 Michael J. Carter
Michael J. Carter Ruth M. Mitchell
Ruth M. Mitchell Patrick M. Meyer Sauteur
Patrick M. Meyer Sauteur Dominic F. Kelly
Dominic F. Kelly Johannes Trück
Johannes Trück
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 June 2017
Sec. B Cell Biology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00630
Despite the availability of advances in molecular diagnostic testing for infectious disease, there is still a need for tools that advance clinical care and public health. Current methods focus on pathogen detection with unprecedented precision, but often lack specificity. In contrast, the host immune response is highly specific for the infecting pathogen. Serological studies are rarely helpful in clinical settings, as they require acute and convalescent antibody testing. However, the B cell response is much more rapid and short-lived, making it an optimal target for determining disease aetiology in patients with infections. The performance of tests that aim to detect circulating antigen-specific antibody-secreting cells (ASCs) has previously been unclear. Test performance is reliant on detecting the presence of ASCs in the peripheral blood. As such, the kinetics of the ASC response to infection, the antigen specificity of the ASC response, and the methods of ASC detection are all critical. In this review, we summarize previous studies that have used techniques to enumerate ASCs during infection. We describe the emergence, peak, and waning of these cells in peripheral blood during infection with a number of bacterial and viral pathogens, as well as malaria infection. We find that the timing of antigen-specific ASC appearance and disappearance is highly conserved across pathogens, with a peak response between day 7 and day 8 of illness and largely absent following day 14 since onset of symptoms. Data show a sensitivity of ~90% and specificity >80% for pathogen detection using ASC-based methods. Overall, the summarised work indicates that ASC-based methods may be very sensitive and highly specific for determining the etiology of infection and have some advantages over current methods. Important areas of research remain, including more accurate definition of the timing of the ASC response to infection, the biological mechanisms underlying variability in its magnitude and the evolution and the B cell receptor in response to immune challenge. Nonetheless, there is potential of the ASC response to infection to be exploited as the basis for novel diagnostic tests to inform clinical care and public health priorities.
The production of pathogen-specific antibody by B lymphocytes (B cells) is of key importance for protection from infection. Naive B cells are activated through interaction with foreign antigen, cognate T cell receptors (TCRs) (1), pattern-recognition receptors (2), and cytokines (3) to form antigen-specific antibody-secreting cells (ASCs; plasmablasts and plasma cells), memory B cells and other subsets (4, 5) (Figure 1). Increasing insights into the cellular aspects of humoral immunity are emerging through a focus on plasmablasts. These are ASCs produced following antigen challenge that transiently circulate through peripheral blood before migrating to secondary lymphoid organs or bone marrow, or undergoing apoptosis. Plasmablasts thus represent an accessible and measurable subset of ASCs only detectable during an acute immune response.
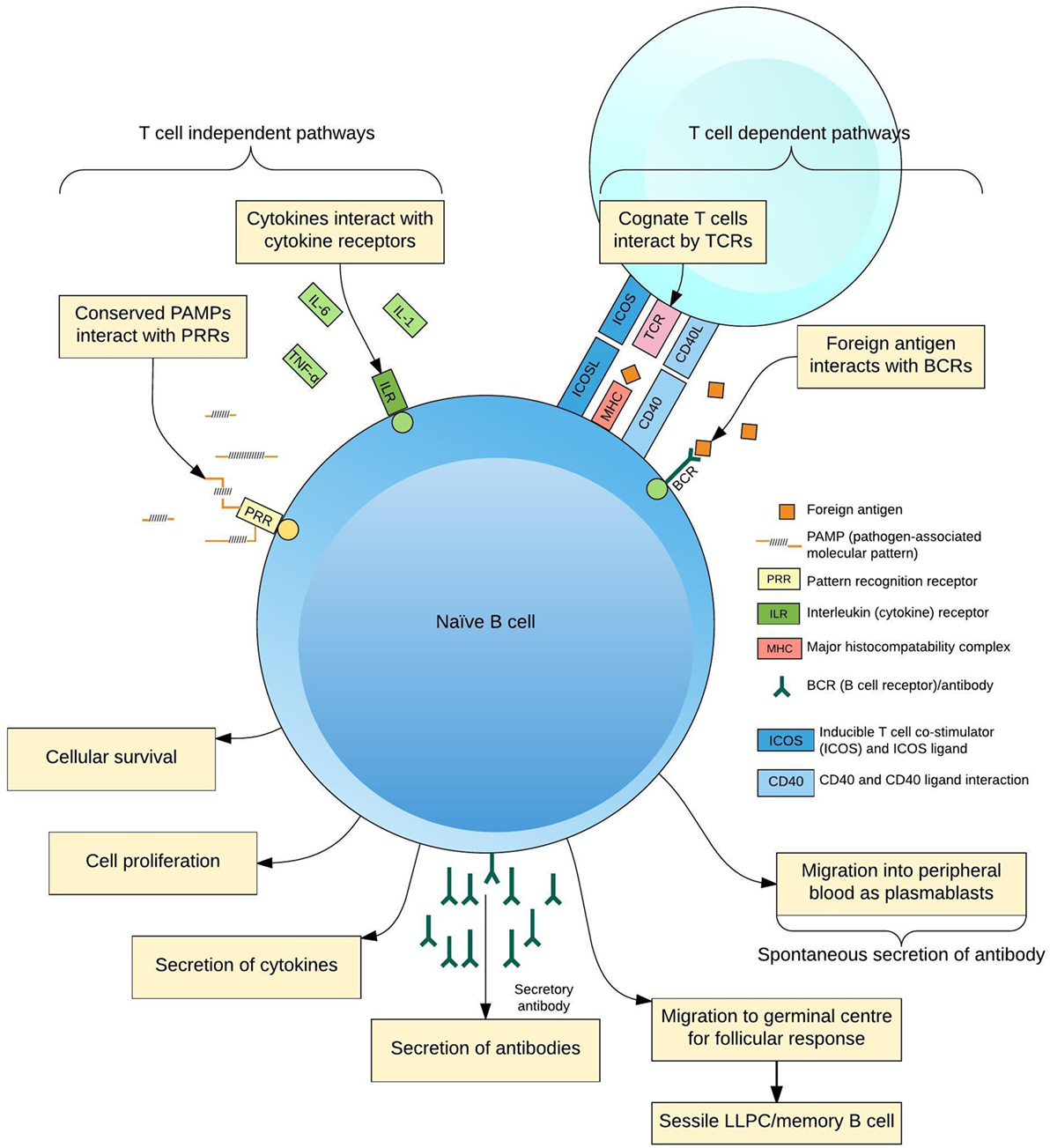
Figure 1. Differentiation of B cells in response to antigen. B cells receive activating signals by at least two pathogen non-specific mechanisms, pattern-recognition receptors (PRRs), and the cytokine context of the immune response; and two antigen-specific mechanisms, interactions with cognate T cells via the T cell receptor (TCR) and other ligands and B cell receptors (BCR)–antigen interaction. Integration of these signals predisposes the fate of the maturing B cell to a certain phenotype for the lifespan of the cell, simplistically resulting in plasmablasts, long-lived plasma cells (LLPCs), and memory B cells.
Measurements of plasmablasts have been used extensively to describe the humoral immune response following vaccination (6). Measurements of plasmablasts may also provide data to inform the humoral response to infection and may translate into novel diagnostic tests. The development of novel diagnostic tests based on the plasmablast response to infection may circumvent difficulties with culture of organisms (usually lack of sensitivity due to difficulty accessing tissue and prior antibiotic administration), serological testing (usually lack of specificity unless a convalescent sample is also taken), or PCR-based methods (often lack of specificity, excepting testing of CSF). Additionally, the B cell receptors (BCRs) of plasmablasts are membrane-bound antibodies. As such, these BCRs could be used to inform vaccine development or to develop therapeutic monoclonal antibodies (7). Such therapeutic antibodies may be particularly important in the context of emerging infections or increasing antimicrobial resistance.
Antibody specificity and isotype and timing of the plasmablast response to infection are key to accurate measurements. However, these data have not previously been systematically reviewed. In this review we describe the data that exist on the timing and magnitude (kinetics) of the antigen-specific B cell responses following infection in humans. Our focus on ASCs (or where possible, the more specific subset of plasmablasts) reflects their importance in the production of antibodies, their dynamic numbers, and their transience in the peripheral blood: aspects of fundamental importance to their clinical applications. Where generated, memory B cells tend to persist at a low frequency; as so, they are not our focus, but have been recently reviewed in detail (8).
Search strategy. We searched MEDLINE with combinations of the following terms “B cell,” “plasmablast,” “plasma cell,” “antibody-secreting cell” AND “infection” AND “kinetics,” “dynamics.” We identified peer-reviewed articles of interest, and conducted further searches for terms identified as important (e.g. “antibodies lymphocyte supernatant,” “ELISpot,” “B cell receptor sequencing” in the context of infection).
Comparison of the ASC response to natural infection across studies is limited by varying definitions of ASC subsets. Here we use the term ASC to include all ASCs as defined by ex vivo assays such as enzyme-linked immunospot assay (ELISpot; below). We use the term plasmablast for ASCs that depict recognized cell surface markers for this B cell subset following immunophenotyping (9). We have eschewed the term “plasma cells” for clarity, since we will not discuss (usually sessile) long-lived plasma cells further. In general, numbers of ASCs are described as number per unit of blood, as a proportion of peripheral blood mononuclear cells (PBMCs), or as a proportion of peripheral blood B cells, as dictated by the techniques available to research teams (10). Antigen-specific ASCs may be further defined as a proportion of total (isotype-specific) ASCs (7), with varying definitions of antigen specificity.
Fluorescence-based (flow) cytometry distinguishes immunophenotypes of cells by the binding of fluorescing monoclonal antibodies to defined cell surface markers. Thus, flow cytometry allows investigators to sort plasmablasts from other PBMCs. Markers of recent proliferation may be used to enrich samples for acutely proliferated plasmablasts, which may enhance specificity for the etiological diagnosis of acute infection (11). Replicability of experiments over time and between laboratories is paramount. As such, optimized panels of reagents for identifying specific populations and automated gating strategies have been developed (12).
ELISpot identifies subsets of cells by the binding of antibody to a chosen membrane-bound antigen. The addition of a substrate causes a color change where bound antibody is present, with the appearance of a spot corresponding to a single antigen-specific ASC (13). ELISpot is thus a highly sensitive technique because individual cells can be easily identified and counted. ELISpot is adaptable and can be applied to populations of cells that are either sorted by flow cytometry, or PBMCs separated from whole blood using density-dependent centrifugation. Although here we describe the detection of antigen-specific ASCs by the use of ELISpot and assay of antibody from lymphocyte supernatant (ALS) (Table 1), ELISpot has also been used for the immunophenotyping of B cell subsets for a variety of non-immunoglobulin markers (14). Unlike flow cytometry, ELISpot is a robust technique and can be used in laboratories in a variety of settings (10). A constitutive limitation to ELISpot, is the need for either PBMCs that have been recently sampled, or PBMCs that have been cryopreserved (15). Since antigen-specific ASCs form only a small proportion of PBMCs, even during the peak of an immune response, the number of antigen-specific ASCs limits the number of antigens that can be assayed. In addition, if fresh PBMCs are used, ELISpot plates must be prepared with predetermined antigens.
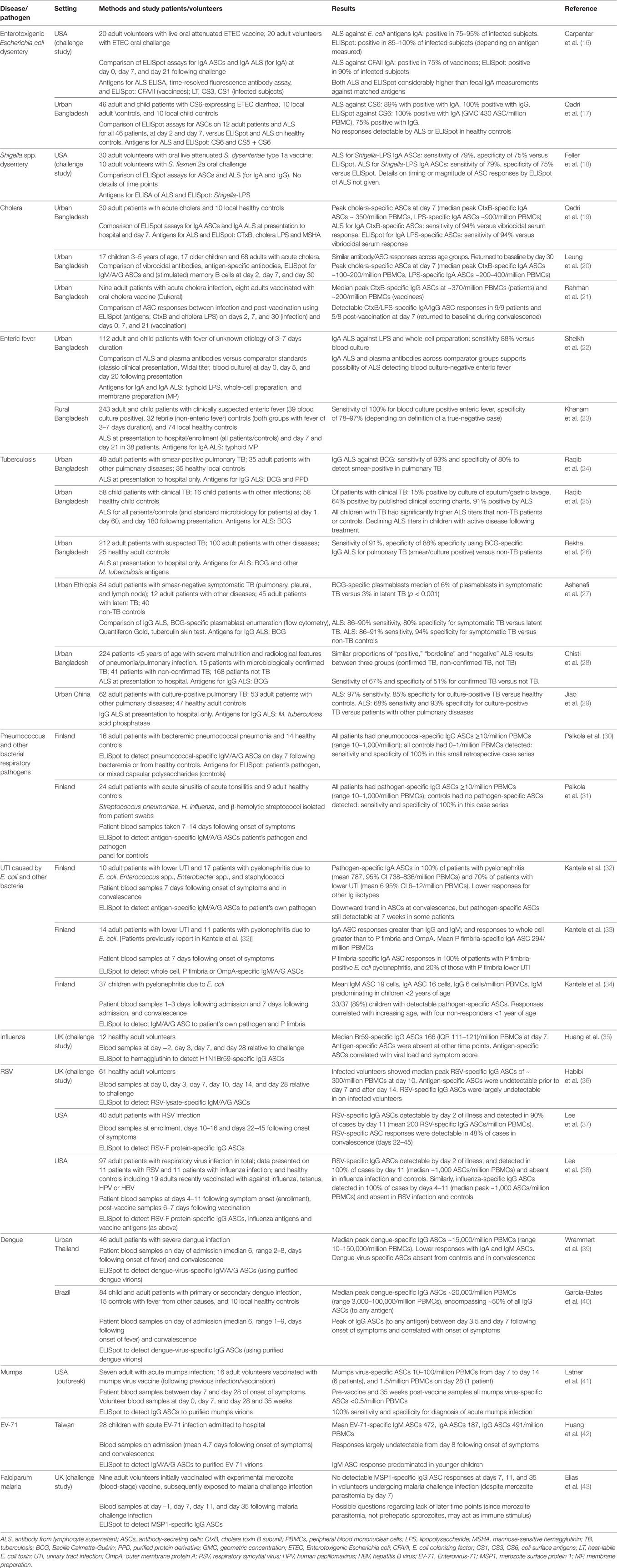
Table 1. Studies investigating the use of ELISpot and ALS for the diagnosis of infection.
Assay of ALS measures the sum of antibodies produced by ASCs during a period of in vitro cell culture. The method is simple: peripheral blood is sampled, and PBMCs are separated by density-dependent centrifugation, washed, and incubated for 24–48 h in cell culture media (44). The resulting supernatant is enriched for antibodies that were secreted during ex vivo incubation by recently activated ASCs. As with ELISpot, this technique has potential to be developed into a diagnostic test for the etiology of acute infections and has already been applied with some success to clinical tuberculosis (26, 27, 45) and enteric fever (22, 23), as well as vaccine responses (16) (Table 1). Although frozen cells can also be used for the ELISpot (15), ALS samples can be more easily frozen and transported to centralized laboratories, allowing responses from many antigens from multiple pathogens to be assayed. This may be of importance given progress in serological techniques (46) and the re-emphasized need for seroepidemiology in the context of emerging infectious diseases (47). Important questions regarding optimal timing of samples relative to onset of symptomatic infection and its sensitivity in comparison to ELISpot, remain.
At present, the use of ALS and ELISpot to detect antigen-specific ASCs for the diagnosis of infection is largely limited to research laboratories, with the exception of an established T cell ELISpot test for tuberculosis [T-spot. TB (48)]. This reflects the need for rigorous standardization of laboratory procedures to detect antigen-specific ASCs (or their secretions) prior to clinical trials or implementation into clinical practice. ALS and ELISpot also require techniques that, although simple, will be unfamiliar to the majority of microbiology technicians such as the separation and washing of PBMCs from fresh blood. ELISpot plates with predetermined antigens for use in low-technology settings (10), or the use of ALS (44), which can be transported relatively simply to reference laboratories for ELISA, have been adapted to these ends.
An alternative to measuring antibodies (and other proteins) produced in response to infection is to measure the upstream transcription of genes across the genome. As such, measurement of the abundance of RNA species in whole blood or PBMCs using gene expression microarrays have been used to define the transcriptome in response to vaccination or infection. Theoretically, this approach is “hypothesis-free” regarding genes that may be differentially regulated in response to immunological challenge and relatively unbiased regarding the detection of genes (49). Additionally, differential gene expression precedes translation of proteins, and therefore may define immune response to infection prior to the detection of antigen-specific ASCs or antibodies in blood. Such gene-level data from microarray experiments can accurately distinguish between bacterial and viral infections at time of presentation to hospital (50–52) and may potentially distinguish within groups of bacterial or viral infections. Gene-level data may also be used to investigate immunological pathways activated by vaccination or colonization/infection (53). However, microarrays are inherently unable to define the hypervariable genes encoding the heavy (IGH) and light (IGL/IGK) chains of the BCR, and do not inform on the antigen specificity of the ASC response. Thus RNA microarrays are unlikely to distinguish between closely related infections or between serotypes of the same infecting pathogen.
The recent development of next-generation sequencing has facilitated investigation of the ASC response to immunization and infection at a fundamental genetic level. Sequencing of genes encoding the BCR has resulted in the successful production of monoclonal antibodies to influenza (7). Further, convergence of sequences encoding the BCR has been described in populations of B cells in the blood of adults following vaccination (54–57). The clinical implications and challenges of these novel techniques are discussed below.
B cell subsets in peripheral blood are dynamic populations. A detailed knowledge of ASC kinetics is therefore paramount when sampling blood to investigate the ASC response to infection (Figure 2). ASC responses to natural infection may be dependent upon host genetic factors (58, 59), the site of antigen presentation (mucosal surfaces, secondary lymphoid organs, or systemic circulation), the type or quantity of antigen to which the host is exposed, and integration of other immune signals (derived from T cells, pattern-recognition receptors, and cytokines). Onset of natural infection is typically difficult to define, particularly since mucosal colonization with a pathogenic organism is likely to be a necessary precursor to many diseases (60–62). Practical limitations on sampling blood from patients with infections have limited the temporal resolution of studies of the ASC response to infection. These challenges are reduced in vaccine studies (6), where a known measure of antigen is administered at a specific time. The broad themes emerging from detailed data on ASC responses to vaccines can therefore be used as a point of comparison with data from the ASC response to infection.
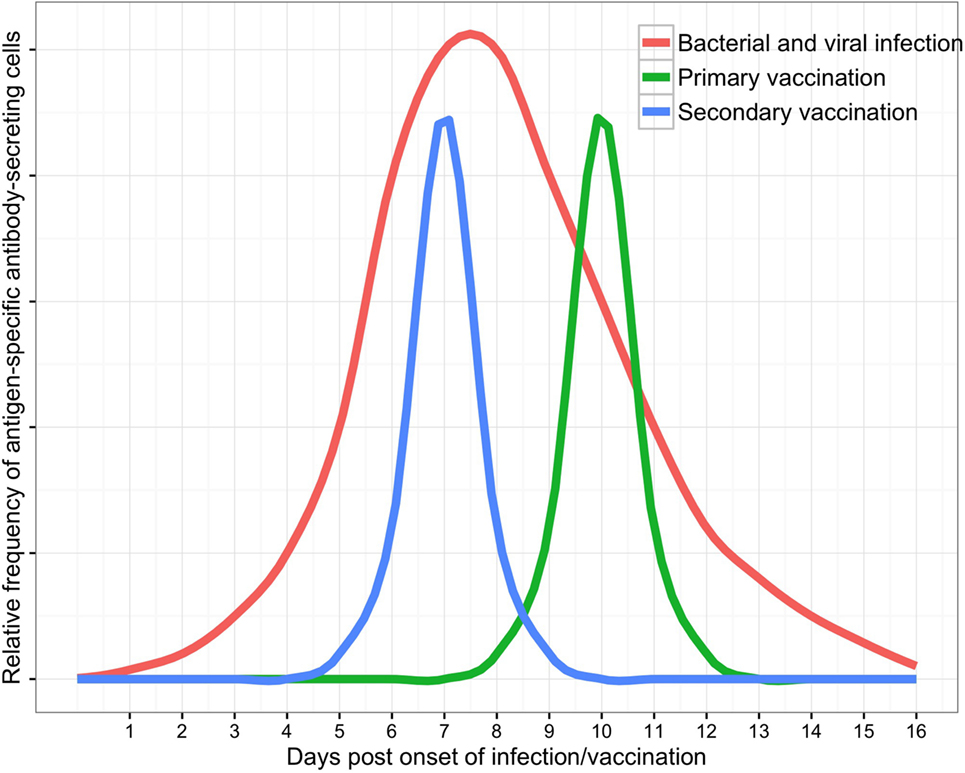
Figure 2. Schematic of antibody-secreting cell responses to infection as measured in peripheral blood (with supporting data and variability discussed in the text).
The timing of the ASC response to vaccination is consistent between most individuals. Vaccination of adult volunteers with polysaccharide–protein conjugate intramuscular (IM) vaccines induced peak antigen-specific ASC responses at 10 days following vaccination of non-primed individuals. In primed individuals, the peak ASC responses occurred earlier, at a median of 7 days following vaccination (6). These adult volunteers had a median frequency of antigen-specific ASCs of ~50% of all ASCs in peripheral blood at peak time points (13, 63–65). Similarly, vaccination with protein IM vaccines induced peak antigen-specific ASC responses at 6–7 days following vaccination in primed adult volunteers. However, the magnitude of IgG antigen-specific responses varied considerably between immunoglobulin subclasses, and between antigens and were highest against tetanus toxoid (66, 67), followed by pertactin (pertussis antigen) (66) and hemagglutinin (influenza antigen) and lowest for diphtheria toxoid (63) and hepatitis B surface protein (56). Moreover, responses varied between individuals in most studies by an order of magnitude.
The timing and magnitude of the ASC response to mucosal vaccines are similar to that induced by IM vaccines and appear conserved between bacterial and viral antigens. Antigen-specific ASC responses to killed oral cholera vaccine (OCV, trade name Dukoral) were measurable in peripheral blood at days 6–7 but not at day 3 or day 28 following vaccination of adults in Bangladesh (21). Similarly, antigen-specific ASC responses to live attenuated influenza vaccine were measurable in blood between day 7 and day 12 following intranasal administration to adults (68). Interestingly, both of these mucosal vaccines induced an antigen-specific IgG ASC response that was 2–3 times higher than the corresponding IgA ASC response, suggesting a systemic immune response to these localized mucosal vaccine challenges.
The timing and magnitude of ASC responses may differ between vaccination (particularly with non-live vaccines) and infection. The details of studies of ASC responses to infection with bacteria, viruses, and malaria are described in Tables 1 and 2. Two studies have directly compared the ASC response to vaccination and infection: enterotoxigenic Escherichia coli (ETEC) vaccine and challenge infection and OCV and natural infection with Vibrio cholerae.
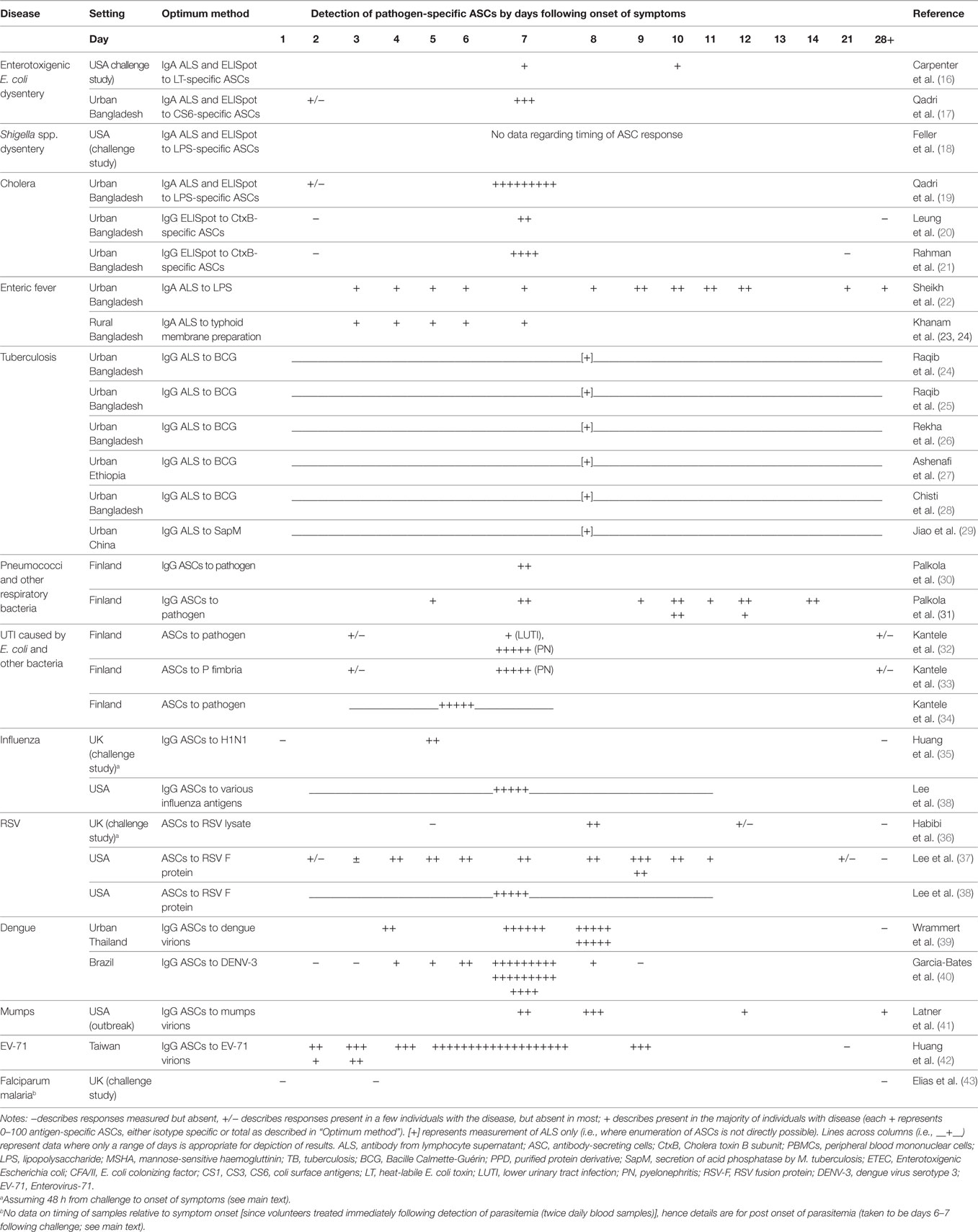
Table 2. Detection of antigen-specific ASCs via ELISpot or ALS by disease/pathogen and time point of sampling relative to onset of symptoms.
Antigen-specific ASC responses to the oral attenuated ETEC vaccine in 20 healthy adult volunteers were compared to antigen-specific ASC responses to challenge with oral non-attenuated ETEC in a further 20 healthy adult volunteers in the USA (16). ELISpot to ETEC vaccine or non-attenuated ETEC antigens was used to assay ASC responses at days 7 and 10. This study also compared the utility of assay of ALS to predict ASC responses. Volunteers who received the ETEC vaccine showed an IgA peak response against anti-colonization factor A II at day 7, which was similar to the IgA peak response against anti-heat-labile toxin in volunteers with non-attenuated ETEC infection. ASC responses to the ETEC vaccine and non-attenuated ETEC infection remained elevated between day 7 and day 10. Assay of IgA ALS showed 75–95% sensitivity versus ELISpot (with a twofold increase from baseline defined as a positive result) for both responders to ETEC vaccine and non-attenuated ETEC infection at day 7. For assay of IgA ALS, sensitivity was reduced at day 10 in comparison to day 7.
Antigen-specific ASC responses to OCV (Dukoral) in eight adults were compared to antigen-specific ASC responses in nine adults with culture-confirmed V. cholerae O1 diarrheal disease (21). Median peak anti-cholera toxin (CtxB)-specific IgG ASC responses were detectable in both groups at day 7 following vaccination or onset of illness. Vaccinees tended to have lower median anti-CtxB-specific IgG ASCs than patients. Median peak anti-LPS-specific IgG ASC responses were also lower in vaccinees than patients. An IgG ASC response was predominant to CtxB, and an IgA ASC response was predominant to LPS in both vaccinees and patients. Flow cytometry demonstrated that ~50% of antigen-specific ASCs of any isotype expressed markers for gut homing plasmablasts. These cholera-specific plasmablasts may thus form the basis for gut-specific immunity following cholera infection.
In general, antigen-specific ASC responses appeared lower in adults following vaccination versus adults with challenge/natural infection, at least for ETEC and cholera. This might be expected if infection induces coactivation of pattern-recognition receptors or produces a pro-ASC cytokine milieu in comparison to vaccination. One might hypothesize that the antigen-specific ASC response to infection might be over a broader timescale than that in response to vaccination. However, these comparative studies had too few time points to be able to detect this.
Other work, primarily from the International Center for Diarrheal Disease Research in Bangladesh, has used ELISpot and assay of ALS to further define the antigen-specific ASC response in cohorts of patients with ETEC, cholera, typhoid, and tuberculosis infections.
The antigen-specific ASC response was assessed in 46 adult patients with ETEC infection and 10 adults and 10 child healthy controls in Bangladesh (17). An IgA ASC response against colonization factor CS6 could be detected as early as 2 days following onset of diarrhea and then increased 30-fold by day 7. Healthy controls had no detectable antigen-specific IgA ASCs. In comparison, CS6-specific IgG ASCs were also detectable at day 2, increased sixfold at day 7, and were undetectable in healthy controls. All patients with ETEC infection showed IgA ASC responses, but only 75% of patients showed IgG ASC responses. Compared to ELISpot, assay of ALS to both Ig isotypes showed that IgA ALS had a sensitivity of 89% for detection of antigen-specific IgA ASC, and IgG ALS had a sensitivity of 100% for antigen-specific IgG ASC responses.
The IgA ASC response to cholera infection was assessed in 30 adult patients with cholera infection and 10 healthy adult controls (19). The antigens CtxB, mannose-sensitive hemagglutinin, and lipopolysaccharide (LPS) were used in ELISpot and assay of ALS. Patients showed large and significant increases in mean IgA ASC responses against all three antigens between day 2 and day 7 following onset of infection and were undetectable in healthy controls. ALS responses correlated well with antigen-specific ASC responses for all Ig isotypes. Similar large increases in concentrations of antigen-specific IgA ALS were shown for patients between day 2 and day 7 following onset of symptoms, and undetectable in healthy controls. Both ELISpot for antigen-specific IgA ASCs and IgA ALS were compared to a gold standard of a fourfold rise in anti-vibriocidal antibodies at day 7 or day 21. Antigen-specific IgA ASCs showed a maximum sensitivity of 94% (anti-LPS), and IgA ALS showed a maximum sensitivity of 94% (anti-CtxB ALS). ASCs were undetectable by both ELISpot and assay of ALS in healthy controls chosen from this cholera-endemic region, demonstrating 100% specificity in this setting.
Recent work has showed that children as young as 3 years of age produce antigen-specific IgA ASC, IgA memory B cell, and plasma IgA and IgG vibriocidal antibodies comparable to adults following cholera infection (20). The OCV Dukoral also provides protection from cholera infection in children between 1 and 4 years of age (69). Infants and young children are disproportionately affected by diarrheal disease and pneumonia (discussed below). Both of these are primarily mucosal infections, of which large proportions are vaccine preventable. Further studies of the antigen-specific IgA and IgG ASC response in this age group are therefore needed.
Infection with Salmonella enterica serovar Typhi or Paratyphi occurs via the faeco-oral route. Unlike cholera, invasion beyond the intestinal mucosa may cause a systemic illness referred to as enteric fever. The antigen-specific ASC response to typhoid/paratyphoid is of considerable interest for the development of diagnostic tests with increased sensitivity in comparison to culture of blood (22, 23). Typhoid-specific ASC responses may also inform the development of new typhoid vaccines (70).
Antigen-specific ASC responses were assessed in 112 adult patients in Bangladesh with suspected enteric fever and 3–7 days of fever at presentation to hospital. ALS assay was done at presentation, day 5 and day 20, against the typhoid antigens LPS, whole-cell preparation, and membrane preparation (MP) (22). Patients were classified into comparator standards for their a priori likelihood of true typhoid infection upon the basis of blood culture results, serum Widal titers, and clinical features, with the addition of a healthy control group. A positive (≥2 standard deviations above the mean of healthy controls) anti-MP IgA ALS was detected in all patients with blood culture-positive typhoid infection. Mean IgA (and IgG) ALS responses at day 5 increased across comparator standards of blood culture-negative patients as the likelihood of true typhoid infection increased. Mean ALS responses had returned to near baseline by day 20.
A further cohort of 243 patients in Bangladesh from 1 to 59 years of age with suspected enteric fever and 3–7 days of fever were assessed for ASC responses with similar ALS assay methods and time points (23). Similarly, patients were classified into comparator standards for their a priori likelihood of true typhoid infection, with the addition of age-matched healthy controls (74 people). As previously, all patients with blood culture-positive typhoid (or paratyphoid) had positive anti-MP IgA ALS responses. Healthy controls all had negative ALS responses. The proportion of patients with positive ALS responses increased as the likelihood of true enteric fever increased across comparator standards. Of the 38 patients with positive anti-MP IgA ALS response, 32 (84%) had converted to a negative response by day 21 following enrollment (days 24–28 of illness). Five of the remaining six patients continued to be bacteremic for typhoid.
Taken together, the data from these cohorts suggest that the assay of anti-MP IgA ALS responses is a sensitive test for the diagnosis of enteric fever. In the absence of a single gold standard, the increasing proportion of positive ALS responses as the likelihood of enteric fever increased in comparator standards suggests that assay of ALS may also be specific for enteric fever.
Tuberculosis is typically a chronic intracellular infection due to Mycobacterium tuberculosis. Protective immune responses against tuberculosis are generally thought to be mainly T cell-mediated (71). A B cell response with anti-tuberculin IgG may prevent reactivation following containment within tuberculous granulomas (72–74), suggesting that both ASCs and other B cell functions are also required for protective immunity. Despite its global importance, vaccination against tuberculosis using Bacille Calmette-Guérin (BCG) has poor efficacy against pulmonary tuberculosis (the most common clinical manifestation). Tuberculosis remains difficult to diagnose, particularly in those living with HIV infection, the malnourished, and young children. The use of the antigen-specific ASC response to tuberculosis to assess novel vaccines and for diagnosis of disease is thus of considerable interest.
Forty-nine adult patients with sputum smear-positive active pulmonary tuberculosis in Bangladesh were assessed by assay of IgG ALS to BCG antigens (24). Patients with active tuberculosis had mean BCG-specific IgG responses that were almost an order of magnitude higher than the comparator standards of patients with other lung diseases (35 patients, with bronchiectasis, lung cancer, lung abscess, and aspergillosis) and healthy controls (35 BCG vaccinated adults). Optimization of the ELISA threshold value for positive ALS responses gave a sensitivity of 92% and specificity of 80%. These findings were validated in a further cohort of 212 adult patients with suspicion of pulmonary tuberculosis and classes of patients with other lung infections and healthy controls as comparator standards (26). Here, numerous M. tuberculosis antigens were assessed for the detection of anti-tuberculosis IgG ALS in ELISAs. Using a threshold value similar to that of the previous study, assay of BCG-specific IgG ALS gave a sensitivity of 90% and specificity of 88% for the diagnosis of active tuberculosis.
Assay of ALS has also been studied for the diagnosis of active pulmonary tuberculosis in children in Bangladesh (25). In this study, children with chronic febrile illnesses aged 11 months to 15 years of age were classified as tuberculosis patients (58 patients) or non-tuberculosis patients (16 patients, in whom tuberculosis was initially considered as a differential diagnosis, but ultimately diagnosed with Hodgkin’s lymphoma, intestinal carcinoma, non-specific lymphadenitis, pneumonia, and pneumonitis). Additionally, 58 age-matched healthy control children were enrolled. BCG was used as the antigen for the detection of anti-tuberculosis ALS. Using an ELISA threshold value similar to that of adult studies, the sensitivity of assay of IgG ALS to BCG antigens at baseline was 91%. In comparison, standard clinical scoring charts over 6 months of follow-up had a sensitivity of 68%. All non-tuberculosis patients and healthy control children had negative ALS responses, suggesting a very high specificity of the ALS assay. Anti-BCG IgG ALS titers had reduced significantly by days 60 and 180 in patients with (now treated) tuberculosis, with the median response less than the positive cutoff value.
A more recent study (28), again from Bangladesh, assessed the use of assay of ALS to BCG antigens for the diagnosis tuberculosis in 224 children admitted to hospital with radiological features of pneumonia and acute severe malnutrition. In this group of patients, using a similar ELISA threshold, sensitivity of assay of ALS was 67% and specificity 45% for the diagnosis of tuberculosis. Time points for blood sampling were not reported. It is unclear whether the relatively unselected nature of this cohort, the timing of sampling relative to treatment, or a reduction in immunity secondary to chronic malnutrition, reduced sensitivity and specificity of assay of ALS in this cohort in comparison to previous studies.
In addition to assay of ALS, ELISpot may also be useful for the diagnosis of tuberculosis. ELISpot was used to enumerate IgA ASCs and memory B cells from the blood of patients with active tuberculosis, adults with latent tuberculosis, and healthy control adults in Uganda (45). Four tuberculosis antigens (ESAT-6, CFP-10, Ag85A, and Ag85b) were used. The most sensitive antigen was ESAT-6. ESAT 6-specific IgG ASCs were twice as high in adults with active tuberculosis compared with latent tuberculosis cases and were undetectable in healthy control adults. Ex vivo stimulation of PBMCs showed memory B cells to be more frequent in the blood of individuals with latent tuberculosis than in patients with active tuberculosis, suggesting a role of memory B cells in controlling tuberculosis disease progression. Of 35 healthy control adults, one had an unusual ASC to memory B cell ratio typical of active tuberculosis. Whether this response correlates with a high risk of developing tuberculosis disease is unknown at present.
A comparison of the utility of assay of ALS and ELISpot for the diagnosis of tuberculosis was made in adult patients with sputum smear-negative (but consequently sputum culture positive) tuberculosis, adults with latent tuberculosis and healthy control adults in Ethiopia (27). Assay of ALS to BCG antigens showed 86% sensitivity and 80% specificity in distinguishing patients with active pulmonary tuberculosis from adults with latent tuberculosis. Assay of ALS distinguished patients with active tuberculosis from healthy control adults with 86% sensitivity and 94% specificity. Assay of ALS was considerably more sensitive and specific than the tuberculin skin test and interferon-gamma release assay. Flow cytometry of PBMCs showed that patients with active tuberculosis had higher BCG-specific IgG-secreting plasmablasts than adults with latent tuberculosis or healthy control adults (6 versus 2% of total IgG-secreting plasmablasts).
The data from studies of tuberculosis suggest that the ASC response is readily detectable in patients at presentation to hospital. Assay of BCG-specific IgG ALS appears to have a high sensitivity and specificity in all cohorts assessed, except in a diverse group of malnourished children presenting with pneumonia. ELISpot and flow cytometry have shown that ASCs are detectable directly from blood. The chronic nature of tuberculosis may induce a relatively stable frequency of ASCs in the circulation. This may facilitate the detection of the tuberculosis-specific ASC response at different time points in patients with tuberculosis disease.
Colonization with Streptococcus pneumoniae, usually in the nasopharynx, is common in children and a prerequisite for pneumococcal disease, but most children colonized do not develop disease (60). Protective immune responses against pneumococci thus involve both prevention of infection (through clearance of nasopharyngeal colonization) and response to infection (should the mucosal surfaces be overwhelmed).
Infants and young children are likely to become colonized with pneumococci on first exposure to a serotype. Salivary IgA against pneumococcal pili proteins is secreted by ASCs in adenoids isolated from healthy children (75). Low antigen-specific salivary IgA from adenoids and serum IgG is associated with pneumococcal colonization, suggesting a protective role for mucosal antibody derived from local ASCs (76). Adults produce a significant increase in anti-capsular pneumococcal IgG to pneumococcal serotypes in response to pneumococcal colonization (77). The extent to which these responses to colonization are detectable as changes in populations of pneumococcus-specific ASCs in peripheral blood in adults or children is unknown.
Pneumococcal pneumonia represents failure to contain pneumococci to the mucosal surface leading to widespread mucosal (alveolar) disease and in some cases bacteremia. A cohort of 16 adult patients in Finland with bacteremic pneumococcal pneumonia all had detectable pneumococcal-specific IgG ASCs in peripheral blood on day 7 following detection of bacteremia (30). For each patient with pneumonia, the ELISpot antigen was the killed invasive pneumococcal isolate detected from the blood of the patient. In all patients, pneumococcal-specific IgG ASCs could be detected. In one of these patients with disease caused by serotype 14, there was also an ELISpot response against a mixture of purified capsular polysaccharides (3, 4, 5, 6B, 7F, 8, 14, 19F, and 23F). IgM and IgA ASCs were detected at lower frequencies. In healthy controls (14 adults), the ELISpot antigens were either the same mix of capsular polysaccharide antigens (n = 8) and/or killed pneumococcal isolates (n = 14). None of the healthy controls showed IgG responses against any of the pneumococcal antigens. Total ASCs (i.e. not only pneumococcal-specific) were 10 times higher in cases than in controls, suggesting a significant polyclonal ASC response.
Pneumococcal-specific ASCs derived from patients with bacteremic pneumococcal pneumonia (n = 16, from the study described above) express a unique set of homing receptors in comparison to pneumococcal-specific ASCs following pneumococcal vaccination (n = 14 with pneumococcal polysaccharide vaccination and n = 11 with pneumococcal conjugate vaccination) (78). The authors suggest that pneumococcal-specific ASCs may therefore be derived from, and home to, the lung mucosa. Work by the same group has recently characterized the antigen-specific ASC response to tonsillitis and sinusitis in 24 adults caused by H. influenzae, S. pneumoniae, S. pyogenes, or other β-hemolytic streptococci (31). Peripheral blood samples were taken 7–14 days following onset of symptoms. These patients all showed detectable IgG ASC responses specific to the causative pathogen, with slightly higher ASC frequencies in sinusitis than tonsillitis.
Taken as a whole, pneumococcal disease induces antigen-specific IgG ASC responses that are detectable in the peripheral blood of adults during illness. These ASC responses are present with or without detectable bacteremia. H. influenzae and non-pneumococcal streptococcal respiratory mucosal infections induce a similar immune response.
Like bacterial pneumonia, UTI is usually a bacterial mucosal infection that may rarely invade to cause bacteremia. However, with appropriate sampling techniques, culture of urine is sensitive and specific for disease etiology in contrast to culture of blood or nasopharyngeal specimens in bacterial pneumonia.
A study of the antigen-specific ASC response to UTI in adults caused by E. coli and other bacterial pathogens found that 10/14 (71%) patients with lower UTI and 17/17 (100%) patients with pyelonephritis had responses to a preparation of the cultured and killed E. coli isolate detectable by ELISpot at 7–8 days following onset of symptoms (32). In pyelonephritis, the mean antigen-specific ASC frequency was sevenfold higher for IgA than IgG. Subsequent studies in adults have confirmed that IgA ASCs predominate in UTI (rather than IgG as in pneumococcal pneumonia), and that ASCs specific for the virulence factor P fimbria are more frequent in E. coli pyelonephritis than uncomplicated lower UTI (33). This has biological plausibility, since P fimbriated E. coli has increased uroepithelial binding and are associated with increased virulence in UTI (79). Following activation in the urinary tract, antigen-specific ASCs express a unique set of homing receptors that may induce migration to regional lymph nodes (34).
A further study of the antigen-specific ASC response to E. coli pyelonephritis in children found a number of differences between the adult and child immune response (80). Here, 33/37 (89%) children aged 1 month to 16 years had responses detectable by ELISpot at either admission or 7 days following admission to hospital. Among all ages, mean antigen-specific ASC frequencies were comparable for IgA and IgM but lower for IgG. Antigen-specific ASC frequency was positively correlated with increasing age, and all four non-responders were infants less than 1 year of age. In contrast to adults, among antigen-specific ASCs, IgM ASCs predominated in children less than 2 years of age, while IgA ASCs predominated in children more than 2 years of age. This suggests that either younger children have a reduced ability to class-switch to IgA ASCs in response to UTI or that a systemic IgM ASC response to UTI is more important in younger children than in older children. Alternatively, older children may have previously been colonized in the urinary tract with E. coli and have a regional memory B cell population primed to secrete IgA. The nuanced differences between younger children and older children and adults will have significant implications for the detection of the ASC response to infection (see “Variation in the B cell response between individuals” section).
Infection with influenza A and B viruses cause moderate upper respiratory tract or gastrointestinal symptoms in most adults but can cause life-threatening pneumonia in individuals of all ages, but particularly at the extremes of age and pregnant women (81). ItsRNA genome evolves rapidly from year to year through a process of antigenic drift, with occasional reassortment of hemagglutinin (H) and neuraminidase (N) antigens, leading to pandemics of novel influenza viruses (most recently the H1N1 strain emerging in 2009) (82).
Human challenge studies have provided data on the immune response to respiratory infection with influenza virus (35, 83, 84). In most adults, challenge with influenza virus leads to rapid viral replication over 2–3 days during which the host is highly infectious. The majority of adults develop symptomatic disease by 2 days following challenge, with symptoms peaking at day 4 following exposure. Viral replication, as measured by viral shedding, is rare beyond 5 days following challenge (approximately 3 days following onset of symptoms) as a result of the induction of innate and adaptive immune responses.
In a nasal influenza challenge study in 12 adults, blood samples were taken at day 2, day 3, day 7, and day 28 relative to challenge (35). PBMCs were sorted for plasmablast markers before influenza-specific IgG ASC frequency was assessed in these cells using ELISpot. Influenza-specific ASCs peaked in peripheral blood at day 7, and were absent at day 2, day 3, and day 28 relative to challenge. Frequency of influenza-specific IgG ASCs in the peripheral blood positively correlated with peak viral load, length of viral shedding, and symptom severity. In summary, data from adult influenza challenge studies of a show the emergence of influenza-specific ASCs between day 3 and day 7 following challenge, as detected by ELISpot.
Analysis of the transcriptome has been used to investigate gene-level changes in response to influenza challenge. In 17 adult participants, the transcriptome was measured at 8–24 hourly intervals for 96 h (4 days) following challenge (84). Strong immune responses at the interface between innate and adaptive immunity (for example, the toll-like receptor pathway) were detected at 48 h, approximately 36 h before peak symptom time. Notably, the upregulation of two genes was able to accurately classify symptomatic from asymptomatic subjects: FGF9 (fibroblast growth factor 9, implicated in lung epithelial repair) and TLN1 (Talin-1, a cytoskeletal protein). Talin-1 is essential for the migration of leukocytes to sites of inflammation, or to lymph nodes, and also stabilizes interactions between leukocytes (such as via the TCR) (85). However, RNA transcripts related to ASC activation and antibody-secretion were not detected within the first 96 h. This is consistent with the absence of influenza-specific ASCs until after at least day 3. Alternatively, hypervariable transcripts (such as those encoding immunoglobulins) may be missed by the use of microarray technology. A more sensitive approach for the detection of transcripts relating to hypervariable immunoglobulin/BCR genes is described below.
The evolving repertoire of plasmablast BCRs (and secreted antibodies) was studied in nine adult patients with moderate to severe pandemic H1N1/2009 influenza infection (86). Only a single time point (typically day 10 following onset of symptoms) was used for blood sampling. Here, flow cytometry and ELISpot demonstrated the presence of influenza-specific IgG plasmablasts, which were absent in unvaccinated healthy volunteers. Single cell capture, PCR amplification and sequencing of the heavy Ig locus (IGH) and light Ig (IGL/IGK) loci from these plasmablasts showed a highly somatically mutated repertoire of BCRs. High somatic hypermutation may be secondary to the derivation of plasmablasts from memory B cells through a recall response. However, in the context of the early pandemic of influenza H1N1/2009, it is likely that a proportion of the somatic hypermutation represents rapid molecular evolution of plasmablast BCRs occurring over the first 10 days of infection. This has significant implications for the hypothetical utility of ELISpot or assay of ALS for the diagnosis of the etiology of respiratory infections. For example, delaying sampling until several days following onset of symptoms might increase sensitivity (due to increased plasmablast numbers) and specificity (due to increasing affinity of BCRs to influenza antigens).
Respiratory syncytial virus is a major global pathogen causing upper and lower respiratory tract infection in young children and causes repeated symptomatic disease in otherwise healthy adults (87). Like influenza, RSV is an RNA virus, predisposing it to rapid evolution under selective pressure due to the inherent error rate when copying the single-stranded RNA genome. As with influenza virus infection, infection-derived immunity to RSV is of short duration (36).
In a nasal RSV challenge study in 61 adult volunteers, blood was sampled at days 0, 3, 7, 10, 14, and 28 post-inoculation (36). Using flow cytometry and ELISpot, IgA and IgG ASCs specific to RSV lysate were detectable at day 10 following challenge and undetectable before day 7 and after day 14.
In a study of 40 adults with natural RSV infection, blood was sampled at admission, days 10–16 following admission and again in convalescence (days 22–45) (37). RSV was detected in the nasopharynx of all patients, and patients shed RSV for a mean of 11.9 days (range 4–30 days). RSV fusion (F)-protein antigen was used for ELISpot. RSV-specific IgG ASC responses were detected using ELISpot in 36/40 (90%) patients within 11 days following onset of symptoms. RSV-specific IgG ASCs were detectable by day 2 and remained detectable until at least day 12. These ASCs were not detected in patients with other respiratory tract infections. However, 7/9 (78%) participants with a second sample at 8–16 days remained positive by ELISpot (>4 standard deviations above the mean for previously studied healthy controls). Of participants with a second sample at 22–45 days, 11/23 (48%) remained positive by ELISpot. Overall, 16/36 (44%) participants had positive responses beyond the last day of RSV shedding. Thus, RSV infection appears to result in antigen shedding of approximately 12 days, with ASC responses prolonged beyond this in many individuals.
A further adult study involving 97 participants assessed the frequency of antigen-specific ASCs during RSV and influenza infection (n = 11 each) as well as in healthy controls including 19 adults recently vaccinated with trivalent influenza vaccine, tetanus toxoid vaccine, human papillomavirus vaccine (HPV) and hepatitis B vaccine (38). Only a single blood sample was taken at enrollment. All RSV-infected patients had RSV-specific IgG ASCs responses detectable with ELISpot at days 2–11 following symptom onset. RSV-specific IgG ASCs were not detected in patients with influenza virus infection or in healthy controls with and without recent vaccinations. Similarly, influenza-specific ASCs were not detected in patients with RSV infection (except in a single-coinfected individual). The specificity of the ASC response to either RSV infection, or influenza infection suggests that ELISpot may be a useful test to distinguish respiratory viral infection from nasopharyngeal carriage. These methods may also be useful to investigate the immune response to RSV with the aim of developing an effective vaccine against this pathogen.
Dengue virus consists of four serotypes that are transmitted by Aedes spp. mosquitoes in the global tropics (88). Most dengue virus infections are asymptomatic or cause mild febrile illness. More rarely, infections can cause life-threatening disease characterized by vascular endothelial damage, cardiovascular shock, and rapid consumption of platelets leading to coagulopathy (severe dengue) (89).
Severe dengue infection appears to be associated with high plasmablast frequencies in vivo. In a cohort of 46 adult patients admitted to hospital with severe dengue infection in Bangkok, dengue-specific IgG plasmablasts were identified by flow cytometry combined with ELISpot (39). These plasmablasts were detectable at levels above that of healthy controls from day 4 following onset of symptoms, and peaked in frequency between day 6 and day 7. Median frequency of dengue-specific IgG plasmablasts was ~10,000 per million PBMCs, representing at least a 1,000-fold increase from healthy adult controls. These plasmablast frequencies are considerably higher than that displayed in other severe viral (and bacterial) infections (90, 91). A further study in South America has confirmed this rapid and massive plasmablast response and found peak plasmablast frequency to be associated with disease severity (40).
In a prospective study of 28 adult patients with severe dengue, the transcriptome showed enrichment for monocyte and macrophage-associated genes in comparison to controls (92). Enrichment for these genes was associated with higher viremia. Monocytes infected with dengue virus secreted proinflammatory cytokines such as interleukin 1 receptor antagonist and induced the differentiation of B cells into plasmablasts in vitro after 6 days. Similar findings have been noted in other cohorts of children and adults (93, 94). Identification of dengue-specific plasmablasts (or their secretions) with ELISpot or ALS may represent a sensitive and specific diagnostic test for dengue infection from day 4 of febrile illness. This may be especially relevant in endemic regions where seropositivity is common in healthy children and adults.
The observations that (a) severe dengue occurs during rapid clearance of the virus, rather than during maximum viremia, (b) severe dengue mainly occurs in secondary infection with a heterotypic dengue virus serotype, and (c) plasmablasts are positively correlated with severity of infection, implicates the host immune response in the pathogenesis of severe dengue (95). Current evidence supports the theory of antibody-dependent enhancement of disease (96): invasion of dengue virions into monocytes (and possibly B cells) through the FCγ receptor is augmented by weakly reactive B memory cell derived antibodies to protein M on the viral surface (97, 98). These virions are rendered hypoimmunogenic (99), resulting in rapid intracellular replication and leading to high viremia in patients with severe dengue disease. Whether the phenotype of severe dengue is caused by the humoral immune response, or by a correlated T cell specific response is undetermined (100). Nonetheless, concerns regarding this phenomenon have hampered the development of a vaccine to dengue virus, further emphasizing the importance of pathogen-specific studies of the B cell response to infections.
Infection with mumps virus causes parotid gland swelling and a moderate febrile illness in the majority of individuals, but may cause meningitis and encephalitis, and oophoritis, orchiditis in girls and boys, respectively. Complications include deafness and sterility (101). Mumps virus infection, or two or more live attenuated vaccinations (typically with measles-mumps-rubella vaccination, MMR), produces long-lasting immunity (102). However, prior vaccination complicates serological diagnosis of mumps infection, and assay of the ASC response to infection with either ELISpot or ALS may be useful in an outbreak situation.
ELISpot was used to assay mumps-specific IgG ASCs in an outbreak of mumps infection in seven previously vaccinated adults in the USA. Blood samples were taken at a single time point between day 7 and day 28 following onset of symptoms. All seven patients had detectable mumps-specific IgG ASCs significantly above a threshold determined from studies of vaccinated individuals. The single patient sampled at day 28 had a mumps-specific IgG ASC count that was just above the threshold. Assay of antigen-specific ASCs may thus be a useful diagnostic test in the context of prior vaccination. Further investigation of the BCR or antibodies derived from these ASCs might also illuminate the underlying reasons for vaccine failure in these individuals.
Enterovirus-71 (EV-71) is an RNA virus that has caused epidemics of herpangina and has been implicated in epidemics of life-threatening encephalitis and multiple organ failure in children in the Asia-Pacific region (103). Biennial epidemics of EV-71 infection occur, mainly in children aged 1–3 years. These epidemiological patterns, similar to pre-vaccine era measles epidemics (104), suggest a protective role for humoral immunity, with epidemics following the accrual of sufficient unprotected infants to allow widespread transmission.
The ASC response to acute EV-71 infection was assayed using ELISpot and flow cytometry on blood from 28 children with laboratory confirmed genotype B EV-71 infection. Blood was sampled once between day 2 and day 11 following symptom onset, and once in convalescence (>19 days following symptom onset) for each child. Acute blood samples showed EV-71-specific IgG and IgM ASCs and lower IgA ASCs. Analysis of ASCs of all Ig isotypes showed a strong ASC antigen-specific response at days 1–3, which further increased ~5-fold at days 4–7, and was almost absent from day 8 onward. Children aged <3 years had a population of EV-71-specific ASCs that almost entirely secreted IgM, while children aged >3 years showed a predominantly IgG ASC response. Enterovirus-specific IgG ASCs may either represent rapid class-switching in older children, or recall memory.
Thus, the ASC response to EV-71 infection in children has a similar kinetic to that described for influenza, RSV, dengue virus, and mumps virus infections. This suggests that there is either a conserved pathway to ASC activation and proliferation or several redundant pathways converging to the same endpoint.
Clinical malaria results from infection with one of four human Plasmodium spp. protozoa, with the majority of severe disease (anemia, acidosis and cerebral malaria) caused by Plasmodium falciparum infection. Naturally acquired immunity to falciparum malaria is generally slowly acquired (amongst survivors) over the course of repeated infections in childhood in areas of endemic transmission (105). Following inoculation, initial invasion of the bloodstream by pre-hepatic sporozoites of P. falciparum does not induce a protective humoral response (106). Only with onset of clinical malaria following dissemination of merozoites is a protective humoral response induced.
In 38 children approximately 5 years of age with clinical malaria in Uganda, plasmablasts were assayed by flow cytometry over 28 days following presentation (107). Plasmablasts were already at peak levels (~6% of peripheral blood B cells) at time of presentation to hospital. These declined by day 7 to ~3% of peripheral blood B cells following treatment for malaria (107). In contrast, memory B cells remained at ~10% of peripheral blood B cells over 28 days. Atypical memory B cells (hypothesized to represent “exhausted” non-function B cells (108)) increased from 10 to 16% of peripheral blood B cells and were positively associated with parasitemia on presentation. However, this study did not estimate the malaria-specific proportion of ASCs (or memory B cells), limiting its generalizability to the studies of the ASC response to infection.
The difficulty of defining onset of malaria infection in areas of endemic transmission (where a large proportion of children may have asymptomatic parasitemia) makes data from malaria challenge studies important. The majority of studies reporting B cell responses to malaria infection have focused on memory B cells and often in the context of malaria vaccination strategies. However, one study quantified both the acutely activated malaria-specific ASC response following vaccination with merozoite surface protein 1 (MSP1) and the ASC response following malaria challenge infection. In this study, 9 healthy adults were enrolled and participated in a phase II trial of MSP1-based vaccination, with blood samples taken at days 5, 7, 8, 9, and 15 following booster vaccination. Immediately following day 15, 8 of 9 volunteers subsequently underwent malaria challenge infection with samples at baseline (day 16 following booster vaccination), day 7, day 11 (4 volunteers), and day 35 following challenge. ELISpot to the MSP1 and apical membrane 1 antigen (AMA1) was used to enumerate ASCs.
In these nine volunteers, MSP1-specific ASC responses post-vaccination were similar in timing (peak at day 8 and absent at day 15) and magnitude to those described for bacterial and viral pathogens above. However, following malaria challenge infection MSP1-specific ASCs were undetectable at all time points measured. This intriguing result suggests that either IgG ASCs induced by malaria challenge are not cognate for MSP1 (unlike ASCs induced by vaccination, and despite this being a dominant antigen for inducing IgG memory B cell responses), or that malaria infection downregulates the normal ASC response to infection, or that the timing of the malaria-specific ASC response differs to that of other acute infections. Given that only blood-stage merozoites induce a significant protective humoral response, and that merozoites emerged at approximately day 7 following challenge, one might expect peak malaria-specific IgG ASC responses to be identified at days 14–17 following challenge (i.e. days 7–10 following emergence of merozoites).
In summary, the ASC response to malaria infection (with an apparent decrease in ASCs during clinical infection) is unlike the responses to bacterial and viral infections (where large increases in ASCs are typical). In studies of natural infection, this phenomenon may be an artifact of delayed presentation of children to healthcare facilities; or the comparatively long time from infection to clinical [approximately 10–14 days from inoculation (105)]. The results of malaria challenge infection suggest, however, that malaria may induce a maladaptive ASC (and possibly B memory cell) response through an unknown mechanism. This would be also in accord with the observed slow generation of immunity to clinical malaria and the loss of immunity to malaria in individuals who migrate out of malaria endemic regions.
Despite the conservation of the ASC response to infection across pathogens (Table 2), a small proportion of individuals do not appear to produce the “normal” ASC response. Rather, some individuals have low or undetectable ASC responses even in confirmed infection. Quite why some individuals exhibit an abnormally low ASC response to the antigens under investigation is unknown. One possibility is that testing only 1–4 antigens in ELISpot or ALS is simply insufficient, and some individuals have an immune response that targets untested pathogen antigens. Such a possibility could be investigated using ALS against a protein array derived from pathogens of interest.
Alternatively, it may be that genetic variation in lymphocytes (109), or in the regulation of the innate and immune response (59), is fundamental in the ASC response to T cell dependent and independent antigens (110, 111). Stochastic mechanisms probably determine B cell fate at the level of the individual cell (4, 112), which may be of importance in B cell memory (where the small number of memory B cells leads to a small repertoire of antibodies/BCRs in the quiescent state). However, given the large number of activated cells, this is unlikely to be of importance in acute infection. Immunological diagnostic tests may also be of limited use in the important group of patients with an impaired immune response (113, 114).
The relevance of abnormally low or high ASC responses to clinical outcome is unclear. As we have described, worsening clinical status in dengue virus infection is correlated with increasing frequency of dengue-specific plasmablasts (40). It may be that plasmablast-mediated pathology is an under-appreciated aspect of a number of diseases. Generally, however, one would expect that lower ASC responses and a hypo-immune state, would correlate with a worse clinical outcome. An immunosuppressed phenotype does appear to correlate with mortality in severe sepsis (53), but to what extent this correlates with changes in B cell and T cell compartments and to what extent this changes during evolving sepsis (115) are topics for future study.
In this review, we have detailed the application of ELISpot and ALS for the study of the ASC response to infection (Table 1). Many of these studies have been done with the goal of developing novel diagnostic tests for a specific pathogen and in patient groups where there is a gold standard against which the results can be compared (e.g. RSV and influenza virus infection). Further assessment of the assay of ASC responses as diagnostic tests for specific pathogens needs to be done. For example, optimizing the antigens to use for detection of ASCs and defining the optimum isotype of BCR for each infection (which may also differ according to the age of child). Most data indicate that the ASC response peaks at approximately 7 days following onset of illness (with the exception of malaria). However, future studies should consider using more frequent time points (e.g. daily samples for duration of hospital admission) in order to provide important detail on the timing of the ASC response.
Assay of antigen-specific ASCs for the diagnosis of the etiology of infection in prospectively enrolled case series is an important future challenge. Pneumonia provides a prototypical example, with multiple potential etiologies. Initial results from the Pneumonia Etiology Research for Child Health (PERCH) project suggest that microbiological diagnostic tests may be insensitive for bacteria (116). Studies of the impact of vaccination against specific pathogens (“vaccine probe studies”), nested within randomized controlled trials, provide epidemiological evidence for the etiology of pneumonia (117–119). Such studies rely on the availability of new vaccines. Alternative paradigms of infectious disease diagnostic testing, based on the highly specific ASC response to infection, or potentially the host transcriptome response to infection (51), are therefore attractive areas of future research.
The data presented here show the ASC response to differ greatly between individuals with similar or identical pathogen stimuli. Variability in the host immune response to infection, particularly with regard to genotype (58) and immune regulation during infection (59), remains an important challenge, with implications both for developing diagnostics based on the immune response and for understanding disease risk associated with this variability. Additionally, some pathogens, such as malaria, Epstein–Barr virus, and others, subvert the B cell-mediated response, resulting in chronic infections (120). Alternatively, pathogens such as dengue virus appear to augment the numerical ASC response to infection, resulting in severe immunopathology (95). Other pathogens, such as Staphylococcus aureus, induce a potent humoral response against secretory proteins thereby biasing the response away from replicating bacteria (121). A detailed understanding therefore of both the heterogeneity of the immune response to a given infection and the varying strategies of pathogens to evade this response will inform the development of diagnostic testing. Such data will also be highly relevant to the development of vaccines and therapeutic monoclonal antibodies.
High-throughput genetic sequencing of the IGH locus and IGL and IGK loci that encode the BCR is now feasible allowing millions of BCR sequences to be determined simultaneously from a single sample (122). Technical challenges remain, including linking IGH and IGL/IGK sequences to represent the BCR on a single ASC and distinguishing genuine hypervariability in transcripts from read-error. Bioinformatic methods must also qualify the constraints on the affinity maturation of the BCR repertoire due to the baseline diversity of BCR sequences [so-called “original antigenic sin” (123)]. However, current data show that, following vaccination with either polysaccharide or protein–polysaccharide conjugate antigens, the detection of BCR transcripts can be used to detect ASCs in PBMCs in an unbiased manner. Further, the plasmablast BCR repertoire undergoes convergent immunological selection to a small number of vaccine epitopes, suggesting that antigen-specific ASCs can be detected (56, 57) The incremental development of a library of sequences that represent BCRs cognate to a variety of vaccine epitopes is therefore significant (54, 124).
If a similar process of convergent selection occurs during infection, then detection of pathogen-specific BCR sequences could define humoral immunity to different pathogen antigens at a fundamental genetic level (55, 122). Such an approach could be used for the etiological diagnosis of infection in a (theoretically) unbiased manner. A further theoretical advantage is an increased sensitivity early in the time course of infection due to PCR amplification of the BCR repertoire (based on primers to conserved regions of the BCR sequences). The successful application of this technique may therefore define etiology of infection in a patient cohort or to develop therapeutic monoclonal antibodies to existing, or emerging pathogens (125, 126).
Monoclonal antibodies may become important in the treatment of infectious, particularly in the context of increasing antimicrobial resistance and emerging infectious diseases. For example, the identification of a monoclonal antibody (FI6v3) that is capable of neutralizing all known human influenza isolates has clear therapeutic potential for patients with severe influenza. This was, however, time-consuming and derived from the screening of over 100,000 plasmablasts (127). Recent advances in enriching antigen-specific plasmablasts from vaccinees is likely to considerably reduce the time and expense required to identify broadly neutralizing antibodies (128).
Identification of broadly neutralizing antibodies to HIV from individuals who are long-term non-progressors to AIDS provides a novel method to protect against HIV infection. The recombinant gene for the broadly neutralizing antibody is engineered into a viral vector that is optimized for persistent transcription and injected intramuscularly (129). In mice models, long-lasting protection against HIV infection can be achieved. This technique, termed vectored immunoprophylaxis, provides a method to immunize individuals with optimized antibodies. It also does not need an intact immune system and may therefore be particularly useful for “vaccination” against HIV in humans (130).
B cell receptors sequencing may also become an alternative molecular method for identifying broadly neutralizing antibodies to a range of infections. When combined with gene editing techniques such as CRISPR-cas9, this could provide a rapid method to produce monoclonal antibodies against conserved pathogen antigens (125, 131). Theoretically, such an approach may be particularly useful to develop novel therapies for prolonged epidemics of acute infectious diseases such as the Ebola virus epidemic in West Africa of 2014–2016 (126).
A number of studies have examined the ASC response to infection across diverse bacterial and viral diseases and in malaria. We present a focused analysis on the timing and magnitud, and the clinical applications of these responses. These studies indicate that ELISpot and ALS may be very sensitive and highly specific methods for determining the etiology of infection and have some advantages over current methods.
The timing of the peripheral blood ASC response to infection is conserved for the bacterial pathogens (V. cholerae, E. coli, S. enterica var Typhi, M. tuberculosis, S. pneumonia, and other bacterial respiratory pathogens) and viral pathogens (influenza viruses, RSV, dengue viruses, mumps virus, and EV-71) for which data are available. Antigen-specific ASCs generally express markers of acute proliferation and are detectable in peripheral blood by approximately day 4 following onset of symptoms, peak in frequency at days 6–8, and are absent from approximately day 11 onward. In contrast to the conserved timing of ASC detection, the magnitude of peak ASC frequency in peripheral blood varies widely between different bacterial and viral pathogens.
At present, data are too few for definitive conclusions. The vagaries of timing of clinical presentation of individuals and the infrequency of blood sampling prohibit a detailed understanding of the ASC response over time (except in a few challenge studies involving a small number of individuals). Variability in the host immune response to infection, particularly with regard to genotype (58) and immune regulation during infection (59) remains an important challenge, with implications both for developing diagnostics based on the immune response and for understanding disease risk associated with this variability. More specifically, details of the ASC response to malaria and other protozoal infections are sparse.
Nonetheless, there is a great potential in the use of antigen-specific ASCs for the etiological diagnosis of infection. Current efforts focus on the broadening and optimization of pathogen detection. This approach, however, is severely hampered by the frequent detection of colonizing organisms and hence difficulties in defining the causative pathogen. Practical further work in this area should include systematic prospective sampling of large number of cases and appropriate controls with a similar clinical phenotype but with a number of potential etiologies. Future studies should also formally integrate data from a variety of current (e.g. clinical features, blood culture) and experimental (e.g. ELISpot, ALS and BCR sequencing) diagnostic methods to estimate the probable etiology of infection in cases. Such data may be useful to assess the burden of disease caused by various pathogens (e.g. in the context of planned vaccine introduction) and ultimately aid the development of rapid diagnostic testing that could inform clinicians working with patients with infectious disease syndromes.
MC helped conceive and plan the review and led the writing of the manuscript. RM helped conceive and plan the review and contributed to the writing of the manuscript. PS contributed to the writing of the manuscript. DK helped conceive and plan the review and contributed to the writing of the manuscript. JT helped conceive and plan the review and contributed to the writing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
MC is supported by a Wellcome Trust Research Training Fellowship (104439/Z/14/Z). DK receives salary support from the NIHR Oxford Biomedical Research Centre.
1. Rajewsky J. Clonal selection and learning in the antibody system. Nature (1996) 381:751–8. doi:10.1038/381751a0
2. Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat Rev Immunol (2012) 12(4):282–94. doi:10.1038/nri3190
3. Benson MJ, Erickson LD, Gleeson MW, Noelle RJ. Affinity of antigen encounter and other early B-cell signals determine B-cell fate. Curr Opin Immunol (2007) 19(3):275–80. doi:10.1016/j.coi.2007.04.009
4. Hasbold J, Corcoran LM, Tarlinton DM, Tangye SG, Hodgkin PD. Evidence from the generation of immunoglobulin G-secreting cells that stochastic mechanisms regulate lymphocyte differentiation. Nat Immunol (2004) 5(1):55–63. doi:10.1038/ni1016
5. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol (2015) 15(3):160–71. doi:10.1038/nri3795
6. Mitchell R, Kelly DF, Pollard AJ, Trück J. Polysaccharide-specific B cell responses to vaccination in humans. Hum Vaccin Immunother (2014) 10(6):1661–8. doi:10.4161/hv.28350
7. Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature (2008) 453(7195):667–71. doi:10.1038/nature06890
8. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol (2015) 15(3):149–59. doi:10.1038/nri3802
9. Moncunill G, Han H, Dobano C, McElrath MJ, De Rosa SC. OMIP-024: pan-leukocyte immunophenotypic characterization of PBMC subsets in human samples. Cytometry A (2014) 85(12):995–8. doi:10.1002/cyto.a.22580
10. Saletti G, Cuburu N, Yang JS, Dey A, Czerkinsky C. Enzyme-linked immunospot assays for direct ex vivo measurement of vaccine-induced human humoral immune responses in blood. Nat Protoc (2013) 8(6):1073–87. doi:10.1038/nprot.2013.058
11. Band VI, Ibegbu C, Kaur SP, Cagle SM, Trible R, Jones CL, et al. Induction of human plasmablasts during infection with antibiotic-resistant nosocomial bacteria. J Antimicrob Chemother (2014) 69(7):1830–3. doi:10.1093/jac/dku047
12. Maecker HT, McCoy JP. FOCIS Human Immunophenotyping Consortium. A model for harmonizing flow cytometry in clinical trials. Nat Immunol (2010) 11(11):975–9. doi:10.1038/ni1110-975
13. Clutterbuck EA, Salt P, Oh S, Marchant A, Beverley P, Pollard AJ. The kinetics and phenotype of the human B-cell response following immunization with a heptavalent pneumococcal-CRM conjugate vaccine. Immunology (2006) 119(3):328–37. doi:10.1111/j.1365-2567.2006.02436.x
14. Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity (1998) 8:363–72. doi:10.1016/S1074-7613(00)80541-5
15. Trück J, Mitchell R, Thompson AJ, Morales-Aza B, Clutterbuck EA, Kelly DF, et al. Effect of cryopreservation of peripheral blood mononuclear cells (PBMCs) on the variability of an antigen-specific memory B cell ELISpot. Hum Vaccin Immunother (2014) 10(8):2490–6. doi:10.4161/hv.29318
16. Carpenter CM, Hall ER, Randall R, McKenzie R, Cassels F, Diaz N, et al. Comparison of the antibody in lymphocyte supernatant (ALS) and ELISPOT assays for detection of mucosal immune responses to antigens of enterotoxigenic Escherichia coli in challenged and vaccinated volunteers. Vaccine (2006) 24(18):3709–18. doi:10.1016/j.vaccine.2005.07.022
17. Qadri F, Ahmed T, Ahmed F, Bhuiyan MS, Mostofa MG, Cassels FJ, et al. Mucosal and systemic immune responses in patients with diarrhea due to CS6-expressing enterotoxigenic Escherichia coli. Infect Immun (2007) 75(5):2269–74. doi:10.1128/IAI.01856-06
18. Feller AJ, McKenzie R, Taylor DN, Woods CC, Grahek SL, Islam D, et al. Comparative evaluation of the antibody in lymphocyte supernatant (ALS) and enzyme-linked immunospot (ELISPOT) assays for measuring mucosal immune responses to Shigella antigens. Vaccine (2011) 29(47):8487–9. doi:10.1016/j.vaccine.2011.09.030
19. Qadri F, Ryan ET, Faruque AS, Ahmed F, Khan AI, Islam MM, et al. Antigen-specific immunoglobulin A antibodies secreted from circulating B cells are an effective marker for recent local immune responses in patients with cholera: comparison to antibody-secreting cell responses and other immunological markers. Infect Immun (2003) 71(8):4808–14. doi:10.1128/IAI.71.8.4808-4814.2003
20. Leung DT, Rahman MA, Mohasin M, Riyadh MA, Patel SM, Alam MM, et al. Comparison of memory B cell, antibody-secreting cell, and plasma antibody responses in young children, older children, and adults with infection caused by Vibrio cholerae O1 El Tor Ogawa in Bangladesh. Clin Vaccine Immunol (2011) 18(8):1317–25. doi:10.1128/CVI.05124-11
21. Rahman A, Rashu R, Bhuiyan TR, Chowdhury F, Khan AI, Islam K, et al. Antibody-secreting cell responses after Vibrio cholerae O1 infection and oral cholera vaccination in adults in Bangladesh. Clin Vaccine Immunol (2013) 20(10):1592–8. doi:10.1128/CVI.00347-13
22. Sheikh A, Bhuiyan MS, Khanam F, Chowdhury F, Saha A, Ahmed D, et al. Salmonella enterica serovar Typhi-specific immunoglobulin A antibody responses in plasma and antibody in lymphocyte supernatant specimens in Bangladeshi patients with suspected typhoid fever. Clin Vaccine Immunol (2009) 16(11):1587. doi:10.1128/CVI.00311-09
23. Khanam F, Sheikh A, Sayeed MA, Bhuiyan MS, Choudhary FK, Salma U, et al. Evaluation of a typhoid/paratyphoid diagnostic assay (TPTest) detecting anti-Salmonella IgA in secretions of peripheral blood lymphocytes in patients in Dhaka, Bangladesh. PLOS Negl Trop Dis (2013) 7(7):e2316. doi:10.1371/journal.pntd.0002316
24. Raqib R, Rahman J, Kamaluddin AK, Kamal SM, Banu FA, Ahmed S, et al. Rapid diagnosis of active tuberculosis by detecting antibodies from lymphocyte secretions. J Infect Dis (2003) 188:364–70. doi:10.1086/376511
25. Raqib R, Mondal D, Karim MA, Chowdhury F, Ahmed S, Luby S, et al. Detection of antibodies secreted from circulating Mycobacterium tuberculosis-specific plasma cells in the diagnosis of pediatric tuberculosis. Clin Vaccine Immunol (2009) 16(4):521–7. doi:10.1128/CVI.00391-08
26. Rekha RS, Kamal SM, Andersen P, Rahim Z, Hoq MI, Ara G, et al. Validation of the ALS assay in adult patients with culture confirmed pulmonary tuberculosis. PLoS One (2011) 6(1):e16425. doi:10.1371/journal.pone.0016425
27. Ashenafi S, Aderaye G, Zewdie M, Raqib R, Bekele A, Magalhaes I, et al. BCG-specific IgG-secreting peripheral plasmablasts as a potential biomarker of active tuberculosis in HIV negative and HIV positive patients. Thorax (2013) 68(3):269–76. doi:10.1136/thoraxjnl-2012-201817
28. Chisti MJ, Salam MA, Raqib R, Banu S, Shahid AS, Shahunja KM, et al. Validity of antibodies in lymphocyte supernatant in diagnosing tuberculosis in severely malnourished children presenting with pneumonia. PLoS One (2015) 10(5):e0126863. doi:10.1371/journal.pone.0126863
29. Jiao J, Wang MS, Yang XG, Wang XF. Evaluation of ALS assay of TB-SA for diagnosis of pulmonary tuberculosis. J Immunoassay Immunochem (2015) 36(2):119–27. doi:10.1080/15321819.2014.908127
30. Palkola NV, Pakkanen SH, Kantele JM, Rossi N, Puohiniemi R, Kantele A. Pathogen-specific circulating plasmablasts in patients with pneumonia. PLoS One (2012) 7(3):e34334. doi:10.1371/journal.pone.0034334
31. Palkola NV, Blomgren K, Pakkanen SH, Puohiniemi R, Kantele JM, Kantele A. Immune defense in upper airways: a single-cell study of pathogen-specific plasmablasts and their migratory potentials in acute sinusitis and tonsillitis. PLoS One (2016) 11(4):e0154594. doi:10.1371/journal.pone.0154594
32. Kantele A, Papunen R, Virtanen E, Möttönen T, Räsänen L, Ala-Kaila K, et al. Antibody-secreting cells in acute urinary tract infection as indicators of local immune response. J Infect Dis (1994) 169:1023–8. doi:10.1093/infdis/169.5.1023
33. Kantele A, Möttönen T, Ala-Kaila K, Arvilommi HS. P fimbria-specific B cell responses in patients with urinary tract infection. J Infect Dis (2003) 188:1885–91. doi:10.1086/380097
34. Kantele AM, Palkola NV, Arvilommi HS, Kantele JM. Distinctive homing profile of pathogen-specific activated lymphocytes in human urinary tract infection. Clin Immunol (2008) 128(3):427–34. doi:10.1016/j.clim.2008.05.003
35. Huang KY, Li CK, Clutterbuck E, Chui C, Wilkinson T, Gilbert A, et al. Virus-specific antibody secreting cell, memory B-cell, and sero-antibody responses in the human influenza challenge model. J Infect Dis (2014) 209(9):1354–61. doi:10.1093/infdis/jit650
36. Habibi MS, Jozwik A, Makris S, Dunning J, Paras A, DeVincenzo JP, et al. Impaired antibody-mediated protection and defective IgA B-cell memory in experimental infection of adults with respiratory syncytial virus. Am J Respir Crit Care Med (2015) 191(9):1040–9. doi:10.1164/rccm.201412-2256OC
37. Lee FE, Falsey AR, Halliley JL, Sanz I, Walsh EE. Circulating antibody-secreting cells during acute respiratory syncytial virus infection in adults. J Infect Dis (2010) 202(11):1659–66. doi:10.1086/657158
38. Lee FE, Halliley JL, Walsh EE, Moscatiello AP, Kmush BL, Falsey AR, et al. Circulating human antibody-secreting cells during vaccinations and respiratory viral infections are characterized by high specificity and lack of bystander effect. J Immunol (2011) 186(9):5514–21. doi:10.4049/jimmunol.1002932
39. Wrammert J, Onlamoon N, Akondy RS, Perng GC, Polsrila K, Chandele A, et al. Rapid and massive virus-specific plasmablast responses during acute dengue virus infection in humans. J Virol (2012) 86(6):2911–8. doi:10.1128/JVI.06075-11
40. Garcia-Bates TM, Cordeiro MT, Nascimento EJ, Smith AP, Soares de Melo KM, McBurney SP, et al. Association between magnitude of the virus-specific plasmablast response and disease severity in dengue patients. J Immunol (2013) 190(1):80–7. doi:10.4049/jimmunol.1103350
41. Latner DR, McGrew M, Williams N, Lowe L, Werman R, Warnock E, et al. Enzyme-linked immunospot assay detection of mumps-specific antibody-secreting B cells as an alternative method of laboratory diagnosis. Clin Vaccine Immunol (2011) 18(1):35–42. doi:10.1128/CVI.00284-10
42. Huang KY, Lin JJ, Chiu CH, Yang S, Tsao KC, Huang YC, et al. A potent virus-specific antibody-secreting cell response to acute enterovirus 71 infection in children. J Infect Dis (2015) 212(5):808–17. doi:10.1093/infdis/jiv094
43. Elias SC, Choudhary P, de Cassan SC, Biswas S, Collins KA, Halstead FD, et al. Analysis of human B-cell responses following ChAd63-MVA MSP1 and AMA1 immunization and controlled malaria infection. Immunology (2014) 141(4):628–44. doi:10.1111/imm.12226
44. Chang HS, Sack DA. Development of a novel in vitro assay (ALS assay) for evaluation of vaccine-induced antibody secretion from circulating mucosal lymphocytes. Clin Diagn Lab Immunol (2001) 8(3):482–8. doi:10.1128/CDLI.8.3.482-488.2001
45. Sebina I, Biraro IA, Dockrell HM, Elliot AM, Cose S. Circulating B-lymphocytes as potential biomarkers of tuberculosis infection activity. PLoS One (2014) 9(9):e106796. doi:10.1371/journal.pone.0106796
46. Xu GJ, Kula T, Xu Q, Li MZ, Vernon SD, Ndung’u T, et al. Comprehensive serological profiling of human populations using a synthetic human virome. Science (2015) 348(6239):aaa0698. doi:10.1126/science.aaa0698
47. Metcalf CJE, Farrar J, Cutts FT, Basta NE, Graham AL, Lessler J, et al. Use of serological surveys to generate key insights into the changing global landscape of infectious disease. Lancet (2016) 388:728–30. doi:10.1016/S0140-6736(16)30164-7
48. Ferrara G, Losi M, D’Amico R, Roversi P, Piro R, Meacci M, et al. Use in routine clinical practice of two commercial blood tests for diagnosis of infection with Mycobacterium tuberculosis: a prospective study. Lancet (2006) 367:1328–34. doi:10.1016/S0140-6736(06)68579-6
49. Schulze A, Downward J. Navigating gene expression using microarrays – a technology review. Nat Cell Biol (2001) 3:E190. doi:10.1038/35087138
50. Tsalik EL, Henao R, Nichols M, Burke T, Ko ER, McClain MT, et al. Host gene expression classifiers diagnose acute respiratory illness aetiology. Sci Transl Med (2016) 8(322):322ra11. doi:10.1126/scitranslmed.aad6873
51. Sweeney TE, Wong HR, Khatri P. Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci Transl Med (2016) 8(346):346ra91. doi:10.1126/scitranslmed.aaf7165
52. Herberg JA, Kaforou M, Wright VJ, Shailes H, Eleftherohorinou H, Hoggart CJ, et al. Diagnostic test accuracy of a 2-transcript host RNA signature for discriminating bacterial vs viral infection in febrile children. JAMA (2016) 316(8):835–45. doi:10.1001/jama.2016.11236
53. Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med (2016) 4:259–71. doi:10.1016/S2213-2600(16)00046-1
54. Trück J, Ramasamy MN, Galson JD, Rance R, Parkhill J, Lunter G, et al. Identification of antigen-specific B cell receptor sequences using public repertoire analysis. J Immunol (2015) 194(1):252–61. doi:10.4049/jimmunol.1401405
55. Galson JD, Trück J, Fowler A, Münz M, Cerundolo V, Pollard AJ, et al. In-depth assessment of within-individual and inter-individual variation in the B cell receptor repertoire. Front Immunol (2015) 6:531. doi:10.3389/fimmu.2015.00531
56. Galson JD, Trück J, Fowler A, Clutterbuck EA, Münz M, Cerundolo V, et al. Analysis of B cell repertoire dynamics following hepatitis B vaccination in humans, and enrichment of vaccine-specific antibody sequences. EBioMedicine (2015) 2(12):2070–9. doi:10.1016/j.ebiom.2015.11.034
57. Galson JD, Trück J, Clutterbuck EA, Fowler A, Cerundolo V, Pollard AJ, et al. B-cell repertoire dynamics after sequential hepatitis B vaccination and evidence for cross-reactive B-cell activation. Genome Med (2016) 8(1):68. doi:10.1186/s13073-016-0337-5
58. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet (2013) 14(9):661–73. doi:10.1038/nrg3502
59. Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science (2014) 343:1118. doi:10.1126/science.1246949
60. Bogaert D, de Groot R, Hermans PWM. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis (2004) 4(3):144–54. doi:10.1016/S1473-3099(04)00938-7
61. Fernandez J, Levine OS, Sanchez J, Balter S, LaClaire L, Feris J, et al. Prevention of Haemophilus influenzae type B colonization by vaccination: correlation with serum anti-capsular IgG concentration. J Infect Dis (2000) 182:1553–6. doi:10.1086/315870
62. MenAfriCar Consortium. Meningococcal carriage in the African meningitis belt. Trop Med Int Health (2013) 18(8):968–78. doi:10.1111/tmi.12125
63. Blanchard Rohner G, Snape MD, Kelly DF, John T, Morant A, Yu LM, et al. The magnitude of the antibody and memory B cell responses during priming with a protein-polysaccharide conjugate vaccine in human infants Is associated with the persistence of antibody and the intensity of booster response. J Immunol (2008) 180(4):2165–73. doi:10.4049/jimmunol.180.4.2165
64. Kelly DF, Snape MD, Perrett KP, Clutterbuck EA, Lewis S, Blanchard Rohner G, et al. Plasma and memory B-cell kinetics in infants following a primary schedule of CRM 197-conjugated serogroup C meningococcal polysaccharide vaccine. Immunology (2009) 127(1):134–43. doi:10.1111/j.1365-2567.2008.02934.x
65. Kelly DF, Snape MD, Clutterbuck EA, Green S, Snowden C, Diggle L, et al. CRM197-conjugated serogroup C meningococcal capsular polysaccharide, but not the native polysaccharide, induces persistent antigen-specific memory B cells. Blood (2006) 108(8):2642–7. doi:10.1182/blood-2006-01-009282
66. Jahnmatz M, Kesa G, Netterlid E, Buisman AM, Thorstensson R, Ahlborg N. Optimization of a human IgG B-cell ELISpot assay for the analysis of vaccine-induced B-cell responses. J Immunol Methods (2013) 391(1–2):50–9. doi:10.1016/j.jim.2013.02.009
67. Qian Y, Wei C, Eun-Hyung Lee F, Campbell J, Halliley J, Lee JA, et al. Elucidation of seventeen human peripheral blood B-cell subsets and quantification of the tetanus response using a density-based method for the automated identification of cell populations in multidimensional flow cytometry data. Cytometry B Clin Cytom (2010) 78(Suppl 1):S69–82. doi:10.1002/cyto.b.20554
68. Sasaki S, Jaimes MC, Holmes TH, Dekker CL, Mahmood K, Kemble GW, et al. Comparison of the influenza virus-specific effector and memory B-cell responses to immunization of children and adults with live attenuated or inactivated influenza virus vaccines. J Virol (2007) 81(1):215–28. doi:10.1128/JVI.01957-06
69. Sur D, Lopez AL, Kanungo S, Paisley A, Manna B, Ali M, et al. Efficacy and safety of a modified killed-whole-cell oral cholera vaccine in India: an interim analysis of a cluster-randomised, double-blind, placebo-controlled trial. Lancet (2009) 374(9702):1694–702. doi:10.1016/S0140-6736(09)61297-6
70. Kirkpatrick BD, Bentley MD, Thern AM, Larsson CJ, Ventrone C, Sreenivasan MV, et al. Comparison of the antibodies in lymphocyte supernatant and antibody-secreting cell assays for measuring intestinal mucosal immune response to a novel oral typhoid vaccine (M01ZH09). Clin Diagn Lab Immunol (2005) 12(9):1127–9. doi:10.1128/CDLI.12.9.1127-1129.2005
71. Seder RA, Hill AVS. Vaccines against intracellular infections requiring cellular immunity. Nature (2000) 406:793–8. doi:10.1038/35021239
72. Feris EJ, Encinales L, Awad C, Stern JN, Tabansky I, Jiménez-Alvarez L, et al. High levels of anti-tuberculin (IgG) antibodies correlate with the blocking of T-cell proliferation in individuals with high exposure to Mycobacterium tuberculosis. Int J Infect Dis (2016) 43:21–4. doi:10.1016/j.ijid.2015.12.004
73. Maglione PJ, Xu J, Chan J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J Immunol (2007) 178(11):7222–34. doi:10.4049/jimmunol.178.11.7222
74. Maglione PJ, Chan J. How B cells shape the immune response against Mycobacterium tuberculosis. Eur J Immunol (2009) 39(3):676–86. doi:10.1002/eji.200839148
75. Zhang Q, Choo S, Finn A. Immune responses to novel pneumococcal proteins pneumolysin, PspA, PsaA, and CbpA in adenoidal B cells from children. Infect Immun (2002) 70(10):5363–9. doi:10.1128/IAI.70.10.5363-5369.2002
76. Zhang Q, Bernatoniene J, Bagrade L, Pollard AJ, Mitchell TJ, Paton JC, et al. Serum and mucosal antibody responses to pneumococcal protein antigens in children: relationships with carriage status. Eur J Immunol (2006) 36(1):46–57. doi:10.1002/eji.200535101
77. Goldblatt D, Hussain M, Andrews N, Ashton L, Virta C, Melegaro A, et al. Antibody responses to nasopharyngeal carriage of Streptococcus pneumoniae in adults: a longitudinal household study. J Infect Dis (2005) 192:387–93. doi:10.1086/431524
78. Palkola NV, Pakkanen SH, Kantele JM, Pakarinen L, Puohiniemi R, Kantele A. Differences in homing potentials of Streptococcus pneumoniae-specific plasmablasts in pneumococcal pneumonia and after pneumococcal polysaccharide and pneumococcal conjugate vaccinations. J Infect Dis (2015) 212(8):1279–87. doi:10.1093/infdis/jiv208
79. Mobley HL, Island MD, Massad G. Virulence determinants of uropathogenic Escherichia coli and Proteus mirabilis. Kidney Int Suppl (1994) 47:S129–36.
80. Kantele A, Palkola N, Arvilommi H, Honkinen O, Jahnukainen T, Mertsola J, et al. Local immune response to upper urinary tract infections in children. Clin Vaccine Immunol (2008) 15(3):412–7. doi:10.1128/CVI.00373-07
81. Nair H, Brooks WA, Katz M, Roca A, Berkley JA, Madhi SA, et al. Global burden of respiratory infections due to seasonal influenza in young children: a systematic review and meta-analysis. Lancet (2011) 378(9807):1917–30. doi:10.1016/S0140-6736(11)61051-9
82. Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe (2010) 7(6):440–51. doi:10.1016/j.chom.2010.05.009
83. Huang Y, Zaas AK, Rao A, Dobigeon N, Woolf PJ, Veldman T, et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet (2011) 7(8):e1002234. doi:10.1371/journal.pgen.1002234
84. Linel P, Wu S, Deng N, Wu H. Dynamic transcriptional signatures and network responses for clinical symptoms in influenza-infected human subjects using systems biology approaches. J Pharmacokinet Pharmacodyn (2014) 41(5):509–21. doi:10.1007/s10928-014-9365-1
85. Calderwood DA, Campbell ID, Critchley DR. Talins and kindlins: partners in integrin-mediated adhesion. Nat Rev Mol Cell Biol (2013) 14(8):503–17. doi:10.1038/nrm3624
86. Wrammert J, Koutsonanos D, Li GM, Edupuganti S, Sui J, Morrissey M, et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med (2011) 208(1):181–93. doi:10.1084/jem.20101352
87. Nair H, Nokes DJ, Gessner BD, Dherani MK, Madhi SA, Singleton RJ, et al. Global burden of acute lower respiratory infection due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet (2010) 375:1545–55. doi:10.1016/S0140-6736(10)60206-1
88. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature (2013) 496(7446):504–7. doi:10.1038/nature12060
89. Simmons CP, Farrar JJ, Nguyen V, Wills B. Dengue. N Engl J Med (2012) 366(15):1432–1432. doi:10.1056/NEJMra1110265
90. Fink K. Origin and function of circulating plasmablasts during acute viral infections. Front Immunol (2012) 3:78. doi:10.3389/fimmu.2012.00078
91. Yam-Puc JC, Cedillo-Barrón L, Aguilar-Medina EM, Ramos-Payán R, Escobar-Gutiérrez A, Flores-Romo L. The cellular bases of antibody responses during dengue virus infection. Front Immunol (2016) 7:218. doi:10.3389/fimmu.2016.00218
92. Kwissa M, Nakaya HI, Onlamoon N, Wrammert J, Villinger F, Perng GC, et al. Dengue virus infection induces expansion of a CD14(+)CD16(+) monocyte population that stimulates plasmablast differentiation. Cell Host Microbe (2014) 16(1):115–27. doi:10.1016/j.chom.2014.06.001
93. Simmons CP, Popper S, Dolocek C, Chau TN, Griffiths M, Dung NT, et al. Patterns of host genome-wide gene transcript abundance in the peripheral blood of patients with acute dengue hemorrhagic fever. J Infect Dis (2007) 195(8):1097–107. doi:10.1086/512162
94. Long HT, Hibberd ML, Hien TT, Dung NM, Van Ngoc T, Farrar J, et al. Patterns of gene transcript abundance in the blood of children with severe or uncomplicated dengue highlight differences in disease evolution and host response to dengue virus infection. J Infect Dis (2009) 199(4):537–46. doi:10.1086/596507
95. Whitehorn J, Simmons CP. The pathogenesis of dengue. Vaccine (2011) 29:7221–8. doi:10.1016/j.vaccine.2011.07.022
96. Halstead SB, O’Rourke EJ. Antibody-enhanced dengue virus infection in primate leukocytes. Nature (1977) 265:739–41. doi:10.1038/265739a0
97. Beltramello M, Williams KL, Simmons CP, Macagno A, Simonelli L, Quyen NT, et al. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe (2010) 8(3):271–83. doi:10.1016/j.chom.2010.08.007
98. Priyamvada L, Cho A, Onlamoon N, Zheng NY, Huang M, Kovalenkov Y, et al. B cell responses during secondary dengue virus infection are dominated by highly cross-reactive, memory-derived plasmablasts. J Virol (2016) 90(12):5574–85. doi:10.1128/JVI.03203-15
99. Dejnirattisai W, Jumnainsong A, Onsirisakul N, Fitton P, Vasanawathana S, Limpitikul W, et al. Cross-reacting antibodies enhance dengue virus infection in humans. Science (2010) 328:745–8. doi:10.1126/science.1185181
100. Mongkolsapaya J, Dejnirattisai W, Xu X, Vasanawathana S, Tangthawornchaikul N, Chairunsri A, et al. Original antigenic sin and apoptosis in the pathogenesis of dengue haemorrhagic fever. Nat Med (2003) 9(7):921–7. doi:10.1038/nm887
101. Hviid A, Rubin S, Mühlemann K. Mumps. Lancet (2008) 371:932–44. doi:10.1016/S0140-6736(08)60419-5
102. Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med (2007) 357:1903–15. doi:10.1056/NEJMoa066092
103. Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis (2010) 10(11):778–90. doi:10.1016/S1473-3099(10)70194-8
104. Grenfell BT, Bjørnstad ON, Kappey J. Travelling waves and spatial hierarchies in measles epidemics. Nature (2001) 414:716–23. doi:10.1038/414716a
105. White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet (2014) 383(9918):723–35. doi:10.1016/S0140-6736(14)60656-5
106. Hoffman SL, Oster CN, Plowe CV, Woollett GR, Beier JC, Chulay JD, et al. Naturally-acquired antibodies to sporozoites do not prevent malaria: vaccine development implications. Science (1987) 237:639–42. doi:10.1126/science.3299709
107. Sullivan RT, Ssewanyana I, Wamala S, Nankya F, Jagannathan P, Tappero JW, et al. B cell sub-types following acute malaria and associations with clinical immunity. Malar J (2016) 15(1):139. doi:10.1186/s12936-016-1190-0
108. Weiss GE, Crompton PD, Li S, Walsh LA, Moir S, Traore B, et al. Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J Immunol (2009) 183(3):2176–82. doi:10.4049/jimmunol.0901297
109. Tsang JS, Schwartzberg PL, Kotliarov Y, Biancotto A, Xie Z, Germain RN, et al. Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell (2014) 157:499–513. doi:10.1016/j.cell.2014.03.031
110. Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature (2005) 438(7066):364–8. doi:10.1038/nature04267
111. Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature (2011) 470(7335):543–7. doi:10.1038/nature09737
112. Duffy KR, Wellard CJ, Markham JF, Zhou JH, Holmberg R, Hawkins ED, et al. Activation-induced B cell fates are selected by intracellular stochastic competition. Science (2012) 335(6066):338–41. doi:10.1126/science.1213230
113. Casanova JL, Abel L. Inborn errors of immunity to infection: the rule rather than the exception. J Exp Med (2005) 202(2):197–201. doi:10.1084/jem.20050854
114. Gaschignard J, Levy C, Chrabieh M, Boisson B, Bost-Bru C, Dauger S, et al. Invasive pneumococcal disease in children can reveal a primary immunodeficiency. Clin Infect Dis (2014) 59(2):244–51. doi:10.1093/cid/ciu274
115. Kwan A, Hubank M, Rashid A, Klein N, Peters MJ. Transcriptional instability during evolving sepsis may limit biomarker based risk stratification. PLoS One (2013) 8(3):e60501. doi:10.1371/journal.pone.0060501
116. Scott JAG. The PERCH integrated analysis and the primary etiology results from the PERCH study. Annual Meeting: American Society of Tropical Medicine and Hygiene. Atlanta, GA, USA (2016).
117. Mulholland EK. Use of vaccine trials to estimate burden of disease. J Health Popul Nutr (2004) 22(3):257–67.
118. O’Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet (2009) 374:893–902. doi:10.1016/S0140-6736(09)61204-6
119. Feikin DR, Scott JAG, Gessner BD. Use of vaccines as probes to define disease burden. Lancet (2014) 383:1762–70. doi:10.1016/S0140-6736(13)61682-7
120. Nothelfer K, Sansonetti PJ, Phalipon A. Pathogen manipulation of B cells: the best defence is a good offence. Nat Rev Microbiol (2015) 13(3):173–84. doi:10.1038/nrmicro3415
121. Pauli NT, Kim HK, Falugi F, Huang M, Dulac J, Henry Dunand C, et al. Staphylococcus aureus infection induces protein A-mediated immune evasion in humans. J Exp Med (2014) 211(12):2331–9. doi:10.1084/jem.20141404
122. Hoehn KB, Fowler A, Lunter G, Pybus OG. The diversity and molecular evolution of B-cell receptors during infection. Mol Biol Evol (2016) 33(5):1147–57. doi:10.1093/molbev/msw015
123. Cobey S, Wilson P, Matsen FAT. The evolution within us. Philos Trans R Soc Lond B Biol Sci (1676) 2015:370.
124. IJspeert H, van Schouwenburg PA, van Zessen D, Pico-Knijnenburg I, Driessen GJ, Stubbs AP, et al. Evaluation of the antigen-experienced B-cell receptor repertoire in healthy children and adults. Front Immunol (2016) 7:410. doi:10.3389/fimmu.2016.00410
125. Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science (2014) 346(6213):1258096. doi:10.1126/science.1258096
126. Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, et al. Isolation of potent neutralising antibodies from a survivor of the 2014 Ebola virus outbreak. Science (2016) 351:1078–83. doi:10.1126/science.aad5788
127. Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science (2011) 333:850–6. doi:10.1126/science.1205669
128. Nakamura G, Chai N, Park S, Chiang N, Lin Z, Chiu H, et al. An in vivo human-plasmablast enrichment technique allows rapid identification of therapeutic influenza A antibodies. Cell Host Microbe (2013) 14:93–103. doi:10.1016/j.chom.2013.06.004
129. Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature (2012) 481(7379):81–4. doi:10.1038/nature10660
130. Dosenovic P, von Boehmer L, Escolano A, Jardine J, Freund NT, Gitlin AD, et al. Immunization for HIV-1 broadly neutralizing antibodies in human Ig knockin mice. Cell (2015) 161(7):1505–15. doi:10.1016/j.cell.2015.06.003
Keywords: B cells, antibody-secreting cells, plasmablasts, adaptive immunity, diagnosis, transcriptomics, monoclonal antibodies, B cell receptor sequencing
Citation: Carter MJ, Mitchell RM, Meyer Sauteur PM, Kelly DF and Trück J (2017) The Antibody-Secreting Cell Response to Infection: Kinetics and Clinical Applications. Front. Immunol. 8:630. doi: 10.3389/fimmu.2017.00630
Received: 06 March 2017; Accepted: 12 May 2017;
Published: 01 June 2017
Edited by:
Deborah K. Dunn-Walters, University of Surrey, United KingdomReviewed by:
Claudio Nicoletti, University of Florence, ItalyCopyright: © 2017 Carter, Mitchell, Meyer Sauteur, Kelly and Trück. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Carter, bWljaGFlbC5qYW1lcy5jYXJ0ZXJAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.