 Jinhua Lu
Jinhua Lu Uday Kishore
Uday Kishore
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 May 2017
Sec. Molecular Innate Immunity
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00592
This article is part of the Research Topic C1q: A Molecular Bridge to Innate and Adaptive Immunity View all 10 articles
Complement C1 is the defining component of the classical pathway. Within the C1qC1r2C1s2 complex, C1q functions as a molecular scaffold for C1r2C1s2 and C1q binding to its ligands activates these two serine proteases. The classic C1q ligands are antigen-bound antibodies and activated C1s cleaves C4 and C2 to initiate the complement cascade. Recent studies suggest broad C1 functions beyond the complement system. C1q binds to the Frizzled receptors to activate C1s, which cleaves lipoprotein receptor-related protein 6 to trigger aging-associated Wnt receptor signaling. C1q binds to apoptotic cells and the activated C1 proteases cleave nuclear antigens. C1s also cleaves MHC class I molecule and potentially numerous other proteins. The diversity of C1q ligands and C1 protease substrates renders C1 complex versatile and modular so that it can adapt to multiple molecular and cellular processes besides the complement system.
In invertebrates, complement takes primitive forms represented only by a few ancestral proteins and lacks the specificity and sophisticated regulatory mechanisms of the modern vertebrate complement system (1–4). In mammals and other higher vertebrates, the complement system is a complex protein network consisting of nearly 30 plasma proteins. Depending on the target ligands, the complement system can be activated via the classical, alternative, or lectin pathway (5, 6). In the case of microbial pathogens, each complement pathway is triggered through a specific mechanism of ligand recognition, and collectively, the three pathways empower this humoral system to defend against a broad range of microorganisms. Like the blood coagulation system, the complement system is orchestrated around serine proteases, which are sequentially activated and then cleave specific downstream complement proteins so as to amplify a cascade of reactions (2, 7, 8). These reactions generate proteolytic or lytic complexes, opsonins, and peptide anaphylatoxins leading to lysis, inflammation, and clearance of opsonized microorganisms (Figure 1) (5, 6). The complement serine proteases exhibit conserved active sites (2). However, these proteases are highly specific for substrate within the complement network, and this appears vital for the directional amplification of each pathway.
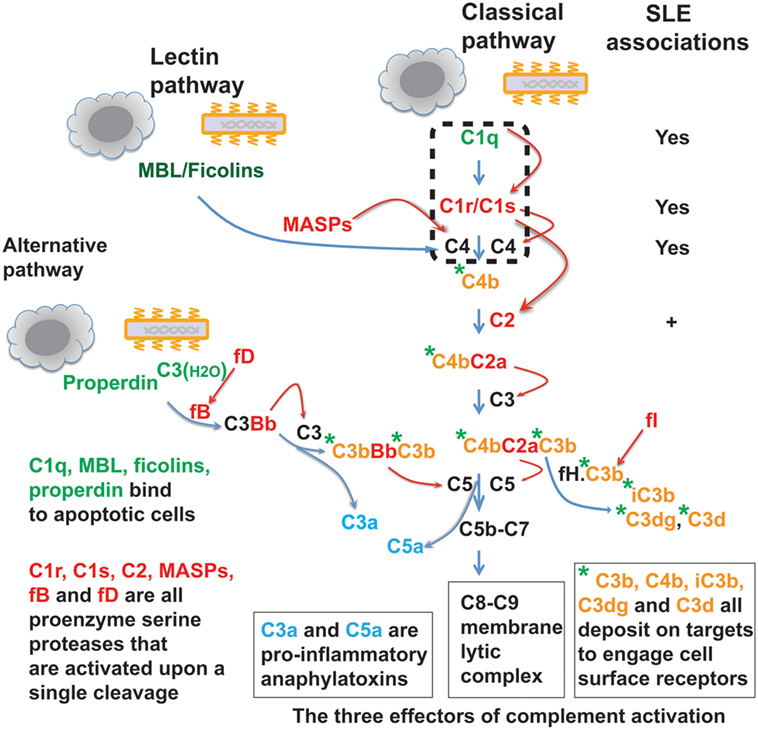
Figure 1. The complement protein and protease network. The complement system operates via three target recognition pathways: the classical, alternative, and lectin pathways. All pathways recognize microorganisms and apoptotic cells and the recognition subcomponents are in green. Upon its triggering via any of the three pathways, the complement acts through three effector reactions: the C8-C9 lytic or membrane attack complex, the soluble C3a and C5a anaphylatoxins (blue color), and surface-bound C3b, C4b, and further proteolytic fragment opsonins (orange color with green asterisk). All three pathways converge at the C3 component and complement reactions are essentially amplified through cascades of serine proteases (red color). MBL, mannose-binding lectin; MASPs, MBL-associated serine proteases; fB, factor B; fD, factor D; fI, factor I; fH, factor H. Homozygous deficiency of C1q, C1r/C1s, or C4 is causally associated with systemic lupus erythematosus (SLE) pathogenesis. Genetic C2 deficiency also increases risk for SLE and some other autoimmune diseases.
The complement system is commonly intended for host defense against microbial infections. Recent data suggest that various non-microbial exogenous and endogenous structures, such as apoptotic cells, may also trigger the complement pathways (Figure 1) (9–15). The effects of complement activation may also be delivered through a segment of the system rather than in its entirety. For example, the C1s protease apparently cleaves non-complement proteins including MHC class I molecule, insulin-like growth factor binding protein 5 (IGFBP5), Wnt receptor, and nuclear autoantigens (16–21). This suggests that, besides its well-defined roles in host defense, the C1 complex functions broadly, e.g., in tissue homeostasis and immune tolerance. In fact, invertebrates also utilize their limited repertoire of complement components to clear damaged cells as well as invading microorganisms (22, 23).
During evolution from invertebrates leading up to higher vertebrates, animals experienced major genomic expansion through gene duplication and recombination, with higher vertebrates acquiring increased complexity in genomic composition, body plans, and physiological processes (24). The expansion of the complement system in higher vertebrates includes at least two aspects: the generation of paralogous complement elements and the formation of a new classical pathway. In invertebrates, ancestral complement elements were only found that were equivalent to the alternative and lectin pathways, including ancestral C3, factor B, mannose-binding lectin (MBL), ficolins, and MBL-associated serine proteases (MASPs) (3, 22). The modern C1 complex, i.e., the C1qC1r2C1s2 pentamer that defines the recognition component of the classical pathway, only appeared from jawed vertebrates when adaptive immunity also emerged.
Complement gene duplication and recombination are evident in higher vertebrates, e.g., factor B/C2, C3/C4/C5, and C6/C7/C8/C9 (3). Evidence that the C1r and C1s genes are relatively modern duplications is also suggested by their close genomic proximity and structural similarity (8, 25). This is even more striking with the three C1q subunit genes, i.e., C1qA, C1qB, and C1qC, which are clustered within a 30-kb genomic region separated by short intergenic sequences (26, 27) (Figure 2). The closest C1q-related protein in invertebrates is encoded by a single gene and the C1q-like protein recognizes carbohydrates rather than immunoglobulins (4). The emergence of the C1 complex or the classical pathway in higher vertebrates, which coincided with the appearance of the adaptive immune system, makes it a “modern” arm of the complement system that responds to antibodies and other self, non-self, and altered self targets.
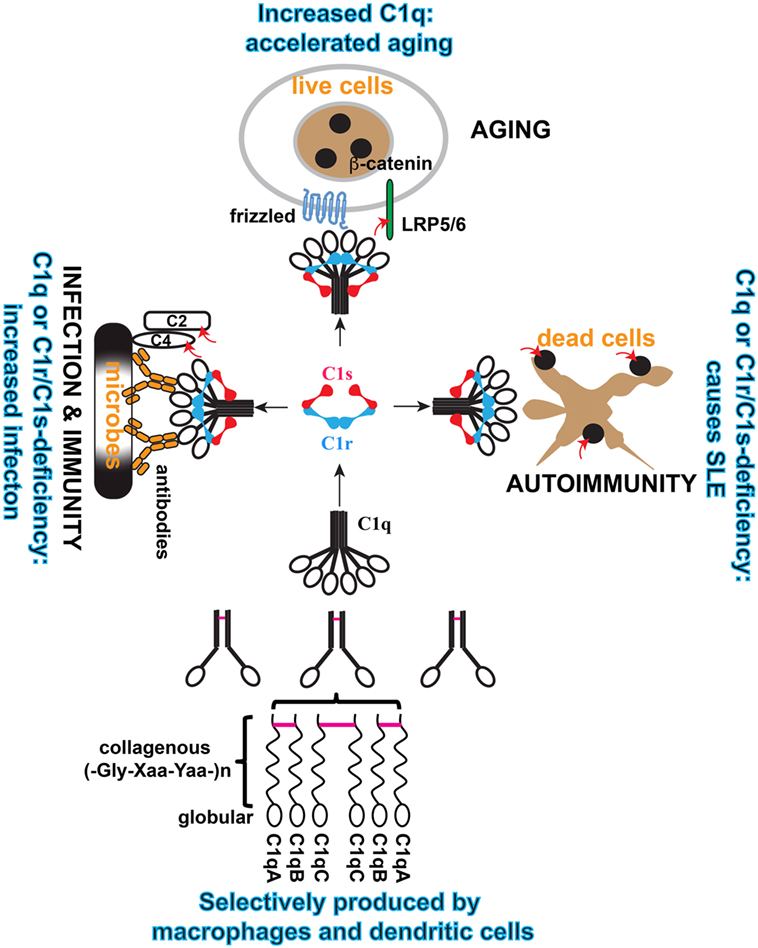
Figure 2. Schematic illustration of the cellular origin of C1q, its assembly, and three distinct modular functions of C1 complex. C1q is an abundant plasma protein. It can be produced broadly in tissues by macrophages and DCs, which also produce C1r/C1s. C1q is formed from three similar but distinct subunits, i.e., A, B, and C, and the order of assembly is illustrated. Inter-subunit disulfide bonds (purple bars) and the collagen-like helices defines the sorting of the three subunits within the C1q polypeptide. In the serum, C1q is associated with the serine protease proenzyme tetramer C1r2C1s2 to form the C1 complex. In this complex, C1q binds to diverse targets that activate the serine proteases and the proteases trigger effector reactions by cleaving specific substrate. Three physiological contexts are highlighted in which the C1 complex is known to play a role. When C1q binds to antibodies on microbial pathogens, the activated C1s cleaves complement C4 and C2 to initiate the proteolytic cascade. When C1q binds to Frizzled, activated C1s cleaves lipoprotein receptor-related protein 6 to cause canonical Wnt signaling and accelerated aging. When C1q binds to apoptotic cells, the activated C1s cleaves apoptotic cellular antigens to reduce autoimmunogenicity. The red arrows indicate C1s cleavage of the specific substrate.
Genetic deficiency has been identified for many complement proteins and, in most cases, this increases susceptibility to infections (28, 29). Deficiency in some complement proteins is also associated with other pathological conditions and particularly strong associations were found between deficiencies in the early components of the complement classical pathway and the autoimmune disease SLE (29–34). The association is especially strong with homozygous C1 and C4 deficiencies. Functionally, C1q binding to ligands causes C1r and then C1s activation and the activated C1s cleaves C4 and then C2 to initiate the further downstream complement cascade (5, 6). C2 deficiency is more prevalent than C1 and C4 deficiencies, but it has substantially less effect and is also associated with other autoimmune diseases (31, 32). However, C1q, C1r/C1s, and C4 deficiencies cause predominantly SLE-like conditions.
In C1 and C4 deficiencies, the disease manifestations also deviate from that found in the larger SLE patient population. Typically, this specific group of SLE patients exhibit early disease onset and equal disease risks from both genders (30, 31, 35). SLE is otherwise a chronic disease that affects predominantly females at childbearing ages (36). How deficiency in each of these intimately related complement proteins, which define the classical pathway (Figure 1), causes SLE remains incompletely understood.
Clinical documentation of SLE disease has existed for more than a century. In 1948, Hargraves pioneered the mechanistic investigation of this disease by reporting the L.E. cell phenomenon, i.e., SLE patient serum caused polymorphnuclear leukocytes to bind or clump around autologous amorphous nuclear materials (37). The serum activity was later attributed to the γ-globulin fraction of the patient serum, presently known as autoantibodies reactive with chromatin or DNA (38–40). A pathogenic role for these autoantibodies became apparent when Tan et al. reported the asymptomatic appearance of anti-DNA autoantibodies, which disappeared during the ensuing disease flare when serum DNA antigen surged to complex with these autoantibodies (41). These autoantibodies are hallmarks in SLE pathogenesis and deposit in tissues leading to inflammatory tissue injury (42–44).
For a large majority of SLE patients, there is no definitive genetic explanation for the disease despite more than 50 SLE risk genes that have been identified (45). Most of these susceptibility genes are not specific for SLE and individually each risk gene has low-to-moderate effect on the disease (32). Known exceptions are genetic deficiencies of the intracellular exonuclease Trex1, and complement C1 and C4 (30–32). How deficiency in each of these complement proteins overrides the complex mechanisms governing host immunity and tolerance to cause this complex autoimmune disease is not fully understood. As anti-nuclear autoantibodies are pathogenic in SLE, understanding how these deficiencies cause anti-nuclear autoimmunity can provide greater insights into the underlying pathogenic mechanisms.
While C1q deficiency causes autoimmunity, its elevated plasma levels signify accelerated aging. Aging is marked by a decline in tissue regeneration and repair, and in the number and dynamics of tissue stem or progenitor cells (46). At the molecular level, one observation is that progenitor cells exhibit elevated Wnt signaling in the aging tissue environment (47, 48). In aged mice, muscle stem cells exhibit increased tendency to fibroblastic differentiation (48). This was found to be conferred by a serum factor(s) in aged mice binding to the Frizzled family of cell surface receptors and causing Wnt receptor signaling (48). This Frizzled-binding protein was identified as C1q (48). Its serum level increased threefold (from 90 to 280 µg/ml) in old mice (20 months) as compared with young mice (2 months) (19).
Mechanistically, C1q binding to the Frizzled receptors causes C1s activation and activated C1s cleaves the Wnt receptor protein low-density lipoprotein receptor-related protein 6 (LRP6) to trigger canonical Wnt receptor signaling (19) (Figure 3). The involvement of C4 and further downstream complement components are not defined. Nonetheless, this emphasizes the rather less-studied aspect of C1-mediated cleavage of proteins outside the complement network. C1s similarly cleaves MHC class I molecule, although the C1q ligands are not defined in this context (16, 17). It appears that activation of the complement classical pathway, which involves C1s cleavage of C4 and C2, is merely one of a number of effector mechanisms downstream of the C1 complex (Figure 3).
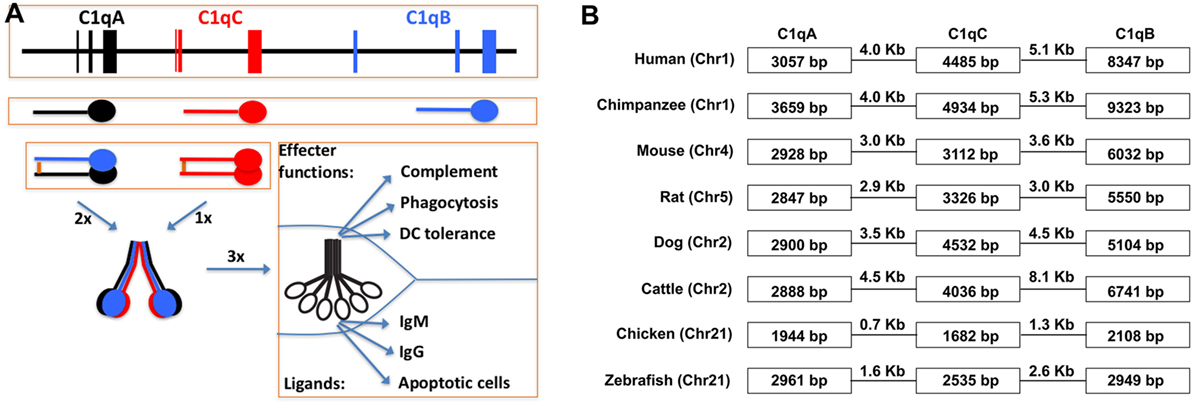
Figure 3. Schematic illustration of C1q gene organization, gene transcription, and multimeric C1q assembly. (A) The three human C1q genes span approximately 25 kb on human chromosome 1. The intergenic regions are 4.0 and 5.1 kb, respectively, which are not distinguishable in size from regular introns in the C1q genes. Each of the three C1q genes contains three exons and the transcribed peptides form disulfide-linked A-B hererodimers and C-C homodimers. Each C chain in the homodimer forms a collagen triple-helix with an A-B heterodimer, and hence, two triple-helices linked by the disulfide bond in the C-C dimer. Three such ABC-CBA twin helices associate non-covalently over the N-terminal ends to form the 18-polypeptide C1q molecule. The gC1q domains are often the ligand-binding sites for C1q and the collagen triple-helices associate with the C1r2C1s2 serine protease tetramer. (B) Conservation of the C1q gene organization in eight different animal species. The three C1q genes in chimpanzee occupied the largest genomic space which is approx. 27 kb. In chickens, the three C1q genes occupied merely 7.7 kb with intergenic sequences of 0.7 and 1.3 kb, respectively.
Besides a distinct decline in tissue regeneration and repair, aging is also characterized by systemic elevation of the inflammatory status (49, 50). In the elderly population, plasma pro-inflammatory cytokines, IL-6 and TNF-α, and the acute phase C-reactive protein (CRP) are chronically elevated. When young (<40 years) and aged (60–81 years) populations were compared in a series of age-related parameters, including muscle mass, plasma C1q, as well as plasma IL-6, TNF-α, and CRP, the young population had clearly lower plasma C1q (80.5 µg/ml) than the aged population (161 µg/ml) (51). Interestingly, after 12 weeks of supervised resistance training intervention, plasma C1q in the elderly group decreased substantially (89.3 µg/ml) with muscle mass being significantly increased, revealing an inverse correlation between plasma C1q level and muscle mass (51). The cause for plasma C1q accumulation in the elderly group and its reduction after training is unclear in this study and a causal relationship between plasma C1q and muscle mass was also not established (51). The overall conclusion was, however, in line with C1q contribution to accelerated aging as reported in mice (19).
The mechanisms for C1 complex function in the context of complement activation and Wnt receptor signaling have been clearly documented. However, mechanistic understanding of its involvement in SLE pathogenesis remains fragmentary (Figure 3). Genetic deficiencies in complement proteins generally increase susceptibility to infections but mostly lack the type of strong association with SLE pathogenesis that is observed with deficiencies of C1 and its immediate substrate C4 (28). This raises the possibility that SLE pathogenesis may be related to a modular C1 activity. Depending on what C1q recognizes, C1 may have effects through the C1r/C1s proteases on various molecular/cellular processes besides the complement system. C1 activation of Wnt receptor signaling is a good example of such a modular activity (19). The degradation of apoptotic cell debris is apparently another process involving a modular C1 complex function (Figure 3) (9).
Since the discovery of C1q binding to apoptotic cells (9), a significant body of work has been published revolving mostly around C1q opsonization of apoptotic cells and its regulation of immune tolerance. First, C1q binding to apoptotic cells opsonizes the cell debris for effective phagocytosis (10). Second, C1q binding contributes to the immunosuppressive nature of apoptotic cells (52, 53). Third, C1q modulates dendritic cell (DC) development to induce more prominent tolerogenic features in these antigen-presenting cells (54, 55). Last, C1q inhibits IFN-α production by DCs induced by SLE autoantibodies in the form of immune complexes (56–58). IFN-α is a SLE-pathogenic cytokine, which causes autoimmunity in patients following its therapeutic administration (59, 60). IFN-α is elevated in those SLE patients who register a chronically elevated signature of IFNα-stimulated gene transcription (61–63). Inhibition of IFN-α induction by C1q potentially contributes to protection against SLE pathogenesis.
Studies that evaluate the role of C1 proteases in these processes are lacking. In fact, how C1r/C1s deficiency also causes SLE has not been investigated. There are two hypotheses that are relevant to explaining how C1 and C4 deficiencies may cause autoimmunity (64, 65). A clearance hypothesis emphasizes on the induction of autoantibodies and autoimmunity by apoptotic cellular debris, which may accumulate due to impaired clearance or excessive cell death (64). A tolerance hypothesis emphasizes on the contribution of complement to promoting self-antigen delivery to primary lymphoid organs for an effective negative selection (65). Considering that C1s cleaves intracellular antigens, it can be highly significant that the C1 complex both opsonizes apoptotic cells through C1q for effective clearance and degrades apoptotic cellular antigens through C1 proteases. Without relying on the rest of the complement system, both processes can reduce the autoantigenicity of apoptotic cell debris.
C1q was initially found to bind to apoptotic blebs, but the spectrum of C1q ligands in apoptotic cells and their contributions to C1q recognition need further delineation (9, 66). C1q appears to bind multiple regions of apoptotic cells (20). In early apoptotic cells, C1q binds to peripheral structures; however, in late apoptotic cells, it binds predominantly to the core nuclear bodies, i.e., the nucleoli (20). With purified nucleoli, C1q not only binds to these nuclear bodies but also causes C1s activation and cleavage of nucleolar proteins, e.g., nucleophosmin-1 (NPM1) and nucleolin (20). Nucleoli are highly immunogenic and contain many autoantigens (67).
This reminds an important aspect in cell apoptosis, i.e., the intrinsic proteolytic/enzymatic dismantling of intracellular structures (68). During cell apoptosis, autoantigens are cleaved and partially inactivated by endogenous proteases (69). It is possible that during late stage apoptosis, exogenous proteases and other enzymes also contribute to the antigen dismantling process. C1q recognizes multiple intracellular regions during apoptosis, including the highly immunogenic nucleoli (20). In cooperation with endogenous proteases, C1 could contribute significantly to the effective protease trimming of dead cells required to prevent their immunogenicity (Figure 4) (70).
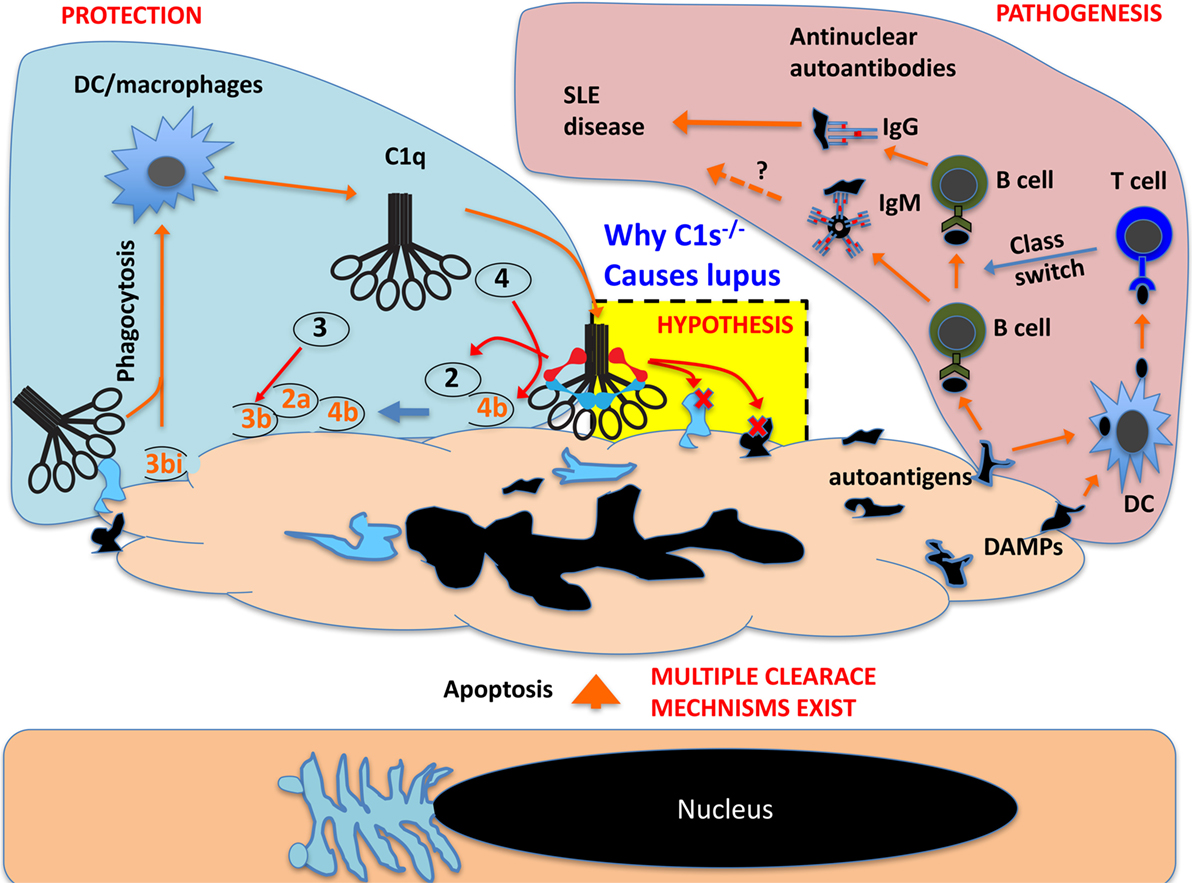
Figure 4. Schematic proposal how C1 deficiency may cause systemic lupus erythematosus pathogenesis. In live cells, the nucleus and other intracellular structures are compartmentalized and excluded from complement recognition. When cells undergo apoptosis, the nucleus and other cellular structures disintegrate and, in late apoptotic cells, these fragments are recognized by C1q, which opsonize apoptotic cells for phagocytosis. This can also cause C1r/C1s activation and the activated C1s could cleave its classic substrate C4 and C2 and produce complement opsonins for phagocytosis. C1s may also cleave numerous exposed nuclear and other cellular proteins that are otherwise autoimmunogenic (autoantigens) and cause B cell production of autoantibodies. C1s may also cleave cellular proteins that are otherwise pro-inflammatory danger-associated molecular patterns (DAMPs) and activate DCs to cause B cell production of pathogenic IgG autoantibodies. C1s may not inactivate all autoantigens but effective inactivation of DAMPs can abrogate class switch of autoantibodies from IgM to pathogenic IgG.
In the complement network, proteases are highly specific and this is essential to the directional propagation of the complement activation (5, 6). Outside the complement network, what other proteins may be cleaved by these proteases are rarely studied. With regard to C1s, it has been known for some time that it cleaves cell surface MHC class I and the secreted IGFBP5 (16–18). More recent addition to the list of non-complement C1s substrates includes LRP6, NPM1, and nucleolin (19, 20). In fact, the substrates of C1s can potentially be numerous based on bioinformatics predictions. Using a library of phage-displayed peptides that were designed based on the classic C1s cleavage sites on C4 and C2, Kerr et al. identified a list of C1s-cleavable peptide variants (21). Based on the conserved peptide framework, a formula was constructed that predicted numerous intracellular proteins as potential C1s substrate (21). NPM1 and nucleolin, which were found to be cleaved by C1 proteases, indeed contained multiple predicted C1s cleavage sites (20). The conjunction of a broad C1s substrateome with a diversity of C1q ligands makes the C1 complex a potentially multifaceted module that can function in a range of biological processes. C1s cleavage of intracellular proteins may be irrelevant to live cells, but this capacity could be important in the context of dead cell debris, reducing autoimmunogenicity by the inactivation of autoantigens and the destruction of danger-associated molecular patterns (DAMPs) (Figure 4). A recent example of this C1 protease function is the demonstrated C1s cleavage and inactivation of HMGB1, which is otherwise a nuclear DAMP (71).
The functional versatility of C1q draws support from the modularity of its structures. C1q is a large, symmetrical, and delicate posttranslational assembly resulting from complex evolutionary innovations. At one stage, the complement system was defined by merely four identifiable components, C1–C4. In 1963, C1 was first separated into three distinct subcomponents, C1q and the two proteases C1r and C1s (72, 73). For C1q, biochemical analysis revealed three types of subunit polypeptides each containing a collagenous (Gly-Xaa-Yaa)n repeating sequence over the N-terminal half (74, 75). Similar collagen-like domains were later found in the N-terminal halves of collectins, ficolins, and some C1q/TNF-related proteins (CTRPs) such as adiponectin and saccular collagen (76–79). The collagenous regions of all these proteins form triple-helices and the C-terminal halves form globular (gC1q) domains that are clustered in three. The triple helices further conjoin at the extreme N-terminal regions to align 3–6 triple-helices in one final assembly (76, 77). In the overall “bundle of tulips” C1q assembly, the gC1q domains are peripherally extended as multivalent binding sites (74, 75). The six triple-helices in C1q form a scaffold for the tetrameric C1r2C1s2 protease complex (80). Binding of C1q to various ligands via the gC1q domain activates the C1r/C1s proteases and C1s triggers effector reactions through cleavage of specific substrate, which, in the complement classical pathway, are C4 and C2.
C1q is distinct from collectins, ficolins, and CTRPs in that it is assembled from more than one type of subunit polypeptide. The other proteins are considered largely homopolymers (76, 77). C1q is an 18-polypeptide macromolecule assembled equally from three similar but distinct subunit peptides, 6 × A, 6 × B, and 6 × C chains (74) (Figure 2). The C1q assembly is partially stabilized by disulfide bonds and, under denaturing conditions, the molecule crumbles into two basic structural identities, an A-B heterodimer and a C-C homodimer that are linked through N-terminal disulfide bonds (Figure 2). One C-C and two A-B dimers form two triple helices over the collagen-like regions (ABC-CBA) and C1q is assembled non-covalently from three such ABC-CBA structures (Figure 2). Therefore, despite the presence of three C1q genes, only one type of C1q is assembled. The collectins, ficolins, and CTRPs are, however, products of single genes (76, 77).
What prevented the formation of three different homopolymeric C1q molecules is not understood. The combination of divergent subunits, i.e., A, B, and C, and their extensive polymerization in C1q offers, besides a scaffold to embrace the C1r2C1s2 tetramer, diversity and multiplicity of binding sites for a broad ligand repertoire. The heterotrimeric congregation of the three globular head modules (ghA, ghB, and ghC) yielding gC1q domain at the C-termini is independent of the N-terminal triple-helix (81). The three different globular head modules in the cluster exhibit differential binding preferences toward known C1q ligands (82, 83).
A dominant source for plasma complement proteins, including C1r and C1s, are hepatocytes in the liver, but C1q is one exception for its extrahepatic origins (84). C1q was initially found produced by macrophages (85). It was later found to be produced by tissue and cultured DCs as well (86, 87). Studies on C1q gene promoters revealed active cis-acting elements for transcription factors PU.1 and IRF8 (26). PU.1 and IRF8, especially PU.1, is a key transcription factor that defines the macrophage and DC lineage of hematopoietic development (88). Tenner and colleagues recently clarified that, in the brain, C1q is also produced by local tissue macrophages, the microglia (89). Therefore, C1q could have evolved first as an effector molecule in macrophages or ancestral phagocytes and its association with the C1r/C1s proteases in the form of C1 complex represents a secondary evolutionary innovation.
Macrophages and DCs populate many tissues and are poorly represented in the blood circulation (90, 91). Monocytes are blood precursors of some tissue macrophages, but these cells only start to produce C1q upon differentiation into macrophages (92). How the broad and heterogeneous tissue origin of C1q and its steady plasma levels are regulated is not fully understood. Tissue macrophages, which orchestrate inflammation and antigen presentation as well as scavenge tissue debris and microorganisms, are responsive to diverse stimuli (93, 94). The complement system is concentrated in the blood and is actively recruited to sites of tissue infections or injuries. The macrophage/DC origin of C1q appears to ensure its steady state tissue distribution. Macrophages also produce C1r/C1s proteases (84). DCs also broadly populate tissues, albeit at a lower density, and also produce C1q, C1r, and C1s (86, 87, 95). This mode of C1q and C1r/C1s production stresses an important C1q or C1 function in sterile tissue homeostasis and other molecular/cellular processes.
Macrophages express a broad repertoire of scavenging and signaling receptors and exhibit a high degree of plasticity in differentiation and activation. This is reflected in the heterogeneity of tissue macrophages in their morphology and effector molecule production (91). As previously summarized, C1q production by macrophages also vary in response to microbial structures, cytokines, hormones, and drugs (66, 96). Overall, microbial structures tend to inhibit C1q production and corticosteroid hormones tend to enhance it (66). With respect to cytokines, IFN-α appears to inhibit C1q production (87), whereas IFN-γ increases C1q production by DCs/macrophages (26, 97). Local and temporal tissue fluctuation in C1q production may not prominently alter plasma C1q levels, but it can impact on local tissue homeostasis, immunity, and tolerance. This can also be of great importance in the microenvironment of tumor, where C1q seems to have a tumor-promoting function (98).
Besides IgG and IgM, many other protein ligands have been identified for C1q (66, 99). These C1q ligands, including soluble, cell surface, normal extracellular matrix, and pathogenic amyloid proteins, often activate C1r/C1s and the complement classical pathway. It has, however, not been addressed whether the activated C1r/C1s proteases also cleave these C1q ligands or proteins near these ligands as they cleave LRP6, IGFBP5, MHC I, NPM1, and nucleolin (16–20). In some pathophysiological contexts, C1q functions were interpreted without specific consideration to its ligands. In the postnatal central nervous system, C1q is localized to synapses and contributes to synapse elimination resembling the disposal of dead cells, which is important for the maturation of neuronal connectivity and functions (100, 101). How C1q causes the selective dismantling of synapses is unclear, but it is tempting to suggest C1q binding to selective neuronal contexts and possible involvement of C1r/C1s-mediated molecular cleavage or cell signaling as observed with the Wnt receptor (19). In excess, the same C1q-mediated synapse elimination could accelerate neurodegeneration related to aging and neuropathology (102, 103).
The scrapie pathogen, prion protein, is another C1q ligand (104, 105). C1q deficiency reduces scrapie pathogenesis (106, 107). To what extent the complement classical pathway may be involved in prion-mediated pathology is incompletely defined, but C4 is apparently activated on prion proteins and C3 depletion also reduced scrapie pathogenesis (104, 107). As C1q, C3, and C4 are all potent opsonins, a prevalent explanation is their involvement in prion transmission from the gut to the central nervous tissues. The role of activated C1r/C1s proteases in scrapie pathogenesis has not been considered.
C1q is also produced in the placenta (108, 109). At this feto–maternal interface, it was shown to mediate trophoblast invasion of the maternal decidua (108). Mechanistically, C1q was found to interact with decidual stroma, to activate trophoblast signaling, and to mediate trophoblast adhesion and migration (108). Whether C1r/C1s might play a role in this context is again unclear.
The complement system is an intimate proteolytic cascade responding to diverse triggering factors. In infections or injuries, the full impact of its activation is realized by three closely related effector reactions: inflammation, opsonization, and lysis (5, 6). The C3a and C5a anaphylatoxins recruit and activate phagocytes and other inflammatory leukocytes at sites of tissue infections or injuries. The membrane attack complexes cause cellular lysis. The C4b, C3b, and the further proteolytic fragments opsonize complement-reacted targets for effective phagocytic clearance (Figure 1). However, this article highlights that C1 complex may function as a module, independent of the rest of the complement network, to participate in other molecular/cellular processes.
Serine proteases are core components of the complement infrastructure and their sequential activation is at the heart of the formation of hierarchical proteolytic or lytic protein complexes. In the context of the complement network, these are highly specific proteases, e.g., C1r only cleaves C1s and C1s only cleaves C4, C2, and C1 inhibitor. The finding that the C1 proteases also cleave a growing list of non-complement proteins, including LRP6, MHC I, IGFBP5, NPM1, and nucleolin, supports a multifaceted, modular function for C1 complex. In this functional C1 module, C1q recognizes targets in various molecular/cellular processes and the C1r/C1s proteases bring about the effects by cleaving substrate in these molecular/cellular processes. Modular functions may also be found in other complement proteases such as factor B and MASPs.
JL initiated the article and contributed to the framework and major details of the final version. UK provided critical and substantial inputs to the overall scope and detailed content. This is a joint project by the two authors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Ken Shortman for critical comments on this manuscript. This work is supported by a Singapore National University Health System seed fund (R-182-000-229-750), a Singapore Ministry of Education Tier 2 grant (MOE2012-T2-2-122), and a Singapore National Medical Research Council Open-funding Individual Research Grant (NMRC/OFIRG/0013/2016).
IGFBP5, insulin-like growth factor-binding protein 5; MBL, mannose-binding lectin; MASP, MBL-associated serine protease; SLE, systemic lupus erythematosus; LRP6, low-density lipoprotein receptor-related protein 6; CRP, C-reactive protein; DC, dendritic cell; NPM1, nucleophosmin-1; CTRP, C1q/TNF-related protein.
1. Farries TC, Atkinson JP. Evolution of the complement system. Immunol Today (1991) 12:295–300. doi:10.1016/0167-5699(91)90002-B
2. Krem MM, Di Cera E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem Sci (2002) 27:67–74. doi:10.1016/S0968-0004(01)02007-2
3. Nonaka M, Kimura A. Genomic view of the evolution of the complement system. Immunogenetics (2006) 58:701–13. doi:10.1007/s00251-006-0142-1
4. Dodds AW, Matsushita M. The phylogeny of the complement system and the origins of the classical pathway. Immunobiology (2007) 212:233–43. doi:10.1016/j.imbio.2006.11.009
6. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344:1058–66. doi:10.1056/NEJM200104053441406
7. Reid KB, Nolan KF, Lijnen HR, Collen D. Proteolytic enzymes in coagulation, fibrinolysis, and complement activation. Introduction. Methods Enzymol (1993) 223:1–9.
8. Sim RB, Tsiftsoglou SA. Proteases of the complement system. Biochem Soc Trans (2004) 32:21–7. doi:10.1042/bst0320021
9. Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol (1997) 158:4525–8.
10. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med (2001) 194:781–95. doi:10.1084/jem.194.6.781
11. Xu W, Berger SP, Trouw LA, de Boer HC, Schlagwein N, Mutsaers C, et al. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol (2008) 180:7613–21. doi:10.4049/jimmunol.180.11.7613
12. Kemper C, Mitchell LM, Zhang L, Hourcade DE. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci U S A (2008) 105:9023–8. doi:10.1073/pnas.0801015105
13. Nauta AJ, Raaschou-Jensen N, Roos A, Daha MR, Madsen HO, Borrias-Essers MC, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol (2003) 33:2853–63. doi:10.1002/eji.200323888
14. Stuart LM, Takahashi K, Shi L, Savill J, Ezekowitz RA. Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol (2005) 174:3220–6. doi:10.4049/jimmunol.174.6.3220
15. Kuraya M, Ming Z, Liu X, Matsushita M, Fujita T. Specific binding of L-ficolin and H-ficolin to apoptotic cells leads to complement activation. Immunobiology (2005) 209:689–97. doi:10.1016/j.imbio.2004.11.001
16. Eriksson H, Nissen MH. Proteolysis of the heavy chain of major histocompatibility complex class I antigens by complement component C1s. Biochim Biophys Acta (1990) 1037:209–15. doi:10.1016/0167-4838(90)90169-G
17. Nissen MH, Roepstorff P, Thim L, Dunbar B, Fothergill JE. Limited proteolysis of beta 2-microglobulin at Lys-58 by complement component C1s. Eur J Biochem (1990) 189:423–9. doi:10.1111/j.1432-1033.1990.tb15505.x
18. Busby WH Jr, Nam TJ, Moralez A, Smith C, Jennings M, Clemmons DR. The complement component C1s is the protease that accounts for cleavage of insulin-like growth factor-binding protein-5 in fibroblast medium. J Biol Chem (2000) 275:37638–44. doi:10.1074/jbc.M006107200
19. Naito AT, Sumida T, Nomura S, Liu ML, Higo T, Nakagawa A, et al. Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell (2012) 149:1298–313. doi:10.1016/j.cell.2012.03.047
20. Cai Y, Teo BH, Yeo JG, Lu J. C1q protein binds to the apoptotic nucleolus and causes C1 protease degradation of nucleolar proteins. J Biol Chem (2015) 290:22570–80. doi:10.1074/jbc.M115.670661
21. Kerr FK, O’Brien G, Quinsey NS, Whisstock JC, Boyd S, de la Banda MG, et al. Elucidation of the substrate specificity of the C1s protease of the classical complement pathway. J Biol Chem (2005) 280:39510–4. doi:10.1074/jbc.M506131200
22. Smith LC, Clow LA, Terwilliger DP. The ancestral complement system in sea urchins. Immunol Rev (2001) 180:16–34. doi:10.1034/j.1600-065X.2001.1800102.x
23. Patthy L. Evolution of blood coagulation and fibrinolysis. Blood Coagul Fibrinolysis (1990) 1:153–66.
24. Donoghue PC, Purnell MA. Genome duplication, extinction and vertebrate evolution. Trends Ecol Evol (2005) 20:312–9. doi:10.1016/j.tree.2005.04.008
25. Kusumoto H, Hirosawa S, Salier JP, Hagen FS, Kurachi K. Human genes for complement components C1r and C1s in a close tail-to-tail arrangement. Proc Natl Acad Sci U S A (1988) 85:7307–11. doi:10.1073/pnas.85.19.7307
26. Chen G, Tan CS, Teh BK, Lu J. Molecular mechanisms for synchronized transcription of three complement C1q subunit genes in dendritic cells and macrophages. J Biol Chem (2011) 286:34941–50. doi:10.1074/jbc.M111.286427
27. Sellar GC, Blake DJ, Reid KB. Characterization and organization of the genes encoding the A-, B- and C-chains of human complement subcomponent C1q. The complete derived amino acid sequence of human C1q. Biochem J (1991) 274(Pt 2):481–90. doi:10.1042/bj2740481
28. Pettigrew HD, Teuber SS, Gershwin ME. Clinical significance of complement deficiencies. Ann N Y Acad Sci (2009) 1173:108–23. doi:10.1111/j.1749-6632.2009.04633.x
29. Skattum L, van Deuren M, van der Poll T, Truedsson L. Complement deficiency states and associated infections. Mol Immunol (2011) 48:1643–55. doi:10.1016/j.molimm.2011.05.001
30. Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol (2000) 76:227–324. doi:10.1016/S0065-2776(01)76021-X
31. Lewis MJ, Botto M. Complement deficiencies in humans and animals: links to autoimmunity. Autoimmunity (2006) 39:367–78. doi:10.1080/08916930600739233
32. Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun (2009) 10:373–9. doi:10.1038/gene.2009.39
33. Petry F, Loos M. Common silent mutations in all types of hereditary complement C1q deficiencies. Immunogenetics (2005) 57:566–71. doi:10.1007/s00251-005-0023-z
34. Lipsker D, Hauptmann G. Cutaneous manifestations of complement deficiencies. Lupus (2010) 19:1096–106. doi:10.1177/0961203310373370
35. Stegert M, Bock M, Trendelenburg M. Clinical presentation of human C1q deficiency: how much of a lupus? Mol Immunol (2015) 67:3–11. doi:10.1016/j.molimm.2015.03.007
36. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med (2008) 358:929–39. doi:10.1056/NEJMra071297
38. Haserick JR, Long R. [Systemic lupus erythematosus preceded by false-positive serologic tests for syphilis: presentation of five cases]. Ann Intern Med (1952) 37:559–65. doi:10.7326/0003-4819-37-3-559
39. Miescher P, Fauconnet M. [Absorption of L. E. factor by isolated cell nuclei]. Experientia (1954) 10:252–3. doi:10.1007/BF02157392
40. Friou GJ. The significance of the lupus globulin; nucleoprotein reaction. Ann Intern Med (1958) 49:866–75. doi:10.7326/0003-4819-49-4-866
41. Tan EM, Schur PH, Carr RI, Kunkel HG. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J Clin Invest (1966) 45:1732–40. doi:10.1172/JCI105479
42. Amoura Z, Piette JC, Bach JF, Koutouzov S. The key role of nucleosomes in lupus. Arthritis Rheum (1999) 42:833–43. doi:10.1002/1529-0131(199905)42:5<833::AID-ANR1>3.0.CO;2-T
43. Dieker JW, van der Vlag J, Berden JH. Triggers for anti-chromatin autoantibody production in SLE. Lupus (2002) 11:856–64. doi:10.1191/0961203302lu307rr
44. Koffler D, Carr RI, Agnello V, Fiezi T, Kunkel HG. Antibodies to polynucleotides: distribution in human serums. Science (1969) 166:1648–9. doi:10.1126/science.166.3913.1648
45. Deng Y, Tsao BP. Advances in lupus genetics and epigenetics. Curr Opin Rheumatol (2014) 26:482–92. doi:10.1097/BOR.0000000000000086
46. Rando TA. Stem cells, ageing and the quest for immortality. Nature (2006) 441:1080–6. doi:10.1038/nature04958
47. Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science (2007) 317:803–6. doi:10.1126/science.1143578
48. Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science (2007) 317:807–10. doi:10.1126/science.1144090
49. Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci (2000) 908:244–54. doi:10.1111/j.1749-6632.2000.tb06651.x
50. Bartlett DB, Firth CM, Phillips AC, Moss P, Baylis D, Syddall H, et al. The age-related increase in low-grade systemic inflammation (inflammaging) is not driven by cytomegalovirus infection. Aging Cell (2012) 11:912–5. doi:10.1111/j.1474-9726.2012.00849.x
51. Watanabe S, Sato K, Hasegawa N, Kurihara T, Matsutani K, Sanada K, et al. Serum C1q as a novel biomarker of sarcopenia in older adults. FASEB J (2015) 29:1003–10. doi:10.1096/fj.14-262154
52. Clarke EV, Weist BM, Walsh CM, Tenner AJ. Complement protein C1q bound to apoptotic cells suppresses human macrophage and dendritic cell-mediated Th17 and Th1 T cell subset proliferation. J Leukoc Biol (2015) 97:147–60. doi:10.1189/jlb.3A0614-278R
53. Chung EY, Kim SJ, Ma XJ. Regulation of cytokine production during phagocytosis of apoptotic cells. Cell Res (2006) 16:154–61. doi:10.1038/sj.cr.7310021
54. Castellano G, Woltman AM, Schlagwein N, Xu W, Schena FP, Daha MR, et al. Immune modulation of human dendritic cells by complement. Eur J Immunol (2007) 37:2803–11. doi:10.1002/eji.200636845
55. Teh BK, Yeo JG, Chern LM, Lu J. C1q regulation of dendritic cell development from monocytes with distinct cytokine production and T cell stimulation. Mol Immunol (2011) 48:1128–38. doi:10.1016/j.molimm.2011.02.006
56. Lood C, Gullstrand B, Truedsson L, Olin AI, Alm GV, Ronnblom L, et al. C1q inhibits immune complex-induced interferon-alpha production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum (2009) 60:3081–90. doi:10.1002/art.24852
57. Santer DM, Hall BE, George TC, Tangsombatvisit S, Liu CL, Arkwright PD, et al. C1q deficiency leads to the defective suppression of IFN-alpha in response to nucleoprotein containing immune complexes. J Immunol (2010) 185:4738–49. doi:10.4049/jimmunol.1001731
58. Bave U, Magnusson M, Eloranta ML, Perers A, Alm GV, Ronnblom L. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J Immunol (2003) 171:3296–302. doi:10.4049/jimmunol.171.6.3296
59. Ronnblom LE, Alm GV, Oberg K. Autoimmune phenomena in patients with malignant carcinoid tumors during interferon-alpha treatment. Acta Oncol (1991) 30:537–40. doi:10.3109/02841869109092414
60. Kalkner KM, Ronnblom L, Karlsson Parra AK, Bengtsson M, Olsson Y, Oberg K. Antibodies against double-stranded DNA and development of polymyositis during treatment with interferon. QJM (1998) 91:393–9. doi:10.1093/qjmed/91.6.393
61. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A (2003) 100:2610–5. doi:10.1073/pnas.0337679100
62. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197:711–23. doi:10.1084/jem.20021553
63. Kirou KA, Lee C, George S, Louca K, Papagiannis IG, Peterson MG, et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum (2004) 50:3958–67. doi:10.1002/art.20798
64. Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol (2004) 22:431–56. doi:10.1146/annurev.immunol.22.012703.104549
65. Carroll MC. A protective role for innate immunity in systemic lupus erythematosus. Nat Rev Immunol (2004) 4:825–31. doi:10.1038/nri1456
66. Lu JH, Teh BK, Wang L, Wang YN, Tan YS, Lai MC, et al. The classical and regulatory functions of C1q in immunity and autoimmunity. Cell Mol Immunol (2008) 5:9–21. doi:10.1038/cmi.2008.2
67. Welting TJ, Raijmakers R, Pruijn GJ. Autoantigenicity of nucleolar complexes. Autoimmun Rev (2003) 2:313–21. doi:10.1016/S1568-9972(03)00029-6
68. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol (2008) 9:231–41. doi:10.1038/nrm2312
69. Casiano CA, Ochs RL, Tan EM. Distinct cleavage products of nuclear proteins in apoptosis and necrosis revealed by autoantibody probes. Cell Death Differ (1998) 5:183–90. doi:10.1038/sj.cdd.4400336
70. Yeo JG, Leong J, Lu J. Complement the cell death. Cell Death Dis (2016) 7:e2465. doi:10.1038/cddis.2016.369
71. Yeo JG, Leong J, Arkachaisri T, Cai Y, Teo BH, Tan JH, et al. Proteolytic inactivation of nuclear alarmin high-mobility group box 1 by complement protease C1s during apoptosis. Cell Death Discov (2016) 2:16069. doi:10.1038/cddiscovery.2016.69
72. Lepow IH, Naff GB, Todd EW, Pensky J, Hinz CF. Chromatographic resolution of the first component of human complement into three activities. J Exp Med (1963) 117:983–1008. doi:10.1084/jem.117.6.983
73. Rapp HJ. Mechanism of immune hemolysis: recognition of two steps in the conversion of EAC’ 1,4,2 to E. Science (1958) 127:234–6. doi:10.1126/science.127.3292.234-a
74. Reid KB, Porter RR. Subunit composition and structure of subcomponent C1q of the first component of human complement. Biochem J (1976) 155:19–23. doi:10.1042/bj1550019
75. Brodsky-Doyle B, Leonard KR, Reid KB. Circular-dichroism and electron-microscopy studies of human subcomponent C1q before and after limited proteolysis by pepsin. Biochem J (1976) 159:279–86. doi:10.1042/bj1590279
76. Lu J, Teh C, Kishore U, Reid KB. Collectins and ficolins: sugar pattern recognition molecules of the mammalian innate immune system. Biochim Biophys Acta (2002) 1572:387–400. doi:10.1016/S0304-4165(02)00320-3
77. Schaffler A, Buechler C. CTRP family: linking immunity to metabolism. Trends Endocrinol Metab (2012) 23:194–204. doi:10.1016/j.tem.2011.12.003
78. Kishore U, Gaboriaud C, Waters P, Shrive AK, Greenhough TJ, Reid KB, et al. C1q and tumor necrosis factor superfamily: modularity and versatility. Trends Immunol (2004) 25:551–61. doi:10.1016/j.it.2004.08.006
79. Ghai R, Waters P, Roumenina LT, Gadjeva M, Kojouharova MS, Reid KB, et al. C1q and its growing family. Immunobiology (2007) 212:253–66. doi:10.1016/j.imbio.2006.11.001
80. Bally I, Rossi V, Lunardi T, Thielens NM, Gaboriaud C, Arlaud GJ. Identification of the C1q-binding sites of human C1r and C1s: a refined three-dimensional model of the C1 complex of complement. J Biol Chem (2009) 284:19340–8. doi:10.1074/jbc.M109.004473
81. Gaboriaud C, Juanhuix J, Gruez A, Lacroix M, Darnault C, Pignol D, et al. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J Biol Chem (2003) 278:46974–82. doi:10.1074/jbc.M307764200
82. Kishore U, Gupta SK, Perdikoulis MV, Kojouharova MS, Urban BC, Reid KB. Modular organization of the carboxyl-terminal, globular head region of human C1q A, B, and C chains. J Immunol (2003) 171:812–20. doi:10.4049/jimmunol.171.2.812
83. Gaboriaud C, Frachet P, Thielens NM, Arlaud GJ. The human c1q globular domain: structure and recognition of non-immune self ligands. Front Immunol (2011) 2:92. doi:10.3389/fimmu.2011.00092
84. Colten HR, Strunk RC, Perlmutter DH, Cole FS. Regulation of complement protein biosynthesis in mononuclear phagocytes. Ciba Found Symp (1986) 118:141–54.
85. Muller W, Hanauske-Abel H, Loos M. Biosynthesis of the first component of complement by human and guinea pig peritoneal macrophages: evidence for an independent production of the C1 subunits. J Immunol (1978) 121:1578–84.
86. Cao W, Bobryshev YV, Lord RS, Oakley RE, Lee SH, Lu J. Dendritic cells in the arterial wall express C1q: potential significance in atherogenesis. Cardiovasc Res (2003) 60:175–86. doi:10.1016/S0008-6363(03)00345-6
87. Castellano G, Woltman AM, Nauta AJ, Roos A, Trouw LA, Seelen MA, et al. Maturation of dendritic cells abrogates C1q production in vivo and in vitro. Blood (2004) 103:3813–20. doi:10.1182/blood-2003-09-3046
88. Nutt SL, Metcalf D, D’Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med (2005) 201:221–31. doi:10.1084/jem.20041535
89. Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, et al. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation (2017) 14:48. doi:10.1186/s12974-017-0814-9
90. Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol (2007) 7:19–30. doi:10.1038/nri1996
91. Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev (2014) 262:36–55. doi:10.1111/imr.12223
92. Lu J, Le Y, Kon OL, Chan J, Lee SH. Biosynthesis of human ficolin, an Escherichia coli-binding protein, by monocytes: comparison with the synthesis of two macrophage-specific proteins, C1q and the mannose receptor. Immunology (1996) 89:289–94. doi:10.1046/j.1365-2567.1996.d01-732.x
93. Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol (2002) 20:825–52. doi:10.1146/annurev.immunol.20.103001.114744
94. Gordon S. The macrophage: past, present and future. Eur J Immunol (2007) 37(Suppl 1):S9–17. doi:10.1002/eji.200737638
95. Li K, Sacks SH, Zhou W. The relative importance of local and systemic complement production in ischaemia, transplantation and other pathologies. Mol Immunol (2007) 44:3866–74. doi:10.1016/j.molimm.2007.06.006
96. Pednekar L, Pathan AA, Paudyal B, Tsolaki AG, Kaur A, Abozaid SM, et al. Analysis of the interaction between globular head modules of human C1q and its candidate receptor gC1qR. Front Immunol (2016) 7:567. doi:10.3389/fimmu.2016.00567
97. Teo BH, Bobryshev YV, Teh BK, Wong SH, Lu J. Complement C1q production by osteoclasts and its regulation of osteoclast development. Biochem J (2012) 447:229–37. doi:10.1042/BJ20120888
98. Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun (2016) 7:10346. doi:10.1038/ncomms10346
99. Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol (2009) 30:83–90. doi:10.1016/j.it.2008.11.003
100. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell (2007) 131:1164–78. doi:10.1016/j.cell.2007.10.036
101. Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A (2010) 107:7975–80. doi:10.1073/pnas.0913449107
102. Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci (2013) 33:13460–74. doi:10.1523/JNEUROSCI.1333-13.2013
103. Takano M, Kawabata S, Komaki Y, Shibata S, Hikishima K, Toyama Y, et al. Inflammatory cascades mediate synapse elimination in spinal cord compression. J Neuroinflammation (2014) 11:40. doi:10.1186/1742-2094-11-40
104. Mitchell DA, Kirby L, Paulin SM, Villiers CL, Sim RB. Prion protein activates and fixes complement directly via the classical pathway: implications for the mechanism of scrapie agent propagation in lymphoid tissue. Mol Immunol (2007) 44:2997–3004. doi:10.1016/j.molimm.2006.12.027
105. Erlich P, Dumestre-Perard C, Ling WL, Lemaire-Vieille C, Schoehn G, Arlaud GJ, et al. Complement protein C1q forms a complex with cytotoxic prion protein oligomers. J Biol Chem (2010) 285:19267–76. doi:10.1074/jbc.M109.071860
106. Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, Zinkernagel RM, et al. Complement facilitates early prion pathogenesis. Nat Med (2001) 7:488–92. doi:10.1038/86567
107. Mabbott NA, Bruce ME, Botto M, Walport MJ, Pepys MB. Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat Med (2001) 7:485–7. doi:10.1038/86562
108. Agostinis C, Bulla R, Tripodo C, Gismondi A, Stabile H, Bossi F, et al. An alternative role of C1q in cell migration and tissue remodeling: contribution to trophoblast invasion and placental development. J Immunol (2010) 185:4420–9. doi:10.4049/jimmunol.0903215
Keywords: complement C1, autoimmunity, aging, infection, inflammation, C1q, macrophage, dendritic cell
Citation: Lu J and Kishore U (2017) C1 Complex: An Adaptable Proteolytic Module for Complement and Non-Complement Functions. Front. Immunol. 8:592. doi: 10.3389/fimmu.2017.00592
Received: 10 February 2017; Accepted: 04 May 2017;
Published: 24 May 2017
Edited by:
Zvi Fishelson, Tel Aviv University, IsraelReviewed by:
Péter Gál, Institute of Enzymology (MTA), HungaryCopyright: © 2017 Lu and Kishore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhua Lu, bWljbHVqaEBudXMuZWR1LnNn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.