 Stephen J. Kuperberg
Stephen J. Kuperberg Raj Wadgaonkar
Raj Wadgaonkar- 1Pulmonary and Critical Care Medicine, Wake Forest University School of Medicine, Winston Salem, NC, United States
- 2SUNY Downstate Medical Center, Brooklyn, NY, United States
Sepsis is not only a significant cause of mortality worldwide but has particularly devastating effects on the central nervous system of survivors. It is therefore crucial to understand the molecular structure, physiology, and events involved in the pathogenesis of sepsis-associated encephalopathy, so that potential therapeutic advances can be achieved. A key determinant to the development of this type of encephalopathy is morphological and functional modification of the blood–brain barrier (BBB), whose function is to protect the CNS from pathogens and toxic threats. Key mediators of pathologic sequelae of sepsis in the brain include cytokines, including TNF-α, and sphingolipids, which are biologically active components of cellular membranes that possess diverse functions. Emerging data demonstrated an essential role for sphingolipids in the pulmonary vascular endothelium. This raises the question of whether endothelial stability in other organs systems such as the CNS may also be mediated by sphingolipids and their receptors. In this review, we will model the structure and vulnerability of the BBB and hypothesize mechanisms for therapeutic stabilization and repair following a confrontation with sepsis-induced inflammation.
Introduction and Overview
Sepsis is a leading cause of morbidity and mortality worldwide. In 2016, consensus guidelines honed the definition of sepsis to refer to “life threatening organ dysfunction caused by a dysregulated host response to infection” (1). If attempts to suppress inflammation and restore perfusion are unsuccessful, septic shock with organ dysfunction may result. Breaching of the blood–brain barrier (BBB) leads to significant alteration of consciousness and reduction in neurocognition. Involvement of the CNS results in sepsis-induced brain dysfunction, which is manifested clinically by a neuropsychiatric continuum starting in an acute confusional state and ultimately coma (2, 3). With the exclusion of drug-induced and other metabolic etiologies, this syndrome is termed “sepsis-associated encephalopathy (SAE)” and is the most common form of encephalopathy occurring in critical care settings (4–7). Although the encephalopathy of sepsis shares several overlapping features with delirium, including its rapid onset and marked deterioration in cognition—delirium may be considered a subgroup, which involves hyperactive and hypoactive changes in awareness and consciousness (8). In the literature, SAE is widely understood as the presence of diffuse cerebral dysfunction in the presence of sepsis but in the absence of CNS infection and other forms of encephalopathy (9, 10). It is usually manifested by disturbances of the sleep–wake cycle, impaired consciousness, mild cognitive dysfunction, overt delirium, and coma (4).
The impact of SAE on public health is significant. It is responsible for short-term morbidity, increased length of hospital stay, long-term physical and cognitive impairment, and poses a large economic burden to health-care systems (11, 12). Mortality in SAE when it occurs as a manifestation multiple organ dysfunction and is estimated at 70% (9). Its psychological toll on families and caregivers cannot be calculated.
In the critically ill patient, SAE is often compounded iatrogenically by sedative-hypnotic use and neuromuscular blockade. Bedside diagnostic tests such as electroencephalography (EEG) and somatosensory-evoked potential are commonly employed to assist with diagnosis (5, 13, 14), while scoring tools such as the Glasgow Coma Scale (7), the CAM-ICU, and Richmond Agitation and Sedation Scale are cost-effective and valuable for measuring dynamic clinical changes (13). Brain MRI may show white matter lesions in the centrum semiovale, multiple ischemic strokes, and decreased in brain volume (9). While MRI may be negative in confirmed SAE, EEG abnormalities may be present even preceding the onset of symptoms in 50% of cases. On EEG, “slowing” in the theta range on EEG is correlated with SAE in the mild stage, where severe cases manifest with delta waves, triphasic waves, and burst suppression (9, 15).
Pathologic States
The pathogenesis of encephalopathy in sepsis is complex and incompletely understood. Microcirculatory dysfunction, underperfusion, and necrosis of peripheral organs yield a systemic inflammatory state involving leukocyte—particularly microglial—activation, lysosomal exocytosis, cytokine release, and free-radical generation (2, 5, 16). Nitric oxide (NO)-mediated oxidative damage of the hippocampus and cerebral cortex was shown in animal models of sepsis, while antioxidant and neuroprotective mediators such as heat shock protein and superoxide dismutase are diminished (2, 10). Neurotransmission is relayed along vagal afferents from the periphery to the nucleus tractus solitarius in the brainstem in a neurally mediated inflammatory reflex (4). Since glial cells and CNS neurons rely on amino acid, glucose, and oxygen metabolism for the production of ATP and neurotransmission, the catabolic conditions of sepsis devastate their bioenergetic capability (17). Our current understanding of the pathogenesis of SAE is multifactorial, involving cytokine effects, mitochondrial dysfunction, neurotransmitter alterations, and hypoxia leading to oxidative damage, ischemia, and cellular death (17). Observations of pathologic findings have included disseminated cerebral microabscesses (18), “multifocal necrotizing leukoencephalopathy” (19, 20), and a reduced functional density of cerebral vessels (21).
Structure and Function of the BBB
Protecting the brain is a physical barrier at its interface with the circulatory and immune systems (22). This separation of the CNS and spinal cord from peripheral organs was first observed by the injection of “vital” dyes by Ehrlich in 1885 and was first labeled “blood–brain barrier” (BBB) by Lewandowsky in 1900. In a landmark in vitro study by Reese and Karnovsky, horseradish peroxidase circulated through peripheral vasculature but did not pass through cerebral endothelial cells into the CNS (23–29). The primary constituent of the BBB is the brain microvascular endothelial cell (BMVEC), which is strategically located in close apposition to perivascular pericytes, astrocyte foot process, and macroglia (25, 28, 30–32). Diffusion is made readily possible by its short distance of only 8–20 μm from CNS neurons (33). The BMVEC is linked to pericytes and astrocytes by a common basement membrane composed of extracellular matrix proteins: collagen, elastin, fibronectin, laminin, and proteoglycans (28, 34). Canaliculi and fenestrae are sparse between these cells—limiting movement of fluids and further preventing capillary leakage (28). Focal adhesions, consisting of transmembrane proteins from the selectin, integrin, and immunoglobulin families tether the BMVEC to the basement membrane (25). Of these, integrins participate in angiogenesis and maintaining vascular integrity (25, 35), while the focal adhesion complex relays mechanical forces from the cytoskeleton to surrounding adhesive and contractile structures (25, 36). Structural support is provided by cellular adhesion molecules (CAMs), which are expressed at the basement membrane’s apical surface, and tight junctions, which bind adjacent endothelial cells, limit diffusion, and paracellular permeability (25, 28, 33). Primary constituents are the transmembrane proteins such as junctional adhesion molecules, claudins, and the adaptor cytoplasmic proteins zonula occludens-1–3 which connect to the actin cytoskeleton and serve as a scaffold as well as mediate cell–cell interactions (25, 33, 35, 37, 38). The high-electrical resistance of 1,500–2,000 Ω/cm2 of tight junctions prevents intracellular and transcellular movement of molecules (39, 40). Neighboring astrocytes and microglia modify tight junction assembly via cytokine release. Additionally, astrocytes exert local influence on barrier development by wnt/B-catenin-mediated signaling (32, 41). Pericytes or vascular smooth muscle cells surround the BBB capillary endothelium and have structural, synthetic, and regulatory function (25, 42). They synthesize proteins of the basement membrane, especially proteoglycans and laminal proteins. Spatially, they cover nearly one-third of its surface area (25, 43) and provide structural integrity to the barrier (25, 44). Extracellular peptidases and nucleosidases lyse proteins and ATP, whereas monoamine oxidase and cytochrome p450 work intracellularly to inactivate neurotoxic compounds (33, 45). Together in a milieu of extracellular matrix, these neighboring cells and structural elements function in coordinated fashion as part of a neurovascular unit (32).
Transport Across the Barrier
The BBB’s ability to maintain homeostasis in the CNS is determined by its ability to govern the means, rate, and regulation of transport of ions, small molecules, immune cells, cytokines, chemokines, and exogenous compounds (32). Under physiologic conditions, nutrients and essential molecules are facilitated entry into the CNS, whereas wastes, toxins, neurologically active agents, and pathogens are excluded from entering (33). Ions, water, and small molecules traverse by paracellular diffusion, whereas larger, hydrophilic compounds such as amino acids and glucose require specific transport systems for transcellular migration (25, 26, 30, 33, 37). As conceptualized in a recent review by Banks, the BBB possesses four essential and independent functions with regard to response to inflammatory and infectious stimuli. Its structural barrier coexists with responder, transporter, and secretor functions—together contributing to homeostatic control of molecular transport (28). “Adsorptive” and “receptor-mediated endocytosis” are principal means of active transport of protein across barrier cells and utilize vesicles. Specific transport proteins exist at the plasma membranes, such as GLUT-1, and ATP-binding cassette (ABC) transporters. p-GP (46) is an ABC transporter that functions as an efflux pump involved in drug delivery, detoxification, and is implicated in mechanisms of drug resistance (25, 37, 47). Also at the endothelial cell membrane, patches of cholesterol and glycosphingolipids known as lipid rafts are produced from intracellular cholesterol-binding proteins caveolin. This process starts with the formation of 60–80 nm invaginations termed caveolae which then form “clathrin-coated pits.” At the BBB, caveolae participate in receptor translocation, vesicular trafficking, and cellular signal transduction such as IL-1β-dependent NF-kB activation (25, 26, 48–52).
Molecular Mechanisms: Complement, Cytokines, and Mechanism of BBB Disruption
Activation of the complement system is critical in the innate immune system’s defense against infection and has been clearly demonstrated in the development of inflammation and neuronal dysfunction that precedes SAE (53–56). Once the complement cascade is activated by endotoxin, C5a acts upon cerebral endothelium, microglia, and brain parenchymal neurons. In studies modeling ischemia–reperfusion injury, C3a and C5a function as leukocyte chemoattractants (57). Endothelial cells and microglia subsequently become activated, secreting TNF-α and IL-B (57, 58), ultimately leading to ROS production, brain edema, and severe CNS neuronal injury (Figure 1).
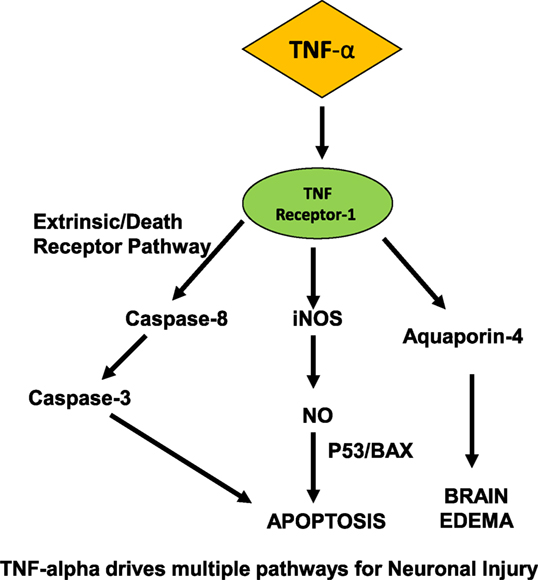
Figure 1. Proposed mechanism for neurocognitive dysfunction in the CNS in sepsis. On the CNS side of blood–brain barrier, TNF-α drives multiple pathways for neuronal injury, induces apoptosis via NO, caspase pathways, and leads to cerebral edema.
During such neuroinflammatory states such as trauma, ischemia, and sepsis, cytokines play a key role in the pathogenesis of injury. Semmler et al. showed that the chemokine MCP-1, and cytokines IL-1-β, TGF-β, and TNF-α were all upregulated in whole brain lysates, along with inducible nitric oxide (iNOS) (59). These findings are pertinent with respect to the effects of sepsis in the CNS, since iNOS mediates oligodendrocyte injury (60), and nitric oxide production was shown to induce apoptosis in astrocytes via BAX- and p-53-dependent pathways (61).
During a septic insult, IL-1β and TNF-α are elevated systemically (62–64). Cytokine interactions modify the barrier via tight junction stability and endothelial permeability (48, 65). Pattern recognition receptors known as Toll Like Receptors (TLR’s) are expressed on cerebral endothelial cells where they function as essential mediators in the response to pathogens and associated proteins, including LPS (66). At the BBB endothelium, TLR-2 is upregulated by TNF-α, and ligand binding at TLR2/6 was shown to mediate tight junction disruption. TNF-α induced changes in the barrier result in depolymerization of actin and the generation of intercellular gaps in the endothelial cytoskeleton (67). Qin et al. (68) demonstrated that an intraperitoneal injection of LPS initiated TNF-α production independently of circulating TNF-α, and that this occurred contemporaneously with microglial activation in substantia nigra, hippocampus, and cortex, while other mediators of inflammation were shown to be increased, such as MCP-1, IL-1β, and NF-kB. O’Carroll et al. (69) recent work supported these findings, showing that in endothelial cell culture, both TNF-α and IL-1β increase expression of leukocyte adhesion molecules, including ICAM-1 and V-CAM-1, chemokines MCP-1 (CCL-2), and RANTES (CCL-5), resulting in barrier dysfunction and decreased transendothelial electrical resistance. TNF-α was also found to increase barrier permeability by activation of protein kinase-6, resulting in cell–cell interactions involving VE-cadherin internalization (70).
In the BBB, TNF-α acts directly on the endothelial capillaries and may also diffuse into brain parenchyma in areas of brain where there is no barrier, such as the circumventricular organs (63). TNF receptor 1, also termed p55, is abundant in the brain and constitutively expressed in astrocytes (53, 63, 71). In inflammatory states, including sepsis, TNF-α binds to TNF-R1 (49). This interaction is facilitated by the cytosolic TNF receptor-associated death domain that recruits TNF receptor-associated factor 2. TNR-receptor associated factor 2 (TRAF-2) is an adaptor protein of TNF receptor that mediates anti-apoptotic signals. Xia et al. (72) showed that signal transduction leading to anti-apoptosis is mediated by sphingolipids, involving the physical interaction between TNF-α, TNF-R, and TRAF-2 (Figure 2). Simultaneously, TNF transport across the endothelial cell membrane can be facilitated by invaginations in the membrane referred to as caveolae that undergo endocytosis at the BBB (48, 49, 73). Intracytoplasmic signaling is mediated through RhoA and RAC, and ultimately transcription is facilitated via NF-kB yielding anti-apoptotic and pro-apoptotic proteins. Apoptosis occurs via activation of the caspase-mediated death receptor pathway (53, 74, 75), in which activated microglia are active participants (76). The interaction between LPS, TNF-α, TNF-R, and the consequences for SAE was elucidated in a murine model of SAE by Alexander et al. (53). The authors found that with comparison to a TNF R1 double knockout population under the same conditions, LPS resulted in a TNFR-1-dependent increase in astrocyte activation and AQP channel-mediated brain edema (53, 61).
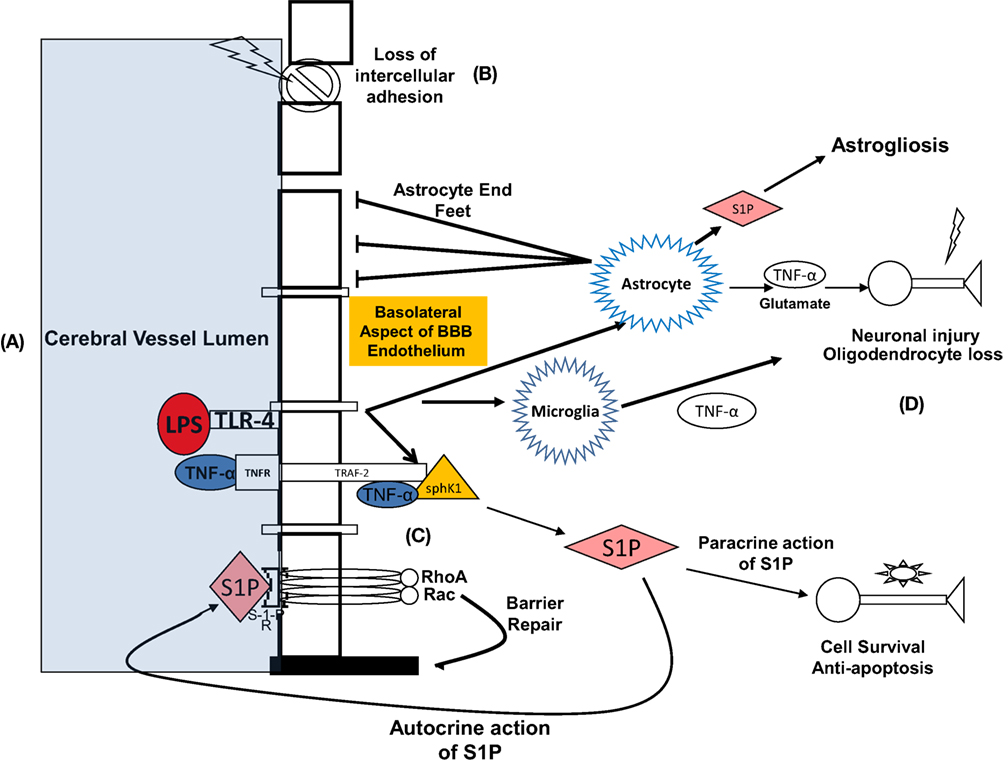
Figure 2. Summary of diverse effects of LPS both during interface with BBB and within CNS: proposed mechanism representing the effects of lipopolysaccharide and TNF-α-at the blood–brain barrier (BBB) and the pathophysiologic sequelae leading to neurocognitive dysfunction. (A) LPS binds to its ligand, toll-like receptor 4 (TLR-4) at the endoluminal surface of brain microvascular endothelial cells. TNF-α-alpha concurrently binds to TNF-α-receptor. (B) Consequently, barrier integrity is lost, and molecular toxins that are normally prevented from entering are now able to migrate to the CNS interstitium, including TNF-α-alpha. TNF-α interacts with glial cells leading to neuronal injury, apoptosis, oligodendrocyte loss, and reactive astrogliosis. Neuronal injury and apoptosis in the hippocampus is a putative mechanism for delirium and protracted neurocognitive deficits. (C) On the basolateral surface of BBB endothelial cells, TRAF-2, TNF-α, and Sphk-1 form a complex that catalyzes the formation of sphingosine-1-phosphate (S-1-P) in the cytosol. S-1-P exits the cell via paracrine function and acts on S-1-P receptor on the luminal surface of the endothelium. Conformational change occurs in the transmembrane domains of the S-1-P receptor, ultimately activating GTPases RhoA and RAC. This results in actin and myosin re-arrangement and re-establishment of BBB integrity. (D) S-1-P also exerts paracrine activity on CNS neurons leading to cellular survival and prevention of apoptosis.
The Essential Role for the Hippocampus in SAE and Pathologic Correlation with Alzheimer Disease (AD)
The hippocampus has been shown to be a key site of involvement for inflammatory mediators and altered synaptic function and plays a key role in the pathogenesis of SAE (Figure 3). LPS administration is strongly linked to the disruption of both memory (77–79) and learning (80). Lynch et al. (81) demonstrated that LPS directly inhibits long-term potentiation in the dentate gyrus of the hippocampus, and Imamura et al. (82) demonstrated a parallel association with IL-1B. Cholinergic neurotransmission is implicated directly in cognition, and its inhibition was shown to be associated with worsening delirium (83). Furthermore, blood brain barrier disruption is associated with hippocampal lesions associated with cognitive impairment (84, 85). Using hippocampal CA1 pyramidal neurons from brain exposed to LPS for 7 days, Hellstrom et al. (86) investigated the mechanism of synaptic dysfunction in a model of LPS neurotoxicity and found that the exposed population had lower membrane resistance, higher action potential threshold, and slower frequency of action potential discharge.
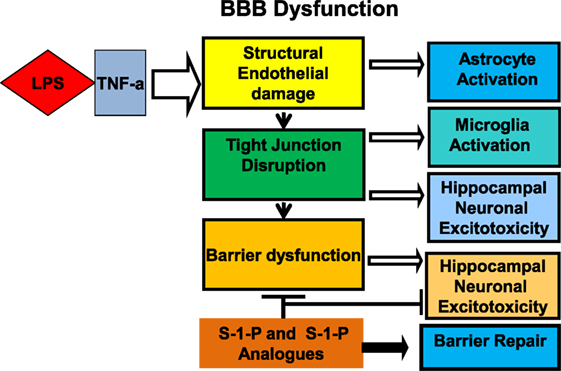
Figure 3. Cross talk between LPS and TNF receptor signaling: The blood–brain barrier (BBB) plays an integral role in the mechanism of neurocognitive injury in sepsis associated encephalopathy. Lipopolysaccharide decreases barrier functional integrity via structural changes in tight junctions and modifications in transendothelial transport. Sphingosine-1-phosphate (S-1-P) or an analogue is proposed to reinforce barrier integrity, potentially attenuating the neurocognitive sequelae of sepsis-associated encephalopathy.
Potential for Sphingolipids as Therapeutic Target for SAE
How can we move from this mechanistic understanding toward therapy for SAE? Management pillars of sepsis include fluid administration and antibiotics, and their early administration may prevent end-organ damage. Recent studies have explored non-pharmacologic interventions such as IVIG, magnesium, steroids, high dose vitamins (87), and monoclonal antibodies (2). For example, after administration of IVIG, rodents that had undergone cecal ligation and perforation had a decreased mortality and decrease BBB permeability with comparison to controls (88). Once they develop, the neuropsychiatric manifestations of SAE remain difficult to treat and are generally limited to supportive care for manifestations of delirium (2). Advances in our understanding of the sphingolipid signaling in the brain (89) have provided a new avenue for the development of therapeutic drug targets in SAE. Sphingolipids are biologically active family of lipids found in cell membranes throughout multiple organ systems. They are essential in mediating vascular permeability, cellular signaling, survival, and apoptosis (90–93). One such sphingolipid, sphingosine-1-phosphate (S-1-P) has been found to play an integral role in angiogenesis and membrane stability, immune cell trafficking, as well as cell proliferation, differentiation, survival, and oncogenesis (94–99). In the “sphingolipid rheostat,” sphingosine is produced when sphingomyelinase catalyzes the production of ceramide from sphingomyelin, after which it is acted upon by ceramidase (90, 96). Sphingosine is phosphorylated to form S-1-P by sphingosine kinase-1 (SphK), a cytosolic 42 kDa lipid kinase with high concentration in the brain, heart, lung, and spleen (90, 96). At the endothelial interface, S-1-P is released by activated platelets (98) and is a ligand for the G-protein-coupled receptors encoded by the endothelial differentiation gene family (100), now termed (S-1-P)1–5. S-1-P also acts as a second messenger, participating in signal transduction (101), such as on oligodendrocytes, where S-1-P is a ligand at the S1P5 receptor. Downstream effects include calcium regulation, cell proliferation, migration, junctional assembly, and prevention of apoptosis (98, 100, 102, 103).
Sphingosine-1-phosphate has a mechanistic and therapeutic role in sepsis-induced endothelial dysfunction and is active on astrocytes, neurons, and glia during inflammatory states (104), as well as the BBB endothelium. Garcia et al. found that vascular endothelial permeability is decreased by the effects of S-1-P (100). When bound to its receptor and coupled to Gi/α, S-1-P promotes signal transduction by kinases p38 MAP kinase and ERK1\2. In murine lung endothelium, this results in an increase in barrier integrity via adherens junction assembly and cytoskeletal cortical actin filament rearrangement and is manifested by increased transendothelial resistance (98, 100, 102, 105). Peng et al. (97) investigated S-1-P’s effects on endothelium in a murine sepsis model. They introduced LPS intraperitoneally, after which dye and albumin extravasation and other markers of transendothelial cell migration were measured in the murine lung. It was observed that vascular leakage and inflammatory cell diapedesis were attenuated significantly both by S-1-P and its analog FTY-720 (97). This work supported previous conclusions (98, 100, 106), indicating that sphingosine1phosphate actively promotes endothelial membrane stability. It was demonstrated that S1P is abundantly produced via phosphorylation by sphingosine kinase in activated platelets, and once released, acts as ligand on sphingosine receptors. Subsequently, GTPases Rho and Rac are activated along with protein kinase C, resulting in both increase in intracellular calcium and transcription of actin resulting in cytoskeletal modification—an essential step given that actin is a critical mediator of barrier stability (100, 107, 108). Both S-1-P and its analog, FTY-720 has reconstitutional activity at the endothelial capillary under conditions of LPS-induced inflammation (97) (Figure 2). Endothelial integrity is strengthened by formation of an actin ring and modification of actin-binding proteins. Our lab (109, 110) demonstrated that inhibition of sphingomyelin synthase (SMS)—and thus sphingomyelin signaling, on lipid rafts in the pulmonary endothelium, resulted in barrier endothelial integrity during LPS-mediated inflammatory insult. After treatment with the SMS inhibitor D609, we observed cytoskeletal rearrangement as evidenced by increased peripheral actin, and interaction of actin and myosin to form a cortical actin ring.
Sphingolipids in the CNS
What we have learned about sepsis-induced modifications of the endothelium in lung may be applicable to other organ systems. Sphingolipids play an active role in central nervous system at the BBB and the cellular level and can be linked to pathological states. For example, under experimental conditions, Cannon et al. (47) showed that the sphingosine analog FTY-720 influences p-glycoprotein-mediated drug uptake at the BBB via a single pathway involving both TNF/TNF-R and sphingosine signaling. In a Parkinson disease model, Martinez et al. (111) demonstrated that in the substantia nigra, dopaminergic neurons are susceptible to caspase-mediated cytotoxicity, and that this pathway is also dependent on both TNF and ceramide. Their study also showed an attenuation of the cytotoxic effect by inhibiting sphingomyelinase. Additionally, Psyzko et al. showed that the oxidative stress response in dopaminergic cells in a PD model was ameliorated by S-1-P, SphK-1, and FTY-720p (112). Sphingolipid signaling in sepsis-induced inflammation in the CNS was investigated by Grin’kina et al. (91) using SphK knockout mice. After intracerebral injection of LPS in the SphK−/− population, significant pathologic changes were noted with comparison to the wild-type population, including an increase in ventricle size, doubling of degree of leukoaraiosis, and increase in white matter rarefactions, together indicating loss of white matter (113). Dysfunction of resident cells of the CNS was significant in the SphK−/− group were reflected by reactive microgliosis, GFAP overexpression (indicating astrocyte activation), and loss of oligodendrocytes (91).
FTY-720, also known as Fingolimod, is currently approved in the management of multiple sclerosis, where it has been well demonstrated to be an immunomodulator hampering T-cell migration in lymphoid tissue (104, 114). It also holds significant potential to utilize the sphingolipid rheostat, with the goal of treating other CNS conditions—including SAE. Specifically, as a sphingosine analog, Fingolimod, acts upon S-1-P receptors to modify endothelium by decreasing its permeability (115). Brinkmann et al. (116) showed that S-1-P prevents VEGF-mediated vascular permeability, and that both S-1-P and phosphorylated FTY-720 strengthened endothelial cell–cell junction assembly. It is also an antagonist at the S-1-P1 receptor on non-lymphoid cells in multiple organ systems and has also been shown to traverse the BBB (104, 117). Further bolstering its utility in SAE is that fact that S-1-P receptors are nearly ubiquitous in the CNS, where they are present on oligodendrocytes, astrocytes, and neurons, functioning as key mediators of normal neural function and repair (104, 118, 119). For example, S-1-P’s actions on oligodendrocytes lead to remyelination (118, 120), and FTY-720 and its metabolite FTY-720-P were shown to prevent excitotoxicity-mediated death in cortical neurons (121). Furthermore, Kanno et al. showed that S-1-P/Sph-K signaling in the hippocampus was associated with enhanced synaptic strength and improved outcomes in memory and learning tasks (122).
In parallel with SAE, AD is manifested by severe cognitive dysfunction, in which neuroinflammation, lipid dysregulation, and plaque formation are well-described mechanisms of development and progression of the disease (123). Ceramide levels are significantly increased in Alzheimer brain (123, 124), which impairs glycolysis, promotes oxidative stress, and ultimately leads to A-beta peptide production (125), implying a critical role for sphingolipids in the pathogenesis. Since severe cognitive dysfunction and the sphingolipid rheostat are shared features of SAE and AD, it is useful to look at the recent preclinical successes of FTY-720 in models of AD as a potential launch point for therapeutic discovery for SAE, especially since the hippocampus as a primary location of pathology for both diseases. In addition, the fact that BBB dysfunction is implicated in both cognitive decline (84) and septic encephalopathy (126) render it a particularly ideal target for FTY-720. For one, there is a relative decrease in S-1-P content in AD affected brain (127), implying that S-1-P agonism may be valuable in treatment. Based on the concept that ceramide levels are inversely proportional to sphingosine in AD (128) plus the potentially neuroprotective property of FTY-720, Asle-Rousta et al. (123) investigated the effectiveness of this compound in a rodent AD model. The authors found that chronic administration of FTY-720 significantly abrogated the A-beta42-induced neuronal loss in the CA1 region of the hippocampus in the study group with comparison to the controls. Aytan et al. (129) showed in transgenic mice that Fingolimod administration decreased amyloid beta plaque density, attenuated microglial activation, and significantly reduced astrocytosis in the hippocampus. Furthermore, Kolahdooz et al. (127) sought to evaluate the ability of two different S-1-P agonists, FTY-720 and SEW7821 on neuroinflammation and LPS-induced memory impairment. The authors found that FTY-720 but not SEW administration significantly decreased LPS-induced memory deficits, and that both agents restored LPS-induced changes in sphingomyelin metabolizing enzymes such as SphK-1.
In summary, the common pathobiologic correlation between SAE and AD together with the success of FTY-720 and its analogs reinforce a promising role for S-1-P in therapeutic development, while further studies in this area are clearly warranted.
Conclusion
Sepsis-associated encephalopathy remains an enigmatic clinical problem despite deep understanding as to the molecular mechanisms of its development. Significant correlations can be made between SAE and AD focusing on the hippocampus, thus providing a mechanistic framework with which we can approach cognitive dysfunction in sepsis. Utilizing our knowledge of sphingolipid rheostat, FTY-720, or an analogous sphingolipid compound may hold the key to stabilizing CNS endothelium and preventing neuroinflammatory injury. If we are successful, we can prevent the often tragic long-term sequelae of SAE including cognitive disability, functional dependency, and chronic institutionalization. The health and quality of life of our aging population is at stake.
Author Contributions
All authors: manuscript writing and revision.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by VA Merit Award.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
2. Adam N, Kandelman S, Mantz J, Chrétien F, Sharshar T. Sepsis-induced brain dysfunction. Expert Rev Anti Infect Ther (2013) 11(2):211–21. doi:10.1586/eri.12.159
3. Ebersoldt M, Sharshar T, Annane D. Sepsis-associated delirium. Intensive Care Med (2007) 33(6):941–50. doi:10.1007/s00134-007-0622-2
4. Sonneville R, Verdonk F, Rauturier C, Klein I, Wolf M, Annane D. Understanding brain dysfunction in sepsis. Ann Int Care (2013) 3:15. doi:10.1186/2110-5820-3-15
5. Papadopoulos MC, Davies DC, Moss RF, Tighe D, Bennett ED. Pathophysiology of septic encephalopathy: a review. Crit Care Med (2000) 28(8):3019–24. doi:10.1097/00003246-200008000-00057
6. Sprung CL, Peduzzi PN, Shatney CH, Schein RM, Wilson MF, Sheagren JN, et al. Impact of encephalopathy on mortality in the sepsis syndrome. The veterans administration systemic sepsis cooperative study group. Crit Care Med (1990) 18(8):801–6. doi:10.1097/00003246-199008000-00001
7. Eidelman LA, Putterman D, Putterman C, Sprung CL. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA (1996) 275(6):470–3. doi:10.1001/jama.1996.03530300054040
8. Neufeld KJ, Thomas C. Delirium: definition, epidemiology, and diagnosis. J Clin Neurophysiol (2013) 30(5):438–42. doi:10.1097/WNP.0b013e3182a73e31
9. Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol (2012) 8(10):557–66. doi:10.1038/nrneurol.2012.183
10. Barichello T, Fortunato JJ, Vitali AM, Feier G, Reinke A, Moreira JC, et al. Oxidative variables in the rat brain after sepsis induced by cecal ligation and perforation. Crit Care Med (2006) 34(3):886–9. doi:10.1097/01.CCM.0000201880.50116.12
11. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA (2010) 304(16):1787Y1794. doi:10.1001/jama.2010.1553
12. Iwashyna TJ, Cooke CR, Wunsch H, Kahn JM. Population burden of long-term survivorship after severe sepsis in older Americans. J Am Geriatr Soc (2012) 60(6):1070–7. doi:10.1111/j.1532-5415.2012.03989.x
13. Iacobone E, Bailly-Salin J, Polito A, Friedman D, Stevens B, Sharshar T. Sepsis-associated encephalopathy and its differential diagnosis. Crit Care Med (2009) 37(10):S331–6. doi:10.1097/CCM.0b013e3181b6ed58
14. Hosokawa K, Gaspard N, Su F, Oddo M, Vincent JL, Taccone FS. Clinical neurophysiological assessment of sepsis-associated brain dysfunction: a systematic review. Crit Care (2014) 18(6):674. doi:10.1186/s13054-014-0674-y
15. Young GB, Bolton CF, Archibald YM, Austin TW, Wells GA. The electroencephalogram in sepsis-associated encephalopathy. J Clin Neurophysiol (1992) 9(1):145–52. doi:10.1097/00004691-199201000-00016
16. Tighe D, Moss R, Boghossian S, Heath M, Chessum B, Bennett ED. Multi-organ damage resulting from experimental faecal peritonitis. Clin Sci (1989) 76:269–76. doi:10.1042/cs0760269
17. Bozza FA, D’Avila JC, Ritter C, Sonneville R, Sharshar T, Dal-Pizzol F. Bioenergetics, mitochondrial dysfunction, and oxidative stress in the pathophysiology of septic encephalopathy. Shock (2013) 39(Suppl 1):10–6. doi:10.1097/SHK.0b013e31828fade1
18. Jackson AC, Gilbert J, Young GB, Bolton CF. The encephalopathy of sepsis. Can J Neurol Sci (1985) 12:303–7. doi:10.1017/S0317167100035381
19. Sharshar T, Polito A, Checinski A, Stevens R. Sepsis-associated encephalopathy – everything starts at a microlevel. Crit Care (2010) 14(5):1999. doi:10.1186/cc9254
20. Sharshar T, Gray F, Poron F, Raphael JC, Gajdos P, Annane D. Multifocal necrotizing leukoencephalopathy in septic shock. Crit Care Med (2002) 30:2371–5. doi:10.1097/00003246-200210000-00031
21. Taccone FS, Su F, De Deyne C, Abdellhai A, Pierrakos C, He X, et al. Sepsis is associated with altered cerebral microcirculation and tissue hypoxia in experimental peritonitis. Crit Care Med (2014) 42(2):e114–22. doi:10.1097/CCM.0b013e3182a641b8
22. Banks WA, Erickson M. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis (2010) 37:26–32. doi:10.1016/j.nbd.2009.07.031
23. Reese TS, Karnovsky MJ. Fine structural localization of a blood–brain barrier to exogenous peroxidase. J Cell Biol (1967) 34:207–17. doi:10.1083/jcb.34.1.207
24. Tsao N, Hsu HP, Wu CM, Liu CC, Lei HY. Tumour necrosis factor-α causes an increase in blood-brain barrier permeability during sepsis. J Med Microbiol (2001) 50:812–21. doi:10.1099/0022-1317-50-9-812
25. Cardoso F, Brites D, Brito M. Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res Rev (2010) 64(2):328–63. doi:10.1016/j.brainresrev.2010.05.003
26. Nico B, Ribatti D. Morphofunctional aspects of the blood-brain barrier. Curr Drug Metab (2012) 13(1):50–60. doi:10.2174/138920012798356970
27. Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab (2012) 32(11):1959–72. doi:10.1038/Jcbfm.2012.126
28. Banks WA. The blood-brain barrier in neuroimmunology: tales of separation and assimilation. Brain Behav Immun (2014) 44:1–8. doi:10.1016/j.bbi.2014.08.007
29. Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev (2005) 57(2):173–85. doi:10.1124/pr.57.2.4
30. Pardridge WM. Blood-brain barrier biology and methodology. J Neurovirol (1999) 5(6):556–556. doi:10.3109/13550289909021285
31. Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis (2004) 16:1–13. doi:10.1016/j.nbd.2003.12.016
32. Abbott NJ, Friedman A. Overview and introduction: the blood-brain barrier in health and disease. Epilepsia (2012) 53(6):1–6. doi:10.1111/j.1528-1167.2012.03696
33. Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci (2006) 7(1):41–53. doi:10.1038/nrn1824
34. Persidksy Y, Ramirez S, Haorah J, Kanmogne G. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimm Pharm (2006) 1(3):223–36. doi:10.1007/s11481-006-9025-3
35. Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol (2002) 38:323–37. doi:10.1016/S1537-1891(02)00200-8
36. Yuan H, Gaber MW, McColgan T, Naimark MD, Kiani MF, Merchant TE. Radiation-induced permeability and leukocyte adhesion in the rat blood-brain barrier: modulation with anti-ICAM-1 antibodies. Brain Res (2003) 969(1–2):59–69. doi:10.1016/S0006-8993(03)02278-9
37. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis (2010) 37(1):13–25. doi:10.1016/j.nbd.2009.07.030
38. Jin L, Nation RL, Li J, Nicolazzo JA. Species-dependent blood-brain barrier disruption of lipopolysaccharide: amelioration by colistin in vitro and in vivo. Antimicrob Agents Chemother (2013) 57(9):4336–42. doi:10.1128/AAC.00765-13
39. Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol (1990) 429:47–62. doi:10.1113/jphysiol.1990.sp018243
40. de Boer AG, Gaillard PJ. Blood-brain barrier dysfunction and recovery. J Neural Transm (2006) 113(4):455–62. doi:10.1007/s00702-005-0375-4
41. Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol (2008) 183:409–17. doi:10.1083/jcb.200806024
42. Obermeier B, Daneman R, Ransohoff R. Development, maintenance and disruption of the blood-brain barrier. Nat Med (2013) 19(12):1584–96. doi:10.1038/nm.3407
43. Kim JA, Tran ND, Li Z, Yang F, Zhou W, Fisher MJ. Brain endothelial hemostasis regulation by pericytes. J Cereb Blood Flow Metab (2006) 26(2):209–17. doi:10.1038/sj.jcbfm.9600181
44. Lai CH, Kuo KH, Leo JM. Critical role of actin in modulating BBB permeability. Brain Res Rev (2005) 50:7–13. doi:10.1016/j.brainresrev.2005.03.007
45. El-Bacha RS, Minn A. Drug metabolizing enzymes in cerebrovascular endothelial cells afford a metabolic protection to the brain. Cell Mol Biol (1999) 45:15–23.
46. Bauer B, Hartz AM, Miller DS. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol Pharmacol (2007) 71(3):667–75. doi:10.1124/mol.106.029512
47. Cannon R, Peart J, Hawkins B, Campos C, Miller D. Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc Natl Acad Sci U S A (2012) 109(39):15930–5. doi:10.1073/pnas.1203534109
48. Pan W, Stone KP, Hsuchou H, Manda VK, Zhang Y, Kastin AJ. Cytokine signaling modulates blood-brain barrier function. Curr Pharm Des (2011) 17(33):3729–40. doi:10.2174/138161211798220918
49. D’Alessio A, Al-Lamki RS, Bradley JR, Pober JS. Caveolae participate in tumor necrosis factor receptor 1 signaling and internalization in a human endothelial cell line. Am J Pathol (2005) 166(4):1273–82. doi:10.1016/S0002-9440(10)62346-2
50. Long M, Huang S-H, Wu C-H, Shackleford GM, Jong A. Lipid raft/caveolae signaling is required for Cryptococcus neoformans invasion into human brain microvascular endothelial cells. J Biomed Sci (2012) 19(1):19. doi:10.1186/1423-0127-19-19
51. Parton R, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol (2007) 8:185–94. doi:10.1038/nrm2122
52. Nag S, Manias JL, Stewart DJ. Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol Appl Neurobiol (2009) 35(4):417–26. doi:10.1111/j.1365-2990.2008.01009.x
53. Alexander J, Jacob A, Cunningham P, Hensley L, Quigg R. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int (2008) 52(3):447–56. doi:10.1016/j.neuron.2007.08.006
54. Jacob A, Brorson JR, Alexander JJ. Septic encephalopathy: inflammation in man and mouse. Neurochem Int (2011) 58(4):472–6. doi:10.1016/j.neuint.2011.01.004
55. Jacob A, Hack B, Chen P, Quigg RJ, Alexander JJ. C5a/CD88 signaling alters blood-brain barrier integrity in lupus through nuclear factor-κB. J Neurochem (2011) 119(5):1041–51. doi:10.1111/j.1471-4159.2011.07490.x
56. Jacob A, Alexander JJ. Complement and blood-brain barrier integrity. Mol Immunol (2014) 61(2):149–52. doi:10.1016/j.molimm.2014.06.039
57. Ducruet AF, Zacharia BE, Hickman ZL, Grobelny BT, Yeh ML, Sosunov SA, et al. The complement cascade as a therapeutic target in intracerebral hemorrhage. Exp Neurol (2009) 219(2):398–403. doi:10.1016/j.expneurol.2009.07.018
58. Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res (2005) 27(3):268–79. doi:10.1179/016164105X25225
59. Semmler A, Hermann S, Mormann F, Weberpals M, Paxian S, Okulla T, et al. Sepsis causes neuroinflammation and concomitant decrease in cerebral metabolism. J Neuroinflamm (2008) 5:38. doi:10.1186/1742-2094-5-38
60. Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U S A (2005) 102(28):9936–41. doi:10.1073/pnas.0502552102
61. Yung HW, Bal-Price AK, Brown GC, Tolkovsky AM. Nitric oxide-induced cell death of cerebrocortical murine astrocytes is mediated through p53- and Bax-dependent pathways. J Neurochem (2004) 89:812–21. doi:10.1111/j.1471-4159.2004.02395.x
62. Cannon JG, Tompkins RG, Gelfand JA, Michie HR, Stanford GG, van der Meer JW, et al. Circulating interleukin-1 and tumor necrosis factor in septic shock and experimental endotoxin fever. J Infect Dis (1990) 161:79–84. doi:10.1093/infdis/161.1.79
63. Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (P55 and P75) in the rat brain: a view from the blood-brain barrier. Neuroscience (1999) 93(4):1449–64. doi:10.1016/S0306-4522(99)00225-0
64. Andreasen A, Krabbe K, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K. Human endotoxemia as model of systemic inflammation. Curr Med Chem (2008) 15:1697–705. doi:10.2174/092986708784872393
65. Bauer HC, Krizbai IA, Traweger A. “You shall not pass” tight junctions of the blood brain barrier. Front Neurosci (2014) 8:392. doi:10.3389/fnins.2014.00392
66. Nagyoszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NT, Haskó J. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int (2010) 57(5):556–64. doi:10.1016/J.Neuint.2010.07.002
67. Goldblum SE, Ding X, Campbell-Washington J. TNFa induces endothelial cell F-actin depolymerization, new actin synthesis, and barrier dysfunction. Am J Physiol (1993) 264:894–905.
68. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia (2007) 55(5):453–62. doi:10.1002/glia.20467
69. O’Carroll SJ, Kho DT, Wiltshire R, Nelson V, Rotimi O, Johnson R, et al. Pro-inflammatory TNFα and IL-1β differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J Neuroinflammation (2015) 12:131. doi:10.1186/s12974-015-0346-0
70. Haines RJ, Beard RS Jr, Wu MH. Protein tyrosine kinase 6 mediates TNFα-induced endothelial barrier dysfunction. Biochem Biophys Res Commun (2015) 456(2):190–6. doi:10.1016/j.bbrc.2014.11.057
71. Dopp JM, Mackenzie-Graham A Jr, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol (1997) 75(1–2):104–12.
72. Xia P, Wang L, Moretti PA, Albanese N, Chai F, Pitson SM. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem (2002) 277(10):7996–8003. doi:10.1074/jbc.M111423200
73. Cheng ZJ, Singh RD, Sharma DK, Holicky EL, Hanada K, Marks DL, et al. Distinct mechanisms of clathrin-independent endocytosis have unique sphingolipid requirements. Mol Biol Cell (2006) 17(7):3197–210. doi:10.1091/mbc.E05-12-1101
75. Yuan J, Yankner BA. Apoptosis in the nervous system. Nature (2000) 407:802–9. doi:10.1038/35037739
76. Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature (2011) 472(7343):319–24. doi:10.1038/nature09788
77. Krabbe KS, Reichenberg A, Yirmiya R, Smed A, Pedersen BK, Bruunsgaard H. Low-dose endotoxemia and human neuropsychological functions. Brain Behav Immun (2005) 19(5):453–60. doi:10.1016/j.bbi.2005.04.010
78. Czerniawski J, Guzowski JF. Acute neuroinflammation impairs context discrimination memory and disrupts pattern separation processes in hippocampus. J Neurosci (2014) 34:12470–80. doi:10.1523/JNEUROSCI.0542-14.2014
79. Hasegawa-Ishii S, Inaba M, Umegaki H, Unno K, Wakabayashi K, Shimada A. Endotoxemia-induced cytokine-mediated responses of hippocampal astrocytes transmitted by cells of the brain-immune interface. Sci Rep (2016) 6:25457. doi:10.1038/srep25457
80. Shaw KN, Commins S, O’Mara SM. Lipopolysaccharide causes deficits in spatial learning in the water maze but not in BDNF expression in the rat dentate gyrus. Behav Brain Res (2001) 124(1):47–54. doi:10.1016/S0166-4328(01)00232-7
81. Lynch AM, Walsh C, Delaney A, Nolan Y, Campbell VA, Lynch MA. Lipopolysaccharide-induced increase in signalling in hippocampus is abrogated by IL-10 – a role for IL-1 beta? J Neurochem (2004) 88(3):635–46. doi:10.1046/j.1471-4159.2003.02157.x
82. Imamura Y, Wang H, Matsumoto N, Muroya T, Shimazaki J, Ogura H, et al. Interleukin-1β causes long-term potentiation deficiency in a mouse model of septic encephalopathy. Neuroscience (2011) 187:63–9. doi:10.1016/j.neuroscience.2011.04.063
83. Semmler A, Frisch C, Debeir T, Ramnanathan M, Okull T, Klockgeteher T. Long-term cognitive impairment, neuronal loss and reduced cortical cholinergic innervation after recovery from sepsis in a rodent model. Exp Neurol (2007) 204(2):733–40. doi:10.1016/j.expneurol.2007.01.003
84. Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron (2015) 85(2):296–302. doi:10.1016/j.neuron.2014.12.032
85. Costantino I. Dangerous leaks: blood-brain barrier woes in the aging hippocampus. Neuron (2015) 85(2):231–3. doi:10.1016/j.neuron.2014.12.056
86. Hellstrom IC, Danik M, Luheshi GN, Williams S. Chronic LPS exposure produces changes in intrinsic membrane properties and a sustained IL-beta-dependent increase in GABAergic inhibition in hippocampal CA1 pyramidal neurons. Hippocampus (2005) 15(5):656–64. doi:10.1002/hipo.20086
87. Marik PE, Khangoora V, Rivera R, Hooper MH, Catravas J. Hydrocortisone, vitamin C and thiamine for the treatment of severe sepsis and septic shock: a retrospective before-after study. Chest (2016). doi:10.1016/j.chest.2016.11.036
88. Esen F, Senturk E, Ozcan PE, Ahishali B, Arican N, Orhan N, et al. Intravenous immunoglobulins prevent the breakdown of the blood-brain barrier in experimentally induced sepsis. Crit Care Med (2012) 40(4):1214–20. doi:10.1097/CCM.0b013e31823779ca
89. Bryan L, Kordula T, Spiegel S, Milstien S. Regulation and function of sphingosine kinases in the brain. Biochim Biophys Acta (2008) 1781(9):459–66. doi:10.1016/j.bbalip.2008.04.008
90. Okada T, Kajimoto T, Jahangeer S, Nakamura S. Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell Signal (2009) 21(1):7–13. doi:10.1016/j.cellsig.2008.07.011
91. Grin’kina NM, Karnabi EE, Damania D, Wadgaonkar S, Muslimov IA, Wadgaonkar R. Sphingosine kinase 1 deficiency exacerbates LPS-induced neuroinflammation. PLoS One (2012) 7(5):e36475. doi:10.1371/journal.pone.0036475
92. Kono M, Allende ML, Proia RL. Sphingosine-1-phosphate regulation of mammalian development. Biochim Biophys Acta (2008) 1781(9):435–41. doi:10.1016/j.bbalip.2008.07.001
93. Pyne S, Pyne N. Sphingosine 1-phosphate signalling in mammalian cells. Biochem J (2000) 349:385–402. doi:10.1042/bj3490385
94. Mendelson K, Evan T, Hla T. Sphingosine-1-phosphate signaling. Development (2014) 141:5–9. doi:10.1242/dev.094805
95. Spiegel S, Milstien S. Sphingosine 1-phosphate, a key cell signaling molecule. J Biol Chem (2002) 277(29):25851–4. doi:10.1074/jbc.R200007200
96. Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta (2006) 1758:2016–26. doi:10.1016/j.bbamem.2006.08.007
97. Peng X, Hassoun P, Sammani S, McVerry B, Burne M, Rabb H, et al. Protective effects of sphingosine-1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med (2004) 169(11):1245–51. doi:10.1164/rccm.200309-1258OC
98. Schaphorst KL, Chiang E, Jacobs KN, Zaiman A, Natarajan V, Wigley F, et al. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier Integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol (2003) 285(1):L258–67. doi:10.1152/ajplung.00311.2002
99. Pitson S, Moretti P, Zebol J, Lynn H, Xia P, Vadas M. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J (2003) 22(20):5491–500. doi:10.1093/emboj/cdg540
100. Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by EDG-dependent cytoskeletal rearrangement. J Clin Invest (2001) 108(5):689–701. doi:10.1172/JCI12450
101. Buccoliero R, Futerman AH. The roles of ceramide and complex sphingolipids in neuronal cell function. Pharmacol Res (2003) 47(5):409–19. doi:10.1016/S1043-6618(03)00049-5
102. Wadgaonkar R, Patel V, Grinkina N, Romano C, Liu J, Zhao Y, et al. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am J Physiol Lung Cell Mol Physiol (2009) 296(4):L603–13. doi:10.1152/ajplung.90357.2008
103. Zheng DM, Kitamura T, Ikejima K, Enomoto N, Yamashina S, Suzuki S, et al. Sphingosine 1-phosphate protects rat liver sinusoidal endothelial cells from ethanol-induced apoptosis: role of intracellular calcium and nitric oxide. Hepatology (2006) 44(5):1278–87. doi:10.1002/hep.21384
104. Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol (2010) 33:91. doi:10.1097/WNF.0b013e3181cbf825
105. Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res (2008) 103(10):1164–72. doi:10.1161/01.RES.0000338501.84810.51
106. Shikata Y, Birukov KG, Garcia JG. S1P induces FA remodeling in human pulmonary endothelial cells: role of Rac, GIT1, FAK, and paxillin. J Appl Physiol (1985) 94(3):1193–203. doi:10.1152/japplphysiol.00690.2002
107. Shasby DM, Shasby S, Sullivan JM, Peach MJ. Role of endothelial cell cytoskeleton in control of endothelial permeability. Circ Res (1982) 51:657–61. doi:10.1161/01.RES.51.5.657
108. Natarajan V, Dudek SM, Jacobson JR, Moreno-Vinasco L, Huang LS, Abassi T, et al. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am J Respir Cell Mol Biol (2013) 49(1):6–17. doi:10.1165/rcmb.2012-0411TR
109. Anjum F, Joshi K, Grinkina N, Gowda S, Cutaia M, Wadgaonkar R. Role of sphingomyelin synthesis in pulmonary endothelial cell cytoskeletal activation and endotoxin-induced lung injury. Am J Respir Cell Mol Biol (2012) 47(1):94–103. doi:10.1165/rcmb.2010-0458OC
110. Gowda S, Yeang C, Wadgaonkar S, Anjum F, Grinkina N, Cutaia M, et al. Sphingomyelin synthase 2 (SMS2) deficiency attenuates LPS-induced lung injury. Am J Physiol Lung Cell Mol Physiol (2011) 300(3):L430–40. doi:10.1152/ajplung.00208.2010
111. Martinez TN, Chen X, Bandyopadhyay S, Merrill AH, Tansey MG. Ceramide sphingolipid signaling mediates tumor necrosis factor (TNF)-dependent toxicity via caspase signaling in dopaminergic neurons. Mol Neurodegener (2012) 13(7):45. doi:10.1186/1750-1326-7-45
112. Pyszko J, Strosznaider J. Sphingosine kinase 1 and sphingosine-1-phosphate in oxidative stress evoked by 1-methyl-4-phenylpyridinium (MPP+) in human dopaminergic neuronal cells. Mol Neurobiol (2014) 50(1):38–48. doi:10.1007/s12035-013-8622-4
114. Gholamnezhadjafari R, Falak R, Tajik N, Aflatoonian R, Ali Keshtkar A, Rezaei A. Effect of FTY720 (fingolimod) on graft survival in renal transplant recipients: a systematic review protocol. BMJ Open (2016) 6(4):e010114. doi:10.1136/bmjopen-2015-010114
115. Sanchez T, Estrada-Hernandez T, Paik JH, Wu MT, Venkataraman K, Brinkmann V, et al. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem (2003) 278:47281–90. doi:10.1074/jbc.M306896200
116. Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, et al. The immune modulator fty720 targets sphingosine 1-phosphate receptors. J Biol Chem (2002) 277:21453–7. doi:10.1074/jbc.C200176200
117. Foster CA, Howard LM, Schweitzer A, Persohn E, Hiestand PC, Balatoni B, et al. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther (2007) 323:469–75. doi:10.1124/jpet.107.127183
118. O’Sullivan S, Dev KK. Sphingosine-1-phosphate receptor therapies: advances in clinical trials for CNS-related diseases. Neuropharmacology (2017) 113(Pt B):597–607. doi:10.1016/j.neuropharm.2016.11.006
119. Groves A, Kihara Y, Chun J. Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci (2013) 328(1–2):9–18. doi:10.1016/j.jns.2013.02.011
120. Miron VE, Jung CG, Kim HJ, Kennedy TE, Soliven B, Antel JP. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol (2008) 63(1):61–71. doi:10.1002/ana.21227
121. Di Menna L, Molinaro G, Di Nuzzo L, Riozzi B, Zappulla C, Pozzilli C, et al. Fingolimod protects cultured cortical neurons against excitotoxic death. Pharmacol Res (2013) 67(1):1–9. doi:10.1016/j.phrs.2012.10.004
122. Kanno T, Nishizaki T, Proia RL, Kajimoto T, Jahangeer S, Okada T, et al. Regulation of synaptic strength by sphingosine 1-phosphate in the hippocampus. Neuroscience (2010) 171(4):973–80. doi:10.1016/j.neuroscience.2010.10.021
123. Asle-Rousta M, Kolahdooz Z, Oryan S, Ahmadiani A, Dargahi L. FTY720 (fingolimod) attenuates beta-amyloid peptide (Aβ42)-induced impairment of spatial learning and memory in rats. J Mol Neurosci (2013) 50(3):524–32. doi:10.1007/s12031-013-9979-6
124. Filippov V, Song MA, Zhang K, Vinters HV, Tung S, Kirsch WM, et al. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J Alzheimers Dis (2012) 29(3):537–47. doi:10.3233/JAD-2011-111202
125. Asle-Rousta M, Kolahdooz Z, Dargahi L, Ahmadiani A, Nasoohi S. Prominence of central sphingosine-1-phosphate receptor-1 in attenuating aβ-induced injury by fingolimod. J Mol Neurosci (2014) 54(4):698–703. doi:10.1007/s12031-014-0423-3
126. Davies DC. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J Anat (2002) 200:639–46. doi:10.1046/j.1469-7580.2002.00047_17.x
127. Kolahdooz Z, Nasoohi S, Asle-Rousta M, Ahmadiani A, Dargahi L. Sphingosin-1-phosphate receptor 1: a potential target to inhibit neuroinflammation and restore the sphingosin-1-phosphate metabolism. Can J Neurol Sci (2015) 42(3):195–202. doi:10.1017/cjn.2015.19
128. He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging (2010) 31(3):398–408. doi:10.1016/j.neurobiolaging.2008.05.010
Keywords: sepsis-associated encephalopathy, lipopolysaccharides, sphingosine, blood–brain barrier, inflammation mediators
Citation: Kuperberg SJ and Wadgaonkar R (2017) Sepsis-Associated Encephalopathy: The Blood–Brain Barrier and the Sphingolipid Rheostat. Front. Immunol. 8:597. doi: 10.3389/fimmu.2017.00597
Received: 03 February 2017; Accepted: 05 May 2017;
Published: 16 June 2017
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Demet Demirkol, Koç University, TurkeyMario Di Napoli, Ospedale San Camillo de Lellis, Italy
Copyright: © 2017 Kuperberg and Wadgaonkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen J. Kuperberg, c3RlcGhlbmt1cGVyYmVyZ21kQGdtYWlsLmNvbQ==;
Raj Wadgaonkar, cmFqLndhZGdhb25rYXJAZG93bnN0YXRlLmVkdQ==