3 glycovariants showed the highest efficacy for CDC, followed by IgG1 (Figure 3). Glycovariants of IgG2 and IgG4 isotypes did not induce CDC. Galactosylation increased CDC mediated by both IgG1 (26% reduction of EC50) and IgG3 (13% reduction of EC50) but did not provide IgG2 and IgG4 with de novo ability to lyse target cells (Figure 3). To investigate the mechanism by which Fc-galactosylation impacts CDC, we determined the C1q binding affinities and kinetics of galactosylated and degalactosylated antibody variants (10). Incubation of CD20-expressing Raji cells in the presence of human serum depleted for C5, an essential component of the complement cascade, which allows to analyze the binding of members of the complement cascade to target cells while preventing cell lysis (10), led to rapid binding of C1q (Figure 4). Fc-galactosylation substantially enhanced the antibodies’ capacity to bind C1q for IgG1 and IgG3 isotypes (Figure 4). These data indicate that the addition of terminal galactose to the Fc-glycan enhances cell-depleting efficacies of human IgG1 and IgG3 isotypes through increased C1q binding.
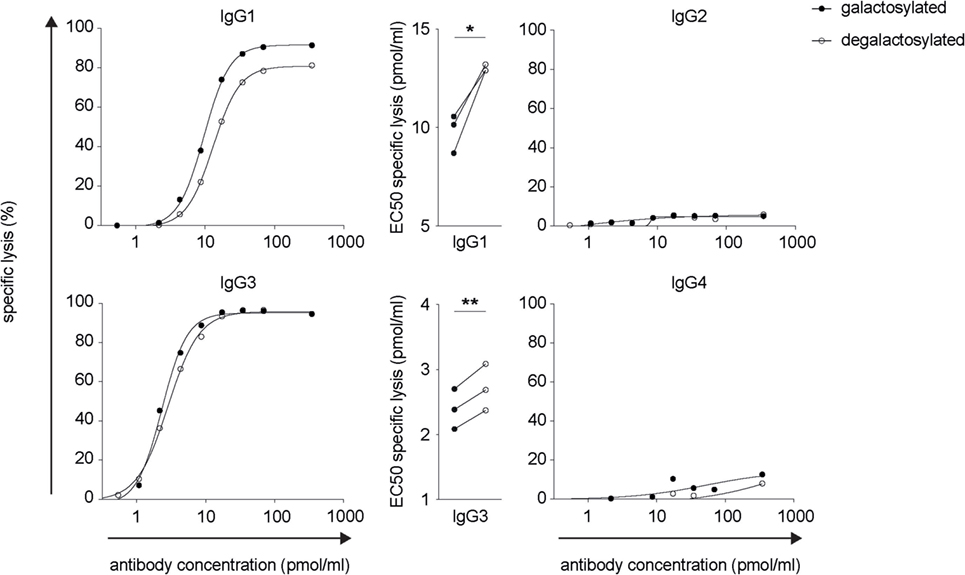
Figure 3. Increased CDC of rituximab-derived immunoglobulin gamma 1 (IgG1) and IgG3 but not IgG2 and IgG4 upon Fc-galactosylation. Complement-dependent lysis of CD20+ target cells in the presence of galactosylated or degalactosylated human anti-CD20 IgG isotypes. Exemplary lysis curves and EC50 values of three independent experiments are shown. Statistical analysis: paired two-tailed Student’s t-test *p < 0.05, **p < 0.01.
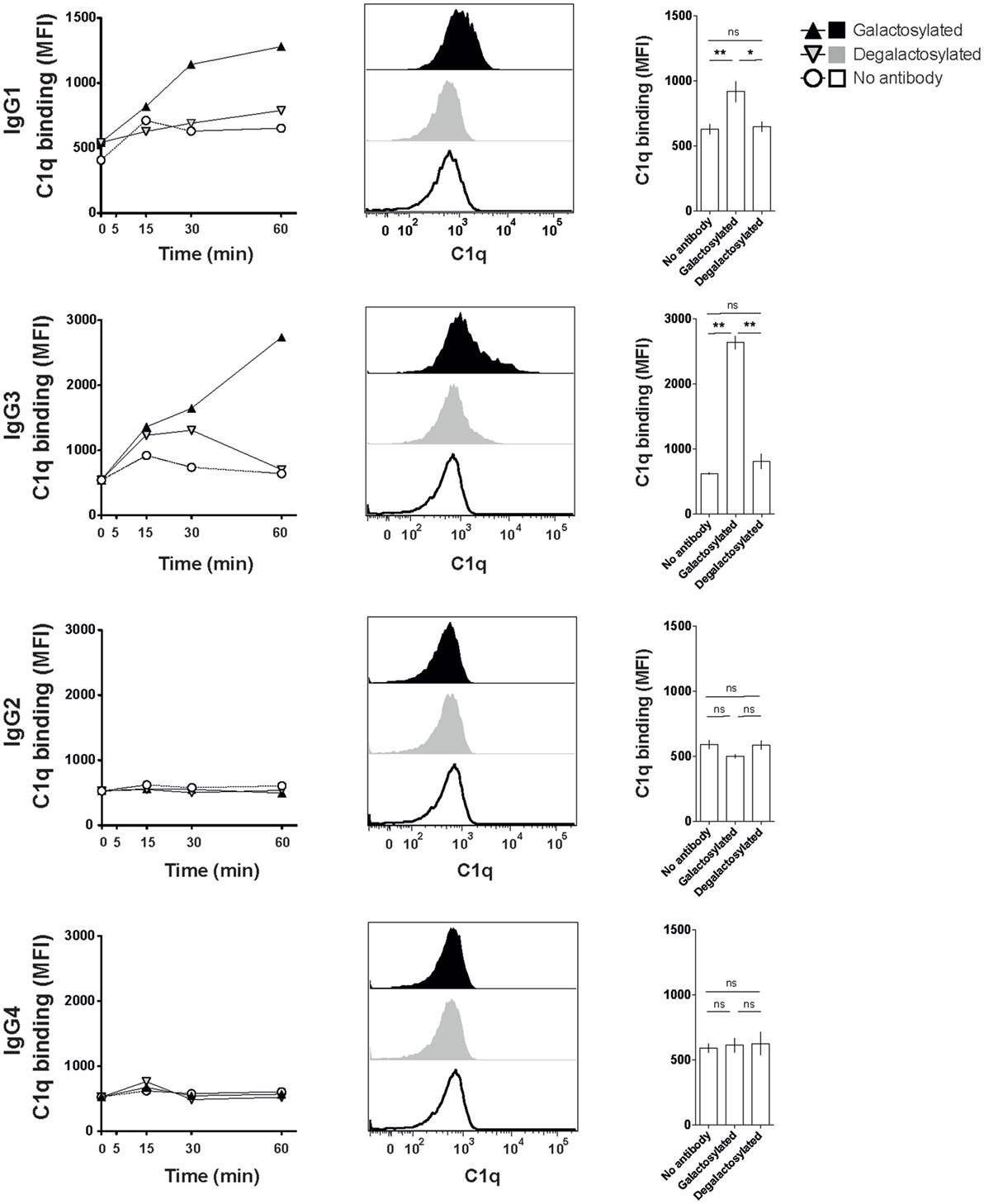
Figure 4. Increased C1q binding of Fc-galactosylated rituximab-derived immunoglobulin gamma 1 (IgG1) and IgG3. Kinetic of C1q binding to CD20+ Raji cells in the presence of galactosylated or degalactosylated glycovariants of human IgG1–4. Exemplary C1q-binding curves (left), flow cytometry histograms of C1q binding after 60 min (center) and statistics at time point 60 min of at least three independent experiments. Statistical analysis: unpaired two-tailed Student’s t-test, mean ± SEM, ns, not significant, *p < 0.05, **p < 0.01, and ***p < 0.001. MFI, median fluorescence intensity.
Rituximab depletes B cells through a combination of CDC and antibody-dependent cell-mediated cytotoxicity (ADCC), which requires antibody binding to the human-activating FcγRIIIa (CD16) (10, 11). Absence of the IgG-Fc core fucose increases binding to FcγRIIIa (8, 12), a finding increasingly being used to improve the efficacy of therapeutic antibodies (13–15). Two studies reported that Fc-galactosylation results in a slight, albeit not statistically significant increase in FcγRIIIa-binding affinity and ADCC activity (16, 17). A more recent study confirmed that afucosylated glycoforms show higher binding affinities for FcγRIIIa, while Fc-galactosylation did not significantly impact FcγRIIIa binding (18). To investigate whether Fc-galactosylation of IgG isotype glycovariants increases binding affinities of IgG isotypes for FcγRIIIa, thereby potentially enhancing the antibodies’ ability to induce ADCC, we titrated IgG glycovariants onto CHO-derived cell lines recombinantly expressing FcγRIIIa. FcγRIIIa binding was strongest for IgG1 and IgG3 isotypes (Figure 5). IgG-Fc galactosylation showed, however, no effect on FcγRIIIa-binding affinities for any of the IgG isotypes tested (Figure 5). Thus, while increasing the antibodies’ affinity for C1q binding and their efficacy to induce CDC, Fc-galactosylation did not change the affinity of human IgG isotypes for FcγRIIIa which, upon ligation, mediates ADCC.
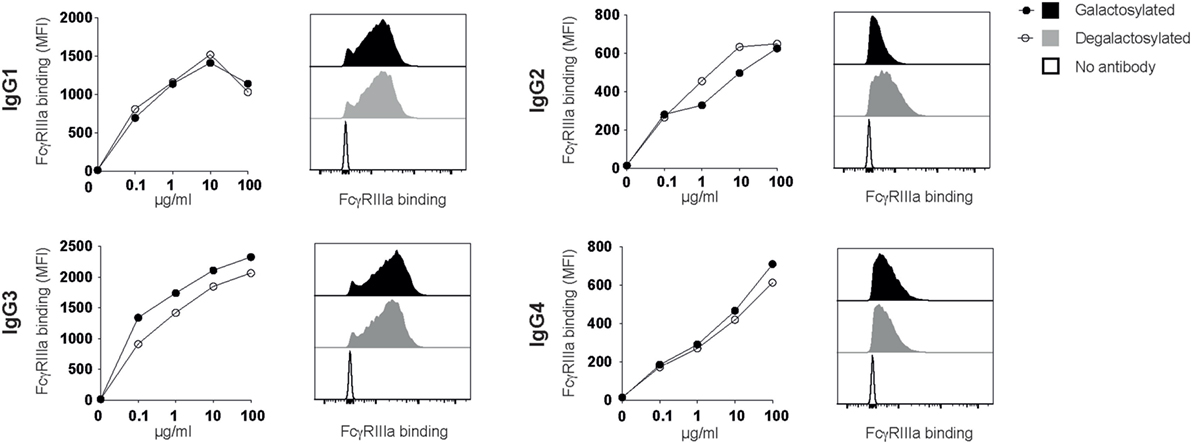
Figure 5. Immunoglobulin gamma (IgG) binding to FcγRIIIa does not require Fc-galactosylation. Binding of galactosylated and degalactosylated IgG isotypes to CHO cells transfected with FcγRIIIa was analyzed by flow cytometry. Binding curves and flow cytometry histograms at 10 µg/ml are shown.
Initiation of the classical complement cascade trough binding of C1q is a potent proinflammatory mechanism by which IgG antibodies trigger immune responses during infection and its deregulation causes tissue damage in a wide array of human inflammatory, degenerative, and autoimmune diseases (19). Our study shows that Fc-galactosylation enhances the efficacy of complement-fixing IgG isotypes to induce CDC through improved binding of C1q.
The antibody-dependent classical complement pathway is initiated if the C1 complex, formed by the multimeric pattern recognition molecule C1q and the modular proteases C1r and C1s, docks on antigen-bound IgG (20). C1q binds to monomeric IgG with very low affinity but antigen-driven antibody clustering allows for the formation of IgG hexamers that bind C1q with high avidity and promote efficient complement activation (21). The N-glycan resides in the CH2 domain, which is required for C1q binding (22). Sites on the surface of human IgG1 that constitute the C1q-binding epicenter are conserved in human IgG isotypes that are deficient in C1q binding and it has therefore been suggested that the composition of the N-glycan might be critical for the antibodies’ conformation and its ability to bind C1q (23). Each biantennary oligosaccharide chain extends one arm toward the CH2–CH3 interface region and the other arm into the space between the CH2 domains, resulting in multiple interactions with the surface of the CH2 domain and the glycan of the opposing CH2 domain, respectively (4). Indeed, optimal C1 activation requires the presence of the Fc-glycan since C1q-mediated effector functions are compromised or lost in aglycosylated or deglycosylated IgGs (24–26). The effect of deglycosylation on reducing C1q binding has recently been attributed to its inhibition of IgG hexamerization via modulation of IgG Fc:Fc interactions rather than reduction of direct C1q-Fc-binding affinities (27). Based on these data and our results, we suggest that Fc-galactosylation modulates Fc:Fc interactions for antigen-bound IgG, thereby improving binding of C1q and increasing the antibodies’ ability to induce classical complement activation and CDC.
We systematically investigated whether Fc-galactosylation facilitates C1q binding and CDC effector functions across all human IgG isotypes. For murine IgG2b and IgG1, it has been demonstrated that addition of terminal galactose increases binding of C1q (28). While presence of terminal galactose enhanced complement activation by CD20 targeting, C1q-fixing human IgG1 and IgG3 isotypes, IgG2 and IgG4 remained deficient in initiating the classical complement cascade indicating that Fc-galactosylation alone is not sufficient for IgG2 and IgG4 to acquire complement-fixing properties.
Rituximab and therapeutic monoclonal antibodies (mAbs) that target tumor cells via ADCC or CDC are approved for the treatment of various cancers (29). B-cell depletion by CD20-targeting antibodies is also widely used for the treatment of autoimmune diseases (30). However, some patients do not sufficiently respond to rituximab therapy (31, 32) and improved versions of B cell depleting antibodies have been developed to increase ADCC activity and improve clinical efficacy (33, 34). Our data indicate that Fc-galactosylation, in addition to the established effect of defucosylation on ADCC, increases target cell cytotoxicity through enhancing CDC. Fc-galactosylation specifically improved binding of C1q to human IgG1 and IgG3 without changing antigen-binding affinities. Our data therefore indicate that Fc-galactosylation should be harnessed in glycoengineering therapeutic antibodies for effective antibody-dependent complement activation and target cell killing.
Human serum complement (Cat. No.: A100; Lot No.: 035997) and C5-depleted human serum (Cat. No.: A501; Lot No.: 031795) were purchased from Quidel® Corporation. Accutase was purchased from StemCell Technologies Inc. (Cat. No.: 07920), Gibco™ RPMI-1640, HEPES (Cat. No.: 52400025), penicillin–streptomycin (P/S; 5,000 U/ml; Cat. No.: 15070063), LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Cat. No.: L34957), LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Cat. No.: L10119), and TO-PRO-3 Stain (Cat. No.: T3605) were purchased from Invitrogen. Fetal calf serum (FCS) was purchased from Sigma-Aldrich (Cat. No.: F7524-500M; Lot No.: 051M3395) and from Bioswisstech (Cat. No.: S0615; Lot No.: 1047D). β1-4 galactosidase (Cat. No.: 345806-50MIU) and UDP-galactose (Cat. No.: 670111-50MG) were purchased from Merck Millipore Corporation, Calbiochem®. Protein-G sepharose was purchased from GE healthcare (Cat. No.: 17-0618-01). Biotinylated Erythrina Cristagalli lectin (Cat. No.: B-1145) and Lens Culinaris Agglutinin (Cat. No.: B-1045) were purchased from Reactolab. Phosphate-buffered saline (PBS; 2.68 mM KCl, 8 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, pH adjusted to 7.3) was produced in-house.
Monoclonal mouse anti-human C1q (Cat. No.: A201) and biotinylated anti-human C1q (Cat. No.: A700) were purchased from Quidel® Corporation. Polyclonal rabbit anti-mouse IgG (H + L)-Alexa Fluor 488 (Cat. No.: A-11059) was purchased from Invitrogen. PE-conjugated streptavidin (Cat. No.: 405203) was purchased from Biolegend.
Raji cells were used as CD20+ target cells to assess CDC. Accordingly, 7 × 104 Raji cells were cultivated in RPMI-1640 containing P/S (50 U/ml) in a humidified incubator (37°C, 5% CO2) in 96-well V-bottom plates. Cells were incubated with titrated concentrations of the respective galactosylated and degalactosylated anti-CD20 antibody isotypes. After 30 min, human serum complement was added to a final concentration of 5% and incubation was continued for another 12 h. Thereafter, cells were washed twice by adding 200 µl cold PBS and centrifugation at 400 × g and 4°C for 5 min. Cells were resuspended in cold PBS and TO-PRO3 stain (final concentration of 200 nM) was added to detect dead cells. All samples were analyzed on a BD FACSCanto-II using FACSDiva v6.1.3 software and FlowJo software v9.3.1 (Tree Star Inc.). Specific lysis was calculated as percent increase in dead cells compared to spontaneous lysis in the absence of an anti-CD20 antibody. EC50 values were calculated via a non-linear regression in GraphPad Prism 5.
A total of 2 × 105 Raji cells were incubated with 10 µg/ml with galactosylated and degalactosylated anti-CD20 antibody isotypes in RPMI-1640 containing 1% P/S (50 U/ml) and 1% C5-depleted human serum for 5, 15, 30, or 60 min in a humidified incubator (37°C, 5% CO2). After incubation with the different anti-CD20 antibody isotypes, cells were washed twice with 200 µl cold PBS, and C1q binding was determined by adding biotinylated anti-C1q antibody (25 µg/ml in PBS) for 1 h on ice followed by washing twice with 200 µl cold PBS and incubation with PE-conjugated streptavidin (1:400 in PBS) and dead cell stain (LIVE/DEAD Fixable Aqua Dead Cell Stain Kit; 1:250 in PBS) for 20 min. After washing twice with 200 µl PBS, samples were analyzed on a BD FACSCanto-II using FACSDiva v6.1.3 software and FlowJo software v9.3.1 (Tree Star Inc.).
Raji cells were maintained in RPMI-1640 containing P/S (50 U/ml) and 10% FCS in a humidified incubator (37°C, 5% CO2). 1.2 × 105 Raji cells and anti-CD20 antibody isotypes and glycovariants were incubated for 25 min on ice. Dead cells were excluded using LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit. Cells were washed twice with 200 µl PBS and anti–mouse IgG (H + L)-Alexa Fluor 488 was added and incubated for 20 min on ice followed by two washing steps with 200 µl cold PBS. All samples were analyzed on a BD FACSCanto-II using FACSDiva v6.1.3 software and FlowJo software v9.3.1 (Tree Star Inc.).
Human FcγRIIIa-expressing CHO cells (35) were kindly provided by Falk Nimmerjahn (Department of Biology, University of Erlangen-Nürnberg, Germany). Cells were maintained in RPMI-1640 containing P/S (50 U/ml) and 10% FCS in a humidified incubator (37°C, 5% CO2). To detect binding of anti-CD20 antibodies, cells were detached using accutase, washed with cold PBS, and 2 × 105 cells were incubated on ice for 30 min with anti-CD20 antibodies. After incubation, cells were washed twice with 200 µl cold PBS and anti-mouse IgG (H + L)-Alexa Fluor 488 was added for 20 min on ice. All samples were washed twice with 200 µl cold PBS and analyzed on a BD FACSCanto-II using FACSDiva v6.1.3 software and FlowJo software v9.3.1 (Tree Star Inc.).
The DNA encoding for the N terminal region of the light chain (amino acids QIVLS until KLEIK) and heavy chain (amino acids QVQLQ until TVSAA) of rituximab (www.drugbank.ca; accession number DB00073) was synthesized by Invitrogen GeneArt gene synthesis. The N terminal region of the light chain was cloned into the multiple cloning site (MCS) of Igκ-AbVec (NCBI GenBank accession number FJ475056.1) using AgeI and BsiWI as previously described (36). To obtain human IgG-1, -2, -3 and -4 heavy chain expression vectors IgG-AbVec (NCBI GenBank accession number FJ475055.1) (36) was modified to encode for human IgG1 (allotype G1m17,1), IgG2 (allotype G2m), IgG3 (allotype G3m), and IgG4 (kindly provided by Lars Hangartner, the Scripps Research Institute, Department of Immunology and Microbial Science, La Jolla, CA, USA) (2, 37, 38). The N terminal region of the rituximab heavy chain was cloned into the MCS using AgeI and SalI as previously described (36). Recombinant monoclonal antibodies were expressed and purified as previously described (10, 32, 39). Briefly, heavy and light chain expression plasmids were cotransfected into a human B cell–epithelial cell fusion cell line (HKB11) supporting high-level recombinant protein expression (40) using calcium phosphate-mediated transfection. After 6 days, antibodies were purified from cell culture supernatants by binding to a protein G column and elution with 0.1 M glycine (pH 2) followed by neutralization with 1 M Tris pH 8.8 and dialysis to PBS. Antibody purity and integrity were confirmed by polyacrylamide gel electrophoresis, coomassie brilliant blue staining and Western blotting with isotype-specific monoclonal antibodies.
Antibody glycovariants were generated as previously described (10, 39). For removal of galactose, antibodies were dialyzed to 50 mM sodium phosphate buffer pH 6.0, 60 mU β1–4 galactosidase was added per mg of antibody and the reaction was incubated for 6 h at room temperature followed by 1 h at 37°C. To add galactose, antibodies were dialyzed to 0.2 M MES pH 6.5 followed by the addition of 10 mM MnCl2, UDP-galactose, 0.02% NaN3, and 5 µg recombinant β1–4 galactosyltransferase [produced in-house (39)]. The reaction was incubated at 37°C for 48 h. Finally, all antibodies were centrifuged for 2 h at 4°C and >20,000 × g to remove aggregates, repurified by gravity-flow protein-G sepharose columns, and dialyzed to PBS. Successful degalactosylation and galactosylation were monitored by lectin-blotting using the galactose-specific lectin from Erythrina Cristagalli.
A p-value of 0.05 or less was defined as statistically significant. For all analyses, Prism software, version 5 (GraphPad Software, Inc.) was used. Specific statistical tests are indicated in the respective figure legends.
IQ and JL conceived the study. JL designed, BP, CK, and IQ designed and performed experiments. PW performed experiments. All the authors cowrote and approved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Lars Hangartner (The Scripps Research Institute, Department of Immunology and Microbial Science, La Jolla, CA, USA) and Michael A. Maurer (Brain Research Institute, University of Zürich) for providing human IgG1–4 expression vectors. We thank Lai-Xi Wang and Tiezheng Li (Department of Chemistry and Biochemistry, University of Maryland, MD, USA) for helpful discussion and reviewing the manuscript.
CK was supported by a scholarship provided by the German Research Foundation (DFG grant KE 1831/1-1) and a Forschungskredit provided by the University of Zurich (FK-14-021). IQ was supported by a DOC scholarship provided by the Austrian Academy of Sciences (ÖAW). JL was supported by the Swiss National Science Foundation (31003A-169664), the Novartis Foundation for medical-biological research, the Sassella Foundation, the Hartmann Müller Foundation, and the Swiss Multiple Sclerosis Society.
1. Shakib F, Stanworth DR. Human IgG subclasses in health and disease. (A review). Part I. Ric Clin Lab (1980) 10:463–79. doi: 10.1007/BF02938793
2. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol (2014) 5:520. doi:10.3389/fimmu.2014.00520
3. Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol (2011) 21:576–82. doi:10.1016/j.sbi.2011.08.005
4. Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol (2003) 325:979–89. doi:10.1016/S0022-2836(02)01250-0
5. Parekh R, Roitt I, Isenberg D, Dwek R, Rademacher T. Age-related galactosylation of the N-linked oligosaccharides of human serum IgG. J Exp Med (1988) 167:1731–6. doi:10.1084/jem.167.5.1731
6. Dekkers G, Plomp R, Koeleman CA, Visser R, von Horsten HH, Sandig V, et al. Multi-level glyco-engineering techniques to generate IgG with defined Fc-glycans. Sci Rep (2016) 6:36964. doi:10.1038/srep36964
7. Bakovic MP, Selman MH, Hoffmann M, Rudan I, Campbell H, Deelder AM, et al. High-throughput IgG Fc N-glycosylation profiling by mass spectrometry of glycopeptides. J Proteome Res (2013) 12:821–31. doi:10.1021/pr300887z
8. Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem (2002) 277:26733–40. doi:10.1074/jbc.M202069200
9. Mizushima T, Yagi H, Takemoto E, Shibata-Koyama M, Isoda Y, Iida S, et al. Structural basis for improved efficacy of therapeutic antibodies on defucosylation of their Fc glycans. Genes Cells (2011) 16:1071–80. doi:10.1111/j.1365-2443.2011.01552.x
10. Quast I, Keller CW, Maurer MA, Giddens JP, Tackenberg B, Wang LX, et al. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J Clin Invest (2015) 125:4160–70. doi:10.1172/JCI82695
12. Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem (2003) 278:3466–73. doi:10.1074/jbc.M210665200
13. Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, et al. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat Biotechnol (2006) 24:210–5. doi:10.1038/nbt1178
14. Yamane-Ohnuki N, Satoh M. Production of therapeutic antibodies with controlled fucosylation. MAbs (2009) 1:230–6. doi:10.4161/mabs.1.3.8328
15. Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci (2009) 30:356–62. doi:10.1016/j.tips.2009.04.007
16. Dashivets T, Thomann M, Rueger P, Knaupp A, Buchner J, Schlothauer T. Multi-angle effector function analysis of human monoclonal IgG glycovariants. PLoS One (2015) 10:e0143520. doi:10.1371/journal.pone.0143520
17. Thomann M, Schlothauer T, Dashivets T, Malik S, Avenal C, Bulau P, et al. In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and ADCC activity. PLoS One (2015) 10:e0134949. doi:10.1371/journal.pone.0134949
18. Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX. Modulating IgG effector function by Fc glycan engineering. Proc Natl Acad Sci U S A (2017) 114(13):3485–90. doi:10.1073/pnas.1702173114
19. Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol (2014) 32:433–59. doi:10.1146/annurev-immunol-032713-120154
20. Mortensen SA, Sander B, Jensen RK, Pedersen JS, Golas MM, Jensenius JC, et al. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc Natl Acad Sci U S A (2017) 114:986–91. doi:10.1073/pnas.1616998114
21. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science (2014) 343:1260–3. doi:10.1126/science.1248943
22. Duncan AR, Winter G. The binding site for C1q on IgG. Nature (1988) 332:738–40. doi:10.1038/332738a0
23. Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, et al. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol (2000) 164:4178–84. doi:10.4049/jimmunol.164.8.4178
24. Allhorn M, Briceno JG, Baudino L, Lood C, Olsson ML, Izui S, et al. The IgG-specific endoglycosidase EndoS inhibits both cellular and complement-mediated autoimmune hemolysis. Blood (2010) 115:5080–8. doi:10.1182/blood-2009-08-239020
25. Yang R, Otten MA, Hellmark T, Collin M, Bjorck L, Zhao MH, et al. Successful treatment of experimental glomerulonephritis with IdeS and EndoS, IgG-degrading streptococcal enzymes. Nephrol Dial Transplant (2010) 25:2479–86. doi:10.1093/ndt/gfq115
26. Jefferis R. Isotype and glycoform selection for antibody therapeutics. Arch Biochem Biophys (2012) 526:159–66. doi:10.1016/j.abb.2012.03.021
27. Wang G, de Jong RN, van den Bremer ET, Beurskens FJ, Labrijn AF, Ugurlar D, et al. Molecular basis of assembly and activation of complement component C1 in complex with immunoglobulin G1 and antigen. Mol Cell (2016) 63:135–45. doi:10.1016/j.molcel.2016.05.016
28. Nimmerjahn F, Anthony RM, Ravetch JV. Agalactosylated IgG antibodies depend on cellular Fc receptors for in vivo activity. Proc Natl Acad Sci U S A (2007) 104:8433–7. doi:10.1073/pnas.0702936104
29. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer (2012) 12:278–87. doi:10.1038/nrc3236
30. Pers YM, Jorgensen C. Perspectives of ofatumumab as CD20 targeted therapy in rheumatoid arthritis and other autoimmune diseases. Immunotherapy (2016) 8:1091–6. doi:10.2217/imt-2016-0003
31. Rezvani AR, Maloney DG. Rituximab resistance. Best Pract Res Clin Haematol (2011) 24:203–16. doi:10.1016/j.beha.2011.02.009
32. Maurer MA, Rakocevic G, Leung CS, Quast I, Lukacisin M, Goebels N, et al. Rituximab induces sustained reduction of pathogenic B cells in patients with peripheral nervous system autoimmunity. J Clin Invest (2012) 122:1393–402. doi:10.1172/JCI58743
33. Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, Theriault J, et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res (2010) 70:4481–9. doi:10.1158/0008-5472.CAN-09-3704
34. Golay J, Da Roit F, Bologna L, Ferrara C, Leusen JH, Rambaldi A, et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood (2013) 122:3482–91. doi:10.1182/blood-2013-05-504043
35. Lux A, Yu X, Scanlan CN, Nimmerjahn F. Impact of immune complex size and glycosylation on IgG binding to human FcgammaRs. J Immunol (2013) 190:4315–23. doi:10.4049/jimmunol.1200501
36. Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, et al. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc (2009) 4:372–84. doi:10.1038/nprot.2009.3
37. Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods (2008) 329:112–24. doi:10.1016/j.jim.2007.09.017
38. Wyrzucki A, Bianchi M, Kohler I, Steck M, Hangartner L. Heterosubtypic antibodies to influenza A virus have limited activity against cell-bound virus but are not impaired by strain-specific serum antibodies. J Virol (2015) 89:3136–44. doi:10.1128/JVI.03069-14
39. Quast I, Maurer MA, Lunemann JD. Generation of IgG-Fc glycovariants using recombinant glycosidases and glycosyltransferases. BioProtoc (2016) 6:e1886. doi:10.21769/BioProtoc.1886
Keywords: c1q, complement system proteins, antibodies, monoclonal, glycan, IgG subclasses
Citation: Peschke B, Keller CW, Weber P, Quast I and Lünemann JD (2017) Fc-Galactosylation of Human Immunoglobulin Gamma Isotypes Improves C1q Binding and Enhances Complement-Dependent Cytotoxicity. Front. Immunol. 8:646. doi: 10.3389/fimmu.2017.00646
Received: 28 March 2017; Accepted: 17 May 2017;
Published: 06 June 2017
Edited by:
Uday Kishore, Brunel University London, United KingdomReviewed by:
Robert Braidwood Sim, University of Leicester, United KingdomCopyright: © 2017 Peschke, Keller, Weber, Quast and Lünemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan D. Lünemann, amFuLmx1ZW5lbWFubkB1emguY2g=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.