
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 18 April 2017
Sec. Primary Immunodeficiencies
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00426
 Samuel C. C. Chiang1†
Samuel C. C. Chiang1† Stephanie M. Wood2†
Stephanie M. Wood2† Bianca Tesi1,3,4
Bianca Tesi1,3,4 Himmet Haluk Akar5
Himmet Haluk Akar5 Waleed Al-Herz6
Waleed Al-Herz6 Sandra Ammann7
Sandra Ammann7 Fatma Burcu Belen8Umran Caliskan9
Fatma Burcu Belen8Umran Caliskan9 Zühre Kaya10Kai Lehmberg11
Zühre Kaya10Kai Lehmberg11 Turkan Patiroglu5Huseyin Tokgoz9Ayşegül Ünüvar12Wendy J. Introne13Jan-Inge Henter3
Turkan Patiroglu5Huseyin Tokgoz9Ayşegül Ünüvar12Wendy J. Introne13Jan-Inge Henter3 Magnus Nordenskjöld4
Magnus Nordenskjöld4 Hans-Gustaf Ljunggren2
Hans-Gustaf Ljunggren2 Marie Meeths3
Marie Meeths3 Stephan Ehl7
Stephan Ehl7 Konrad Krzewski14
Konrad Krzewski14 Yenan T. Bryceson1,15*
Yenan T. Bryceson1,15*
Chediak–Higashi syndrome (CHS) is caused by autosomal recessive mutations in LYST, resulting in enlarged lysosomal compartments in multiple cell types. CHS patients display oculocutaneous albinism and may develop life-threatening hemophagocytic lymphohistiocytosis (HLH). While NK cell-mediated cytotoxicity has been reported to be uniformly defective, variable defects in T cell-mediated cytotoxicity has been observed. The latter has been linked to the degree of HLH susceptibility. Since the discrepancies in NK cell- and T cell-mediated cellular cytotoxicity might result from differences in regulation of cytotoxic granule release, we here evaluated perforin-containing secretory lysosome size and number in freshly isolated lymphocytes from CHS patients and furthermore compared their exocytic capacities. Whereas NK cells from CHS patients generally contained a single, gigantic perforin-containing granule, cytotoxic T cells predominantly contained several smaller granules. Nonetheless, in a cohort of 21 CHS patients, cytotoxic T cell and NK cell granule exocytosis were similarly impaired upon activating receptor stimulation. Mechanistically, polarization of cytotoxic granules was defective in cytotoxic lymphocytes from CHS patients, with EEA1, a marker of early endosomes, mislocalizing to lysosomal structures. The results leads to the conclusion that lysosome enlargement corresponds to loss of distinct organelle identity in the endocytic pathway, which on a subcellular level more adversely affects NK cells than T cells. Hence, vesicular size or numbers do not per se dictate the impairment of lysosomal exocytosis in the two cell types studied.
Chediak–Higashi syndrome (CHS, MIM 214500) is an autosomal recessive disease associated with mutations in LYST (1–3). LYST encompasses 55 exons and encodes an evolutionary conserved, ubiquitously expressed cytosolic protein of 3801 amino acids. CHS is characterized by oculocutaneous albinism and not seldom development of hemophagocytic lymphohistiocytosis (HLH), the latter a life-threatening hyperinflammatory syndrome (4, 5). However, age at onset of HLH is highly variable. Up to 20% of patients do not develop HLH. CHS patients may otherwise present with lymphoma (6, 7). Neurological deficits often appear in adolescence and may progress even after hematopoietic stem cell transplantation (8). An ability to predict clinical outcome based on genotype or diagnostic criteria, in particular with respect to HLH development, would be of value for the clinical management of CHS patients.
Orthologs of human LYST have been implicated in regulation of protein sorting and endosomal vesicle fusion (9–11). In humans, the defining characteristic of CHS is giant lysosomes and lysosome-related organelles (12). While this does not seem to impair the physiology of most cell types (12), functional defects are apparent in cells that perform lysosomal secretion, including melanocytes, lymphocytes, platelets, MHC class II-expressing antigen presenting cells, and glial cells, resulting in oculocutaneous albinism and prolonged bleeding (13). In lymphocytes, these defects lead to impaired cytotoxicity, a trait that has been closely linked to the development of HLH (7, 14–16). Development of lymphoma and an overall increased risk of cancer have also been reported in individuals with impaired lymphocyte cytotoxicity, including CHS patients (17–19). Mouse models have provided insights to the pathogenesis of CHS. In souris mice, which have a splice-site frameshift mutation in intron 27 of Lyst, development of HLH is seen upon LCMV infection (20). In contrast, beige mice, which have a 3-nucleotide in-frame deletion at the C-terminal encoding region of Lyst, do not develop HLH upon similar infection (1–3). A study encompassing analyses of mouse models as well as human CHS patients indicated that the risk of HLH development in CHS is determined by subtle differences in cytotoxic T lymphocyte (CTL) function, while NK cell cytotoxic function was generally more severely and uniformly compromised (20). While that study did not find clear genotype–phenotype correlations, other clinical studies have suggested genotype–phenotype correlations that may explain CHS severity (5).
To elucidate possible differences in T cell- and NK cell-mediated regulation of cytotoxic granule release in CHS, we here performed a detailed comparison of the characteristics and exocytic capacity of perforin-containing secretory lysosomes in cytotoxic lymphocyte subsets freshly isolated from CHS patients.
Genetic analysis was subsequently performed as described below. As controls, peripheral blood was obtained from the Karolinska University Hospital blood bank.
Genomic DNA was isolated from the peripheral blood according to standard procedures. HLH-associated genes, including LYST, were sequenced as described previously (21). Mutations identified by high-throughput sequencing were verified by Sanger sequencing. Primers, PCR conditions, and sequencing reaction conditions are available on request.
Peripheral blood was collected in heparin tubes and processed within 48 h of collection. Peripheral blood mononuclear cells (PBMC) were obtained by density gradient centrifugation (Lymphoprep; Axis-Shield) and resuspended in complete medium (RPMI 1640 medium with 10% FBS, l-glutamine, penicillin, and streptomycin; all Hyclone). For some experiments, NK cells were isolated from PBMC by negative selection (Miltenyi Biotec), maintained in complete medium, and used within 2 days of isolation. These cells were determined by flow cytometry to be more than 95% pure CD3−CD56+ NK cells. The human erythroleukemia cell line K562 and mouse mastocytoma cell line P815 (American Type Culture Collection) were maintained in complete medium.
For cell staining and flow cytometry, fluorochrome-conjugated antibodies to CD3 (clone S4.1; Invitrogen), CD4 (S3.5; Invitrogen), CD8 (RPA-T8; BioLegend), CD16 (3G8; BD Biosciences), CD45 (30-F11; Invitrogen), CD56 (NCAM16.2; BD Biosciences), CD57 (HCD57; BioLegend), CD107a (H4A3; BD Biosciences), perforin (δG9; BD Biosciences), granzyme A (CB9; BD Biosciences), and granzyme B (GB11; BD Biosciences), were used. Fluorochrome-conjugated IgG1 and IgG2b (MOPC-21 and 27-35; BD Biosciences) isotype control antibodies were also used in addition to a fixable live/dead cell marker (Invitrogen). Mouse anti-CD3 (S4.1) and purified anti-CD16 (3G8) mAbs were used for redirected ADCC. For confocal microscopy, mouse monoclonal antibodies to α-tubulin (236-10501; Invitrogen), mannose-6-phosphate receptor (MEM-238; Invitrogen), and perforin (δG9; BioLegend) mAbs were used. Rabbit polyclonal antibodies used were Rab27a, Stx11, and Munc13-4 (all Protein Technologies Group), EEA1 (Cell Signaling Technologies), WASP-interacting protein (WIP, Santa-Cruz), and Cathepsin D (Upstate). Secondary antibodies were donkey anti-mouse and donkey anti-rabbit (both Invitrogen). DNA was labeled with DAPI and actin with phalloidin (both Invitrogen).
Fifty microliters of fresh whole blood were stained in Trucount tubes (BD Biosciences) according to manufacturers instructions with fluorescent conjugated antibodies as listed above to distinguish various leukocyte and lymphocyte populations.
Cells were either evaluated without pre-stimulation or after 36–48 h of stimulation with 500 IU/ml interleukin (IL)-2 (Proleukin, Novartis). For quantification of cytotoxic granule exocytosis, PBMC were mixed with target cells, with 2.5 μg/ml activating mAb where indicated as published (22). Cells were incubated for 2 h at 37°C. For intracellular staining, PBMC were surface stained with fluorochrome-conjugated antibodies as indicated. Cells were then fixed (Cytofix; BD Biosciences), followed by intracellular staining (Cytoperm; BD Biosciences). Isotype control antibodies were used as negative control. For assessment of functional responses, freshly isolated or IL-2-stimulated PBMC were incubated alone, with K562 cells, P815 cells, or P815 cells with either anti-CD16 or anti-CD3 antibody. After stimulation, the lymphocytes were surface stained with antibodies as indicated. All flow cytometry data were acquired on an LSRFortessa (BD Biosciences) and the resulting data were analyzed with FlowJo v9.9 software (TreeStar) and Prism Version 5.0 software (GraphPad). For analyses of cytotoxic granule constituent expression, at least 100 gated cytotoxic lymphocytes were analyzed.
Standard 4-h 51Cr-release assays were performed for NK cells as previously described (23). Briefly, 4 × 104 51Cr-labeled K562 target cells were incubated with peripheral blood effector cells in 200 μl of complete medium in 96-well V-bottom plates at various concentrations. Experiments were performed in triplicate, and effector to target cell ratios ranged from 100 to 0.3. The supernatants were measured for 51Cr release on a gamma-counter (Cobra Auto-Gamma, Packard). Lytic units at 25% target cell lysis were calculated as previously described (24). Cytotoxic T cell cytotoxicity requiring purified CD3+CD8+CD57+ cells was not performed due to the limited volumes of blood obtained from patients (22).
Freshly isolated NK cells or CTL, resting or stimulated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA) (Calbiochem) and 0.5 mM ionomycin (Sigma-Aldrich), or co-incubated with K562 cells were spun down in glass-bottom plates (Matrical, Brooks). After 15 min, cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 0.5% saponin in PBS, and blocked in PBS with 5% FBS, 0.1% BSA-c (Aurion), and 2% normal donkey serum (Jackson ImmunoResearch). Images were acquired on a confocal microscope (Nikon A1R) with a 63× oil objective, using NIS Elements Software, and analyzed using ImageJ (Research Service Branch, National Institutes of Health, Bethesda, MD) or PAD (Digital Cell Imaging Laboratories, Belgium). To quantify the size and number of lytic granules, mean fluorescence intensity, as well as distances between granules, immune synapses (IS) and the microtubule-organizing center (MTOC), images of fixed cells with were analyzed using the PAD software. Object detection based on perforin or CD107a labeling was performed automatically and checked manually. MTOC XY co-ordinates were selected manually based on α-tubulin labeling, and the center of the IS was entered based on phalloidin labeling and phase contrast images. Mean and standard deviation (SD) per patient were then graphed using Prism.
Statistical analysis was performed using Prism software (version 5, GraphPad Software, Inc.), as specified.
In characterizing this cohort of CHS patients, we identified biallelic mutations in LYST in a total of 21 patients from 14 unrelated families (Table 1). While the majority of identified LYST mutations have been previously described (20, 21, 25–30), LYST c.3938delA (p.Q1313RfsX4) in family 9, and c.265insA (p.R886TfsX5) and c.5601delA (p.K1867NfsX11) in family 12 are novel. With the exception of LYST p.A1454N (patient 19) and p.G408R (families 17 and 18), which were present in compound heterozygous form, all mutations were nonsense or frameshift mutations predicted to result in truncated LYST protein. Clinically, 20 out of 21 patients had oculocutaneous albinism, 11 out of 21 patients developed HLH, and 12 out of 21 patients suffered neurological sequelae, including nystagmus, convulsions, and abnormal MRI, The six adult patients who had not developed HLH displayed albinism and neurological symptoms. Only three patients were transplanted, all successfully.
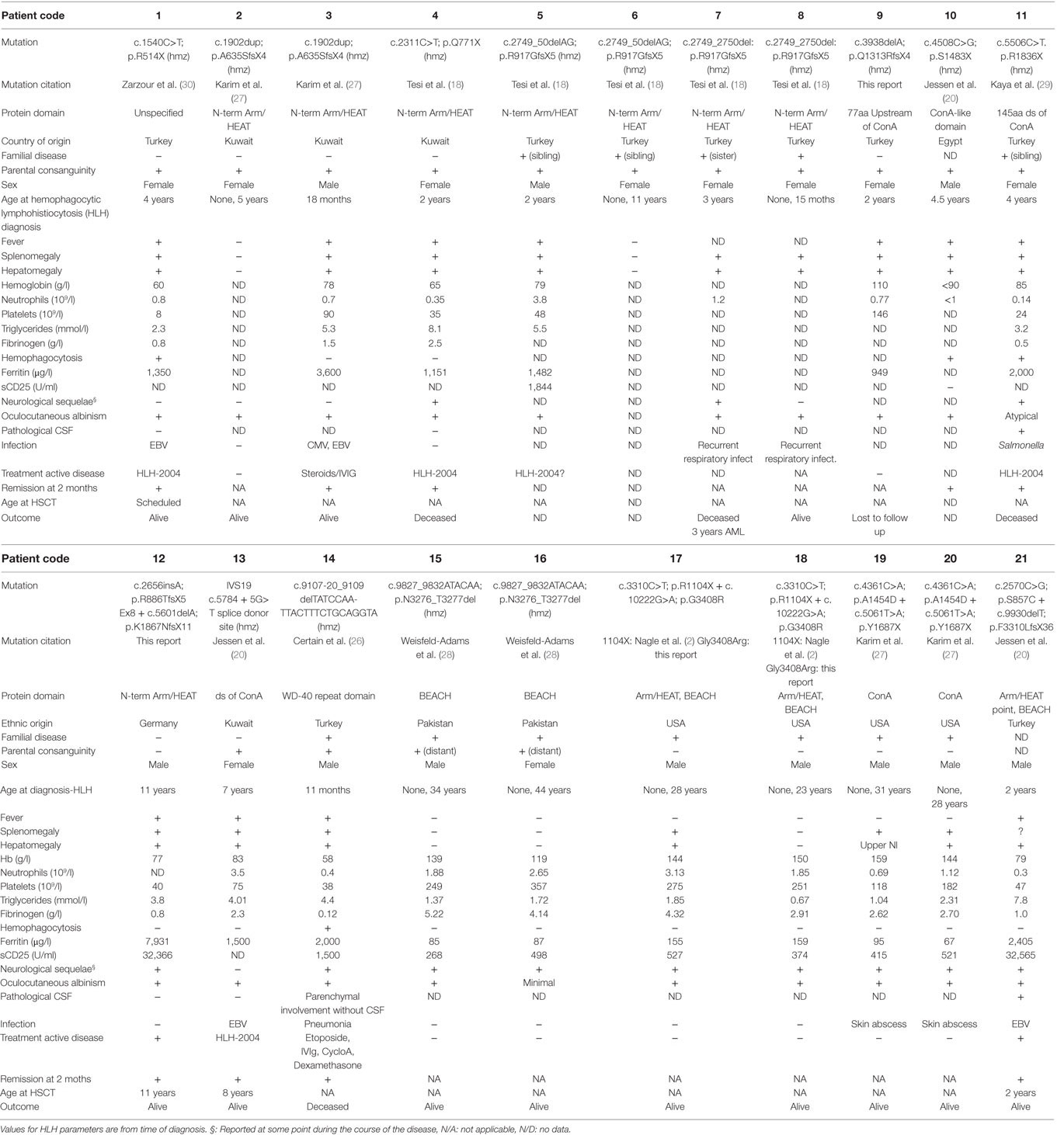
Table 1. Clinical and laboratory findings in patients with LYST mutations.
Peripheral blood from the 21 CHS patients with defined mutations in LYST were analyzed with respect to numbers and also stratified according to age at onset of HLH (Table 1). To grossly assess whether the CHS patient cytotoxic lymphocytes developed and differentiated normally, leukocyte subsets were enumerated in whole blood from 13 patients. In this respect, we examined PBMC counts of patients and compared them with healthy related (family) and unrelated (transport control) samples. CHS patient leukocyte numbers were similar to numbers observed in healthy individuals (Figure 1A). More specifically, neutrophil numbers were significantly decreased in CHS patients (Figure 1B), whereas monocyte cell numbers tended to be higher in CHS patients (Figure 1C), and lymphocyte numbers were significantly elevated in CHS patients as compared to healthy controls (Figure 1D). Overall B cell and T cell numbers were elevated above both control groups (Figures 1E–G), while the CD8+ T cell subset was not specifically elevated (Figure 1H). With respect to cytotoxic lymphocytes, CTL (CD8+CD57+ T cells) and NK cell numbers were not significantly elevated in CHS patients (Figures 1I,J), in contrast with a previous report that indicated somewhat elevated HNK-1+ (CD57+) cell numbers in CHS patients (31). The severity of lymphocytosis, consisting of elevated B cell and CD4+ T cell numbers, and neutropenia was most severe in patients with early onset HLH, and least severe in patients with no HLH (data not shown).
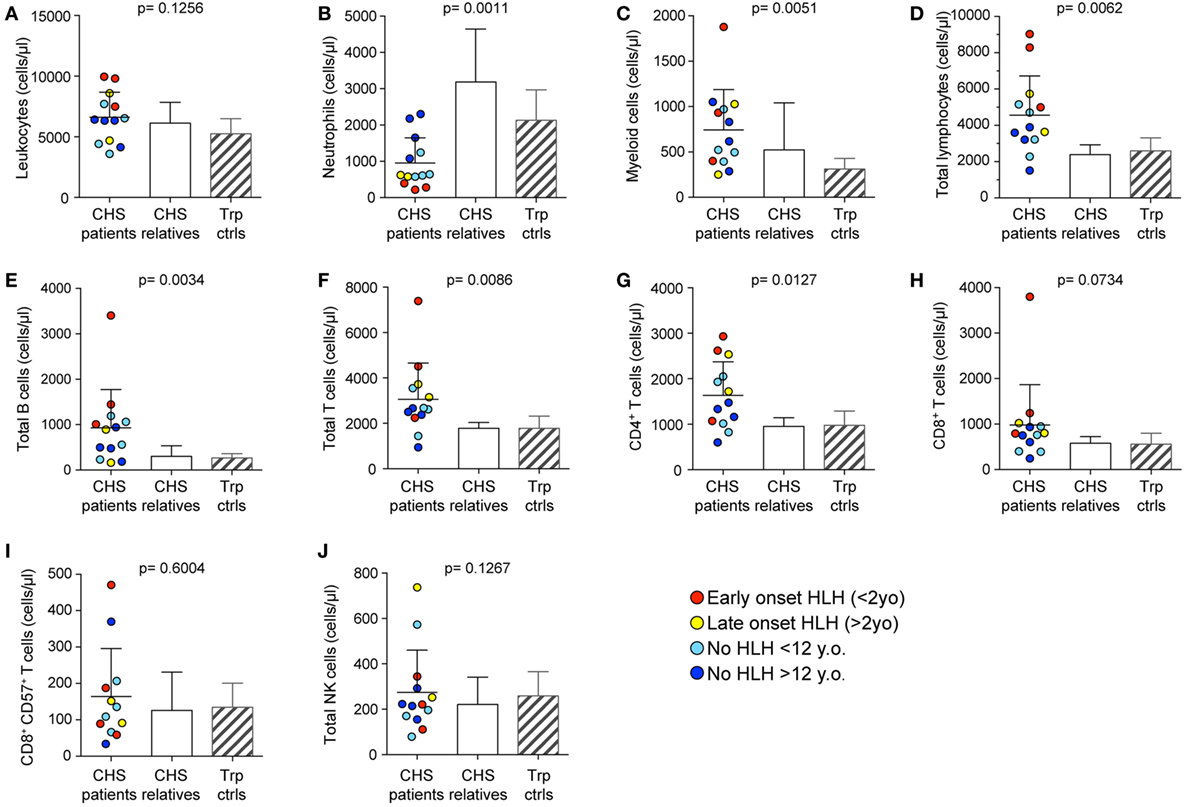
Figure 1. Peripheral blood cell numbers in Chediak–Higashi syndrome (CHS) patients. (A) Total leukocyte, (B) neutrophil, (C) monocyte, (D) lymphocyte, (E) CD19+ B cell, (F) CD3+ T cell, (G) CD3+CD4+ T cell, (H) CD3+CD8+ T cell, (I) cytotoxic CD3+CD8+CD57+ T cell, and (J) bulk CD3−CD56+ NK cell numbers were enumerated in peripheral blood from 13 CHS patients. The patients are color-coded according to whether they presented with early-onset hemophagocytic lymphohistiocytosis (HLH) (<2 years), late-onset HLH (>2 years), or no HLH diagnosis, as indicated. Patient cell numbers were compared to those of healthy relatives and transport controls, as indicated. Columns depict mean values, bars indicate SD. Non-parametric one-way ANOVA Kruskal–Wallis tests are reported as exact p values.
To examine whether CHS patient cytotoxic lymphocytes contained sufficient cytotoxic cargo proteins to induce target cell killing, we labeled freshly isolated PBMC with fluorochrome-conjugated antibodies to surface lineage and differentiation markers as well as to intracellular cytotoxic granule proteins and analyzed cells by flow cytometry (Figure 2A). The median fluorescence intensity of CD107a (lysosomal-associated membrane protein 1), perforin, granzyme A, and granzyme B was measured in CD3+CD8+CD57+ CTL (Figures 2B–E) and CD3−CD56dim NK cell (Figures 2F–I) subsets in 18 and 20 patients, respectively. Values from patients, as well as familial and transport controls were normalized against local controls. Intracellular CD107a expression was slightly reduced, whereas perforin expression in CTL and NK cells varied considerably among patients. Granzyme A and B expression was elevated in patient CTL and NK cells. No correlation between expression of cytotoxic granule constituents and HLH diagnosis or age was observed (Figure 2). Thus, consistent with published analyses on 14-day cultured CHS CTL clones that indicated normal perforin and granzyme expression and processing (32), freshly isolated CHS patient cytotoxic lymphocytes expressed normal levels of cytotoxic granule constituents. If anything, levels of perforin and granzymes A and B were elevated in CHS patient lymphocytes, as observed in other familial HLH patients with mutations in genes required for lymphocyte cytotoxic granule exocytosis (23, 33). Of note, granzymes play pleiotropic roles in immune responses, not restricted to induction of target cell death (34).
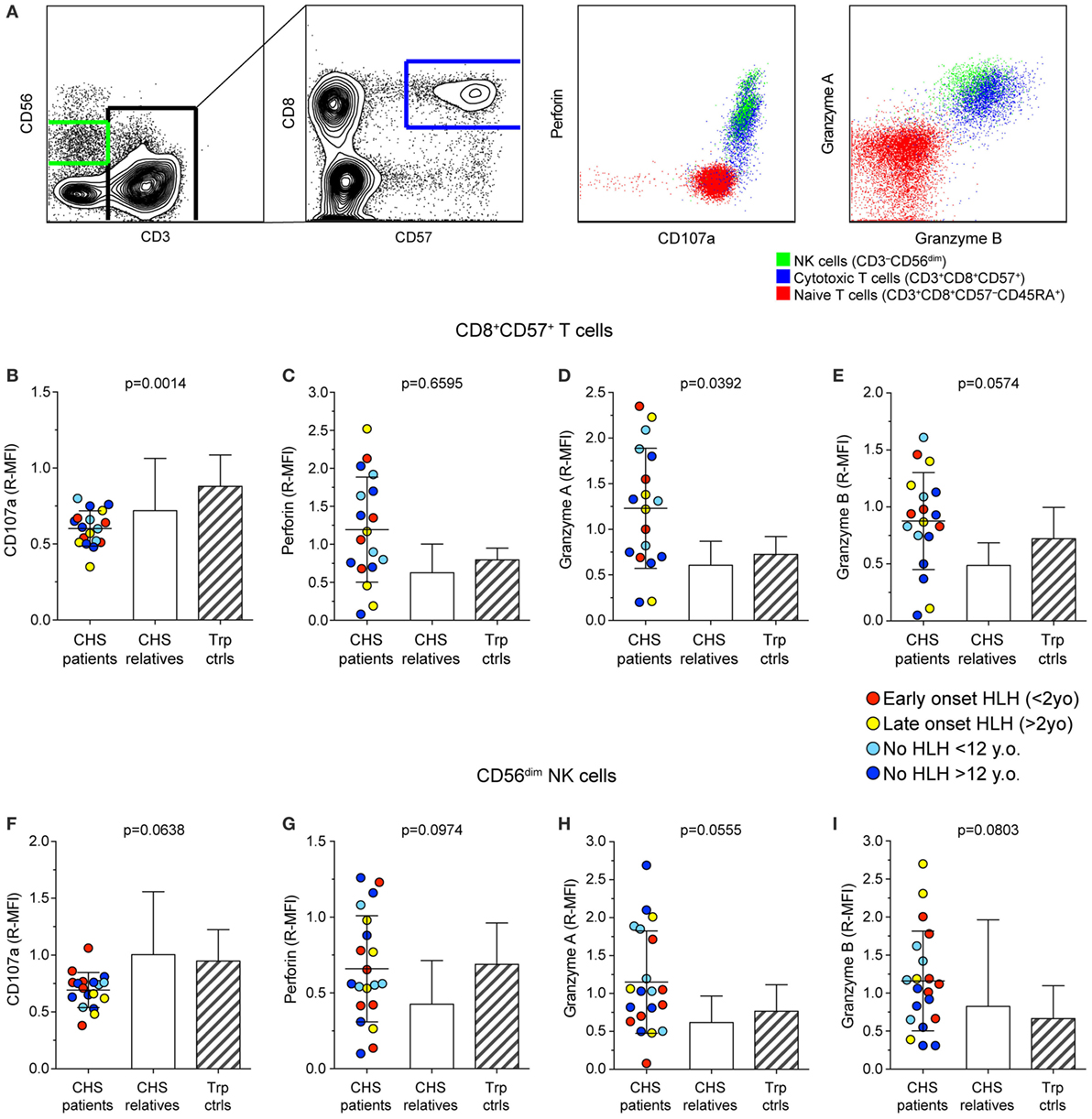
Figure 2. Expression of granule constituent proteins in cytotoxic lymphocyte subsets of Chediak–Higashi syndrome (CHS) patients. Gating strategy and representative dot plots of various lysosomal markers are shown in (A). Peripheral blood mononuclear cells were labeled for intracellular granule constituent proteins as well as surface proteins to distinguish [(B–E); n = 18 patients] CD3+CD8+CD57+ cytotoxic T cell and [(F–I); n = 20 patients] CD3−CD56dim NK cells, as indicated. Plots depict relative median fluorescent intensities of (B,F) CD107a, (C,G) perforin, (D,H) granzyme A, and (E,I) granzyme B in CHS patients, healthy relatives and transport controls, as indicated. The patients are color-coded according to whether they presented with early-onset HLH (<2 years), late-onset HLH (>2 years), or no HLH diagnosis, as indicated. Columns depict mean values, bars indicate SD. Non-parametric one-way ANOVA Kruskal–Wallis tests are reported as exact p values.
Lysosomes from all cell types are enlarged in CHS patients and mouse models carrying Lyst mutations (35). We assessed lysosomal size and number in freshly isolated NK cells and CTL from our cohort of CHS patients by immunofluorescence staining of perforin and CD107a and analyses by confocal microscopy. The number of perforin (Figures 3A,B) and CD107a (Figure 3C) objects and their diameters (Figures 3D–F) among CTL (defined as CD3+perforin+ cells) or NK cell in patients and healthy controls (both familial and unrelated transport samples combined) were determined, providing estimates of perforin dense core numbers and diameters, as well as diameters of the outer, CD107a-coated cytotoxic granule limiting membrane (36). It should be noted that it has previously been reported that CD57+ NK cells show a particularly striking phenotype, with a single large cytotoxic granule (31), but lysosomal size and numbers have not been systematically compared between CTL and NK cells. In our cohort, the three patients (patients 1, 2, and 3) with the most 5′ LYST nonsense mutations had just one, enlarged perforin-containing vesicle in 81, 98, and 100% of NK cells, respectively. NK cells from 17 patients in total were examined and found to have 1–2 perforin-containing vesicles per cell (1.3 vesicles on average), whereas CTLs from seven patients had 1–4 perforin-containing vesicles per cell (3.6 vesicles on average). As such, relative to NK cells, T cells from CHS patients had significantly more cytotoxic granules (Figure 3A). In contrast, both NK cells and CTL from healthy controls averaged approximately seven perforin-containing vesicles per cell (Figure 3A). By comparison, CHS patients showed more numerous CD107a-delimited vesicles in NK cells compared to CTL (Figure 3C). Reflecting enlarged cytotoxic granules, in the three patients with the earliest LYST nonsense mutations, the mean NK cell perforin object diameter was 1.1 μm (0.81 ± 0.20, mean ± SD, for all 17 patients examined), with a CD107a object diameter of up to 1.3 μm (0.82 ± 0.23 μm), significantly larger than perforin (0.33 ± 0.04 μm) and CD107a (0.49 ± 0.13 μm) object diameters in normal NK cells (Figures 3D–F). CTL from the same patients contained more than one perforin object of a smaller size (0.50 ± 0.15 μm) relative to CD107a objects (0.54 ± 0.12 μm), albeit significantly larger than control CTL perforin (0.31 ± 0.03 μm) and CD107a (0.42 ± 0.72 μm) object diameters. The larger diameter of CD107a relative to perforin objects likely reflects staining of perforin in dense cores, whereas CD107a is confined to outer, delimiting membranes (36). In several patients, lysosomal morphology was also measured following 20 min of stimulation with target cells or PMA and ionomycin without any discernable difference observed in size or number of cytotoxic granules per cell (data not shown). Altogether, in spite of overall perforin expression being higher in NK cells relative to CTL (22), CHS patient NK cells most often had one giant perforin-containing cytotoxic granule as opposed to smaller and more numerous cytotoxic granules in CTL.
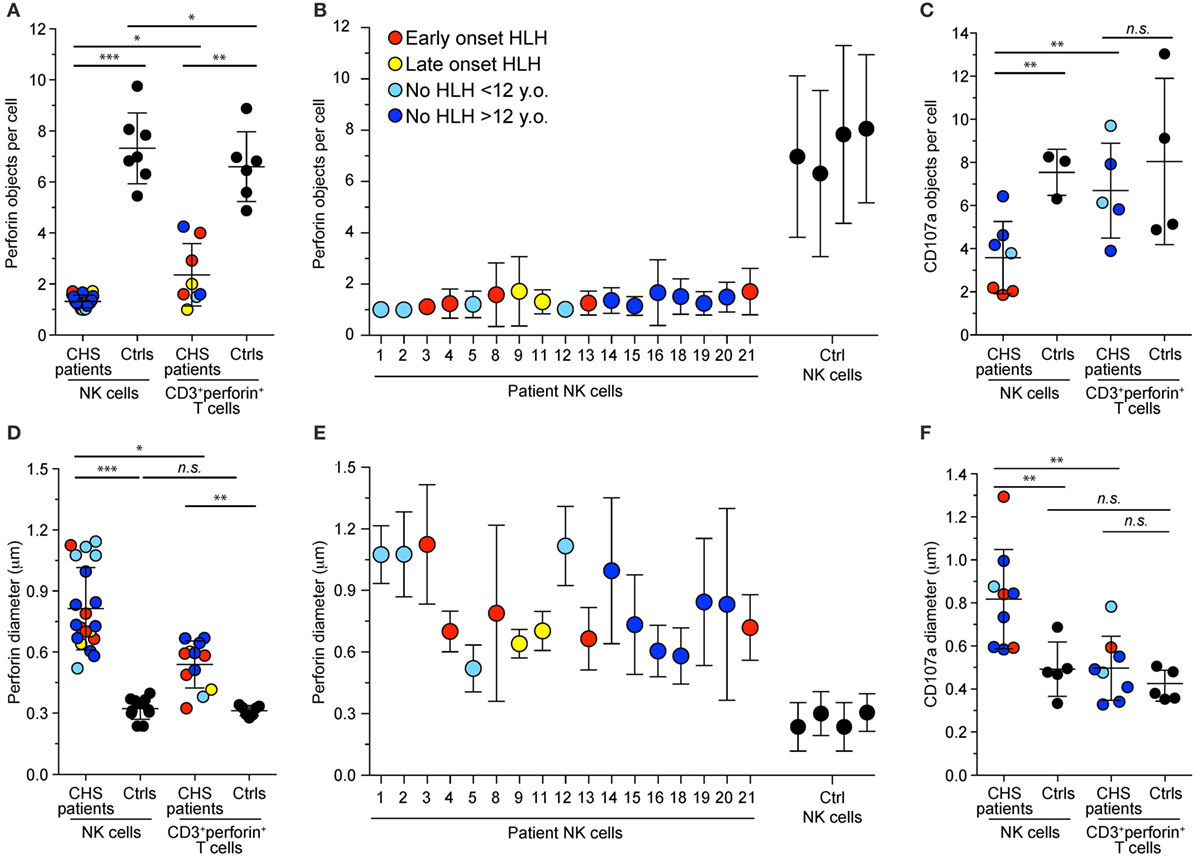
Figure 3. Cytotoxic granule numbers and size in NK and T cells of Chediak–Higashi syndrome (CHS) patients. Freshly isolated, blood-derived NK cells and CD3+perforin+ T cells were labeled intracellularly with antibodies to perforin and CD107a. Plots depict the (A–C) number and (D–E) diameter of (A,B,D,E) perforin or (C,F) CD107a granules per NK cell or cytotoxic T lymphocyte from individual CHS patients and healthy controls. Values represent the average of multiple cells in each individual with mean and SD indicated for (A,C,D,F) cumulated individuals or for (B,E) each patient and control individually. Symbols represent mean values and bars SD. With respect to perforin stainings, an average of 69 (range 4–510) CHS patient and 176 (range 35–536) control NK cells and 7 (range 2–14) CHS patient and 144 (range 35–309) control T cells and ere quantified. The patients are color-coded according to whether they presented with early-onset hemophagocytic lymphohistiocytosis (HLH) (<2 years), late-onset HLH (>2 years), or no HLH diagnosis, as indicated. Non-parametric Mann–Whitney U tests were performed.
For release of cytotoxic granule content and target cell killing, perforin-containing granules traverse the actin cytoskeleton (37, 38). Smaller cytotoxic granules in CTL might, therefore, enable more efficient traversation and exocytosis. Based on this notion and previous findings of more granule exocytosis by in vitro stimulated CTL as compared to freshly isolated NK cells (20), we thus assessed whether freshly isolated or stimulated CTL from CHS patients might display more cytotoxic granule exocytosis than NK cell counterparts. Lysis of K562 target cells by freshly isolated PBMC was defective compared with controls and below the limit set as pathologic for a diagnosis of HLH in all except patients 6 and 15 (Figure 4A) (39). Culturing healthy control PBMC in IL-2 for 36–48 h increased NK cytotoxicity against K562 cells. CHS patient NK cell cytotoxicity also improved with IL-2, as previously shown (40), but not to the level seen in familial controls (Figure 4B), or patients with STX11 mutations (14, 23).
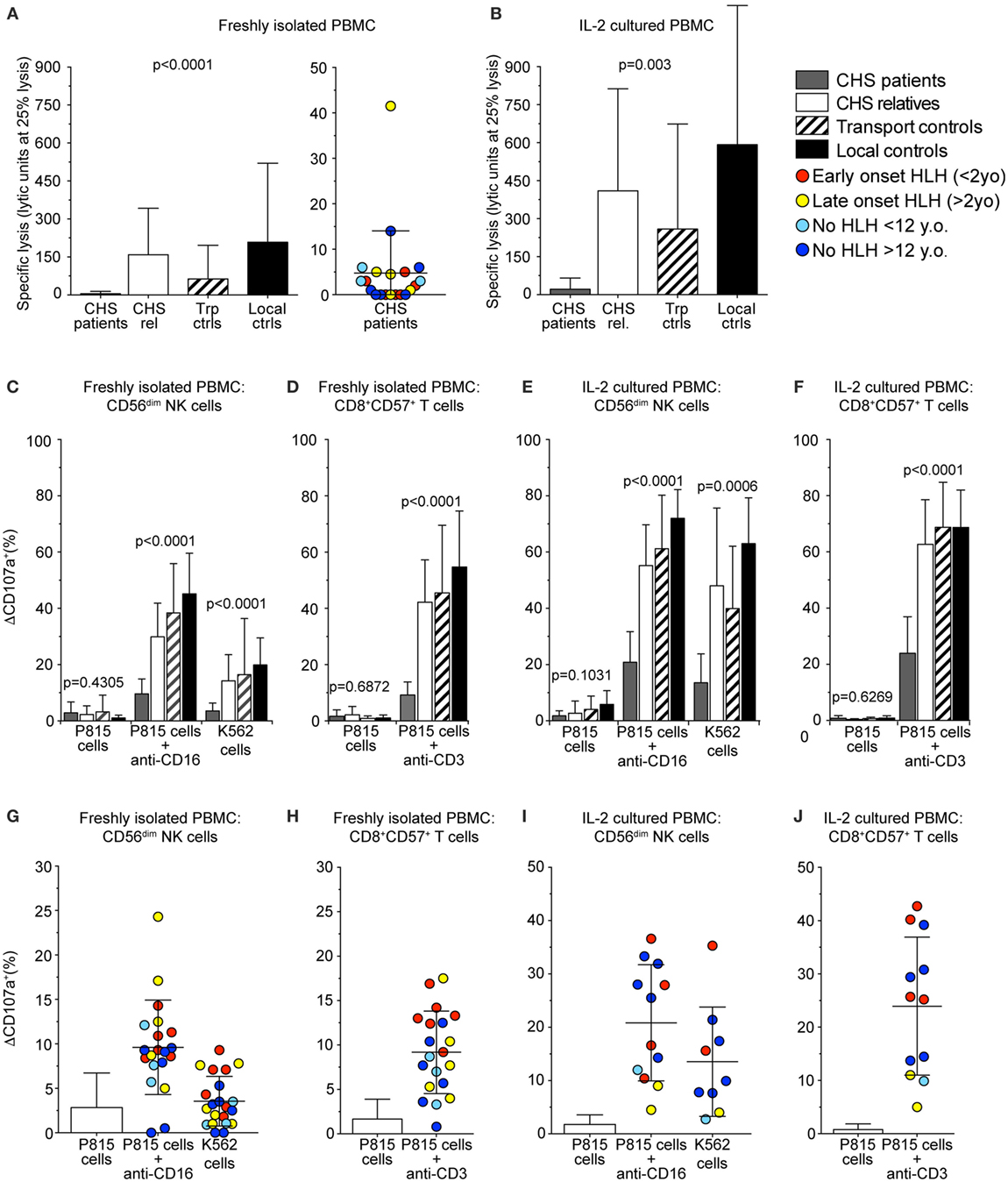
Figure 4. Chediak–Higashi syndrome (CHS) patient NK and T cells have defective cytotoxicity and exocytosis. Freshly isolated peripheral blood mononuclear cells (PBMCs) or PBMCs stimulated with interleukin (IL)-2 for 36–48 h from healthy family members, transport controls, and local Swedish controls were evaluated for cytotoxicity and exocytosis toward target cells. (A,B) Plots depict NK cell-mediated K562 target cell lysis by (A) freshly isolated or (B) IL-2 stimulated PBMC. (C–J) CD107a surface expression was assessed by flow cytometry after stimulation of PBMC with P815 cells alone, P815 cells supplemented with anti-CD16 or anti-CD3 antibody, or K562 cells, as indicated. Plots depict induced CD107a surface expression as quantified on gated (C,E,G,I) CD56dim NK cells or (D,F,H,J) CD8+CD57+ CTL, (C,D,G,H) freshly isolated, or (E,F,I,J) stimulated with IL-2. (A–F) Columns represent mean values and bars SD. (G–J) Symbols represent mean values and bars SD. The patients are color-coded according to whether they presented with early-onset hemophagocytic lymphohistiocytosis (HLH) (<2 years), late-onset HLH (>2 years), or no HLH diagnosis, as indicated. Non-parametric one-way ANOVA Kruskal–Wallis tests are reported as exact p values.
Cytotoxic lymphocyte exocytosis was assessed in PBMC co-incubated with either K562 or P815 cells added anti-CD16 antibody to trigger NK cells, or P815 cells added anti-CD3 to trigger CTL (Figures 4C–J). Exocytosis induced by K562 cells was defective (ΔCD107a+ < 5%) in CD3−CD56+ NK cells (ΔCD107a+ 3.5 ± 2.8%; mean ± SD) in 15 of 20 CHS patients (Figures 4C,G). Exocytosis induced by P815 cells added anti-CD16 was reduced in CD3−CD56+ NK cells from CHS patients (ΔCD107a+ 9.6 ± 5.3%), relative to healthy controls (ΔCD107a+ 45.2 ± 14.4%), though higher than against K562 cells (Figures 4C,G). Exocytosis induced by P815 cells added anti-CD3 was defective in CD3+CD8+CD57+ CTL (ΔCD107a+ 9.2 ± 4.6%), relative to healthy controls (ΔCD107a+ 54.7 ± 19.9%, Figures 4D,H). No correlation was observed between age of onset of HLH and the degree of impairment of fresh or short-term stimulated NK cell or CTL degranulation (not shown). Neither did the severity of the mutation correlate with exocytosis in freshly isolated nor IL-2 activated NK or T cells (Figures 4E,F,I,J), as was previously observed (20). Thus, contrary to our expectations, our assays did not uncover evidence supporting the notion of differential impairment of cytotoxic granule exocytosis in CTL versus NK cells from CHS patients.
In order to assess cytotoxic IS formation and granule polarization, freshly isolated NK cells from two CHS patients were mixed with K562 targets and allowed to form conjugates, which were then analyzed by microscopy. Healthy control NK cells conjugated to K562 target cells polarized perforin granules and accumulated actin and WIP, a key mediator of cytotoxic granule polarization and cellular cytotoxicity (41), at the mature IS (Figure 5A). NK cells from CHS patients were able to form conjugates with K562 targets to a similar extent as healthy controls and showed accumulated actin and WIP at the synapse, demonstrating that a mature IS was formed (Figures 5B–D). Strikingly, CHS NK cells failed to polarize their perforin granules to the mature IS (Figures 5E–G). The distance between the perforin granule and the IS in CHS NK cells was markedly increased compared with healthy donors (Figure 5E; 3.7 ± 0.42 μm, mean ± SD, versus 2.4 ± 0.86 μm, respectively). Similarly, the average distance of the MTOC to the synapse also was increased in LYST-deficient NK cells relative to controls (Figure 5F; 3.9 ± 0.38 μm in CHS patients versus 1.7 ± 0.69 μm in controls). Furthermore, the distance of the granules to the MTOC was also increased in LYST-deficient NK cells relative to controls (Figure 5G; 2.9 ± 0.15 versus 1.6 ± 0.97 μm, respectively). This indicates that in freshly isolated, LYST-deficient NK cells, cytotoxic granule coalescence and MTOC trafficking, early events in cytotoxic granule secretion, are severely impaired. Together, these results substantiate observations in IL-2 cultured NK cells and T cells from CHS patients, where normal IS formation yet impaired cytotoxic granule trafficking has been reported (25, 42).
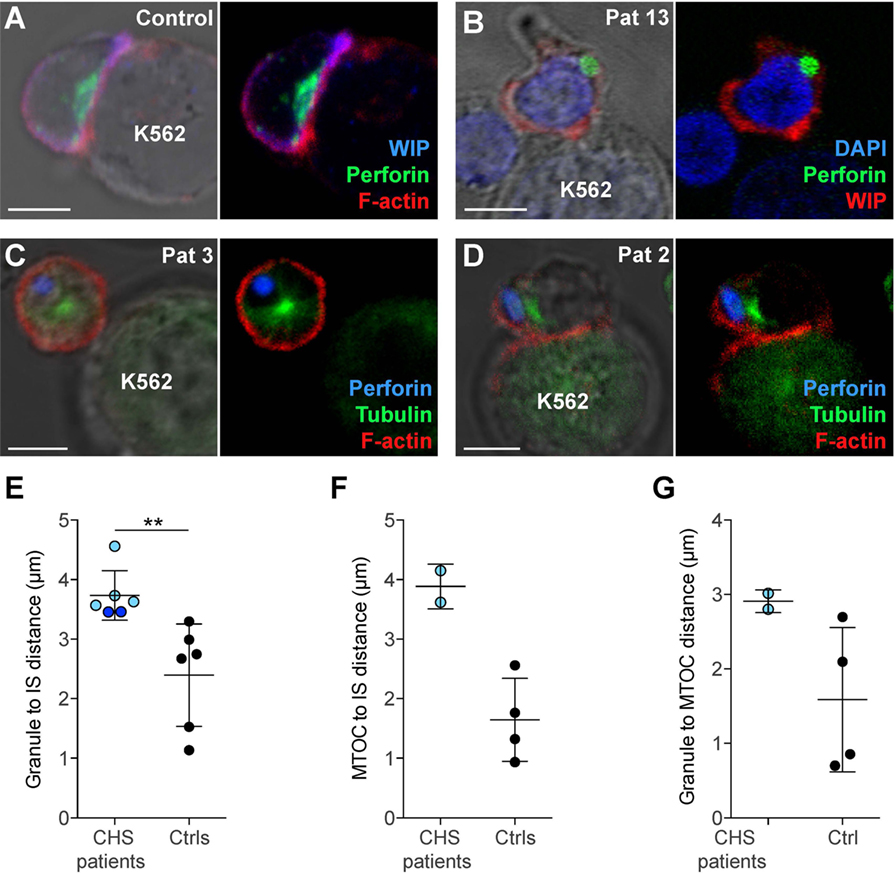
Figure 5. Chediak–Higashi syndrome (CHS) patient NK cells form conjugates with target cells, but granules and microtubule-organizing center (MTOC) do not efficiently polarize to the immune synapse (IS). (A) A representative NK cell from a healthy donor conjugated to a K562 target cell, labeled with antibodies to WASP-interacting protein (WIP) and perforin as well as phalloidin to detect F-actin, as indicated. (B) A representative NK cell from a CHS patient conjugated to a K562 cell, labeled with antibodies to WIP and perforin, in addition to DAPI to discern the nucleus, as indicated. (C–D) NK cells from CHS patients conjugated to K562 cells, labeled with antibodies to perforin and tubulin as well as phalloidin to detect F-actin, as indicated. (A–D) Scale bars indicate 5 μm. Distances were measured between (E) MTOC and IS, (F) granules and IS (center of the contact between NK cell and target cell), or (G) granule and MTOC. (E–G) Each symbol represents average values from multiple cells from individual patients and controls, with overall mean and SD indicated. (E) 45 (range 9–150) CHS patient and 266 (range 54–488) control NK cells and (F,G) 17 (range 16–18) CHS patient and 269 (range 201–325) control NK cells were quantified.
In order to further characterize the enlarged granules in CHS patient cells, freshly isolated NK cells were labeled for proteins to demarcate cytotoxic granules, late endosomes and early endosomes (Figure 6). Perforin and/or CD107a were used to label the giant lysosomal compartment. A hole was sometimes observed in the center of the perforin positive organelle in CHS NK cells, as previously noted in CTL (Figure 6D) (42, 43). The late endosomal/lysosomal enzyme cathepsin D localized to the lumen of the enlarged cytotoxic granules in CHS patient NK cells (Figure 6A) and has previously been observed to be processed and localized correctly in CHS cultured CTLs, as expected from the largely normal lysosomal function in most cell types of CHS patients (32, 43). However, NK cell cytotoxic granules from CHS patients were negative for mannose-6-phosphate receptor (M6PR, Figure 6A), which transfers some lysosomal enzymes, including granzymes and serglycin, from the trans-Golgi network to late endosomes (44, 45). M6PR partially colocalized with perforin granules in healthy freshly isolated NK cells. Notably, the cytotoxic granule exocytosis regulators Rab27a and Munc13-4 were found to decorate the cytotoxic granule limiting membrane in freshly isolated, unstimulated CHS NK cells (Figures 6B,C), contrasting healthy donor cells where Rab27a and Munc13-4 co-localization is specifically induced by signals from activating receptors (46, 47). Moreover, intriguingly, freshly isolated CHS NK cells and CD3+perforin+ cells showed EEA1 labeling of the enlarged lytic granules in all 13 patients examined (Figures 6E–H). Taken together, the present results extend earlier findings of mislocalized early and late endosomal markers on CHS lysosomes in cultured NK and T cells to freshly isolated cytotoxic lymphocytes.
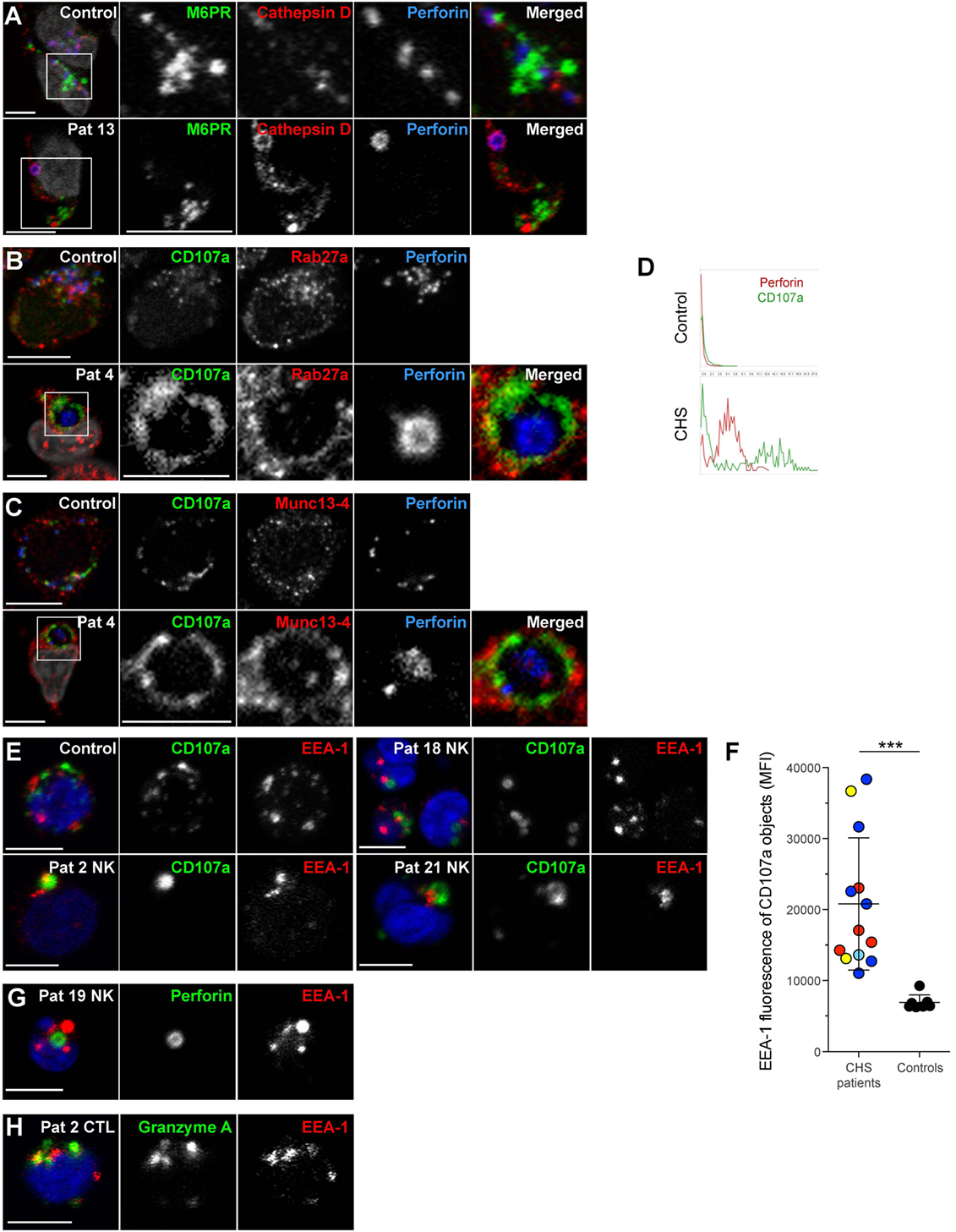
Figure 6. Chediak–Higashi syndrome (CHS) NK cell granules are positive for Munc13-4, Rab27a, CD107a, cathepsin D, and EEA1, but not mannose-6-phosphate receptor. Freshly isolated NK cells were labeled for perforin, DAPI and (A) M6PR and cathepsin D, (B) CD107a and Rab27a, or (C) CD107a and Munc13-4. (D) Traces show perforin and CD107a staining along a transect of a representative cytotoxic granule in a NK cell from (top panel) healthy control individual as well as (bottom panel) CHS patient, respectively. (E) Freshly isolated NK cells from a healthy control and three CHS patients with various LYST mutation positions were labeled for CD107a, EEA1, and DAPI, as indicated. (F) EEA1 median fluorescence intensity (MFI) within CD107a-positive objects was quantified in healthy control and CHS patient NK cells. On average 90 (range 8–187) CHS patient and 85 (range 42–135) control NK cells were quantified. (G) Freshly isolated NK cells from a CHS patient labeled for perforin, EEA1, and DAPI, as indicated. (H) Freshly isolated T cells from a CHS patient labeled for granzyme A, EEA1, and DAPI, as indicated. In micrographs, scale bars indicate 5 μm.
Here, we studied a cohort of 21 CHS patients, evaluating cytotoxic T cell and NK cell lysosome morphology, exocytosis and clinical presentation while questioning whether any of these parameters correlated with each other or with specific LYST mutations. In addition, we studied vesicular markers with respect to cytotoxic granule identity, revealing novel aberrances in protein localization and sorting.
A key clinical challenge with CHS is to predict the likelihood of developing an accelerated phase of disease culminating in HLH. In our cohort, six adults with some degree of oculocutaneous albinism and neurological sequelae but no HLH episodes have at least one LYST allele of >3,000 amino acids which includes the ARM/HEAT, ConA-like and PH-like domains. The relatively mild disease in these adult patients thus suggests some residual LYST function in immune cells. In the absence of an antibody to LYST protein, it is unclear whether truncating mutations throughout the LYST gene result in partially functional LYST protein fragments, or whether such mutant proteins are degraded. In our assays, these patients displayed equally disrupted cytotoxic granule morphology and exocytosis as compared to patients that suffered an accelerated phase. Moreover, we did not find any correlations between mutation position of truncation mutants and cytotoxic granule morphology or lysosomal exocytosis. Notably, two early-onset HLH patients carried mutations toward the C-terminus of LYST, one in homozygous form (patient 14) and another in a compound heterozygous form (a point mutation and one truncation in the BEACH domain, patient 21). Our cohort also included two siblings discordant for presentation of HLH. Thus, patients with identical mutations may present with highly divergent clinical pictures (26), and the location of mutations in LYST cannot reliably predict development of HLH.
Jessen and colleagues previously described a stronger impairment of antigen-specific T cell exocytosis in souris relative to beige mice, which have an exon 27 splice site versus an exon 54, C-terminal single amino acid deletion in Lyst, respectively (20). While NK cells displayed uniformly defective exocytosis following engagement of YAC-1 cells, the degree of impaired T cell exocytosis and cytotoxicity corresponded to development of HLH in souris, but not beige mice. Congruently, in a cohort of 12 CHS patients, Jessen and colleagues found that late or no onset of HLH in CHS patients correlated with less impaired cytotoxicity of day 7 phytohemagglutinin (PHA)/IL-2 blast T cells, whereas fresh NK cell exocytosis was abnormal in all patients and, therefore, not predictive (20). This could indicate a differential role of LYST in the two cytotoxic lymphocyte populations. We, therefore, carefully compared the morphology and function of NK cells and CD8+CD57+ T cells, corresponding to bona fide antigen-experienced, cytotoxic T cells, in our cohort of CHS patients in order to determine any morphological or functional correlations. The results revealed that lymphocyte counts in patients tended to be elevated. Cytotoxic granule protein contents were comparable or elevated, though intracellular CD107a levels were reduced as compared to healthy controls. Remarkably, exocytosis was equally impaired among CTL and NK cells, from both HLH and non-HLH CHS patients, irrespective of cells being unstimulated or stimulated for 48 h with IL-2. In our analyses, the patients with the highest levels of exocytosis and recovery of function with IL-2 stimulation were those that had developed HLH. Contrasting the human T cell data from Jessen et al. (20), examining bulk PHA/IL-2 activated CD8+ T cells, our degranulation assays of fresh or short-term IL-2 stimulated CTL cells thus did not predict the severity of clinical outcome from CTL exocytosis. It is possible that differences in the stimulation protocol and the experimental read-out can explain these discrepancies. Our assays were robust, and revealed a significant increase in exocytosis upon strong receptor or cytokine-mediated activation of both CTL and NK cells.
Of note, patient 7 had over 10 LU NK cell cytotoxicity, which corresponded to the highest NK cell and CD8+CD57+ T cell exocytosis among the CHS patients (and within the normal range for healthy pediatric patients), although her sister (patient 8) displayed lower NK cell and CTL exocytosis as well as NK cell-mediated cytotoxicity. Patient 7 was sampled during an HLH flare, which can result in transiently elevated lymphocyte activity in FHL patients due to hypercytokinemia (48). Unfortunately, this patient succumbed to an accelerated phase of HLH at the age of 3 years. This observation serves as an additional note of caution with respect to interpreting increased cytotoxic lymphocyte exocytosis as a possible predictor of milder CHS.
The process of cytotoxic granule exocytosis requires formation of an IS, instigating DAG-gradient directed MTOC recruitment, trafficking of perforin-containing granules along microtubules to the MTOC, polarization of MTOC to the IS, granule migration along microtubules, and through an actin meshwork to finally fuse with the plasma membrane (49). CHS lymphocytes and other cell types contain normal numbers and lengths of assembled microtubules (50). Transport of CHS lysosomes along microtubules in vitro is possible, indicating correct localization of microtubule-interacting proteins; however, trafficking of such enlarged lysosomes in the cytoplasm may be spatially constrained (12, 35). Substantiating findings in CTL clones and IL-2 cultured NK cells (25, 42), we found that MTOC recruitment to the IS was impaired in freshly isolated CHS NK cells. Defective exocytosis, therefore, is apparently not solely due to enlarged granule size, as patients with LYST mutations that result in smaller granules did not demonstrate proportionately improved transport to the synapse or exocytosis. Moreover, we found that CHS patient CTL cytotoxic granules were on average less than half the diameter of NK cell granules, yet the exocytosis defect was equally severe in both cell types. These findings may suggest differences in regulation of lysosomal fusion and fission in NK cells versus CTL. Notably, melanosome secretion is also defective in CHS patients, a process that is not a polarized secretion event and requires granule movement only away from the MTOC.
Recently, defective granule exocytosis by CHS cytotoxic lymphocytes has been attributed to disrupted vesicular identity (25, 42). Unlike healthy NK cell cytotoxic granules, enlarged CHS granules did not contain M6PR. M6PR localized to secretory lysosomes in CD8+ CTL clones during early stages of culture, while granule biogenesis is underway, but not after 7 days of culture (32). In CHS B cell lines, M6PR is mis-sorted to enlarged lysosomes rather than the trans-Golgi network and late endosomes (13). Our data indicate that circulating CHS NK cells contain mature granules that no longer require delivery of nascent protein from the Golgi complex. Cytotoxic granule exocytosis is mediated by machinery including Munc13-4 and Rab27a, proteins recruited to cytotoxic granules only upon activation signals in both CTL and NK cells (46, 47). Substantiating findings in CTL clones and IL-2 cultured NK cells (25, 42), we found that enlarged CHS granules co-localized with Munc13-4 and Rab27a in freshly isolated NK cells. It is not clear if mistargeting of these facilitators of granule exocytosis can result in defective exocytosis. Overexpression of Munc13-4, Rab27a, and Slp3 could rescue the secretion defect in CHS CTL (42). Intriguingly, freshly isolated CHS NK cells and CD3+ cells showed that EEA1, an early endosome marker that regulates vesicle trafficking and fusion (51), overlapped with the enlarged cytotoxic granules in all 13 patients examined. Our observations contrast those of Sepulveda and colleagues (42), who did not find EEA1 (early endosome marker) colocalization with cytotoxic granules in CHS CTL clones, though Rab7 (late endosome marker) and Rab11 (recycling endosome marker) both mislocalized to granules. This discrepancy may reflect a difference between unstimulated lymphocytes freshly isolated from blood and CTL clones cultured in cytokines over several weeks. In non-immune cell lines, siRNA to LYST induces enlarged lysosomes but did not affect the size of early endosomes or result in localization of early endosome proteins to enlarged lysosomes (52). The presence of LYST during organelle biogenesis prior to the siRNA treatment may preserve early endosome morphology, or there may be a cell type-specific defect in immune cells or cells with specialized secretory lysosomes.
Both an excess of fusion and absence of fission have been hypothesized as the cause of the enlarged CHS lysosomes. Our data indicate that not only is there an excess of fusion between compartments that would normally fuse in the course of lysosome formation (53), but mistargeted fusion of early endosomes with these later compartments occurs, and this is not corrected by fission of membranes containing identifier of early endosomes. Interestingly, EEA1 did not coat the entire cytotoxic granule, but was confined to one portion of the granule, indicating a partial loss of organelle identity, possibly as proteins cluster but fail to fission off. As CHS NK cells had on average one enlarged cytotoxic granule relative to three enlarged cytotoxic granules in CTL, there may be differences in the regulation of cytotoxic granule biogenesis and fusion in different cytotoxic lymphocyte subsets. The molecular basis for such differences warrants further investigations.
In conclusion, we detail cytotoxic granule morphology and vesicular identity in cytotoxic lymphocyte subsets freshly isolated from peripheral blood. Our analyses uncover several aberrances with respect to CHS cytotoxic granule identity, as well as intriguing morphological differences between CTL and NK cells. Moreover, our results demonstrate similar impairments in CHS CTL and NK cell exocytosis, despite a more severe defect in granule morphology in NK cells. In terms of predicting susceptibility to developing HLH, it thus appears that differences between CTL and NK cell exocytosis may not be predictive. It is, however, possible that differences in exocytosis by activated cytotoxic lymphocyte may provide some guidance (26). Moreover, less severe missense mutations generally also appear associated with a lower susceptibility to develop HLH.
This study was approved by The Regional Ethics Review Board in Stockholm (approval number 2006/228-31/3, 2006/229-31/3, 2006/230-31/3, 2008/1689-32). Peripheral blood from patients with suspected CHS was obtained with their/their parents’ written informed consent in accordance with the Declaration of Helsinki.
SC performed functional assays and wrote the manuscript; SMW performed microscopy and wrote the manuscript; BT performed genetic analyses; HA, WA-H, SA, FB, UC, ZK, KL, TP, HT, AU, and WI provided samples, clinical data, and cared for the patients; J-IH, MN, H-GL, MM, SE, and KK provided supervision and critically reviewed the manuscript; YB designed the study, interpreted results, and wrote the manuscript. All authors approve the final manuscript version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement no. 311335, Swedish Research Council, Norwegian Research Council, Swedish Foundation for Strategic Research, Wallenberg Foundation, Swedish Cancer Foundation, Swedish Childhood Cancer Foundation, as well as the Stockholm County Council and Karolinska Institutet Center for Innovative Medicine (to YB); by the Swedish Research Council, Swedish Cancer Foundation, Swedish Childhood Cancer Foundation, and the Stockholm County Council (to J-IH); and by the BMBF (BMBF 01 EO 0803) and the DFG (SFB1160, TP1) (to SE).
1. Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature (1996) 382:262–5. doi: 10.1038/382262a0
2. Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet (1996) 14:307–11. doi:10.1038/ng1196-307
3. Perou CM, Moore KJ, Nagle DL, Misumi DJ, Woolf EA, McGrail SH, et al. Identification of the murine beige gene by YAC complementation and positional cloning. Nat Genet (1996) 13:303–8. doi:10.1038/ng0796-303
4. Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab (1999) 68:283–303. doi:10.1006/mgme.1999.2927
5. Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet (2002) 108:16–22. doi:10.1002/ajmg.10184.abs
6. Dent PB, Fish LA, White LG, Good RA. Chediak-Higashi syndrome. Observations on the nature of the associated malignancy. Lab Invest (1966) 15:1634–42.
7. Roder JC, Haliotis T, Klein M, Korec S, Jett JR, Ortaldo J, et al. A new immunodeficiency disorder in humans involving NK cells. Nature (1980) 284:553–5. doi:10.1038/284553a0
8. Tardieu M, Lacroix C, Neven B, Bordigoni P, de Saint Basile G, Blanche S, et al. Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome. Blood (2005) 106:40–2. doi:10.1182/blood-2005-01-0319
9. Falkenstein K, De Lozanne A. Dictyostelium LvsB has a regulatory role in endosomal vesicle fusion. J Cell Sci (2014) 127:4356–67. doi:10.1242/jcs.138123
10. Shiflett SL, Vaughn MB, Huynh D, Kaplan J, Ward DM. Bph1p, the Saccharomyces cerevisiae homologue of CHS1/beige, functions in cell wall formation and protein sorting. Traffic (2004) 5:700–10. doi:10.1111/j.1600-0854.2004.00213.x
11. Barrett A, Hermann GJ. A caenorhabditis elegans homologue of LYST functions in endosome and lysosome-related organelle biogenesis. Traffic (2016) 17:515–35. doi:10.1111/tra.12381
12. Perou CM, Leslie JD, Green W, Li L, Ward DM, Kaplan J. The Beige/Chediak-Higashi syndrome gene encodes a widely expressed cytosolic protein. J Biol Chem (1997) 272:29790–4. doi:10.1074/jbc.272.47.29790
13. Faigle W, Raposo G, Tenza D, Pinet V, Vogt AB, Kropshofer H, et al. Deficient peptide loading and MHC class II endosomal sorting in a human genetic immunodeficiency disease: the Chediak-Higashi syndrome. J Cell Biol (1998) 141:1121–34. doi:10.1083/jcb.141.5.1121
14. Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood (2012) 119:2754–63. doi:10.1182/blood-2011-08-374199
15. de Saint Basile G, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol (2010) 10:568–79. doi:10.1038/nri2803
16. Meeths M, Chiang SC, Löfstedt A, Müller ML, Tesi B, Henter JI, et al. Pathophysiology and spectrum of diseases caused by defects in lymphocyte cytotoxicity. Exp Cell Res (2014) 325:10–7. doi:10.1016/j.yexcr.2014.03.014
17. Chia J, Yeo KP, Whisstock JC, Dunstone MA, Trapani JA, Voskoboinik I. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci U S A (2009) 106:9809–14. doi:10.1073/pnas.0903815106
18. Tesi B, Chiang SC, El-Ghoneimy D, Hussein AA, Langenskiöld C, Wali R, et al. Spectrum of atypical clinical presentations in patients with biallelic PRF1 missense mutations. Pediatr Blood Cancer (2015) 62:2094–100. doi:10.1002/pbc.25646
19. Löfstedt A, Chiang SC, Onelöv E, Bryceson YT, Meeths M, Henter JI. Cancer risk in relatives of patients with a primary disorder of lymphocyte cytotoxicity: a retrospective cohort study. Lancet Haematol (2015) 2:e536–42. doi:10.1016/S2352-3026(15)00223-9
20. Jessen B, Maul-Pavicic A, Ufheil H, Vraetz T, Enders A, Lehmberg K, et al. Subtle differences in CTL cytotoxicity determine susceptibility to hemophagocytic lymphohistiocytosis in mice and humans with Chediak-Higashi syndrome. Blood (2011) 118:4620–9. doi:10.1182/blood-2011-05-356113
21. Tesi B, Lagerstedt-Robinson K, Chiang SC, Ben Bdira E, Abboud M, Belen B, et al. Targeted high-throughput sequencing for genetic diagnostics of hemophagocytic lymphohistiocytosis. Genome Med (2015) 7:130. doi:10.1186/s13073-015-0244-1
22. Chiang SC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood (2013) 121:1345–56. doi:10.1182/blood-2012-07-442558
23. Bryceson YT, Rudd E, Zheng C, Edner J, Ma D, Wood SM, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood (2007) 110:1906–15. doi:10.1182/blood-2007-02-074468
24. Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med (2005) 202:1001–12. doi:10.1084/jem.20051143
25. Gil-Krzewska A, Wood SM, Murakami Y, Nguyen V, Chiang SC, Cullinane AR, et al. Chediak-Higashi syndrome: lysosomal trafficking regulator domains regulate exocytosis of lytic granules but not cytokine secretion by natural killer cells. J Allergy Clin Immunol (2016) 137:1165–77. doi:10.1016/j.jaci.2015.08.039
26. Certain S, Barrat F, Pastural E, Le Deist F, Goyo-Rivas J, Jabado N, et al. Protein truncation test of LYST reveals heterogenous mutations in patients with Chediak-Higashi syndrome. Blood (2000) 95:979–83.
27. Karim MA, Nagle DL, Kandil HH, Bürger J, Moore KJ, Spritz RA. Mutations in the Chediak-Higashi syndrome gene (CHS1) indicate requirement for the complete 3801 amino acid CHS protein. Hum Mol Genet (1997) 6:1087–9. doi:10.1093/hmg/6.7.1087
28. Weisfeld-Adams JD, Mehta L, Rucker JC, Dembitzer FR, Szporn A, Lublin FD, et al. Atypical Chediak-Higashi syndrome with attenuated phenotype: three adult siblings homozygous for a novel LYST deletion and with neurodegenerative disease. Orphanet J Rare Dis (2013) 8:46. doi:10.1186/1750-1172-8-46
29. Kaya Z, Ehl S, Albayrak M, Maul-Pavicic A, Schwarz K, Kocak U, et al. A novel single point mutation of the LYST gene in two siblings with different phenotypic features of Chediak Higashi syndrome. Pediatr Blood Cancer (2011) 56:1136–9. doi:10.1002/pbc.22878
30. Zarzour W, Kleta R, Frangoul H, Suwannarat P, Jeong A, Kim SY, et al. Two novel CHS1 (LYST) mutations: clinical correlations in an infant with Chediak-Higashi syndrome. Mol Genet Metab (2005) 85:125–32. doi:10.1016/j.ymgme.2005.02.011
31. Abo T, Roder JC, Abo W, Cooper MD, Balch CM. Natural killer (HNK-1+) cells in Chediak-Higashi patients are present in normal numbers but are abnormal in function and morphology. J Clin Invest (1982) 70:193–7. doi:10.1172/JCI110592
32. Baetz K, Isaaz S, Griffiths GM. Loss of cytotoxic T lymphocyte function in Chediak-Higashi syndrome arises from a secretory defect that prevents lytic granule exocytosis. J Immunol (1995) 154:6122–31.
33. Mellor-Heineke S, Villanueva J, Jordan MB, Marsh R, Zhang K, Bleesing JJ, et al. Elevated granzyme B in cytotoxic lymphocytes is a signature of immune activation in hemophagocytic lymphohistiocytosis. Front Immunol (2013) 4:72. doi:10.3389/fimmu.2013.00072
34. Susanto O, Trapani JA, Brasacchio D. Controversies in granzyme biology. Tissue Antigens (2012) 80:477–87. doi:10.1111/tan.12014
35. Stinchcombe JC, Griffiths GM. Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol (2007) 23:495–517. doi:10.1146/annurev.cellbio.23.090506.123521
36. Peters PJ, Borst J, Oorschot V, Fukuda M, Krähenbühl O, Tschopp J, et al. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med (1991) 173:1099–109. doi:10.1084/jem.173.5.1099
37. Rak GD, Mace EM, Banerjee PP, Svitkina T, Orange JS. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol (2011) 9:e1001151. doi:10.1371/journal.pbio.1001151
38. Brown AC, Oddos S, Dobbie IM, Alakoskela JM, Parton RM, Eissmann P, et al. Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy. PLoS Biol (2011) 9:e1001152. doi:10.1371/journal.pbio.1001152
39. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48:124–31. doi:10.1002/pbc.21039
40. Targan SR, Oseas R. The “lazy” NK cells of Chediak-Higashi syndrome. J Immunol (1983) 130:2671–4.
41. Krzewski K, Chen X, Strominger JL. WIP is essential for lytic granule polarization and NK cell cytotoxicity. Proc Natl Acad Sci U S A (2008) 105:2568–73. doi:10.1073/pnas.0711593105
42. Sepulveda FE, Burgess A, Heiligenstein X, Goudin N, Ménager MM, Romao M, et al. LYST controls the biogenesis of the endosomal compartment required for secretory lysosome function. Traffic (2015) 16:191–203. doi:10.1111/tra.12244
43. Stinchcombe JC, Page LJ, Griffiths GM. Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and Chediak Higashi syndrome patients. Traffic (2000) 1:435–44. doi:10.1034/j.1600-0854.2000.010508.x
44. Lemansky P, Fester I, Smolenova E, Uhländer C, Hasilik A. The cation-independent mannose 6-phosphate receptor is involved in lysosomal delivery of serglycin. J Leukoc Biol (2007) 81:1149–58. doi:10.1189/jlb.0806520
45. Griffiths GM. Protein sorting and secretion during CTL killing. Semin Immunol (1997) 9:109–15. doi:10.1006/smim.1997.0059
46. Wood SM, Meeths M, Chiang SC, Bechensteen AG, Boelens JJ, Heilmann C, et al. Different NK cell-activating receptors preferentially recruit Rab27a or Munc13-4 to perforin-containing granules for cytotoxicity. Blood (2009) 114:4117–27. doi:10.1182/blood-2009-06-225359
47. Ménager MM, Ménasché G, Romao M, Knapnougel P, Ho CH, Garfa M, et al. Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13-4. Nat Immunol (2007) 8:257–67. doi:10.1038/ni1431
48. Meeths M, Entesarian M, Al-Herz W, Chiang SC, Wood SM, Al-Ateeqi W, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis (FHL) type 5 patients with mutations in STXBP2. Blood (2010) 116:2635–43. doi:10.1182/blood-2010-05-282541
49. Mace EM, Dongre P, Hsu HT, Sinha P, James AM, Mann SS, et al. Cell biological steps and checkpoints in accessing NK cell cytotoxicity. Immunol Cell Biol (2014) 92:245–55. doi:10.1038/icb.2013.96
50. Pryzwansky KB, Schliwa M, Boxer LA. Microtubule organization of unstimulated and stimulated adherent human neutrophils in Chediak-Higashi syndrome. Blood (1985) 66:1398–403.
51. Schink KO, Tan KW, Stenmark H. Phosphoinositides in control of membrane dynamics. Annu Rev Cell Dev Biol (2016) 32:143–71. doi:10.1146/annurev-cellbio-111315-125349
52. Holland P, Torgersen ML, Sandvig K, Simonsen A. LYST affects lysosome size and quantity, but not trafficking or degradation through autophagy or endocytosis. Traffic (2014) 15:1390–405. doi:10.1111/tra.12227
Keywords: Chediak–Higashi syndrome, NK cells, cytotoxic T cells, exocytosis, lytic granule polarization, lysosomes, endosomes
Citation: Chiang SCC, Wood SM, Tesi B, Akar HH, Al-Herz W, Ammann S, Belen FB, Caliskan U, Kaya Z, Lehmberg K, Patiroglu T, Tokgoz H, Ünüvar A, Introne WJ, Henter J-I, Nordenskjöld M, Ljunggren H-G, Meeths M, Ehl S, Krzewski K and Bryceson YT (2017) Differences in Granule Morphology yet Equally Impaired Exocytosis among Cytotoxic T Cells and NK Cells from Chediak–Higashi Syndrome Patients. Front. Immunol. 8:426. doi: 10.3389/fimmu.2017.00426
Received: 04 January 2017; Accepted: 27 March 2017;
Published: 18 April 2017
Edited by:
Isabelle Meyts, KU Leuven, BelgiumReviewed by:
Emily Mace, Baylor College of Medicine, USACopyright: © 2017 Chiang, Wood, Tesi, Akar, Al-Herz, Ammann, Belen, Caliskan, Kaya, Lehmberg, Patiroglu, Tokgoz, Ünüvar, Introne, Henter, Nordenskjöld, Ljunggren, Meeths, Ehl, Krzewski and Bryceson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yenan T. Bryceson, eWVuYW4uYnJ5Y2Vzb25Aa2kuc2U=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.