
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 April 2017
Sec. Inflammation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00475
This article is part of the Research Topic Danger signals triggering immune response and inflammation View all 17 articles
 Brad Griesenauer1,2,3
Brad Griesenauer1,2,3 Sophie Paczesny1,2,3*
Sophie Paczesny1,2,3*
Il1rl1 (also known as ST2) is a member of the IL-1 superfamily, and its only known ligand is IL-33. ST2 exists in two forms as splice variants: a soluble form (sST2), which acts as a decoy receptor, sequesters free IL-33, and does not signal, and a membrane-bound form (ST2), which activates the MyD88/NF-κB signaling pathway to enhance mast cell, Th2, regulatory T cell (Treg), and innate lymphoid cell type 2 functions. sST2 levels are increased in patients with active inflammatory bowel disease, acute cardiac and small bowel transplant allograft rejection, colon and gastric cancers, gut mucosal damage during viral infection, pulmonary disease, heart disease, and graft-versus-host disease. Recently, sST2 has been shown to be secreted by intestinal pro-inflammatory T cells during gut inflammation; on the contrary, protective ST2-expressing Tregs are decreased, implicating that ST2/IL-33 signaling may play an important role in intestinal disease. This review will focus on what is known on its signaling during various inflammatory disease states and highlight potential avenues to intervene in ST2/IL-33 signaling as treatment options.
In 1989, the Il1rl1 gene product, which has also been given the alias ST2 and defined as the IL-33 receptor, as it binds to IL-33, was discovered (1, 2). It belongs to the IL-1-receptor superfamily. The literature has been misnaming ST2 as “suppressor of tumorigenicity 2,” when in fact the original name was “growth stimulation expressed gene 2” (2) and has recently been renamed by the original discoverer, Shin-ichi Tominaga, as “serum stimulation-2” (3), as it was first discovered to function as a mediator of type 2 inflammatory responses (4). IL1RL1 is located on chromosome 2q12.1 in humans, while the gene “suppressor of tumorigenicity 2,” also called ST2, is located on chromosome 11p14.3-p12 in humans. In this review, we will call ST2 as the IL-33 receptor or Ilr1rl gene product.
ST2 has two main splice variants due to differential promoter binding: a membrane-bound form (ST2), which promotes NF-κB signaling, and a soluble form (sST2), which prevents its signaling. It was not until 2005 that the ligand for ST2, the cytokine IL-33, was identified through database searching for genes homologous to other IL-1 superfamily members (5, 6). IL-33 has been identified as a mediator of various inflammatory diseases such as asthma, cardiovascular diseases, and allergic diseases (6). Besides being secreted, IL-33 can be found in the nucleus of human high endothelial venules (7), lung airway epithelium, keratinocytes, fibroblastic reticular cells, and some epithelial cells of the stomach and salivary glands (8). Due to the presence of a N-terminal domain nuclear localization sequence and a homeodomain-like helix-turn-helix motif, IL-33 is able to bind heterochromatin, potentially giving IL-33 transcriptional regulatory capacity (7).
Dysregulation of ST2/IL-33 signaling and sST2 production have been implicated in a variety of inflammatory diseases such as cardiac disease (9–12), inflammatory bowel disease (IBD) (13–16), graft-versus-host disease (GVHD) (17–24), small bowel transplant rejection (25), and type 2 diabetes (26–29). The purpose of this review is to highlight the function of both ST2 and sST2, ST2/IL-33 in regard to different immune cells, and sST2 production and ST2 signaling in inflammatory diseases.
The ST2 gene is located on human chromosome 2q12.1 and is approximately 40 kb long. Homologs of ST2 are found in the genomes of mouse, rat, and fruit fly. ST2 has four splice isoforms from a single transcript dependent on the promoter being used: ST2, a membrane receptor; sST2, a soluble factor; ST2V, a variant form of ST2; and ST2LV, another variant form of ST2, which are differentially regulated through alternative promoter binding (30–32). Little is known about ST2V other than it is expressed highly in gastrointestinal organs (33). ST2LV lacks the transmembrane domain found in ST2; is secreted by eye, heart, lung, and liver tissues; and is found during later stages of embryogenesis (34). Other information on ST2LV is currently lacking.
By cloning the Il1rl1 gene in rat and sequencing sST2 and ST2 cDNAs, it was found that sST2 and ST2 have different exon 1 sequences (30). Mapping the promoter regions for Il1rl1 showed that the transcription start site for sST2 is in a proximal promoter region while the transcription start site for ST2 is in a distal promoter region, 15 kb upstream from the sST2 proximal promoter (30) (Figure 1). Three to four GATA transcription factors have been identified at the distal promoter region within 1,001 bp, two of which were conserved between human and mouse Il1rl1 genes (32, 35). These GATA elements binding to the distal promoter lead to ST2 expression. The transcription factor PU.1 also binds to the distal promoter near the GATA elements in both human mast cells and basophils (36). PU.1 and GATA2 cooperatively transactivate the distal ST2 promoter inducing expression of ST2, but not sST2 (36). Loss of PU.1 significantly decreased ST2 expression (36). Conversely, a PMA-responsive element has been found near the proximal promoter region of ST2 in the mouse fibroblast line NIH 3T3 (37). Similarly, activated human fibroblast line TM12, which only uses the proximal promoter for Il1rl1 transcription, led to sST2 expression (32). These data further suggest that the distal promoter is used to transcribe ST2 and the proximal promoter is used to transcribe sST2. To verify these results and find other transcription factors important in ST2 and sST2 expressions, ChIP-seq experiments should be performed.

Figure 1. Different promoter usage dictates ST2 and sST2 expressions. ST2 consists of two main splice isoforms: ST2 and sST2. These isoforms are splice variants of each other regulated by alternative promoter bindings, the distal promoter for ST2, and the proximal promoter for sST2. Exon 1 varies between ST2 and sST2 depending on the promoter being bound. In immune cells, GATA1, GATA2, and PU.1 have been shown to bind to the distal promoter. The proximal promoter has not been well studied; it is thought that a PMA-responsive element induced sST2 transcription (37).
ST2 was first found in serum-stimulated BALB/c-3T3 cells in the presence of cycloheximide (38). It contains an extracellular domain, which binds IL-33 with the help of IL-1 receptor accessory protein (IL-1RAP), a transmembrane domain, and an intercellular domain called a Toll/interleukin-1 receptor (TIR) domain. Due to the presence of the TIR domain, ST2 has been classified as a member of the IL-1 receptor superfamily. ST2 is expressed on cardiomyocytes (39) and a large variety of immune cells, including T conventional cells, particularly type 2 (40), regulatory T cells (Tregs) (41), innate helper 2 cells [innate lymphoid cell type 2 (ILC2)] (42), M2 polarized macrophages (43), mast cells (44), eosinophils (45), basophils (46), neutrophils (46), NK (47), and iNKT cells (47). Signaling through ST2 in immune cells induces type 2 and Treg immune responses, IgE production, and eosinophilia (5, 40–42, 48).
sST2 protein lacks the transmembrane and cytoplasmic domains contained on ST2 and contains a unique nine amino acid C-terminal sequence (35). In vitro, sST2 production has been shown to be enhanced by pro-inflammatory cytokines (IL-1β and TNF-α) in human lung epithelial cells and cardiac myocytes. In humans, sST2 can be not only produced spontaneously by cells in the lung, kidney, heart, and small intestine (49) but also produced after activation with IL-33 in mast cells (50) or anti-CD3/anti-CD28 in both CD4 and CD8 conventional T cells (51). In a murine GVHD model, it has recently been shown that intestinal Th17 and Tc17 cells produced large amounts of sST2 following alloreactivity (51). This enhanced sST2 presence has been shown to inhibit the production of the type 2 cytokines IL-4 and IL-5 but not the type 1 cytokine IFN-γ (52).
Upon IL-33 binding, the membrane-anchored ST2 forms a heterodimer along with IL-1RAP (53, 54) leading to the dimerization of the TIR domain. This leads to the recruitment of the TIR domain binding protein MyD88 and subsequent IL-1R-associated kinase activation, which can activate MAP kinases and NF-κB pathways (Figure 2) (5, 6). In regards to ST2/IL-33 signaling, how ST2/IL-33 signals specifically to either the MAPK or NF-κB is currently unclear. However, downstream events of ST2 do seem to occur differentially, as TRAF6 is required for NF-κB activation and induction of type 2 cytokines but TRAF6 is not needed for IL-33-induced ERK (a MAPK protein) activation (55). How TRAF6-independent activation of ERK occurs after IL-33 binding ST2 is currently unknown.
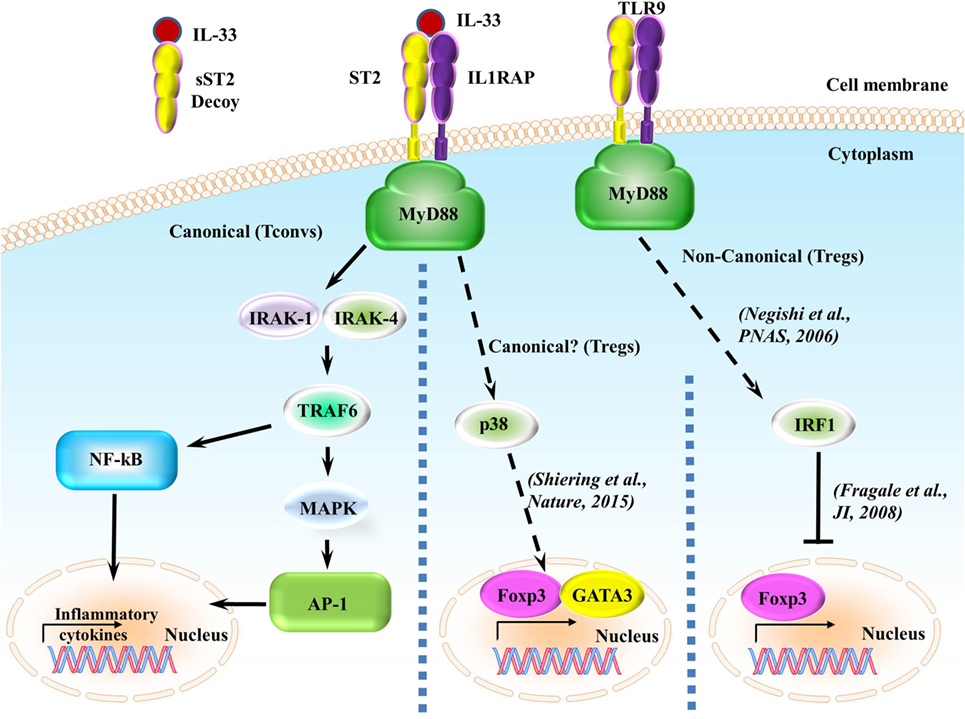
Figure 2. ST2/IL-33 signaling pathway. IL-33 either binds to the ST2/IL-1 receptor accessory protein (IL-1RAP) heterodimer, recruiting MyD88 to its intracellular domain, or the sST2 decoy receptor, which does not signal. MyD88 binding recruits IL-1R-associated kinase (IRAK) and TRAF6, leading to either the NF-κB or AP-1 pathway being activated. NF-κB and AP-1 activations promote inflammatory cytokine expressions. On regulatory T cells (Tregs), ST2/IL-33 signaling has been shown to promote Foxp3 and GATA3 expressions, while also promoting Treg function and expansion through enhancing TGF-β1-mediated differentiation though a p38-dependent mechanism. It has recently been shown that IFN regulatory factor (IRF) 1, which can be activated through MyD88 signaling, can inhibit Tregs by binding to the Foxp3 promoter and preventing Foxp3 transcription.
A recent report has shown that signaling through ST2/IL-33 in colonic Tregs helps to promote Foxp3 and GATA3 expressions while also promoting Treg function through enhancing TGF-β1-mediated differentiation (41). This enhancement is caused by phosphorylation of GATA3, which recruits more GATA3 and RNA polymerase II to the Foxp3 promoter (41). GATA3 binds to the ST2 promoter, enhancing ST2 on the surface of both Th2 cells (56, 57) and Tregs (41, 57). IL-33 has been shown to drive NF-κB and p38 signaling in Tregs, leading to the selective expansion of ST2+ Tregs (58). As this effect is observed in Tregs in a non-diseased setting, independent of outside inflammatory responses, we believe that the ST2/IL-33-GATA3-Foxp3 pathway to be canonical. Conversely, in a non-canonical MyD88-dependent pathway (59), IFN regulatory factor (IRF) 1 signaling can inhibit Tregs by binding to the Foxp3 promoter and preventing Foxp3 transcription in murine T cells (60); however, this signaling leading to IRF1 activation through MyD88 has only been shown to be induced using CpG-B, a TLR9 agonist and a pathway independent from ST2/IL-33 (59). Whether ST2/IL-33 can activate IRF1 in a MyD88-dependent pathway and whether this ST2/IL-33-IRF1 activation can affect Treg function are currently unknown. We have highlighted the different ST2 signaling pathways in Figure 2.
Unlike IL-1RAP, the single immunoglobulin domain IL-1R-related molecule (SIGIRR or TIR8) SIGIRR can form a complex with ST2 upon IL-33 stimulation and can inhibit ST2/IL-33-mediated signaling both in vitro and in vivo (6, 61). IL-33 binding to ST2 has also been shown to negatively regulate ST2 through ST2 polyubiquitination, internalization, and degradation (62).
sST2 acts as a decoy receptor to sequester free IL-33, preventing ST2/IL-33 signaling. This was shown using a thymoma cell line transfected to express ST2, but not sST2, in the presence of added IL-33. When these thymoma cells were pretreated with sST2, they showed suppressed NF-κB activity (63). Another group used IL-33-treated cardiomyocytes and observed blocked prohypertrophic effects of angiotensin II or phenylephrines in the presence of sST2 (64). Blocking NF-κB signaling in lung alveolar epithelial cells and cardiac myocytes with the specific NF-κB inhibitor CAPE prevented sST2 production by these cells (49). In a human endotoxin model, healthy donors injected with LPS (2 ng/kg) had increased sST2 in their plasma within 24 h of injection (49). Fibroblast growth factor 2 enhanced sST2 production in the human breast adenocarcinoma cell line MCF-7 through MEK/ERK signaling (65). Lysophosphatidic acid has also been shown to increase sST2 production by human bronchial epithelial cells in an NF-κB- or JNK-dependent manner (66). Enhanced sST2 plasma circulation has been correlated with pulmonary fibrosis (67), acute myocardial infarction (39), subclinical brain injury and stroke (68), celiac disease (69), gastric cancer (70), HBV-related acute-on-chronic liver failure (71), HIV progression (72), and GVHD (17–24).
During cell stress or damage, IL-33 is released in either a full length or a cleaved form. Unlike IL-1β, however, IL-33 is not cleaved via caspase-1, and cleavage is not necessary for secretion nor biological activity of released IL-33, further suggesting its role as an alarmin (73, 74). Surprisingly, caspase-1, caspase-3, or caspase-7 processing actually leads to IL-33 inactivation (75, 76). Inactivation of IL-33 via caspases is therefore thought to alleviate the immune response, rather than enhance it. Other proteins are able to cleave IL-33, such as the neutrophil serine proteases cathepsin G and elastase, mast cell-derived serine proteases, tryptase, and chymase, and these proteins, unlike caspases, increase the biological activity of cleaved IL-33 10–30 times compared to that of full-length IL-33 (74, 77, 78).
IL-33 is expressed mainly by non-hematopoietic cells, including endothelial cells, adipocytes, fibroblasts, and intestinal and bronchial epithelial cells (8, 79, 80); however, some hematopoietic cells like dendritic cells (DCs) have also been show to express IL-33 when activated (5). In many non-hematopoietic tissues, IL-33 is constitutively expressed. Constitutive expression of IL-33 in epithelial cells suggests that IL-33 is used as an alarmin in response to infection or injury (8). Further suggesting IL-33 as an alarmin, IL-33 is released by damaged or necrotic cells (8), leading to activation of the immune system through ST2/IL-33 signaling (8, 81).
IL-33 can be found in the nucleus due to a nuclear localization sequence in the N-terminus, leading to binding of heterochromatin in the nucleus (7). Nuclear IL-33 can bind directly to NF-κB, sequestering it and preventing NF-κB signaling in HEK293RI cells, causing a downregulation of pro-inflammatory signaling (82). Further evidence of IL-33 having the ability to repress gene transcription is described because there is a structural similarity between a part of the IL-33 protein and the Kaposi sarcoma herpes virus motif latency-associated nuclear antigen (82). This mimicry allows IL-33 to bind to the H2A–H2B chromatin dimer and regulate the compaction of chromatin through nucleosome–nucleosome interactions. Recent discoveries have shown that nuclear IL-33 can bind to multiple sites in the promoter regions of ST2 in human endothelial cells and that knockdown of IL-33 increased sST2 levels (83). Loss of the nuclear localization domain of IL-33 led to non-resolving lethal inflammation (84). However, IL-33−/− mice fail to develop autoimmune disease, and no one has shown whether nuclear IL-33 has been found in immune cells. These results indicate that nuclear IL-33 could act as a moderator of inflammation, but more evidence is needed to confirm the extent of the ability of nuclear IL-33 to moderate inflammation.
ST2 was first shown both in vitro and ex vivo to be preferentially expressed on murine Th2 cells (Figure 3; Table 1) expressing predominantly IL-4, IL-5, or IL-10, but not IFN-γ or IL-2 (40, 85). Its expression is independent of IL-4, IL-5, and IL-10, as loss of any of these cytokines does not affect ST2 expression on Th2 cells (40). ST2 expression on Th2 cells is dependent on GATA3 signaling (86) and is enhanced by IL-6, IL-1, TNF-α, and IL-5 (4). Given that ST2 expression in Th2 cells is independent of IL-4 and dependent on GATA3 signaling, it makes sense that ST2 expression occurs late during Th2 differentiation (4). IL-33 stimulation of Th2 cells in vitro increased the amount of IL-5 and IL-13 produced (5). Antigen-specific ST2+ Th2 cells were shown to produce more IL-5 and IL-13 compared to non-antigen-specific Th cells and ST2−/− Th2 cells (87). Interestingly, IL-33 polarization of antigen-stimulated murine and human naïve CD4+ T cells leads to high IL-5 production but no IL-4 production, independent of GATA3 and STAT6 induction but dependent on MAPK and NF-κB signaling (88, 89). Adoptive transfer of these cells into naïve IL-4−/− mice still triggered airway inflammation (88). In vivo administration of IL-33 led to an increase in the number of lymphocytes circulating in the blood and increased type 2 cytokine secretions in the thymus, spleen, liver, and lung (5). IL-33 has also been shown to be a chemoattractant for Th2 cells, as adoptive transfer of Th2 cells into ST2 knockout (KO) followed by IL-33 administration into the footpad of these mice led to the accumulation of the transferred Th2 cells (90). Loss of ST2 on Th2 during infection with the helminthic parasite Nippostrongylus brasiliensis did not affect Th2-mediated clearance of the infection nor was recruitment of Th2 cells in a murine model of asthma dependent on ST2, indicating that ST2 is not necessary for Th2 function (91). Recently, it was shown that human and murine Th2 cells do not produce sST2 in vitro (51).
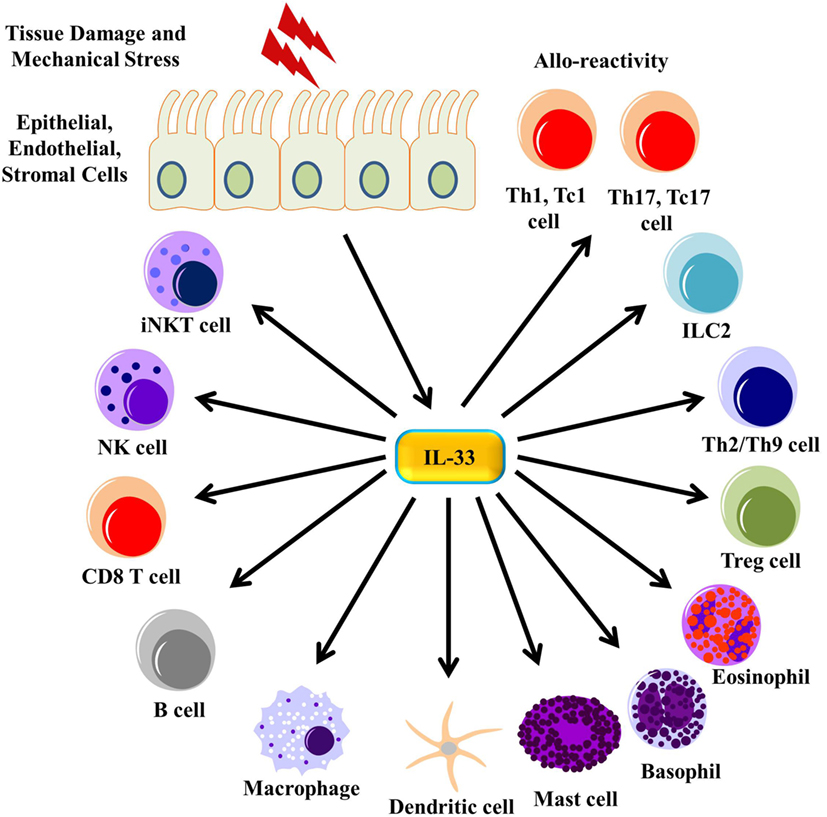
Figure 3. IL-33 signaling on immune cells. Tissue damage and mechanical stress to epithelial, endothelial, and stromal cells lead to the release of IL-33 from these cells. IL-33 then signals through many different immune cells, enhancing their function.

Table 1. ST2 and sST2 expressions and their regulation in immune cells.
IL-9-producing Th9 cells are the newest T cell subset to be described, polarized through TGF-β and IL-4 signaling (92, 93). When used separately on naïve T cells, TGF-β alone would cause Treg development, while IL-4 would induce Th2 cell differentiation. It has been found that the PU.1 gene is a Th9-specific transcription factor, which could induce IL-9 production in cells under Th2- or Th9-stimulating condition in vitro (94). Human or mouse PU.1-deficient T cells have diminished IL-9 production. Furthermore, IRF4 binds directly to the IL-9 promoter and is required for the development of Th9 cells, just like PU.1 (95). However, unlike PU.1, IRF4 is also required for the development of other Th cell subsets, including Th2 and Th17 cells (96, 97). Studies have shown that Th9 cells primarily secrete IL-9 to mediate the immune response in several diseases, such as asthma, autoimmune diseases, and parasitic infections (98), and IL-9 is associated with impaired Th1 immune response in patients with tuberculosis (99). Treatment of in vitro polarized human Th2 cells with TGF-β and IL-33 increases their IL-9 and ST2 expressions (100, 101).
ST2/IL-33 signaling in Tregs was first suggested to enhance their protective ability in an experimental colitis model in which IL-33 treatment ameliorated colonic tissue injury and colitis symptoms (41). IL-33 was shown to increase both ST2 and Foxp3 levels and expand Tregs in mice with colitis. ST2/IL-33 signaling in Tregs has also been shown to increase Treg frequency and decrease IL-17 and IFN-γ productions in an experimental autoimmune encephalomyelitis (EAE) model (102, 103). ST2+ Treg expansion is helped by IL-33 signaling in DCs, as IL-33 has been shown to stimulate DC production of IL-2, which selectively expands ST2+ Tregs (104). In a model of GVHD, treatment of mice daily with IL-33 from 10 days pretransplantation to day 4 posttransplantation enhanced the frequency of ST2+ Tregs, which persisted after irradiation, leading to disease amelioration through prevention of T conventional cell accumulation in target GVHD organs (58). Treatment of mice receiving a heart transplant with IL-33 has been shown to prolong graft survival through increase of Treg and myeloid suppressor-derived cell numbers (105, 106). Similarly, mice treated with IL-33 after skin transplantation had increased Treg numbers in the graft, decreased IFN-γ and IL-17 production, increased IL-10 production, and increased skin graft survival (107). This group also showed that ST2/IL-33 signaling can convert Foxp3− CD4 cells into Foxp3+ CD4 Tregs in the periphery. We have shown that in a murine model of allogeneic hematopoietic stem cell transplantation (allo-HCT), transplanting ST2 KO Tregs with wild-type (WT) T conventional cells worsens GVHD compared to mice receiving WT T conventional cells and Tregs (51), further indicating the enhanced suppressive effect of ST2+ Tregs. Conversely to the enhanced protective effect of Tregs through ST2/IL-33, it has been reported that IRF1 is downstream of MyD88 (108) and negatively regulates Foxp3 transcription (60, 108), although whether ST2/IL-33 signaling increases IRF1 expression, leading to decreased Treg function, has yet to be studied. Given that ST2/IL-33 has been shown to enhance, rather than impair Treg function, upregulation of IRF1 through MyD88 signaling is probably independent of ST2/IL-33. These data show that IL-33 signaling on Tregs increases their immunomodulatory function and could be further studied for their potential clinical benefits in a variety of diseases.
Innate lymphoid cells type 2 were first discovered in the mouse and human mesenteries and found to be lineage marker negative, c-Kit positive, Sca-1 positive, IL-7Rα positive, and ST2 positive (42, 109). These cells have been shown to play a protective role against helminth infection and regulate metabolic homeostasis (110). In humans ST2+, ILC2s were later found in the lung and gut (111), and these ILC2s produced IL-5 and IL-13. During ILC2 activation, ST2 is upregulated in a GATA3- and Gfi1-dependent manner (112, 113). Treatment of Rag2 KO mice with IL-33 induced IL-5 and IL-13 production, whereas Rag2 and common gamma chain double KOs, which still have mast cells and basophils (both of which express ST2 and secrete type 2 cytokines), did not increase IL-5 or IL-13 production, indicating that this increase is due to ILC2 stimulation with IL-33 (42). ST2/IL-33 signaling enhancement was shown to expand ILC2s in vivo (42, 114). This group also found that ILC2s are major producers of type 2 cytokines after Nippostrongylus brasiliensis infection. It was also shown using the N. brasiliensis infection model that loss of both IL-33 and IL-25 signaling on ILC2s completely abrogated the early response against this infection due to impaired expansion of ILC2s and lack of IL-13 production, and adoptive transfer of WT ILC2s rescued this phenotype (42). During lung inflammation, ILC2s produce IL-9 (115), and IL-33 can promote cytokine production by ILC2s (116). Recently, it was shown that in a murine eosinophilic airway inflammation model that T-bet regulates IL-9 production by IL-33-stimulated ILC2s (117). ST2/IL-33 signaling in ILC2s is also important for protection against lung infection, as blocking ST2 signaling during influenza infection in mice lowered ILC2 frequency and number in the lung and resulted in diminished lung function, loss of airway epithelial integrity, and impaired respiratory tissue remodeling (118). Histological examination of influenza-infected lungs from anti-ST2-treated mice showed severe damage similar to that seen in a similar experiment where ILC2s were depleted (118). ILC2s have been recently reported to home to the skin in humans, where activation induces upregulation of ST2 (116). ST2/IL-33 signaling of ILC2s in the murine skin has been shown to not only promote atopic dermatitis (AD)-like inflammation (116, 119) but also promote skin wound repair (120). However, overstimulation of ILC2s with IL-33 during tissue remodeling of the liver after chemical injury promoted liver fibrosis (121). Also, signaling through ST2/IL-33 on ILC2s during breast cancer has been shown to promote breast cancer growth and metastasis (122). These data indicate that beneficial or harmful ST2/IL-33 stimulation in ILC2s is dependent on certain disease states.
CD8 T cells have been shown to either express ST2 or produce sST2 (51, 123, 124). Although CD8 T cells express low levels of ST2, loss of either IL-33 or ST2 impaired the CD8 T cell response to LCMV infection (124). ST2/IL-33 signaling has also been shown to enhance CD8 T cell antitumor activity (125). During GVHD, however, IL-33 treatment during peak inflammation significantly increased GVHD severity and mortality in part through increased expansion of Tc1 cells (126). Given that IL-33 can increase type 1 responses when IL-12 levels are high (127), IL-33 treatment during peak inflammation was deleterious in this case.
ST2 has been shown to be expressed on B1 B cells but not B2 B cells, leading to enhanced proliferation capacity and IgM, IL-5, and IL-13 productions both in vitro and in vivo; neutralizing IL-5 almost completely abolished this effect (128). Recent studies have also shown that IL-33 treatment in mice increases circulating IL-10-producing B cells that are neither conventional B1 nor B2 B cells (129). Adoptive transfer of these IL-33-treated, IL-10-producing B cells prevented spontaneous colitis in IL-10−/− mice without affecting Treg frequency (129).
ST2/IL-33 signaling in murine iNKT cells causes their expansion and activation (130). Mice treated with IL-33 had twice as many iNKT cells in the spleen and liver compared to untreated mice (130). Unexpectedly, ST2 signaling in iNKT cells induced IFN-γ instead of IL-4 upon TCR engagement, which synergized in the presence of IL-12 (47, 130). This effect was also seen in Vα24+ human iNKT cells (47). NK cells constitutively express ST2, and ST2/IL-33 signaling increases IFN-γ levels synergistically with IL-12 (47, 130). Loss of ST2 in Ly49H+ NK cells did not affect their development but did impair their ability to expand and protect against MCMV (131). These data have yet to translate to human disease.
Although much of the research on ST2/IL-33 signaling in T conventional cells has been devoted to type 2 signaling, recent studies have come out looking at ST2/IL-33 signaling in type 1- and type 17-mediated diseases. Blockade of IL-33 with 200 µg anti-IL-33 every other day from day 0 until day 18 post-MOG35–55 injection during MOG-induced EAE ameliorated the disease in part through decreased IL-17 and IFN-γ productions, and treatment of 50 µg/kg IL-33 during this same time course enhanced IL-17 and IFN-γ productions (102). However, the amount of IL-33 given here is not physiological, so caution must be advised when interpreting these data. Conversely, another group using the same EAE model found that treatment with 1 µg IL-33 daily from day 12 to day 20 after immunization reduced IL-17 and IFN-γ productions and alleviated the disease (103). Seemingly, timing of ST2/IL-33 signaling affects response, perhaps through differing environments. In a murine model of collagen-induced arthritis, treatment with anti-ST2 antibody reduced both IFN-γ and IL-17 productions (132). In a murine model of rheumatoid arthritis, treatment with an sST2-Fc fusion protein attenuated disease and decreased production of IFN-γ, TNF-α, and IL-6 (133). Recently, we were the first to show that both murine and human Th1 and Th17 cells produce sST2 in vitro and in vivo after allo-HCT (51). Blocking ST2 with a blocking antibody in vivo decreased sST2 production in intestinal T cells 10 days after allo-HCT while maintaining ST2. Recipients of ST2−/− T cells, compared to WT T cells, showed lower frequencies of T cells expressing the Th1 transcription factor T-bet and the Th17 transcription factor RORγt and their associated cytokines IFN-γ and IL-17, respectively, while increasing the expressions of the Th2 transcription factor GATA3 and the Treg transcription factor Foxp3 and their associated cytokines IL-4 and IL-10, respectively (51). Importantly, anti-ST2 treatment did not lead to loss of immunomodulatory ST2+ Tregs but rather maintained them in the intestine. On the basis of our findings, we have suggested that increased sST2 production affects the normal balance of pathogenic Th1/Th17 cells and immunomodulatory Th2/Treg cells by promoting the Th1/Th17 response and dampening the ST2-mediated Th2/Treg response through sequestering IL-33 (51).
We were also the first to demonstrate that CD8 T cells, particularly Tc1 and Tc17 cells but not Tc2 cells, produce significant amounts of sST2 in vitro and after allo-HCT due to alloreactivity (51). sST2 secretion by donor T cells significantly increased as GVHD progressed. Similar to CD4 T cells, blocking ST2 with a blocking antibody decreased sST2 production by Tc1 and Tc17 cells in vivo after allo-HCT (51). Our data indicate that sST2 secretion by Tc1 and Tc17 cells sequester free IL-33, preventing ST2/IL-33-mediated Th2/Treg responses. In patients with early HIV infection, sST2 levels were strongly correlated with CD8 T cell count and their expressions of the activation markers HLA-DR and CD38 (72). However, it is not known if sST2 was produced from the CD8 T cells themselves or if sST2 is only a marker of gut damage and disease progression. While our study was the first to show that preventing sST2 secretion from CD8 T cells prevented disease pathogenesis, further studies are warranted to determine their role in other disease pathogeneses.
Macrophages, mast cells, basophils, eosinophils, and DCs all have been shown to express ST2 (43–46, 134). IL-33 amplifies the expression of M2 markers on murine macrophages (43, 135). Bone-derived human macrophages have been shown to constitutively express both ST2 and sST2; however, skewing these macrophages toward an M2 phenotype using IL-4 and IL-13 increased the expression of ST2 while not affecting sST2 expression (136). ST2/IL-33 signaling has been shown to enhance the activation of macrophages by upregulating the LPS receptor components TLR4 and MD2, soluble CD14, and MyD88 (135).
ST2/IL-33 signaling on both murine and human mast cells has been shown to promote their survival through upregulation of B-cell lymphoma-X large in the peritoneum (137). ST2/IL-33 signaling also promotes mast cell activation and maturation, as IL-33 treatment of CD34+ mast cell precursors accelerated their maturation in vitro and induced GM-CSF, IL-5, IL-13, CXCL8, CCL17, CCL22, and CCL2 secretions (138, 139). These cytokine and chemokine secretions may be NFAT and AP-1 signaling dependent (140). It is well documented that mast cells can produce a variety of type 2 cytokines after ST2 signaling (141–143); however, ST2/IL-33 signaling on mast cells during airway inflammation has also been shown to promote a Th17 response (144).
ST2/IL-33 signaling promotes not only type 2 cytokine secretions such as IL-4 and IL-13 but also IL-8 in synergy with IL-3 or Fcε receptor activation on basophils (145). Basophils can also release sST2 after activation via IL-3 and C5a or anti-FcεRIα antibody, while IL-33 prevents sST2 release (145). IL-33 induces the degranulation of eosinophils and production of superoxide (45); controls their responsiveness to Siglec 8 (146); and increases IL-13, TGF-β, CCL3, CCL17, and CCL24 in the lungs during airway inflammation (147). Treatment with anti-ST2 antibodies prevented the upregulation of CD11b and decreased survival of eosinophils (148).
Dendritic cells express low basal levels of ST2 on their cell surfaces (134); however, activation of DCs with rapamycin strongly upregulates ST2 through autocrine IL-1β signaling (149). Treatment of DCs with IL-33 has been shown to increase surface levels of MHC-II, CD40, CD80, CD86, OX40L, and CCR7 (134, 150, 151). ST2/IL-33 signaling in DCs also increases their productions of IL-4, IL-5, IL-13, CCL17, TNF-α, and IL-1β (150). In the presence of naïve CD4+ T cells, IL-33-activated DCs induce IL-5 and IL-13 but not IL-4 and IFN-γ from the T cells (134, 151). Interestingly, sST2 has also been shown to be internalized by DCs, suggesting a non-canonical method of action for sST2 (152). It is currently unknown whether sST2 can be internalized by other immune cells. IL-33-activated murine DCs have recently been shown to be required for in vitro and in vivo expansion of ST2+ Tregs through DC IL-2 production (104), which could be used for therapeutic benefit against inflammatory diseases through expansion of Tregs both in vitro and in vivo. ST2 expression on host hematopoietic cells, including DCs, and non-hematopoietic cells was not implicated in the severity of GVHD as recipient ST2 KO bone marrow chimeras did not modify GVHD severity (51).
While ST2 has been shown to be present on neutrophils (46, 153), not much is known about the role of ST2 on neutrophils. It has been shown that IL-33-treated murine and human neutrophils do not downregulate CXCR2 induced by the activation of TLR4 through the inhibition of GRK2 (153). IL-33 injected into the ears of mice induced neutrophil recruitment to the skin (154); however, it is not clear if ST2/IL-33 signaling on the neutrophils directly led to their migration.
It is believed that IBD starts with a dysregulated immune response to either food or commensal gut bacteria, leading to the production of pro-inflammatory cytokines such as TNF-α, IL-6, IL-1, and IL-8. Expression of these cytokines along with chemokine release leads to attraction of T cells, specifically type 1 T cells, to the intestines. Continual damage of the gut mucosa by these type 1 cells and other immune cells such as macrophages, neutrophils, and DCs leads to the release of various alarmins and other proteins. sST2 was found to be significantly increased in both the gut mucosa and the serum in both patients and experimental models of IBD (13–16). However, in IBD patients, ST2 expression remained similar to that of healthy patients (13). In the lamina propria of active ulcerative colitis (UC) patients, ST2 predominately came from CD11b+ and CD4+ cells (14). These findings suggest that increased sST2 production by lymphocytes or the gut mucosa could either lead to development of IBD, particularly UC, or that these proteins are markers for disease severity.
ST2/IL-33 signaling has been shown to enhance epithelial proliferation and mucus production in the gut (5), suggesting that the increase in IL-33 in the colonic mucosa in active UC could be beneficial. However, in multiple mouse models of IBD, use of ST2 KO mice led to amelioration of IBD compared to WT controls. These results were verified using an IL-33 KO. Using bone marrow chimeras, it was shown that ST2 signaling in non-hematopoietic cells was responsible for IBD. This was due to ST2/IL-33 signaling impairing epithelial barrier function and delayed wound healing. Lack of ST2 signaling in hematopoietic cells did not prevent UC development. A ST2 blocking antibody confirmed the findings from the KO experiments (155). Crohn’s disease (CD), however, shows opposite results from UC. In a trinitrobenzene sulfonic acid-induced IBD model, which mimics the pathology of human CD, administration of recombinant IL-33 (rIL-33) into mice ameliorated colonic tissue injury and clinical symptoms (156). Protection was shown to be through upregulation of type 2 cytokines, Foxp3+ Tregs, and CD103 DCs, which promote Treg development. In patient colons with active IBD, Treg levels in the lamina propria are increased compared to healthy controls and function normally (157, 158). It has recently been shown that colonic Tregs preferentially express ST2 and that signaling through ST2/IL-33 promotes both Treg accumulation and maintenance in the intestine and enhances their protective function (41). However, treatment with rIL-33 to promote Treg-mediated protection may be time dependent, as rIL-33 treatment at onset of a DSS-induced colitis model exacerbated disease severity. rIL-33 treatment during recovery or chronic phases ameliorated DSS-induced colitis (159). Given these data, selective treatment of ST2+ Tregs with IL-33 could provide therapeutic benefits.
Graft-versus-host disease is a common occurrence in patients who undergo allo-HCT as treatment for both malignant and non-malignant diseases of the blood and bone marrow. The pathogenesis of GVHD has been well documented and is now thought to occur in three steps: (1) activation of antigen-presenting cells caused by tissue damage from the conditioning regimen leading to the release of pro-inflammatory cytokines and danger signals, (2) alloactivation of donor T cells leading to their proliferation and differentiation into type 1 and type 17 T cells, and (3) tissue destruction by alloreactive T cells through release of cytolytic molecules leading to donor cell apoptosis, mainly in the mucosal tissues (160). Discovering prognostic and diagnostic biomarkers for GVHD has been successful with sST2 being one of the most validated to date (17–24). Blocking sST2 with a blocking antibody during the peritransplant period decreased GVHD morbidity and mortality in both minor histocompatibility and humanized murine models (Figure 4). Importantly, the ST2-blocking antibody, which inhibits the full length of ST2 and not specifically sST2, maintained protective ST2-expressing T cells while also not impairing the graft-versus-leukemia activity (51), suggesting that addition of anti-ST2 or a ST2 small molecule inhibitor could show efficacy in reducing GVHD-related morbidity and mortality in patients. Using IL-33 as a treatment seems to be time dependent, as injections with IL-33 during the peak inflammatory response in a murine model led to increased morbidity and mortality in mice due to increased migration and increased pro-inflammatory cytokine production (126). IL-33 treatment preconditioning, however, increased the number of ST2+ Tregs, which persisted after irradiation in a murine model. This led to decreased GVHD severity and mortality. Adoptive transfer of ST2+ versus ST2− Tregs showed that GVHD protection is increased by ST2+ and not ST2− Tregs (58). Given that IL-33 is pleiotropic, IL-33 treatment for GVHD seems to be dependent on both timing and the state of inflammation present.
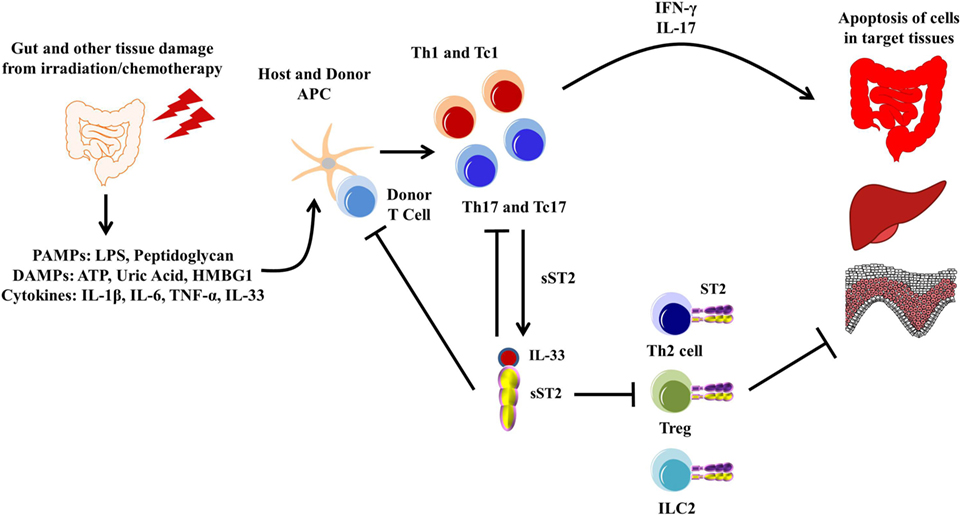
Figure 4. Pathogenesis of graft-versus-host disease (GVHD). The gut and other issues are damaged during irradiation or chemotherapy, leading to the release of various DAMPs, PAMPs, and cytokines, including IL-33. These DAMPs, PAMPs, and cytokines activate both host and donor antigen-presenting cells (APCs), which then activate the donor T cells. The APCs are also secreting various cytokines that promotes T cell differentiation toward type 1 and type 17 responses. These activated type 1 and type 17 T cells are able to secrete various pro-inflammatory cytokines, leading to apoptosis of healthy tissue, mainly in the gut, liver, and skin, which can be exacerbated by free IL-33. Furthermore, sST2 is produced by both type 1 and type 17 T cells, and while this may sequester free IL-33 from the type 1 and type 17 T cells, sST2 can also prevent the potential beneficial effects from ST2/IL-33 signaling in Th2 cells, Tregs, and lymphoid cell type 2 (ILC2s).
ST2/IL-33 signaling has been implicated in protection from various infections, which could impact the gut. Studies have shown that treatment of mice with rIL-33 led to epithelial cell hyperplasia in the gut along with infiltration of eosinophils and mononuclear cells in the lamina propria (42, 161). These effects are thought to be mediated by IL-13, which becomes overexpressed after IL-33 treatment (5). Treatment of mice with IL-33 after Trichuris muris infection increased parasite clearance through increased Th2 cytokine response (161). Other infections that can impact the gut, including Toxoplasma gondii (162), Leptospira (163), and Pseudomonas aeruginosa (164), have shown that the loss of ST2 or high sST2 levels led to higher morbidity and mortality with increased Th1 cytokine profiles. Recent studies have shown that gut epithelial barrier dysfunction and immune activation independently predict mortality during treated HIV infection (165). A later study showed that patients during the early stage of HIV infection, defined as being within 180 days of the date of infection, had higher levels of sST2 in their plasma and was highly correlated with CD8 T cell count and levels of gut mucosal damage, but not with viral load or CD4 T cell count (72).
sST2 increase has also been implicated during small bowel transplant rejection (25). Patients who had rejection of small bowel transplants had higher serum levels of sST2 during rejection compared to that during rejection-free time points and that rejection increased allograft ST2 expression. An increase in sST2 in the allograft was predicted by Pathway and Network Analysis caused by TNF-α and IL-1β signaling (25). However, these data do not implicate sST2 as a mediator of disease but rather a biomarker of occurring transplant rejection.
Asthma is characterized by varying levels of airway hyperresponsiveness, mucus secretion, bronchoconstriction, and chronic inflammation, affecting 300 million people worldwide (166). ST2/IL-33 signaling in mast cells, ILC2s, eosinophils, basophils, Th2, and Th9 cells drive allergic asthma (141–143, 147, 167–170). Activating ST2 signaling in bone marrow-derived mast cells and basophils in vitro shows strong type 2 cytokine production, including IL-4 and IL-13 (171). Treatment of mice with IL-33 alone induced airway hyperresponsiveness and goblet cell hyperplasia through IL-4, IL-5, and IL-13 induction in the lungs (172). RAG2−/− mice treated with IL-33 also induce this phenotype, implicating the importance of the innate immune system in generating airway inflammation (171). IL-33 has also been shown to be a chemoattractant for Th2 cells, as injection of IL-33 into the footpad of ST2−/− mice, which were adoptively transferred with WT polarized Th2 cells led to local accumulation of the transferred Th2 cells (90). After ovalbumin (OVA)-induced acute allergic lung inflammation, levels of IL-33, ST2, and sST2 are significantly increased in the lung (172). While activation of the innate immune system in part through ST2/IL-33 signaling establishes airway inflammation, ST2+ T cells maintain this inflammation. Indeed, injecting mice with a ST2 blocking antibody during the resolution phase after OVA-induced allergic inflammation, when Th2 cells but not eosinophils are present in the lung, reduced airway hyperresponsiveness and mucus production (173). Loss of both IL-4 and IL-5 during OVA-induced airway inflammation did not abolish airway hyperreactivity, which was abolished only after anti-CD4 treatment (174), suggesting that ST2/IL-33 signaling in CD4 T cells may be critical for efficient antigen-induced airway inflammation (175). Blockade of either IL-33 or addition of sST2 before OVA-induced allergic airway inflammation showed reduced total cell counts and eosinophil counts in the bronchoalveolar lavage fluid and decreased IL-4, IL-5, and IL-13 (40, 52, 176).
During idiopathic pulmonary fibrosis, patients with stable disease versus healthy controls had similar levels of serum sST2; however, during exacerbations of fibrosis, there was over a sixfold increase in serum sST2 in the exacerbation group compared to both healthy controls and stable disease groups, which correlated with measurements of inflammation (67). Patients with acute respiratory distress syndrome had significantly higher sST2 levels at day 0, which were associated with worse prognosis and mortality (177). Serum levels of sST2 were also increased in patients with chronic obstructive pulmonary disease compared to control patients (178). Patients with moderate or severe H1N1 influenza infection also had significantly increased serum sST2 compared to patients with mild H1N1 infection or rhinovirus infection (179). Idiopathic pneumonia syndrome (IPS) occurs in 5–10% of patients who undergo allo-HCT. In a cohort of 42 patients with IPS without infection, sST2 was significantly higher in the serum of patients with IPS compared to healthy controls and patients with human rhinovirus and parainfluenza occurring at approximately the same time after transplantation (180).
Atopic dermatitis is a Th2-driven disease and patients with AD show higher IgE levels and eosinophilia in the skin and blood (181). ST2/IL-33 signaling has been recently implicated in AD, as transgenic mice expressing IL-33 under the keratin 14 promoter had spontaneous AD-like inflammation (119). This led to increased IL-5 producing ILC2s. Mice receiving topically applied OVA, house dust mites, or staphylococcal enterotoxin B led to upregulation of both sST2 and IL-33 expressions (182). Patients with AD have higher IL-33 expression levels in their skin lesions compared to healthy controls (182). However, in a small cohort of 71 adults and 61 children with AD, serum sST2 levels were not significantly elevated compared to adult controls, in contrast to sST2 expression in patient skin lesions (182, 183). Currently, phase I–II clinical trials are being conducted using novel anti-IL-33 antibodies.
Unlike AD, psoriasis is driven by Th1 and Th17 cytokines (184). However, psoriatic skin still shows increased IL-33 expression compared to healthy skin in patients (154). ST2−/− mice had reduced cutaneous inflammatory responses compared to WT mice in a phorbol ester-induced murine model of psoriasis (154).
Patients with vitiligo are characterized by the disappearance of their melanocytes. In lesions of patients with vitiligo, both ST2 and IL-33 levels were increased, and serum levels of IL-33 were increased (185). As ST2 signaling in psoriasis and vitiligo is relatively new, not much else has been published as of yet.
Scleroderma is characterized by the fibrosis and hardening of the skin and connective tissues, measured by the modified Rodnan skin score (MRSS) test (186). In a two cohort study, serum sST2 levels were increased in patients with scleroderma compared to healthy controls, which, when combined with Spondin-1, best described longitudinal change in MRSS, using mixed linear models (187). This was validated using three other independent cohorts (187).
Chronic GVHD can affect multiple organs, with skin involvement being one of the most common. In a large study of chronic GVHD, plasma samples were collected from patients at day +100 post-allo-HCT. A four-biomarker panel, which included sST2, correlated strongly with chronic GVHD diagnosis, severity, and non-relapse mortality (21).
sST2 was found to be upregulated after mechanically stimulating cardiomyocytes and stimulating with IL-1β (39). Inducing myocardial infarction via coronary artery ligation increased sST2 in the serum of mice compared to unoperated controls (39). This observation was also seen in patients, as those who suffered myocardial infarction had elevated serum sST2 levels 1 day postevent (39). In a cohort of over 800 patients with acute ST-elevation myocardial infarction, sST2 levels 1 day postevent correlated with 30-day mortality independent of age, blood pressure, heart rate, infarct territory, and time from symptom onset to treatment (11). sST2 levels of patients with non-ischemic congestive heart failure at time of entry to the study also correlated with both brain natriuretic peptide levels (BNP), which is routinely used in the clinic and serum noradrenaline levels (12). This study also found that changes in serum sST2 were an independent predictor of mortality. sST2 levels are correlated with impaired epicardial coronary flow and risk of death or congestive heart failure within 30 days of presentation, independent of BNP (188). These data show the value of sST2 as a biomarker in cardiac diseases.
Functional analysis of ST2/IL-33 signaling and sST2 production has shown that treatment of cultured rat neonatal cardiomyocytes with rIL-33 blocked angiotensin II or phenylephrine induced hypertrophy, while addition of sST2 or blocking of ST2 with an antibody reversed this effect (64). When using ST2−/− or WT mice to look at in vivo response to pressure overload by transverse aortic constriction, ST2−/− mice had more left ventricular (LV) hypertrophy, more chamber dilation, reduced fractional shortening, more fibrosis, and impaired survival compared with WT mice (64). Treatment of WT mice with rIL-33 reduced fibrosis and hypertrophy and increased survival in WT mice (64). This reduction in damage when treating with IL-33 may be due to inhibited apoptosis in cardiomyocytes (189).
In an atherosclerosis model in which mice deficient for the ApoE protein fed a high-fat diet, treatment with rIL-33 reduced aortic atherosclerotic plaque development and increased levels of type 2 cytokines in the serum (190), which have an atheroprotective effect. Mice treated with sST2 developed significantly larger atherosclerotic plaques (190). These data indicate that ST2/IL-33 signaling may have a protective effect, while sST2 plays a deleterious role in cardiac diseases.
Accumulation of visceral adipose tissue (VAT) due to obesity leads to inflammation, insulin resistance, and development of type 2 diabetes (191), leading to the reduction and function of Tregs in the VAT (192), which have been shown to be enriched for ST2 expression (193, 194). IL-33 is critical for the development and maintenance of these VAT Tregs (194). In vitro culturing of murine adipocytes with IL-33 induced IL-5 and IL-13 production, decreased expression of genes associated with adipogenesis and lipid metabolism, and reduced lipid storage (195). ST2−/− mice fed a high-fat diet had increased body weight and fat mass and impaired glucose regulation and insulin secretion compared to high-fat diet WT controls (195). IL-33 treatment to genetically obese diabetic mice led to reduced adiposity, lower fasting glucose levels, improved glucose and insulin tolerance, accumulation of Th2 cells and M2 macrophages in their adipose tissue, and increased the proportion of ST2+ Tregs in the VAT (193, 195). However, ST2/IL-33 signaling may only help obesity-related insulin resistance. An age-associated insulin resistance model showed that fat-resident Treg depletion protected against insulin resistance, and these findings were confirmed using an anti-ST2 antibody (196).
ST2 and IL-33 are produced by white adipose tissue and in preadipocyte and adipocyte cell cultures in humans (79), while sST2 expression has been shown to be increased in omental and subcutaneous human adipose tissues (197). In a large, population-based study, sST2 levels in the plasma of patients strongly correlated with markers of diabetes, after adjusting for age and gender (28). In another study separating 525 patients into normal, prediabetic, and diabetic groups, plasma sST2 levels were only significantly increased in the diabetic group compared to prediabetic and normal groups (29). In a multicenter, cross-sectional study of 180 patients with metabolic syndrome with normal LV ejection fraction, LV mass index was independently associated with serum sST2 concentrations. Increased sST2 associated with increased likelihood of LV hypertrophy and increased systolic blood pressure (198). New-onset posttransplantation diabetes mellitus (PTDM) is a common occurrence after allo-HCT. Serum sST2 levels from three cohorts collected at engraftment and day 30 showed elevated sST2 levels at both time points and that high sST2 levels predicted PTDM and non-relapse mortality, independent of conditioning and high-grade GVHD (27). These data suggest that high sST2 levels correlate with obesity and type 2 diabetes and metabolic complications even when sST2 is already elevated by alloreactivity.
The clinical usefulness of targeting either sST2 excess of secretion or ST2/IL-33 excess signaling or use of sST2 as a biomarker for diseases has been a hot topic in the last few years, as shown by the increase in translational studies devoted to ST2/IL-33 and sST2. Manipulation of ST2/IL-33 signaling or blocking sST2 secretion or sequestration of IL-33 is highly disease dependent. Several new antibodies that inhibit IL-33 binding to ST2 are currently being tested in phase I–II clinical trials for patients with asthma and chronic obstructive pulmonary disease. Using either an antibody or small molecule inhibitor is an attractive option for therapeutics targeting sST2 in CD, GVHD, or heart disease, while ideally maintaining ST2. However, given the involvement of ST2/IL-33 in a multitude of processes, caution must be afforded.
sST2 usefulness as a clinical biomarker has been studied extensively in both cardiac and allo-HCT patients, showing both prognostic and diagnostic value (11, 12, 17–24, 27, 39, 187, 188). sST2 levels are also increased in patients suffering from intestinal (13–16) and metabolic diseases (27, 28, 197, 198); however, the data from these studies so far are correlative and have not passed the qualification for biomarkers that can be used in clinic (199).
ST2/IL-33 signaling in immune cells has recently become a hot target of study. This signaling helps to activate T cells, ILC2s, DCs, B cells, mast cells, basophils, eosinophils, and other immune cells. Most of the work has shown that ST2/IL-33 signaling enhances the type 2 response, although recent studies have shown how ST2/IL-33 signaling enhances the immunomodulatory effects of Tregs. T cells have also been recently shown to produce sST2, which was once thought to be produced only by non-hematopoietic cells. ST2/IL-33 signaling in Tregs, ILC2s, and IL-10-producing B cells protects against inflammation, while sST2 can act either as a biomarker or can play a role in a variety of diseases by sequestering IL-33 and preventing ST2/IL-33 signaling. However, ST2/IL-33 signaling can also lead to progression of various lung and skin diseases such as asthma and AD. Given the complexity between ST2/IL-33 signaling and timing during the immune response and the importance of ST2/IL-33 in various organ systems, several questions and challenges remain. When does ST2/IL-33 signaling affect Treg response more so than the inflammatory response in various diseases? Which mediators can enhance ST2 expression on immunomodulatory cells? Which mediators can reduce or promote sST2 production during disease? A better understanding of the impact of ST2/IL-33 and sST2 during disease and how ST2 and sST2 targeting could affect different organ systems will be critical for the development of therapeutics.
Both BG and SP devised, wrote, and revised the manuscript.
SP has a patent on “Methods of detection of graft-versus-host disease” licensed to Viracor-IBT Laboratories. Otherwise, the other author has no other relevant conflicts of interest to declare.
The authors are supported by grants from the National Cancer Institute (R01CA168814), the National Institute of Diabetes and Digestive and Kidney Diseases (T32DK007519), and the Leukemia & Lymphoma Society (1293-15).
1. Werenskiold AK, Hoffmann S, Klemenz R. Induction of a mitogen-responsive gene after expression of the Ha-ras oncogene in NIH 3T3 fibroblasts. Mol Cell Biol (1989) 9(11):5207–14. doi:10.1128/MCB.9.11.5207
2. Tominaga S. A putative protein of a growth specific cDNA from BALB/c-3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Lett (1989) 258(2):301–4. doi:10.1016/0014-5793(89)81679-5
3. Tominaga S-I, Ohta S, Tago K. Soluble form of the ST2 gene product exhibits growth promoting activity in NIH-3T3 cells. Biochem Biophys Rep (2016) 5:8–15. doi:10.1016/j.bbrep.2015.11.020
4. Meisel C, Bonhagen K, Lohning M, Coyle AJ, Gutierrez-Ramos JC, Radbruch A, et al. Regulation and function of T1/ST2 expression on CD4+ T cells: induction of type 2 cytokine production by T1/ST2 cross-linking. J Immunol (2001) 166(5):3143–50. doi:10.4049/jimmunol.166.5.3143
5. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity (2005) 23(5):479–90. doi:10.1016/j.immuni.2005.09.015
6. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol (2016) 16(11):676–89. doi:10.1038/nri.2016.95
7. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A (2007) 104(1):282–7. doi:10.1073/pnas.0606854104
8. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ’alarmin’? PLoS One (2008) 3(10):e3331. doi:10.1371/journal.pone.0003331
9. Caselli C. Inflammation in cardiac disease: focus on Interleukin-33/ST2 pathway. Inflamm Cell Signal (2014) 1(2):e149. doi:10.14800/ics.149
10. Ciccone MM, Cortese F, Gesualdo M, Riccardi R, Di Nunzio D, Moncelli M, et al. A novel cardiac bio-marker: ST2: a review. Molecules (2013) 18(12):15314–28. doi:10.3390/molecules181215314
11. Shimpo M, Morrow DA, Weinberg EO, Sabatine MS, Murphy SA, Antman EM, et al. Serum levels of the interleukin-1 receptor family member ST2 predict mortality and clinical outcome in acute myocardial infarction. Circulation (2004) 109(18):2186–90. doi:10.1161/01.CIR.0000127958.21003.5A
12. Weinberg EO, Shimpo M, Hurwitz S, Tominaga S, Rouleau JL, Lee RT. Identification of serum soluble ST2 receptor as a novel heart failure biomarker. Circulation (2003) 107(5):721–6. doi:10.1161/01.CIR.0000047274.66749.FE
13. Beltran CJ, Nunez LE, Diaz-Jimenez D, Farfan N, Candia E, Heine C, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis (2010) 16(7):1097–107. doi:10.1002/ibd.21175
14. Pastorelli L, Garg RR, Hoang SB, Spina L, Mattioli B, Scarpa M, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A (2010) 107(17):8017–22. doi:10.1073/pnas.0912678107
15. Diaz-Jimenez D, Nunez LE, Beltran CJ, Candia E, Suazo C, Alvarez-Lobos M, et al. Soluble ST2: a new and promising activity marker in ulcerative colitis. World J Gastroenterol (2011) 17(17):2181–90. doi:10.3748/wjg.v17.i17.2181
16. Diaz-Jimenez D, De la Fuente M, Dubois-Camacho K, Landskron G, Fuentes J, Perez T, et al. Soluble ST2 is a sensitive clinical marker of ulcerative colitis evolution. BMC Gastroenterol (2016) 16:103. doi:10.1186/s12876-016-0520-6
17. Vander Lugt MT, Braun TM, Hanash S, Ritz J, Ho VT, Antin JH, et al. ST2 as a marker for risk of therapy-resistant graft-versus-host disease and death. N Engl J Med (2013) 369(6):529–39. doi:10.1056/NEJMoa1213299
18. Ponce DM, Hilden P, Mumaw C, Devlin SM, Lubin M, Giralt S, et al. High day 28 ST2 levels predict for acute graft-versus-host disease and transplant-related mortality after cord blood transplantation. Blood (2015) 125(1):199–205. doi:10.1182/blood-2014-06-584789
19. McDonald GB, Tabellini L, Storer BE, Lawler RL, Martin PJ, Hansen JA. Plasma biomarkers of acute GVHD and nonrelapse mortality: predictive value of measurements before GVHD onset and treatment. Blood (2015) 126(1):113–20. doi:10.1182/blood-2015-03-636753
20. Levine JE, Braun TM, Harris AC, Holler E, Taylor A, Miller H, et al. A prognostic score for acute graft-versus-host disease based on biomarkers: a multicenter study. Lancet Haematol (2015) 2(1):e21–9. doi:10.1016/S2352-3026(14)00035-0
21. Yu J, Storer BE, Kushekhar K, Abu Zaid M, Zhang Q, Gafken PR, et al. Biomarker panel for chronic graft-versus-host disease. J Clin Oncol (2016) 34(22):2583–90. doi:10.1200/JCO.2015.65.9615
22. Abu Zaid M, Wu J, Wu C, Logan BR, Yu J, Cutler C, et al. Plasma biomarkers of risk for death in a multicenter phase 3 trial with uniform transplant characteristics post-allogeneic HCT. Blood (2017) 129(2):162–70. doi:10.1182/blood-2016-08-735324
23. Hartwell MJ, Ozbek U, Holler E, Renteria AS, Major-Monfried H, Reddy P, et al. An early-biomarker algorithm predicts lethal graft-versus-host disease and survival. JCI Insight (2017) 2(3):e89798. doi:10.1172/jci.insight.89798
24. Kanakry CG, Bakoyannis G, Perkins SM, McCurdy SR, Vulic A, Warren EH, et al. Plasma-derived proteomic biomarkers in HLA-haploidentical or HLA-matched bone marrow transplantation using post-transplantation cyclophosphamide. Haematologica (2017). doi:10.3324/haematol.2016.152322
25. Mathews LR, Lott JM, Isse K, Lesniak A, Landsittel D, Demetris AJ, et al. Elevated ST2 distinguishes incidences of pediatric heart and small bowel transplant rejection. Am J Transplant (2016) 16(3):938–50. doi:10.1111/ajt.13542
26. Pei C, Barbour M, Fairlie-Clarke KJ, Allan D, Mu R, Jiang HR. Emerging role of interleukin-33 in autoimmune diseases. Immunology (2014) 141(1):9–17. doi:10.1111/imm.12174
27. Johnpulle RA, Paczesny S, Jung DK, Daguindau E, Jagasia MH, Savani BN, et al. Metabolic complications precede alloreactivity and are characterized by changes in suppression of tumorigenicity 2 signaling. Biol Blood Marrow Transplant (2017) 23(3):529–32. doi:10.1016/j.bbmt.2016.12.627
28. Miller AM, Purves D, McConnachie A, Asquith DL, Batty GD, Burns H, et al. Soluble ST2 associates with diabetes but not established cardiovascular risk factors: a new inflammatory pathway of relevance to diabetes? PLoS One (2012) 7(10):e47830. doi:10.1371/journal.pone.0047830
29. Lin YH, Zhang RC, Hou LB, Wang KJ, Ye ZN, Huang T, et al. Distribution and clinical association of plasma soluble ST2 during the development of type 2 diabetes. Diabetes Res Clin Pract (2016) 118:140–5. doi:10.1016/j.diabres.2016.06.006
30. Bergers G, Reikerstorfer A, Braselmann S, Graninger P, Busslinger M. Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. EMBO J (1994) 13(5):1176–88.
31. Thomassen E, Kothny G, Haas S, Danescu J, Hultner L, Dormer P, et al. Role of cell type-specific promoters in the developmental regulation of T1, an interleukin 1 receptor homologue. Cell Growth Differ (1995) 6(2):179–84.
32. Iwahana H, Yanagisawa K, Ito-Kosaka A, Kuroiwa K, Tago K, Komatsu N, et al. Different promoter usage and multiple transcription initiation sites of the interleukin-1 receptor-related human ST2 gene in UT-7 and TM12 cells. Eur J Biochem (1999) 264(2):397–406. doi:10.1046/j.1432-1327.1999.00615.x
33. Tago K, Noda T, Hayakawa M, Iwahana H, Yanagisawa K, Yashiro T, et al. Tissue distribution and subcellular localization of a variant form of the human ST2 gene product, ST2V. Biochem Biophys Res Commun (2001) 285(5):1377–83. doi:10.1006/bbrc.2001.5306
34. Iwahana H, Hayakawa M, Kuroiwa K, Tago K, Yanagisawa K, Noji S, et al. Molecular cloning of the chicken ST2 gene and a novel variant form of the ST2 gene product, ST2LV. Biochim Biophys Acta (2004) 1681(1):1–14. doi:10.1016/j.bbaexp.2004.08.013
35. Gachter T, Werenskiold AK, Klemenz R. Transcription of the interleukin-1 receptor-related T1 gene is initiated at different promoters in mast cells and fibroblasts. J Biol Chem (1996) 271(1):124–9. doi:10.1074/jbc.271.1.124
36. Baba Y, Maeda K, Yashiro T, Inage E, Niyonsaba F, Hara M, et al. Involvement of PU.1 in mast cell/basophil-specific function of the human IL1RL1/ST2 promoter. Allergol Int (2012) 61(3):461–7. doi:10.2332/allergolint.12-OA-0424
37. Trub T, Kalousek MB, Frohli E, Klemenz R. Growth factor-mediated induction of the delayed early gene T1 depends on a 12-O-tetradecanoylphorbol 13-acetate-responsive element located 3.6 kb upstream of the transcription initiation site. Proc Natl Acad Sci U S A (1994) 91(9):3896–900. doi:10.1073/pnas.91.9.3896
38. Yanagisawa K, Takagi T, Tsukamoto T, Tetsuka T, Tominaga S. Presence of a novel primary response gene ST2L, encoding a product highly similar to the interleukin 1 receptor type 1. FEBS Lett (1993) 318(1):83–7. doi:10.1016/0014-5793(93)81333-U
39. Weinberg EO, Shimpo M, De Keulenaer GW, MacGillivray C, Tominaga S, Solomon SD, et al. Expression and regulation of ST2, an interleukin-1 receptor family member, in cardiomyocytes and myocardial infarction. Circulation (2002) 106(23):2961–6. doi:10.1161/01.CIR.0000038705.69871.D9
40. Lohning M, Stroehmann A, Coyle AJ, Grogan JL, Lin S, Gutierrez-Ramos JC, et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci U S A (1998) 95(12):6930–5. doi:10.1073/pnas.95.12.6930
41. Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature (2014) 513(7519):564–8. doi:10.1038/nature13577
42. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464(7293):1367–70. doi:10.1038/nature08900
43. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol (2009) 183(10):6469–77. doi:10.4049/jimmunol.0901575
44. Moritz DR, Rodewald HR, Gheyselinck J, Klemenz R. The IL-1 receptor-related T1 antigen is expressed on immature and mature mast cells and on fetal blood mast cell progenitors. J Immunol (1998) 161(9):4866–74.
45. Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol (2008) 121(6):1484–90. doi:10.1016/j.jaci.2008.04.005
46. Suzukawa M, Iikura M, Koketsu R, Nagase H, Tamura C, Komiya A, et al. An IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J Immunol (2008) 181(9):5981–9. doi:10.4049/jimmunol.181.9.5981
47. Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol (2008) 20(8):1019–30. doi:10.1093/intimm/dxn060
48. Komai-Koma M, Brombacher F, Pushparaj PN, Arendse B, McSharry C, Alexander J, et al. Interleukin-33 amplifies IgE synthesis and triggers mast cell degranulation via interleukin-4 in naive mice. Allergy (2012) 67(9):1118–26. doi:10.1111/j.1398-9995.2012.02859.x
49. Mildner M, Storka A, Lichtenauer M, Mlitz V, Ghannadan M, Hoetzenecker K, et al. Primary sources and immunological prerequisites for sST2 secretion in humans. Cardiovasc Res (2010) 87(4):769–77. doi:10.1093/cvr/cvq104
50. Bandara G, Beaven MA, Olivera A, Gilfillan AM, Metcalfe DD. Activated mast cells synthesize and release soluble ST2-a decoy receptor for IL-33. Eur J Immunol (2015) 45(11):3034–44. doi:10.1002/eji.201545501
51. Zhang J, Ramadan AM, Griesenauer B, Li W, Turner MJ, Liu C, et al. ST2 blockade reduces sST2-producing T cells while maintaining protective mST2-expressing T cells during graft-versus-host disease. Sci Transl Med (2015) 7(308):308ra160. doi:10.1126/scitranslmed.aab0166
52. Oshikawa K, Yanagisawa K, Tominaga S, Sugiyama Y. Expression and function of the ST2 gene in a murine model of allergic airway inflammation. Clin Exp Allergy (2002) 32(10):1520–6. doi:10.1046/j.1365-2745.2002.01494.x
53. Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol (2007) 179(4):2551–5. doi:10.4049/jimmunol.179.4.2551
54. Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci U S A (2007) 104(47):18660–5. doi:10.1073/pnas.0705939104
55. Funakoshi-Tago M, Tago K, Hayakawa M, Tominaga S, Ohshio T, Sonoda Y, et al. TRAF6 is a critical signal transducer in IL-33 signaling pathway. Cell Signal (2008) 20(9):1679–86. doi:10.1016/j.cellsig.2008.05.013
56. Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity (2011) 35(2):299–311. doi:10.1016/j.immuni.2011.08.007
57. Peine M, Marek RM, Lohning M. IL-33 in T cell differentiation, function, and immune homeostasis. Trends Immunol (2016) 37(5):321–33. doi:10.1016/j.it.2016.03.007
58. Matta BM, Reichenbach DK, Zhang X, Mathews L, Koehn BH, Dwyer GK, et al. Peri-alloHCT IL-33 administration expands recipient T-regulatory cells that protect mice against acute GVHD. Blood (2016) 128(3):427–39. doi:10.1182/blood-2015-12-684142
59. Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, et al. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in toll-like receptor-dependent gene induction program. Proc Natl Acad Sci U S A (2006) 103(41):15136–41. doi:10.1073/pnas.0607181103
60. Fragale A, Gabriele L, Stellacci E, Borghi P, Perrotti E, Ilari R, et al. IFN regulatory factor-1 negatively regulates CD4+ CD25+ regulatory T cell differentiation by repressing Foxp3 expression. J Immunol (2008) 181(3):1673–82. doi:10.4049/jimmunol.181.3.1673
61. Bulek K, Swaidani S, Qin J, Lu Y, Gulen MF, Herjan T, et al. The essential role of single Ig IL-1 receptor-related molecule/Toll IL-1R8 in regulation of Th2 immune response. J Immunol (2009) 182(5):2601–9. doi:10.4049/jimmunol.0802729
62. Zhao J, Wei J, Mialki RK, Mallampalli DF, Chen BB, Coon T, et al. F-box protein FBXL19-mediated ubiquitination and degradation of the receptor for IL-33 limits pulmonary inflammation. Nat Immunol (2012) 13(7):651–8. doi:10.1038/ni.2341
63. Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem (2007) 282(36):26369–80. doi:10.1074/jbc.M704916200
64. Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest (2007) 117(6):1538–49. doi:10.1172/JCI30634
65. Kunze MM, Benz F, Brauss TF, Lampe S, Weigand JE, Braun J, et al. sST2 translation is regulated by FGF2 via an hnRNP A1-mediated IRES-dependent mechanism. Biochim Biophys Acta (2016) 1859(7):848–59. doi:10.1016/j.bbagrm.2016.05.005
66. Zhao J, Chen Q, Li H, Myerburg M, Spannhake EW, Natarajan V, et al. Lysophosphatidic acid increases soluble ST2 expression in mouse lung and human bronchial epithelial cells. Cell Signal (2012) 24(1):77–85. doi:10.1016/j.cellsig.2011.08.004
67. Tajima S, Oshikawa K, Tominaga S, Sugiyama Y. The increase in serum soluble ST2 protein upon acute exacerbation of idiopathic pulmonary fibrosis. Chest (2003) 124(4):1206–14. doi:10.1378/chest.124.4.1206
68. Andersson C, Preis SR, Beiser A, DeCarli C, Wollert KC, Wang TJ, et al. Associations of circulating growth differentiation factor-15 and ST2 concentrations with subclinical vascular brain injury and incident stroke. Stroke (2015) 46(9):2568–75. doi:10.1161/STROKEAHA.115.009026
69. Lopez-Casado MA, Lorite P, Palomeque T, Torres MI. Potential role of the IL-33/ST2 axis in celiac disease. Cell Mol Immunol (2017) 14(3):285–92. doi:10.1038/cmi.2015.85
70. Bergis D, Kassis V, Radeke HH. High plasma sST2 levels in gastric cancer and their association with metastatic disease. Cancer Biomark (2016) 16(1):117–25. doi:10.3233/CBM-150547
71. Lei Z, Mo Z, Zhu J, Pang X, Zheng X, Wu Z, et al. Soluble ST2 plasma concentrations predict mortality in HBV-related acute-on-chronic liver failure. Mediators Inflamm (2015) 2015:535938. doi:10.1155/2015/535938
72. Mehraj V, Jenabian MA, Ponte R, Lebouche B, Costiniuk C, Thomas R, et al. The plasma levels of soluble ST2 as a marker of gut mucosal damage in early HIV infection. AIDS (2016) 30(10):1617–27. doi:10.1097/QAD.0000000000001105
73. Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem (2009) 284(29):19420–6. doi:10.1074/jbc.M901744200
74. Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A (2012) 109(5):1673–8. doi:10.1073/pnas.1115884109
75. Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U S A (2009) 106(22):9021–6. doi:10.1073/pnas.0812690106
76. Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity (2009) 31(1):84–98. doi:10.1016/j.immuni.2009.05.007
77. Lefrancais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci U S A (2014) 111(43):15502–7. doi:10.1073/pnas.1410700111
78. Waern I, Lundequist A, Pejler G, Wernersson S. Mast cell chymase modulates IL-33 levels and controls allergic sensitization in dust-mite induced airway inflammation. Mucosal Immunol (2013) 6(5):911–20. doi:10.1038/mi.2012.129
79. Wood IS, Wang B, Trayhurn P. IL-33, a recently identified interleukin-1 gene family member, is expressed in human adipocytes. Biochem Biophys Res Commun (2009) 384(1):105–9. doi:10.1016/j.bbrc.2009.04.081
80. Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol (2012) 188(7):3488–95. doi:10.4049/jimmunol.1101977
81. Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol (2010) 10(2):103–10. doi:10.1038/nri2692
82. Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, et al. The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J Immunol (2011) 187(4):1609–16. doi:10.4049/jimmunol.1003080
83. Shao D, Perros F, Caramori G, Meng C, Dormuller P, Chou PC, et al. Nuclear IL-33 regulates soluble ST2 receptor and IL-6 expression in primary human arterial endothelial cells and is decreased in idiopathic pulmonary arterial hypertension. Biochem Biophys Res Commun (2014) 451(1):8–14. doi:10.1016/j.bbrc.2014.06.111
84. Bessa J, Meyer CA, de Vera Mudry MC, Schlicht S, Smith SH, Iglesias A, et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun (2014) 55:33–41. doi:10.1016/j.jaut.2014.02.012
85. Xu D, Chan W, Leung BP, Huang F, Wheeler R, Piedrafita D, et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med (1998) 187:787–94. doi:10.1084/jem.187.5.787
86. Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A (2009) 106(32):13463–8. doi:10.1073/pnas.0906988106
87. Piehler D, Grahnert A, Eschke M, Richter T, Kohler G, Stenzel W, et al. T1/ST2 promotes T helper 2 cell activation and polyfunctionality in bronchopulmonary mycosis. Mucosal Immunol (2013) 6(2):405–14. doi:10.1038/mi.2012.84
88. Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol (2008) 181(7):4780–90. doi:10.4049/jimmunol.181.11.8170
89. Yang Z, Sun R, Grinchuk V, Fernandez-Blanco JA, Notari L, Bohl JA, et al. IL-33-induced alterations in murine intestinal function and cytokine responses are MyD88, STAT6, and IL-13 dependent. Am J Physiol Gastrointest Liver Physiol (2013) 304(4):G381–9. doi:10.1152/ajpgi.00357.2012
90. Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol (2007) 37(10):2779–86. doi:10.1002/eji.200737547
91. Hoshino K, Kashiwamura S, Kuribayashi K, Kodama T, Tsujimura T, Nakanishi K, et al. The absence of interleukin 1 receptor-related T1/ST2 does not affect T helper cell type 2 development and its effector function. J Exp Med (1999) 190(10):1541–8. doi:10.1084/jem.190.10.1541
92. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta ’reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol (2008) 9(12):1341–6. doi:10.1038/ni.1659
93. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol (2008) 9(12):1347–55. doi:10.1038/ni.1677
94. Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol (2010) 11(6):527–34. doi:10.1038/ni.1867
95. Staudt V, Bothur E, Klein M, Lingnau K, Reuter S, Grebe N, et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity (2010) 33(2):192–202. doi:10.1016/j.immuni.2010.07.014
96. Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol (2007) 8(9):958–66. doi:10.1038/ni1500
97. Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med (2002) 195(8):1003–12. doi:10.1084/jem.20011128
98. Schmitt E, Klein M, Bopp T. Th9 cells, new players in adaptive immunity. Trends Immunol (2014) 35(2):61–8. doi:10.1016/j.it.2013.10.004
99. Wu B, Huang C, Kato-Maeda M, Hopewell PC, Daley CL, Krensky AM, et al. IL-9 is associated with an impaired Th1 immune response in patients with tuberculosis. Clin Immunol (2008) 126(2):202–10. doi:10.1016/j.clim.2007.09.009
100. Blom L, Poulsen BC, Jensen BM, Hansen A, Poulsen LK. IL-33 induces IL-9 production in human CD4+ T cells and basophils. PLoS One (2011) 6(7):e21695. doi:10.1371/journal.pone.0021695
101. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol (2014) 15(7):676–86. doi:10.1038/ni.2920
102. Li M, Li Y, Liu X, Gao X, Wang Y. IL-33 blockade suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. J Neuroimmunol (2012) 247(1–2):25–31. doi:10.1016/j.jneuroim.2012.03.016
103. Jiang HR, Milovanovic M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur J Immunol (2012) 42(7):1804–14. doi:10.1002/eji.201141947
104. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol (2014) 193(8):4010–20. doi:10.4049/jimmunol.1400481
105. Brunner SM, Schiechl G, Falk W, Schlitt HJ, Geissler EK, Fichtner-Feigl S. Interleukin-33 prolongs allograft survival during chronic cardiac rejection. Transpl Int (2011) 24(10):1027–39. doi:10.1111/j.1432-2277.2011.01306.x
106. Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol (2011) 187(9):4598–610. doi:10.4049/jimmunol.1100519
107. Gajardo T, Morales RA, Campos-Mora M, Campos-Acuna J, Pino-Lagos K. Exogenous interleukin-33 targets myeloid-derived suppressor cells and generates periphery-induced Foxp3(+) regulatory T cells in skin-transplanted mice. Immunology (2015) 146(1):81–8. doi:10.1111/imm.12483
108. Lal G, Yin N, Xu J, Lin M, Schroppel S, Ding Y, et al. Distinct inflammatory signals have physiologically divergent effects on epigenetic regulation of Foxp3 expression and Treg function. Am J Transplant (2011) 11(2):203–14. doi:10.1111/j.1600-6143.2010.03389.x
109. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature (2010) 463(7280):540–4. doi:10.1038/nature08636
110. Stehle C, Saikali P, Romagnani C. Putting the brakes on ILC2 cells. Nat Immunol (2016) 17(1):43–4. doi:10.1038/ni.3353
111. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol (2011) 12(11):1055–62. doi:10.1038/ni.2104
112. Yagi R, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, et al. The transcription factor GATA3 is critical for the development of all IL-7Ralpha-expressing innate lymphoid cells. Immunity (2014) 40(3):378–88. doi:10.1016/j.immuni.2014.01.012
113. Spooner CJ, Lesch J, Yan D, Khan AA, Abbas A, Ramirez-Carrozzi V, et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol (2013) 14(12):1229–36. doi:10.1038/ni.2743
114. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A (2010) 107(25):11489–94. doi:10.1073/pnas.1003988107
115. Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol (2011) 12(11):1071–7. doi:10.1038/ni.2133
116. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med (2013) 210(13):2939–50. doi:10.1084/jem.20130351
117. Matsuki A, Takatori H, Makita S, Yokota M, Tamachi T, Suto A, et al. T-bet inhibits innate lymphoid cell-mediated eosinophilic airway inflammation by suppressing IL-9 production. J Allergy Clin Immunol (2017) 139(4):1355–67.e6. doi:10.1016/j.jaci.2016.08.022
118. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12(11):1045–54. doi:10.1031/ni.2131
119. Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci U S A (2013) 110(34):13921–6. doi:10.1073/pnas.1307321110
120. Rak GD, Osborne LC, Siracusa MC, Kim BS, Wang K, Bayat A, et al. IL-33-dependent group 2 innate lymphoid cells promote cutaneous wound healing. J Invest Dermatol (2016) 136(2):487–96. doi:10.1038/JID.2015.406
121. McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity (2013) 39(2):357–71. doi:10.1016/j.immuni.2013.07.018
122. Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, et al. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer (2014) 134(7):1669–82. doi:10.1002/ijc.28481
123. Yang Q, Li G, Zhu Y, Liu L, Chen E, Turnquist H, et al. IL-33 synergizes with TCR and IL-12 signaling to promote the effector function of CD8+ T cells. Eur J Immunol (2011) 41(11):3351–60. doi:10.1002/eji.201141629
124. Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science (2012) 335(6071):984–9. doi:10.1126/science.1215418
125. Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol (2015) 194(1):438–45. doi:10.4049/jimmunol.1401344
126. Reichenbach DK, Schwarze V, Matta BM, Tkachev V, Lieberknecht E, Liu Q, et al. The IL-33/ST2 axis augments effector T-cell responses during acute GVHD. Blood (2015) 125(20):3183–92. doi:10.1182/blood-2014-10-606830
127. Komai-Koma M, Wang E, Kurowska-Stolarska M, Li D, McSharry C, Xu D. Interleukin-33 promoting Th1 lymphocyte differentiation dependents on IL-12. Immunobiology (2016) 221(3):412–7. doi:10.1016/j.imbio.2015.11.013
128. Komai-Koma M, Gilchrist DS, McKenzie AN, Goodyear CS, Xu D, Liew FY. IL-33 activates B1 cells and exacerbates contact sensitivity. J Immunol (2011) 186(4):2584–91. doi:10.4049/jimmunol.1002103
129. Sattler S, Ling GS, Xu D, Hussaarts L, Romaine A, Zhao H, et al. IL-10-producing regulatory B cells induced by IL-33 (Breg(IL-33)) effectively attenuate mucosal inflammatory responses in the gut. J Autoimmun (2014) 50:107–22. doi:10.1016/j.jaut.2014.01.032
130. Bourgeois E, Van LP, Samson M, Diem S, Barra A, Roga S, et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur J Immunol (2009) 39(4):1046–55. doi:10.1002/eji.200838575
131. Nabekura T, Girard JP, Lanier LL. IL-33 receptor ST2 amplifies the expansion of NK cells and enhances host defense during mouse cytomegalovirus infection. J Immunol (2015) 194(12):5948–52. doi:10.4049/jimmunol.1500424
132. Palmer G, Talabot-Ayer D, Lamacchia C, Toy D, Seemayer CA, Viatte S, et al. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum (2009) 60(3):738–49. doi:10.1002/art.24305
133. Leung BP, Xu D, Culshaw S, McInnes IB, Liew FY. A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J Immunol (2004) 173(1):145–50. doi:10.4049/jimmunol.173.1.145
134. Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, Kita H. IL-33-activated dendritic cells induce an atypical TH2-type response. J Allergy Clin Immunol (2009) 123(5):1047–54. doi:10.1016/j.jaci.2009.02.026
135. Espinassous Q, Garcia-de-Paco E, Garcia-Verdugo I, Synguelakis M, von Aulock S, Sallenave JM, et al. IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. J Immunol (2009) 183(2):1446–55. doi:10.4049/jimmunol.0803067
136. Joshi AD, Oak SR, Hartigan AJ, Finn WG, Kunkel SL, Duffy KE, et al. Interleukin-33 contributes to both M1 and M2 chemokine marker expression in human macrophages. BMC Immunol (2010) 11:52. doi:10.1186/1471-2172-11-52
137. Wang JX, Kaieda S, Ameri S, Fishgal N, Dwyer D, Dellinger A, et al. IL-33/ST2 axis promotes mast cell survival via BCLXL. Proc Natl Acad Sci U S A (2014) 111(28):10281–6. doi:10.1073/pnas.1404182111
138. Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: the ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol (2007) 179(4):2051–4. doi:10.4049/jimmunol.179.4.2051
139. Sabatino G, Nicoletti M, Neri G, Saggini A, Rosati M, Conti F, et al. Impact of IL-9 and IL-33 in mast cells. J Biol Regul Homeost Agents (2012) 26(4):577–86.
140. Andrade MV, Iwaki S, Ropert C, Gazzinelli RT, Cunha-Melo JR, Beaven MA. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur J Immunol (2011) 41(3):760–72. doi:10.1002/eji.201040718
141. Amin K. The role of mast cells in allergic inflammation. Respir Med (2012) 106(1):9–14. doi:10.1016/j.rmed.2011.09.007
142. Sawaguchi M, Tanaka S, Nakatani Y, Harada Y, Mukai K, Matsunaga Y, et al. Role of mast cells and basophils in IgE responses and in allergic airway hyperresponsiveness. J Immunol (2012) 188(4):1809–18. doi:10.4049/jimmunol.1101746
143. Saluja R, Khan M, Church MK, Maurer M. The role of IL-33 and mast cells in allergy and inflammation. Clin Transl Allergy (2015) 5:33. doi:10.1186/s13601-015-0076-5
144. Cho KA, Suh JW, Sohn JH, Park JW, Lee H, Kang JL, et al. IL-33 induces Th17-mediated airway inflammation via mast cells in ovalbumin-challenged mice. Am J Physiol Lung Cell Mol Physiol (2012) 302(4):L429–40. doi:10.1152/ajplung.00252.2011
145. Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood (2009) 113(7):1526–34. doi:10.1182/blood-2008-05-157818
146. Na HJ, Hudson SA, Bochner BS. IL-33 enhances Siglec-8 mediated apoptosis of human eosinophils. Cytokine (2012) 57(1):169–74. doi:10.1016/j.cyto.2011.10.007
147. Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol (2010) 185(6):3472–80. doi:10.4049/jimmunol.1000730
148. Suzukawa M, Koketsu R, Iikura M, Nakae S, Matsumoto K, Nagase H, et al. Interleukin-33 enhances adhesion, CD11b expression and survival in human eosinophils. Lab Invest (2008) 88(11):1245–53. doi:10.1038/labinvest.2008.82
149. Turnquist HR, Sumpter TL, Tsung A, Zahorchak AF, Nakao A, Nau GJ, et al. IL-1beta-driven ST2L expression promotes maturation resistance in rapamycin-conditioned dendritic cells. J Immunol (2008) 181(1):62–72. doi:10.4049/jimmunol.181.1.62
150. Su Z, Lin J, Lu F, Zhang X, Zhang L, Gandhi NB, et al. Potential autocrine regulation of interleukin-33/ST2 signaling of dendritic cells in allergic inflammation. Mucosal Immunol (2013) 6(5):921–30. doi:10.1038/mi.2012.130
151. Besnard AG, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur J Immunol (2011) 41(6):1675–86. doi:10.1002/eji.201041033
152. Nagata A, Takezako N, Tamemoto H, Ohto-Ozaki H, Ohta S, Tominaga S, et al. Soluble ST2 protein inhibits LPS stimulation on monocyte-derived dendritic cells. Cell Mol Immunol (2012) 9(5):399–409. doi:10.1038/cmi.2012.29
153. Alves-Filho JC, Sonego F, Souto FO, Freitas A, Verri WA Jr, Auxiliadora-Martins M, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med (2010) 16(6):708–12. doi:10.1038/nm.2156
154. Hueber AJ, Alves-Filho JC, Asquith DL, Michels C, Millar NL, Reilly JH, et al. IL-33 induces skin inflammation with mast cell and neutrophil activation. Eur J Immunol (2011) 41(8):2229–37. doi:10.1002/eji.201041360
155. Sedhom MA, Pichery M, Murdoch JR, Foligne B, Ortega N, Normand S, et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut (2013) 62(12):1714–23. doi:10.1136/gutjnl-2011-301785
156. Duan L, Chen J, Zhang H, Yang H, Zhu P, Xiong A, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol Med (2012) 18:753–61. doi:10.2119/molmed.2011.00428
157. Makita S, Kanai T, Oshima S, Uraushihara K, Totsuka T, Sawada T, et al. CD4+CD25bright T cells in human intestinal lamina propria as regulatory cells. J Immunol (2004) 173(5):3119–30. doi:10.4049/jimmunol.173.5.3119
158. Holmen N, Lundgren A, Lundin S, Bergin AM, Rudin A, Sjovall H, et al. Functional CD4+CD25high regulatory T cells are enriched in the colonic mucosa of patients with active ulcerative colitis and increase with disease activity. Inflamm Bowel Dis (2006) 12(6):447–56. doi:10.1097/00054725-200606000-00003
159. Grobeta P, Doser K, Falk W, Obermeier F, Hofmann C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm Bowel Dis (2012) 18(10):1900–9. doi:10.1002/ibd.22900
160. Ferrara JL, Cooke KR, Teshima T. The pathophysiology of acute graft-versus-host disease. Int J Hematol (2003) 78(3):181–7. doi:10.1007/BF02983793
161. Humphreys NE, Xu D, Hepworth MR, Liew FY, Grencis RK. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J Immunol (2008) 180(4):2443–9. doi:10.4049/jimmunol.180.4.2443
162. Jones LA, Roberts F, Nickdel MB, Brombacher F, McKenzie AN, Henriquez FL, et al. IL-33 receptor (T1/ST2) signalling is necessary to prevent the development of encephalitis in mice infected with Toxoplasma gondii. Eur J Immunol (2010) 40(2):426–36. doi:10.1002/eji.200939705
163. Wagenaar JF, Gasem MH, Goris MG, Leeflang M, Hartskeerl RA, van der Poll T, et al. Soluble ST2 levels are associated with bleeding in patients with severe Leptospirosis. PLoS Negl Trop Dis (2009) 3(6):e453. doi:10.1371/journal.pntd.0000453
164. Huang X, Du W, Barrett RP, Hazlett LD. ST2 is essential for Th2 responsiveness and resistance to pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci (2007) 48(10):4626–33. doi:10.1167/iovs.07-0316
165. Hunt PW, Sinclair E, Rodriguez B, Shive C, Clagett B, Funderburg N, et al. Gut epithelial barrier dysfunction and innate immune activation predict mortality in treated HIV infection. J Infect Dis (2014) 210(8):1228–38. doi:10.1093/infdis/jiu238
166. Olin JT, Wechsler ME. Asthma: pathogenesis and novel drugs for treatment. BMJ (2014) 349:g5517. doi:10.1136/bmj.g5517
167. Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol (2013) 132(4):933–41. doi:10.1016/j.jaci.2013.05.012
168. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage-CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol (2012) 188(3):1503–13. doi:10.4049/jimmunol.1102832
169. Ying S, Humbert M, Barkans J, Corrigan CJ, Pfister R, Menz G, et al. Expression of IL-4 and IL-5 mRNA and protein product by CD4+ and CD8+ T cells, eosinophils, and mast cells in bronchial biopsies obtained from atopic and nonatopic (intrinsic) asthmatics. J Immunol (1997) 158(7):3539–44.
170. Sehra S, Yao W, Nguyen ET, Glosson-Byers NL, Akhtar N, Zhou B, et al. TH9 cells are required for tissue mast cell accumulation during allergic inflammation. J Allergy Clin Immunol (2015) 136(2):433–40.e1. doi:10.1016/j.jaci.2015.01.021
171. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol (2008) 20(6):791–800. doi:10.1093/intimm/dxn037
172. Louten J, Rankin AL, Li Y, Murphy EE, Beaumont M, Moon C, et al. Endogenous IL-33 enhances Th2 cytokine production and T-cell responses during allergic airway inflammation. Int Immunol (2011) 23(5):307–15. doi:10.1093/intimm/dxr006
173. Kearley J, Buckland KF, Mathie SA, Lloyd CM. Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2-IL-33 pathway. Am J Respir Crit Care Med (2009) 179(9):772–81. doi:10.1164/rccm.200805-666OC
174. Hogan SP, Matthaei KI, Young JM, Koskinen A, Young IG, Foster PS. A novel T cell-regulated mechanism modulating allergen-induced airways hyperreactivity in BALB/c mice independently of IL-4 and IL-5. J Immunol (1998) 161(3):1501–9.
175. Coyle AJ, Lloyd C, Tian J, Nguyen T, Erikkson C, Wang L, et al. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J Exp Med (1999) 190(7):895–902. doi:10.1084/jem.190.7.895
176. Lee HY, Rhee CK, Kang JY, Byun JH, Choi JY, Kim SJ, et al. Blockade of IL-33/ST2 ameliorates airway inflammation in a murine model of allergic asthma. Exp Lung Res (2014) 40(2):66–76. doi:10.3109/01902148.2013.870261
177. Bajwa EK, Volk JA, Christiani DC, Harris RS, Matthay MA, Thompson BT, et al. Prognostic and diagnostic value of plasma soluble suppression of tumorigenicity-2 concentrations in acute respiratory distress syndrome. Crit Care Med (2013) 41(11):2521–31. doi:10.1097/CCM.0b013e3182978f91
178. Xia J, Zhao J, Shang J, Li M, Zeng Z, Wang J, et al. Increased IL-33 expression in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol (2015) 308(7):L619–27. doi:10.1152/ajplung.00305.2014
179. Bradley-Stewart A, Jolly L, Adamson W, Gunson R, Frew-Gillespie C, Templeton K, et al. Cytokine responses in patients with mild or severe influenza A(H1N1)pdm09. J Clin Virol (2013) 58(1):100–7. doi:10.1016/j.jcv.2013.05.011
180. Seo S, Yu J, Jenkins IC, Stevens-Ayers TL, Kuypers JM, Huang M-L, et al. Biomarkers for idiopathic pneumonia syndrome (IPS) after hematopoietic cell transplantation (HCT): comparison with viral infectious pneumonia. Biol Blood Marrow Transplant (2016) 22(Suppl 3):S73–4. doi:10.1016/j.bbmt.2015.11.366
182. Savinko T, Matikainen S, Saarialho-Kere U, Lehto M, Wang G, Lehtimaki S, et al. IL-33 and ST2 in atopic dermatitis: expression profiles and modulation by triggering factors. J Invest Dermatol (2012) 132(5):1392–400. doi:10.1038/jid.2011.446
183. Nygaard U, Hvid M, Johansen C, Buchner M, Folster-Holst R, Deleuran M, et al. TSLP, IL-31, IL-33 and sST2 are new biomarkers in endophenotypic profiling of adult and childhood atopic dermatitis. J Eur Acad Dermatol Venereol (2016) 30(11):1930–8. doi:10.1111/jdv.13679
184. Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol (2008) 128(5):1207–11. doi:10.1038/sj.jid.5701213
185. Li P, Ma H, Han D, Mou K. Interleukin-33 affects cytokine production by keratinocytes in vitiligo. Clin Exp Dermatol (2015) 40(2):163–70. doi:10.1111/ced.12464
186. Lafyatis R. Application of biomarkers to clinical trials in systemic sclerosis. Curr Rheumatol Rep (2012) 14(1):47–55. doi:10.1007/s11926-011-0216-4
187. Rice LM, Mantero JC, Stifano G, Ziemek J, Simms RW, Gordon J, et al. A proteome-derived longitudinal pharmacodynamic biomarker for diffuse systemic sclerosis skin. J Invest Dermatol (2017) 137(1):62–70. doi:10.1016/j.jid.2016.08.027
188. Sabatine MS, Cannon CP, Gibson CM, Lopez-Sendon JL, Montalescot G, Theroux P, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med (2005) 352(12):1179–89. doi:10.1056/NEJMoa050522
189. Seki K, Sanada S, Kudinova AY, Steinhauser ML, Handa V, Gannon J, et al. Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ Heart Fail (2009) 2(6):684–91. doi:10.1161/CIRCHEARTFAILURE.109.873240
190. Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, et al. IL-33 reduces the development of atherosclerosis. J Exp Med (2008) 205(2):339–46. doi:10.1084/jem.20071868
191. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest (2011) 121(6):2111–7. doi:10.1172/JCI57132
192. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15(8):930–9. doi:10.1038/nm.2002
193. Han JM, Wu D, Denroche HC, Yao Y, Verchere CB, Levings MK. IL-33 reverses an obesity-induced deficit in visceral adipose tissue ST2+ T regulatory cells and ameliorates adipose tissue inflammation and insulin resistance. J Immunol (2015) 194(10):4777–83. doi:10.4049/jimmunol.1500020
194. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol (2015) 16(3):276–85. doi:10.1038/ni0515-544d
195. Miller AM, Asquith DL, Hueber AJ, Anderson LA, Holmes WM, McKenzie AN, et al. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res (2010) 107(5):650–8. doi:10.1161/CIRCRESAHA.110.218867
196. Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, et al. Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature (2015) 528(7580):137–41. doi:10.1038/nature16151
197. Zeyda M, Wernly B, Demyanets S, Kaun C, Hammerle M, Hantusch B, et al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int J Obes (2013) 37(5):658–65. doi:10.1038/ijo.2012.118
198. Celic V, Majstorovic A, Pencic-Popovic B, Sljivic A, Lopez-Andres N, Roy I, et al. Soluble ST2 levels and left ventricular structure and function in patients with metabolic syndrome. Ann Lab Med (2016) 36(6):542–9. doi:10.3343/alm.2016.36.6.542
199. Paczesny S, Hakim FT, Pidala J, Cooke KR, Lathrop J, Griffith LM, et al. National Institutes of Health Consensus Development Project on criteria for clinical trials in chronic graft-versus-host disease: III. The 2014 Biomarker Working Group Report. Biol Blood Marrow Transplant (2015) 21(5):780–92. doi:10.1016/j.bbmt.2015.01.003
Keywords: IL-33, ST2, IL1RL1, graft-versus-host disease, cardiac diseases, lung diseases
Citation: Griesenauer B and Paczesny S (2017) The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 8:475. doi: 10.3389/fimmu.2017.00475
Received: 23 November 2016; Accepted: 05 April 2017;
Published: 24 April 2017
Edited by:
Philippe Saas, Etablissement Français du Sang BFC, FranceReviewed by:
Heth Roderick Turnquist, University of Pittsburgh, USACopyright: © 2017 Griesenauer and Paczesny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Paczesny, c29waHBhY3pAaXUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.