 Mihály Józsi
Mihály Józsi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 18 May 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00571
This article is part of the Research Topic Mechanisms of Activation and Evasion of The Complement System by Pathogens View all 13 articles
Human-pathogenic microbes possess various means to avoid destruction by our immune system. These include interactions with the host complement system that may facilitate pathogen entry into cells and tissues, expression of molecules that defuse the effector complement components and complexes, and acquisition of host complement inhibitors to downregulate complement activity on the surface of the pathogen. A growing number of pathogenic microorganisms have acquired the ability to bind the complement inhibitor factor H (FH) from body fluids and thus hijack its host protecting function. In addition to FH, binding of FH-related (FHR) proteins was also demonstrated for several microbes. Initial studies assumed that these proteins are complement inhibitors similar to FH. However, recent evidence suggests that FHR proteins may rather enhance complement activation both directly and also by competing with the inhibitor FH for binding to certain ligands and surfaces. This mini review focuses on the role of the main alternative pathway regulator FH in host–pathogen interactions, as well as on the emerging role of the FHR proteins as enhancers of complement activation.
Innate and adaptive immune mechanisms work in a collaborative manner to effectively eliminate invading microorganisms and develop immune memory. In turn, pathogenic microbes have acquired various means during their co-evolution with their host organisms to evade host immune responses. The complement system, a major humoral arm of innate immunity, includes ~40 plasma and cell membrane-anchored proteins that act in a cascade-like manner to opsonize microbes and facilitate their phagocytosis, activate cellular responses, initiate inflammation, or directly lyse certain microbes by punching holes into them (1). Complement can be activated by three major pathways, the classical, the lectin, and the alternative pathway. The recognition molecules of the pathways initiate activation by interacting with enzymatically active components that propagate the cascade and generate active complement fragments and complexes that mediate the biological effects of the system (Figure 1A) (2).
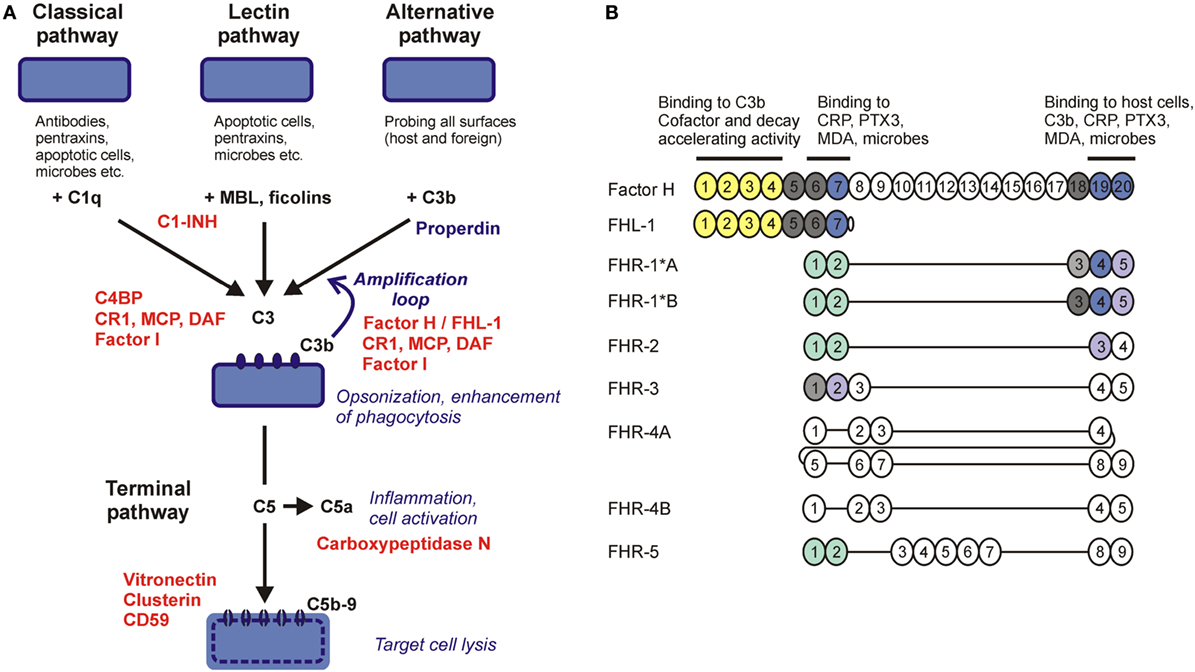
Figure 1. Complement pathways and the human factor H (FH) protein family. (A) Schematic overview of the major complement activation and regulation pathways. Molecules acting as complement inhibitors are shown in red. (B) The five human FH-related (FHR) proteins retained domains homologous to complement control protein domains 6–9 and 18–20 of FH (showed by vertical alignment). Colors indicate domains identical between FH and FHRs; light shades indicate high sequence similarity (>80% identity) but not complete identity. The domains marked green are closely related to each other but only distantly to FH and mediate dimerization of FHR-1, FHR-2, and FHR-5. Functional sites in FH are shown by horizontal lines. FH-like protein 1 (FHL-1) is a splice variant of FH.
Because complement is a powerful system to facilitate destruction of microbes or other target cells, host cells and tissues are protected by various combinations of fluid phase and membrane complement regulatory proteins that fine tune and/or block the activation steps of the complement cascade, restrict activation in both time and space, and prevent the potential deleterious effects of full-blown, excessive activation (Figure 1A) (3). Most complement regulatory proteins are negative regulators, i.e., inhibitors of the various activation steps, including the soluble regulators C1-inhibitor, C4b-binding protein, factor H (FH), vitronectin and clusterin, and the membrane-anchored regulators complement receptor type 1, membrane cofactor protein, decay accelerating factor, and CD59. Properdin is a positive regulator of complement activation. Recently, the FH-related (FHR) proteins have emerged as additional positive regulators that promote activation of the system, particularly the alternative pathway (4, 5).
Factor H is a conserved plasma glycoprotein that inhibits the alternative pathway and the amplification loop (6, 7). By binding to C3b, a major cleavage product of the central complement component C3, FH prevents assembly of the C3bBb alternative pathway C3 convertase enzyme, facilitates the decay of the convertase if already formed by displacing bound Bb from C3b (decay accelerating activity), and acts as a cofactor for the plasma serine protease factor I that then cleaves C3b into the inactive form iC3b (cofactor activity). Interaction of FH with C3b also allows for regulating the C5 convertases.
Factor H is composed of 20 individually folding complement control protein (CCP) domains. The complement regulatory activities of FH are mediated by the N-terminal CCP1–4 domains, which harbor a C3b-binding site (8). CCP7 contains binding sites for certain ligands including glycosaminoglycans on host cellular surfaces, pentraxins, and malondialdehyde (MDA) epitopes generated by lipid peroxidation. The C-terminal CCP19–20 domains harbor binding sites for C3b/C3d, pentraxins, and sialic acid/glycosaminoglycans, and thus anchor FH on host surfaces under complement attack (i.e., with deposited C3b) (9–11). This allows FH for restriction of complement activation on host cells and also on non-cellular surfaces lacking membrane complement regulators, such as basement membranes. Thus, FH has an important function in self–non-self discrimination by recognizing specific host surfaces (12, 13).
The FH-like protein 1 (FHL-1) is derived from an alternative transcript of the CFH gene, and includes the seven N-terminal CCPs of FH plus four amino acids at its C-terminal end. FHL-1 shares with FH complement inhibiting and ligand-binding capacities associated with these domains but may display functional differences, as well, that need to be more precisely defined in the future (14).
In humans, five CFHR genes are found adjacent to the CFH gene and code for five distinct FHR proteins. These proteins have structural homology to FH; however, they lack domains homologous to CCPs 1–4 of FH that are responsible for the complement inhibiting activity (Figure 1B). Initial studies on FHRs investigated their complement inhibiting capacity, and some form of—generally weak—activity was indeed described for all of them. FHR-1 was reported to inhibit C5 and the terminal pathway (15), FHR-2 was to inhibit the alternative pathway C3 convertase and activation of the terminal pathway (16), FHR-3 and FHR-4 were to enhance the cofactor activity of FH (17), FHR-3 was also to possess cofactor activity on its own (18), and FHR-5 was to display weak cofactor activity and inhibit the C3 convertase in fluid phase (19). However, some of these reported activities were not confirmed by other studies, e.g., the terminal pathway inhibition by FHR-1 (20–22). In general, FHR proteins appear to lack significant complement inhibitory activity (4), but further studies are needed to clarify if any of the FHRs possess some form of such activity. Because FHRs were shown to interact with C3b, they may modulate C3b degradation by competing out FH, but may also interfere with the assembly and/or activity of the C3b containing convertase enzymes (i.e., the alternative pathway C3 convertase and the C5 convertases), as suggested for FHR-5 and FHR-2.
The conserved domains of the FHR proteins are homologous to CCPs 6–9 and 18–20 of FH (Figure 1B). Because CCPs 6–7 and 19–20 of FH mediate interactions of the complement regulator with C3b, the pentraxins C-reactive protein (CRP) and pentraxin 3 (PTX3), MDA epitopes, host cells, and basement membranes, due to the potentially overlapping ligand-binding capacity associated with the homologous domains, FHRs could interfere with FH functions through competition (23). Recent data suggest that, contrary to previous assumptions, a major role of the FHR proteins is to recognize and bind certain ligands, surfaces and cells, and thus act as competitive inhibitors of FH.
CCPs 1–2 of FHR-1, FHR-2, and FHR-5 were found to mediate dimerization of these proteins, thus increasing their avidity for surface-bound C3b and resulting in increased competition with FH, termed complement deregulation. Disease-associated mutants of these proteins with duplicated dimerization domains result in enhanced alternative pathway activation by diminishing FH binding to surface-bound C3b (21, 24, 25). FHR-5 can also compete with FH for binding to CRP, PTX3, and extracellular matrix, resulting in enhancement of complement activation (26). Altogether, these recent data support a major role for the FHRs in modulating alternative pathway activation as antagonists of FH.
In addition, FHR-4 was shown to activate the alternative pathway by binding C3b and allowing the assembly of an active C3bBb convertase, and also to promote classical pathway activation via its interaction with CRP (27–29). Similarly, FHR-5 was demonstrated to enhance alternative pathway activation by C3b binding (26).
Why would this enhanced complement activation be useful for us as hosts and what does that mean in the context of infectious disease? Host FH is sequestered by pathogenic microbes, facilitating serum/complement resistance (Figure 2A). This can be an important step in evading first-line immune defense and aids dissemination of microbes and colonization of host niches. FHRs, in turn, were suggested to be decoys that due to their overlapping ligand spectrum with FH may displace this complement inhibitor from the surface of microbes, and may also fine tune complement activation under physiological conditions, e.g., on altered self (4). Thus, FHRs may increase opsonization of microbes, dying cells, and cellular debris, and help the resolution of inflammation (Figure 2A). Notably, most FH-binding microbial proteins also bind within those FH domains that are conserved among the FHR proteins (4, 30).
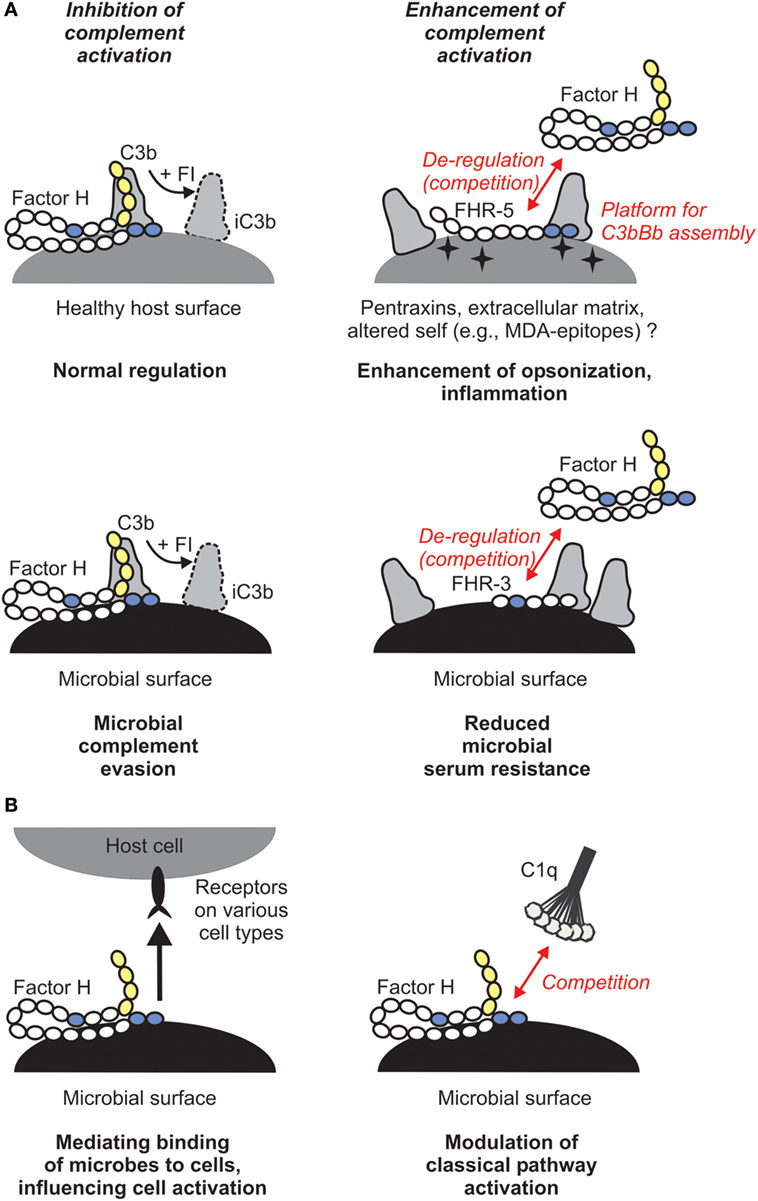
Figure 2. Role of the human factor H (FH) protein family in microbial immune evasion. (A) In addition to its role as plasma complement inhibitor, FH recognizes and binds to host surfaces and protects them from complement attack. Several microbes exploit this mechanism and recruit FH to their surface in order to escape from the complement system. FH-related (FHR) proteins may bind to certain host ligands or altered host surfaces that are exposed during inflammation or tissue damage (such as pentraxins, extracellular matrix proteins, or oxidative modifications of lipids) and displace FH, resulting in increased opsonization. FHRs may act as decoys and compete with FH for binding to microbial proteins. For example, FHR-3 was described to inhibit binding of FH to fHbp of Neisseria meningitidis. (B) FH was described to have additional functions. By simultaneously binding to certain microbes and receptors (such as CR3) on host cells, it may facilitate uptake of the microbe by immune cells and modulate cell activation, or facilitate entry of microbes into epithelial cells (left panel). FH was also shown to inhibit binding of C1q to apoptotic cells and E. coli and thus may modulate classical pathway activation and opsonization (right panel).
Various classes of microbial pathogens were shown to bind human FH; these were reviewed in detail elsewhere [see, e.g., Ref. (30, 31)]. Instead of providing an ever-growing list of such microorganisms, this mini review aims to highlight general patterns (to which exceptions may exist) regarding the relevance of FH binding to microbes, and critically evaluate available literature, by discussing selected representative examples.
Overall, binding FH (or FHL-1) from body fluids is thought to be of advantage for pathogenic microbes in their survival in the host (Figure 2A). Prominent examples include the OspE protein of Borrelia burgdorferi (32), Sbi of Staphylococcus aureus (33), PspC of Streptococcus pneumoniae (34), and fHbp of Neisseria meningitidis (35). Sialylated Neisseria gonorrhoeae binds FH and provides an example of pathogen mimicry of host glycans (36, 31). Apparently, numerous and otherwise unrelated microbial proteins target the same conserved domains of FH, which thus involve pathogen- and host–ligand-binding sites. Such a common microbe binding site was determined and characterized in CCP20 of FH recently (37). Microbes thus can misdirect the self-recognition domains and mimic host ligands/surfaces (38).
Selective binding of FH is one of the reasons of host restriction of certain infections: human FH is preferentially bound by, e.g., group A streptococci (39), N. meningitidis (38, 40), N. gonorrhoeae (41), and non-typeable Haemophilus influenzae (42). By contrast, bacteria that infect various hosts, such as B. burgdorferi, bind FH from several species (43).
The importance of FH binding for bacterial survival is well documented for N. meningitidis, and fHbp is one of the components of N. meningitidis serogroup B vaccine (35, 38, 44, 45). In other cases, the role of FH as being beneficial for the microbe is controversial. The hypervariable region of several M proteins of Streptococcus pyogenes binds FH, which was attributed to downregulate opsonization and promote phagocytic resistance of the pathogen (46). Later studies, on the other hand, found no clear benefit of FH binding in resisting killing in a whole blood model or in an in vivo infection model (47). The used strains and models may influence this; recently, in a human FH transgenic mouse increased virulence of the S. pyogenes strain AP1 (which expresses protein H) was observed (39). Similarly, while several borrelial proteins with FH-binding capacity have been described (48), in some cases they may be dispensable for virulence (49).
Furthermore, some microorganisms were shown to degrade FH (50–52). This appears counterproductive because cleaved FH then loses its ability to inhibit complement activation (51). However, microbes may gain advantage from a more inflammatory micro-environment (53, 54) or, because their proteases could also cleave complement factors necessary for the propagation of the cascade (55), the functional inactivation of FH may not cause significant disadvantage in complement resistance. In addition, the kinetics of inactivation may allow sufficient regulation by FH. In any case, this issue needs further clarification.
Besides its role in the regulation of the alternative pathway, FH was also shown to compete with C1q for binding to lipid A component of LPS, and also to the surface of the E. coli strain TG1, pointing to the possibility that in certain cases FH may modulate the activity of the classical pathway (Figure 2B) (56, 57). This potentially important aspect needs to be further studied.
In addition, by binding to receptors on cells, FH can mediate microbe–host cell interactions (Figure 2B). In this non-canonical role, FH was described to act as a bridging molecule between complement receptor 3 (CR3; CD11b/CD18) and pathogens, and helping either pathogen entry into host cells or the antimicrobial response of the host cells (51, 58–62). Such scenarios were described for FH bound to S. pneumoniae, N. gonorrhoeae, and Candida albicans (58–61). FH bound on C. albicans was shown to facilitate the adhesion, phagocytosis and antifungal responses by neutrophilic granulocytes, such as increased lactoferrin and reactive oxygen species production (61). FH can also enhance the response of macrophages when exposed to C. albicans (51).
Thus, while for most studied microbes binding of FH (and in some cases also that of FHL-1) and the ability of FH/FHL-1 to act as a cofactor for C3b cleavage when bound on the surface or on certain microbial ligands were shown in vitro, direct evidence that demonstrates a relevant role of certain FH/FHR-binding proteins in serum resistance is less substantial. It is important to define the relative contribution of such potential virulence factors to microbial survival in serum and in animal models of infectious diseases. Novel technologies and model organisms may help clarifying to which extent specific FH-binding proteins contribute to the survival of pathogens. Studying non-pathogenic strains for FH binding and activity in parallel would likely be also informative.
Interaction of FHR proteins with microbes (63) has not yet been extensively studied; particularly, functional studies are scarce. This is related to our limited knowledge on these proteins, as discussed above. However, some important observations suggest that FHRs could emerge during evolution as decoys that counteract the sequestration of FH from host body fluids (4). Notably, in FHRs the conserved domains are homologous to those of FH that mediate binding of FH to various ligands/surfaces, both self and non-self, thus FHRs likely share the capacity to bind microbes. FHRs are also described in several non-human species, including mice, rats, and fish; these FHRs also lack the complement regulatory domains of FH and differ in number and domain composition from their human counterparts, there are no clear direct homologs (64–68).
In most cases, FHR-1 binding to microbes and microbial proteins that otherwise bind FH (and in some cases also FHL-1) was demonstrated, such as for several borrelial proteins (48, 69, 70), Leptospira interrogans (71), S. aureus (33), Pseudomonas aeruginosa (72), N. gonorrhoeae (60), Plasmodium falciparum (73), C. albicans (61), and Aspergillus fumigatus (74). So far, in most reports, no functional role for FHR-1 when associated/bound to microbes was demonstrated; in most cases, it was merely assumed that FHR-1 inhibits complement terminal pathway based on the report of Heinen et al. (15). In the case of the streptococcal Scl1 protein, FHR-1 was shown to inhibit terminal pathway activation (75). Even so, FHR-1 was shown not to influence bacterial opsonization and survival in the case of B. burgdorferi (70).
FHR-1, FHR-2, and FHR-5 bind to B. burgdorferi. Functional analysis, however, could not demonstrate a contribution of the FHR proteins to serum resistance of this microbe (70). On the other hand, FHR-1 bound on C. albicans was shown to facilitate interaction with human neutrophils and promote neutrophil antimicrobial responses (61).
Fusobacterium necrophorum binds FH, FHL-1, FHR-1, and FHR-4. Various strains were compared, and a weakly FH-binding strain showed increased C3b and terminal C5b-9 complex deposition on its surface, and decreased survival in human serum, compared with strains that bind FH stronger. The role of FHR-1 and FHR-4 was not addressed (76). FHR-4 also binds to C. albicans, but the functional relevance of this interaction is unclear (61). In both cases, the FHR-4A isoform (77) bound from serum, which shows increased C3b binding compared with FHR-4B, and activates the alternative pathway (29). Further studies need to assess its potential role in enhancing opsonization.
Direct evidence for an important role in infectious disease was described for FHR-3. A genome-wide disease-association study linked the CFHR3 gene to N. meningitidis infection (78). A following functional study found that FHR-3 binds to this pathogen and competes with FH for binding to fHbp of N. meningitidis, thus acts as a competitive inhibitor of FH and enhances complement activation (Figure 2A). FHR-3 and FH bind with similar, nanomolar affinities to fHbp, but relative affinities differ between fHbp variants. Altogether, the genes of both the human host (by determining FH/FHR-3 levels) and the pathogen (by determining fHbp variants, e.g., that preferentially bind FH) influence disease susceptibility (79).
Additional indirect evidence supports such a role of the FHRs. For example, increased FHR concentrations were described in the middle-ear effusion fluid of patients with otitis media with effusion (80). In the zebrafish, FHR expression was found to be upregulated by LPS, indicating a role for them as acute phase proteins (68). These and other data (81) indicate that FHRs may be upregulated during infection or inflammation.
Although the role of FH in complement evasion is of medical importance for some microbes, further aspects of binding of this regulator need to be elucidated, such as the relevance of mediating cellular interactions and regulation of the classical pathway. The role of the FHR proteins is still poorly understood. While they emerge as positive complement regulators via competition with FH and by directly activating the alternative pathway through C3b binding, important questions include (1) the relative concentrations and their regulation, (2) affinity differences toward specific ligands, (3) functional redundancy among them, and (4) clarification of proposed and still unknown complement inhibitory capacity. Further studies will help to evaluate their role in host–pathogen interactions, identify novel vaccine candidates, and may also address the potential therapeutic use of FHR proteins in infectious diseases.
MJ prepared the text and the figures.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author’s work on the function of human factor H family proteins is supported by the grants from the Hungarian Academy of Sciences (Lendület program, grant no. LP2012-43), the National Research, Development and Innovation Office (OTKA grant no. K 109055), the MedInProt, and the Kidneeds Foundation (IA, USA).
1. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi:10.1038/ni.1923
2. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi:10.3389/fimmu.2015.00262
3. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol (2009) 9(10):729–40. doi:10.1038/nri2620
4. Józsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, Rodríguez de Córdoba S. Factor H-related proteins determine complement-activating surfaces. Trends Immunol (2015) 36(6):374–84. doi:10.1016/j.it.2015.04.008
5. Medjeral-Thomas N, Pickering MC. The complement factor H-related proteins. Immunol Rev (2016) 274(1):191–201. doi:10.1111/imr.12477
6. Kopp A, Hebecker M, Svobodová E, Józsi M. Factor H: a complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules (2012) 2(1):46–75. doi:10.3390/biom2010046
7. Parente R, Clark SJ, Inforzato A, Day AJ. Complement factor H in host defense and immune evasion. Cell Mol Life Sci (2016) 74(9):1605–24. doi:10.1007/s00018-016-2418-4
8. Wu J, Wu YQ, Ricklin D, Janssen BJ, Lambris JD, Gros P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat Immunol (2009) 10(7):728–33. doi:10.1038/ni.1755
9. Kajander T, Lehtinen MJ, Hyvärinen S, Bhattacharjee A, Leung E, Isenman DE, et al. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci U S A (2011) 108(7):2897–902. doi:10.1073/pnas.1017087108
10. Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, et al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol (2011) 18(4):463–70. doi:10.1038/nsmb.2018
11. Schmidt CQ, Herbert AP, Kavanagh D, Gandy C, Fenton CJ, Blaum BS, et al. A new map of glycosaminoglycan and C3b binding sites on factor H. J Immunol (2008) 181(4):2610–9. doi:10.4049/jimmunol.181.4.2610
12. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrín D, Stehle T. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol (2015) 11(1):77–82. doi:10.1038/nchembio.1696
13. Meri S. Self-nonself discrimination by the complement system. FEBS Lett (2016) 590(15):2418–34. doi:10.1002/1873-3468.12284
14. Clark SJ, Ridge LA, Herbert AP, Hakobyan S, Mulloy B, Lennon R, et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol (2013) 190(5):2049–57. doi:10.4049/jimmunol.1201751
15. Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, et al. Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood (2009) 114(12):2439–47. doi:10.1182/blood-2009-02-205641
16. Eberhardt HU, Buhlmann D, Hortschansky P, Chen Q, Böhm S, Kemper MJ, et al. Human factor H-related protein 2 (CFHR2) regulates complement activation. PLoS One (2013) 8(11):e78617. doi:10.1371/journal.pone.0078617
17. Hellwage J, Jokiranta TS, Koistinen V, Vaarala O, Meri S, Zipfel PF. Functional properties of complement factor H-related proteins FHR-3 and FHR-4: binding to the C3d region of C3b and differential regulation by heparin. FEBS Lett (1999) 462(3):345–52. doi:10.1016/S0014-5793(99)01554-9
18. Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet (2010) 19(23):4694–704. doi:10.1093/hmg/ddq399
19. McRae JL, Duthy TG, Griggs KM, Ormsby RJ, Cowan PJ, Cromer BA, et al. Human factor H-related protein 5 has cofactor activity, inhibits C3 convertase activity, binds heparin and C-reactive protein, and associates with lipoprotein. J Immunol (2005) 174(10):6250–6. doi:10.4049/jimmunol.174.10.6250
20. Strobel S, Abarrategui-Garrido C, Fariza-Requejo E, Seeberger H, Sánchez-Corral P, Józsi M. Factor H-related protein 1 neutralizes anti-factor H autoantibodies in autoimmune hemolytic uremic syndrome. Kidney Int (2011) 80(4):397–404. doi:10.1038/ki.2011.152
21. Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci U S A (2013) 110(12):4685–90. doi:10.1073/pnas.1219260110
22. Mészáros T, Csincsi ÁI, Uzonyi B, Hebecker M, Fülöp TG, Erdei A, et al. Factor H inhibits complement activation induced by liposomal and micellar drugs and the therapeutic antibody rituximab in vitro. Nanomedicine (2016) 12(4):1023–31. doi:10.1016/j.nano.2015.11.019
23. Józsi M, Zipfel PF. Factor H family proteins and human diseases. Trends Immunol (2008) 29(8):380–7. doi:10.1016/j.it.2008.04.008
24. Tortajada A, Yébenes H, Abarrategui-Garrido C, Anter J, García-Fernández JM, Martínez-Barricarte R, et al. C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation. J Clin Invest (2013) 123(6):2434–46. doi:10.1172/JCI68280
25. Chen Q, Wiesener M, Eberhardt HU, Hartmann A, Uzonyi B, Kirschfink M, et al. Complement factor H-related hybrid protein deregulates complement in dense deposit disease. J Clin Invest (2014) 124(1):145–55. doi:10.1172/JCI71866
26. Csincsi ÁI, Kopp A, Zöldi M, Bánlaki Z, Uzonyi B, Hebecker M, et al. Factor H-related protein 5 interacts with pentraxin 3 and the extracellular matrix and modulates complement activation. J Immunol (2015) 194(10):4963–73. doi:10.4049/jimmunol.1403121
27. Mihlan M, Hebecker M, Dahse HM, Hälbich S, Huber-Lang M, Dahse R, et al. Human complement factor H-related protein 4 binds and recruits native pentameric C-reactive protein to necrotic cells. Mol Immunol (2009) 46(3):335–44. doi:10.1016/j.molimm.2008.10.029
28. Hebecker M, Okemefuna AI, Perkins SJ, Mihlan M, Huber-Lang M, Józsi M. Molecular basis of C-reactive protein binding and modulation of complement activation by factor H-related protein 4. Mol Immunol (2010) 47(6):1347–55. doi:10.1016/j.molimm.2009.12.005
29. Hebecker M, Józsi M. Factor H-related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase via its interaction with C3b protein. J Biol Chem (2012) 287(23):19528–36. doi:10.1074/jbc.M112.364471
30. Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol (2008) 6(2):132–42. doi:10.1038/nrmicro1824
31. Ram S, Shaughnessy J, DeOliveira RB, Lewis LA, Gulati S, Rice PA. Utilizing complement evasion strategies to design complement-based antibacterial immunotherapeutics: lessons from the pathogenic Neisseriae. Immunobiology (2016) 221(10):1110–23. doi:10.1016/j.imbio.2016.05.016
32. Hellwage J, Meri T, Heikkilä T, Alitalo A, Panelius J, Lahdenne P, et al. The complement regulator factor H binds to the surface protein OspE of Borrelia burgdorferi. J Biol Chem (2001) 276(11):8427–35. doi:10.1074/jbc.M007994200
33. Haupt K, Reuter M, van den Elsen J, Burman J, Hälbich S, Richter J, et al. The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement factor H and C3b. PLoS Pathog (2008) 4(12):e1000250. doi:10.1371/journal.ppat.1000250
34. Janulczyk R, Iannelli F, Sjoholm AG, Pozzi G, Bjorck L. Hic, a novel surface protein of Streptococcus pneumoniae that interferes with complement function. J Biol Chem (2000) 275(47):37257–63. doi:10.1074/jbc.M004572200
35. Pizza M, Donnelly J, Rappuoli R. Factor H-binding protein, a unique meningococcal vaccine antigen. Vaccine (2008) 26(Suppl 8):I46–8. doi:10.1016/j.vaccine.2008.11.068
36. Ram S, Sharma AK, Simpson SD, Gulati S, McQuillen DP, Pangburn MK, et al. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J Exp Med (1998) 187(5):743–52. doi:10.1084/jem.187.5.743
37. Meri T, Amdahl H, Lehtinen MJ, Hyvärinen S, McDowell JV, Bhattacharjee A, et al. Microbes bind complement inhibitor factor H via a common site. PLoS Pathog (2013) 9(4):e1003308. doi:10.1371/journal.ppat.1003308
38. Schneider MC, Prosser BE, Caesar JJ, Kugelberg E, Li S, Zhang Q, et al. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature (2009) 458(7240):890–3. doi:10.1038/nature07769
39. Ermert D, Shaughnessy J, Joeris T, Kaplan J, Pang CJ, Kurt-Jones EA, et al. Virulence of group A streptococci is enhanced by human complement inhibitors. PLoS Pathog (2015) 11(7):e1005043. doi:10.1371/journal.ppat.1005043
40. Granoff DM, Welsch JA, Ram S. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect Immun (2009) 77(2):764–9. doi:10.1128/IAI.01191-08
41. Ngampasutadol J, Ram S, Gulati S, Agarwal S, Li C, Visintin A, et al. Human factor H interacts selectively with Neisseria gonorrhoeae and results in species-specific complement evasion. J Immunol (2008) 180(5):3426–35. doi:10.4049/jimmunol.180.5.3426
42. Langereis JD, de Jonge MI, Weiser JN. Binding of human factor H to outer membrane protein P5 of non-typeable Haemophilus influenzae contributes to complement resistance. Mol Microbiol (2014) 94(1):89–106. doi:10.1111/mmi.12741
43. Hovis KM, Tran E, Sundy CM, Buckles E, McDowell JV, Marconi RT. Selective binding of Borrelia burgdorferi OspE paralogs to factor H and serum proteins from diverse animals: possible expansion of the role of OspE in Lyme disease pathogenesis. Infect Immun (2006) 74(3):1967–72. doi:10.1128/IAI.74.3.1967-1972.2006
44. Schneider MC, Exley RM, Chan H, Feavers I, Kang YH, Sim RB, et al. Functional significance of factor H binding to Neisseria meningitidis. J Immunol (2006) 176(12):7566–75. doi:10.4049/jimmunol.176.12.7566
45. Seib KL, Serruto D, Oriente F, Delany I, Adu-Bobie J, Veggi D, et al. Factor H-binding protein is important for meningococcal survival in human whole blood and serum and in the presence of the antimicrobial peptide LL-37. Infect Immun (2009) 77(1):292–9. doi:10.1128/IAI.01071-08
46. Horstmann RD, Sievertsen HJ, Knobloch J, Fischetti VA. Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proc Natl Acad Sci U S A (1988) 85(5):1657–61. doi:10.1073/pnas.85.5.1657
47. Gustafsson MC, Lannergård J, Nilsson OR, Kristensen BM, Olsen JE, Harris CL, et al. Factor H binds to the hypervariable region of many Streptococcus pyogenes M proteins but does not promote phagocytosis resistance or acute virulence. PLoS Pathog (2013) 9(4):e1003323. doi:10.1371/journal.ppat.1003323
48. Kraiczy P. Hide and seek: how lyme disease spirochetes overcome complement attack. Front Immunol (2016) 7:385. doi:10.3389/fimmu.2016.00385
49. Fine LM, Miller DP, Mallory KL, Tegels BK, Earnhart CG, Marconi RT. The Borrelia hermsii factor H binding protein FhbA is not required for infectivity in mice or for resistance to human complement in vitro. Infect Immun (2014) 82(8):3324–32. doi:10.1128/IAI.01892-14
50. McDowell JV, Huang B, Fenno JC, Marconi RT. Analysis of a unique interaction between the complement regulatory protein factor H and the periodontal pathogen Treponema denticola. Infect Immun (2009) 77(4):1417–25. doi:10.1128/IAI.01544-08
51. Svoboda E, Schneider AE, Sándor N, Lermann U, Staib P, Kremlitzka M, et al. Secreted aspartic protease 2 of Candida albicans inactivates factor H and the macrophage factor H-receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18). Immunol Lett (2015) 168(1):13–21. doi:10.1016/j.imlet.2015.08.009
52. Riva R, Korhonen TK, Meri S. The outer membrane protease PgtE of Salmonella enterica interferes with the alternative complement pathway by cleaving factors B and H. Front Microbiol (2015) 6:63. doi:10.3389/fmicb.2015.00063
53. Miller DP, Bell JK, McDowell JV, Conrad DH, Burgner JW, Héroux A, et al. Structure of factor H-binding protein B (FhbB) of the periopathogen, Treponema denticola: insights into progression of periodontal disease. J Biol Chem (2012) 287(16):12715–22. doi:10.1074/jbc.M112.339721
54. Miller DP, McDowell JV, Bell JK, Goetting-Minesky MP, Fenno JC, Marconi RT. Analysis of the complement sensitivity of oral treponemes and the potential influence of FH binding, FH cleavage and dentilisin activity on the pathogenesis of periodontal disease. Mol Oral Microbiol (2014) 29(5):194–207. doi:10.1111/omi.12054
55. Gropp K, Schild L, Schindler S, Hube B, Zipfel PF, Skerka C. The yeast Candida albicans evades human complement attack by secretion of aspartic proteases. Mol Immunol (2009) 47(2–3):465–75. doi:10.1016/j.molimm.2009.08.019
56. Tan LA, Yang AC, Kishore U, Sim RB. Interactions of complement proteins C1q and factor H with lipid A and Escherichia coli: further evidence that factor H regulates the classical complement pathway. Protein Cell (2011) 2(4):320–32. doi:10.1007/s13238-011-1029-y
57. Kishore U, Sim RB. Factor H as a regulator of the classical pathway activation. Immunobiology (2012) 217(2):162–8. doi:10.1016/j.imbio.2011.07.024
58. Hammerschmidt S, Agarwal V, Kunert A, Haelbich S, Skerka C, Zipfel PF. The host immune regulator factor H interacts via two contact sites with the PspC protein of Streptococcus pneumoniae and mediates adhesion to host epithelial cells. J Immunol (2007) 178(9):5848–58. doi:10.4049/jimmunol.178.9.5848
59. Agarwal V, Asmat TM, Luo S, Jensch I, Zipfel PF, Hammerschmidt S. Complement regulator factor H mediates a two-step uptake of Streptococcus pneumoniae by human cells. J Biol Chem (2010) 285(30):23486–95. doi:10.1074/jbc.M110.142703
60. Agarwal S, Ram S, Ngampasutadol J, Gulati S, Zipfel PF, Rice PA. Factor H facilitates adherence of Neisseria gonorrhoeae to complement receptor 3 on eukaryotic cells. J Immunol (2010) 185(7):4344–53. doi:10.4049/jimmunol.0904191
61. Losse J, Zipfel PF, Józsi M. Factor H and factor H-related protein 1 bind to human neutrophils via complement receptor 3, mediate attachment to Candida albicans, and enhance neutrophil antimicrobial activity. J Immunol (2010) 184(2):912–21. doi:10.4049/jimmunol.0901702
62. Schneider AE, Sándor N, Kárpáti É, Józsi M. Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol Immunol (2016) 72:37–48. doi:10.1016/j.molimm.2016.02.011
63. Zipfel PF, Skerka C, Hellwage J, Jokiranta ST, Meri S, Brade V, et al. Factor H family proteins: on complement, microbes and human diseases. Biochem Soc Trans (2002) 30(Pt 6):971–8. doi:10.1042/bst0300971
64. Vik DP, Muñoz-Cánoves P, Kozono H, Martin LG, Tack BF, Chaplin DD. Identification and sequence analysis of four complement factor H-related transcripts in mouse liver. J Biol Chem (1990) 265(6):3193–201.
65. Ren G, Doshi M, Hack BK, Alexander JJ, Quigg RJ. Isolation and characterization of a novel rat factor H-related protein that is up-regulated in glomeruli under complement attack. J Biol Chem (2002) 277(50):48351–8. doi:10.1074/jbc.M205135200
66. Hellwage J, Eberle F, Babuke T, Seeberger H, Richter H, Kunert A, et al. Two factor H-related proteins from the mouse: expression analysis and functional characterization. Immunogenetics (2006) 58(11):883–93. doi:10.1007/s00251-006-0153-y
67. Pouw RB, Vredevoogd DW, Kuijpers TW, Wouters D. Of mice and men: the factor H protein family and complement regulation. Mol Immunol (2015) 67(1):12–20. doi:10.1016/j.molimm.2015.03.011
68. Sun G, Li H, Wang Y, Zhang B, Zhang S. Zebrafish complement factor H and its related genes: identification, evolution, and expression. Funct Integr Genomics (2010) 10(4):577–87. doi:10.1007/s10142-010-0182-3
69. Haupt K, Kraiczy P, Wallich R, Brade V, Skerka C, Zipfel PF. Binding of human factor H-related protein 1 to serum-resistant Borrelia burgdorferi is mediated by borrelial complement regulator-acquiring surface proteins. J Infect Dis (2007) 196(1):124–33. doi:10.1086/518509
70. Siegel C, Hallström T, Skerka C, Eberhardt H, Uzonyi B, Beckhaus T, et al. Complement factor H-related proteins CFHR2 and CFHR5 represent novel ligands for the infection-associated CRASP proteins of Borrelia burgdorferi. PLoS One (2010) 5(10):e13519. doi:10.1371/journal.pone.0013519
71. Castiblanco-Valencia MM, Fraga TR, Silva LB, Monaris D, Abreu PA, Strobel S, et al. Leptospiral immunoglobulin-like proteins interact with human complement regulators factor H, FHL-1, FHR-1, and C4BP. J Infect Dis (2012) 205(6):995–1004. doi:10.1093/infdis/jir875
72. Kunert A, Losse J, Gruszin C, Hühn M, Kaendler K, Mikkat S, et al. Immune evasion of the human pathogen Pseudomonas aeruginosa: elongation factor Tuf is a factor H and plasminogen binding protein. J Immunol (2007) 179(5):2979–88. doi:10.4049/jimmunol.179.5.2979
73. Rosa TF, Flammersfeld A, Ngwa CJ, Kiesow M, Fischer R, Zipfel PF, et al. The Plasmodium falciparum blood stages acquire factor H family proteins to evade destruction by human complement. Cell Microbiol (2016) 18(4):573–90. doi:10.1111/cmi.12535
74. Behnsen J, Hartmann A, Schmaler J, Gehrke A, Brakhage AA, Zipfel PF. The opportunistic human pathogenic fungus Aspergillus fumigatus evades the host complement system. Infect Immun (2008) 76(2):820–7. doi:10.1128/IAI.01037-07
75. Reuter M, Caswell CC, Lukomski S, Zipfel PF. Binding of the human complement regulators CFHR1 and factor H by streptococcal collagen-like protein 1 (Scl1) via their conserved C termini allows control of the complement cascade at multiple levels. J Biol Chem (2010) 285(49):38473–85. doi:10.1074/jbc.M110.143727
76. Friberg N, Carlson P, Kentala E, Mattila PS, Kuusela P, Meri S, et al. Factor H binding as a complement evasion mechanism for an anaerobic pathogen, Fusobacterium necrophorum. J Immunol (2008) 181(12):8624–32. doi:10.4049/jimmunol.181.12.8624
77. Józsi M, Richter H, Löschmann I, Skerka C, Buck F, Beisiegel U, et al. FHR-4A: a new factor H-related protein is encoded by the human FHR-4 gene. Eur J Hum Genet (2005) 13(3):321–9. doi:10.1038/sj.ejhg.5201324
78. Davila S, Wright VJ, Khor CC, Sim KS, Binder A, Breunis WB, et al. Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat Genet (2010) 42(9):772–6. doi:10.1038/ng.640
79. Caesar JJ, Lavender H, Ward PN, Exley RM, Eaton J, Chittock E, et al. Competition between antagonistic complement factors for a single protein on N. meningitidis rules disease susceptibility. Elife (2014) 3:e04008. doi:10.7554/eLife.04008
80. Närkiö-Mäkelä M, Hellwage J, Tahkokallio O, Meri S. Complement-regulator factor H and related proteins in otitis media with effusion. Clin Immunol (2001) 100(1):118–26. doi:10.1006/clim.2001.5043
Keywords: complement deregulation, complement evasion, microbial virulence, factor H, factor H-related, opsonization
Citation: Józsi M (2017) Factor H Family Proteins in Complement Evasion of Microorganisms. Front. Immunol. 8:571. doi: 10.3389/fimmu.2017.00571
Received: 13 March 2017; Accepted: 28 April 2017;
Published: 18 May 2017
Edited by:
Allan Zajac, University of Alabama at Birmingham, United StatesReviewed by:
Michael Kirschfink, Heidelberg University, GermanyCopyright: © 2017 Józsi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mihály Józsi, bWloYWx5LmpvenNpQGdteC5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.