 Ivan K. Chinn1,2,3*†
Ivan K. Chinn1,2,3*† Robert P. Sanders4†
Robert P. Sanders4† Asbjørg Stray-Pedersen5,6,7
Asbjørg Stray-Pedersen5,6,7 Zeynep H. Coban-Akdemir7,8
Zeynep H. Coban-Akdemir7,8 Vy Hong-Diep Kim9Harjit Dadi9,10Chaim M. Roifman9,10
Vy Hong-Diep Kim9Harjit Dadi9,10Chaim M. Roifman9,10 Troy Quigg4James R. Lupski1,7,8,11
Troy Quigg4James R. Lupski1,7,8,11 Jordan S. Orange1,2,3
Jordan S. Orange1,2,3 I. Celine Hanson1,2
I. Celine Hanson1,2
- 1Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA
- 2Section of Immunology, Allergy, and Rheumatology, Texas Children’s Hospital, Houston, TX, USA
- 3Center for Human Immunobiology, Texas Children’s Hospital, Houston, TX, USA
- 4Texas Transplant Institute, Methodist Hospital, San Antonio, TX, USA
- 5Norwegian National Unit for Newborn Screening, Department of Pediatric and Adolescent Medicine, Oslo University Hospital, Oslo, Norway
- 6Institute of Clinical Medicine, University of Oslo, Oslo, Norway
- 7Baylor-Hopkins Center for Mendelian Genomics, Baylor College of Medicine, Houston, TX, USA
- 8Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA
- 9Division of Immunology and Allergy, Department of Pediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
- 10Canadian Centre for Primary Immunodeficiency, The Jeffrey Model Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
- 11Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, USA
With the advent of high-throughput genomic sequencing techniques, novel genetic etiologies are being uncovered for previously unexplained Mendelian phenotypes, and the underlying genetic architecture of disease is being unraveled. Although most of these “mendelizing” disease traits represent phenotypes caused by single-gene defects, a percentage of patients have blended phenotypes caused by pathogenic variants in multiple genes. We describe an adult patient with susceptibility to bacterial, herpesviral, and fungal infections. Immunologic defects included CD8+ T cell lymphopenia, decreased T cell proliferative responses to mitogens, hypogammaglobulinemia, and radiation sensitivity. Whole-exome sequencing revealed compound heterozygous variants in ZAP70. Biallelic mutations in ZAP70 are known to produce a spectrum of immune deficiency that includes the T cell abnormalities observed in this patient. Analyses for variants in genes associated with radiation sensitivity identified the presence of a homozygous RNF168 variant of unknown significance. RNF168 deficiency causes radiosensitivity, immunodeficiency, dysmorphic features, and learning difficulties syndrome and may account for the radiation sensitivity. Thus, the patient was found to have a novel blended phenotype associated with multilocus genomic variation: i.e., separate and distinct genetic defects. These findings further illustrate the clinical utility of applying genomic testing in patients with primary immunodeficiency diseases.
Introduction
Clinical Presentation
This individual has been reported within a larger cohort study (subject 27.1), and we now describe the detailed clinical and genomic information concerning the case (1). The patient is a 30-year-old female of Mexican-American descent who presented with a clinical history of susceptibility to infections that suggested the presence of an underlying primary immunodeficiency disease (PIDD). She developed recurrent otitis media and upper respiratory tract infections starting at 3 months of age, and from 3 years of age she had difficulty with severe, recurrent warts. Repeated episodes of pneumonia began at the age of 7 years, resulting in chronic lung disease and bronchiectasis. At the age of 10 years, she developed chickenpox, which was noted to be more severe than in her sisters, who had concurrent symptoms. At the age of 11 years, she was diagnosed with cryptococcal meningitis. She was treated with fluconazole for several weeks as an inpatient and 4 months as an outpatient. Upon discontinuation of treatment, severe, recurrent episodes of meningitis rapidly ensued, resulting in hydrocephalus and ventriculo-peritoneal (VP) shunt placement. This complication was associated with damage of her vision, rendering her legally blind. The patient was then placed on daily fluconazole and intravenous immunoglobulin (IVIG) prophylaxes and subsequently demonstrated persistent serum cryptococcal antigen positivity but no clinical disease or further life-threatening illnesses. In adult life, she developed recurrent human papilloma virus (HPV)-associated oral and cutaneous lesions. She also developed low-grade squamous intraepithelial lesions of the cervix that were attributed to HPV infection. The cervical dysplasia required localized surgical resection. Due to poor response of the other HPV-associated lesions to topical antiviral therapy, interferon-gamma (IFN-γ) treatment was initiated, producing resolution of the oral disease. The cutaneous problems persisted, however. Many of these lesions transformed to squamous cell carcinomas, causing disfigurement due to numerous resections and destructive topical treatments. Treatment for the progressive skin disease included an option for combined surgery and radiotherapy, which was deferred due to the absence of a specific immunologic diagnosis.
Laboratory Test Results
At the time of the cryptococcal meningitis diagnosis, the patient was evaluated by an immunologist outside of our institutions and diagnosed with combined immunodeficiency characterized by low serum IgG levels (Table 1) and T cell deficiency. DNA PCR testing excluded HIV infection. Although she was subsequently placed and maintained on IVIG replacement, no further immunologic testing was performed until she was transferred to our care at 21 years of age. At that time, she began to receive regular immunologic evaluations, and the T cell defects were observed to have persisted (Table 1). Immunophenotyping demonstrated variable T cell lymphopenia, predominantly involving the CD8+ subset (2). T cell proliferative responses were measured using isolated peripheral blood mononuclear cells cultured in microwell plates loaded with 10-fold dilutions of mitogens or specific antigens. The responses were enumerated in terms of counts per minute (cpm) of tritiated thymidine incorporation. These tests showed decreased T cell activity toward mitogens, particularly at lower concentrations. The responses to antigens were very low, as well, but could not be interpreted due to lack of recent immunization (on IgG supplementation) or exposure (to Candida). Evaluation of natural killer (NK) cell function showed abnormal CD107a mobilization, suggesting poor NK cell degranulation (Table 1).
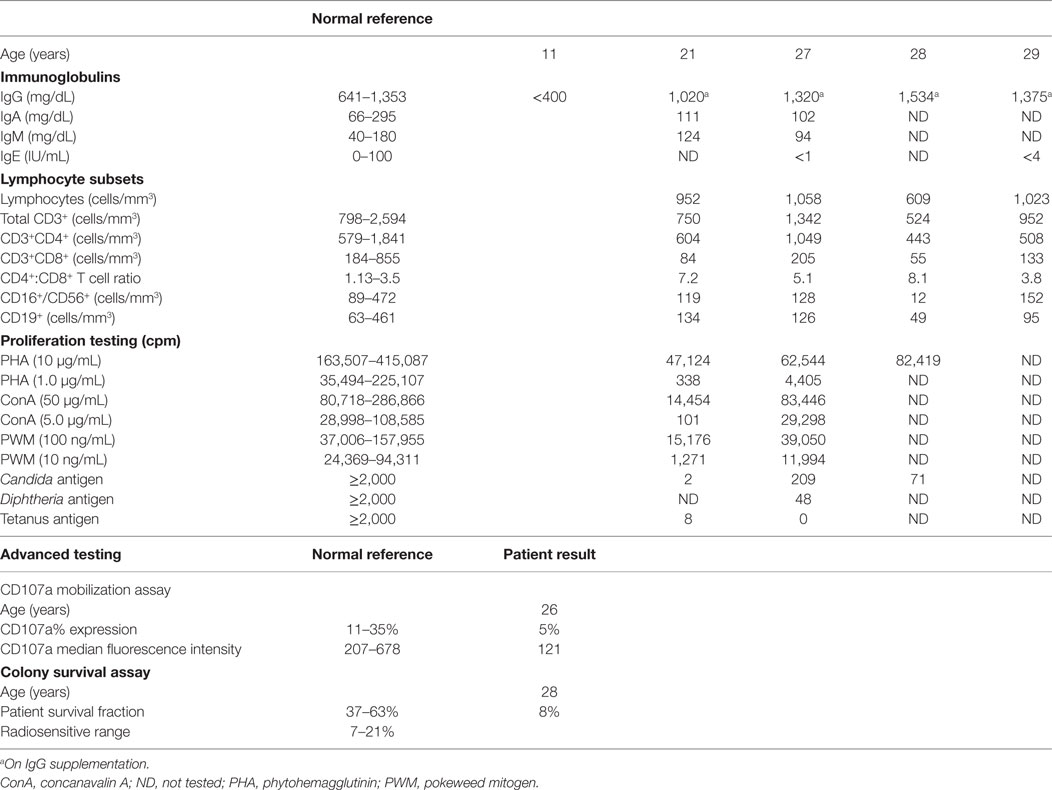
Table 1. Immunologic studies in a patient with ZAP70 and RNF168 variants.
At 28 years of age (Table 1), as part of the ongoing immunologic evaluation, the patient was tested for a DNA repair disorder in a College of American Pathologists accredited and Clinical Laboratory Improvement Amendments certified laboratory (Diagnostic Molecular Pathology and DNA Repair Clinical Testing Laboratory, University of California at Los Angeles) (3, 4). Briefly, peripheral blood lymphocytes were immortalized in vitro with Epstein-Barr virus. Once a persistent lymphoblastoid cell culture (LCL) was established, various concentrations of cells were plated in 96-well plates. Some of these plates were irradiated with 1 Gy, while others were kept as controls. After 2 weeks of culture, the number of surviving colonies in each plate was enumerated to determine a survival fraction. Positive and negative controls were included with each testing batch. The test revealed the presence of significant radiation sensitivity (8% survival fraction for patient LCLs), a percentage typically seen in patients with ataxia-telangiectasia.
Several diagnostic possibilities were considered at this point. The CD8+ T cell lymphopenia and decreased T cell proliferative responses to mitogens suggested the presence of zeta chain-associated protein of 70 kiloDaltons (ZAP70) deficiency, although the presence of hypomorphic mutations in severe combined immunodeficiency disease (SCID)-causing genes or a novel genetic defect could not be immediately excluded as possible explanations (5–8). Although ZAP70 deficiency could account for the T cell defects present in the patient, it is not known to cause radiation sensitivity, and the protein is not associated with any DNA repair pathways. On the other hand, mutations in several SCID-causing genes (i.e., DCLRE1C, LIG4, PRKDC, and NHEJ1) are known to cause radiosensitivity and could produce a unifying diagnosis. Meanwhile, other genes associated with radiosensitivity and immune deficiency merited consideration and included ATM, NBN, MRE11A, RAD50, APTX, BLM, DKC1, and RNF168 (9). In fact, several of these genetic defects have been categorized together to form the clinical entity known as XCIND syndrome, which is characterized by X-ray hypersensitivity, cancer susceptibility, immunodeficiency, neurological abnormality, and double-strand DNA breakage (10–12). Upon further testing, however, the patient was determined to have normal protein levels of ataxia-telangiectasia mutated (ATM); nibrin; meiotic recombination 11, S. cerevisiae homolog of, A (MRE11); RAD50; DNA ligase 4; and aprataxin by western blotting. Normal enzymatic kinase activity of the ATM protein was also confirmed.
Thus, written informed consent was obtained from the patient and her family members to participate in a Baylor College of Medicine Institutional Review Board approved protocol for whole-exome sequencing (WES) as part of the Baylor-Hopkins Center for Mendelian Genomics project at Baylor College of Medicine (Houston, TX, USA). The patient and family members also provided written informed consent to have their clinical and genetic information published in medical or scientific journals. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. WES was performed by the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) using genomic DNA extracted from whole blood. Sequencing was performed with greater than 90% coverage at a read-depth of 20× or greater. Stratified disease-associated variants were confirmed by Sanger sequencing. Methodology, CORE design, and variant selection have been previously described (1, 13).
Analysis of the exome data revealed the presence of two novel compound heterozygous missense variants in ZAP70, one in exon 12 [c.1505C>T (NM_001079), p.P502L] and the other in exon 6 [c.733G>A (NM_001079), p.G245R], that could explain the immunologic defects present in the patient (Table 2). Familial cosegregation of the variants with phenotype was confirmed by Sanger sequencing (Figure 1). The mother was a heterozygous carrier of the exon 12 variant, and the father was a heterozygous carrier of the exon 6 variant. Two healthy sisters were also tested, and one was found to be a heterozygous carrier of the exon 12 variant. Both variants affect highly conserved residues and lead to amino acid changes that are predicted to be damaging by Combined Annotation Dependent Depletion (CADD) score (34 for c.1505C>T and 33 for c.733G>A) (14–16). No other variants were observed that could provide a suitable explanation for the T cell defects (Table S1 in Supplementary Material).
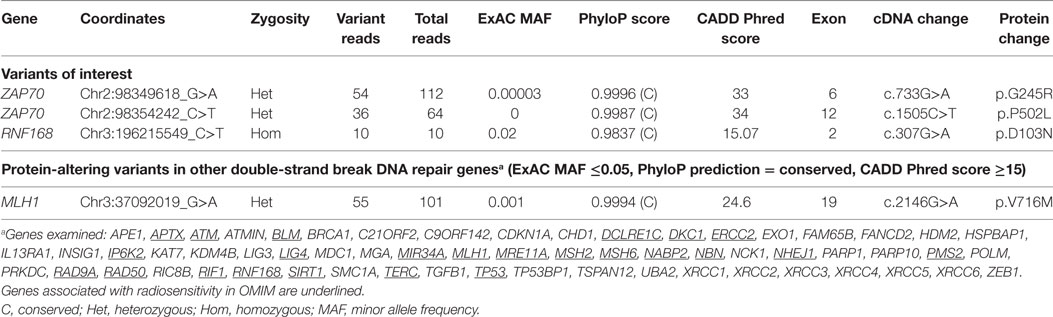
Table 2. Variants relevant to phenotype.
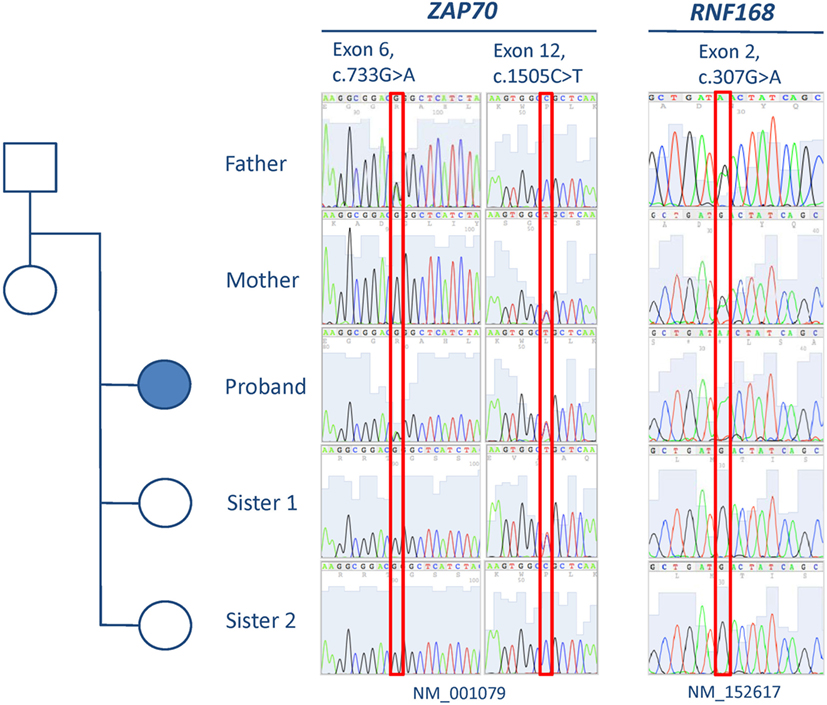
Figure 1. Pedigree and results from Sanger sequencing of exons 6 and 12 of ZAP70 and exon 2 of RNF168.
Subsequent investigations of lymphocytes isolated from the patient supported the presence of ZAP70 deficiency. In vitro proliferation was depressed at 10% [stimulation index (SI) = 18] of control responses (SI = 196) to phytohemagglutinin. Proliferation after stimulation with anti-CD3 antibody was similarly impaired (patient SI = 18, control SI = 156). The response was partially rescued by incubation with phorbol 12-myristate 13-acetate and ionomycin, as previously observed in other patients with ZAP70 deficiency (5). T cell receptor excision circle levels were also extremely low (47 per 0.5 µg of DNA, normal >400) (17). Reduced ZAP70 protein expression was demonstrated in patient lymphocytes by immunoblotting (Figure 2) (18). Phosphorylation of ZAP70 was diminished after co-incubation of patient lymphocytes with anti-CD3 antibody (results not shown), although it remains unclear whether this finding resulted from the overall decreased ZAP70 expression, inability to auto-phosphorylate, or both.
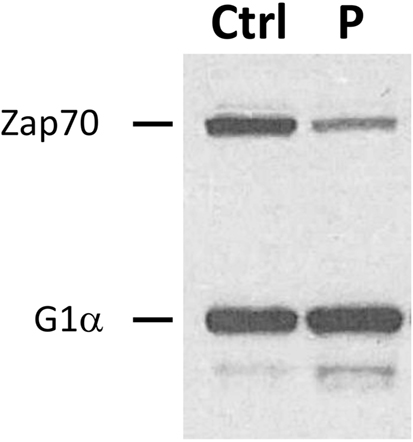
Figure 2. Reduced expression of ZAP70 protein. Immunoblotting was performed to detect ZAP70 in lysates obtained from patient (P) and control (Ctrl) peripheral blood lymphocytes. Simultaneous assessment of G1α expression is shown as a loading control.
Further analyses were then performed to identify a genetic cause for the radiation sensitivity. We examined the 21,536 variants identified by WES for 61 genes (Table 2) encoding molecules known or anticipated to be associated with a cellular phenotype of radiosensitivity containing protein-altering (truncating, splicing, or non-synonymous) changes. Non-synonymous variants were considered if predicted to be conserved (by PhyloP) and damaging (CADD score ≥15) (14, 16). Only one variant with an ExAC minor allelic frequency (MAF) below 0.01 was identified in this manner: a single heterozygous missense change in MLH1 [c.2146G>A (NM_000249), p.V716M]. While biallelic MLH1 mutations are associated with slight radiosensitivity in cultured skin fibroblasts (Mendelian Inheritance in Man #276300), single allelic variants in the gene are not known to produce this phenotype. As a result and because polymorphisms have been reported to be associated with radiosensitivity, we expanded the analyses to identify variants in the same 61 genes with MAFs below 0.05 (19–22). This analysis revealed only the presence of a homozygous missense variant in exon 2 of RNF168 [c.307G>A, (NM_152617), p.D103N] (Figure 1). Both parents were confirmed to be heterozygous carriers of the RNF168 variant, but neither of the two siblings received a copy of the altered gene (Figure 1).
Background
History
ZAP70 deficiency was first reported in 1994 (5, 23, 24). ZAP70 plays a critical role in T cell receptor signal transduction (5, 25). As a result, biallelic mutations of ZAP70 are known to produce both abnormal thymic development of T cells and defective T cell function (5, 26). Because ZAP70 is highly expressed in CD8+ T cells and NK cells, loss of function can lead to recurrent and severe viral infections.
Radiosensitivity, immunodeficiency, dysmorphic features, and learning difficulties (RIDDLE) syndrome (Mendelian Inheritance in Man #611943) was first described in 2007 (27). The condition was ultimately determined to be caused by biallelic mutations in RNF168 (27, 28). RNF168 is a ubiquitin-ligase ring-finger protein that plays an important role in the ubiquitin-dependent DNA damage response (9, 29). After recognition of a double-stranded DNA break by ATM, MRE11, RAD50, and nibrin, RNF168 is recruited to the site by mono- or di-ubiquitinated histone H2A motifs. In addition, in combination with UBC13, it also directly ubiquitinates H2A during this process. RNF168 then extends the histone modifications, forming polyubiquitin chains to recruit BRCA1 and 53BP1, which facilitate DNA repair.
Review of Similar Cases
ZAP70 deficiency results in a spectrum of immune deficiency or dysregulation ranging from atopy, autoimmunity, and late-onset combined immunodeficiency to severe, combined T and B cell deficiency from birth (6, 24, 30, 31). Many mutations have been identified in the gene, impacting the protein in several domains (Figure S1 in Supplementary Material, as reported in the Human Gene Mutation Database and Mendelian Inheritance in Man). Complete deficiency of protein expression results in severe immune deficiency that results in marked susceptibility to infections within the first few months of life (26). Hypomorphic mutations produce overall T cell lymphopenia, reduced T cell function, and later-onset presentation of clinical disease, as seen in our patient (26, 31). In all cases, CD8+ T cell lymphopenia is present.
Two patients with RNF168 deficiency have been reported to date (27, 32). In the first, immune deficiency was characterized by low serum IgG levels (27). The other patient had IgA deficiency but normal serum IgG levels (32). Both patients exhibited infectious susceptibility in a similar manner to our patient. In RNF168-deficient mice, the immunologic defects are more pronounced (33). The first patient was reported to have learning difficulties, but the second had normal intelligence. Our patient currently lives with her parents but functions independently. She recently completed a bachelor’s degree in art and psychology. Both previously reported individuals also had short stature, motor deficits or ataxia, dysmorphic facial features or microcephaly, and radiosensitivity. Of these additional features, our patient exhibited only radiosensitivity. That said, her severe radiation hypersensitivity suggests that the detected RNF168 variant may be phenotypically relevant and impactful. The mutations in the 2 previously reported patients are highly deleterious (compound heterozygous frameshift changes leading to premature stop codons in the first patient and a homozygous p.R131X mutation in the second, see Figure S1 in Supplementary Material), perhaps accounting for the severe phenotypes. The less damaging homozygous missense variant in our patient may produce a milder phenotype consisting only of radiation sensitivity.
Discussion
Diagnosis
Recent advances in our understanding of the genetics and pathophysiology of immune deficiency have shown that mutations in critical pathways in T cell development and function can result in a broad spectrum of clinical phenotypes. The presented patient is, to our knowledge, not only the oldest reported patient with newly diagnosed ZAP70 deficiency but also expresses a unique blended clinical phenotype and genotype with mutations linked to two distinct immune deficiency disorders. Diagnosis of ZAP70 deficiency in adulthood is unusual, and it remains unclear whether the homozygous RNF168 variant may have altered the classic presentation and course of ZAP70 deficiency in manners that we do not yet understand. Vice versa, the possibility that ZAP70 deficiency may have affected the biological impact of the RNF168 homozygous variant cannot be excluded. The relatively high allelic frequency of this RNF168 variant (0.02 in the ExAC database), especially among the Hispanic population (0.19), may suggest that radiosensitivity represents a clinical phenotype that occurs more commonly than recognized, perhaps because it is rarely tested. Indeed, it has been recognized that 20–40% of apparently healthy individuals have increased radiosensitivity and impaired DNA damage repair when screened for these abnormalities (34–37). Thus, it seems possible that homozygous carriers of the RNF168 c.307G>A variant typically possess radiation sensitivity that is not clinically apparent. As such, this variant may have clinical implications, especially if it represents a cancer susceptibility gene defect. Meanwhile, ZAP70 is known to be expressed in B cells, although its function in these cells remains poorly understood (38–40). If it has a role in B cell survival, particularly after a critical insult, such as ionizing radiation-induced DNA damage, loss of ZAP70 function could result in augmented radiation sensitivity, as assessed by the clinical in vitro DNA repair disorder assay. This hypothesis merits further future investigations.
Treatment
With the identification of the variants in ZAP70 and RNF168, the patient has been referred for evaluation for potentially curative allogeneic hematopoietic stem cell transplantation (HSCT). Elements favoring this approach include the poor quality of life (due to the skin problems and significant medical support consisting of IgG supplementation; antibiotic, antifungal, and antiviral therapy or prophylaxis; and IFN-γ for HPV infection) and continued oncologic risk. On the other hand, this patient has survived into the third decade of life. While allogeneic HSCT is recommended to occur as early as possible for children with ZAP70 deficiency to prevent fatal infectious complications, the current age of the patient makes the potential survival benefit from allogeneic HSCT less clear (41). In addition, she has considerable transplantation risks, including suppressed HPV and cryptococcal infections, absence of a fully HLA-matched donor, active skin inflammation that may increase the risk of skin graft-versus-host disease, radiation sensitivity, and advanced age.
Concluding Remarks
In summary, we report the oldest individual diagnosed with ZAP70-related primary immune deficiency. In addition, through WES, we were able to identify the presence of potentially pathogenic mutant alleles at two loci, resulting in a blended phenotype. Blended phenotypes have been reported to occur in about 5–7% of individuals tested by WES with a higher incidence (11%) in patients with PIDDs (1, 42–45). In this patient, WES demonstrated compound heterozygosity for two novel mutations in ZAP70 and homozygosity for a missense variant RNF168, which may confer radiation sensitivity. The finding of sequence alterations in two distinct disease genes with a blended clinical phenotype of combined immunodeficiency and radiation hypersensitivity has not been reported in the literature. This case highlights the importance of examining for mutations in multiple loci beyond focusing upon single likely candidate genes. It strongly suggests the utility of WES and comprehensive and genomic testing for patients with PIDDs, particularly for patients who exhibit atypical clinical features, to exclude the presence of additional molecular defects that may have implications concerning various approaches for therapy (1). As such, multilocus genomic variation and the coexistence of two underlying genotypic abnormalities in this individual also draw attention to the clinical complexity of managing patients with such distinct or overlapping molecular phenotypes (45).
Ethics Statement
Written informed consent was obtained from the patient and her family members to participate in a Baylor College of Medicine Institutional Review Board approved protocol for WES as part of the Baylor-Hopkins Center for Mendelian Genomics project at Baylor College of Medicine (Houston, TX, USA). The patient and family members also provided written informed consent to have their clinical and genetic information published in medical or scientific journals. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Author Contributions
IC collected data, performed genetic data analyses, and wrote the manuscript. RS collected clinical data and prepared a draft of the manuscript. AS-P collected and analyzed genetic data. ZC-A provided bioinformatics for genetic data analyses. VK collected samples. HD performed biological testing. CR provided supervision of research. TQ collected clinical data and prepared a draft of the manuscript. JL provided WES and genomic and genetic data analyses and wrote portions of the manuscript. JO provided supervision of the clinical care and research. IH collected clinical and immunologic data and wrote portions of the manuscript.
Conflict of Interest Statement
Baylor College of Medicine (BCM) and Miraca Holdings, Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics Laboratories (BG), which performs clinical exome sequencing. JL derives support through a professional services agreement between BCM and BG. JL also serves on the Scientific Advisory Board of the BG. JL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, has stock options in Lasergen, Inc., and is a coinventor of US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. None of the remaining authors have potential conflicts of interest.
Acknowledgments
We would like to acknowledge Carlos R. Bachier for excellent clinical care of the patient. We also appreciate Jennifer E. Posey for kind review of the manuscript during preparation. Our gratitude goes to the affected patient and her family for their participation with informed consent in all studies conducted to elucidate her genetic diagnoses.
Funding
This work was supported by the following: NIH-NIAID Grants R01AI067946 and R01 AI120989 to JO, NIH-NHGRI/NHLBI Grant UM1HG006542 to JL, NIH-NINDS Grant R01NS058529 to JL, and the Jeffrey Modell Foundation Translational Research Program Grant to IC.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00576/full#supplementary-material.
Figure S1. Known mutations in the (A) ZAP70 and (B) RNF168 proteins, as reported in the Human Gene Mutation and Mendelian Inheritance in Man databases. Amino acid positions are marked above and mutations are shown below each protein using dotted lines. (A) ZAP70 protein domains are labeled within the shaded boxes. Patient mutations are designated by arrows above the protein. Note that the figure does not include the known IVS7 c.837 + 121G>A, IVS12 c.1624-11G>A, and c. 836_837delAT mutations. (B) RNF168 protein domains are labeled above the amino acid positions. Abbreviations: RING, really interesting new gene finger; LRM, LR motif; MIU, motif interacting with ubiquitin; UMI, ubiquitin interacting motif- and MIU-related ubiquitin binding domain. Patient mutations are designated by arrows below the protein.
Table S1. Other proband variants identified by WES.
References
1. Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy ClinImmunol (2017) 139(1):232–45. doi: 10.1016/j.jaci.2016.05.042
2. Fleisher TA, Bosco Oliveira J. Functional and molecular evaluation of lymphocytes. J Allergy ClinImmunol (2004) 114(2):227–34. doi:10.1016/j.jaci.2004.06.001
3. Huo YK, Wang Z, Hong J-H, Chessa L, McBride WH, Perlman SL, et al. Radiosensitivity of ataxia-telangiectasia, X-linked agammaglobulinemia, and related syndromes using a modified colony survival assay. Cancer Res (1994) 54(10):2544–7.
4. Sun X, Becker-Catania SG, Chun HH, Hwang MJ, Huo Y, Wang Z, et al. Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. J Pediatr (2002) 140(6):724–31. doi:10.1067/mpd.2002.123879
5. Arpaia E, Shahar M, Dadi H, Cohen A, Rolfman CM. Defective T cell receptor signaling and CD8+ thymic selection in humans lacking Zap-70 kinase. Cell (1994) 76(5):947–58. doi:10.1016/0092-8674(94)90368-9
6. Turul T, Tezcan I, Artac H, de Bruin-Versteeg S, Barendregt BH, Reisli I, et al. Clinical heterogeneity can hamper the diagnosis of patients with ZAP70 deficiency. Eur J Pediatr (2008) 168(1):87–93. doi:10.1007/s00431-008-0718-x
7. Mazer B, Harbeck RJ, Franklin R, Schwinzer R, Kubo R, Hayward A, et al. Phenotypic features of selective T cell deficiency characterized by absence of CD8+T lymphocytes and undetectable mRNA for ZAP-70 kinase. Clin Immunol Immunopathol (1997) 84(2):129–38. doi:10.1006/clin.1997.4365
8. Chinn IK, Shearer WT. Severe combined immunodeficiency disorders. Immunol Allergy Clin North Am (2015) 35(4):671–94. doi:10.1016/j.iac.2015.07.002
9. Blundred RM, Stewart GS. DNA double-strand break repair, immunodeficiency and the RIDDLE syndrome. Expert Rev Clin Immunol (2011) 7(2):169–85. doi:10.1586/eci.10.93
10. Gatti RA, Boder E, Good RA. Immunodeficiency, radiosensitivity, and the XCIND syndrome. Immunol Res (2007) 38(1):87–101. doi:10.1007/s12026-007-0018-y
11. Nahas SA, Gatti RA. DNA double strand break repair defects, primary immunodeficiency disorders, and ‘radiosensitivity’. Curr Opin Allergy Clin Immunol (2009) 9(6):510–6. doi:10.1097/ACI.0b013e328332be17
12. Mizutani S, Takagi M. XCIND as a genetic disease of X-irradiation hypersensitivity and cancer susceptibility. Int J Hematol (2013) 97(1):37–42. doi:10.1007/s12185-012-1240-5
13. Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge MN, Jhangiani S, Buhay CJ, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Med (2013) 5(6):57. doi:10.1186/gm461
14. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res (2010) 20(1):110–21. doi:10.1101/gr.097857.109
15. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet (2014) 46(3):310–5. doi:10.1038/ng.2892
16. Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, Belkadi A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy ClinImmunol (2016) 138(4):957–69. doi:10.1016/j.jaci.2016.08.003
17. Roifman CM, Dadi H, Somech R, Nahum A, Sharfe N. Characterization of ζ-associated protein, 70 kd (ZAP70)-deficient human lymphocytes. J Allergy ClinImmunol (2010) 126(6):1226.e–33.e. doi:10.1016/j.jaci.2010.07.029
18. Dadi HK, Simon AJ, Roifman CM. Effect of CD3δ deficiency on maturation of α/β and γ/δ T-cell lineages in severe combined immunodeficiency. N Engl J Med (2003) 349(19):1821–8. doi:10.1056/NEJMoa031178
19. Smirnov DA, Brady L, Halasa K, Morley M, Solomon S, Cheung VG. Genetic variation in radiation-induced cell death. Genome Res (2012) 22(2):332–9. doi:10.1101/gr.122044.111
20. Hornhardt S, Rößler U, Sauter W, Rosenberger A, Illig T, Bickeböller H, et al. Genetic factors in individual radiation sensitivity. DNA Repair (2014) 16:54–65. doi:10.1016/j.dnarep.2014.02.001
21. Fuentes-Raspall MJ, Caragol I, Alonso C, Ramón y Cajal T, Fisas D, Seoane A, et al. Apoptosis for prediction of radiotherapy late toxicity: lymphocyte subset sensitivity and potential effect of TP53 Arg72Pro polymorphism. Apoptosis (2015) 20(3):371–82. doi:10.1007/s10495-014-1056-2
22. Alsbeih G, Al-Meer RS, Al-Harbi N, Bin Judia S, Al-Buhairi M, Venturina NQ, et al. Gender bias in individual radiosensitivity and the association with genetic polymorphic variations. Radiother Oncol (2016) 119(2):236–43. doi:10.1016/j.radonc.2016.02.034
23. Chan AC, Kadlecek TA, Elder ME, Filipovich AH, Kuo WL, Iwashima M, et al. ZAP-70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science (1994) 264(5165):1599. doi:10.1126/science.8202713
24. Elder ME, Lin D, Clever J, Chan AC, Hope TJ, Weiss A, et al. Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science (1994) 264(5165):1596–9. doi:10.1126/science.8202712
25. Chan AC, Iwashima M, Turck CW, Weiss A. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell (1992) 71(4):649–62. doi:10.1016/0092-8674(92)90598-7
26. Fischer A, Picard C, Chemin K, Dogniaux S, le Deist F, Hivroz C. ZAP70: a master regulator of adaptive immunity. Semin Immunopathol (2010) 32(2):107–16. doi:10.1007/s00281-010-0196-x
27. Stewart GS, Stankovic T, Byrd PJ, Wechsler T, Miller ES, Huissoon A, et al. RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proc Natl Acad Sci U S A (2007) 104(43):16910–5. doi:10.1073/pnas.0708408104
28. Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell (2009) 136(3):420–34. doi:10.1016/j.cell.2008.12.042
29. Hiom K. DNA repair: a riddle at a double-strand break. Curr Biol (2009) 19(8):R331–3. doi:10.1016/j.cub.2009.03.024
30. Rosenberg SL, Larkin A. ZAP70-related severe combined immunodeficiency. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews. Seattle, WA: University of Washington (2014). p. 1993–2015.
31. Picard C, Dogniaux S, Chemin K, Maciorowski Z, Lim A, Mazerolles F, et al. Hypomorphic mutation of ZAP70 in human results in a late onset immunodeficiency and no autoimmunity. Eur J Immunol (2009) 39(7):1966–76. doi:10.1002/eji.200939385
32. Devgan SS, Sanal O, Doil C, Nakamura K, Nahas SA, Pettijohn K, et al. Homozygous deficiency of ubiquitin-ligase ring-finger protein RNF168 mimics the radiosensitivity syndrome of ataxia-telangiectasia. Cell Death Differ (2011) 18(9):1500–6. doi:10.1038/cdd.2011.18
33. Bohgaki T, Bohgaki M, Cardoso R, Panier S, Zeegers D, Li L, et al. Genomic instability, defective spermatogenesis, immunodeficiency, and cancer in a mouse model of the riddle syndrome. PLoS Genet (2011) 7(4):e1001381. doi:10.1371/journal.pgen.1001381
34. Marcon F, Andreoli C, Rossi S, Verdina A, Galati R, Crebelli R. Assessment of individual sensitivity to ionizing radiation and DNA repair efficiency in a healthy population. Mutat Res (2003) 541(1–2):1–8. doi:10.1016/S1383-5718(03)00171-2
35. Sigurdson AJ, Stram DO. Genetic predisposition to radiation-related cancer and potential implications for risk assessment. Ann ICRP (2012) 41(3–4):108–16. doi:10.1016/j.icrp.2012.06.030
36. Kato TA, Wilson PF, Nagasaw H, Peng Y, Weil MM, Little JB, et al. Variations in radiosensitivity among individuals: a potential impact on risk assessment? Health Phys (2009) 97(5):470–80. doi:10.1097/HP.0b013e3181b08eee
37. Hu JJ, Smith TR, Miller MS, Lohman K, Case LD. Genetic regulation of ionizing radiation sensitivity and breast cancer risk. Environ Mol Mutagen (2002) 39(2–3):208–15. doi:10.1002/em.10058
38. Anbazhagan K, Rabbind Singh A, Isabelle P, Stella I, Céline A-DM, Bissac E, et al. Human pre-B cell receptor signal transduction: evidence for distinct roles of PI3kinase and MAP-kinase signalling pathways. Immun Inflamm Dis (2013) 1(1):26–36. doi:10.1002/iid3.4
39. Fedele AL, Tolusso B, Gremese E, Bosello SL, Carbonella A, Canestri S, et al. Memory B cell subsets and plasmablasts are lower in early than in long-standing rheumatoid arthritis. BMC Immunol (2014) 15(1):28. doi:10.1186/s12865-014-0028-1
40. Michelutti A, Gremese E, Morassi F, Petricca L, Arena V, Tolusso B, et al. B-cell subsets in the joint compartments of seropositive and seronegative rheumatoid arthritis (RA) and no-RA arthritides express memory markers and ZAP70 and characterize the aggregate pattern irrespectively of the autoantibody status. Mol Med (2011) 17(9–10):901–9. doi:10.2119/molmed.2011.00034
41. Kim VH-D, Murguia L, Schechter T, Grunebaum E, Roifman CM. Emergency treatment for ζ chain-associated protein of 70 kDa (ZAP70) deficiency. J Allergy ClinImmunol (2013) 131(4):1233–5. doi:10.1016/j.jaci.2012.09.020
42. Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med (2013) 369(16):1502–11. doi:10.1056/NEJMoa1306555
43. Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA (2014) 312(18):1870–9. doi:10.1001/jama.2014.14601
44. Posey JE, Rosenfeld JA, James RA, Bainbridge M, Niu Z, Wang X, et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet Med (2016) 18(7):678–85. doi:10.1038/gim.2015.142
Keywords: primary immunodeficiency, ZAP70, RNF168, radiosensitivity, immunodeficiency, dysmorphic features, and learning difficulties syndrome, whole-exome sequencing, blended phenotype
Citation: Chinn IK, Sanders RP, Stray-Pedersen A, Coban-Akdemir ZH, Kim VH-D, Dadi H, Roifman CM, Quigg T, Lupski JR, Orange JS and Hanson IC (2017) Novel Combined Immune Deficiency and Radiation Sensitivity Blended Phenotype in an Adult with Biallelic Variations in ZAP70 and RNF168. Front. Immunol. 8:576. doi: 10.3389/fimmu.2017.00576
Received: 06 February 2017; Accepted: 01 May 2017;
Published: 26 May 2017
Edited by:
Andrew Gennery, Newcastle University, UKReviewed by:
Filomeen Haerynck, Ghent University, BelgiumKohsuke Imai, Tokyo Medical and Dental University, Japan
Copyright: © 2017 Chinn, Sanders, Stray-Pedersen, Coban-Akdemir, Kim, Dadi, Roifman, Quigg, Lupski, Orange and Hanson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivan K. Chinn, Y2hpbm5AYmNtLmVkdQ==
†These authors have contributed equally to this work.