 Jeremy V. Camp
Jeremy V. Camp Colleen B. Jonsson
Colleen B. Jonsson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 May 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00550
This article is part of the Research Topic Neutrophils and Microbial Pathogens: The Never Ending Battle View all 23 articles
Neutrophils are immune cells that are well known to be present during many types of lung diseases associated with acute respiratory distress syndrome (ARDS) and may contribute to acute lung injury. Neutrophils are poorly studied with respect to viral infection, and specifically to respiratory viral disease. Influenza A virus (IAV) infection is the cause of a respiratory disease that poses a significant global public health concern. Influenza disease presents as a relatively mild and self-limiting although highly pathogenic forms exist. Neutrophils increase in the respiratory tract during infection with mild seasonal IAV, moderate and severe epidemic IAV infection, and emerging highly pathogenic avian influenza (HPAI). During severe influenza pneumonia and HPAI infection, the number of neutrophils in the lower respiratory tract is correlated with disease severity. Thus, comparative analyses of the relationship between IAV infection and neutrophils provide insights into the relative contribution of host and viral factors that contribute to disease severity. Herein, we review the contribution of neutrophils to IAV disease pathogenesis and to other respiratory virus infections.
Neutrophils are a type of polymorphonuclear granulocyte that differentiate from myeloblasts in the bone marrow to comprise approximately 60% of the circulating blood leukocytes (1). The formation of intracellular granules (azurophilic granules, specific granules, gelatinase granules, and secretory vesicles) and the morphologically characteristic segmentation of nuclei occur during the terminal differentiation process into neutrophils (1). Neutrophils are often considered professional bacteria-responsive immune cells: they express bacteria-specific receptors (e.g., formylated peptide receptors or certain toll-like receptors, “TLRs”) and their granules have anti-bacterial or bacteriostatic properties. Currently, their role in viral infection has received very little scientific attention (2).
Neutrophils are present during many types of lung diseases associated with acute respiratory distress syndrome (ARDS) and may contribute to acute lung injury (3–12). The lung has a global inflammatory response to infection regardless of etiology, and this response includes the infiltration of neutrophils and macrophages in response to chemotactic signaling which originates in the lung (3, 5, 12–20). These phagocytic cells leave circulation and hone to sites within the infected airways where they may deploy potent effector functions to control disease (Figure 1) in response to pathogen associated molecular patterns (PAMPs) and inflammatory cytokines and chemokines (21, 22). In the case of viral infections, the type I interferons (IFN) and IFN-stimulated genes (ISGs) signal an appropriate immune response (23–26). Lethal infections may result from insufficient information or incorrect information about the specific cause(s) of infection, thereby signaling inappropriate (incorrect or excessive) immune responses (27). Neutrophils, as first-responders to many forms of airway infection, may be a keystone species in determining viral disease outcome; however, neutrophils are poorly studied with respect to viral infection and specifically to respiratory viral disease.
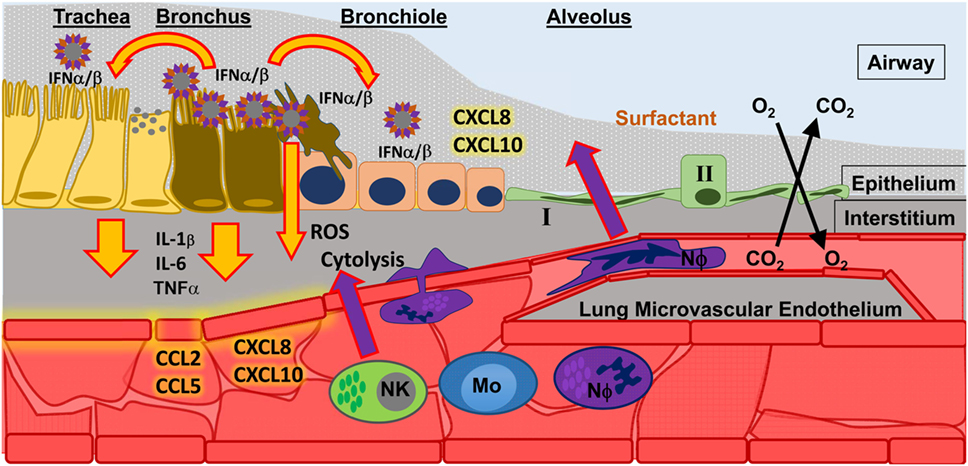
Figure 1. Influenza A virus (IAV) infection in the upper respiratory tract. Infection of epithelial cells in the bronchus results in the release of type I interferons (IFN α/β) which signal to nearby cells. The result of IFN α/β signaling is the release of pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNFα) that signal to endothelial cells, which help spread inflammatory signals (chemokines, such as CCL2, CCL5, CXCL8, CXCL10) throughout the blood to recruit innate immune cells to the site of infection. Recruited innate immune cells [such as natural killer cells (NK); monocytes (Mo); and neutrophils (NΦ)] must interact with activated endothelium to leave the blood stream and migrate toward the site of infection. There they can perform effector functions to control infection, such as releasing reactive oxygen species (ROS) and directly killing infected cells (cytolysis).
Much research has focused on the role of neutrophils in severe versus mild respiratory disease, as well as their role in bacterial infections. It is, therefore, important to establish a general role of neutrophils in respiratory virus infection to provide a groundwork into more specific questions (e.g., are neutrophils capable of catering to a virus-specific response?). Herein, the evidence for the presence and activities of neutrophils during respiratory viral infection is reviewed. Having established spatio-temporal aspects of the neutrophil response to influenza A virus (IAV), the potential antiviral function(s) of neutrophils during acute virus infection as well as recovery from infection are discussed. The discussion will focus on IAV and neutrophil activities during the course of a “typical” flu infection: activation, migration, and effector functions in situ (Figure 1). IAV are particularly well-suited for the study of neutrophils in viral respiratory disease, since they are well-studied in humans and animal models, and it is well-established that infection with specific viral variants (i.e., genetic point mutations) alter the course of disease from mild to severe (28). More recently, specific IAV viral variants that affect pathogenicity have been linked to alterations of the neutrophil response (29). Thus, a comparison of the neutrophil response between disease phenotypes of a single virus species (Influenza A virus) may elucidate a role for neutrophils in the viral microenvironment. Herein, we review evidence of neutrophil responses during the course of disease in various IAV phenotypes in animal models of infection, as well as comparing these responses to what is known about neutrophil responses during bacterial infection of the airways.
Influenza A virus poses a concern for global public health due to emergence of strains with increased human transmission and/or increased pathology (30–35). In 2009, a novel virus type, influenza A(H1N1)pdm09 IAV, emerged with an increased transmission rate and greater disease, i.e., moderate to severe pathology relative to seasonal human IAV (36–50). Clinical isolates of influenza A(H1N1)pdm09 have relatively little genetic variability yet cause variable clinical outcomes from moderate to severe pathology, including ARDS (39, 41, 47, 48, 51). Therefore, they are well-suited to understand host and viral contributions to IAV pathogenesis. A detailed discussion of the viral replicative cycle is beyond this review, yet excellent reviews are plentiful [e.g., Ref. (52–54)]. IAV is a well-studied model for virus infection in laboratory animals, such as mice and ferrets, and much is known about the contributions of viral and host determinants to severe disease (55–58). Retrospective and experimental infection studies routinely demonstrate common occurrences in the formation of severe IAV [including severe influenza pneumonia (SIP) and ARDS] in humans, ferrets, and mice; these include increased cytokine secretions in the lung, diffuse alveolar damage (“DAD,” bronchointerstitial pneumonia in veterinary pathology), and neutrophilic infiltration (29, 55, 58–66).
In general, IAV infection is an excellent model to investigate the respiratory system’s immune response to viral infection, specifically the pathway leading to severe pneumonia and/or ARDS. Influenza disease is commonly relatively mild and self-limiting, although highly pathogenic forms exist (42, 59, 67–72). The major complication from IAV infection is the formation of SIP which may develop into ARDS (59, 65, 67, 68, 70–73). The reason(s) why infection with IAV may lead to severe viral pneumonia and ARDS is poorly understood, but is thought to involve both host and viral factors. The respective and combined contributions of the host innate immune response and viral factors to the timing and severity of SIP are poorly understood. Neutrophils are present in the respiratory tract during infection with mild seasonal IAV, SIP, and highly pathogenic avian influenza viruses [“HPAI,” which includes avian influenza A (H5N1) virus] (50, 51, 65, 67, 68, 74–76). During SIP and HPAI infection, an increase in the number of neutrophils in the lower respiratory tract (LRT) is correlated with disease severity (50, 51, 65, 67, 68, 76).
Although clinical pathology suggests that a spectrum of disease results from IAV infection, there are at least three disease “phenotypes” caused by infection with IAV, listed by increasing case fatality rate: a mild upper respiratory tract (URT) infection, a SIP which can lead to ARDS, and a LRT infection which can lead to hypercytokinemia. The virological basis for disease phenotype is related to adaptations to mammals—most important are receptor specificity and efficiency of replication—and the major mechanisms have been defined through the use of experimental animal models (30, 32, 33, 57, 77–80). An “ideal” viral infection (i.e., one that is successful for the virus and non-lethal for the host) may be considered a balance between virus replication and an immune response necessary to promote viral shedding, typical of mild seasonal (“epidemic”) IAV. In general, emergent IAV, directly or indirectly from avian enzootic cycles, have increased pathology in humans, the most fatal form of which is a syndrome of complete immune dysregulation (65, 69, 70, 81–84). IAV is genetically highly variable, and mechanisms for increased disease severity are multifactorial, involving host and viral factors.
The majority of yearly, seasonal IAV infections in the world cause a relatively mild, self-limiting URT disease. Influenza disease is characterized by an abrupt onset fever, myalgia, and malaise, with symptoms similar to other URT infections, such as sneezing, coryza, and rhinorrhea (67, 85). Symptoms can last anywhere from 1–5 days and are clinically indistinguishable from other “flu”-like illnesses, including bacterial and viral infections that cause the common cold [e.g., Streptococcus pneumoniae, Haemophilus influenzae, human rhinovirus (HRV) infection, Human respiratory syncytial virus (hRSV) infection, and coronavirus infection] (85–88). Experimental infection of humans with IAV suggests that the virus is mainly restricted to the URT, although sampling the LRT is difficult (67, 68, 87, 89). While fever typically begins 2 days postinfection, virus is shed from the URT in nasal secretions as quickly as 24 h postinfection, allowing efficient transmission prior to symptom onset and continues until 4–5 days postinfection (86, 87, 89) (Figure 2). Rhinorrhea is coincident with neutrophilic rhinitis and shedding of necrotic nasal epithelium (67, 90, 91). Surprisingly, the LRT seems to be involved in uncomplicated IAV infection, although this observation is frequently overlooked or unaddressed in studies (68, 92). In humans, local and systemic concentrations of IL6, CXCL8/IL8, and MCP1/CCL2 correlate with increased disease severity (i.e., symptom severity and increased virus shedding) (87–89, 93) (Figure 2).
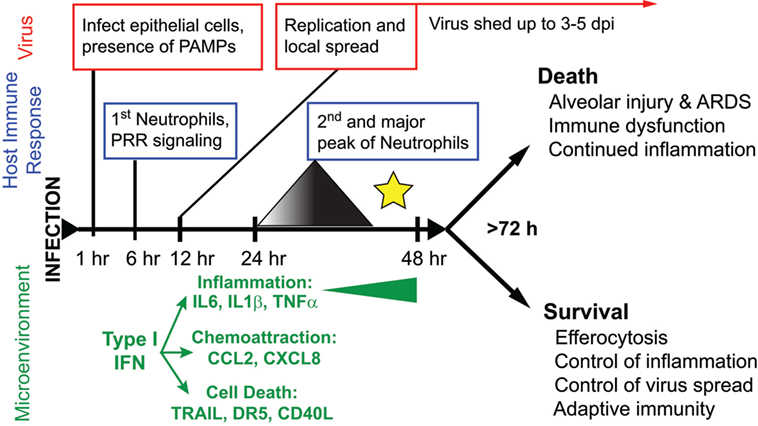
Figure 2. The course of disease following influenza A virus (IAV) infection. A timeline depicting major events in the viral replication cycle (red), the host immune response (blue), and the effects on the host tissue environment (green) during an IAV infection of the airways. A star marks the critical point for the formation of severe disease versus recovery from infection. This review posits that at this timepoint, coincident with a second wave of increasing neutrophilia and inflammation, the outcome of disease is determined.
Influenza A(H1N1)pdm09 virus spread quickly throughout the globe, much like previous pandemic viruses, such as the 1918 H1N1 “Spanish flu” IAV. Humans infected with influenza A(H1N1)pdm09 virus also presented with typical flu-like symptoms (e.g., fever, cough); however, there was an increased number of cases presenting with dyspnea, respiratory distress, and pneumonia (36–38, 40–48, 50, 94, 95). Additionally, retrospective assessments show a proportionately greater number of adolescents and adults with severe disease compared to typical seasonal influenza, and patients with comorbidities, such as obesity and asthma, were at higher risk of severe infection (51, 96–98). In general, the virus causes infection of URT, as well as bronchitis and bronchiolitis, and a high proportion of cases presented with severe disease in the form of viral pneumonia (42, 51, 96). Histopathologic changes in autopsies revealed extensive cytonecrosis, desquamation, and inflammatory infiltration of the bronchus and trachea, mild to severe necrotizing bronchiolitis (42, 50, 51, 99). The primary pathologic finding of SIP was sporadic to DAD with hyaline membrane formation, edema, and occasionally hemorrhage (42, 50, 51, 99). As is typical of influenza infections, some patients experienced bacterial coinfection although this was not in a majority of patients, including those dying from ARDS (42, 48, 50, 51, 93, 99–103). This may distinguish the influenza A(H1N1)pdm09 virus from the 1918 H1N1 IAV, for which bacterial superinfection was determined to cause a majority of the deaths (104, 105), although this may more accurately reflect improved hygiene and standard of care. As discussed below, studies of the reconstructed 1918 H1N1 IAV using animal models suggest that this virus was highly pathogenic irrespective of secondary bacterial pneumonia (62, 106–108).
Across many cohorts of clinical patients, serum concentration of IL6, CCL2, and CXCL8 were significantly elevated in severe cases of influenza A(H1N1)pdm09 viral pneumonia compared to patients with other confirmed illnesses including seasonal IAV, milder forms of influenza A(H1N1)pdm09 virus, bacterial pneumonia, or other viral respiratory infection [hRSV, HRV, human adenovirus (hAdv)] (93, 100–102, 109). These cytokines and chemokines remained elevated over time (up to 6 days following hospital admission) in cases of severe influenza A(H1N1)pdm09 viral pneumonia, whereas they decreased as patients recovered from seasonal and mild influenza A(H1N1)pdm09 virus infection (93, 100). In severe cases of influenza A(H1N1)pdm09 virus infection, decreased type I IFN and ISG production was occasionally noticed compared to adult patients with seasonal IAV infection (93). The influenza A(H1N1)pdm09 viruses have received much scrutiny, and a large dataset of the genetics and pathogenic phenotypes of virus isolates exists in human and animal models. The H1N1 subtype IAV are highly important viruses due to their pandemic potential, as supported by the historical record (62, 68, 110). Some influenza A(H1N1)pdm09 viruses can infect the LRT in humans and in the ferret animal model, which makes them excellent laboratory viruses to investigate the involvement of the LRT in pathogenesis, specifically in the development of severe disease (42, 111–115). Their overall genetic similarity makes for excellent comparison studies between natural clinical isolates, and reverse genetics systems exist to study molecular pathogenicity (41, 95, 113, 116–126).
Avian IAV has been recorded sporadically entering the human population over the last 20 years, beginning with an H5N1 subtype virus which emerged in China in 1997 (70, 82, 127, 128). It is likely that this avian influenza A (H5N1) virus and other emergent strains result from contact with infected domestic poultry that are infected with HPAI (82). The disease caused by avian influenza A (H5N1) virus is characterized by DAD, alveolar necrosis, and alveolar hemorrhage [human disease, including pathology, is reviewed in Ref. (83)]. There is evidence of viremia and systemic spread; IAV antigen has been detected in the trachea, bronchi, and alveolar pneumocytes (69, 76), as well as infrequently in the brain and gastric epithelium (59, 76, 83). The innate cellular immune response in the lungs was characterized by an increase in inter-alveolar macrophages/histiocytes (59, 65, 69, 71, 81, 129) and only moderate infiltration of lymphocytes and neutrophils in the few patients that were analyzed postmortem (69, 76). Systemically, patient serum had high concentrations of CXCL10, CCL2, IL6, IL8, and IL10 compared to matched-control patients with seasonal H3 and H1 IAV, and these concentrations were correlated with viral load in throat (59, 65, 69). In lethal cases, the result of infection and immune dysregulation led to multiple organ failure (e.g., kidney tubular inflammation, necrotic lesions in brain, impaired liver function) and abnormal clotting. Reactive histiocytes undergoing hemophagocytosis were frequently found in bone marrow and lungs of patients, which is indicative of diseases involving hypercytokinemia (59, 65, 70, 81, 83, 130).
Other events involving avian IAV transmission to humans are known and are often associated with veterinary or other animal workers; for example, a 2004 case of avian influenza A (H7N7) virus infection in a veterinarian in Europe showed severe fatal pneumonia and DAD (131). It was reported that 1 L of serosanguineous fluid was drained from his chest upon autopsy. In 2013, another avian IAV emerged in Southeast Asia; this time an avian influenza A (H7N9) virus (71, 72, 129, 132). The histopathology was similar to avian influenza A (H5N1) virus: severe pneumonia, DAD, and epithelial necrosis were common features of infection with both viruses (71, 72). Therefore, it seems the typical presentation of human patients infected with either virus includes high levels of CXCL10, CCL2, IL-6, and CXCL8 in the plasma, peripheral blood leukopenia, and lung neutrophilia (65, 69, 81, 129, 130, 132), and this is also similar to experimental infection of laboratory animal models (111, 133). There were slightly more bacterial coinfections in cases of H7N9 compared to H5N1 avian IAV (71, 72, 81). In a direct comparison, serum from patients with infected with avian influenza A (H5N1) virus had higher concentrations of IFNα and IFNγ in the blood and lower levels of IL8, whereas the opposite was true for patients with avian influenza A (H7N9) virus (134). Similarly, CXCL9 and CXCL10 were higher in patients with avian influenza A (H5N1) virus, whereas CCL4 concentrations were higher in patients with avian influenza A (H7N9) virus (134). Infection with either virus resulted in higher blood C-reactive protein (CRP) (129). Variability between patients may account for the apparent discrepancies between specific immune responses. Therefore, experimental infection of laboratory animals removes individual variability and provides a clear picture of general disease progression, with important caveats for their comparison to human disease as discussed in more detail in the next section (58, 135).
Neutrophils are increased in the lungs and blood after infection with pathogenic IAV in mice, humans, and ferrets (28, 136, 137). Cell depletion studies have demonstrated that neutrophils are necessary for recovery from severe, but not mild, IAV infection (29, 138, 139). Studies in mice show that neutrophils have effects during both early and late stages of disease (140). As discussed in detail below, initial pathogen sensing through various pathogen recognition receptors (PRRs) stimulates inflammatory signals from resident macrophages to initiate neutrophil chemotaxis to the infected airways (Figure 1). For example, TLR7 recognition of IAV dsRNA- and Myd88-mediated release of TNFα and CCL3 by mononuclear cells is important for neutrophil recruitment to the site of infection (141, 142). Transgenic mice have been used to study the contribution of specific cytokines and chemokines to inflammation following IAV infection, particularly as this relates to “hypercytokinemia,” and are summarized in Tables S1–S4 in Supplementary Material. The signals from the infected lung are propagated systemically by endothelial cells, which recruit and tether neutrophils. The importance of endothelial signaling in the development of severe disease has been shown recently using sphingosine-1 phosphate agonist to prevent severe disease in animal models of both influenza virus and respiratory syncytial virus (143, 144). The complex interactions governing neutrophil extravasation, migration through the interstitium, and crossing the alveolar epithelium are well known in relation to many forms of ALI and the development of ARDS with the exception of conditions surrounding viral infection, although mechanistically they should be quite similar (3–5, 7, 145, 146).
In the ferret model, the migration of neutrophils to the lungs occurs in two distinct waves: a first wave within hours of challenge, peaking after 24 h then decreasing; and a second wave that increases over time until disease resolution or death (111) (Figure 2). In the ferret model, we have shown that the neutrophils become concentrated at specific foci in the lungs coincident with influenza-positive epithelium and the expression of chemoattractant chemokine genes (22). Neutrophil chemotaxis in humans is thought to be mediated by many factors, such as the chemokine CXCL8, cytokines IL-1 and TNFα, and complement C5a (145, 147, 148). During both mild and severe IAV disease, patients show increased blood CRP and activation of C5a (149), as well as increased secretion of CXCL8, TNFα, and IL-1 in nasal washes, which correlate with disease severity (87, 89, 93, 150–152). In the mouse model of influenza virus infection, chemical reduction of C5a during IAV infection reduced lung neutrophilia (153). Similarly, knockout mice deficient in the inflammasome pathway or mice not expressing cytokines, such as IL-1b and IL-6, have decreased neutrophil activation and migration to the lungs during IAV infection (Table S1 in Supplementary Material) (154–156). Mice do not possess CXCL8, but CXCL1 and CXCL2 have equivalent functions. Neutrophils contribute CXCL2 to the IAV-infected mouse lung to further stimulate neutrophil recruitment (157). More recently, it was shown that removing a CXCL1 repressor (Setdb2) does not increase recruitment of neutrophils to the lungs of mice infected with IAV PR8, rather it reduces the ability to respond to bacterial superinfection (158). In addition, it was shown that another ISG, CXCL10, operates on a unique subset of CXCR3+ neutrophils present during mouse IAV infection in an autocrine manner, increasing chemotaxis, oxidative burst, and enhancing inflammation (157) (Tables S3 and S4 in Supplementary Material). Finally, aryl hydrocarbon receptor is somehow linked to increases in NO and neutrophilia in the lungs of IAV-infected mice independently of known neutrophil chemoattractants or mechanisms of neutrophil extravasation (159–162).
In cell coculture, human neutrophils were seen to interact specifically with IAV-infected cells (163), although the nature of this interaction in the infected lung is unknown. Neutrophils are phagocytic cells, and their methods for sensing extracellular pathogens rely on TLRs (2, 147). Stimulation of neutrophils through cell surface TLRs has been recorded to promote cytokine secretion (CXCL8 and TNFα via NFκB and AP-1), formation of reactive oxygen species (ROS), phagocytosis, granule secretion, neutrophil extracellular trap (NET) formation, and migration (145, 147, 148). Human neutrophils highly express nucleic acid-detecting TLRs, specifically endosomal TLR8 (164, 165), but do not express nor respond to activators of TLR3 or TLR7 (164–166). [Interestingly, TLR3−/− mice have increased neutrophilia and fewer macrophages in the lungs, yet have increased survival after infection with IAV (167, 168).] TLR4 is required for LPS-induced neutrophil migration to the lung (169) and can stimulate immunostimulatory responses via TRIF adaptors (170); however, TLR4-stimulation does not lead to the production of type I IFN in neutrophils (166, 171).
Several innate immune effector proteins with opsonizing functions that are present in airway mucosae are known to interact with both IAV and neutrophils. Surfactant protein D, a lung collectin, is an innate immune defense against a variety of viruses, opsonizing the viruses for phagocytosis by neutrophils, which in turn causes the production of ROS (172–178). Human neutrophil defensins are short basic peptides released from neutrophil granules during inflammation (145, 178, 179). They have been shown to interact with IAV, reducing infectivity, and promote neutrophil phagocytosis and clearance of IAV (178–182). Defensins may also buffer the oxidative burst from neutrophils that follows from phagocytosis of viruses that have been opsonized by surfactant protein D (178–180). One study indicated that neutrophils do not interact with immunoglobulin-bound IAV (183); however, another showed that protective anti-IAV antibody therapy only protected mice in the presence of neutrophils (184). The hypothesis that IAV pathogenicity can be partially explained by infection with viral variants that can evade opsonization by innate immune effectors is attractive and deserves further study (176).
Neutrophils express sialic acid receptors and may become infected with IAV (74, 185, 186). Neutrophils infected with IAV have increased apoptosis, but infection does not result in the production of virus (186). Infection of human neutrophils with IAV treated at 56°C to denature the viral replicase but not HA suggested that infection alone, but not replication, is sufficient to stimulate the release of CXCL8 and CCL4 in human neutrophils (187). Neutrophils infected with IAV have rapid upregulation (<9 hpi) of type I IFN pathways, including cytoplasmic PRRs, IFNβ, and ISGs (186), which is counter to the long-held dogma that neutrophils were incapable of gene expression. This is may be due to RIG-like receptors (RLRs) sensing of viral dsRNA, as neutrophils transfected with poly(I:C) (a viral RNA mimic) have a similar response (166). Additionally, neutrophils express nod-like receptors but it is unclear how these interact with IAV infection (188, 189). In general, inflammasome and pro-IL1 activation following IAV infection is poorly understood (154, 155, 190, 191). However, studies of IAV infection using caspase-1, IL-1β, or IL-1R transgenic mice show modulation of neutrophil infiltration and pathology and suggest that it is an IAV subtype-dependent effect (154, 191–195) (Tables S1 and S2 in Supplementary Material). Furthermore, it has been demonstrated that the HA of some IAV isolates suppresses neutrophil activation, providing further evidence for IAV subtype-dependent effects on neutrophils (185, 186, 196).
Activation of TLRs and RLRs trigger degranulation and the expression of surface CD11b (166), which pairs with CD18 to form the “Mac-1” integrin dimer that binds collagen (197). This facilitates migration through tissues, and release of gelatinase or collagenase (MMP-2 and MMP-9) from neutrophils assist in clearing connective tissue from the path. At the site of infection, neutrophils release microbial effectors [reviewed in Ref. (145, 198)]. Neutrophils develop granules sequentially (azurophilic, specific, gelatinase, secretory) and secrete granules in the reverse order (145). Secretory and gelatinase granules are released shortly after endothelial transmigration and contain membrane proteins essential for movement [extracellular matrix (ECM)-binding integrins] and pathogen recognition [immunoglobulin (FcR) and complement receptors] (145). Specific and azurophilic granules contain tissue-destroying enzymes and antimicrobial proteins. For example, neutrophil myeloperoxidase (MPO) may contribute to lung injury during IAV infection (199); however, it may have direct antiviral effects on IAV (200). An investigation found no difference between IAV infection of a wild-type and neutrophil elastase knockout mouse, measuring lung function, chemokine secretion, and neutrophil recruitment (201) (Table S5 in Supplementary Material). The contents of the granules can be secreted to destroy ECM (such as MMP-9) or directed toward phagosomes to destroy engulfed microbes. The production of hypochlorous acid (HOCl) is the main oxidant used in phagosomal killing, and its production is dependent on the generation of ROS by the neutrophils. Interestingly, infection by IAV was related to the inhibition of phagosomal killing of bacteria (196).
Neutrophil degranulation primes neutrophils for ROS generation by mobilizing NADPH oxidase components to the plasma membrane (145) and exocytosis of MPO. IAV infection causes the generation of ROS in neutrophils (202). Oxidative burst is thought to have direct microbial effects; however, the direct effect on IAV has not been published (203). IAV infection benefits from the presence of ROS in the environment (204, 205), yet IAV also suppresses NADPH oxidase activity within infected phagocytes (206, 207). Many have investigated the effects of ROS and NO on lung inflammation during IAV infection and found that reduction of oxidative stress in the form of both ROS and NO alieves IAV-dependent lung injury (206, 208–213) (Table S5 in Supplementary Material). For example, oxidized lipids in the lung environment may trigger TLR4, activating immune cells and contributing to increased lung injury (214–216).
Neutrophils undergo a form of programmed cell death called netosis, in which NETs are formed (217). NETs are extracellular strands DNA wrapped in histones and enriched in neutrophil effector proteins (e.g., neutrophil elastase and MPO) (218). NETs have the effect of killing many pathogens, including bacteria (146), fungi (219), protozoans (220), and more recently viruses (221). NETs are becoming the focus of study in autoimmune disease atherosclerosis, since they damage endothelium (222–224). Recently, it was shown that hantavirus stimulates NET production during infection, which leads to the generation of autoantibodies and may provide a mechanism for the hemorrhagic fever caused by Old World hantaviruses (225). NETs are typically found with histones, MPO, and neutrophil elastase, and the effect is to isolate the effects of these molecules directly onto the pathogen surface with a “sticky” NET of nucleic acid. NETs contribute to acute lung injury and alveolar capillary damage during IAV infection (139). Yet, very little is known about the relationship between NETs and viral infection in viral disease pathogenesis (226).
In summary, neutrophils are capable of recognizing viruses via PRRs as either opsonized virions or via endosomal TLR. Although the signaling cascade differs from other phagocytic cells, neutrophils are capable of responding to viral PAMPs with respiratory burst, degranulation of proteases and cytokines, and/or netosis. It is not clear if these responses are effective against influenza virus; in fact, evidence exists that suggest influenza viruses may take advantage of the inflammatory environment. More importantly, it is not clear if there are differences in response to different influenza virus subtypes or strains. As is true for many respiratory etiologies, neutrophil responses must be balanced during influenza virus infection to adequately control of inflammation while promoting pro-immune responses. The timing of neutrophils during disease progression correlates with a key point in divergent disease outcomes (Figure 2), and neutrophils may act both globably and locally at foci of infection (22, 111). Thus, neutrophils are focused in the airways at critical timepoints following infection and therefore balancing their potent inflammatory effector functions may determine disease outcome.
As discussed above, there appears to be a correlation between the timing and location of IAV infection and the action of neutrophils, but evidence directly linking these phenomena together remains overall circumstantial. In contrast, during bacterial pneumonia there is direct evidence of the importance of neutrophils in disease: bacterial PAMPs directly upregulate neutrophil activating and chemoattractant chemokines, bacteria have defined anti-neutrophil functions, and some bacteria, e.g., Mycobacterium tuberculosis, rely on neutrophils to establish their granulomatous niche (6, 227, 228). Similarly, we suggest that viruses may interact with respiratory cells to create a viral microenvironmental niche. To further substantiate a link between neutrophils and virus infection, evidence for the role of neutrophils in selected viral respiratory diseases is summarized in Tables 1 and 2. Discussion below focuses on common patterns of neutrophil responses in severe and mild forms of respiratory viral infection.
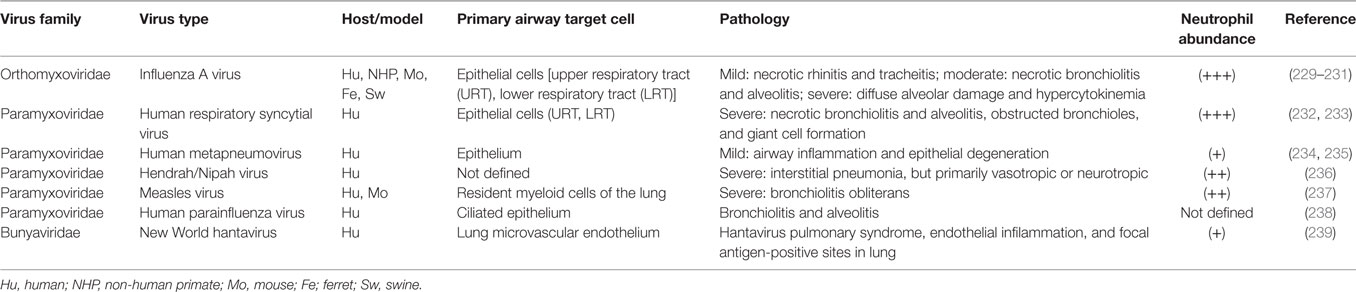
Table 1. (−) Sense RNA respiratory viruses that cause increased neutrophil infiltration during infection.
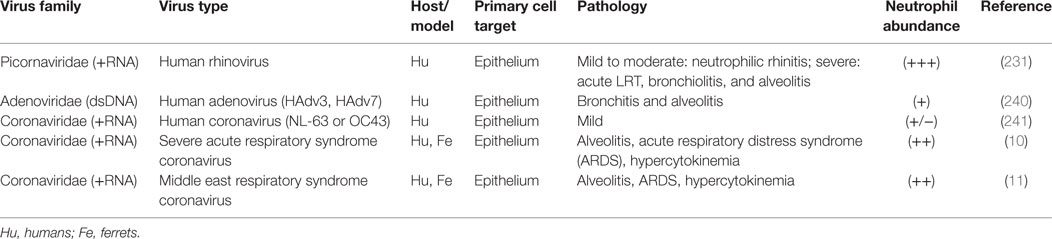
Table 2. (+) Sense RNA and DNA respiratory viruses that cause increased neutrophil infiltration during infection.
For both viral and bacterial etiologies, the most severe clinical complications result from infection of the LRT. Infection of the LRT by viruses, such as human parainfluenza viruses, influenza A(H1N1)pdm09, HPAI, New World hantavirus infections (causing hantavirus pulmonary syndrome), Severe acute respiratory syndrome-related coronavirus (SARS-Cov), and Middle East respiratory syndrome coronavirus (MERS-CoV), are all associated with neutrophilic infiltration at sites of infection to various degrees and may develop into ARDS (9–11, 19, 60, 131, 242–245). Clinically defined, ARDS has three phases; and most patents die within the first phase, the acute or “exudative” phase [reviewed in Ref. (12–14)]. This phase is characterized by an increased immune response with high production of pro-inflammatory cytokines and chemokines, increased neutrophil infiltration and accumulation in the alveoli, and disruption of the alveolar epithelial–capillary barrier, which leads to increased vascular permeability and edema (13). The distinctive role for lung neutrophil infiltration in viral infection is summarized as follows: some, but not all, viruses that infect the LRT result in clinically defined ARDS, and lung neutrophil infiltration is associated with viruses that do and do not lead to ARDS (3, 5, 9, 10, 60, 131, 199, 242–244, 246, 247). In ARDS, there are data that directly support the role of neutrophils as both beneficial and detrimental (3, 13, 14). Perhaps, there are general factors (host or virus) that lead to a common antiviral response of neutrophils.
In contrast to emergent highly pathogenic respiratory viruses, notable “mild” human respiratory viruses also involve increased neutrophils at the site of infection (e.g., hRSV). As expected, infection with these viruses is typically associated with the increase of neutrophil chemoattractant chemokines. For example, infection with HRV is a well-studied virus for which there are several studies on the link between neutrophils and disease (229–231, 248) (Table 2). HRV virions enter nasal epithelial cells via endocytosis, yet, unlike influenza, infection does not cause major damage to the nasal epithelium (249–251). Neutrophilic rhinitis, increased vascular permeability, and mucus hypersecretion are the key pathological features of HRV infection (229–231, 249), and infected epithelium seems to be the source of large amounts of neutrophil chemotactic molecules, particularly CXCL8 and kinins (229, 248, 252, 253). Interestingly, in vitro studies have shown that viral recognition of HRV shares features with hRSV, but is somewhat different than with IAV (254–256). It has been established that there are virus-specific and cell-specific differences in sensing RNA viruses via primarily TLR- and/or RLR-pathways (and even in a preference for RIG-I versus MDA5), yet these pathways may have similar general endpoints, such as chemokine and cytokine signaling (255, 257–259).
Finally, a key question is whether virus-induced cytopathy drives neutrophilia or whether it is the result of host response to viral infection. Infections with influenza A(H1N1)pdm09 virus, HPAI, SARS-CoV, and MERS-CoV are thought to cause acute lung injury which results in ARDS; characterized by excessive damage to the alveolar epithelium and involving the infiltration of neutrophils (67, 68) (Tables 1 and 2). However, less pathogenic strains such as HRV infections do not cause significant damage to the respiratory mucosa, yet neutrophils are present (249, 250). Conversely, viruses that cause moderate, focal cytopathy in the lungs, for example seasonal IAV and hRSV, are known to cause neutrophilic infiltration (250, 252, 253). Therefore, neutrophils are not necessarily associated with direct cytopathic effect nor are they exclusively associated with severe disease.
Despite many years of searching, there is no single reliable biomarker to indicate a bacterial versus viral infection (although CRP is a good candidate). This is surprising, given the otherwise significant fundamental differences between the biology of these two types of pathogens; differences which are reflected in general immune responses and begin with pathogen detection. Signaling through TLR on the plasma membrane versus endosomal or cytoplasmic PRRs is controlled by complex intracellular adaptor proteins [reviewed in Ref. (260)]. For example, the complexities of signaling allow the characteristically bacteria-specific TLR4 to signal the upregulation of immunostimulatory type I IFN characteristic of a virus infection (170). As evidenced above, many viral infections associated with neutrophil infiltration have RNA genomes. Host cells detect RNA viruses primarily through RLR as well as TLR, whereas bacteria rely on a different group of PRRs to detect extracellular PAMPs (255, 261–263). Interestingly, the lung microenvironment to hRSV has been shown to be different from IAV, specifically in the presence of IL-4 (264, 265). It is thought that this is driven by the presence of alternatively activated macrophages during RSV infection (266). It is not known if this directs differences in neutrophil chemoattraction, yet IL-4 is known to drive a Th2 (“bacterial” or antibody-biased) immune response (267). In sum, surprisingly little is clinically different between the innate immune responses to viral versus bacterial infection; however, perhaps comparative studies that focus on neutrophils can uncover virus-specific responses.
In general, immune activation pathways that involve the activation of NFκB lead to the secretion of neutrophil chemotactic chemokines [reviewed in Ref. (268)]. This is heavily driven by PAMP recognition and activation pathways, and during a viral infection type I IFNs and ISGs are the unique elements in the virus-inflamed lung environment [reviewed in Ref. (24–26, 269, 270)]. This single difference between viral and bacterial infections could have drastic effects on the actions of neutrophils once in the lung. Moreover, without this information (e.g., pathogenic viruses that suppress type I IFN) neutrophils may respond to inflammation in an inefficient way, potentially with pathologic effects (23, 186, 256, 271–277). Neutrophils are known to respond to IAV and type I IFN by upregulation of ISGs (186). In systemic lupus erythematosus, neutrophils may be a large contributor of type I IFN (278, 279). During increased inflammation, left-shifted or immature neutrophils emerge from the bone marrow—a classic sign for sepsis, but also known to be present during some severe viral respiratory diseases (e.g., HPS) (239). It is known that immature neutrophils do neither express IFNα/β receptor nor many other cytokine receptors (273). It is unknown what affect this would have during increased inflammation during pathogenic influenza infection, although their role in other inflammatory conditions suggests it may affect their functions (271, 273, 280, 281).
All forms of respiratory infection require resolution of the infection and inflammation. IFNs are essential components of initiating sterilizing immunity to virus infection via the adaptive immune system (i.e., resolution of infection), at which point the resolution of inflammation can effectively proceed (25, 26, 282). Although the mechanisms are poorly understood, through their direct antiviral actions and indirect actions on the lung microenvironment (e.g., efferocytosis of apoptotic neutrophils by macrophages), neutrophils have the ability to influence outcomes toward successful resolution as well as toward the formation of ARDS (3, 5, 12, 13, 21, 74, 166, 184, 198, 199, 283–285). Thus, there is evidence that the role of neutrophils in viral infections of the respiratory system is not limited to inflammation, but likely includes recovery from infection and the initiation of adaptive immunity (74, 138, 284). Apart from initiating adaptive immunity, resolution of inflammation may be partially regulated by secretion of IL-1RA and chemokine-destroying factors by recruited macrophages (286, 287). Additionally, it has been proposed that efferocytosis of apoptotic neutrophils is a key step in resolution of inflammation (288–290), and occurs in the lung during bacterial pneumonia (291). It is unclear if this happens during IAV infection or infection with other respiratory viruses.
Factors that prolong the life span of neutrophils in the lungs increase the probability that they may contribute to immunopathology. IL-6 and G-CSF are immune mediators present in the lung during infection and are known to prolong survival of neutrophils in mouse lungs following IAV infection (292). Both neutrophils and macrophages are known to phagocytose apoptotic epithelial cells in mouse lungs during IAV infection (283). The cells may be recruited via chemokines or damage receptors. For example, necrotic IAV-infected epithelial cells are a source of CXCL8 (293), which attract neutrophils to dying cells. In addition, neutrophils can detect DAMPs such as S100A9 (294). It has been shown that extracellular S100A9 is abundant during IAV infection in mice (295). Antibody-mediated neutralization of S100A9 decreased lung inflammation in mice and improved disease outcome (295). Apart from potential tissue-destroying effects of neutrophil proteases, the presence of NETs may induce even more inflammation in the lungs (21, 139). Thus, there are limited data supporting directly malevolent actions of neutrophils (Table S5 in Supplementary Material), yet factors that increase their presence and prolong their survival in the lung are correlated with increased disease severity.
There are substantial data that suggest neutrophils are a part of a viral response to infection. Neutrophils are among the first responders to IAV infection in the lung, and they remain in great numbers throughout the development of ARDS. Although neutrophils are an important component of the general response to infection in the respiratory system, as is discussed herein, neutrophils are capable of recognizing viruses (via viral PAMPs), responding to viruses with specific effector functions, and may be instrumental in determining disease outcome. Evidence exists to support the hypothesis that neutrophils respond specifically to the focal nature of viral infection, and they act to influence this microenvironment via their virus-specific effector functions. Factors that influence successful recovery from respiratory viral infection (versus lethal outcome) are complex and both host- and virus-specific. However, a better understanding of the role neutrophils, previously underappreciated with respect to viral infections, will reveal important information about disease outcome. Many questions remain before it is determined the part neutrophils play in mild and severe disease.
Both authors contributed to the formulation of this scientific research topic. JC wrote the manuscript as part of his Ph.D. dissertation under the mentorship of CJ (296).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful for the helpful discussions provided by Silvia M. Uriarte, Ph.D. in pursuit of this topic of research.
Financial support was provided in part by the Commonwealth of Kentucky as a Clinical and Translational Science Pilot Project Program at the University of Louisville to CJ. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00550/full#supplementary-material.
1. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev (2016) 273:11–28. doi: 10.1111/imr.12440
2. Galani IE, Andreakos E. Neutrophils in viral infections: current concepts and caveats. J Leukoc Biol (2015) 98:557–64. doi:10.1189/jlb.4VMR1114-555R
3. Abraham E. Neutrophils and acute lung injury. Crit Care Med (2003) 31:S195–9. doi:10.1097/01.CCM.0000057843.47705.E8
4. Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev (2003) 83:309–36. doi:10.1152/physrev.00023.2002
5. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med (2011) 17:293–307. doi:10.2119/molmed.2010.00138
6. Lowe DM, Redford PS, Wilkinson RJ, O’Garra A, Martineau AR. Neutrophils in tuberculosis: friend or foe? Trends Immunol (2012) 33:14–25. doi:10.1016/j.it.2011.10.003
7. McDonald B, Kubes P. Cellular and molecular choreography of neutrophil recruitment to sites of sterile inflammation. J Mol Med (Berl) (2011) 89:1079–88. doi:10.1007/s00109-011-0784-9
8. Smith PK, Wang SZ, Dowling KD, Forsyth KD. Leucocyte populations in respiratory syncytial virus-induced bronchiolitis. J Paediatr Child Health (2001) 37:146–51. doi:10.1046/j.1440-1754.2001.00618.x
9. Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY, et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet (2003) 361:1773–8. doi:10.1016/S0140-6736(03)13413-7
10. Tse GM, To KF, Chan PK, Lo AW, Ng KC, Wu A, et al. Pulmonary pathological features in coronavirus associated severe acute respiratory syndrome (SARS). J Clin Pathol (2004) 57:260–5. doi:10.1136/jcp.2003.013276
11. van den Brand JM, Smits SL, Haagmans BL. Pathogenesis of Middle East respiratory syndrome coronavirus. J Pathol (2015) 235:175–84. doi:10.1002/path.4458
12. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest (2012) 122:2731–40. doi:10.1172/JCI60331
13. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med (2000) 342:1334–49. doi:10.1056/NEJM200005043421806
14. Abraham E, Matthay MA, Dinarello CA, Vincent JL, Cohen J, Opal SM, et al. Consensus conference definitions for sepsis, septic shock, acute lung injury, and acute respiratory distress syndrome: time for a reevaluation. Crit Care Med (2000) 28:232–5. doi:10.1097/00003246-200001000-00039
15. Aherne W, Bird T, Court SD, Gardner PS, McQuillin J. Pathological changes in virus infections of the lower respiratory tract in children. J Clin Pathol (1970) 23:7–18. doi:10.1136/jcp.23.1.7
16. Craig A, Mai J, Cai S, Jeyaseelan S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun (2009) 77:568–75. doi:10.1128/IAI.00832-08
17. Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol (2011) 2:65. doi:10.3389/fimmu.2011.00065
18. Lien DC, Wagner WW Jr, Capen RL, Haslett C, Hanson WL, Hofmeister SE, et al. Physiological neutrophil sequestration in the lung: visual evidence for localization in capillaries. J Appl Physiol (1985) (1987) 62:1236–43.
19. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med (2005) 353:1685–93. doi:10.1056/NEJMoa050333
20. Reutershan J, Basit A, Galkina EV, Ley K. Sequential recruitment of neutrophils into lung and bronchoalveolar lavage fluid in LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol (2005) 289:L807–15. doi:10.1152/ajplung.00477.2004
21. Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe (2013) 13:169–80. doi:10.1016/j.chom.2013.01.005
22. Camp JV, Bagci U, Chu YK, Squier B, Fraig M, Uriarte SM, et al. Lower respiratory tract infection of the ferret by 2009 H1N1 pandemic influenza A virus triggers biphasic, systemic, and local recruitment of neutrophils. J Virol (2015) 89:8733–48. doi:10.1128/JVI.00817-15
23. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun (2014) 5:3864. doi:10.1038/ncomms4864
24. Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science (2006) 312:879–82. doi:10.1126/science.1125676
25. Le Bon A, Tough DF. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol (2002) 14:432–6. doi:10.1016/S0952-7915(02)00354-0
26. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity (2006) 25:373–81. doi:10.1016/j.immuni.2006.08.007
27. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev (2012) 76:16–32. doi:10.1128/MMBR.05015-11
28. Watanabe T, Tisoncik-Go J, Tchitchek N, Watanabe S, Benecke AG, Katze MG, et al. 1918 Influenza virus hemagglutinin (HA) and the viral RNA polymerase complex enhance viral pathogenicity, but only HA induces aberrant host responses in mice. J Virol (2013) 87:5239–54. doi:10.1128/JVI.02753-12
29. Vidy A, Maisonnasse P, Da Costa B, Delmas B, Chevalier C, Le Goffic R. The influenza virus protein PB1-F2 increases viral pathogenesis through neutrophil recruitment and NK cells inhibition. PLoS One (2016) 11:e0165361. doi:10.1371/journal.pone.0165361
30. Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev (1992) 56:152–79.
31. Webster RG. Predictions for future human influenza pandemics. J Infect Dis (1997) 176(Suppl 1):S14–9. doi:10.1086/514168
32. Webster RG. Influenza virus: transmission between species and relevance to emergence of the next human pandemic. Arch Virol Suppl (1997) 13:105–13.
33. Webster RG, Shortridge KF, Kawaoka Y. Influenza: interspecies transmission and emergence of new pandemics. FEMS Immunol Med Microbiol (1997) 18:275–9. doi:10.1111/j.1574-695X.1997.tb01056.x
34. Webster RG. Influenza: an emerging disease. Emerg Infect Dis (1998) 4:436–41. doi:10.3201/eid0403.980325
35. Cox NJ, Subbarao K. Global epidemiology of influenza: past and present. Annu Rev Med (2000) 51:407–21. doi:10.1146/annurev.med.51.1.407
36. CDC. Outbreak of swine-origin influenza A (H1N1) virus infection – Mexico, March-April 2009. MMWR Morb Mortal Wkly Rep (2009) 58:467–70.
37. Cao B, Li XW, Mao Y, Wang J, Lu HZ, Chen YS, et al. Clinical features of the initial cases of 2009 pandemic influenza A (H1N1) virus infection in China. N Engl J Med (2009) 361:2507–17. doi:10.1056/NEJMoa0906612
38. Donaldson LJ, Rutter PD, Ellis BM, Greaves FE, Mytton OT, Pebody RG, et al. Mortality from pandemic A/H1N1 2009 influenza in England: public health surveillance study. BMJ (2009) 339:b5213. doi:10.1136/bmj.b5213
39. Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science (2009) 325:197–201. doi:10.1126/science.1176225
40. Jain S, Kamimoto L, Bramley AM, Schmitz AM, Benoit SR, Louie J, et al. Hospitalized patients with 2009 H1N1 influenza in the United States, April-June 2009. N Engl J Med (2009) 361:1935–44. doi:10.1056/NEJMoa0906695
41. Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature (2009) 459:1122–5. doi:10.1038/nature08182
42. Gill JR, Sheng ZM, Ely SF, Guinee DG, Beasley MB, Suh J, et al. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Arch Pathol Lab Med (2010) 134:235–43. doi:10.1043/1543-2165-134.2.235
43. Libster R, Bugna J, Coviello S, Hijano DR, Dunaiewsky M, Reynoso N, et al. Pediatric hospitalizations associated with 2009 pandemic influenza A (H1N1) in Argentina. N Engl J Med (2010) 362:45–55. doi:10.1056/NEJMoa0907673
44. Agarwal PP, Cinti S, Kazerooni EA. Chest radiographic and CT findings in novel swine-origin influenza A (H1N1) virus (S-OIV) infection. AJR Am J Roentgenol (2009) 193:1488–93. doi:10.2214/AJR.09.3599
45. Mollura DJ, Asnis DS, Crupi RS, Conetta R, Feigin DS, Bray M, et al. Imaging findings in a fatal case of pandemic swine-origin influenza A (H1N1). AJR Am J Roentgenol (2009) 193:1500–3. doi:10.2214/AJR.09.3365
46. Neumann G, Noda T, Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature (2009) 459:931–9. doi:10.1038/nature08157
47. Peiris JS, Poon LL, Guan Y. Emergence of a novel swine-origin influenza A virus (S-OIV) H1N1 virus in humans. J Clin Virol (2009) 45:169–73. doi:10.1016/j.jcv.2009.06.006
48. Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, Hernandez M, Quinones-Falconi F, Bautista E, et al. Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. N Engl J Med (2009) 361:680–9. doi:10.1056/NEJMoa0904252
49. Woo PC, Tung ET, Chan KH, Lau CC, Lau SK, Yuen KY. Cytokine profiles induced by the novel swine-origin influenza A/H1N1 virus: implications for treatment strategies. J Infect Dis (2010) 201:346–53. doi:10.1086/649785
50. Calore EE, Uip DE, Perez NM. Pathology of the swine-origin influenza A (H1N1) flu. Pathol Res Pract (2011) 207:86–90. doi:10.1016/j.prp.2010.11.003
51. Shieh WJ, Blau DM, Denison AM, Deleon-Carnes M, Adem P, Bhatnagar J, et al. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am J Pathol (2010) 177:166–75. doi:10.2353/ajpath.2010.100115
52. Fukuyama S, Kawaoka Y. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr Opin Immunol (2011) 23:481–6. doi:10.1016/j.coi.2011.07.016
53. Medina RA, Garcia-Sastre A. Influenza A viruses: new research developments. Nat Rev Microbiol (2011) 9:590–603. doi:10.1038/nrmicro2613
54. Tscherne DM, Garcia-Sastre A. Virulence determinants of pandemic influenza viruses. J Clin Invest (2011) 121:6–13. doi:10.1172/JCI44947
55. Belser JA, Katz JM, Tumpey TM. The ferret as a model organism to study influenza A virus infection. Dis Model Mech (2011) 4(5):575–9. doi:10.1242/dmm.007823
56. Smith H, Sweet C. Lessons for human influenza from pathogenicity studies with ferrets. Rev Infect Dis (1988) 10:56–75. doi:10.1093/clinids/10.1.56
57. Barnard DL. Animal models for the study of influenza pathogenesis and therapy. Antiviral Res (2009) 82:A110–22. doi:10.1016/j.antiviral.2008.12.014
58. Belser JA, Szretter KJ, Katz JM, Tumpey TM. Use of animal models to understand the pandemic potential of highly pathogenic avian influenza viruses. Adv Virus Res (2009) 73:55–97. doi:10.1016/S0065-3527(09)73002-7
59. de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med (2006) 12:1203–7. doi:10.1038/nm1477
60. van den Brand JM, Haagmans BL, van Riel D, Osterhaus AD, Kuiken T. The pathology and pathogenesis of experimental severe acute respiratory syndrome and influenza in animal models. J Comp Pathol (2014) 151:83–112. doi:10.1016/j.jcpa.2014.01.004
61. van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, et al. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol (2007) 171:1215–23. doi:10.2353/ajpath.2007.070248
62. Watanabe T, Kawaoka Y. Pathogenesis of the 1918 pandemic influenza virus. PLoS Pathog (2011) 7:e1001218. doi:10.1371/journal.ppat.1001218
63. Cameron CM, Cameron MJ, Bermejo-Martin JF, Ran L, Xu L, Turner PV, et al. Gene expression analysis of host innate immune responses during lethal H5N1 infection in ferrets. J Virol (2008) 82:11308–17. doi:10.1128/JVI.00691-08
64. Lu X, Tumpey TM, Morken T, Zaki SR, Cox NJ, Katz JM. A mouse model for the evaluation of pathogenesis and immunity to influenza A (H5N1) viruses isolated from humans. J Virol (1999) 73:5903–11.
65. To KF, Chan PK, Chan KF, Lee WK, Lam WY, Wong KF, et al. Pathology of fatal human infection associated with avian influenza A H5N1 virus. J Med Virol (2001) 63:242–6. doi:10.1002/1096-9071(200103)63:3<242::AID-JMV1007>3.0.CO;2-N
66. Zitzow LA, Rowe T, Morken T, Shieh WJ, Zaki S, Katz JM. Pathogenesis of avian influenza A (H5N1) viruses in ferrets. J Virol (2002) 76:4420–9. doi:10.1128/JVI.76.9.4420-4429.2002
67. Kuiken T, Taubenberger JK. Pathology of human influenza revisited. Vaccine (2008) 26(Suppl 4):D59–66. doi:10.1016/j.vaccine.2008.07.025
68. Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol (2008) 3:499–522. doi:10.1146/annurev.pathmechdis.3.121806.154316
69. Peiris JS, Yu WC, Leung CW, Cheung CY, Ng WF, Nicholls JM, et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet (2004) 363:617–9. doi:10.1016/S0140-6736(04)15595-5
70. Yuen KY, Chan PK, Peiris M, Tsang DN, Que TL, Shortridge KF, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet (1998) 351:467–71. doi:10.1016/S0140-6736(98)01182-9
71. Gao HN, Lu HZ, Cao B, Du B, Shang H, Gan JH, et al. Clinical findings in 111 cases of influenza A (H7N9) virus infection. N Engl J Med (2013) 368:2277–85. doi:10.1056/NEJMoa1305584
72. Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med (2013) 368:1888–97. doi:10.1056/NEJMoa1304459
73. Rello J, Pop-Vicas A. Clinical review: primary influenza viral pneumonia. Crit Care (2009) 13:235. doi:10.1186/cc8183
74. Hufford MM, Richardson G, Zhou H, Manicassamy B, Garcia-Sastre A, Enelow RI, et al. Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8(+) T cells. PLoS One (2012) 7:e46581. doi:10.1371/journal.pone.0046581
75. Sprenger H, Meyer RG, Kaufmann A, Bussfeld D, Rischkowsky E, Gemsa D. Selective induction of monocyte and not neutrophil-attracting chemokines after influenza A virus infection. J Exp Med (1996) 184:1191–6. doi:10.1084/jem.184.3.1191
76. Gu J, Xie Z, Gao Z, Liu J, Korteweg C, Ye J, et al. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet (2007) 370:1137–45. doi:10.1016/S0140-6736(07)61515-3
77. Chandrasekaran A, Srinivasan A, Raman R, Viswanathan K, Raguram S, Tumpey TM, et al. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotechnol (2008) 26:107–13. doi:10.1038/nbt1375
78. Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature (2012) 486:420–8. doi:10.1038/nature10831
79. de Graaf M, Fouchier RA. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J (2014) 33:823–41. doi:10.1002/embj.201387442
80. Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, et al. Airborne transmission of influenza A/H5N1 virus between ferrets. Science (2012) 336:1534–41. doi:10.1126/science.1213362
81. Chan PK. Outbreak of avian influenza A(H5N1) virus infection in Hong Kong in 1997. Clin Infect Dis (2002) 34(Suppl 2):S58–64. doi:10.1086/338820
82. Claas EC, Osterhaus AD, van Beek R, De Jong JC, Rimmelzwaan GF, Senne DA, et al. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet (1998) 351:472–7. doi:10.1016/S0140-6736(97)11212-0
83. Korteweg C, Gu J. Pathology, molecular biology, and pathogenesis of avian influenza A (H5N1) infection in humans. Am J Pathol (2008) 172:1155–70. doi:10.2353/ajpath.2008.070791
84. Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL, et al. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med (2007) 13:147–9. doi:10.1038/nm1529
85. Hilleman MR. Realities and enigmas of human viral influenza: pathogenesis, epidemiology and control. Vaccine (2002) 20:3068–87. doi:10.1016/S0264-410X(02)00254-2
86. Carrat F, Vergu E, Ferguson NM, Lemaitre M, Cauchemez S, Leach S, et al. Time lines of infection and disease in human influenza: a review of volunteer challenge studies. Am J Epidemiol (2008) 167:775–85. doi:10.1093/aje/kwm375
87. Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J Clin Invest (1998) 101:643–9. doi:10.1172/JCI1355
88. Skoner DP, Gentile DA, Patel A, Doyle WJ. Evidence for cytokine mediation of disease expression in adults experimentally infected with influenza A virus. J Infect Dis (1999) 180:10–4. doi:10.1086/314823
89. Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol (2001) 64:262–8. doi:10.1002/jmv.1045
90. Francis T, Stuart-Harris CH. Studies on the nasal histology of epidemic influenza virus infection in the ferret: I. The development and repair of the nasal lesion. J Exp Med (1938) 68:789–802. doi:10.1084/jem.68.6.789
91. Haff RF, Schriver PW, Stewart RC. Pathogenesis of influenza in ferrets: nasal manifestations of disease. Br J Exp Pathol (1966) 47:435–44.
92. Walsh JJ, Dietlein LF, Low FN, Burch GE, Mogabgab WJ. Bronchotracheal response in human influenza. Type A, Asian strain, as studied by light and electron microscopic examination of bronchoscopic biopsies. Arch Intern Med (1961) 108:376–88. doi:10.1001/archinte.1961.03620090048006
93. Lee N, Wong CK, Chan PK, Chan MC, Wong RY, Lun SW, et al. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS One (2011) 6:e26050. doi:10.1371/journal.pone.0026050
94. Glinsky GV. Genomic analysis of pandemic (H1N1) 2009 reveals association of increasing disease severity with emergence of novel hemagglutinin mutations. Cell Cycle (2010) 9:958–70. doi:10.4161/cc.9.5.10913
95. Baillie GJ, Galiano M, Agapow PM, Myers R, Chiam R, Gall A, et al. Evolutionary dynamics of local pandemic H1N1/2009 influenza virus lineages revealed by whole-genome analysis. J Virol (2012) 86:11–8. doi:10.1128/JVI.05347-11
96. Zarychanski R, Stuart TL, Kumar A, Doucette S, Elliott L, Kettner J, et al. Correlates of severe disease in patients with 2009 pandemic influenza (H1N1) virus infection. CMAJ (2010) 182:257–64. doi:10.1503/cmaj.091884
97. Louie JK, Acosta M, Winter K, Jean C, Gavali S, Schechter R, et al. Factors associated with death or hospitalization due to pandemic 2009 influenza A(H1N1) infection in California. JAMA (2009) 302:1896–902. doi:10.1001/jama.2009.1583
98. Nguyen-Van-Tam JS, Openshaw PJ, Hashim A, Gadd EM, Lim WS, Semple MG, et al. Risk factors for hospitalisation and poor outcome with pandemic A/H1N1 influenza: United Kingdom first wave (May-September 2009). Thorax (2010) 65:645–51. doi:10.1136/thx.2010.135210
99. Rosen DG, Lopez AE, Anzalone ML, Wolf DA, Derrick SM, Florez LF, et al. Postmortem findings in eight cases of influenza A/H1N1. Mod Pathol (2010) 23:1449–57. doi:10.1038/modpathol.2010.148
100. Hagau N, Slavcovici A, Gonganau DN, Oltean S, Dirzu DS, Brezoszki ES, et al. Clinical aspects and cytokine response in severe H1N1 influenza A virus infection. Crit Care (2010) 14:R203. doi:10.1186/cc9324
101. Kim YH, Kim JE, Hyun MC. Cytokine response in pediatric patients with pandemic influenza H1N1 2009 virus infection and pneumonia: comparison with pediatric pneumonia without H1N1 2009 infection. Pediatr Pulmonol (2011) 46:1233–9. doi:10.1002/ppul.21496
102. Takano T, Tajiri H, Kashiwagi Y, Kimura S, Kawashima H. Cytokine and chemokine response in children with the 2009 pandemic influenza A (H1N1) virus infection. Eur J Clin Microbiol Infect Dis (2011) 30:117–20. doi:10.1007/s10096-010-1041-9
103. To KK, Hung IF, Li IW, Lee KL, Koo CK, Yan WW, et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin Infect Dis (2010) 50:850–9. doi:10.1086/650581
104. Stevens J, Blixt O, Glaser L, Taubenberger JK, Palese P, Paulson JC, et al. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol Biol (2006) 355:1143–55. doi:10.1016/j.jmb.2005.11.002
105. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis (2008) 198:962–70. doi:10.1086/591708
106. Memoli MJ, Tumpey TM, Jagger BW, Dugan VG, Sheng ZM, Qi L, et al. An early ’classical’ swine H1N1 influenza virus shows similar pathogenicity to the 1918 pandemic virus in ferrets and mice. Virology (2009) 393:338–45. doi:10.1016/j.virol.2009.08.021
107. Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature (2007) 445:319–23. doi:10.1038/nature05495
108. Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science (2005) 310:77–80. doi:10.1126/science.1119392
109. Wang SM, Liao YT, Hu YS, Ho TS, Shen CF, Wang JR, et al. Immunophenotype expressions and cytokine profiles of influenza A H1N1 virus infection in pediatric patients in 2009. Dis Markers (2014) 2014:195453. doi:10.1155/2014/195453
111. van den Brand JM, Stittelaar KJ, van Amerongen G, Reperant L, de Waal L, Osterhaus AD, et al. Comparison of temporal and spatial dynamics of seasonal H3N2, pandemic H1N1 and highly pathogenic avian influenza H5N1 virus infections in ferrets. PLoS One (2012) 7:e42343. doi:10.1371/journal.pone.0042343
112. van den Brand JM, Stittelaar KJ, van Amerongen G, Rimmelzwaan GF, Simon J, de Wit E, et al. Severity of pneumonia due to new H1N1 influenza virus in ferrets is intermediate between that due to seasonal H1N1 virus and highly pathogenic avian influenza H5N1 virus. J Infect Dis (2010) 201:993–9. doi:10.1086/651132
113. Belser JA, Jayaraman A, Raman R, Pappas C, Zeng H, Cox NJ, et al. Effect of D222G mutation in the hemagglutinin protein on receptor binding, pathogenesis and transmissibility of the 2009 pandemic H1N1 influenza virus. PLoS One (2011) 6:e25091. doi:10.1371/journal.pone.0025091
114. Maines TR, Belser JA, Gustin KM, van Hoeven N, Zeng H, Svitek N, et al. Local innate immune responses and influenza virus transmission and virulence in ferrets. J Infect Dis (2012) 205:474–85. doi:10.1093/infdis/jir768
115. Maines TR, Jayaraman A, Belser JA, Wadford DA, Pappas C, Zeng H, et al. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science (2009) 325:484–7. doi:10.1126/science.1177238
116. Neumann G, Ozawa M, Kawaoka Y. Reverse genetics of influenza viruses. Methods Mol Biol (2012) 865:193–206. doi:10.1007/978-1-61779-621-0_12
117. Abed Y, Pizzorno A, Hamelin ME, Leung A, Joubert P, Couture C, et al. The 2009 pandemic H1N1 D222G hemagglutinin mutation alters receptor specificity and increases virulence in mice but not in ferrets. J Infect Dis (2011) 204:1008–16. doi:10.1093/infdis/jir483
118. Camp JV, Chu YK, Chung DH, McAllister RC, Adcock RS, Gerlach RL, et al. Phenotypic differences in virulence and immune response in closely related clinical isolates of influenza A 2009 H1N1 pandemic viruses in mice. PLoS One (2013) 8:e56602. doi:10.1371/journal.pone.0056602
119. Chutinimitkul S, Herfst S, Steel J, Lowen AC, Ye J, van Riel D, et al. Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J Virol (2010) 84:11802–13. doi:10.1128/JVI.01136-10
120. Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature (2009) 460:1021–5. doi:10.1038/nature08260
121. Jhung MA, Swerdlow D, Olsen SJ, Jernigan D, Biggerstaff M, Kamimoto L, et al. Epidemiology of 2009 pandemic influenza A (H1N1) in the United States. Clin Infect Dis (2011) 52(Suppl 1):S13–26. doi:10.1093/cid/ciq008
122. Melidou A, Gioula G, Exindari M, Chatzidimitriou D, Diza E, Malisiovas N. Molecular and phylogenetic analysis of the haemagglutinin gene of pandemic influenza H1N1 2009 viruses associated with severe and fatal infections. Virus Res (2010) 151:192–9. doi:10.1016/j.virusres.2010.05.005
123. Meunier I, Embury-Hyatt C, Stebner S, Gray M, Bastien N, Li Y, et al. Virulence differences of closely related pandemic 2009 H1N1 isolates correlate with increased inflammatory responses in ferrets. Virology (2012) 422:125–31. doi:10.1016/j.virol.2011.10.018
124. Otte A, Gabriel G. 2009 pandemic H1N1 influenza A virus strains display differential pathogenicity in C57BL/6J but not BALB/c mice. Virulence (2011) 2:563–6. doi:10.4161/viru.2.6.18148
125. Safronetz D, Rockx B, Feldmann F, Belisle SE, Palermo RE, Brining D, et al. Pandemic swine-origin H1N1 influenza A virus isolates show heterogeneous virulence in macaques. J Virol (2011) 85:1214–23. doi:10.1128/JVI.01848-10
126. Ye J, Sorrell EM, Cai Y, Shao H, Xu K, Pena L, et al. Variations in the hemagglutinin of the 2009 H1N1 pandemic virus: potential for strains with altered virulence phenotype? PLoS Pathog (2010) 6:e1001145. doi:10.1371/journal.ppat.1001145
127. Gubareva LV, McCullers JA, Bethell RC, Webster RG. Characterization of influenza A/HongKong/156/97 (H5N1) virus in a mouse model and protective effect of zanamivir on H5N1 infection in mice. J Infect Dis (1998) 178:1592–6. doi:10.1086/314515
128. Shortridge KF, Zhou NN, Guan Y, Gao P, Ito T, Kawaoka Y, et al. Characterization of avian H5N1 influenza viruses from poultry in Hong Kong. Virology (1998) 252:331–42. doi:10.1006/viro.1998.9488
129. Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, et al. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet (2013) 381:1916–25. doi:10.1016/S0140-6736(13)60903-4
130. Wong SS, Yuen KY. Avian influenza virus infections in humans. Chest (2006) 129:156–68. doi:10.1378/chest.129.1.156
131. Fouchier RA, Schneeberger PM, Rozendaal FW, Broekman JM, Kemink SA, Munster V, et al. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc Natl Acad Sci U S A (2004) 101:1356–61. doi:10.1073/pnas.0308352100
132. Chen E, Chen Y, Fu L, Chen Z, Gong Z, Mao H, et al. Human infection with avian influenza A(H7N9) virus re-emerges in China in winter 2013. Euro Surveill (2013) 18:20616. doi:10.2807/1560-7917.ES2013.18.43.20616
133. Shinya K, Gao Y, Cilloniz C, Suzuki Y, Fujie M, Deng G, et al. Integrated clinical, pathologic, virologic, and transcriptomic analysis of H5N1 influenza virus-induced viral pneumonia in the rhesus macaque. J Virol (2012) 86:6055–66. doi:10.1128/JVI.00365-12
134. Zhou J, Wang D, Gao R, Zhao B, Song J, Qi X, et al. Biological features of novel avian influenza A (H7N9) virus. Nature (2013) 499:500–3. doi:10.1038/nature12379
135. Bouvier NM, Lowen AC. Animal models for influenza virus pathogenesis and transmission. Viruses (2010) 2:1530–63. doi:10.3390/v20801530
136. Long JP, Kotur MS, Stark GV, Warren RL, Kasoji M, Craft JL, et al. Accumulation of CD11b(+)Gr-1(+) cells in the lung, blood and bone marrow of mice infected with highly pathogenic H5N1 and H1N1 influenza viruses. Arch Virol (2013) 158:1305–22. doi:10.1007/s00705-012-1593-3
137. Zhu H, Wang D, Kelvin DJ, Li L, Zheng Z, Yoon SW, et al. Infectivity, transmission, and pathology of human-isolated H7N9 influenza virus in ferrets and pigs. Science (2013) 341:183–6. doi:10.1126/science.1239844
138. Tate MD, Ioannidis LJ, Croker B, Brown LE, Brooks AG, Reading PC. The role of neutrophils during mild and severe influenza virus infections of mice. PLoS One (2011) 6:e17618. doi:10.1371/journal.pone.0017618
139. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol (2011) 179:199–210. doi:10.1016/j.ajpath.2011.03.013
140. Tate MD, Brooks AG, Reading PC. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir Res (2008) 9:57. doi:10.1186/1465-9921-9-57
141. Bradley LM, Douglass MF, Chatterjee D, Akira S, Baaten BJ. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog (2012) 8:e1002641. doi:10.1371/journal.ppat.1002641
142. Seo SU, Kwon HJ, Song JH, Byun YH, Seong BL, Kawai T, et al. MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. J Virol (2010) 84:12713–22. doi:10.1128/JVI.01675-10
143. Walsh KB, Teijaro JR, Brock LG, Fremgen DM, Collins PL, Rosen H, et al. Animal model of respiratory syncytial virus: CD8+ T cells cause a cytokine storm that is chemically tractable by sphingosine-1-phosphate 1 receptor agonist therapy. J Virol (2014) 88:6281–93. doi:10.1128/JVI.00464-14
144. Oldstone MB, Rosen H. Cytokine storm plays a direct role in the morbidity and mortality from influenza virus infection and is chemically treatable with a single sphingosine-1-phosphate agonist molecule. Curr Top Microbiol Immunol (2014) 378:129–47. doi:10.1007/978-3-319-05879-5_6
145. Borregaard N, Sorensen OE, Theilgaard-Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol (2007) 28:340–5. doi:10.1016/j.it.2007.06.002
146. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi:10.1126/science.1092385
147. Prince LR, Whyte MK, Sabroe I, Parker LC. The role of TLRs in neutrophil activation. Curr Opin Pharmacol (2011) 11:397–403. doi:10.1016/j.coph.2011.06.007
148. Thomas CJ, Schroder K. Pattern recognition receptor function in neutrophils. Trends Immunol (2013) 34:317–28. doi:10.1016/j.it.2013.02.008
149. Bjornson AB, Mellencamp MA, Schiff GM. Complement is activated in the upper respiratory tract during influenza virus infection. Am Rev Respir Dis (1991) 143:1062–6. doi:10.1164/ajrccm/143.5_Pt_1.1062
150. Fritz RS, Hayden FG, Calfee DP, Cass LM, Peng AW, Alvord WG, et al. Nasal cytokine and chemokine responses in experimental influenza A virus infection: results of a placebo-controlled trial of intravenous zanamivir treatment. J Infect Dis (1999) 180:586–93. doi:10.1086/314938
151. Lee NL. Role of cytokines and chemokines in severe and complicated influenza infections. Hong Kong Med J (2009) 15(Suppl 8):38–41.
152. Lee N, Chan PK, Wong CK, Wong KT, Choi KW, Joynt GM, et al. Viral clearance and inflammatory response patterns in adults hospitalized for pandemic 2009 influenza A(H1N1) virus pneumonia. Antivir Ther (2011) 16:237–47. doi:10.3851/IMP1722
153. Garcia CC, Weston-Davies W, Russo RC, Tavares LP, Rachid MA, Alves-Filho JC, et al. Complement C5 activation during influenza A infection in mice contributes to neutrophil recruitment and lung injury. PLoS One (2013) 8:e64443. doi:10.1371/journal.pone.0064443
154. Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol (2005) 79:6441–8. doi:10.1128/JVI.79.10.6441-6448.2005
155. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med (2009) 206:79–87. doi:10.1084/jem.20081667
156. Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, et al. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol (2009) 183:5301–10. doi:10.4049/jimmunol.0900995
157. Ichikawa A, Kuba K, Morita M, Chida S, Tezuka H, Hara H, et al. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am J Respir Crit Care Med (2013) 187:65–77. doi:10.1164/rccm.201203-0508OC
158. Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminathan S, Bosnjak B, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol (2015) 16:67–74. doi:10.1038/ni.3046
159. Wheeler JL, Martin KC, Lawrence BP. Novel cellular targets of AhR underlie alterations in neutrophilic inflammation and inducible nitric oxide synthase expression during influenza virus infection. J Immunol (2013) 190:659–68. doi:10.4049/jimmunol.1201341
160. Teske S, Bohn AA, Hogaboam JP, Lawrence BP. Aryl hydrocarbon receptor targets pathways extrinsic to bone marrow cells to enhance neutrophil recruitment during influenza virus infection. Toxicol Sci (2008) 102:89–99. doi:10.1093/toxsci/kfm282
161. Neff-LaFord H, Teske S, Bushnell TP, Lawrence BP. Aryl hydrocarbon receptor activation during influenza virus infection unveils a novel pathway of IFN-gamma production by phagocytic cells. J Immunol (2007) 179:247–55. doi:10.4049/jimmunol.179.1.247
162. Teske S, Bohn AA, Regal JF, Neumiller JJ, Lawrence BP. Activation of the aryl hydrocarbon receptor increases pulmonary neutrophilia and diminishes host resistance to influenza A virus. Am J Physiol Lung Cell Mol Physiol (2005) 289:L111–24. doi:10.1152/ajplung.00318.2004
163. Ratcliffe D, Migliorisi G, Cramer E. Translocation of influenza virus by migrating neutrophils. Cell Mol Biol (1992) 38:63–70.
164. Hayashi F, Means TK, Luster AD. Toll-like receptors stimulate human neutrophil function. Blood (2003) 102:2660–9. doi:10.1182/blood-2003-04-1078
165. Hattermann K, Picard S, Borgeat M, Leclerc P, Pouliot M, Borgeat P. The toll-like receptor 7/8-ligand resiquimod (R-848) primes human neutrophils for leukotriene B4, prostaglandin E2 and platelet-activating factor biosynthesis. FASEB J (2007) 21:1575–85. doi:10.1096/fj.06-7457com
166. Tamassia N, Le Moigne V, Rossato M, Donini M, McCartney S, Calzetti F, et al. Activation of an immunoregulatory and antiviral gene expression program in poly(I:C)-transfected human neutrophils. J Immunol (2008) 181:6563–73. doi:10.4049/jimmunol.181.9.6563
167. Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, et al. Detrimental contribution of the toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog (2006) 2:e53. doi:10.1371/journal.ppat.0020053
168. Leung YH, Nicholls JM, Ho CK, Sia SF, Mok CK, Valkenburg SA, et al. Highly pathogenic avian influenza A H5N1 and pandemic H1N1 virus infections have different phenotypes in toll-like receptor 3 knockout mice. J Gen Virol (2014) 95:1870–9. doi:10.1099/vir.0.066258-0
169. Andonegui G, Goyert SM, Kubes P. Lipopolysaccharide-induced leukocyte-endothelial cell interactions: a role for CD14 versus toll-like receptor 4 within microvessels. J Immunol (2002) 169:2111–9. doi:10.4049/jimmunol.169.4.2111
170. Kolb JP, Casella CR, SenGupta S, Chilton PM, Mitchell TC. Type I interferon signaling contributes to the bias that toll-like receptor 4 exhibits for signaling mediated by the adaptor protein TRIF. Sci Signal (2014) 7:ra108. doi:10.1126/scisignal.2005442
171. van Bruggen R, Drewniak A, Tool AT, Jansen M, van Houdt M, Geissler J, et al. Toll-like receptor responses in IRAK-4-deficient neutrophils. J Innate Immun (2010) 2:280–7. doi:10.1159/000268288
172. Hartshorn K, Chang D, Rust K, White M, Heuser J, Crouch E. Interactions of recombinant human pulmonary surfactant protein D and SP-D multimers with influenza A. Am J Physiol (1996) 271:L753–62.
173. Hartshorn KL, Crouch EC, White MR, Eggleton P, Tauber AI, Chang D, et al. Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J Clin Invest (1994) 94:311–9. doi:10.1172/JCI117323
174. Hartshorn KL, White MR, Voelker DR, Coburn J, Zaner K, Crouch EC. Mechanism of binding of surfactant protein D to influenza A viruses: importance of binding to haemagglutinin to antiviral activity. Biochem J (2000) 351(Pt 2):449–58. doi:10.1042/bj3510449
175. LeVine AM, Whitsett JA, Hartshorn KL, Crouch EC, Korfhagen TR. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J Immunol (2001) 167:5868–73. doi:10.4049/jimmunol.167.10.5868
176. Qi L, Kash JC, Dugan VG, Jagger BW, Lau YF, Sheng ZM, et al. The ability of pandemic influenza virus hemagglutinins to induce lower respiratory pathology is associated with decreased surfactant protein D binding. Virology (2011) 412:426–34. doi:10.1016/j.virol.2011.01.029
177. van Eijk M, Bruinsma L, Hartshorn KL, White MR, Rynkiewicz MJ, Seaton BA, et al. Introduction of N-linked glycans in the lectin domain of surfactant protein D: impact on interactions with influenza A viruses. J Biol Chem (2011) 286:20137–51. doi:10.1074/jbc.M111.224469
178. Hartshorn KL, White MR, Tecle T, Holmskov U, Crouch EC. Innate defense against influenza A virus: activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J Immunol (2006) 176:6962–72. doi:10.4049/jimmunol.176.11.6962
179. Tecle T, White MR, Gantz D, Crouch EC, Hartshorn KL. Human neutrophil defensins increase neutrophil uptake of influenza A virus and bacteria and modify virus-induced respiratory burst responses. J Immunol (2007) 178:8046–52. doi:10.4049/jimmunol.178.12.8046
180. Doss M, White MR, Tecle T, Gantz D, Crouch EC, Jung G, et al. Interactions of alpha-, beta-, and theta-defensins with influenza A virus and surfactant protein D. J Immunol (2009) 182:7878–87. doi:10.4049/jimmunol.0804049
181. Salvatore M, Garcia-Sastre A, Ruchala P, Lehrer RI, Chang T, Klotman ME. alpha-Defensin inhibits influenza virus replication by cell-mediated mechanism(s). J Infect Dis (2007) 196:835–43. doi:10.1086/521027
182. Daher KA, Selsted ME, Lehrer RI. Direct inactivation of viruses by human granulocyte defensins. J Virol (1986) 60:1068–74.
183. Ratcliffe DR, Michl J, Cramer EB. Neutrophils do not bind to or phagocytize human immune complexes formed with influenza virus. Blood (1993) 82:1639–46.
184. Fujisawa H. Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J Virol (2008) 82:2772–83. doi:10.1128/JVI.01210-07
185. Hartshorn KL, Liou LS, White MR, Kazhdan MM, Tauber JL, Tauber AI. Neutrophil deactivation by influenza A virus. Role of hemagglutinin binding to specific sialic acid-bearing cellular proteins. J Immunol (1995) 154:3952–60.
186. Ivan FX, Tan KS, Phoon MC, Engelward BP, Welsch RE, Rajapakse JC, et al. Neutrophils infected with highly virulent influenza H3N2 virus exhibit augmented early cell death and rapid induction of type I interferon signaling pathways. Genomics (2013) 101:101–12. doi:10.1016/j.ygeno.2012.11.008
187. Wang JP, Bowen GN, Padden C, Cerny A, Finberg RW, Newburger PE, et al. Toll-like receptor-mediated activation of neutrophils by influenza A virus. Blood (2008) 112:2028–34. doi:10.1182/blood-2008-01-132860
188. Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity (2011) 34:854–65. doi:10.1016/j.immuni.2011.03.026
189. Jaworska J, Coulombe F, Downey J, Tzelepis F, Shalaby K, Tattoli I, et al. NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc Natl Acad Sci U S A (2014) 111:E2110–9. doi:10.1073/pnas.1322118111
190. Stasakova J, Ferko B, Kittel C, Sereinig S, Romanova J, Katinger H, et al. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J Gen Virol (2005) 86:185–95. doi:10.1099/vir.0.80422-0
191. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity (2009) 30:556–65. doi:10.1016/j.immuni.2009.02.005
192. Belisle SE, Tisoncik JR, Korth MJ, Carter VS, Proll SC, Swayne DE, et al. Genomic profiling of tumor necrosis factor alpha (TNF-alpha) receptor and interleukin-1 receptor knockout mice reveals a link between TNF-alpha signaling and increased severity of 1918 pandemic influenza virus infection. J Virol (2010) 84:12576–88. doi:10.1128/JVI.01310-10
193. Huang CH, Chen CJ, Yen CT, Yu CP, Huang PN, Kuo RL, et al. Caspase-1 deficient mice are more susceptible to influenza A virus infection with PA variation. J Infect Dis (2013) 208:1898–905. doi:10.1093/infdis/jit381
194. Perrone LA, Szretter KJ, Katz JM, Mizgerd JP, Tumpey TM. Mice lacking both TNF and IL-1 receptors exhibit reduced lung inflammation and delay in onset of death following infection with a highly virulent H5N1 virus. J Infect Dis (2010) 202:1161–70. doi:10.1086/656365
195. Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR, et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol (2007) 81:2736–44. doi:10.1128/JVI.02336-06
196. Abramson JS, Lewis JC, Lyles DS, Heller KA, Mills EL, Bass DA. Inhibition of neutrophil lysosome-phagosome fusion associated with influenza virus infection in vitro. Role in depressed bactericidal activity. J Clin Invest (1982) 69:1393–7. doi:10.1172/JCI110580
197. Walzog B, Schuppan D, Heimpel C, Hafezi-Moghadam A, Gaehtgens P, Ley K. The leukocyte integrin Mac-1 (CD11b/CD18) contributes to binding of human granulocytes to collagen. Exp Cell Res (1995) 218:28–38. doi:10.1006/excr.1995.1127
198. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6:173–82. doi:10.1038/nri1785
199. Sugamata R, Dobashi H, Nagao T, Yamamoto K, Nakajima N, Sato Y, et al. Contribution of neutrophil-derived myeloperoxidase in the early phase of fulminant acute respiratory distress syndrome induced by influenza virus infection. Microbiol Immunol (2012) 56:171–82. doi:10.1111/j.1348-0421.2011.00424.x
200. Yamamoto K, Miyoshi-Koshio T, Utsuki Y, Mizuno S, Suzuki K. Virucidal activity and viral protein modification by myeloperoxidase: a candidate for defense factor of human polymorphonuclear leukocytes against influenza virus infection. J Infect Dis (1991) 164:8–14. doi:10.1093/infdis/164.1.8
201. Foong RE, Sly PD, Larcombe AN, Zosky GR. No role for neutrophil elastase in influenza-induced cellular recruitment, cytokine production or airway hyperresponsiveness in mice. Respir Physiol Neurobiol (2010) 173:164–70. doi:10.1016/j.resp.2010.08.003
202. Abramson JS, Mills EL, Giebink GS, Quie PG. Depression of monocyte and polymorphonuclear leukocyte oxidative metabolism and bactericidal capacity by influenza A virus. Infect Immun (1982) 35:350–5.
203. Schwarz KB. Oxidative stress during viral infection: a review. Free Radic Biol Med (1996) 21:641–9. doi:10.1016/0891-5849(96)00131-1
204. Knobil K, Choi AM, Weigand GW, Jacoby DB. Role of oxidants in influenza virus-induced gene expression. Am J Physiol (1998) 274:L134–42.
205. Lazrak A, Iles KE, Liu G, Noah DL, Noah JW, Matalon S. Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J (2009) 23:3829–42. doi:10.1096/fj.09-135590
206. Snelgrove RJ, Edwards L, Rae AJ, Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur J Immunol (2006) 36:1364–73. doi:10.1002/eji.200635977
207. Cooper JA Jr, Carcelen R, Culbreth R. Effects of influenza A nucleoprotein on polymorphonuclear neutrophil function. J Infect Dis (1996) 173:279–84. doi:10.1093/infdis/173.2.279
208. Buffinton GD, Christen S, Peterhans E, Stocker R. Oxidative stress in lungs of mice infected with influenza A virus. Free Radic Res Commun (1992) 16:99–110. doi:10.3109/10715769209049163
209. Perrone LA, Belser JA, Wadford DA, Katz JM, Tumpey TM. Inducible nitric oxide contributes to viral pathogenesis following highly pathogenic influenza virus infection in mice. J Infect Dis (2013) 207:1576–84. doi:10.1093/infdis/jit062
210. Suliman HB, Ryan LK, Bishop L, Folz RJ. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am J Physiol Lung Cell Mol Physiol (2001) 280:L69–78.
211. Vlahos R, Stambas J, Bozinovski S, Broughton BR, Drummond GR, Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog (2011) 7:e1001271. doi:10.1371/journal.ppat.1001271
212. Zhang WJ, Wei H, Tien YT, Frei B. Genetic ablation of phagocytic NADPH oxidase in mice limits TNFalpha-induced inflammation in the lungs but not other tissues. Free Radic Biol Med (2011) 50:1517–25. doi:10.1016/j.freeradbiomed.2011.02.027
213. Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng YM, et al. Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc Natl Acad Sci U S A (1996) 93:2448–53. doi:10.1073/pnas.93.6.2448
214. Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell (2008) 133:235–49. doi:10.1016/j.cell.2008.02.043
215. Nhu QM, Shirey K, Teijaro JR, Farber DL, Netzel-Arnett S, Antalis TM, et al. Novel signaling interactions between proteinase-activated receptor 2 and toll-like receptors in vitro and in vivo. Mucosal Immunol (2010) 3:29–39. doi:10.1038/mi.2009.120
216. Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature (2013) 497:498–502. doi:10.1038/nature12118
217. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176:231–41. doi:10.1083/jcb.200606027
218. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol (2012) 189:2689–95. doi:10.4049/jimmunol.1201719
219. Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol (2006) 8:668–76. doi:10.1111/j.1462-5822.2005.00659.x
220. Guimaraes-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao-Silva F, et al. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci U S A (2009) 106:6748–53. doi:10.1073/pnas.0900226106
221. Agraz-Cibrian JM, Giraldo DM, Mary FM, Urcuqui-Inchima S. Understanding the molecular mechanisms of NETs and their role in antiviral innate immunity. Virus Res (2017) 228:124–33. doi:10.1016/j.virusres.2016.11.033
222. Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15:623–5. doi:10.1038/nm.1959
223. Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, Vitkov L, et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med (2010) 16:1018–23. doi:10.1038/nm.2209
224. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A (2010) 107:9813–8. doi:10.1073/pnas.0909927107
225. Raftery MJ, Lalwani P, Krautkrmer E, Peters T, Scharffetter-Kochanek K, Kruger R, et al. beta2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J Exp Med (2014) 211:1485–97. doi:10.1084/jem.20131092
226. Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One (2011) 6:e22043. doi:10.1371/journal.pone.0022043
227. Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun (2005) 73:1744–53. doi:10.1128/IAI.73.3.1744-1753.2005
228. Seiler P, Aichele P, Bandermann S, Hauser AE, Lu B, Gerard NP, et al. Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur J Immunol (2003) 33:2676–86. doi:10.1002/eji.200323956
229. Kennedy JL, Turner RB, Braciale T, Heymann PW, Borish L. Pathogenesis of rhinovirus infection. Curr Opin Virol (2012) 2:287–93. doi:10.1016/j.coviro.2012.03.008
230. Levandowski RA, Weaver CW, Jackson GG. Nasal-secretion leukocyte populations determined by flow cytometry during acute rhinovirus infection. J Med Virol (1988) 25:423–32. doi:10.1002/jmv.1890250406
231. Turner RB. The role of neutrophils in the pathogenesis of rhinovirus infections. Pediatr Infect Dis J (1990) 9:832–5. doi:10.1097/00006454-199011000-00011
232. Domachowske JB, Rosenberg HF. Respiratory syncytial virus infection: immune response, immunopathogenesis, and treatment. Clin Microbiol Rev (1999) 12:298–309.
233. Pickles RJ, DeVincenzo JP. Respiratory syncytial virus (RSV) and its propensity for causing bronchiolitis. J Pathol (2015) 235:266–76. doi:10.1002/path.4462
234. Schildgen V, van den Hoogen B, Fouchier R, Tripp RA, Alvarez R, Manoha C, et al. Human metapneumovirus: lessons learned over the first decade. Clin Microbiol Rev (2011) 24:734–54. doi:10.1128/CMR.00015-11
235. Vargas SO, Kozakewich HP, Perez-Atayde AR, McAdam AJ. Pathology of human metapneumovirus infection: insights into the pathogenesis of a newly identified respiratory virus. Pediatr Dev Pathol (2004) 7:478–86; discussion 421. doi:10.1007/s10024-004-1011-2
236. Hooper P, Zaki S, Daniels P, Middleton D. Comparative pathology of the diseases caused by Hendra and Nipah viruses. Microbes Infect (2001) 3:315–22. doi:10.1016/S1286-4579(01)01385-5
237. Koh YY, Jung da E, Koh JY, Kim JY, Yoo Y, Kim CK. Bronchoalveolar cellularity and interleukin-8 levels in measles bronchiolitis obliterans. Chest (2007) 131:1454–60. doi:10.1378/chest.06-0188
238. Downham MA, Gardner PS, McQuillin J, Ferris JA. Role of respiratory viruses in childhood mortality. Br Med J (1975) 1:235–9. doi:10.1136/bmj.1.5952.235
239. Zaki SR, Greer PW, Coffield LM, Goldsmith CS, Nolte KB, Foucar K, et al. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am J Pathol (1995) 146:552–79.
240. Wu W, Booth JL, Duggan ES, Patel KB, Coggeshall KM, Metcalf JP. Human lung innate immune cytokine response to adenovirus type 7. J Gen Virol (2010) 91:1155–63. doi:10.1099/vir.0.017905-0
241. Pyrc K, Berkhout B, van der Hoek L. The novel human coronaviruses NL63 and HKU1. J Virol (2007) 81:3051–7. doi:10.1128/JVI.01466-06
242. Moolenaar RL, Dalton C, Lipman HB, Umland ET, Gallaher M, Duchin JS, et al. Clinical features that differentiate hantavirus pulmonary syndrome from three other acute respiratory illnesses. Clin Infect Dis (1995) 21:643–9. doi:10.1093/clinids/21.3.643
243. Smits SL, van den Brand JM, de Lang A, Leijten LM, van Ijcken WF, van Amerongen G, et al. Distinct severe acute respiratory syndrome coronavirus-induced acute lung injury pathways in two different nonhuman primate species. J Virol (2011) 85:4234–45. doi:10.1128/JVI.02395-10
244. Schomacker H, Schaap-Nutt A, Collins PL, Schmidt AC. Pathogenesis of acute respiratory illness caused by human parainfluenza viruses. Curr Opin Virol (2012) 2:294–9. doi:10.1016/j.coviro.2012.02.001
245. Jiang Y, Xu J, Zhou C, Wu Z, Zhong S, Liu J, et al. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am J Respir Crit Care Med (2005) 171:850–7. doi:10.1164/rccm.200407-857OC
246. Bataki EL, Evans GS, Everard ML. Respiratory syncytial virus and neutrophil activation. Clin Exp Immunol (2005) 140:470–7. doi:10.1111/j.1365-2249.2005.02780.x
247. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi:10.1038/nri3399
248. van Kempen M, Bachert C, Van Cauwenberge P. An update on the pathophysiology of rhinovirus upper respiratory tract infections. Rhinology (1999) 37:97–103.
249. Winther B. Effects on the nasal mucosa of upper respiratory viruses (common cold). Dan Med Bull (1994) 41:193–204.
250. Winther B, Gwaltney JM Jr, Mygind N, Hendley JO. Viral-induced rhinitis. Am J Rhinol (1998) 12:17–20. doi:10.2500/105065898782102954
251. Racaniello V. Picornaviridae: the viruses and their replication. 5th ed. In: Knipe DM, Howley PM, editors. Fields Virology. (Vol. 1), Philadelphia, PA: Lippincott Williams and Wilkins (2007). p. 795–838.
252. Gern JE, Busse WW. Association of rhinovirus infections with asthma. Clin Microbiol Rev (1999) 12:9–18.
253. Gern JE, Martin MS, Anklam KA, Shen K, Roberg KA, Carlson-Dakes KT, et al. Relationships among specific viral pathogens, virus-induced interleukin-8, and respiratory symptoms in infancy. Pediatr Allergy Immunol (2002) 13:386–93. doi:10.1034/j.1399-3038.2002.01093.x
254. Murawski MR, Bowen GN, Cerny AM, Anderson LJ, Haynes LM, Tripp RA, et al. Respiratory syncytial virus activates innate immunity through toll-like receptor 2. J Virol (2009) 83:1492–500. doi:10.1128/JVI.00671-08
255. Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol (2008) 82:335–45. doi:10.1128/JVI.01080-07
256. Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M Jr, Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol (2007) 81:514–24. doi:10.1128/JVI.01265-06
257. Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity (2005) 23:19–28. doi:10.1016/j.immuni.2005.04.010
258. Koyama S, Ishii KJ, Kumar H, Tanimoto T, Coban C, Uematsu S, et al. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J Immunol (2007) 179:4711–20. doi:10.4049/jimmunol.179.7.4711
259. Kelly JT, Busse WW. Host immune responses to rhinovirus: mechanisms in asthma. J Allergy Clin Immunol (2008) 122:671–82; quiz 683–74. doi:10.1016/j.jaci.2008.08.013
260. Honda K, Taniguchi T. IRFs: master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol (2006) 6:644–58. doi:10.1038/nri1900
261. Dixit E, Kagan JC. Intracellular pathogen detection by RIG-I-like receptors. Adv Immunol (2013) 117:99–125. doi:10.1016/B978-0-12-410524-9.00004-9
262. Zinman G, Brower-Sinning R, Emeche CH, Ernst J, Huang GT, Mahony S, et al. Large scale comparison of innate responses to viral and bacterial pathogens in mouse and macaque. PLoS One (2011) 6:e22401. doi:10.1371/journal.pone.0022401
263. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303:1529–31. doi:10.1126/science.1093616
264. Byeon JH, Lee JC, Choi IS, Yoo Y, Park SH, Choung JT. Comparison of cytokine responses in nasopharyngeal aspirates from children with viral lower respiratory tract infections. Acta Paediatr (2009) 98:725–30. doi:10.1111/j.1651-2227.2008.01208.x
265. Sung RY, Hui SH, Wong CK, Lam CW, Yin J. A comparison of cytokine responses in respiratory syncytial virus and influenza A infections in infants. Eur J Pediatr (2001) 160:117–22. doi:10.1007/s004310000676
266. Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JC, et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol (2010) 3:291–300. doi:10.1038/mi.2010.6
267. Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell (1994) 76:241–51. doi:10.1016/0092-8674(94)90332-8
268. Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol (2004) 25:280–8. doi:10.1016/j.it.2004.03.008
269. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem (2007) 282:20059–63. doi:10.1074/jbc.R700016200
270. Borden EC, Williams BR. Interferon-stimulated genes and their protein products: what and how? J Interferon Cytokine Res (2011) 31:1–4. doi:10.1089/jir.2010.0129
271. Furuya Y, Steiner D, Metzger DW. Does type I interferon limit protective neutrophil responses during pulmonary Francisella tularensis infection? Front Immunol (2014) 5:355. doi:10.3389/fimmu.2014.00355
272. Garcia-Sastre A. Induction and evasion of type I interferon responses by influenza viruses. Virus Res (2011) 162:12–8. doi:10.1016/j.virusres.2011.10.017
273. Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, et al. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem (2004) 279:44123–32. doi:10.1074/jbc.M405883200
274. Meunier I, von Messling V. NS1-mediated delay of type I interferon induction contributes to influenza A virulence in ferrets. J Gen Virol (2011) 92:1635–44. doi:10.1099/vir.0.032193-0
275. Seo SU, Kwon HJ, Ko HJ, Byun YH, Seong BL, Uematsu S, et al. Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog (2011) 7:e1001304. doi:10.1371/journal.ppat.1001304
276. Svitek N, Rudd PA, Obojes K, Pillet S, von Messling V. Severe seasonal influenza in ferrets correlates with reduced interferon and increased IL-6 induction. Virology (2008) 376:53–9. doi:10.1016/j.virol.2008.02.035
277. Wang X, Li M, Zheng H, Muster T, Palese P, Beg AA, et al. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol (2000) 74:11566–73. doi:10.1128/JVI.74.24.11566-11573.2000
278. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra20. doi:10.1126/scitranslmed.3001201
279. Decker P. Neutrophils and interferon-alpha-producing cells: who produces interferon in lupus? Arthritis Res Ther (2011) 13:118. doi:10.1186/ar3345
280. Stock AT, Smith JM, Carbone FR. Type I IFN suppresses Cxcr2 driven neutrophil recruitment into the sensory ganglia during viral infection. J Exp Med (2014) 211:751–9. doi:10.1084/jem.20132183
281. Xin L, Vargas-Inchaustegui DA, Raimer SS, Kelly BC, Hu J, Zhu L, et al. Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J Immunol (2010) 184:7047–56. doi:10.4049/jimmunol.0903273
282. Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM, et al. Type I IFN modulates innate and specific antiviral immunity. J Immunol (2000) 164:4220–8. doi:10.4049/jimmunol.164.8.4220
283. Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol (2007) 178:2448–57. doi:10.4049/jimmunol.178.4.2448
284. Tate MD, Brooks AG, Reading PC, Mintern JD. Neutrophils sustain effective CD8(+) T-cell responses in the respiratory tract following influenza infection. Immunol Cell Biol (2012) 90:197–205. doi:10.1038/icb.2011.26
285. Tate MD, Deng YM, Jones JE, Anderson GP, Brooks AG, Reading PC. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol (2009) 183:7441–50. doi:10.4049/jimmunol.0902497
286. Herold S, Tabar TS, Janssen H, Hoegner K, Cabanski M, Lewe-Schlosser P, et al. Exudate macrophages attenuate lung injury by the release of IL-1 receptor antagonist in gram-negative pneumonia. Am J Respir Crit Care Med (2011) 183:1380–90. doi:10.1164/rccm.201009-1431OC
287. Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood (2008) 112:3455–64. doi:10.1182/blood-2007-12-129080
288. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101:890–8. doi:10.1172/JCI1112
289. El Kebir D, Filep JG. Targeting neutrophil apoptosis for enhancing the resolution of inflammation. Cells (2013) 2:330–48. doi:10.3390/cells2020330
290. Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol (2000) 164:1663–7. doi:10.4049/jimmunol.164.4.1663
291. Poe SL, Arora M, Oriss TB, Yarlagadda M, Isse K, Khare A, et al. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol (2013) 6:189–99. doi:10.1038/mi.2012.62
292. Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, et al. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol (2012) 5:258–66. doi:10.1038/mi.2012.2
293. Arndt U, Wennemuth G, Barth P, Nain M, Al-Abed Y, Meinhardt A, et al. Release of macrophage migration inhibitory factor and CXCL8/interleukin-8 from lung epithelial cells rendered necrotic by influenza A virus infection. J Virol (2002) 76:9298–306. doi:10.1128/JVI.76.18.9298-9306.2002
294. Simard JC, Girard D, Tessier PA. Induction of neutrophil degranulation by S100A9 via a MAPK-dependent mechanism. J Leukoc Biol (2010) 87:905–14. doi:10.1189/jlb.1009676
295. Tsai SY, Segovia JA, Chang TH, Morris IR, Berton MT, Tessier PA, et al. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog (2014) 10:e1003848. doi:10.1371/journal.ppat.1003848
Keywords: neutrophil, influenza, acute respiratory distress syndrome, respiratory virus, viral microenvironment
Citation: Camp JV and Jonsson CB (2017) A Role for Neutrophils in Viral Respiratory Disease. Front. Immunol. 8:550. doi: 10.3389/fimmu.2017.00550
Received: 03 December 2016; Accepted: 24 April 2017;
Published: 12 May 2017
Edited by:
Lee-Ann H. Allen, University of Iowa, USAReviewed by:
Elsa Anes, Universidade de Lisboa, PortugalCopyright: © 2017 Camp and Jonsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Colleen B. Jonsson, Y2pvbnNzb25AdXRrLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.