 Andrew S. Flies
Andrew S. Flies Nicholas B. Blackburn
Nicholas B. Blackburn Alan Bruce Lyons
Alan Bruce Lyons John D. Hayball
John D. Hayball Gregory M. Woods
Gregory M. Woods- 1Menzies Institute for Medical Research, University of Tasmania, Hobart, TAS, Australia
- 2Department of Pharmacy and Medical Sciences, University of South Australia, Adelaide, SA, Australia
- 3School of Medicine, South Texas Diabetes and Obesity Institute, University of Texas Rio Grande Valley, Brownsville, TX, USA
- 4School of Medicine, University of Tasmania, Hobart, TAS, Australia
- 5Discipline of Obstetrics and Gynaecology, School of Medicine, Robinson Research Institute, The University of Adelaide, Adelaide, SA, Australia
Immune checkpoint molecules function as a system of checks and balances that enhance or inhibit immune responses to infectious agents, foreign tissues, and cancerous cells. Immunotherapies that target immune checkpoint molecules, particularly the inhibitory molecules programmed cell death 1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), have revolutionized human oncology in recent years, yet little is known about these key immune signaling molecules in species other than primates and rodents. The Tasmanian devil facial tumor disease is caused by transmissible cancers that have resulted in a massive decline in the wild Tasmanian devil population. We have recently demonstrated that the inhibitory checkpoint molecule PD-L1 is upregulated on Tasmanian devil (Sarcophilus harrisii) facial tumor cells in response to the interferon-gamma cytokine. As this could play a role in immune evasion by tumor cells, we performed a thorough comparative analysis of checkpoint molecule protein sequences among Tasmanian devils and eight other species. We report that many of the key signaling motifs and ligand-binding sites in the checkpoint molecules are highly conserved across the estimated 162 million years of evolution since the last common ancestor of placental and non-placental mammals. Specifically, we discovered that the CTLA-4 (MYPPPY) ligand-binding motif and the CTLA-4 (GVYVKM) inhibitory domain are completely conserved across all nine species used in our comparative analysis, suggesting that the function of CTLA-4 is likely conserved in these species. We also found that cysteine residues for intra- and intermolecular disulfide bonds were also highly conserved. For instance, all 20 cysteine residues involved in disulfide bonds in the human 4-1BB molecule were also present in devil 4-1BB. Although many key sequences were conserved, we have also identified immunoreceptor tyrosine-based inhibitory motifs (ITIMs) and immunoreceptor tyrosine-based switch motifs (ITSMs) in genes and protein domains that have not been previously reported in any species. This checkpoint molecule analysis and review of salient features for each of the molecules presented here can serve as road map for the development of a Tasmanian devil facial tumor disease immunotherapy. Finally, the strategies can be used as a guide for veterinarians, ecologists, and other researchers willing to venture into the nascent field of wild immunology.
Introduction
The immune system plays a vital role in protecting an animal from pathogens and cancers through various immunosurveillance and effector mechanisms. The pathogenic cells and/or microbes that escape or neutralize host immunity produce more surviving descendants that are in turn more effective at evading immune responses. The architecture of the vertebrate immune system and the mechanisms by which immune responses are regulated are the result of this long and complex evolutionary arms race. This arms race drives continuous evolution of the immune system, but many defenses have persisted throughout this long-history and are conserved across speciation events.
Immune checkpoint molecules function as a complex system of checks and balances that promote or inhibit immune responses. Checkpoint molecules likely evolved to precisely balance the need to control pathogens and cancer with the need to maintain tolerance to healthy tissues and the microbiota (Figure 1). However, the delicate balance of tolerance and immunity can be upset by pathogens and tumor cells that subvert inhibitory pathways to evade host immune defenses.
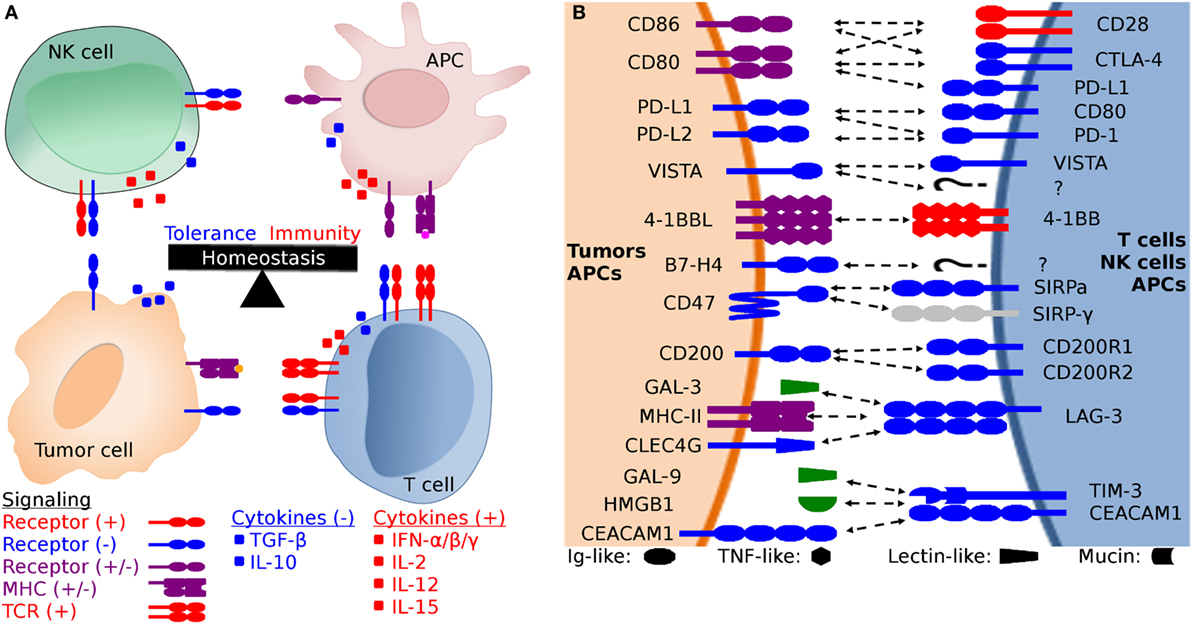
Figure 1. Generalized schematic of checkpoint molecule structure and expression, and the balance between stimulatory and inhibitory signaling. (A) The net balance of inhibitory (−, blue) and stimulatory (+, red) signals regulates antitumor immune responses. The tumor microenvironment is dynamic and is molded by inhibitory and stimulatory signals both in the microenvironment and distant tissues (i.e., lymph nodes). Development of cytotoxic effector T cells and NK cells is regulated by the balance between inhibitory and stimulatory signaling, including both receptor:coreceptor interactions and cytokines. Both receptor and cytokine expression are also regulated by that balance of stimulatory and inhibitory signals, creating feedback loops that evolved to eliminate pathogens and cancer while simultaneously limiting collateral damage. (B) Expanded view of potential receptor:coreceptor interactions between T cells, NK cells, antigen-presenting cells (APCs), and tumor cells. The graphic here demonstrates some of the best characterized pathways, with a particular focus on T cell and NK cell interactions with tumor cells and APCs. Note that many of the ligands depicted here are expressed on several cell types and interactions with coreceptors and ligands can result in different functional outcomes depending on the cell types and microenvironment. For example, CD47 is expressed on all cells, but the pathway of primary interest for transmissible tumors is for the ability of tumor cell-expressed CD47 to inhibit phagocytosis via binding to SIRP-α on APCs. In addition to binding to SIRP-α, CD47 also binds to SIRP-β, SIRP-γ, THBS1, and THBS2, but we have only shown the CD47:SIRP-α interaction here to keep the figure relatively simple. Both V-domain Ig suppressor of T cell activation (VISTA) and B7-H4 have unknown coreceptors (labeled as “?” in the diagram) whose function has been studied using fusion proteins, but the genes for the coreceptors have yet to be identified.
Checkpoint molecule immunotherapy has recently surged onto the world stage, but this field emerged over three decades of basic immunobiological research, primarily focused on T cells. In general, activation of T cells requires signaling through the T cell receptor (TCR) (signal 1) and cosignaling through CD28 (signal 2), a constitutively expressed disulfide-linked homodimer on the surface of naïve T cells (1–3). Signal one is derived from interactions of the TCR with its cognate major histocompatibility complex (MHC) peptide-complex (4). Signal two is derived from CD28 binding with either CD80 (B7-1) or CD86 (B7-2), primarily expressed by dendritic cells (DCs) and other antigen-presenting cells (APCs), such as macrophages and B cells. TCR signaling in the absence of CD28 cosignaling can result in T cell anergy (5). Following activation of T cells, the inhibitory molecule cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is upregulated on the cell surface (6). CTLA-4 has a higher affinity for both CD80 and CD86 than does CD28 (7–9), and thus CD28 cosignaling is suppressed by CTLA-4 outcompeting CD28 for CD80 and CD86 binding. The result is a downregulation of the T cell response and loss of T cell function.
Immunotherapies targeting the CTLA-4 checkpoint molecule have opened new doors in the realm of human oncology. Two antagonistic anti-CTLA-4 mAbs, ipilimumab (IgG1 isotype) and tremelimumab (IgG2 isotype), that block interactions between CTLA-4 and its coreceptors CD80 and CD86 have been clinically approved for advanced stage cancers. The exact mechanisms by which anti-CTLA-4 works are not completely understood. Accumulating evidence suggests that anti-CTLA-4 works primarily in the secondary lymphoid organs by releasing pre-existing T cells from inhibitory signaling to target tumor neoantigens and by depleting Tregs in the tumor microenvironment (10, 11).
Despite the promising early results of anti-CTLA-4 mAb monotherapy in human clinical trials, objective response rates remained low in most cases. Improvements have been observed when anti-CTLA-4 was paired with other treatments, such as the chemotherapy drug dacarbazine (12). However, the most promising outcomes have been observed when anti-CTLA-4 mAb is paired with blockade of the programmed cell death 1 (PDCD1, PD-1) protein (13–15).
Anti-PD-1 has been the most successful checkpoint blocking monoclonal antibody in treating human cancer, in monotherapies or in combination therapies. Anti-CTLA-4 and anti-PD-1 combination therapies have achieved unprecedented objective response rates of nearly 60% in patients with stage III and IV melanoma (14, 15). PD-1 immunotherapies have focused largely on stimulating T cell antitumor responses by blocking binding of PD-1 to its ligands PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273). PD-1 blockade abrogates the inhibitory effects of PD-1 ligation mediated by the immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) in the PD-1 cytoplasmic tail (16–18). In addition to blocking the inhibitory effects of PD-1, blockade of PD-L1 ligation can render tumor cells less resistant to apoptosis (19). PD-L1 and PD-L2 proteins are largely absent in normal tissues, but PD-L1 expression is elevated in more than 20 types of human cancer [reviewed in Ref. (20)]. Furthermore, many cancers and healthy cells upregulate PD-L1 expression in response to inflammatory cytokines, such as interferon-gamma (IFN-γ).
In humans, PD-L1 expression in the tumor microenvironment is associated with an inhibited T cell antitumor response and reduced patient survival [reviewed in Ref. (20)]. PD-L1 expression in the tumor microenvironment plays an important role in shaping a dysfunctional immune response through reprogramming of APCs, regulatory T cells, and expression of inhibitory molecules. PD-L2 is not routinely detected on cancer cells, but is commonly expressed by DCs, which led to the alternative name of B7-DC (21). A second coreceptor for PD-L2, repulsive guidance molecule b, has recently been discovered and has been implicated in respiratory immune tolerance (22).
Orthologs of CTLA-4 and PD-1 and their coreceptors have been found in nearly all mammals and birds studied to date (23, 24), but comparatively little is known about the potential for checkpoint molecule immunotherapy in the field of veterinary medicine or in the realm of wildlife disease. Initial investigations into the function of CTLA-4, PD-1, and PD-L1 have been undertaken for canids (25–30), felids (31), and bovids (32–35), and results suggest that the CTLA-4 and PD-1/PD-L1 expression patterns and function are similar among these placental mammals.
An area where checkpoint molecule immunology could make a wildlife conservation impact is in the unusual cases of transmissible tumors. These transmissible tumors represent a unique immunological enigma because they can also simultaneously be considered infections and allografts. Disease progression, transmission, and the host immune response can only be properly understood when these tumors are considered in this complex tumor–infection–allograft context.
The Tasmanian devil facial tumor (DFT) was identified in 1996, and in 2006 the etiologic agent was found to be cancer cells that are transmitted between biting devils (36, 37). At the time of discovery, the only other known naturally transmissible tumors were the canine (Canis lupus familiaris) transmissible venereal tumor (38, 39) and a set of related Syrian hamster (Mesocricetus auratus) transmissible sarcomas (40, 41). In recent years, additional transmissible tumors have been discovered in soft-shell clams and other bivalves (42, 43). Remarkably, in 2015, we discovered a second transmissible devil facial tumor (DFT2) in wild Tasmanian devils (referred to as devils hereafter) that is genetically distinct from the original devil facial tumor (DFT1) (44).
Previous research has shown that reduced cell surface expression of MHC-I plays a major role in DFT1 immune evasion, but that IFN-γ can upregulate beta-2 microglobulin (β2m), ergo MHC-I expression (45). This immune evasion mechanism has been described in many types of cancer, and begs the question as to why the MHC-negative DFT cells are not targeted by NK cells in line with the “missing-self” hypothesis (46). Studies have demonstrated the NK cell genes are present in devils and that devil NK-like cells can mount an antitumor response (47–49). Despite the complimentary mechanisms of cytotoxic T cells and NK cells, immune responses against the DFTs are rare (50), suggesting that additional immune evasion mechanisms could be present.
Recent vaccination efforts have demonstrated that IFN-γ-stimulated DFT cells can induce antitumor responses and tumor regressions in a subset of cases (51). However, initial forays into checkpoint molecule immunology of the DFT disease has shown that both DFT1 and DFT2 also upregulate PD-L1 upon exposure to IFN-γ (52), suggesting that anti-DFT immunotherapies could be enhanced by neutralizing the PD-1 inhibitory pathway. Given that the immune system has many redundant mechanisms for identifying and killing infectious agents and tumors, it is likely that the transmissible tumors employ additional methods beyond MHC-I downregulation and PD-L1 upregulation for evading host immune defenses and facilitating tumor transmission.
We have performed a comparative analysis of the protein sequences of key immune checkpoint molecules in the three mammalian species known to harbor naturally transmissible cancers (devils, dogs, and hamsters) and six other mammals (Table 1). We have not included the analysis of PD-1, PD-L1, and PD-L2 here, as we have previously published this information (52). Understanding the broad role of checkpoint molecules in these complex diseases has the potential to shed light on fundamental aspects of immune evasion, transplant tolerance and rejection, and infectious disease; this is logically relevant to both human and veterinary medicine. This checkpoint molecule analysis and review of salient features for each of the molecules presented here can serve as road map for the development of a DFT immunotherapy and as a guide for veterinarians, ecologists, and other researchers willing to venture into the nascent field of wild immunology.

Table 1. Names and identifiers for species used in comparative sequence analysis.
Materials and Methods
Species
We included nine species in our comparative analysis of checkpoint molecules. Humans and mice were included because checkpoint molecules have been best characterized in these species. We have included the little brown bat (Myotis lucifugus) in our comparative analyses because like devils, the species has recently undergone a dramatic population crash due to disease, and understanding bat immunology has become increasingly important due to the ability of bats to serve as reservoirs for viral pathogens (e.g., rabies, ebola, SARS). We have also included cats (Felis catus) and cattle (Bos taurus) due to their widespread distribution as important domestic animals. See Table 1 for a full list of species names and abbreviations used.
Sequences
DNA sequences for all genes analyzed here were collected from GenBank and Ensembl and reference numbers for all sequences are available in Table S1 in Supplementary Material (53). All reference sequences current as of 30-Jan-2017, including the most current devil genome (Devil_ref v7.0). The complete coding sequence for the V-domain Ig suppressor of T cell activation (VISTA), CD47, and TIM-3 genes was not available from either GenBank or Ensembl, so sequences were obtained from a de novo transcriptome assembly (Table S2 in Supplementary Material) described below. The full coding sequences were submitted to GenBank with accession numbers: KY856749 (VISTA), KY856750 (CD47), and KY856751 (TIM-3). Where more than one sequence was present we generally chose GenBank transcript X1, unless preliminary alignments suggested that an alternative transcript was a better match to the other species used here. BLAST was used to find additional potential matching sequences for each gene (54). The available reference sequences for several genes that have not been characterized in their respective species contained elongated 5′ regions upstream of the likely start codon, so we manually truncated the amino acid sequences depicted in the alignment figures for the following genes: opossum CD80 and VISTA, bat CD86 and CD47, and hamster B7-H4.
RNA Sequencing (RNA-seq)
RNA sequencing data were generated as part of a previous study (55). Briefly, RNA was extracted from peripheral blood mononuclear cells (PBMCs) obtained from two disease-free devils. Illumina single-end 100 bp RNA-seq was conducted by the Australian Genome Research Facility. Sequencing generated 82,609,965 and 82,254,904 sequences, respectively. Fastq reads for each sample were assessed with FastQC version 0.11.5 (56), with both samples having acceptable per base sequence quality measurements. Genome-independent de novo assembly of the RNA-seq data was completed using the Trinity platform version 2.2.0 (57, 58) with default settings applied. This generated a reference transcriptome of assembled transcripts collectively from the two samples. The de novo transcriptome assembly was queried with BLASTN version 2.4.0+ (59) with coding sequences of the target genes obtained from Ensembl (53), database version 87.7, and UCSC Genome Browser (60). This identified transcript sequences from the assembled transcriptome of our target genes to use in comparative alignment analysis. Further information on this analysis is detailed in the Data Sheet S1 in Supplementary Material. Access to the raw fastq sequence data, Trinity assembled de novo transcriptome, and BLASTN results are detailed in the Data Sheet S1 in Supplementary Material.
Protein Sequence Alignments
Protein sequences were aligned with CLC sequence viewer using a progressive alignment algorithm with gap open cost = 5, gap extension cost = 1, end gap cost = free (61, 62). Pairwise protein sequence identities between devils and other species were determined using the default parameters on the Clustal Omega web server (63). We used the RasMol coloring scheme to highlight traditional amino acid properties in all sequence alignments, which aids in interpretation of amino differences across species (64).
Analysis of Protein Sequences
All sequences were analyzed using the ExPASy server ProtParam tool to predict molecular weight and extinction coefficients (65). The eukaryotic linear motif (ELM) tool was used to predict key signaling motifs (66). The Simple Modular Architecture Research Tool (SMART) and the Phobius web server (67) were used to predict Ig-like domains and membrane topology (68). SignalP was used to confirm predicted signal peptides (69). Disulfide bonds and other unique features of each protein that were annotated on UniProt (70) were used to predict features in devil protein sequences.
Results
The CTLA-4 and CD28 pathways are perhaps the most studied of the checkpoint molecules, but functional testing in species other than mice and humans is limited. A battery of tools (i.e., recombinant proteins, monoclonal antibodies) and time are needed for proper testing of checkpoint molecule function, but much information and insight into function can be gained by analysis of protein sequences. For each molecule analyzed, we begin with a description of the general structure (i.e., Ig-like superfamily domains) and proceed to more nuanced details. This provides insight into potential functions of the molecules and can provide direction for designing critical experiments prior to the embarking on the time-consuming process of empirical testing of molecular function. See Table S3 in Supplementary Material for details of checkpoint molecule structure.
CTLA-4, CD28, CD80, and CD86
Devil CTLA-4 and CD28 are predicted to be 25 and 28 kDa, respectively, type-I transmembrane proteins with a single Ig-like V-type extracellular domain (ECD) (Figure 1). Protein sequence identity for CTLA-4 between devils and opossums was 86% and ranged from 63 to 69% between devils and placental mammals (Figure 2; Table 2). Percent identity for CD28 among species was lower than for CTLA-4, with 72% between devils and opossums and ranging from 54 to 57% between devils and placental mammals (Figure 3). Devil CTLA-4 had approximately 29% amino acid sequence identity with CD28. The CTLA-4 and CD28 coreceptors CD80 and CD86 are predicted to both be 35 kDa type-I transmembrane proteins that contain Ig-like V-type and an Ig-like C2-type domains. Devil CD80 and CD86 share only 21% sequence identity with each other, and the identity across the species analyzed here ranges from 34 to 55% and 32–49% for CD80 (Figure 4; Table 2) and CD86 (Figure 5; Table 2), respectively; the percent identity for CD80 and CD86 across species is the lowest of any of the checkpoint molecules compared here. All intra- and intermolecular disulfide bonds that have been identified in humans for CTLA-4, CD28, CD80, and CD86 are likely present in all the orthologs analyzed here, as we observed 100% conservation of cysteine residues at these sites in all species tested (Figures 2–5).

Figure 2. Alignment of cytotoxic T-lymphocyte-associated protein 4 reference genes for nine species. The predicted signal peptides (SigP), extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD) are demarcated with bars. “d” above the alignment represents predicted disulfide bonds. MYPPPY indicates the conserved CD80- and CD86-binding site. GVYVKM indicates a conserved cytoplasmic inhibitory domain and TRAF indicates a putative TRAF6-binding site. The black bars graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is shown in the bottom right corner of the alignment.
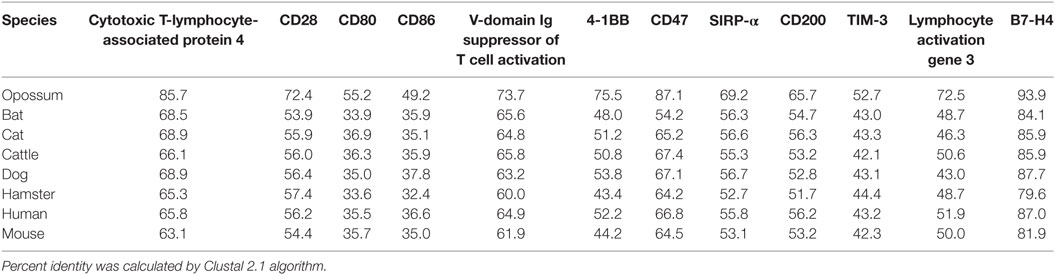
Table 2. Protein sequence percent identity between checkpoint molecules in devils and eight other species.

Figure 3. Alignment of CD28 reference genes for nine species. The predicted signal peptides (SigP), extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD) are demarcated with bars. “d” above the alignment represents predicted disulfide bonds. MYPPPY indicates the conserved CD80- and CD86-binding site, and SDYMNM and PYAP indicate key phosphorylation sites involved with cell proliferation and activation. The black bar graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is shown in the bottom right corner of the alignment.
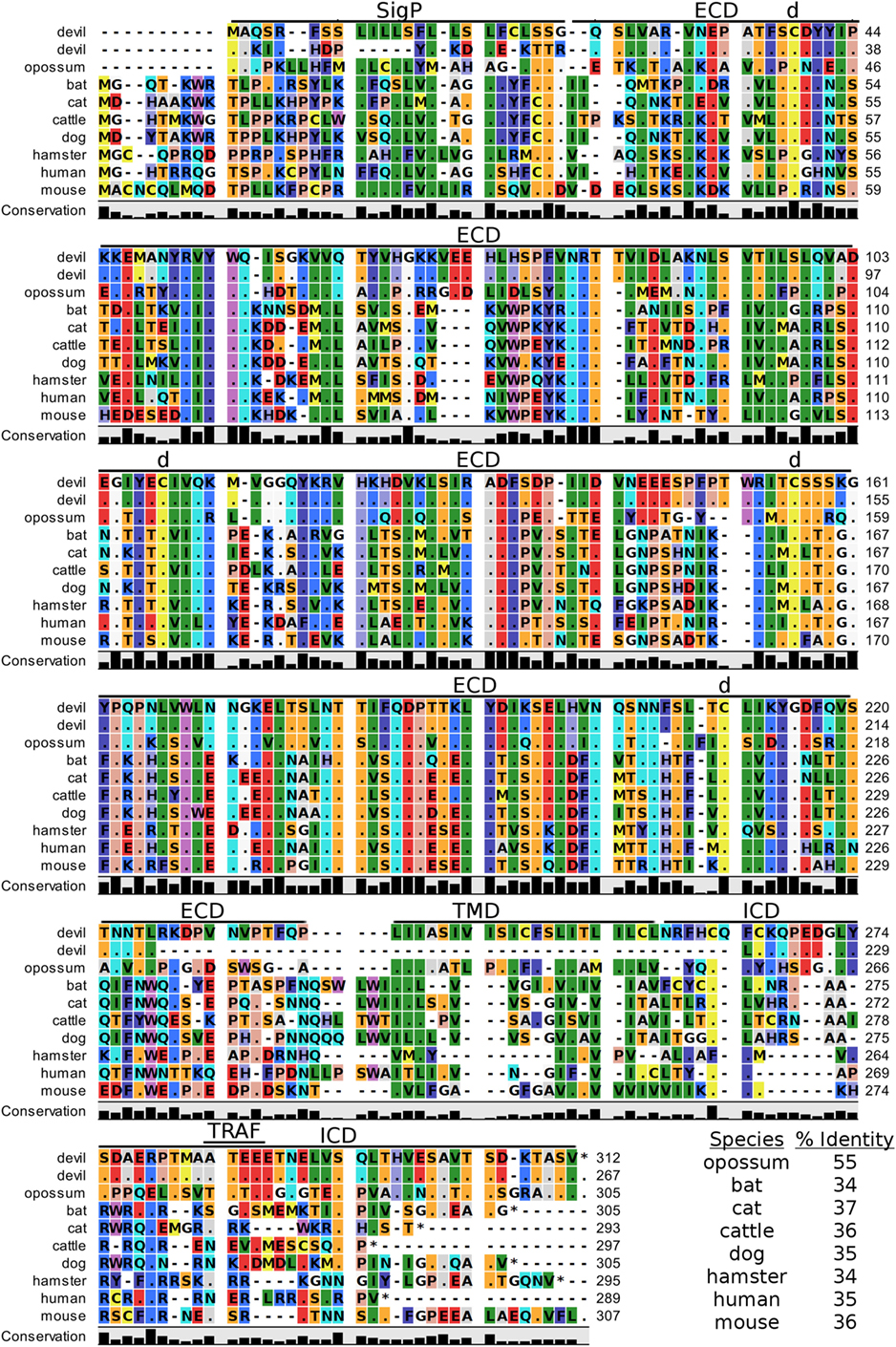
Figure 4. Alignment of CD80 reference genes for nine species. The predicted signal peptides (SigP), extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD) are demarcated with bars. The uppermost devil CD80 sequence is the full-length sequence and the lower devil CD80 is the potential secreted CD80 sequence. “d” above the alignment represents predicted disulfide bonds. TRAF indicates a putative TRAF1/2-binding site. The black bar graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is shown in the bottom right corner of the alignment.

Figure 5. Alignment of CD86 reference genes for nine species. The predicted signal peptides (SigP), extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD) are demarcated with bars. “d” above the alignment represents predicted disulfide bonds. TRAF indicates a putative TRAF6-binding site. The black bar graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is shown in the bottom right corner of the alignment. See Table 2 for the sequence identity between devils and other species.
Key Ligand-Binding Regions Conserved in Devil CTLA-4 and CD28
Our analysis shows that devil CTLA-4 and CD28 both contain the MYPPPY motif that is necessary for binding of CTLA-4 and CD28 to CD80 and CD86 in humans and mice (Figures 2 and 3) (7). Of the 18 combined CTLA-4 and CD28 sequences aligned here, only cattle CD28 did not contain the CD28MYPPPY motif (71); cattle had a CD28LYPPPY sequence, which matches the CD28LYPPPY and CTLA-4LYPPPY that has been previously reported in pigs (Sus scrofa) and decreases affinity of porcine CTLA-4 for human CD80 and CD86 (35, 72). Additionally, all species also contain the CTLAGVYVKM inhibitory domain. Except for bats, all species also contain the key CD28SDYMNM and CD28PYAP motifs are critical for intracellular signaling that promotes T cell proliferation and survival via the CD28–PI3K–AKT [reviewed in Ref. (73)]. Bats have a CD28M228L substitution in the CD28SDYMNM motif.
Potential Unique Signaling Motifs Identified
The two marsupials examined contain a putative TRAF6-binding domain in CTLA-4 proteins that was not present in any of the placental mammals. We identified a putative ITIM in predicted the ECD of dog CD80VKYGDL and a TRAF1/2-binding site in the devil CD80ATEEE cytoplasmic domain (Figure 4). Our analysis predicted a TRAF6-binding site in the cytoplasmic tail of devil CD86GHPKERDEV, a motif not predicted for CD86 in other species (Figure 5). Interestingly, the ELM tool predicted a putative ITIM domain in the second Ig-like segment of the human CD86IEYDGI ECD. To our knowledge, this putative ITIM has not been previously reported, and thus the potential inhibitory capacity of this motif is unknown.
V-Domain Ig Suppressor of T Cell Activation
Devil VISTA (PD-1H, C10orf54, Gi24, DD1α, Dies1) is a type-I transmembrane protein in the Ig superfamily predicted to be 33 kDa. The full coding sequence for this gene was previously not available in the devil genome (Devil_ref v7.0), but our de novo RNA transcriptome assembly uncovered a full coding devil VISTA transcript. Devil VISTA shares 60–74% sequence identity at the protein level with the other species analyzed here (Figure 6; Table 2). The ELM tool predicted a TRAF1/2-binding site in the ECD of devil VISTASIQE.
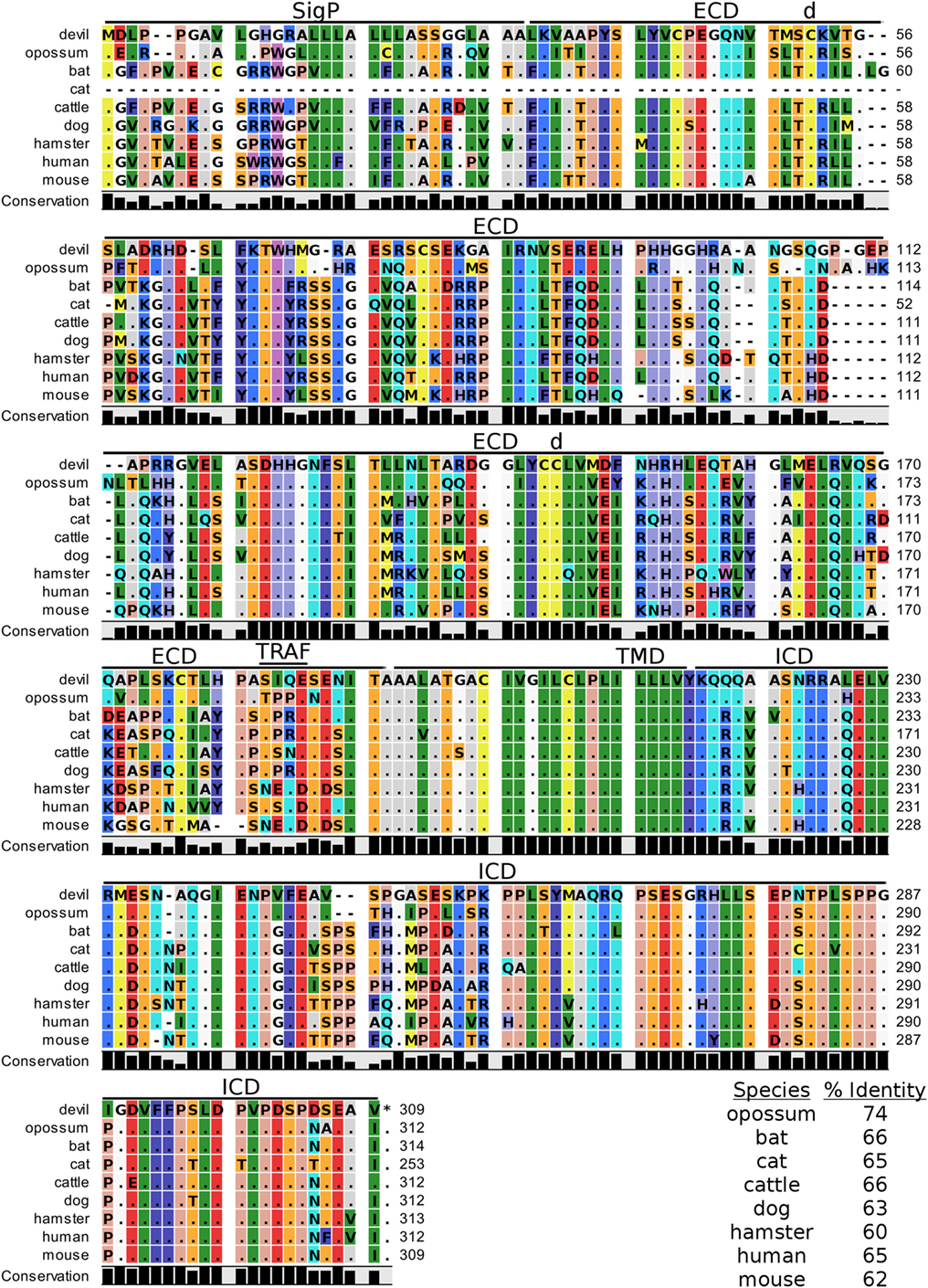
Figure 6. Alignment of V-domain Ig suppressor of T cell activation reference genes for nine species. The predicted signal peptides (SigP), extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD) are demarcated with bars. “d” above the alignment represents predicted disulfide bonds and “TRAF” indicates a potential TRAF1/2-binding site. The black bar graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is shown in the bottom right corner of the alignment.
4-1BB
4-1BB (TNFRSF9, CD137) is a predicted 31 kDa member of the tumor necrosis factor (TNF) receptor family expressed predominantly as a type-I transmembrane homodimer (74, 75). 4-1BB ligand (4-1BBL, TNFSF9, CD137L) is a TNF family member of 20 kDa, expressed as a type II transmembrane homotrimer (Figure 1). SMART analysis of devil 4-1BB identified two TNFR-Cys repeat domains, whereas human 4-1BB contains four TNFR-Cys repeats (Figure 7). All cysteine repeats in the four human TNFR-Cys domains were conserved in devils, suggesting that devil 4-1BB might also contain four TNFR-Cys domains despite not being predicted by the SMART program. Indeed, all 20 cysteine residues involved in disulfide bonds in human 4-1BB were also present in devil 4-1BB. Overall sequence identity among species compared here ranged from 43 to 75%, with mouse and hamster 4-1BB having the lowest percent identities with devil 4-1BB (Table 2).
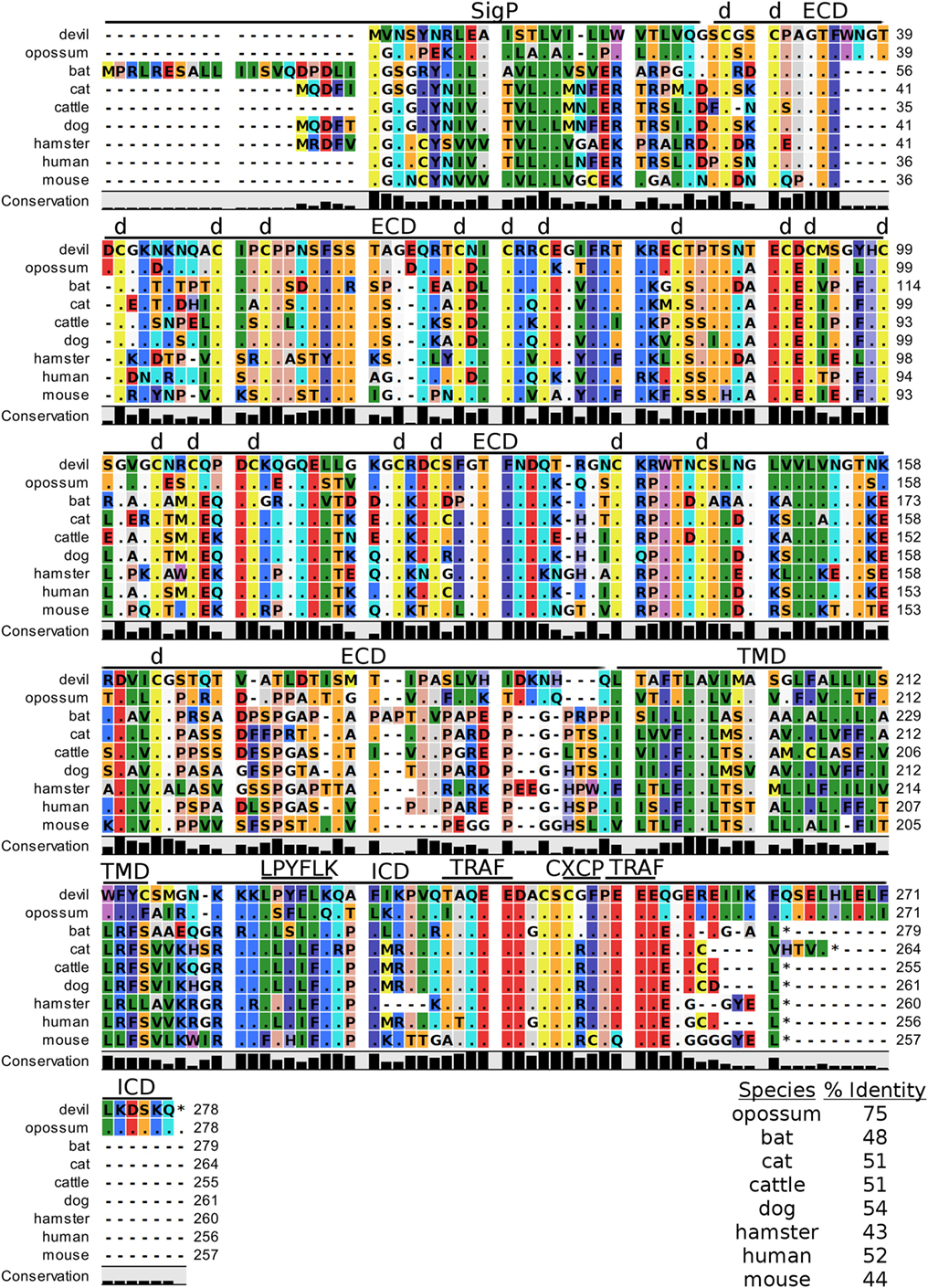
Figure 7. Alignment of 4-1BB reference genes for nine species. The predicted signal peptides (SigP), extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD) are demarcated with bars. “d” above the alignment represents predicted disulfide bonds. Two potential TRAF1/2-binding sites are demarcated by “TRAF.” “CXCP” marks a motif associated with optimal T cell response in mice, and “LPYFLK” denotes a potential inhibitory domain in devils. The black bar graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is shown in the bottom right corner of the alignment.
We identified a putative inhibitory domain in devil 4-1BBLPYFLK based on alignment with a potential inhibitory domain in human 4-1BBLLYIFK (76, 77). The putative inhibitory domain resembles an ITIM domain but lacks the I, L, or V in the terminal amino acid position. To date, this potential inhibitory domain has only been reported in humans, but several species analyzed here have similar motifs. Additionally, mice have a 4-1BBCXCP motif in the cytoplasmic domain that facilitates binding of CD4 and CD8 molecules to p56lck, a protein tyrosine kinase that is important for optimal T cell responses (78, 79); of the nine species compared here, mice are the only species that have the 4-1BBCXCP sequence. Our analysis using the ELM tool predicted two TRAF1/2 ligand-binding sites (devil 4-1BBTAQEE, 4-1BBPEEE), which align with the known TRAF1/2-binding sites in human and mouse 4-1BB (80).
CD47
Human CD47, also known as integrin-associated protein for its role in cell adhesion, is a cell surface protein that contains five transmembrane segments and a single Ig-like V-type domain (81, 82). The most up-to-date gene sequences for devil CD47 in Ensembl (ENSSHAT00000014546.1) did not contain a full coding sequence (i.e., no start codon) and the GenBank (XM_012544496) gene had an extended 5′ region that did not align well with other species. Our de novo RNA transcriptome assembly uncovered a full coding transcript for CD47 from devil PBMCs that aligned well with other species analyzed here. Our analysis suggests that the 34 kDa devil CD47 also contains five transmembrane segments and a single Ig-like domain (Figure 8). The four cysteines needed for the disulfide bonds reported in human CD47 are conserved in all species examined here, and overall sequence identity across all species compared here ranges from 54 to 87% (Table 2). Curiously, the ELM tool predicted an ITIM in the devil CD47LKYHVV ECD. All other species except bats and hamsters also contain the putative ITIM domains, but the ELM tool classified the ITIM as an excluded motif due to the putative ITIM being located in ECD of the protein. We also identified a putative TRAF2-binding site in the third devil intracellular domain of CD47KAVEE that was conserved across all species examined here except bats.
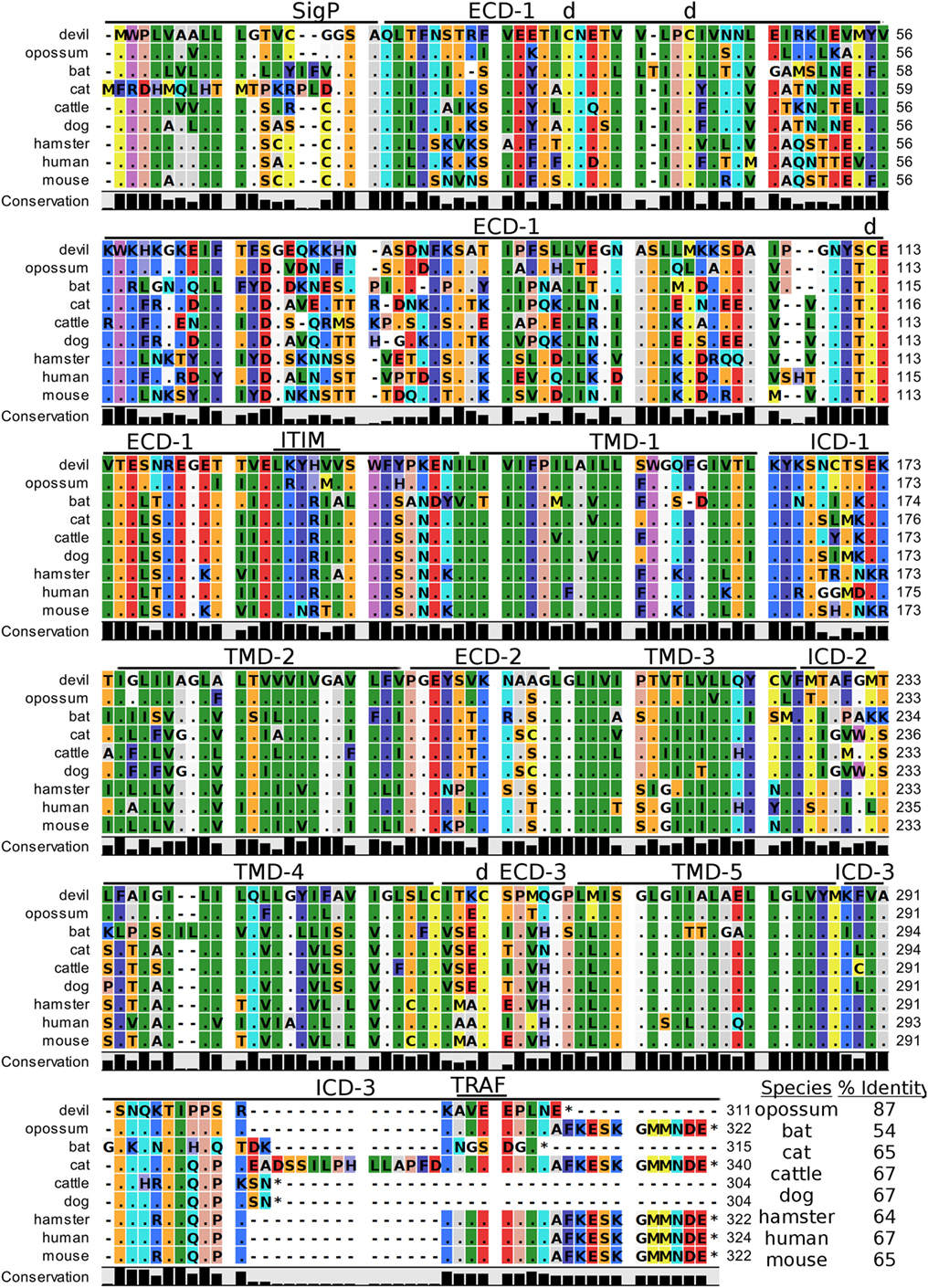
Figure 8. Alignment of CD47 reference genes for nine species. The predicted signal peptides (SigP), extracellular domains (ECDs), transmembrane domains (TMDs), and intracellular domain (ICD) are demarcated with bars. “d” above the alignment represents predicted disulfide bonds. “TRAF” indicates a potential TRAF1/2-binding site and immunoreceptor tyrosine-based inhibitory motif (ITIM) marks a putative ITIM motif predicted in the ECD. The black bar graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is shown in the bottom right corner of the alignment.
SIRP-α
Devil SIRP-α is a predicted 56 kDa protein. Human SIRP-α binds to CD47 as either a monomer or a dimer (83, 84) and forms three intramolecular disulfide bonds. All six of the cysteines that form intramolecular disulfide bonds in humans are conserved in devils (Figure S1 in Supplementary Material). The percent sequence identity for SIRP-α ranged from 53 to 69%. Analysis of devil SIRP-α revealed two putative ITIM and one ITSM domain. The ITIMITYADL shares 100% identity with all species except cattle, and the ITIMLTYADL is 100% conserved across all species. The devil ITSMSQTEYASI matches 5/7 amino acids with all other species; to our knowledge no ITSM has previously been reported in SIRP-α. We identified two potential SIRP-β1 orthologs in devils (ENSSHAT00000016950, XM_012553634.1) that contained ITIM motifs (SIRP-β1VSYNLI) in the ECD. The putative SIRP-β1 (ENSSHAT00000016950) ITIM aligned with an SIRP-β1VSYNIT sequence in SIRP-α that contains the key tyrosine residue for ITIMs but did not contain the canonical 3′ I/V/L.
CD200
Devil CD200 is predicted to be a 31 kDa type-I transmembrane protein that shares 52–66% sequence identity across the species compared here (Figure 9; Table 2). The cysteine residues that form intramolecular and intermolecular disulfide bonds necessary for binding as either a monomer or homodimer to CD200 receptor (CD200R) are 100% conserved across all species examined here (Figure 9) (85). Additionally, the Ig-like V-type and Ig-like C2-type domains of human and mouse CD200 were identified by the SMART tool. Many key motifs, including endocytic sorting signals, and TRAF2, SH2, and SH3 binding motifs, are present in all species compared here. Our analysis using ELM predicts a potential ITSM only in devil CD200. However, the devil CD200TLQE TRAF1/2-binding site and CD200NKTEYVVI ITSM are both located in the predicted ECD of devil CD200, so empirical testing is needed to determine if these are functional sites. Although not analyzed in depth here, CD200R and CD200R-like proteins were identified in the nine species compared here. The primary reference sequence for each species contained the key CD200RNPLY motif (86), which has been shown to be a key motif for CD200R inhibitory function (87).
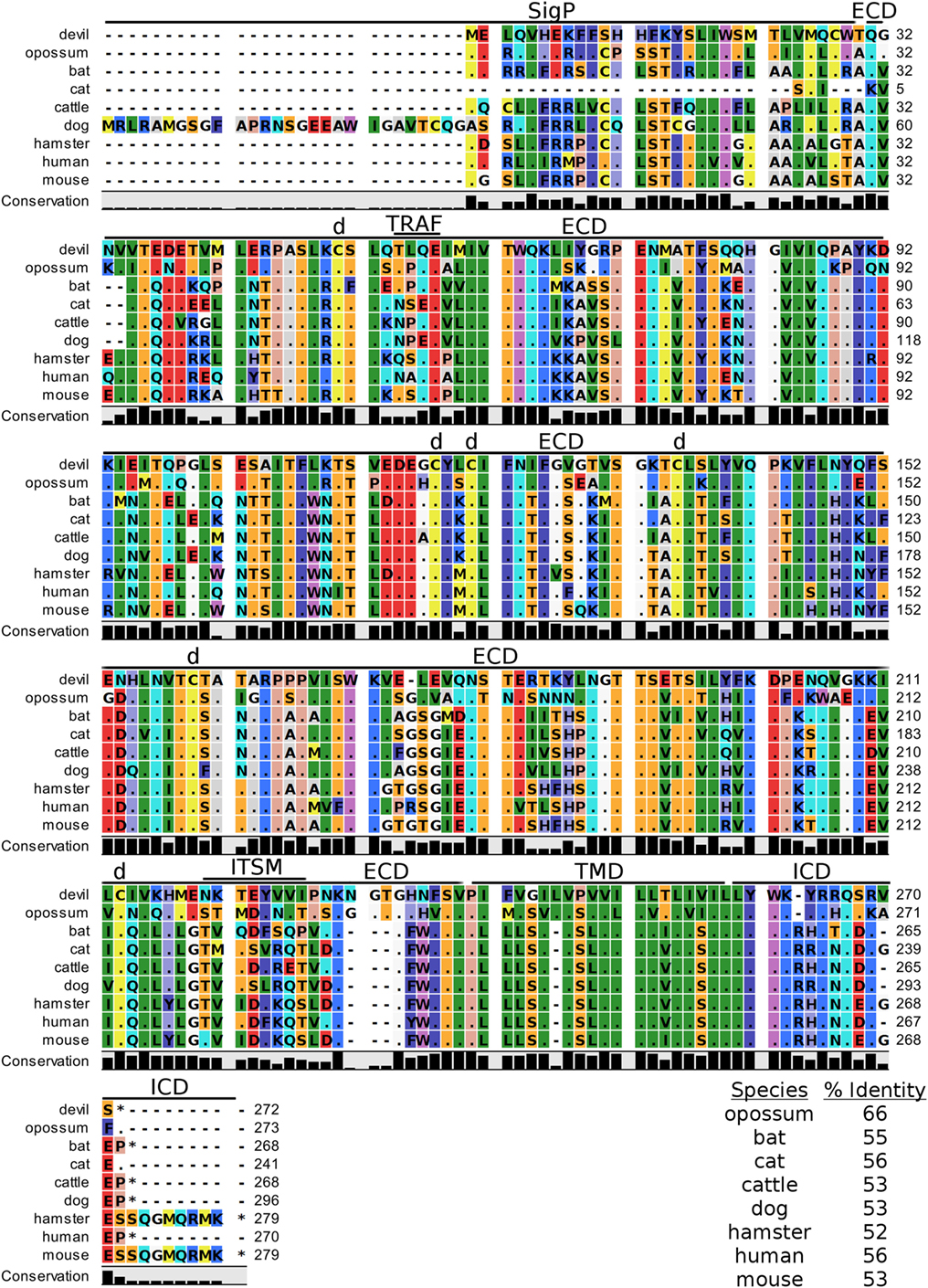
Figure 9. Alignment of CD200 reference genes for nine species. The predicted signal peptides (SigP), extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD) are demarcated with bars. “d” above the alignment represents predicted disulfide bonds. “TRAF” indicates a potential TRAF1/2-binding site and “ITSM” represents a putative immunoreceptor tyrosine-based switch motif (ITSM). The black bar graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is shown in the bottom right corner of the alignment.
TIM-3
TIM-3, also known as T-cell immunoglobulin and mucin domain-containing protein 3, hepatitis A virus cellular receptor 2, and CD366, is a surface protein expressed on T cells, Tregs, monocytes, macrophages, and DCs in mice and humans [reviewed in Ref. (88)]. The most up-to-date gene sequences for devil TIM-3 in Ensembl (ENSSHAT00000018107.1) and GenBank (XM_012552117.1) had truncated 3′ region of the coding sequence, and the Ensembl gene had an extended 5′ region that did not align with other species. Our de novo RNA transcriptome assembly uncovered a full coding devil TIM-3 transcript from devil PBMCs that aligned well with other species analyzed here. Analysis of the devil TIM-3 predicted sequence suggests that it is a 24 kDa protein with a Ig-like V-type domain and mucin domain in the extracellular region of the protein (Figure 10). Percent amino acid sequence identity between devils and the other eight species analyzed here ranges from 42 to 53%, which is the lowest identity for any molecule analyzed here except for CD80 and CD86 (Table 2). Despite this relatively low conservation of protein sequence, many of the key TIM-3 features identified in mice and humans appear to be present in devil TIM-3.
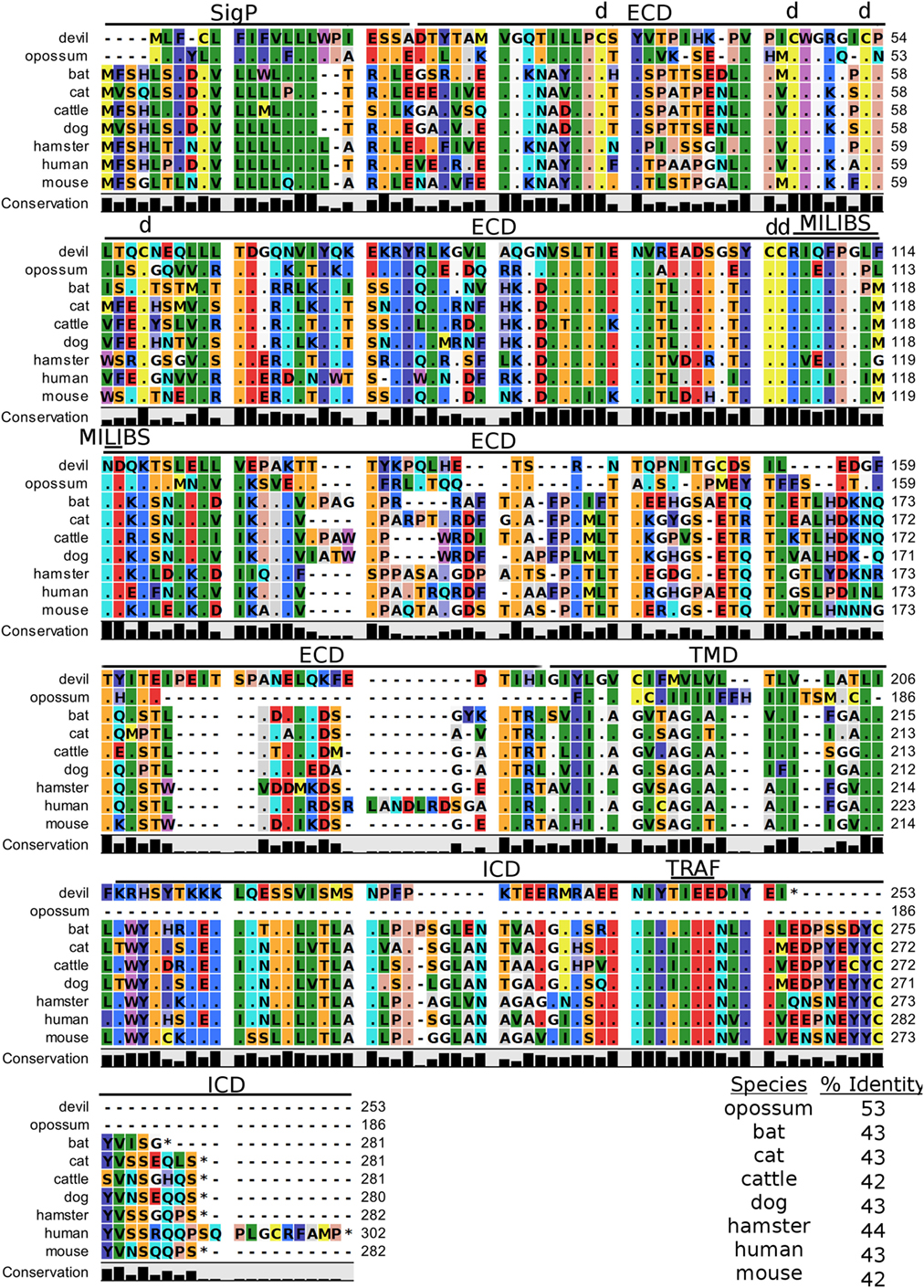
Figure 10. Alignment of TIM-3 reference genes for nine species. The predicted signal peptides (SigP), extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD) are demarcated with bars. “d” above the alignment represents predicted disulfide bonds. “TRAF” indicates a potential TRAF1/2-binding site and “MILIBS” demarcates a conserved metal ion-dependent ligand-binding site (MILIBS) found in phosphatidylserine on the surface of apoptotic cells. The black bar graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is shown in the bottom right corner of the alignment.
A unique feature of human TIM-3 is the double cleft which facilitates binding to multiple ligands (89, 90). The six cysteines that form three intramolecular disulfide bonds in the Ig-like region of human TIM-3 (89) are all present in devil TIM-3 (Figure 10), suggesting that devil TIM-3 folding and structure might be similar to human TIM-3. Analysis of human TIM-3 sequences identified five key amino acids for the two ligand-binding clefts; four of the five key amino acids were conserved in devil TIM-3. All species analyzed here also have two critical tyrosine residues in the TIM-3 cytoplasmic domain that align with mouse TIM-3Y256 and TIM-3Y263 (Figure 10), which are phosphorylated following ligation by GAL-9 and are associated with increased NFAT/AP-1 and NF-κB activity (91). A TIM-3TIEE TRAF1/2-binding site is located between these tyrosine residues in all species except for cats, dogs, and cattle (Figure 10). Also, mouse TIM-1, TIM-3, and TIM-4 all bind to phosphatidylserine on the surface of apoptotic cells via a conserved metal ion-dependent ligand-binding site (MILIBS). The mouse TIM-3RIQFPGLMND MILIBS motif is 100% conserved in cats, dogs, and cattle TIM-3, whereas devils have a phenylalanine instead of a methionine in this motif. Whether this TIM-3M114F affects phosphatidylserine binding and clearance of apoptotic cells in devils is unknown.
Lymphocyte Activation Gene 3
In humans and mice, LAG-3 (CD223) is an inhibitory type-I transmembrane protein that is structurally homologous to CD4 (92). Human and mice LAG-3 each contain one Ig-like V-type and three Ig-like C-type domains, whereas devil LAG-3 is predicted to be a 54 kDa protein with three Ig-like domains. However, a 100 amino acid (LAG3330–429) region downstream of the three predicted Ig-like domains could contain a fourth Ig-like domain that was not predicted by the SMART prediction tool (68, 93). Devil LAG-3 percent sequence identity with the other eight species analyzed here ranges from 43 to 73% (Table 2). In humans and mice, the LAG-3KIEELE (Figure S2 in Supplementary Material) motif in the cytoplasmic domain is critical for downstream inhibitory signaling interactions, but this exact sequence is not present in the other species analyzed here. The critical lysine (LAG-3K468) in this motif is present in devil (LAG-3KAEEME) and all other mammals tested here, with the exception of opossums (LAG-3IAEQME).
B7-H4
Devil V-set domain-containing T-cell activation inhibitor 1, or more commonly referred to as B7 homolog 4 (B7-H4), is a 30 kDa type-I membrane protein that contains two Ig-like V-type domains in the extracellular region (Figure 11). Devil B7-H4 protein shares 87% sequence identity with human B7-H4 and ranges from 80 to 94% for the other seven species analyzed here (Table 2). Based on homology to human and mouse B7-H4 and analysis of devil B7-H4 using cNLS Mapper (score of 3.4) (94, 95), it is likely that devil B7-H4KKRGH contains a bipartite nuclear localization signal (NLS). Interestingly, B7-H4 has only two amino acids in the cytoplasmic domain when expressed as a surface protein.
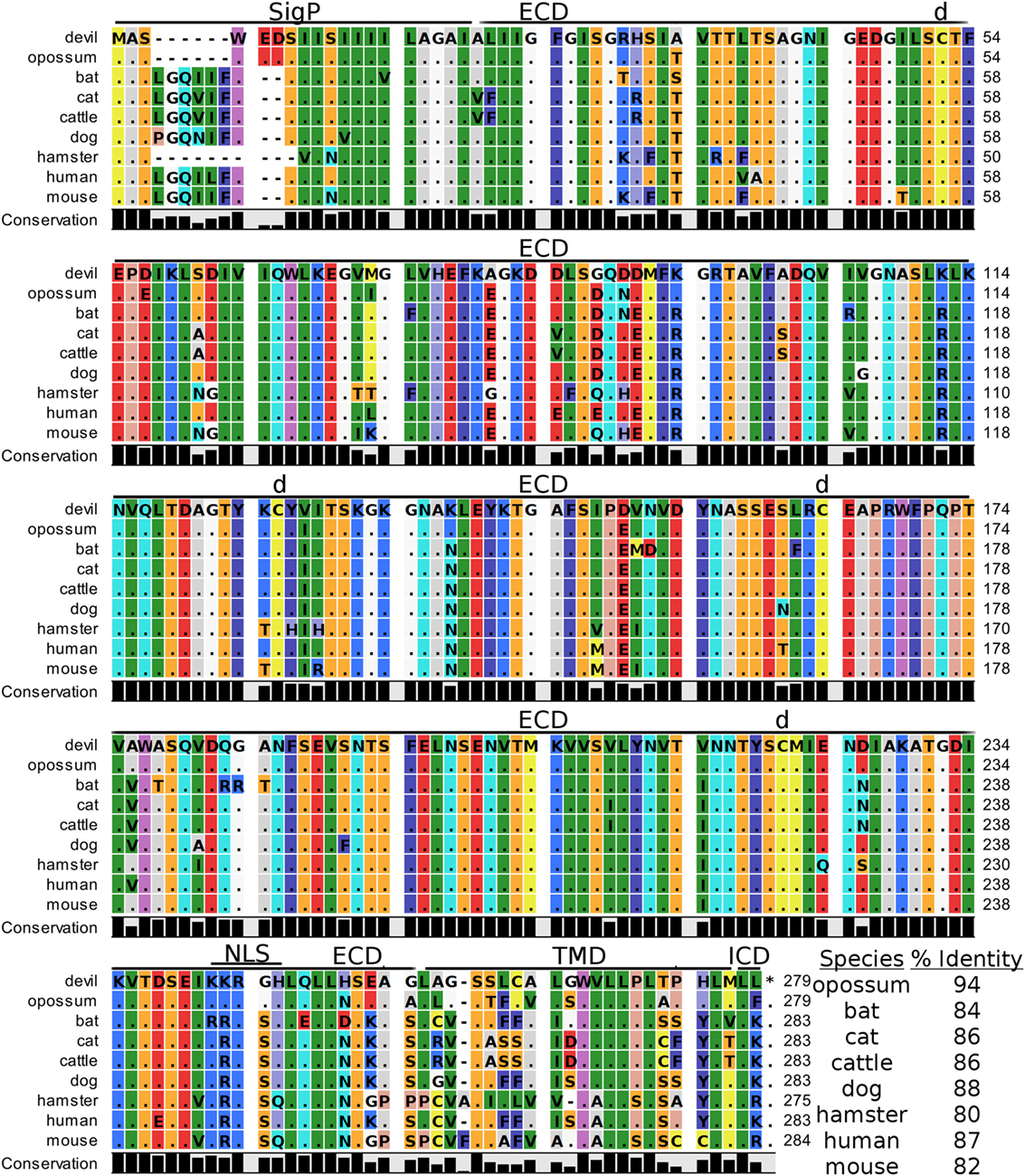
Figure 11. Alignment of V-set domain-containing T-cell activation inhibitor 1 reference genes for nine species. The predicted signal peptides (SigP), extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD) are demarcated with bars. “d” above the alignment represents predicted disulfide bonds. NLS indicates a predicted nuclear localization signal. The black bar graphs below the alignment represent the conservation of amino acids across all nine species. The percent amino acid sequence identity between devils and other species is show in the bottom right corner of the alignment.
Discussion
Evolutionary Conservation of Key Protein Regions
Despite an estimated 162 million years of evolution since the last common ancestor of placental and non-placental mammals, most of the checkpoint molecules we analyzed have orthologs in Tasmanian devils. Importantly, many of the key signaling motifs and ligand-binding sites are highly conserved in the genes analyzed, particularly cysteine residues for intra- and intermolecular disulfide bonds and transmembrane and intracellular tyrosine residues for receptor tyrosine kinases. The strong conservation of the key protein regions suggests that inhibitory and stimulatory function of these molecules, which is relatively well-documented in humans and mice, is conserved in devils. Importantly, by following established procedures and focusing on the checkpoint molecules that have proven most clinically effective in humans, we can rapidly characterize key checkpoint molecules in devils and incorporate potent immunomodulatory agents into our DFT disease vaccine-development efforts.
The Web of CD28–CTLA-4 Cosignaling Pathways
Our analysis shows for the first time that the key CTLA-4MYPPPY and CD28MYPPPY are conserved in devils. Given that the MYPPPY motif present in chickens facilitates binding to mammalian CD80 and that a single leucine for methionine substitution in pig CTLA-4LYPPPY is sufficient to disrupt binding of porcine CTLA-4 to human CD80 (35, 72, 96), the evolutionary conservation of CTLA-4MYPPPY and CD28MYPPPY in devils suggests that the competitive binding of CTLA-4 and CD28 for CD80/86 should be present in devils.
In addition to the conserved binding motifs, the key CD28SDYMNN stimulatory domain (97) and CTLAGVYVKM inhibitory domains are also conserved in devils, suggesting that the complementary stimulatory and inhibitory functions of CD28 and CTLA-4 are present. The CTLA-4GVYVKM motif, in association with T-cell receptor-interacting molecule (TRIM), regulates surface expression of CTLA-4 (98). The TRIM-mediated recycling of CTLA-4 from the surface provides the means for cell-extrinsic inhibitory function by trans-endocytosis of CD80/86 (99). By trans-endocytosing, or in other words stripping CD80/86 from the surface of an opposing cell (i.e., APCs) and degrading the CD80/86 molecules in the CTLA-4-expressing cell, the potential of costimulation via CD80/86 binding to CD28 is eliminated. Whether trans-endocytosis or trogocytosis (100) occurs in devils is unknown. It could be readily tested by developing CD80 transgenic cell lines that have a fluorescent protein, such as green fluorescent protein (GFP), fused to the 3′ tail of CD80. Flow cytometric or immunofluorescence observations of GFP transferred from the transgenic cell line to the CTLA-4 expressing cells would demonstrate that this inhibitory pathway is conserved in devils.
The complex interactions involving CTLA-4 are further complicated by some anti-CTLA-4 monoclonal antibody effects that are mediated through Fc receptor signaling (11, 101, 102). Tregs expressing high levels of CTLA-4 can be killed via antibody-dependent cell-mediated cytotoxicity, thus reducing the inhibition of antitumor immunity. This could be a useful tactic for increasing T cell responses against neoantigens in DFT cells, but little is known about IgG isotypes and Fc receptors in devils (103). To our knowledge only a single IgG subclass has been identified in marsupials (103), and the number and type of Fc receptors are unknown. Investigations into devil isotypes and Fc receptors are needed to aid development of potential monoclonal antibody immunotherapies for devils. Additionally, we identified a potential TRAF1/2-binding site in devil CD80 and a TRAF6-binding site in devil CD86. TRAF6 is associated with NF-κB activation and production of IL-6 and IL-8, whereas TRAF2 is associated with IκB-kinases. The binding site for TRAF2 but not TRAF6 is required CD40-mediated antibody class switching (104–106). Specific targeting of CD80 or CD86 by monoclonal antibodies or soluble ligands could help to determine if additional IgG subclasses exist in devils and to understand the potential role of IL-6 and IL-8 in immune evasion by DFT cells.
In addition to binding to both CTLA-4 and CD28 in humans and mice, CD80 binds to PD-L1 with a greater affinity than to CD28, but less than affinity of CD80 for CTLA and PD-L1 for PD-1 (107, 108). In humans, overexpression of CD80 blocks PD-L1 expression by sequestering PD-L1 in the cytoplasm while simultaneously providing costimulation (109). Additional pleiotropic effects of CD80 include its ability to stimulate NK cells through both CD28-dependent and -independent pathways (110–112). As DFT1 cells are poorly immunogenic and do not express MHC-I, the ability to stimulate NK cells using CD80 could provide immunotherapeutic potential.
Soluble CD80 can also effectively block PD-1:PD-L1 interactions and provide costimulation (113, 114). Interestingly, in addition to the full-length devil CD80, we identified a devil CD80 splice variant that lacks a transmembrane domain (TMD) and likely codes for soluble version of this key checkpoint molecule. Humans are also known to express soluble CD80 and soluble PD-L1, and high plasma levels of PD-L1 are associated with poor prognosis in B cell lymphomas (115–117). The anti-PD-L1 monoclonal antibodies we have previously developed could be used to screen devil plasma for soluble PD-L1 and possibly serve as a prognostic test for DFT disease.
Devil 4-1BB Function Is Predicted to Resemble Human 4-1BB Function
Despite the potential for enhanced immunotherapies targeting the 4-1BB/L pathway, actual progress has been slow, in part due to differences between mice and humans. 4-1BB costimulation in both mice and humans leads to enhanced T cell-mediated immunity, but the effects are contrasting in human and mouse NK cells. In mice, 4-1BB stimulation leads to enhanced NK cell effector mechanisms, but in humans 4-1BB signaling inhibits NK effector activity and reverse signaling through 4-1BBL on tumor cells can induce IL-10 and TNF-α (77, 118). This is potentially mediated by a putative inhibitory motif 4-1BBLLYIFK in the human cytoplasmic domain that resembles an ITIM domain (76, 77). Interestingly, devils have a similar putative 4-1BBLPYFLK inhibitory domain that includes the key tyrosine residue, but opossum 4-1BB does not contain the putative inhibitory motif (4-1BBLSFLLQ). Comparative testing of opossum and devil 4-1BB should be able to determine the inhibitory capacity of these motifs. Devil 4-1BB also contains conserved TRAF1/2-binding sites that in humans are necessary for recruiting TRAF1 and TRAF2 and inducing expression of pro-survival genes, including Bcl-2, Bcl-XL, and Bfl-1 and decreasing expression of Bim, a pro-apoptotic molecule (119–121). Altogether, devil 4-1BB has key features that more closely resemble human 4-1BB, and thus devil 4-1BB might stimulate T cell function but inhibit NK cell function.
As expression of 4-1BB on T cells is largely dependent on activation state, combining agonistic anti-4-1BB immunotherapies with stimulatory cytokines presents an opportunity for promoting immune memory. For instance, CD28- or IL-15-mediated upregulation of 4-1BB combined with agonistic anti-4-1BB monoclonal antibodies or recombinant 4-1BBL could promote antigen-independent survival and expansion of memory CD8 T cells (122, 123); this could be critical for long-term memory against repeated exposure to DFT cells in the wild. Also of particular relevance to the DFT disease is that blockade of 4-1BB:4-1BBL pathways leads to prolonged allograft survival for heart (124), skin (124), eye (125), and intestinal (126) tissues in other species.
To Eat or Not to Eat: Phagocytic Checkpoints
CD47
CD47 functions as an identifier of “self” tissue and is expressed on nearly all human and mouse cells. It has become known as the “don’t eat me” signal due to its ability to inhibit phagocytosis following ligation of SIRP-α (CD172a) on phagocytic cells (i.e., APCs, macrophages) (127). Of particular relevance to transmissible cancers, which are both allografts and cancer, CD47 appears to play a crucial role in transplant tolerance. The potent inhibitory signaling capacity of CD47 was clearly demonstrated in a xenotransplant model in which phagocytosis of pig cells by human macrophages was abrogated by transfecting the pig cells with human CD47 (128, 129). Several other studies have confirmed the importance of the CD47:SIRP-α interaction in graft and xenotransplant tolerance (129–132). Interestingly, the intraspecies CD47:SIRP-α interaction and function seems to be conserved in most species, but interspecies compatibility of CD47:SIRP-α is low (133). This suggests that CD47:SIRP-α binding could be a barrier to interspecies transmission of tumors. The closest extant relatives of devils, the Eastern quoll (Dasyurus viverrinus) and spotted-tail quoll (Dasyurus maculatus), are also a conservation priority, so understanding the role of CD47 in preventing interspecies transmission could prove useful for conservation of quolls.
DFT1 and DFT2 are not xenotransplants in devils, but the CD47:SIRP-α interaction could be a key mechanism of inhibiting phagocytosis and allo-responses against the DFT cells. Interestingly, in rats and mice, CD47 has been implicated in myelin phagocytosis through modulation of CD47:SIRP-α interactions (134) This is an immune evasion pathway that may have been exploited by DFT1, which originated from a Schwann cell and expresses high levels of myelin (37). Despite the nearly ubiquitous expression of CD47 by all cell types, expression levels are highly variable and can be modulated by inflammation (135, 136). High levels of CD47 found on many types of human cancer, including leukemia (137–139), lymphoma (140), and several types of solid tumor cancers (141, 142) and are often associated with poor prognosis. Importantly, blockade of CD47:SIRP-α interactions stimulated enhanced antitumor responses in several mouse models of human cancer (141, 143, 144), and trials of anti-CD47 antagonistic monoclonal antibodies are underway in humans. The MYC oncogene transcription factor binds to the promotors of both CD47 and PD-L1 and expression of both CD47 and PD-L1 can be downregulated by targeting MYC (145), presenting an opportunity to simultaneously target both PD-L1 and CD47.
A drawback of the anti-CD47 monoclonal antibody treatment is that CD47 blockade can lead to anemia due to rapid depletion of red blood cells. However, this risk could potentially be minimized by injection of anti-CD47 mAbs or SIRP-α fusion proteins directly into accessible DFTs (i.e., tumors in the mouth and face) (146). In addition to facilitating cancer cell immune evasion, CD47 also affects T cell and NK cell homeostasis (147, 148). CD47 is suggested to facilitate longevity by reducing clearance by phagocytes (135). Blockade of CD47 could thus decrease memory cell longevity and increase turnover of T cells and NK cells and could be detrimental to long-term DFT immunity in wild devils that could be re-exposed to DFT.
V-Domain Ig Suppressor of T Cell Activation
In contrast to the “don’t eat me” signal from CD47 ligation, VISTA can function as an “eat me” signal via intercellular homophilic binding that affects clearance of apoptotic cells and immune surveillance by macrophages and T cells (149, 150). VISTA expression is upregulated in response to genotoxic stress in a p53-dependent manner, and homophilic VISTA interactions both in cis and trans among tumor cells, APCs, and T cells may facilitate tumor cell escape from immune surveillance (149). No polymorphisms in devil p53 have been reported (151), but given the chromosomal rearrangements and greater than 17,000 somatic mutation reported in DFT1 cells (152, 153), it seems probable that p53 would be at least occasionally activated in DFT cells and could induce upregulation of tolerogenic VISTA signaling.
The tolerogenic role of VISTA in humans and mice and the strong conservation of VISTA across the nine species analyzed here (only B7-H4 and CTLA-4 were more conserved) suggest that VISTA may play an important evolutionary role in maintaining the balance between tolerance and immunity in mammals. Despite high sequence identity, the ELM tool unexpectedly predicted a putative TRAF1/2 (VISTASIQE) binding site in the ECD of devil VISTA. TRAF binding sites are most commonly found in TNF receptors, such as CD27, CD40, OX40, and 4-1BB.
Interestingly, two independent research groups first identified VISTA, but each reported contrasting functional effects of anti-VISTA monoclonal antibodies. One study reported that mouse VISTA inhibited T cell proliferation and cytokine production in vitro, that antagonistic anti-VISTA monoclonal antibody treatment exacerbated experimental autoimmune encephalomyelitis, and that VISTA overexpression on tumor cells led to reduced antitumor immunity in mice (154). By contrast, a single dose of agonistic anti-VISTA monoclonal antibody completely abrogated graft-versus-host disease in a partial and complete allogenic hematopoietic cell transplantation in mice with mismatched MHC between donor and host (155). This is of particular relevance to DFT, which in most cases likely has mismatched MHC, so blockade of VISTA could prove useful for breaking tolerance to DFT cells. In addition to the role of VISTA in transplant tolerance, early studies suggest that VISTA could play a more general role in immune escape by tumor cells. Several groups have independently demonstrated that VISTA-deficient mice have enhanced antitumor immunity compared to wild-type mice (156, 157). These effects are independent of PD-1 but are amplified in VISTA/PD-1 double knockout mice (158).
TIM-3
Like VISTA, TIM-3 plays a role in phagocytosis of apoptotic cells and T cell immunity [reviewed in Ref. (88)]. Ligation of TIM-3 by phosphatidylserine on apoptotic cells induces an “eat me” signal (159–161). The interaction of TIM-3 with its coreceptors has been associated with diverse functional outcomes, including both inhibitory and stimulatory functions. TIM-3 also binds to secreted and surface-expressed galectin-9 (GAL-9, LGALS9) and negatively regulates Th1 immunity and IFN-γ production (162, 163). As DFT1 cells do not naturally express MHC-I or PD-L1 but upregulate both MHC-I and PD-L1 in response to IFN-γ, the potential role of TIM-3 in IFN-γ production warrants further investigation. TIM-3 and PD-1 are often coexpressed on exhausted CD8+ T cells (164) in mice and exhausted TILs in mice and humans (165–168).
In contrast to the inhibitory function of TIM-3 on adaptive T cells, several types of innate immune cells can be activated by TIM-3 ligation (169). Ligation of TIM-3 on mouse and human DCs promotes secretion of inflammatory cytokines (169). TIM-3 is upregulated by TGF-β on tumor-associated macrophages (TAMs) of the M2 phenotype and ligation of TIM-3 leads to increased NF-κB and IL-6 production and is associated with reduced survival in human hepatocellular carcinoma (170). TIM-3 also binds to the pleiotropic alarmin HMGB1 and suppresses antitumor immunity via interfering with the ability of HMGB1 to transport tumor-derived DNA to the endosomes of DCs (171). As large DFTs often become ulcerated and/or necrotic, it will be interesting to see if HMGB1 or other alarmins are expressed.
In regard to receptor structure, TIM-3 is unique due to the formation of both cis- and trans-heterodimers with carcinoembryonic antigen cell adhesion molecule 1. The cis heterodimerization is critical for stable surface expression of TIM-3 and its inhibitory function (172, 173). Together, the conserved disulfide bonds and ligand-binding amino acids, suggest that devil TIM-3 function might be similar to humans and mice. Several of the most widely used cell lines in laboratory research, including Chinese hamster ovary cells, Jurkat RMA-S human lymphoma cells, and 3T3 mouse fibroblast cells, express natural ligands that bind to TIM-3 tetramers (89). This presents the opportunity for the use of cross-reactive reagents for devils and other non-traditional study species, but as a cautionary note, it also has the potential to cause confusing results in attempts to understand checkpoint molecule interactions.
Paired Inhibitory and Stimulatory Receptors
CD47 binds to several coreceptors, including SIRP-γ, THSB1, and THSB2, which induce either activation or inhibition signals (174, 175). An additional SIRP-family protein, SIRP-β1, does not bind to CD47 and no ligand for SIRP-β1 has been identified to date in humans (176). The SIRP-family proteins are hypothesized to be paired receptors that have evolved to reduce pathogen exploitation of conserved motifs found in inhibitory receptors, essentially a decoy target system that facilitates pathogen control. Pathogens that trigger inhibitory signals via conserved motifs in inhibitory receptors would thus also trigger activation signals when exploiting the similar motifs in the activating receptor (177, 178). Analysis of devil SIRP-α revealed two predicted cytoplasmic ITIM and one ITSM inhibitory domains, suggesting the inhibitory capacity of the CD47:SIRP-α interaction is conserved in devils and could be an immunotherapy target. The devil SIRP-α ITSM was not detected in any other species, so empirical testing will be needed to clarify the potential for targeting the CD47:SIRP-α pathway in devils.
Like the SIRP-family proteins, members of the CD200R family are hypothesized to have evolved as paired activating and inhibitory receptors. Specifically, herpesviruses, cytomegaloviruses, pox viruses, and adenoviruses inhibit macrophage activation via CD200 mimics that bind to CD200R-family members (179–181). In recent years, CD200 has received greater interest as an inhibitory checkpoint molecule associated with antitumor immunity. CD200 is expressed on a wide range of tumors, including lymphomas (182), myelomas (183), leukemias (184), and gliomas (185), and CD200 expression is correlated with expression of epithelial cancer stem cell markers on prostate, brain, breast, and colon cancers (186, 187).
Expression of CD200 on tumor cells has been associated with impaired NK cell function, increased frequencies of Tregs, suppression of memory T cells, decreased CTL activity, and induction of tolerance to allografts (184, 188, 189). Interestingly, CD200 expression has also been associated with Th2-skewed cytokine profiles and prolonged allograft survival (190–193). The transplantable EMT6 mouse breast cancer line expresses low levels of CD200 in vitro, but upregulates CD200 following inoculation into immunocompetent mice (194); this is similar to PD-L1, which is often expressed at low levels in vitro, but rapidly upregulated in immune active environments. Given the ongoing transmission of DFT for at least 20 years, immunohistochemical analysis of archived DFT tissue samples should determine expression levels of CD200 in the tumor microenvironment, and understanding this potential inhibitory pathway could help direct vaccine-development efforts.
Like devil PD-1, devil CD200 and SIRP-α are predicted to contain ITSM domains capable of regulating TCR signal transduction. ITSMs have been reported for both PD-1 and SIRP-α, but to our knowledge this is the first report of an ITSM in CD200. Interestingly, the CD200 ITSM was predicted only for devils and not any of the other eight species analyzed here, although it is possible that other isoforms of CD200 exist. There have only been a limited number molecules that have been predicted to contain ITSMs: BTLA; 2B4 (CD244); Siglec7; Siglec9; SLAMF1 (CD150); Ly9 (SLAMF3); CD84 (SLAMF5); SLAMF6 (CD352); and SLAMF7 (195, 196). Paired inhibitory and stimulatory receptors are hypothesized to have evolved in response to exploitation of inhibitory receptors by pathogens. Thus, pathogen motifs that utilize the inhibitory receptor will also trigger the stimulatory signal, so it is interesting that the three of the families that contain ITSMs are also predicted to be paired-receptor families (SIRP, CD200, Siglecs). The original ITSM was identified in CD150 and was named “switch-motif” due to its ability to switch from recruitment of SHP-2 to SHIP in the presence of SH2D1A (17, 197). This flexible switching aspect of the paired receptors could be an additional evolved response to exploitation of inhibitory receptors by common pathogens.
The Unexplored Function of ECD ITIMs and ITSMs
The short cytoplasmic tail of CD200 and known soluble forms in humans suggests that the primary inhibition through CD200 is mediated via ligation of CD200Rs. However, as mentioned above we identified a potential inhibitory ITSM in the extracellular region of CD200NKTEYVVI. Interestingly, our analysis predicted putative inhibitory motifs in the extracellular region of several checkpoint molecules: devil CD47LKYHVV ITIM near the border of the first extracellular and TMDs; devil SIRP-β1VSYNLI extracellular ITIM; dog CD80VKYGDL extracellular ITIM; and human CD86IEYDGI extracellular ITIM.
To our knowledge, the function of ITIMs and ITSMs has only been explored in intracellular regions of checkpoint molecules. However, despite most checkpoint molecules being considered transmembrane proteins, the majority of checkpoint molecules covered in this analysis are only surface expressed in particular contexts. For instance, CTLA-4 protein can be detected in the cytoplasm prior to T cell activation, but then is transiently moved to the cell surface following activation. Trans-endocytosis of ITIM-containing CD80 (dog) or CD86 (human) could allow the ECD ITIMs to affect intracellular signaling. Also, CD80, CD86, CD200, CD47, all of which were identified to contain extracellular inhibitory domains, are part of multi-receptor or paired-receptor interactions and the potential role of ECD ITIMs and ITSMs may have been overlooked. This is potentially an understudied aspect of molecular and cellular immunology that warrants further investigation.
The Potential for LAG-3 As an Adjuvant
Although human LAG-3 has a higher affinity for MHC-II than CD4, the inhibitory effects of LAG-3 in humans and mice are thought to be mediated primarily via negative regulation of TCR:CD3 signal transduction rather than competition for MHC-II binding (198, 199). In addition to binding to MHC-II, LAG-3 also binds to C-type lectin domain family 4 member G (CLEC4G, LSECTin) and galectin 3 (Gal-3, LGALS3). In mice, Gal-3 expression can suppress CD8 antitumor responses and expansion of plasmacytoid DCs (200). Tumor cell expression of Gal-3 has also been associated with poor prognosis in non-small-cell lung cancer (201), Hodgkin lymphoma (202), and acute myeloid leukemia (203), but the opposite effect has also been reported for breast (204) and gastric (205) cancer.
Of particular interest for devils is the potential for LAG-3 to facilitate anti-DFT immunity. Immunization of mice with LAG-3+ tumor cells, or irradiated tumor cells along with soluble LAG-3-Ig resulted in tumor regression or reduced tumor growth (206). In addition, soluble LAG-3-Ig binding to MHC-II reliably induced DC maturation, which is indicated by upregulation of MHC-II, costimulatory molecules CD40, CD80, CD83, CD86, and IL-12 and TNF-α cytokines (207, 208). In allogenic bone marrow transplant models, LAG-3 blockade abrogates CD8 T cell tolerance to tissue transplants (209), which suggests that LAG-3 blockade could be a target for inducing CD8 T cell responses to allogenic DFT cells. Additionally, because LAG-3 signaling is bidirectional, DFT expressed Gal-3 could inhibit T and NK cell responses via direct Gal-3:LAG-3 interactions. Finally, LAG-3 and PD-1 are coexpressed in exhausted T cells in viral infection and on TILs, and blockade of both LAG-3 and PD-1 has been demonstrated to have synergistic effects in mouse models (33, 210–212). Altogether, there is a strong potential for LAG-3 to stimulate anti-DFT immunity due to the potential for overexpression of LAG-3 on tumor cells and/or administration of soluble LAG-3-Ig to serve as an adjuvant, and to use monoclonal antibodies that block LAG-3 on T cells to reduce T cell inhibition.
Strong Conservation of B7-H4 Protein across All Species
Despite more than a decade of searching, the identity of the B7-H4 coreceptor remains unknown. Further complicating the functional role of B7-H4 protein is that it is commonly expressed in the cytoplasm and nucleus of cancer cells, and nuclear localization of B7-H4 is associated with cell cycle progression and proliferation of cancer cells (95). B7-H4 is upregulated to the cell surface on T and B cells by several mitogens (213, 214) and on monocytes, TAMs, and DCs in response to IL-6 and IL-10 (215, 216).
Despite the receptor for B7-H4 being unknown, the inhibitory function of B7-H4 in regard to T cell responses and apoptosis has been well-documented (213, 214, 217). Binding of B7-H4-Ig to the putative coreceptor inhibits T cell activation, which can be abrogated by monoclonal blockade of B7-H4 (213, 214). B7-H4 protein has been detected on breast, lung, ovarian, prostate, renal cell, and uterine endometrioid cancer cells and is associated with decreased survival [reviewed in Ref. (20, 218)]. The high sequence identity of B7-H4 between humans and devils (87%) suggests that B7-H4 function might be similar in devils, and thus could be used as a prognostic indicator for DFT in addition to the potential for anti-B7-H4 monoclonal antibody immunotherapy.
BTLA and TIGIT Inhibitory Genes Not Detected in Devils
The molecules described above demonstrated strong homology to other mammals, but another key inhibitory molecule in humans and mice, BTLA4, has not yet been identified in devils. Multiple BTLAs have been identified in bony fish (Teleosts), but have not been identified in amphibians or aves (24). Our search of GenBank and Ensembl and also our de novo assembly of a transcriptome from devil PBMCs failed to identify a BTLA gene and full coding sequence for the TIGIT gene in devils. Several TIGIT coreceptors are present in the devil genome and three TIGIT transcript variants reported in the opossum genome. Given that TIGIT expression appears to be tightly regulated in humans and mice (219), we believe it likely that a functional TIGIT gene will eventually be detected in devils through targeted laboratory methods (i.e., RACE PCR).
Conclusion
In addition to the highly conserved motifs in several molecules, we documented strong conservation of cysteine residues for intra- and intermolecular bonds. These remarkable similarities suggest that immunotherapies that have revolutionized human oncology might be translatable into immunotherapies for the DFT disease. The wealth of data on checkpoint molecule expression and function in humans should facilitate understanding of the key immune evasion pathways employed by the DFTs and allow development of vaccines that target these key pathways. However, given the general paucity of reagents for non-traditional study species, innovative new techniques may be needed to assess the potential roles of the putative signaling motifs, and more generally to assess checkpoint molecule function in species other than humans, mice, and rats.
Investigations into the function of checkpoint molecules in devils, dogs, and hamsters may provide insight into the transmissibility of these tumors and uncover potential immunotherapy targets relevant to both human and veterinary medicine. Additionally, our analysis has spawned new questions about the role of inhibitory ITIM and ITSM motifs and paired receptors that could be of highly relevant generally—not only to human medicine. Further insight into the evolution of the immune system might be gained by studying the role of checkpoint molecules in natural disease systems, such as white-nose syndrome in bats and chytridiomycosis in amphibians. Diversifying immunological studies into non-traditional species may facilitate a broader understanding of key immune defense pathways and elucidate general principles that are unlikely to be found when studying only a small number of model organisms.
Author Contributions
AF collected, analyzed, and interpreted the data; prepared the figures; and wrote the manuscript. NB performed the de novo transcriptome assembly, provided intellectual input, and provided critical feedback on the manuscript. AL and GW provided intellectual input on immunological processes and assisted in the drafting of relevant sections of the manuscript. JH provided intellectual input into the overarching framework of the study, preclinical insight into feasibility of immunotherapeutically targeting immune checkpoint inhibition in species other than humans and mice, and critical detailed review of the manuscript.
Conflict of Interest Statement
AF and GW received funding from an Entrepreneurs’ Programme Research Connections grant with Nexvet Australia Pty. Ltd. (RC50680). Nexvet is a global clinical-stage biopharmaceutical company focused on transforming the therapeutic market for companion animals by developing and commercializing novel biologic therapies. The other authors declare no conflict of interest.
Acknowledgments
The authors thank David Gearing and Sam Busfield (Nexvet Australia Pty. Ltd.) for critical review of the manuscript. The authors also would like to thank Alexander Kreiss, Amanda Patchett, Cesar Tovar, Georgina Kalodimos, Jocelyn Darby, Patrick Lennard, Chrissie Ong, and Ruth Pye for assistance in the lab and comments on research and/or manuscript, Elizabeth Murchison for advice in regard to the devil genome, Jac Charlesworth for help with data archiving, and Emily Flies for her ongoing research advice. The authors wish to thank Ginny Ralph and DPIPWE devil keepers who provided care for the devils used in these experiments.
Funding
Research support was provided by the Australian Research Council (ARC Linkage grant # LP0989727 and ARC Discovery grant # DP130100715) to GW, Morris Animal Foundation (# D14ZO-410), Sansom Institute Small Grants Scheme, University of Tasmania Foundation Dr Eric Guiler Tasmanian Devil Research Grant through funds raised by the Save the Tasmanian Devil Appeal (2013, 2015), and Entrepreneurs’ Programme—Research Connections grant with Nexvet Australia Pty. Ltd. (RC50680) to AF.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00513/full#supplementary-material.
References
1. Aruffo A, Seed B. Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. Proc Natl Acad Sci U S A (1987) 84:8573–7. doi:10.1073/pnas.84.23.8573
2. Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter JA. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med (1991) 173:721–30. doi:10.1084/jem.173.3.721
3. Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol (1991) 147:2461–6.
4. Janeway CA, Travers P, Walport M, Shlomchik M. Immunobiology: The Immune System in Health and Disease. 6th ed. New York, NY: Garland Science Publishing (2001).
5. Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature (1992) 356:607–9. doi:10.1038/356607a0
6. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily – CTLA-4. Nature (1987) 328:267–70. doi:10.1038/328267a0
7. Balzano C, Buonavista N, Rouvier E, Golstein P. CTLA-4 and CD28: similar proteins, neighbouring genes. Int J Cancer Suppl (1992) 7:28–32.
8. Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature (2001) 410:608–11. doi:10.1038/35069118
9. Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature (2001) 410:604–8. doi:10.1038/35069112
10. Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med (2014) 6:254ra128. doi:10.1126/scitranslmed.3008918
11. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520:373–7. doi:10.1038/nature14292
12. Ribas A, Kefford R, Marshall MA, Punt CJA, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol (2013) 31:616–22. doi:10.1200/JCO.2012.44.6112
13. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369:122–33. doi:10.1056/NEJMoa1302369
14. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373:23–34. doi:10.1056/NEJMoa1504030
15. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med (2015) 372:2006–17. doi:10.1056/NEJMoa1414428
16. Chatterjee P, Patsoukis N, Freeman GJ, Boussiotis VA. Distinct roles of PD-1 ITSM and ITIM in regulating interactions with SHP-2, ZAP-70 and Lck, and PD-1-mediated inhibitory function. Blood (2013) 122:191.
17. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol (2004) 173:945–54. doi:10.4049/jimmunol.173.2.945
18. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett (2004) 574:37–41. doi:10.1016/j.febslet.2004.07.083
19. Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood (2008) 111:3635–43. doi:10.1182/blood-2007-11-123141
20. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol (2008) 8:467–77. doi:10.1038/nri2326
21. Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med (2001) 193:839–46. doi:10.1084/jem.193.7.839
22. Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med (2014) 211:943–59. doi:10.1084/jem.20130790
23. Hansen JD, Du Pasquier L, Lefranc MP, Lopez V, Benmansour A, Boudinot P. The B7 family of immunoregulatory receptors: a comparative and evolutionary perspective. Mol Immunol (2009) 46:457–72. doi:10.1016/j.molimm.2008.10.007
24. Bernard D, Hansen JD, Du Pasquier L, Lefranc MP, Benmansour A, Boudinot P. Costimulatory receptors in jawed vertebrates: conserved CD28, odd CTLA4 and multiple BTLAs. Dev Comp Immunol (2007) 31:255–71. doi:10.1016/j.dci.2006.06.003
25. Esch KJ, Juelsgaard R, Martinez PA, Jones DE, Petersen CA. Programmed death 1-mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J Immunol (2013) 191(11):5542–50. doi:10.4049/jimmunol.1301810
26. Maekawa N, Konnai S, Ikebuchi R, Okagawa T, Adachi M, Takagi S, et al. Expression of PD-L1 on canine tumor cells and enhancement of IFN-g production from tumor-infiltrating cells by PD-L1 blockade. PLoS One (2014) 9:e98415. doi:10.1371/journal.pone.0098415
27. Hartley G, Faulhaber E, Caldwell A, Coy J, Kurihara J, Guth A, et al. Immune regulation of canine tumour and macrophage PD-L1 expression. Vet Comp Oncol (2016). doi:10.1111/vco.12197
28. Regan D, Guth A, Coy J, Dow S. Cancer immunotherapy in veterinary medicine: current options and new developments. Vet J (2015) 207:1–9. doi:10.1016/j.tvjl.2015.10.008
29. Khatlani TS, Ma Z, Okuda M, Onishi T. Molecular cloning and sequencing of canine T-cell costimulatory molecule (CD28). Vet Immunol Immunopathol (2001) 78:341–8. doi:10.1016/S0165-2427(01)00238-0
30. Shosu K, Sakurai M, Inoue K, Nakagawa T, Sakai H, Morimoto M, et al. Programmed cell death ligand 1 expression in canine cancer. In Vivo (2016) 30:195–204.
31. Folkl A, Wen X, Kuczynski E, Clark ME, Bienzle D. Feline programmed death and its ligand: characterization and changes with feline immunodeficiency virus infection. Vet Immunol Immunopathol (2010) 134:107–14. doi:10.1016/j.vetimm.2009.10.019
32. Ikebuchi R, Konnai S, Shirai T, Sunden Y, Murata S, Onuma M, et al. Increase of cells expressing PD-L1 in bovine leukemia virus infection and enhancement of anti-viral immune responses in vitro via PD-L1 blockade. Vet Res (2011) 42:103–17. doi:10.1186/1297-9716-42-103
33. Okagawa T, Konnai S, Deringer JR, Ueti MW, Scoles GA, Murata S, et al. Cooperation of PD-1 and LAG-3 contributes to T-cell exhaustion in Anaplasma marginale-infected cattle. Infect Immun (2016) 84:2779–90. doi:10.1128/IAI.00278-16
34. Ikebuchi R, Konnai S, Okagawa T, Yokoyama K, Nakajima C, Suzuki Y, et al. Blockade of bovine PD-1 increases T cell function and inhibits bovine leukemia virus expression in B cells in vitro. Vet Res (2013) 44:1–14. doi:10.1186/1297-9716-44-59
35. Parsons KR, Young JR, Collins BA, Howard CJ. Cattle CTLA-4, CD28 and chicken CD28 bind CD86: MYPPPY is not conserved in cattle CD28. Immunogenetics (1996) 43:388–91. doi:10.1007/s002510050080
36. Pearse A-M, Swift K. Allograft theory: transmission of devil facial-tumour disease. Nature (2006) 439:549. doi:10.1038/439549a
37. Murchison EP, Tovar C, Hsu A, Bender HS, Kheradpour P, Rebbeck CA, et al. The Tasmanian devil transcriptome reveals Schwann cell origins of a clonally transmissible cancer. Science (2010) 327:84–7. doi:10.1126/science.1180616
38. Murgia C, Pritchard JK, Kim SY, Fassati A, Weiss RA. Clonal origin and evolution of a transmissible cancer. Cell (2006) 126:477–87. doi:10.1016/j.cell.2006.05.051
39. vonHoldt BM, Ostrander EA. The singular history of a canine transmissible tumor. Cell (2006) 126:445–7. doi:10.1016/j.cell.2006.07.016
40. Ashbel R. Spontaneous transmissible tumours in the Syrian hamster. Nature (1945) 155:607–607. doi:10.1038/155607b0
41. Brindley DC, Banfield WG. A contagious tumor of the hamster. J Natl Cancer Inst (1961) 26:949–57. doi:10.1093/jnci/26.4.949
42. Metzger MJ, Villalba A, Carballal MJ, Iglesias D, Sherry J, Reinisch C, et al. Widespread transmission of independent cancer lineages within multiple bivalve species. Nature (2016) 534:705–9. doi:10.1038/nature18599
43. Metzger MJ, Reinisch C, Sherry J, Goff SP. Horizontal transmission of clonal cancer cells causes leukemia in soft-shell clams. Cell (2015) 161:255–63. doi:10.1016/j.cell.2015.02.042
44. Pye RJ, Pemberton D, Tovar C, Tubio JMC, Dun KA, Fox S, et al. A second transmissible cancer in Tasmanian devils. Proc Natl Acad Sci U S A (2016) 113:201519691. doi:10.1073/pnas.1519691113
45. Siddle HV, Kreiss A, Tovar C, Yuen CK, Cheng Y, Belov K, et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc Natl Acad Sci U S A (2013) 110:5103–8. doi:10.1073/pnas.1219920110
46. Ljunggren HG, Karre K. In search of the missing self: MHC molecules and NK cell recognition. Immunol Today (1990) 11:237–44.
47. Brown GK, Kreiss A, Lyons AB, Woods GM. Natural killer cell mediated cytotoxic responses in the Tasmanian devil. PLoS One (2011) 6:e24475. doi:10.1371/journal.pone.0024475
48. Brown GK, Tovar C, Cooray AA, Kreiss A, Darby J, Murphy JM, et al. Mitogen-activated Tasmanian devil blood mononuclear cells kill devil facial tumour disease cells. Immunol Cell Biol (2016) 94:673–9. doi:10.1038/icb.2016.38
49. Van Der Kraan LE, Wong ESW, Lo N, Ujvari B, Belov K. Identification of natural killer cell receptor genes in the genome of the marsupial Tasmanian devil (Sarcophilus harrisii). Immunogenetics (2013) 65:25–35. doi:10.1007/s00251-012-0643-z
50. Pye R, Hamede R, Siddle HV, Caldwell A, Knowles GW, Swift K, et al. Demonstration of immune responses against devil facial tumour disease in wild Tasmanian devils. Biol Lett (2016) 12:20160553. doi:10.1098/rsbl.2016.0553
51. Tovar C, Pye RJ, Kreiss A, Cheng Y, Brown GK, Darby J, et al. Regression of devil facial tumour disease following immunotherapy in immunised Tasmanian devils. Sci Rep (2017) 7:43827. doi:10.1038/srep43827
52. Flies AS, Lyons AB, Corcoran LM, Papenfuss AT, Murphy JM, Knowles GW, et al. PD-L1 is not constitutively expressed on Tasmanian devil facial tumor cells but is strongly upregulated in response to IFN-γ and can be expressed in the tumor microenvironment. Front Immunol (2016) 7:581. doi:10.3389/fimmu.2016.00581
53. Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, et al. Ensembl 2017. Nucleic Acids Res (2017) 45:D635–42. doi:10.1093/nar/gkw1104
54. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol (1990) 215:403–10. doi:10.1016/S0022-2836(05)80360-2
55. Patchett AL, Latham R, Brettingham-Moore KH, Tovar C, Lyons AB, Woods GM. Toll-like receptor signaling is functional in immune cells of the endangered Tasmanian devil. Dev Comp Immunol (2015) 53:123–33. doi:10.1016/j.dci.2015.07.003
56. Andrews S, Babraham B. FastQC A Quality Control Tool for High Throughput Sequence Data. (2010). Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
57. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol (2011) 29:644–52. doi:10.1038/nbt.1883
58. Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc (2013) 8:1494–512. doi:10.1038/nprot.2013.084
59. Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol (2000) 7:203–14. doi:10.1089/10665270050081478
60. James Kent W, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res (2002) 12:996–1006. doi:10.1101/gr.229102
61. Knudsen B, Knudsen T, Flensborg M, Sandmann H, Heltzen M, Andersen A, et al. CLC Sequence Viewer. (2012). Available from: http://www.clcbio.com
62. Feng DF, Doolittle RF. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol Evol (1987) 25:351–60. doi:10.1007/BF02603120
63. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol (2011) 7:539. doi:10.1038/msb.2011.75
64. Goodsell DS. Representing structural information with RasMol. In: Baxevanis AD, editor. Current Protocols in Bioinformatics. Hoboken, NJ: John Wiley and Sons, Inc. (2005). p. 5.4.1–5.4.23.
65. Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res (2003) 31:3784–8. doi:10.1093/nar/gkg563
66. Dinkel H, Van Roey K, Michael S, Davey NE, Weatheritt RJ, Born D, et al. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res (2014) 42:D259–66. doi:10.1093/nar/gkt1047
67. Käll L, Krogh A, Sonnhammer ELL. Advantages of combined transmembrane topology and signal peptide prediction – the Phobius web server. Nucleic Acids Res (2007) 35:W429–32. doi:10.1093/nar/gkm256
68. Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A (1998) 95:5857–64. doi:10.1073/pnas.95.11.5857
69. Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods (2011) 8:785–6. doi:10.1038/nmeth.1701
70. The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res (2014) 43:D204–12. doi:10.1093/nar/gku989
71. Metzler WJ, Bajorath J, Fenderson W, Shaw SY, Constantine KL, Naemura J, et al. Solution structure of human CTLA-4 and delineation of a CD80/CD86 binding site conserved in CD28. Nat Struct Biol (1997) 4:527–31. doi:10.1038/nsb0797-527
72. Vaughan AN, Malde P, Rogers NJ, Jackson IM, Lechler RI, Dorling A. Porcine CTLA4-Ig lacks a MYPPPY motif, binds inefficiently to human B7 and specifically suppresses human CD4+ T cell responses costimulated by pig but not human B7. J Immunol (2000) 165:3175–81. doi:10.4049/jimmunol.165.6.3175
73. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13:227–42. doi:10.1038/nri3405
74. Schwarz H, Tuckwell J, Lotz M. A receptor induced by lymphocyte activation (ILA): a new member of the human nerve-growth-factor/tumor-necrosis-factor receptor family. Gene (1993) 134:295–8. doi:10.1016/0378-1119(93)90110-O
75. Pollok KE, Kim Y-J, Zhou Z, Hurtado J, Kim KK, Pickard RT, et al. Inducible T cell antigen 4-1BB. Analysis of expression and function. J Immunol (1993) 150:771–81.
76. Marvel J, Walzer T. CD137 in NK cells. Blood (2010) 115:2987–8. doi:10.1182/blood-2010-01-261404
77. Baessler T, Charton JE, Schmiedel BJ, Gru F, Krusch M, Wacker A, et al. CD137 ligand mediates opposite effects in human and mouse NK cells and impairs NK-cell reactivity against human acute myeloid leukemia cells. Blood (2010) 115:3058–69. doi:10.1182/blood-2009-06-227934.An
78. Barao I. The TNF receptor-ligands 4-1BB-4-1BBL and GITR-GITRL in NK cell responses. Front Immunol (2012) 3:402. doi:10.3389/fimmu.2012.00402
79. Kim YJ, Pollok KE, Zhou Z, Shaw A, Bohlen JB, Fraser M, et al. Novel T cell antigen 4-1BB associates with the protein tyrosine kinase p56lck1. J Immunol (1993) 151:1255–62.
80. Arch RH, Thompson CB. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol Cell Biol (1998) 18:558–65. doi:10.1016/j.bulcan.2015.03.022
81. Brown E, Hooper L, Ho T, Gresham H. Integrin-associated protein: a 50-kD plasma membrane antigen physically and functionally associated with integrins. J Cell Biol (1990) 111:2785–94. doi:10.1083/jcb.111.6.2785
82. Campbell IG, Freemont PS, Foulkes W, Trowsdale J. An ovarian tumor marker with homology to vaccinia virus contains an IgV-like region and multiple transmembrane domains advances in brief an ovarian tumor marker with homology to vaccinia virus contains an IgV-like region and multiple transmembrane domains. Cancer Res (1992) 52:5416–20.
83. Lee WY, Weber DA, Laur O, Stowell SR, McCall I, Andargachew R, et al. The role of cis dimerization of signal regulatory protein α (SIRPα) in binding to CD47. J Biol Chem (2010) 285:37953–63. doi:10.1074/jbc.M110.180018
84. Weiskopf K, Ring AM, Ho CCM, Volkmer J-P, Levin AM, Volkmer AK, et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science (2013) 341:88–91. doi:10.1126/science.1238856
85. Hatherley D, Cherwinski HM, Moshref M, Barclay AN. Recombinant CD200 protein does not bind activating proteins closely related to CD200 receptor. J Immunol (2005) 175:2469–74. doi:10.4049/jimmunol.175.4.2469
86. Prigent SA, Gullick WJ. Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J (1994) 13:2831–41.
87. Zhang S, Phillips JH. Identification of tyrosine residues crucial for CD200R-mediated inhibition of mast cell activation. J Leukoc Biol (2005) 79:363–8. doi:10.1189/jlb.0705398
88. Le Mercier I, Lines JL, Noelle RJ. Beyond CTLA-4 and PD-1, the generation Z of negative checkpoint regulators. Front Immunol (2015) 6:418. doi:10.3389/fimmu.2015.00418
89. Cao E, Zang X, Ramagopal UA, Mukhopadhaya A, Fedorov A, Fedorov E, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity (2007) 26:311–21. doi:10.1016/j.immuni.2007.01.016
90. Anderson AC, Xiao S, Kuchroo VK. Tim protein structures reveal a unique face for ligand binding. Immunity (2007) 26:273–5. doi:10.1016/j.immuni.2007.03.004
91. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of tim-3 to T-cell receptor signaling pathways. Mol Cell Biol (2011) 31:3963–74. doi:10.1128/MCB.05297-11
92. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171:1393–405. doi:10.1084/jem.171.5.1393
93. Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res (2015) 43:D257–60. doi:10.1093/nar/gku949
94. Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A (2009) 106:10171–6. doi:10.1073/pnas.0900604106
95. Zhang L, Wu H, Lu D, Li G, Sun C, Song H, et al. The costimulatory molecule B7-H4 promote tumor progression and cell proliferation through translocating into nucleus. Oncogene (2013) 32(46):5347–58. doi:10.1038/onc.2012.600
96. O’Regan MN, Parsons KR, Tregaskes CA, Young JR. A chicken homologue of the co-stimulating molecule CD80 which binds to mammalian CTLA-4. Immunogenetics (1999) 49:68–71. doi:10.1007/s002510050464
97. Yin L, Schneider H, Rudd CE. Short cytoplasmic SDYMNM segment of CD28 is sufficient to convert CTLA-4 to a positive signaling receptor. J Leukoc Biol (2003) 73:178–82. doi:10.1189/jlb.0702365
98. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev (2009) 229:12–26. doi:10.1111/j.1600-065X.2009.00770.x
99. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell extrinsic function of CTLA-4. Science (2011) 332:600–3. doi:10.1126/science.1202947
100. Gu P, Fang Gao J, D’Souza CA, Kowalczyk A, Chou K-Y, Zhang L. Trogocytosis of CD80 and CD86 by induced regulatory T cells. Cell Mol Immunol (2012) 9:136–46. doi:10.1038/cmi.2011.62
101. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210:1695–710. doi:10.1084/jem.20130579
102. Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol (2014) 92:475–80. doi:10.1038/icb.2014.26
103. Sun Y, Wei Z, Li N, Zhao Y. A comparative overview of immunoglobulin genes and the generation of their diversity in tetrapods. Dev Comp Immunol (2012) 39:103–9. doi:10.1016/j.dci.2012.02.008
104. Zhang W, Zhang X, Wu X-L, He L-S, Zeng X-F, Crammer AC, et al. Competition between TRAF2 and TRAF6 regulates NF-kappaB activation in human B lymphocytes. Chin Med Sci J (2010) 25:1–12. doi:10.1016/S1001-9294(10)60013-2
105. Jabara HH, Laouini D, Tsitsikov E, Mizoguchi E, Bhan AK, Castigli E, et al. The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity (2002) 17:265–76. doi:10.1016/S1074-7613(02)00394-1
106. Purkerson JM, Smith RS, Pollock SJ, Phipps RP. The TRAF6, but not the TRAF2/3, binding domain of CD40 is required for cytokine production in human lung fibroblasts. Eur J Immunol (2005) 35:2920–8. doi:10.1002/eji.200526219
107. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity (2007) 27:111–22. doi:10.1016/j.immuni.2007.05.016
108. Butte MJ, Peña-Cruz V, Kim M-J, Freeman GJ, Sharpe AH. Interaction of human PD-L1 and B7-1. Mol Immunol (2008) 45:3567–72. doi:10.1016/j.molimm.2008.05.014
109. Haile ST, Bosch JJ, Agu NI, Zeender AM, Somasundaram P, Srivastava MK, et al. Tumor cell programmed death ligand 1-mediated T cell suppression is overcome by coexpression of CD80. J Immunol (2011) 186:6822–9. doi:10.4049/jimmunol.1003682
110. Wilson JL, Charo J, Martín-Fontecha A, Dellabona P, Casorati G, Chambers BJ, et al. NK cell triggering by the human costimulatory molecules CD80 and CD86. J Immunol (1999) 163:4207–12.
111. Chambers BJ, Salcedo M, Ljunggren HG. Triggering of natural killer cells by the costimulatory molecule CD80 (B7-1). Immunity (1996) 5:311–7. doi:10.1016/S1074-7613(00)80257-5
112. Kocak E, Abdessalam SF, May K, Martin EW, Liu Y. CD28 is not required for B7-mediated costimulation of Nk cell proliferation in response to tumor challenge. J Surg Res (2003) 114:301. doi:10.1016/j.jss.2003.08.101
113. Haile ST, Dalal SP, Clements V, Tamada K, Ostrand-Rosenberg S. Soluble CD80 restores T cell activation and overcomes tumor cell programmed death ligand 1-mediated immune suppression. J Immunol (2013) 191:2829–36. doi:10.4049/jimmunol.1202777
114. Haile ST, Horn LA, Ostrand-Rosenberg S. A soluble form of CD80 enhances antitumor immunity by neutralizing programmed death ligand-1 and simultaneously providing costimulation. Cancer Immunol Res (2014) 2:610–5. doi:10.1158/2326-6066.CIR-13-0204
115. Kakoulidou M, Giscombe R, Zhao X, Lefvert AK, Wang X. Human soluble CD80 is generated by alternative splicing, and recombinant soluble CD80 binds to CD28 and CD152 influencing T-cell activation. Scand J Immunol (2007) 66:529–37. doi:10.1111/j.1365-3083.2007.02009.x
116. Rossille D, Azzaoui I, Feldman AL, Maurer MJ, Labouré G, Parrens M, et al. Soluble programmed death-ligand 1 as a prognostic biomarker for overall survival in patients with diffuse large B-cell lymphoma: a replication study and combined analysis of 508 patients. Leukemia (2016) 31:988–91. doi:10.1038/leu.2016.385
117. Rossille D, Gressier M, Damotte D, Pangault C, Semana G, Le Gouill S, et al. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: results from a French multicenter clinical trial. Leukemia (2014) 28:2367–75. doi:10.1038/leu.2014.137
118. Navabi S s, Doroudchi M, Tashnizi AH, Habibagahi M. Natural killer cell functional activity after 4-1BB costimulation. Inflammation (2015) 38:1181–90. doi:10.1007/s10753-014-0082-0
119. Ward-Kavanagh LK, Lin WW, Šedý JR, Ware CF. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity (2016) 44:1005–19. doi:10.1016/j.immuni.2016.04.019
120. Mbanwi AN, Watts TH. Costimulatory TNFR family members in control of viral infection: outstanding questions. Semin Immunol (2014) 26:210–9. doi:10.1016/j.smim.2014.05.001
121. Wortzman ME, Clouthier DL, Mcpherson AJ, Lin GHY, Watts TH. The contextual role of TNFR family members in CD8+ T-cell control of viral infections. Immunol Rev (2013) 255:125–48. doi:10.1111/imr.12086
122. Zhu Y, Zhu G, Luo L, Flies AS, Chen L. CD137 stimulation delivers an antigen-independent growth signal for T lymphocytes with memory phenotype. Blood (2007) 109:4882–9. doi:10.1182/blood-2006-10-043463
123. Pulle G, Vidric M, Watts TH. IL-15-dependent induction of 4-1BB promotes antigen-independent CD8 memory T cell survival. J Immunol (2006) 176:2739–48. doi:10.4049/jimmunol.176.5.2739
124. Cho HR, Kwon B, Yagita H, La S, Lee EA, Kim JE, et al. Blockade of 4-1BB (CD137)/4-1BB ligand interactions increases allograft survival. Transpl Int (2004) 17:351–61. doi:10.1111/j.1432-2277.2004.tb00454.x
125. Asai T, Choi BK, Kwon PM, Kim WY, Kim JD, Vinay DS, et al. Blockade of the 4-1BB (CD137)/4-1BBL and/or CD28/CD80/CD86 costimulatory pathways promotes corneal allograft survival in mice. Immunology (2007) 121:349–58. doi:10.1111/j.1365-2567.2007.02581.x
126. Wang J, Guo Z, Dong Y, Kim O, Hart J, Adams A, et al. Role of 4-1BB in allograft rejection mediated by CD8+ T cells. Am J Transpl (2003) 3:543–51. doi:10.1034/j.1600-6143.2003.00088.x
127. Seiffert BM, Cant C, Chen Z, Rappold I, Brugger W, Kanz L, et al. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood (1999) 94:3633–44.
128. Ide K, Wang H, Tahara H, Liu J, Wang X, Asahara T, et al. Role for CD47-SIRPalpha signaling in xenograft rejection by macrophages. Proc Natl Acad Sci U S A (2007) 104:5062–6. doi:10.1073/pnas.0609661104
129. Wang C, Wang H, Ide K, Wang Y, van Rooijen N, Ohdan H, et al. Human CD47 expression permits survival of porcine cells in immunodeficient mice that express SIRPa capable of binding to human CD47. Cell Transplant (2011) 20:1915–20. doi:10.3727/096368911X566253
130. Wang Y, Wang H, Wang S, Fu Y, Yang YG. Survival and function of CD47-deficient thymic grafts in mice. Xenotransplantation (2010) 17:160–5. doi:10.1111/j.1399-3089.2010.00578.x
131. Wang H, Verhalen J, Madariaga ML, Xiang S, Wang S, Lan P, et al. Attenuation of phagocytosis of xenogeneic cells by manipulating CD47. Blood (2007) 109:836–42. doi:10.1182/blood-2006-04-019794
132. Waern JM, Yuan Q, Rüdrich U, Becker PD, Schulze K, Strick-Marchand H, et al. Ectopic expression of murine CD47 minimizes macrophage rejection of human hepatocyte xenografts in immunodeficient mice. Hepatology (2012) 56:1479–88. doi:10.1002/hep.25816
133. Subramanian S, Boder ET, Discher DE. Phylogenetic divergence of CD47 interactions with human signal regulatory protein alpha reveals locus of species specificity: implications for the binding site. J Biol Chem (2007) 282:1805–18. doi:10.1074/jbc.M603923200
134. Gitik M, Liraz-Zaltsman S, Oldenborg P-A, Reichert F, Rotshenker S. Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPα (signal regulatory protein-α) on phagocytes. J Neuroinflammation (2011) 8:24. doi:10.1186/1742-2094-8-24
135. Van VQ, Raymond M, Baba N, Rubio M, Wakahara K, Susin SA, et al. CD47high expression on CD4 effectors identifies functional long-lived memory T cell progenitors. J Immunol (2012) 188:4249–55. doi:10.4049/jimmunol.1102702
136. Baba N, Van VQ, Wakahara K, Rubio M, Fortin G, Panzini B, et al. CD47 fusion protein targets CD172a+ cells in Crohn’s disease and dampens the production of IL-1β and TNF. J Exp Med (2013) 210:1251–63. doi:10.1084/jem.20122037
137. Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res (2011) 71:1374–84. doi:10.1158/0008-5472.CAN-10-2238
138. Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell (2009) 138:271–85. doi:10.1016/j.cell.2009.05.046
139. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell (2009) 138:286–99. doi:10.1016/j.cell.2009.05.045
140. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell (2010) 142:699–713. doi:10.1016/j.cell.2010.07.044
141. Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A (2012) 109:6662–7. doi:10.1073/pnas.1121623109
142. Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med (2010) 2:63ra94. doi:10.1126/scitranslmed.3001375
143. Tseng D, Volkmer J-P, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A (2013) 110:11103–8. doi:10.1073/pnas.1305569110
144. Chao MP, Tang C, Pachynski RK, Chin R, Majeti R, Weissman IL. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood (2011) 118:4890–901. doi:10.1182/blood-2011-02-338020
145. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (2016) 352:227–31. doi:10.1126/science.aac9935
146. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi:10.1016/j.coi.2014.01.004
147. Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, et al. Functional CD47/signal regulatory protein alpha (SIRP(alpha)) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. Proc Natl Acad Sci U S A (2011) 108:13224–9. doi:10.1073/pnas.1101398108
148. Sockolosky JT, Dougan M, Ingram JR, Ho CCM, Kauke MJ, Almo SC, et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci U S A (2016) 113:E2646–54. doi:10.1073/pnas.1604268113
149. Yoon KW, Byun S, Kwon E, Hwang S-Y, Chu K, Hiraki M, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science (2015) 349:1261669. doi:10.1126/science.1261669
150. Muñoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol (2016) 16:741–50. doi:10.1038/nri.2016.99
151. Miller W, Hayes VM, Ratan A, Petersen DC, Wittekindt NE, Miller J, et al. Genetic diversity and population structure of the endangered marsupial Sarcophilus harrisii (Tasmanian devil). Proc Natl Acad Sci U S A (2011) 108:12348–53. doi:10.1073/pnas.1102838108
152. Deakin JE, Bender HS, Pearse AM, Rens W, O’Brien PCM, Ferguson-Smith MA, et al. Genomic restructuring in the Tasmanian devil facial tumour: chromosome painting and gene mapping provide clues to evolution of a transmissible tumour. PLoS Genet (2012) 8:e1002483. doi:10.1371/journal.pgen.1002483
153. Murchison EP, Schulz-Trieglaff OB, Ning Z, Alexandrov LB, Bauer MJ, Fu B, et al. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell (2012) 148:780–91. doi:10.1016/j.cell.2011.11.065
154. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208:577–92. doi:10.1084/jem.20100619
155. Flies DB, Wang S, Xu H, Chen L. Cutting edge: a monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol (2011) 187:1537–41. doi:10.4049/jimmunol.1100660
156. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res (2014) 74:1933–44. doi:10.1158/0008-5472.CAN-13-1506
157. Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. J Clin Invest (2014) 124:1966–75. doi:10.1172/JCI74589
158. Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A (2015) 112:6682–7. doi:10.1073/pnas.1420370112
159. Kobayashi N, Karisola P, Peña-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity (2007) 27:927–40. doi:10.1016/j.immuni.2007.11.011
160. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim Y-LE, Lee H-H, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol (2010) 184:1918–30. doi:10.4049/jimmunol.0903059
161. Savill J, Gregory C. Apoptotic PS to phagocyte TIM-4: eat me. Immunity (2007) 27:830–2. doi:10.1016/j.immuni.2007.12.002
162. Oomizu S, Arikawa T, Niki T, Kadowaki T, Ueno M, Nishi N, et al. Cell surface galectin-9 expressing Th cells regulate Th17 and Foxp3+ Treg development by galectin-9 secretion. PLoS One (2012) 7:e48574. doi:10.1371/journal.pone.0048574
163. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6:1245–52. doi:10.1038/ni1271
164. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117:4501–10. doi:10.1182/blood-2010-10-310425
165. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One (2012) 7:e30676. doi:10.1371/journal.pone.0030676
166. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207:2187–94. doi:10.1084/jem.20100643
167. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of TIm-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207:2175–86. doi:10.1084/jem.20100637
168. Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest (2014) 124:2425–40. doi:10.1172/JCI66375
169. Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of tissue inflammation by the immune receptor tim-3 expressed on innate immune cells. Science (2007) 318:1141–3. doi:10.1126/science.1148536
170. Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut (2015) 64:1593–604. doi:10.1136/gutjnl-2014-307671
171. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13:832–42. doi:10.1038/ni.2376
172. Huang Y-H, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517:386–90. doi:10.1038/nature13848
173. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi:10.1016/j.immuni.2016.05.001
174. Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol (2001) 11:130–5. doi:10.1016/S0962-8924(00)01906-1
175. Brooke G, Holbrook JD, Brown MH, Barclay AN. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J Immunol (2004) 173:2562–70. doi:10.4049/jimmunol.173.4.2562
176. Barclay AN, van den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol (2014) 32:25–50. doi:10.1146/annurev-immunol-032713-120142
177. Hatherley D, Graham SC, Turner J, Harlos K, Stuart DI, Barclay AN. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol Cell (2008) 31:266–77. doi:10.1016/j.molcel.2008.05.026
178. Hatherley D, Graham SC, Harlos K, Stuart DI, Barclay AN. Structure of signal regulatory protein alpha: a link to antigen receptor evolution. J Biol Chem (2009) 284:26613. doi:10.1074/jbc.M109.017566
179. Foster-Cuevas M, Wright GJ, Puklavec MJ, Brown MH, Barclay AN. Human herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J Virol (2004) 78:7667–76. doi:10.1128/JVI.78.14.7667-7676.2004
180. Foster-Cuevas M, Westerholt T, Ahmed M, Brown MH, Barclay AN, Voigt S. Cytomegalovirus e127 protein interacts with the inhibitory CD200 receptor. J Virol (2011) 85:6055–9. doi:10.1128/JVI.00064-11
181. Voigt S, Sandford GR, Hayward GS, Burns WH. The English strain of rat cytomegalovirus (CMV) contains a novel captured CD200 (vOX2) gene and a spliced CC chemokine upstream from the major immediate-early region: further evidence for a separate evolutionary lineage from that of rat CMV Maastricht. J Gen Virol (2005) 86:263–74. doi:10.1099/vir.0.80539-0
182. Dorfman DM, Shahsafaei A, Alonso MA. Utility of CD200 immunostaining in the diagnosis of primary mediastinal large B cell lymphoma: comparison with MAL, CD23, and other markers. Mod Pathol (2012) 25:1637–43. doi:10.1038/modpathol.2012.129
183. Tazawa S, Shiozawa E, Homma M, Arai N, Kabasawa N, Kawaguchi Y, et al. CD200 expression on plasma cell myeloma cells is associated with the efficacies of bortezomib, lenalidomide and thalidomide. J Clin Exp Hematop (2015) 55:1–5. doi:10.3960/jslrt.55.121
184. Coles S, Hills RK, Wang ECY, Burnett AK, Man ST, Darley RL, et al. Expression of CD200 on AML blasts directly suppresses memory T-cell function. Leukemia (2012) 26:2148–51. doi:10.1038/leu.2012.77
185. Moertel CL, Xia J, LaRue R, Waldron NN, Andersen BM, Prins RM, et al. CD200 in CNS tumor-induced immunosuppression: the role for CD200 pathway blockade in targeted immunotherapy. J Immunother cancer (2014) 2:46. doi:10.1186/s40425-014-0046-9
186. Kawasaki BT, Mistree T, Hurt EM, Kalathur M, Farrar WL. Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochem Biophys Res Commun (2007) 364:778–82. doi:10.1016/j.bbrc.2007.10.067
187. Kawasaki BT, Farrar WL. Cancer stem cells, CD200 and immunoevasion. Trends Immunol (2008) 29:464–8. doi:10.1016/j.it.2008.07.005
188. Coles SJ, Wang ECY, Man S, Hills RK, Burnett AK, Tonks A, et al. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia (2011) 25:792–9. doi:10.1038/leu.2011.1
189. Coles S, Hills RK, Wang ECY, Burnett AK, Man ST, Darley RL, et al. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells [Letter]. Leukemia (2012) 26:2146–8. doi:10.1038/leu.2012.75
190. Yu K, Gorczynski RM. Persistence of gene expression profile in CD200 transgenic skin allografts is associated with graft survival on retransplantation to normal recipients. Transplantation (2012) 94:36–42. doi:10.1097/TP.0b013e318257ad5c
191. Gorczynski L, Chen Z, Hu J, Kai Y, Lei J, Ramakrishna V, et al. Evidence that an OX-2-positive cell can inhibit the stimulation of type 1 cytokine production by bone marrow-derived B7-1 (and B7-2)-positive dendritic cells. J Immunol (1999) 162:774–81.
192. Gorczynski RM, Chen Z, Khatri I, Yu K. Graft-infiltrating cells expressing a CD200 transgene prolong allogeneic skin graft survival in association with local increases in Foxp3 +Treg and mast cells. Transpl Immunol (2011) 25:187–93. doi:10.1016/j.trim.2011.07.006
193. Gorczynski RM, Chen Z, He W, Khatri I, Sun Y, Yu K, et al. Expression of a CD200 transgene is necessary for induction but not maintenance of tolerance to cardiac and skin allografts. J Immunol (2009) 183:1560–8. doi:10.4049/jimmunol.0900200
194. Gorczynski RM, Chen Z, Diao J, Khatri I, Wong K, Yu K, et al. Breast cancer cell CD200 expression regulates immune response to EMT6 tumor cells in mice. Breast Cancer Res Treat (2010) 123:405–15. doi:10.1007/s10549-009-0667-8
195. Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol (2012) 188:2957–65. doi:10.4049/jimmunol.1100038
196. Davis SJ, van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol (2006) 7:803–9. doi:10.1038/ni1369
197. Shlapatska LM, Mikhalap SV, Berdova AG, Zelensky OM, Yun TJ, Nichols KE, et al. CD150 association with either the SH2-containing inositol phosphatase or the SH2-containing protein tyrosine phosphatase is regulated by the adaptor protein SH2D1A. J Immunol (2001) 166:5480–7. doi:10.4049/jimmunol.166.9.5480
198. Hannier S, Triebel F. The MHC class II ligand lymphocyte activation gene-3 is co-distributed with CD8 and CD3-TCR molecules after their engagement by mAb or peptide-MHC class I complexes. Int Immunol (1999) 11:1745–52. doi:10.1093/INTIMM/11.11.1745
199. Hannier S, Tournier M, Bismuth G, Triebel F. CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J Immunol (1998) 161:4058–65.
200. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res (2015) 3:412–23. doi:10.1158/2326-6066.CIR-14-0150
201. Yoshimura A, Gemma A, Hosoya Y, Komaki E, Hosomi Y, Okano T, et al. Increased expression of the LGALS3 (galectin 3) gene in human non-small-cell lung cancer. Genes Chromosomes Cancer (2003) 37:159–64. doi:10.1002/gcc.10205
202. Koh YW, Jung SJ, Park C-S, Yoon DH, Suh C, Huh J. LGALS3 as a prognostic factor for classical Hodgkin’s lymphoma. Mod Pathol (2014) 27:1338–44. doi:10.1038/modpathol.2014.38
203. Cheng C, Hou H, Lee M, Liu C, Jhuang J, Lai Y, et al. Prognostic factor for overall survival in patients with acute myeloid higher bone marrow LGALS3 expression is an independent unfavorable prognostic factor for overall survival in patients with acute myeloid leukemia. Blood (2013) 121:3172–80. doi:10.1182/blood-2012-07-443762
204. Senkus E, Kyriakides S, Ohno S, Penault-Llorca F, Poortmans P, Rutgers E, et al. Primary breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol (2015) 26:v8–30. doi:10.1093/annonc/mdv298
205. Okada K, Shimura T, Suehiro T, Mochiki E, Kuwano H. Reduced galectin-3 expression is an indicator of unfavorable prognosis in gastric cancer. Anticancer Res (2006) 26:1369–76.
206. Prigent P, El Mir S, Dréano M, Triebel F. Lymphocyte activation gene-3 induces tumor regression and antitumor immune responses. Eur J Immunol (1999) 29:3867–76. doi:10.1002/(SICI)1521-4141(199912)29:12<3867::AID-IMMU3867>3.3.CO;2-5
207. Andreae S, Piras F, Burdin N, Triebel F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223). J Immunol (2002) 168:3874–80. doi:10.4049/jimmunol.168.8.3874
208. Andreae S, Buisson S, Triebel F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood (2003) 102:2130–7. doi:10.1182/blood-2003-01-0273
209. Lucas CL, Workman CJ, Beyaz S, LoCascio S, Zhao G, Vignali DAA, et al. LAG-3, TGF-B, and cell-intrinsic PD-1 inhibitory pathways contribute to CD8 but not CD4 T-cell tolerance induced by allogeneic BMT with anti-CD40L. Blood (2011) 117:5532–40. doi:10.1182/blood-2010-11-318675
210. Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen H-R, Pyle KJ, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol (2009) 182:6659–69. doi:10.4049/jimmunol.0804211
211. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest (2007) 117:3383–92. doi:10.1172/JCI31184
212. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72:917–27. doi:10.1158/0008-5472.CAN-11-1620
213. Sica GL, Choi IH, Zhu G, Tamada K, Wang SD, Tamura H, et al. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity (2003) 18:849–61. doi:10.1016/S1074-7613(03)00152-3
214. Prasad DVR, Richards S, Mai XM, Dong C. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity (2003) 18:863–73. doi:10.1016/S1074-7613(03)00147-X
215. Kryczek I, Wei S, Zou L, Zhu G, Mottram P, Xu H, et al. Cutting edge: induction of B7-H4 on APCs through IL-10: novel suppressive mode for regulatory T cells. J Immunol (2006) 177:40–4. doi:10.4049/jimmunol.177.1.40
216. Kryczek I, Wei S, Zhu G, Myers L, Mottram P, Cheng P, et al. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res (2007) 67:8900–5. doi:10.1158/0008-5472.CAN-07-1866
217. Choi I-H, Zhu G, Sica GL, Strome SE, Cheville JC, Lau JS, et al. Genomic organization and expression analysis of B7-H4, an immune inhibitory molecule of the B7 family. J Immunol (2003) 171:4650–4. doi:10.4049/jimmunol.171.9.4650
218. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity (2016) 44:955–72. doi:10.1016/j.immuni.2016.05.002
Keywords: devil, transmissible tumor, cosignaling immunotherapy, checkpoint blockade, wild immunity, allograft, transplant rejection, evolution
Citation: Flies AS, Blackburn NB, Lyons AB, Hayball JD and Woods GM (2017) Comparative Analysis of Immune Checkpoint Molecules and Their Potential Role in the Transmissible Tasmanian Devil Facial Tumor Disease. Front. Immunol. 8:513. doi: 10.3389/fimmu.2017.00513
Received: 04 March 2017; Accepted: 18 April 2017;
Published: 03 May 2017
Edited by:
Evan W. Newell, Singapore Immunology Network (A*STAR), SingaporeReviewed by:
Daniel Olive, Institut national de la santé et de la recherche médicale (INSERM), FranceHaidong Dong, Mayo Clinic Minnesota, USA
Copyright: © 2017 Flies, Blackburn, Lyons, Hayball and Woods. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew S. Flies, YW5keS5mbGllc0B1dGFzLmVkdS5hdQ==