
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 11 April 2017
Sec. Molecular Innate Immunity
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00330
This article is part of the Research Topic Immunometabolic Regulations in Adaptive and Innate Immune Cells Shapes and Re-Directs Host Immunity View all 12 articles
 Nikolaos Patsoukis1,2†
Nikolaos Patsoukis1,2† Jessica D. Weaver1,2†
Jessica D. Weaver1,2† Laura Strauss1,2Christoph Herbel1,2
Laura Strauss1,2Christoph Herbel1,2 Pankaj Seth3,4
Pankaj Seth3,4 Vassiliki A. Boussiotis1,2,4*
Vassiliki A. Boussiotis1,2,4*
Host immunity provides wide spectrum protection that serves to eradicate pathogens and cancer cells, while maintaining self-tolerance and immunological homeostasis. Ligation of the T cell receptor (TCR) by antigen activates signaling pathways that coordinately induce aerobic glycolysis, mitochondrial activity, anabolic metabolism, and T effector cell differentiation. Activation of PI3K, Akt, and mTOR triggers the switch to anabolic metabolism by inducing transcription factors such as Myc and HIF1, and the glucose transporter Glut1, which is pivotal for the increase of glucose uptake after T cell activation. Activation of MAPK signaling is required for glucose and glutamine utilization, whereas activation of AMPK is critical for energy balance and metabolic fitness of T effector and memory cells. Coinhibitory receptors target TCR-proximal signaling and generation of second messengers. Imbalanced activation of such signaling pathways leads to diminished rates of aerobic glycolysis and impaired mitochondrial function resulting in defective anabolic metabolism and altered T cell differentiation. The coinhibitory receptors mediate distinct and synergistic effects on the activation of signaling pathways thereby modifying metabolic programs of activated T cells and resulting in altered immune functions. Understanding and therapeutic targeting of metabolic programs impacted by coinhibitory receptors might have significant clinical implications for the treatment of chronic infections, cancer, and autoimmune diseases.
When the T cell receptor (TCR) is engaged, tyrosine phosphorylation of the TCR-associated CD3 chains recruits kinases and scaffold proteins leading to the formation of a supramolecular complex that promotes activation of signaling cascades, generation of second messengers, and initiation of transcriptional events, which lead to T cell differentiation programs (1). These signaling pathways synergistically promote glycolysis and anabolic metabolism to support T cell clonal expansion and effector cell generation (2–4). It is now well understood that metabolic mediators function as intermediates between these signaling events and the functional outcome of T cell activation. Costimulatory receptors, engaged simultaneously with the TCR, have a major impact on signaling events and a decisive role in the differentiation program of T cells. Thus, their role in altering the immunometabolic programming mediated by TCR-mediated activation is unequivocal (5–7).
Our understanding about the functional role of costimulation has evolved from the two-signal model proposed by Lafferty and Cunningham to explain the activation of naïve T cells (8, 9). Although T cell costimulatory pathways were envisioned as stimulators of T cell responses by that model, it is now clear that both stimulatory (costimulatory) and inhibitory (coinhibitory) second signals exist and mediate their impact not only in naïve (TN) but also in effector (TEFF), memory (TM), and regulatory (TREG) T cells (10–12). These receptors are key regulators of T cell activation, tolerance, and exhaustion, and therapeutic modulation of costimulatory and coinhibitory pathways provides effective new treatment strategies in cancer, autoimmunity, infectious diseases, and allogeneic transplantation (13–15).
The first costimulatory receptor CD28 and the first coinhibitory receptor CTLA-4 and their shared ligands CD80 (B7-1) and CD86 (B7-2) constitute the best-characterized pathway, which serves as a paradigm for other costimulatory and coinhibitory pathways. The massive lymphoproliferation and organ infiltration by activated T cells in the CTLA-4-deficient mice (16, 17) was the first indication that coinhibitory receptors have a mandatory role in the regulation of T cell tolerance. Today, many additional costimulatory and coinhibitory receptors have been identified, and their role in the induction and maintenance of T cell tolerance has been established (Figure 1). These pathways fall into two major families: the Ig superfamily, which includes the B7–CD28, TIM, CD226–TIGIT–CD96 families as well as LAG-3, and the TNF–TNF receptor superfamily (18–20). Coinhibitory receptors provide a balance on the activation and expansion of antigen-specific T cells upon encounter with antigen but also regulate T cell tolerance by restraining the initial activation of naïve self-reactive T cells and/or responses of harmful self-reactive T cells. Coinhibitory pathways also regulate the generation and function of thymic-derived TREG and TREG generated at peripheral sites (21, 22). Ligands for various coinhibitory receptors are expressed on antigen-presenting cells (APCs) but also in non-hematopoietic cells (18–20). The expression of coinhibitory ligands on non-hematopoietic cells has a key role for the maintenance of tissue tolerance by suppressing the expansion and function of self-reactive T cells. Importantly, tumors and infectious agents that cause chronic infections exploit these natural tolerance mechanisms to evade surveillance and attack by the immune system (13, 23).
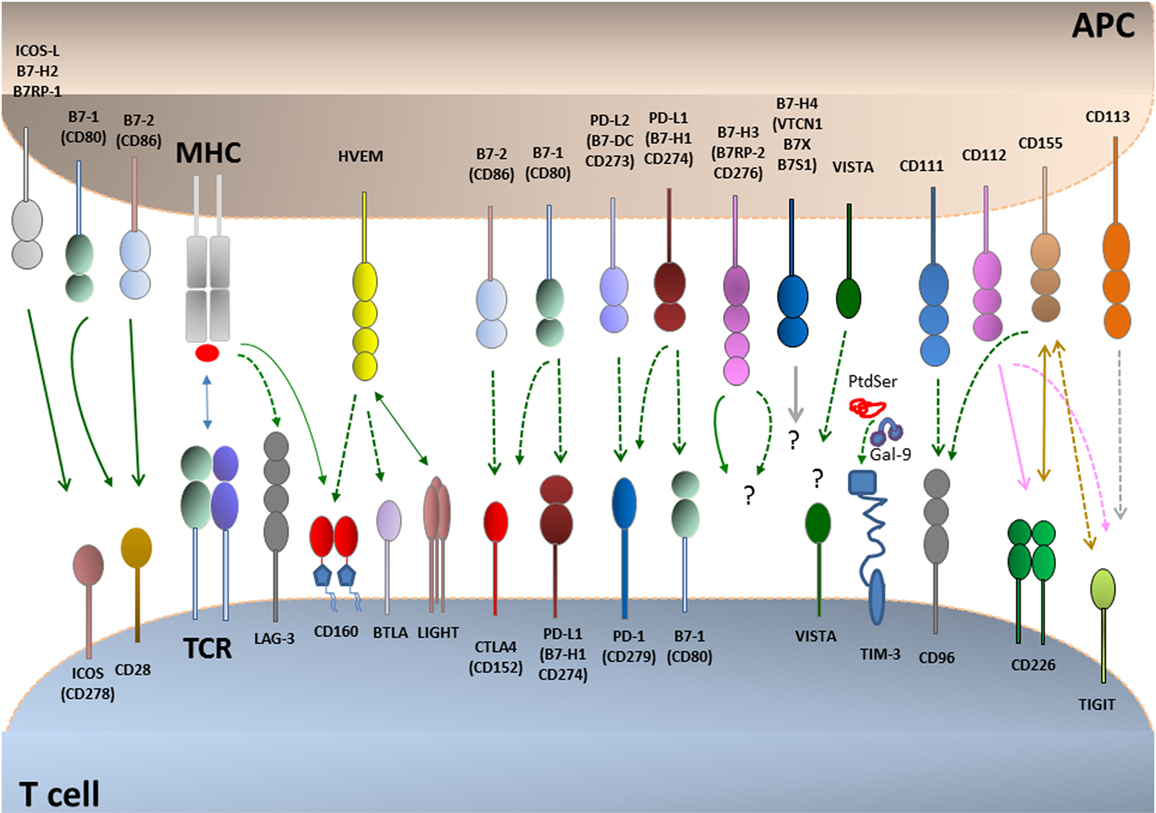
Figure 1. Coinhibitory pathways. T cell activation is initiated by recognition of antigens presented by antigen-presenting cells (APCs) to the T cell receptor (TCR)–CD3 complex in the presence of CD28 costimulation. CD28 is the prototype costimulatory receptor and interacts with CD80 and CD86. Many coinhibitory pathways are upregulated upon T cell activation and can attenuate TCR and costimulatory signals. Coinhibitory pathways in the B7–CD28 family control responses of naive, effector, regulatory, memory, and exhausted T cells. Ligands for coinhibitory receptors are expressed on APCs, non-hematopoietic cells, tumors, and some of them also on T cells. The receptors for B7-H3 and B7-H4 and their effects remain unclear. Binding ligands for VISTA have not been identified. Continuous lines indicate interactions mediating stimulatory effects, and dotted lines indicate interactions mediating inhibitory effects.
Coinhibitory receptors have a major impact on the differentiation fate of T cells as well as on their ability to proliferate. These functional endpoints are regulated by T cell metabolic reprogramming (24, 25). Therefore, altering metabolic reprogramming might be a key mechanism by which coinhibitory receptors modify T cell differentiation and function. Because various coinhibitory receptors mediate distinct effects on the activation of signaling pathways downstream of the TCR, the role of coinhibitory receptors on altering the metabolic programs of T cells is also anticipated to be different. Identification and targeting the specific immunometabolic pathways relayed by coinhibitory receptors might have significant clinical implications. For example, selective receptor targeting might be employed in order to achieve specific desired modifications in the metabolic programs and functional fates of T cells. In this review, first, we will discuss the current understanding of the mechanisms underlying metabolic programs induced during T cell differentiation and how these are affected by cytotoxic lymphocyte antigen-4 (CTLA-4) and programmed death-1 (PD-1), the two most clinically relevant coinhibitory receptors thus far, for which therapeutic targeting has achieved major success in cancer treatment. Next, we will review briefly other inhibitory pathways, and we will discuss lymphocyte activation gene-3 (LAG-3), T cell-immunoglobulin–mucin domain 3 (TIM-3), and T cell immunoglobulin and ITIM domain (TIGIT), which are additional promising therapeutic targets. Finally, we will briefly discuss the CD160/B and T lymphocyte attenuator (BTLA)/LIGHT/herpesvirus entry mediator (HVEM) pathway, which has the rare property to engage members of the Ig superfamily and the TNF receptor superfamily and to deliver both coinhibitory and costimulatory signals (Please see glossary in Supplementary Material).
Similar to the metabolism of other non-proliferating cells, naïve T cells depend on catabolic metabolism to meet their energetic needs, relying predominantly on oxidative phosphorylation (OXPHOS) to generate ATP (26). Extrinsic IL-7 survival signal maintains naïve T cell quiescence, a mechanism that relies on tuberous sclerosis (TSC) function to keep mTOR activation in check (27–29). Upon antigen encounter, T cells switch to anabolic metabolism to meet the increased metabolic demands of biomass accumulation, proliferation, and biosynthesis of mediators of effector functions (Figures 2 and 3A). Signaling via the TCR is sufficient to upregulate expression of the glucose transporter Glut1 via Myc (30). However, the metabolic reprogramming accompanying the differentiation into TEFF requires costimulation. Signaling via CD28 costimulation activates the PI3K/Akt/mTOR pathway, which is crucial to activate aerobic glycolysis required for T cell differentiation and function (6, 31, 32). mTOR has a key role in regulating metabolism coupling nutrient availability to cell growth and division (33, 34).
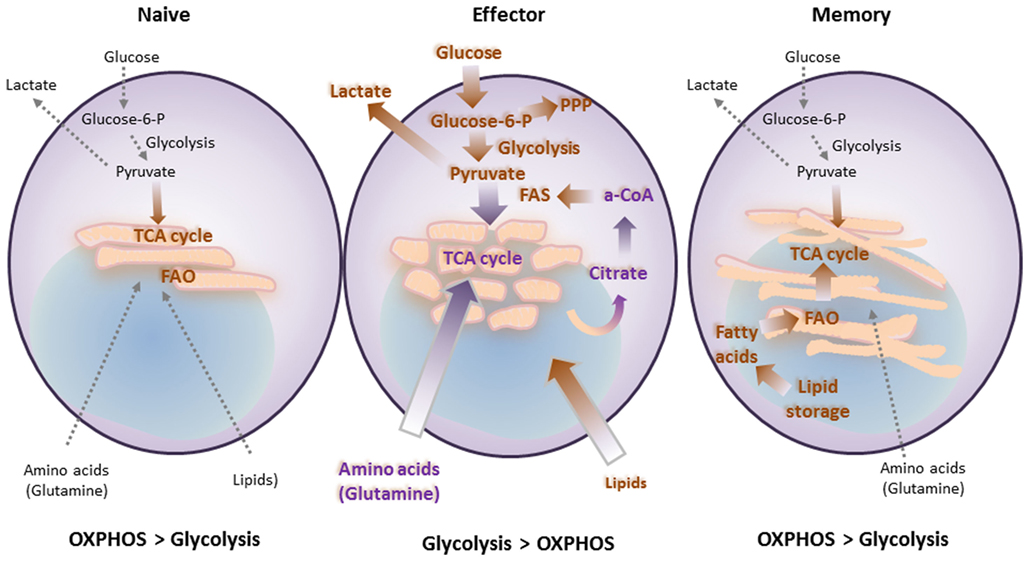
Figure 2. T cell differentiation is accompanied by metabolic changes, which are affected by costimulatory and coinhibitory receptors. Naïve T cells function in antigenic surveillance and do not proliferate. This requires minimal energetic and biosynthetic activity, which is represented by a metabolically quiescent state, and is accompanied by minimal nutrient uptake. Their only energy-demanding processes are ion homeostasis, membrane integrity, and movement. The primary ATP sources are oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) to fuel the low energy demand. Upon antigen encounter T cells differentiate into effector cells. This process is accompanied by metabolic changes, which are required to fulfill their new (effector) functions and rapid proliferation. Uptake of nutrients is enhanced. Glucose is the main nutrient used for energy and for generation of biosynthetic precursors. These changes combined with increased glutaminolysis and a high degree of protein, lipid, and nucleic acid synthesis support cell growth and proliferation. These metabolic changes coincide with mitochondria fission. Memory T cells do not proliferate and thus have minimal biosynthesis and nutrient uptake. However, they have increased spare respiratory capacity, which supports their ability to rapidly proliferate upon re-encounter of antigen. This cellular fate includes another metabolic adaption, which supports metabolic switch to FAO via increased carnitine palmitoyltransferase 1A. These metabolic and energetic changes are supported by fusion of mitochondria.
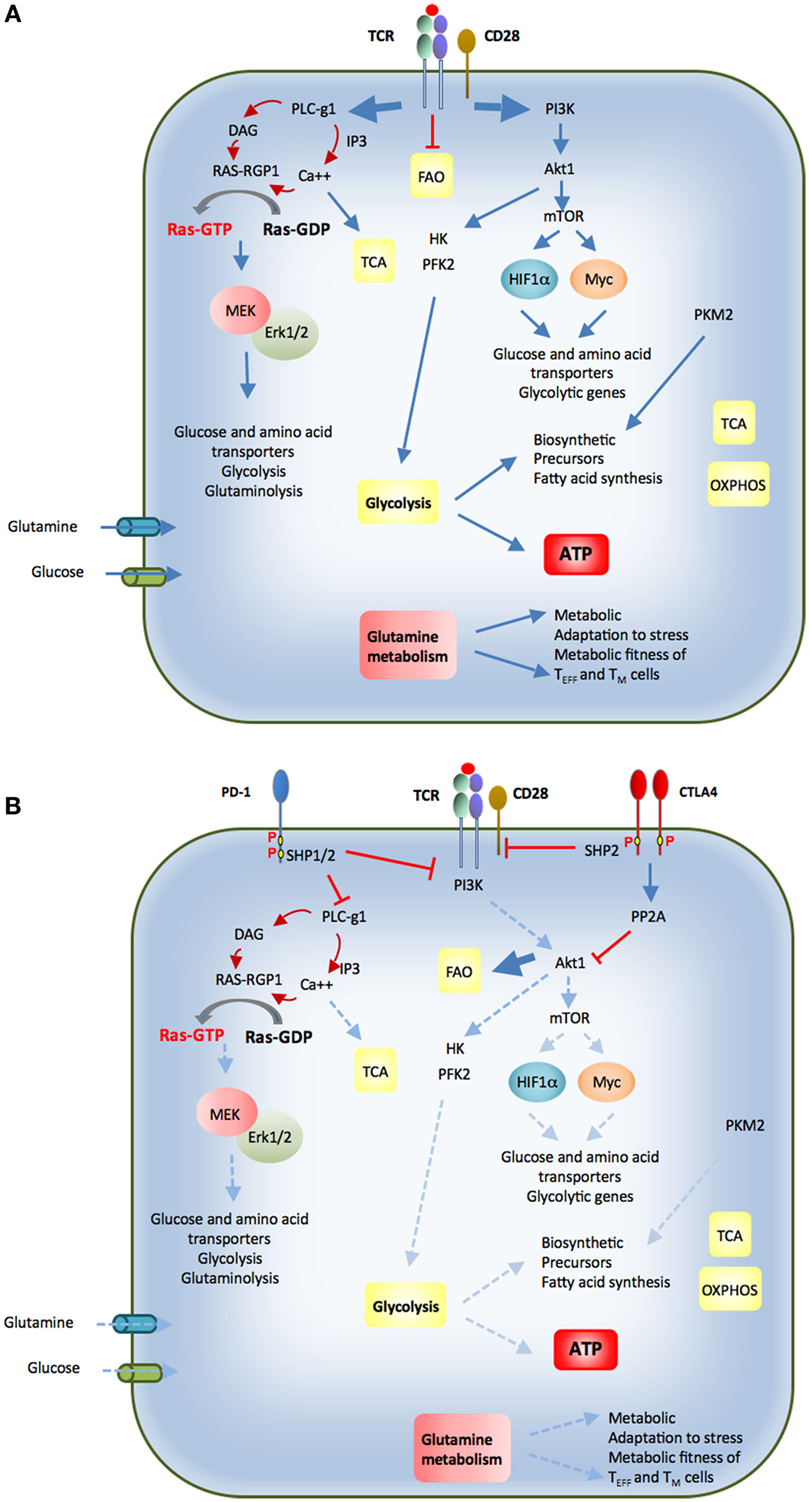
Figure 3. (A) Upon antigen encounter T cells differentiate into effector cells. Antigen binding to the T cell receptor (TCR) and coactivation by CD28 inhibit fatty acid oxidation (FAO) and activate PI3K-Akt. This activation triggers glycolytic enzymes HK and PFK2. Additionally, mTOR signaling is turned on, which enhances expression of glycolytic genes, glucose, and amino acid transporters via activation of transcription factors HIF1α and Myc. Activation of PLC-γ1 and generation of second messengers result in activation of Ras and MEK/Erk pathway, which is required for expression of nutrient transporters and nutrient utilization. Calcium release activates calcium-dependent mitochondrial dehydrogenases, which activate the TCA cycle. Effector T cells also switch from balanced PKM1 and PKM2 expression to increased and predominant expression of PKM2, which promotes generation of biosynthetic precursors. These events promote glucose and glutamine uptake, increased glycolysis and glutaminolysis combined with a high degree of protein, lipid, and nucleic acid synthesis to support cell growth and proliferation. CD28 costimulation is required for activation of the signaling pathways that support these metabolic changes. (B) Cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed death-1 (PD-1) coinhibitory receptors are expressed in activated T cells. Via the recruitment of phosphatases, both these coinhibitory receptors attenuate the signaling events mediated by ligation of the TCR by antigen and have a mandatory role in the metabolic changes required for optimal T cell activation, function, and differentiation. CTLA-4 opposes the effects of CD28 costimulation and can inhibit potent TCR-mediated signals. PD-1 inhibits weak but not strong TCR signals. The imbalanced activation of these signaling pathways alters the metabolic reprogramming of T cells and their differentiation fate (see text for details).
After pathogen clearance, antigen-specific T cells undergo contraction, and most TEFF cells undergo cell death. A small subset survives and goes on to become TM cells. TM cells exhibit a distinctive increase in mitochondrial mass and higher spare respiratory capacity (SRC) (35). This is a consequence of fusion in the mitochondria of TM cells, which configures associations of the electron transport chain and favors OXPHOS (36). In contrast to TEFF, which rely predominantly on glycolytic metabolism, TM rely on mitochondrial fatty acid oxidation (FAO) of de novo synthesized FA, rather than uptake of lipids (37). During TCR engagement with APCs, T cells can divide asymmetrically. It has been determined that the APC-proximal daughter generated during cell division is more likely to become a TEFF while the APC-distal daughter cell is more likely to differentiate into a TM (38). In addition to its role in promoting glycolysis, asymmetric Myc sorting between the daughter cells plays a role in the distribution of amino acid content and transporters as well as mTORC1. This results in altered metabolism and thus altered proliferation and differentiation that may give rise to effector versus memory fates of the daughter cells (39). This is likely not the only mechanism to generate TM cells, as other studies have shown that TEFF cells are the ones that can give rise to TM (40, 41). Enforcing the metabolic switch to FAO by enhancing AMPK or by inhibiting mTOR results in increased numbers of TM cells (42, 43). In mammalian cells, mTOR forms functionally distinct complexes—mTORC1 and mTORC2. Raptor and Rictor are the core adaptor subunits of mTORC1 and mTORC2, respectively. Interestingly, T cells deficient in Rictor show enhanced CD8 memory formation with enhanced recall responses without dampening effector functions (44).
Since the activation of T cells requires accompanying metabolic reprogramming for differentiation, it is no surprise that metabolism drives the differentiation of CD4 T helper (Th) subsets as well. Th1, Th2, Th17, and TREG cells are the best metabolically defined thus far. Initial evidence showing their metabolic distinction came when suppression of mTOR by rapamycin prevented TEFF proliferation and instead promoted TREG generation, even in Th17 polarizing conditions (45). Genetic deletion of mTOR in T cells confirmed the results of pharmacological inhibition and caused production of only TREG, illustrating the obligatory role of mTOR for TEFF production (46). Furthermore, TEFF show a strong preference for glycolytic rather than mitochondrial metabolism, whereas TREG rely more on the oxidation of lipids (47).
Th17 cells are particularly dependent on glycolysis. Although Myc is dominant over the hypoxia-inducible factor 1α (HIF1α) in regulating differentiation of naïve T cells into TEFF cells (30), HIF1α has a key role in the metabolic switch to aerobic glycolysis that influences the balance of Th17/TREG in favor of Th17 cells. Genetic deletion of HIF1α in mice or blocking glycolysis with 2-deoxyglucose showed reduced Th17 and enhanced TREG development (48). HIF1α in coordination with RORγt and p300 promotes the development of Th17 cells while concurrently weakening the development of TREG cells by promoting proteasomal degradation of FoxP3 (49). Furthermore, differentiation of Th1 and Th17 cells is selectively regulated by mTORC1 while Th2 differentiation is regulated by mTORC2 (50).
It should be noted that glycolytic enzymes themselves might also have an active role in regulating TEFF cell function. For example, it has been proposed that when glycolytic rates are low in Th1 CD4+ T cells, glyceraldehyde phosphate dehydrogenase (GAPDH) binds to and suppresses IFN-γ mRNA translation (51). In effector memory CD8+ T cells, glycolysis was observed to impact chromatin remodeling in the promoter region of the IFN-γ gene, thus facilitating gene transcription (52). In addition, naïve T cells express both isoforms of the glycolytic enzyme pyruvate kinase, PKM1 and PKM2, which catalyzes the conversion of phosphoenolpyruvate to pyruvate. After activation, the M2 isoform rapidly accumulates and is the dominant form expressed in TEFF cells (53, 54). The M2 isoform is less efficient, but this skews glycolysis toward biosynthetic pathways and may give cells a growth advantage during rapid proliferation (55). Because coinhibitory receptors are induced after T cell activation, a process that is dependent on glycolysis, there might be a correlation between the expression and function of glycolytic enzymes and coinhibitory receptors. These findings provide evidence for an intimate causative link among TCR-mediated signaling, T cell metabolism, and T cell differentiation.
Signaling pathways activated by engagement of the TCR and costimulatory receptors and CD28, the prototype costimulatory receptor, activates the PI3K/Akt/mTOR and MEK/Erk MAPK pathways, and their simultaneous activation is required to promote T cell growth, division, and cytokine production (56) (Figure 3A). Autocrine and paracrine cytokine signaling loops induce further PI3K and MAPK activation, together with JAK/STAT signaling (57, 58). PI3K catalyzes the production of the second messenger phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3], leading to the activation of several downstream kinases, including Akt, its most prominent effector. In naïve T cells, active Akt leads to increased Glut1 surface expression, improved coupling of HKII to the mitochondria, and increased rates of glycolysis, which correlates with proliferative capacity (6, 7, 53). Akt also promotes aerobic glycolysis by direct phosphorylation and activation of glycolytic enzymes, such as PFK2 (59), by regulating Glut1 trafficking, and by inducing several transcription factors (60). Akt can activate mTORC1, a key driver of anabolic metabolism (61).
MAP kinases have a central role in metabolic regulation during T cell activation by affecting both glucose and glutamine uptake and metabolism (2, 62). One target of MAPK with a direct role on metabolic regulation is HIF1α. MAPK promotes the ability of HIF1α to activate transcription and expression of many glycolytic genes, while mediating suppression of genes involved in OXPHOS (63). HIF1α levels are also upregulated by PI(3,4,5)P3 signaling through mTOR (61) providing a mechanistic explanation for the requirement of simultaneous activation of PI3K and MAPK pathways for optimal effector T cell expansion (56). The critical role of HIF1α in T cell differentiation and function is underlined by the observation that T cells with constitutively elevated HIF1α display sustained increase of aerobic glycolysis and constitutively maintain effector function (64).
After TCR and costimulatory receptor engagement, T cell activation is maintained by sustained signaling from IL-2 and other cytokines acting on common gamma chain (γc) cytokine receptor complexes (65). This effect of IL-2 leads to glycolysis and anabolic metabolism by activating transcriptional upregulation and trafficking of Glut1. This is mediated by Jak-STAT5 signaling (66) and also by activation of PI(3,4,5)P3/Akt (57).
T cell receptor signaling pathways also lead to the generation of second messengers, diacylglycerol (DAG), and calcium. Calcium flux from intracellular stores stimulates the calcium-activated mitochondrial dehydrogenases that drive the TCA cycle (67). In addition, calcium flux downstream of the TCR causes a short-term phosphorylation of AMPK mediated by CAMKK (68). AMPK acts as an important metabolic checkpoint to control cellular anabolic pathways and to induce energy-conserving mechanisms under conditions of nutrient shortage or cellular stress. AMPK activation early during the initiation of T cell stimulation may facilitate transient increase of OXPHOS before the cells switch their metabolism toward glycolysis thereby supporting the energy demands at this early T cell activation stage. Mechanistically, AMPK activation is induced by the upstream kinase LKB1 when the cellular levels of ATP are diminished and the levels of AMP and ADP are increased in response to physiological stress (69). During T cell activation, in addition to regulation of energy, AMPK also provides metabolic fitness to the differentiating TEFF and TM cells (70). After activation, AMPK phosphorylates several substrates involved in the metabolism of glucose, lipid, and proteins, in autophagy, cell cycling, and cell growth (71). Activation of the TSC1/TSC2 complex by AMPK can negatively regulate the mTOR pathway (72, 73) not only by inhibiting mTOR activation via Rheb (74), a member of the Ras family of GTPases, but also by mediating phosphorylation of the mTORC1 partner Raptor (75).
The key signaling pathways that are activated by antigen encounter are targets of costimulatory and coinhibitory receptors (Figure 3B). Costimulatory receptors amplify TCR-mediated signaling and impact T cell metabolic reprogramming. For example, CD28-mediated costimulation mediates transcriptional upregulation of the glucose receptor Glut 1 (6) and the glutamine receptors Snat1 and Snat2 (62) after TCR ligation. Costimulation also amplifies MAPK and PI3K/Akt activation, and the simultaneous engagement of these signaling pathways is mandatory for cell cycle progression and cytokine production (56). Conversely, coinhibitory receptors oppose the signaling and functional effects induced by TCR and costimulatory pathways, thereby limiting T cell expansion and cytokine production and altering the ability of T cells to differentiate into TEFF and TM cells. Importantly, coinhibitory receptors do not induce global signal inhibition but rather selective targeting of signaling, resulting in imbalanced activation of pathways that are normally engaged by TCR-plus-costimulation. As a consequence, the implications of coinhibitory pathways on the metabolic and functional programs of T cells might significantly vary, dependent on the signaling targets of these inhibitory receptors.
The coinhibitory receptor CTLA-4 is a central negative regulator of T cell activation. CTLA-4 is a homolog of CD28 and binds to the same ligands, CD80 (B7-1) and CD86 (B7-2), but with higher affinity (76). CTLA-4 is induced in CD4+ and CD8+ T cells after activation (77) but is constitutively expressed on TREG cells, because it is a direct transcriptional target of Foxp3 (22). Crosslinking of the CTLA-4 simultaneously with TCR and CD28 inhibits cell cycle progression (78) and suppresses IL-2 production leading to proliferative arrest (79, 80). Engagement of CTLA-4 can induce an anergic phenotype similar to that observed in response to TCR stimulation alone (78) establishing the notion that CTLA-4 might function to counteract CD28-mediated costimulation. Notably, the inhibitory functions of CTLA-4 on CD4+ T cells appear to be more important for the prevention of autoimmunity as CTLA-4-deficient CD8+ T cells are incapable of inducing autoimmune pathology in the absence of CD4+ T cells (81, 82). This may be due to the fact that CTLA-4 expression in CD4+ TREG is indispensable for the maintenance of TREG suppressive function (22). However, direct CTLA-4-mediated inhibition of CD8+ T cells may be particularly important in the effector/memory CD8+ T cells in which CTLA-4 controls secondary responses (83, 84).
CTLA-4 controls T cell activation by selectively reversing CD28-mediated costimulation (79, 85). Because CTLA-4 is the high-affinity ligand for CD80 and CD86, CTLA-4 might inhibit activation by competing with CD28 for interaction with CD80 and CD86. It can also downregulate the expression of CD80 and CD86 expression on APCs (22) or remove these costimulatory ligands from APCs through transendocytosis and, as a consequence, reduce CD28 engagement (86). After binding to CD80/CD86, CTLA-4 may also alter the properties of dendritic cells by inducing the tryptophan-degrading enzyme IDO, a known T cell inhibitor (87).
CTLA-4 also inhibits T cell activation through cell-intrinsic signaling mechanisms (88). The cytoplasmic tail of CTLA-4 interacts with several molecules that are key components in TCR and CD28-mediated signaling pathways (Figure 3B). TCR/CD28-mediated stimulation induces activation of several kinases, including Lck, Fyn, Lyn, Rlk, and Jak2, which are capable of phosphorylating Y165 and Y182 of the CTLA-4 cytoplasmic domain (89–92). Phosphorylation at Y165 creates a docking site for the protein tyrosine phosphatase SHP-2, which subsequently inhibits proximal TCR signaling via dephosphorylation of the TCRζ chain, LAT, and the Ras regulator p52SHC (93, 94). CTLA-4 interferes with the formation of lipid rafts, TCR:ZAP70 microclusters, and the central supramolecular activation complex, each of which plays important roles in T cell activation (89, 95, 96). CTLA-4 has also been identified to interact with the serine/threonine phosphatase PP2A, leading to inhibition of Akt (97, 98).
The effects of CTLA-4 on depleting CD80 and CD86 costimulatory ligands and inhibiting TCR signaling pathways have major implications on regulation of metabolic reprogramming by cell-intrinsic mechanisms. By dampening CD28-mediated signaling, which has a dominant role in regulating Glut1 expression and HKII activation (5, 6), CTLA-4 directly targets a key regulator of glucose transport, affecting intracellular glucose availability and glycolysis. Furthermore, by decreasing calcium release, which is involved in the activation of the TCA cycle (67), CTLA-4 can suppress not only glucose uptake and metabolism but also oxidation of other metabolites in the TCA cycle. Furthermore, CTLA-4 inhibits production of IL-2 (79, 80), by impairing the activation of and nuclear accumulation of AP-1, NFAT, and NF-κB (99, 100). Because IL-2 serves as an autocrine mechanism that activates and amplifies T cell metabolic reprogramming toward a glycolytic phenotype (57), inhibition of IL-2 is an additional mechanism by which this coinhibitory receptor shuts off the activation of metabolism-related expansion and differentiation of T cells downstream of the TCR and CD28.
A unique feature of CTLA-4 biology is related to its intracellular localization and trafficking. Specifically, the majority of CTLA-4 resides within intracellular vesicles of the trans-Golgi network and endosomal compartments (101, 102). In resting T cells, a small amount of CTLA-4 protein cycles continuously from the Golgi apparatus to the cell surface, followed by rapid endocytosis and lysosomal degradation (103). This intracellular trafficking is mediated by the interaction of the cytoplasmic tail of CTLA-4 with the clathrin-associated adaptors AP-1 and AP-2/AP50 (104, 105). During TCR ligation, CTLA-4 protein expression is increased, and CTLA-4-containing intracellular vesicles relocalize to the immune synapse (102). The TCR-proximal kinases Lck and ZAP70 phosphorylate the cytoplasmic tail of CTLA-4 at Y165. This event leads to disruption of the CTLA-4:AP-2 association and retention of CTLA-4 at the cell surface in the immune synapse (106). The more potent the TCR-mediated stimulation, the more CTLA-4 accumulates at the immune synapse. By this mechanism, CTLA-4 provides a dynamic inhibitory signal, which is fine-tuned by the intensity of the TCR signal (107). The continuous surface recycling and endocytosis of CTLA-4 in T cells and its rapid stabilization on the cell surface after T cell activation might be responsible for the potent inhibitory effect of CTLA-4, which is mediated early upon initiation of T cell activation for maintenance of an immune quiescent phenotype. Consistent with the biochemical evidence that CTLA-4 might rapidly inhibit TCR and CD28-mediated signaling, gene expression studies indicate that the net outcome of CTLA-4 ligation is the suppression of activation transcripts downstream of the TCR without specific targeting on metabolism-related genes (108).
PD-1 is a transmembrane coinhibitory receptor that controls T cell activation, exhaustion, tolerance, and resolution of inflammation. PD-1 appears within 24 h of T cell activation, declines with the clearance of antigen, and reappears in TM cells (109, 110). NFATc1, IRF9, and Notch promote PD-1 transcription (111–113), providing evidence that PD-1 expression requires not only TCR-proximal signaling but also generation of second messengers. When T cells are exposed to antigen for prolonged periods of time (as with chronic infection or cancer), the level of PD-1 expression remains high, and T cells undergo various changes in transcription factor expression, which alter their differentiation program and guide the development of a distinct state termed “exhaustion.” It should be noted that this functional state is not mediated exclusively by PD-1. Under these conditions, exhausted T (TEX) cells express multiple other coinhibitory receptors in addition to PD-1, making them susceptible to inhibition by multiple checkpoint pathways. Depending on the number of coinhibitory receptors expressed, TEX cells display a different level of functional incompetence (114, 115). PD-1 can also be expressed on TREG and T follicular regulatory cells (21, 116), as well as on natural killer (NK), NKT, B cells, macrophages, and some DC subsets during immune activation and chronic inflammation (20, 110, 117).
PD-1 has two known ligands, programmed death-ligand 1 (PD-L1; also known as CD274 and B7-H1) and programmed death-ligand 2 (also known as CD273 and B7-DC) (20, 118). PD-1 ligands are expressed on APCs and other hematopoietic cells but also on non-hematopoietic cell types and are induced or upregulated by proinflammatory cytokines such as type I and type II interferons, TNF-α, VEGF, and GM-CSF. The induction of PD-1 ligands by proinflammatory signals might serve as a negative feedback mechanism that downregulates effector T cell activity, thereby protecting tissues from excessive immune damage. These mechanisms are exploited by several pathogens and tumors to avoid attack from the immune system (13, 23).
Binding of PD-1 ligands to PD-1 results in tyrosine phosphorylation of the PD-1 cytoplasmic domain and recruitment of the tyrosine phosphatase SHP-2 (119). These events correlate with attenuated signals downstream of the TCR, decreased T cell activation and impaired cytokine production (Figure 3B). Signaling through PD-1 inhibits PI3K activity (120, 121). The main negative regulator of the PI3K/Akt pathway is PTEN, which is a lipid phosphatase, dephosphorylating PI(3,4,5)P3 at the 3′ position to generate PI(4,5)P2 (122). By limiting the amount of PI(3,4,5)P3, PTEN opposes the activation of PI3K and therefore diminishes the pro-survival and proliferative signals mediated by PI3K/Akt (123). PD-1 activates PTEN phosphatase by inhibiting its inactivating phosphorylation by CK2 (120), leading to diminished activation of Akt and decreased expression of critical transcription factors such as Tbx21 (T-bet), Eomes, and Gata3, which are involved in the function of effector cells. PD-1 ligation also inhibits Ras-MEK-ERK pathway signaling, possibly via SHP-2-regulated dephosphorylation of PLCγ1 (124), which is required for Ras activation via calcium- and DAG-dependent activation of Ras-GRP1 (125). Thus, PD-1 targets many pathways that have a decisive role in metabolic reprogramming of T cells after antigen encounter.
As a consequence of these signaling events, after PD-1 engagement, T cells have significantly diminished capacity to activate glycolysis, have reduced respiration, a function that depends on mitochondria, and also have altered lipid metabolism characterized by suppression of fatty acid synthesis and elevated FAO (126). Subsequently, T cells are unable to become effectors, a fate that requires engagement in glycolysis as well as potent mitochondrial function (51). Instead, PD-1 can promote the differentiation of TREG cells, which suppress TEFF functions (21). Because TREG differentiation depends on FAO and lack of glycolysis (47), it is possible that the effects of PD-1 on T cell metabolic reprogramming are responsible for the switch of the T cell functional fate from TEFF to TREG differentiation. Such effects on immunometabolic programs may also have a causative role in the control of peripheral tolerance by PD-1 because PD-1 signaling inhibits aerobic glycolysis, which is required for Th1 and Th17 cell differentiation (51) and metabolically reprograms T cells from aerobic glycolysis to FAO (126), which promotes the generation of TREG cells. Notably, the downregulation of Akt, mTOR, and S6K, which is induced by PD-1, has been previously identified as a requirement for the development of peripheral TREG cells (127). Moreover, PD-1 ligation in TEFF cells might reprogram TEFF cells from aerobic glycolysis and FAS to catabolic lipid metabolism and FAO. By selectively inducing metabolic reprogramming of TEFF cells from glycolysis to FAO, PD-1 can guide differentiation of TEFF cells to TREG to terminate the ongoing immune responses and to TM cells to maintain the T cell repertoire as differentiation of both these cell programs depends on FAO (37, 47).
By targeting metabolic reprogramming, the PD-1:PD-L1 pathway might also alter metabolism in the tumor microenvironment. Changes in metabolism of T cells and tumor cells will have a central role in antitumor immune responses. Highly glycolytic tumor cells reduce glucose levels in the tumor microenvironment, thereby reducing the ability of tumor-infiltrating lymphocytes (TILs) to mount effector antitumor immune responses. Moreover, ligation of PD-1 by PD-L1 expressed by tumor or APC of the tumor microenvironment shuts off glycolysis and mitochondrial function of TIL leading to impaired effector function. Simultaneously, by this mechanism, TREG cells are preferentially generated in the tumor microenvironment leading to progressive immunosuppression through additional TEFF cell-extrinsic mechanisms.
In addition to CTLA-4 and PD-1, multiple other coinhibitory molecules exist including LAG-3, TIM-3, TIGIT, CD160, BTLA, B7-H3, B7-H4, VISTA, CD244, HHLA2, and BTNL2 (Figure 1). Several of these coinhibitory receptors are expressed on dysfunctional T cells similarly to PD-1. T cell subsets coexpressing multiple coinhibitory receptors are more dysfunctional than those expressing each coinhibitory receptor alone. The coexpression of coinhibitory receptors has important clinical relevance because combined blockade with PD-1 significantly enhances therapeutic benefit.
LAG-3 is a CD4 homolog that binds MHC class II molecules with higher affinity than CD4 (128). Among T cells, LAG-3 is expressed on activated CD4+ and CD8+ T cells, as well as in thymic and peripherally induced TREG (129). It is also expressed in NK cells, NKT cells, B cells, and plasmacytoid dendritic cells. LAG-3 negatively regulates T cell proliferation and homeostasis (130, 131). The KIEELE motif of the cytoplasmic domain of LAG-3 is involved in its inhibitory function (132), but the downstream signaling pathways mediating the inhibitory effect have not been identified. LAG-3 regulates peripheral T cell tolerance by controlling self-reactive effector cells and TREG (133–135). Mice lacking both LAG-3 and PD-1 rapidly develop lethal, systemic autoimmunity (136), providing evidence that these two pathways control T cell tolerance in a synergistic manner.
T cells in chronic viral infections and tumors often coexpress PD-1 and LAG-3. Furthermore, consistent with the synergistic effect in T cell autoimmunity induced by combined deficiency of PD-1 and LAG-3, combined blockade of PD-1 and LAG-3 in the lymphocytic choriomeningitis virus (LCMV) chronic infection model or in tumor models has a greater therapeutic benefit than blockade of each coinhibitory receptor alone. In this regard, LAG-3 and PD-1 blockade together can induce almost complete tumor regression in mice with established MC38 or SA1N tumors, in contrast to only ~15% efficacy when only one of the two receptors is blocked (137). Similarly, in TILs from patients with ovarian cancer, simultaneous LAG-3 and PD-1 blockade restores responsiveness of NY-ESO-1-specific LAG3+/PD-1+ CD8+ T cells to a greater extent than blockade of each coinhibitory receptor alone. Thus, although it remains unclear how LAG-3 affects signaling pathways regulated by TCR, the functional synergy between PD-1 and LAG-3 suggests that LAG-3 might target pathways not affected by PD-1 and, as a consequence, combined signals mediated by these coinhibitory receptors result in more profound impairment of TEFF cell function.
Recent studies have shown that LAG-3 is cleaved within the short connecting peptide between the membrane proximal D4 domain and the transmembrane domain, which is mediated by ADAM family metalloproteases, resulting in the release of a soluble form of LAG-3 (sLAG-3) in vitro and in vivo (138). LAG-3 expression and function might be linked to oxidative signals generated by metabolic reprogramming of activated T cells. Treatment with an antioxidant superoxide dismutase (SOD)-mimetic compound prevented type I diabetes in mouse models by inhibiting metalloprotease activity resulting in maintenance of LAG-3 surface expression at levels adequate to attenuate TCR-mediated Th1 cell activation and effector function (139). It has also been determined that LAG-3−/− splenocytes exhibit enhanced SRC, which correlates with increased mitochondrial mass and expression of the mitochondrial biogenesis transcription factor TFAM and mitochondrial complexes I–IV (140). Moreover, after suboptimal stimulation with Con-A, LAG-3−/− splenocytes demonstrated greater upregulation of glycolysis-associated genes together with elevated expression of Myc and mTOR signaling (140), which are crucial for driving aerobic glycolysis. Thus, LAG-3 might regulate T cell exhaustion in conjunction with PD-1 because LAG-3 and PD-1 signals converge into regulating glycolytic metabolism and mitochondrial function both of which are involved in the differentiation of TEFF cells.
TIM-3 was initially identified at the cell surface of IFN-γ-producing CD4+ Th1 and CD8+ cytotoxic T cells (141). In addition, TIM-3 is expressed in TREG cells and innate immune cells such as DCs, NK cells, and monocytes. The discovery of TIM-3 led to the identification of the TIM family of genes (142), and examination of TIM-3 function with TIM-3-deficient mice, blocking antibodies, and TIM-3-Ig fusion protein suggest that TIM-3 family members function as negative regulators of T cell responses (141, 143, 144). Several ligands for TIM-3 have been identified, including C-type lectin galectin-9, phosphatidylserine (PtdSer), HMGB1, and CEACAM-1 (145–147). The mechanism of TIM-3-mediated inhibitory function differs from that of other coinhibitory receptors because TIM-3 has the unique ability to induce PtdSer-mediated recognition and phagocytosis. It has been postulated that blocking TIM-3 alters the immune response induced by recognition of dying tumor cells (148).
TIM-3 does not have a classical signaling motif in its cytoplasmic tail but contains five conserved tyrosine residues. Y256 and Y263 can be phosphorylated by Src kinases (149) or ITK (150) and are involved in the binding of Bat3 (HLA-B associated transcript 3), p85 PI3K, Fyn, and/or Lck (149, 151). When TIM-3 is not bound to its ligands, Bat3 is bound to TIM-3 and blocks SH2 domain-binding sites in TIM-3 tail and recruits the catalytically active form of Lck that preserves and promotes T cell signaling. In contrast, in the absence of Bat3, interaction of TIM-3 tail with the catalytically inactive form of Lck pY505 increases (151). Galectin-9 and CEACAM-1 binding to TIM-3 leads to Y256 and Y263 phosphorylation and release of Bat3 from the TIM-3 tail, thus promoting TIM-3-mediated T cell inhibition by allowing binding of Src kinases, preferentially Fyn (147, 151). Interestingly, Fyn and Bat3 compete for the same region on the TIM-3 tail. Although it is clear from these studies that the cytoplasmic tail of TIM-3 has the potential to interact with multiple components of the TCR complex, the precise mechanisms via which TIM-3 alters T cell signaling remains to be elucidated.
It has been determined that TIM-3+ T cells from HIV-infected patients display defective stimulation-induced phosphorylation of Stat5, ERK1/2, and p38 but have higher basal phosphorylation of these pathways (152). These CD8+TIM-3+ dysfunctional T cells lacked PD-1 expression suggesting that TIM-3 might be an alternative exhaustion-related inhibitory receptor and biomarker. However, when coexpressed with PD-1, TIM-3 marks the most dysfunctional/exhausted population in patients with chronic viral infections (153–155). Similarly, coexpression of TIM-3 with PD-1 also marks the most dysfunctional TEX CD8+ cells in cancer (156).
Because TIM-3 targets key metabolism-related signaling pathways, such as PI3K/Akt/mTOR (157), and is highly expressed on dysfunctional or TEX cells in chronic infections (152–155, 158) and TILs in various types of cancers (159–161), it is possible that TIM-3-mediated effects alter metabolism of T effector cells in a manner similar to PD-1. Alternatively, the additive and synergistic effect of combined PD-1 and TIM-3 blockade in the functional re-invigoration of TEX cells might be explained by a selective role of TIM-3 on metabolic programs that are not affected by PD-1.
The coinhibitory receptor TIGIT (also called Vsig9, Vstm3, or WUCAM) is an immunoglobulin superfamily member (162, 163). It belongs to a group of stimulatory and inhibitory receptors and ligands that bind to these receptors (164). TIGIT and the costimulatory receptor CD226 (DNAM-1) share the ligands CD155 and CD112 with TIGIT as their high-affinity receptor. TIGIT also binds to CD113 whereas CD155 also binds to the inhibitory receptor CD96 (Figure 1). CD155 and CD112 are expressed on APCs and on several cancers, whereas CD155 is also expressed on several non-hematopoietic cells. TIGIT is induced after T cell activation and is expressed in TEFF, TM, TREG, and NK cells. TIGIT can exert immunosuppressive effects by various mechanisms. By engaging CD155 on dendritic cells, TIGIT can inhibit IL-12 production by dendritic cells, thereby preventing Th1 differentiation and functional responses (165). TIGIT also can mediate cell-intrinsic inhibitory effects on T cells and can inhibit priming of CD4+ and CD8+ T cells (166). Thus, TIGIT can promote T cell tolerance by controlling both the activation of self-reactive T cells and the function of TREG (167).
TIGIT cytoplasmic tail has an ITIM motif and an immunoglobulin tail tyrosine (ITT)-like motif (164). The ITT-like motif plays a critical role in inhibitory signaling as binding of CD155 to TIGIT induces phosphorylation of the ITT motif and recruitment of SHIP1, which correlates with inhibition of NF-κB pathway activation. TIGIT is expressed on CD8+ TILs either alone or together with PD-1 in various human cancer types (168). In mouse cancer models, in contrast to minimal effects of blockade of TIGIT or PD-L1 alone, combined blockade of TIGIT and PD-L1 can induce substantial tumor regression and improved survival by enhancing the function of CD8+ TILs (169). Similarly, synergistic effects of combined PD-L1 and TIGIT blockade have been observed in the LCMV chronic viral infection model to reverse T cell exhaustion and enhance viral control (169). These findings suggest functional synergies between PD-1 and TIGIT and support exploration of combined TIGIT and PD-1 blockade for re-invigoration of immune responses.
CD160 was discovered in an effort to identify NK cell-specific receptors (170) and contains a single IgV-like domain and a glycosyl-phosphatidylinositol (GPI)-anchor (171). CD160 interacts with classical and non-classical MHC class I molecules with weak affinity (172). CD160 is expressed in secondary lymphoid organs, intestinal lymphocytes, and peripheral CD8 and NK cells (171). Although originally thought to be involved in the cytolytic activity of CD8+ T cells, CD160 was subsequently identified as one of the coinhibitory receptors expressed in CD8+ TEX cells. In fact, together with PD-1, CTLA-4, LAG-3, and CD224, CD160 is one of the coinhibitory receptors that are progressively upregulated as the CD8+ cell effector function declines during the differentiation to TEX cells (115). Increasing numbers of CD160+ T cells are detected during HIV infection and are higher in patients who have optimal control of HIV (173) providing, once more, evidence about the active role of TEX cells in the containment of chronic viral infections.
B and T lymphocyte attenuator was identified as another coinhibitory receptor of the Ig superfamily, related to CTLA-4 and PD-1 (174, 175). The highest BTLA level is detected in B cells but is also expressed on T cells, DCs, and macrophages (174, 175). In contrast to other coinhibitory receptors, BTLA is moderately expressed on naïve T cells, transiently upregulated upon antigenic stimulation, and downregulated in activated T cells (176). BTLA is highly expressed in anergic T cells generated in vivo (177). Although BTLA has a different expression pattern from other coinhibitory receptors, extensive studies have showed that BTLA has a direct inhibitory effect on T cell proliferation and cytokine production. In vitro stimulation of BTLA-deficient T cells resulted in enhanced proliferation in response to anti-CD3 (174, 175). Conversely, retroviral overexpression of BTLA in DO11.10 cells suppressed anti-CD3-mediated production of IL-2 (175). BTLA-deficient mice develop elevated autoantibodies and organ infiltration by activated CD4+ T cells indicating that BTLA has an active role in T cell tolerance (178). Consistent with a role of BTLA in maintaining peripheral tolerance, BTLA-deficient mice have increased susceptibility to experimental autoimmune encephalitis, whereas induction of oral tolerance is also compromised (175, 179).
A common ligand for CD160 and BTLA is the HVEM, which is a member of the tumor necrosis factor receptor superfamily, also known as TNFR superfamily 14 (180, 181). As mentioned above, the interaction of BTLA, which belongs to the Ig superfamily, with HVEM, which is a member of the TNF receptor superfamily, is a rare example of cross talk between members of different families of coinhibitory receptors and ligands. HVEM also interacts with another TNFR-associated factor-associated receptor, TNF receptor-like molecule 2, or LIGHT receptor (182). HVEM was initially discovered as a molecule that interacts with viral glycoprotein D to facilitate entry of herpes simplex virus (183). It is expressed in hematopoietic cells and nearly all parenchymal tissues with highest expression in lung, liver, and kidney, and lower expression in brain, pancreas, heart, placenta, and skeletal muscle (183). HVEM is expressed in high levels in naïve T cells, is transiently decreased after activation and is upregulated at the late activation stage (180, 184). Interaction of HVEM with LIGHT induces T cell proliferation whereas blockade of LIGHT: HVEM interaction inhibits responses of activated T cells (182). Thus, the CD160/BTLA/LIGHT/HVEM pathway mediates coinhibitory signals through the engagement of CD160 and BTLA with HVEM but promotes T cell activation by engagement of LIGHT. The distinct functions of HVEM ligands are mediated via distinct binding sites on HVEM. Specifically, HVEM has three cysteine-rich domains (CRDs) and a fourth CRD that is less typical and has only two of the three disulfide bonds (18, 183). The CRD1 is required for coinhibitory binding of HVEM with CD160 and BTLA but has no role in the costimulatory binding with LIGHT (180, 181). Deletion of CRD1 can convert the function of HVEM from a coinhibitory receptor, which seems to be its net effect, into a solely costimulatory receptor (181).
Little is known about the signaling pathways engaged by HVEM downstream of BTLA and CD160. BTLA contains two conserved ITIMs in its cytoplasmic tail (129), which are phosphorylated on Y274 and Y299 upon BTLA ligation and result in recruitment of SHP-1 and SHP-2 (18, 174, 175, 185). CD160, a GPI-anchor protein, is localized at the lipid rafts. By this localization, CD160 can regulate activation of key components of the TCR-proximal signaling machinery when engaged in cytolytic function (186). However, the mechanism via which CD160 inhibits T cell responses remains elusive. Currently, there is no information of how CD160 and BTLA might affect T cell immunometabolic programs. The involvement of BTLA in the maintenance of self-tolerance, the role of CD160 as coinhibitory receptor on TEX cells, and their reciprocal expression in naïve versus activated T cells strongly suggest that together CD160 and BTLA must play an important role in the fine tuning of metabolic programs that determine T cell fate and function upon encounter of self-antigens, pathogens, or tumor antigens. Furthermore, the ability of HVEM to switch its properties from being a mediator of inhibitory signals via interaction with CD160 and BTLA to being a T cell costimulator via interaction with LIGHT indicates that the CD160/BTLA/LIGHT/HVEM pathway might regulate immunometabolic plasticity that rapidly switches T cell differentiation programs and guides distinct functional outcomes.
The extensive studies regarding the expression and function of coinhibitory receptors provide currently emerging evidence that these receptors have synergistic roles in suppressing T cell function and provide evidence for their non-redundant roles in inhibiting the responses of activated T cells. Although the evolutional purpose for the coexpression of multiple coinhibitory receptors is not clearly understood, several observations provide challenging hypotheses. It is possible that coinhibitory receptors are expressed in distinct combinations on T cell subsets depending on the type of activation signals that such T cells receive, which might depend on the engagement of distinct costimulatory receptors or on the activation of autocrine and paracrine cytokine loops. For example, expression and recruitment of CTLA-4 to the immunological synapse depend on the strength of the TCR signal (107). It is also possible that ligands for the different coinhibitory receptors are differentially expressed in various tissues or tissue resident APC and, as a consequence, distinct coinhibitory receptors control T cell responses in distinct microenvironments. For example, parenchymal tissues and organs express ligands for PD-1 but not for CTLA-4 (20). As nutrients and metabolites provide important environmental cues with decisive role in T cell activation (187), it is also possible that distinct metabolic programs of various microenvironments selectively promote the expression and function of different coinhibitory receptors. For example, protection from oxidative damage mediated by reactive oxygen species (ROS) by an SOD-mimetic compound, resulted in elevated expression of LAG-3 in T cells and inhibition of TCR-mediated Th1 cell activation and effector function (139). Such metabolism-driven events might also differentially regulate the expression of coinhibitory receptors in T cell subsets (53).
Currently, there is little known about the metabolic repercussions of coinhibitory receptors on T cells and the metabolic consequences by targeting these immune checkpoint pathways in pathogen-specific and tumor-specific T cells. The immunometabolic effects mediated by the most clinically relevant coinhibitory receptors, CTLA-4 and PD-1, are starting to be elucidated. Laboratory studies have provided evidence that inhibitory checkpoint pathways can alter the metabolic program of T cells (126, 188). PD-1 engagement makes T cells unable to engage in glycolysis, glutaminolysis, or amino acid metabolism but increases FAO (126). This effect is due to inhibition of transport and utilization of glucose and glutamine. In contrast, PD-1 promotes FAO of endogenous lipids by inhibiting the lipid oxidation PI3K pathway, resulting in increased expression of carnitine palmitoyltransferase 1A (CPT1A), the rate-limiting enzyme of FAO, which plays an important role in the utilization of fatty acids as an energy source. In parallel, PD-1 abrogates the induction of fatty acid synthase and, as a consequence, decreases anabolic lipid metabolism and biosynthesis of lipids, which normally occurs during T cell activation.
Compared to T cells activated without PD-1 ligation, activated T cells receiving PD-1 signals have lower extracellular acidification rate (ECAR), an indicator of glycolysis, and lower oxygen consumption rate, an indicator of OXPHOS. These findings indicate that T cells receiving PD-1 signals are rather metabolically quiescent and preferentially use OXPHOS over glycolysis as indicated by the higher OCR/ECAR ratio. As fat-based metabolism is associated with longevity (189), the augmentation of FAO may explain the longevity of T cells, which engage the PD-1 pathway, in patients with chronic infections and cancer and their ability to be reinvigorated by blockade of PD-1. The role of PD-1:PD-L1 pathway in restricting glucose metabolism in vivo was also reported in a study of graft-versus-host disease in mice, where recipients of allogenic bone marrow deficient in PD-L1 had higher levels of Glut1 and lactate production (190).
Because of the increased FAO and the elevated OCR/ECAR ratio, it is possible that PD-1-mediated inhibitory effects in T cells might be related to a more oxidative environment. Although depletion of the antioxidant glutathione (GSH) was observed in T cells stimulated with or without PD-1 ligation, consistent with the expected increase of ROS during TCR-mediated activation (191, 192), PD-1 ligation resulted in more pronounced decrease in the levels of reduced GSH. However, under these conditions, T cells had higher levels of cysteine-GSH disulfide and ophthtalmate, a GSH-like product that is synthesized by the same enzymes (126). These changes might be suggestive of a more oxidative environment in T cells receiving TCR signals in conjunction with PD-1 ligation. However, several other tentative mechanisms might be implicated in these observations. In alloreactive T cells after allogeneic bone marrow transplantation, PD-1 expression has been correlated with an oxidative environment, ROS production, and higher susceptibility of these cells to metabolic inhibition by F1F0-ATP synthase complex inhibitors (193). Thus, expression and function of PD-1 and possibly other coinhibitory receptors might be linked with the T cell oxidative state, which can serve as a fine-tuning system of cellular differentiation.
A key mediator of oxidative detoxification is the PPARγ coactivator-1a (PGC-1α) (194, 195). Induction of PGC-1α during caloric restriction mediates selective cell adaptations that provide modifications permissive for cell survival and function under increased OXPHOS. Because PGC-1α is regulated by mTOR (196), which is downstream of PI3K/Akt pathway that is targeted by PD-1 (120, 121), it was hypothesized that by dysregulating PGC-1α expression and function, PD-1 might impair oxidative detoxification. Moreover, different subsets of TEX cells might have distinct metabolic and bioenergetics properties such as mitochondrial biogenesis, which might correlate with glycolysis, SRC, CPT1A, and PGC-1α (126). Consistent with this hypothesis, studies in chronic LCMV infection, which leads to the generation of various subsets of TEX cells, showed that the development of intermediate PD-1 (PD-1int) TEX cells correlated with reduced glucose uptake and glycolysis, dysregulated mitochondrial bioenergetics, and suppressed PGC-1α expression. PGC-1α expression was enhanced in the absence of PD-1, and forced expression of PGC-1α partially reverted some of the metabolic derangements of PD-1Int—but not in PD-1Hi—TEX cells and improved effector function (197). Similarly to the observations of in vitro stimulated human T cells (126), transcriptional analysis during TEX development in response to LCMV infection revealed enrichment of genes involved in OXPHOS, particularly CPT1A (197). While these results point to a strong connection between PD-1-mediated metabolic reprogramming and induction of T cell exhaustion, it should be pointed out that similar bioenergetics mechanisms of T cell exhaustion in vivo mediated by chronic infection and cancer can also occur in a PD-1-independent manner (198, 199). Thus, other coinhibitory receptors might mediate similar immunometabolic effects, thereby resulting in differentiation of functionally impaired T cells.
As mentioned above, PD-1 together with several other coinhibitory receptors coregulate T cell exhaustion during cancer and chronic infections. Pathogen-specific CD8+ T cells in mouse models and humans with chronic viral infections as well as TILs from mouse tumor models and patients with cancer can coexpress multiple repressors, including PD-1, LAG-3, TIM-3, TIGIT, and others. The pattern and number of coinhibitory receptors expressed on the same cell can dramatically influence the severity of exhaustion (114, 115). T cell exhaustion is an active process and can lead to measurable consequences on T cell function. Studies have shown that distinct subsets of TEX cells exist, which have different potentials for recovery after PD-1 blockade. Although TEX cells with PD-1int expression can be reinvigorated by PD-1 blockade, TEX cells with high PD-1 (PD-1high) expression cannot (115). These distinct subsets of TEX cells may also have different metabolic and bioenergetics properties depending on the nature of coinhibitory receptors expressed and engaged on their surface. Identification of the unique bioenergetics profiles of TEX subsets might provide new tools to identify the level of exhaustion mediated by the combined and synergistic effects of coinhibitory receptors. It might also provide novel targets to reverse exhaustion and might serve as meaningful biomarkers to assess response to checkpoint inhibitor therapies.
In contrast to the effects of PD-1 in T cell metabolic reprogramming, CTLA-4 inhibits glycolysis without augmenting CPT1A and FAO, suggesting that CTLA-4 maintains immune quiescence by preserving the metabolic profile of non-stimulated cells (126). RNA-seq of TILs from progressor sarcoma tumors in mice that were either treated with control, anti-CTLA-4, anti-PD-1 blocking antibodies, or a combination of anti-CTLA-4 and anti-PD-1 antibodies revealed differential gene enrichment. PD-1 blocking showed enrichment predominantly in metabolic genes, while CTLA-4 blocking showed enrichment in cell cycle and effector memory genes. The combination of PD-1 and CTLA-4 antibody treatment revealed synergism of metabolic and T cell effector genes that correlated with the strongest enhancement of effector molecules (108). The distinct properties of CTLA-4 and PD-1 in affecting T cell metabolic reprogramming might be related to their differential ability to oppose signals mediated by the TCR. CTLA-4 mediates a potent inhibition of T cell activation, and CTLA-4 recruitment at the immune synapse is regulated by the strength of TCR signal (107). In contrast, PD-1 inhibits weak but not strong TCR signals (200). As a consequence, TCR-mediated biochemical events might be impaired but not fully abrogated (124, 201). Notably, the strength of PD-1 signaling also differentially affects TEFF cell function (201). This rheostat effect of PD-1 on TCR signaling that likely depends on the level of PD-1 expression might mediate fine tuning of metabolic reprogramming leading to distinct T cell functional fates. The different immunometabolic implications of CTLA-4 and PD-1 in T cells, serve as a paradigm that provides evidence for the purpose of multiple coinhibitory receptors, which likely have unique roles in regulating immunometabolic programs and functional commitment of T cells. Mechanistic understanding of the immunometabolic effects of coinhibitory receptors will shed light not only to the state of T cell functional impairment and exhaustion in chronic infections and cancer but also to the metabolic regulation of T cell tolerance.
It has become increasingly understood that coordinated metabolic switches in T cells and innate immune cells modulate cellular activities and contribute to the progression of cancer and chronic infections. Metabolic communication among T cells, innate immune cells, and cancer might contribute to immunometabolic regulations and fine tuning of the activation and antitumor responses of T cells and myeloid cells (202) (Figure 4). In this regard, immunometabolic regulations mediated by coinhibitory receptors can impact T cell immune responses due to direct T cell-intrinsic signaling and by altering the metabolic properties and the differentiation program of innate immune cells (203). Not only the coinhibitory receptor ligands but also the coinhibitory receptors themselves are also present in various types of innate immune cells in addition to T cells. Intriguingly, it was recently reported that the PD-1: PD-L1 axis is implicated in immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia (203). In that system, triggering PD-1 checkpoint on monocytes hampers glycolysis and phagocytosis. Conversely, disrupting PD-1: PD-L1 signaling reverses these immune metabolic dysfunctions. Taken together, these findings imply a novel metabolic interplay between cancer cells and monocytes and that blocking PD-1: PD-L1 might restore metabolic together with antitumor activity of monocytes/macrophages.
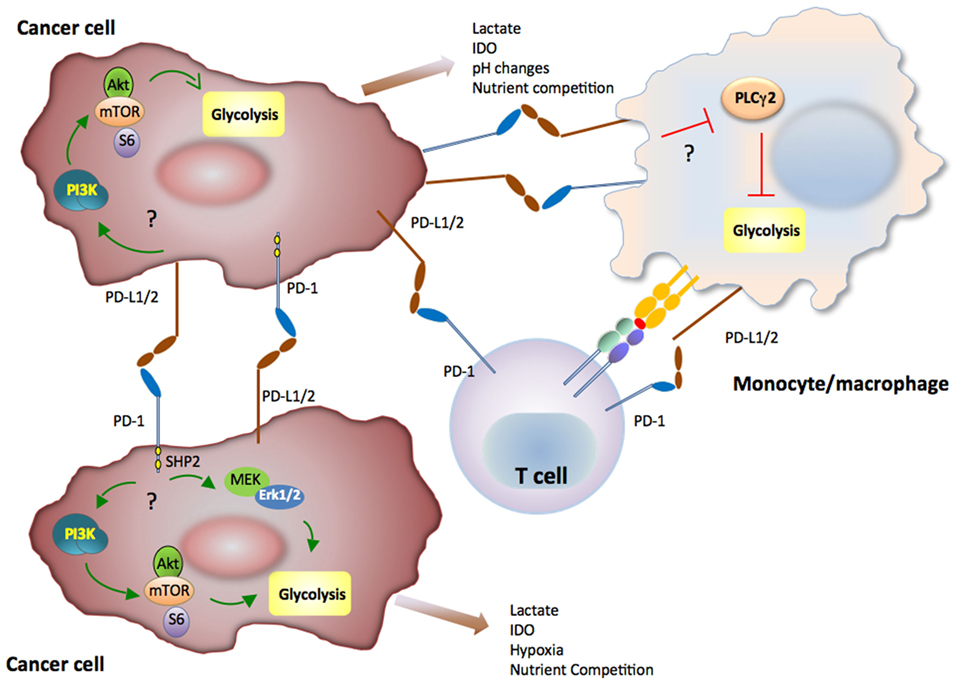
Figure 4. Immunometabolic regulations mediated by coinhibitory receptors in innate immune cells and cancer can impact T cell immune responses. Metabolic signaling mediated by the programmed death-1 (PD-1):programmed death-ligand 1 (PD-L1) pathway in cancer or myeloid cells of the tumor microenvironment might subsequently alter T cell metabolic reprogramming and immune function due to nutrient competition and by generating metabolic products with detrimental effects on T cell immune function. PD-1 and PD-L1 might activate glycolysis in cancer cells, whereas PD-1 ligation might inhibit glycolysis of monocytes/macrophages in the context of cancer.
In a sarcoma tumor model, it was reported that expression of PD-L1 on cancer cells was associated with cell-intrinsic signaling via the PI3K/Akt pathway and mTOR, leading to expression of glycolysis genes and enhanced glycolytic metabolism (204). Although it is unclear whether PD-L1 can trigger reverse signals to tumor cells by its short cytoplasmic tail that does not have obvious signaling motifs, PD-L1 functions as an inhibitory receptor that protects cancer cells from immune-mediated cancer cell destruction and Fas-mediated killing (205). Because cancer cells have elevated activation of the PI3K/Akt pathway and are highly glycolytic, expression of PD-L1 might result in concomitant increase of PI3K/Akt activation and elevated rate of tumor-intrinsic glycolysis as a consequence of improved survival. It has also been observed that subpopulations of certain human and murine melanoma cell lines and subpopulations of malignant cells from patients with melanomas express PD-1 (206, 207). However, in contrast to T cells, in which PD-1 ligation inhibits the activation of PI3K/Akt, MAPK, and mTOR pathways, ligation of PD-1 in melanoma cells was reported to activate these pathways and to promote expression of glycolytic enzymes, which correlate with tumor growth (206). To date, such finding of cancer cell-intrinsic growth-promoting effects of PD-1 have not been validated in tumor models and primary cancer cells. It is also unclear whether PD-L1 naturally expressed on tumor cells can trigger activation of glycolytic metabolism in cancer cells and whether such effect might be associated with tumor growth. However, together these findings (203, 204, 206) suggest that cell-intrinsic metabolic signaling mediated by the PD-1:PD-L1 pathway in cancer or myeloid cells of the tumor microenvironment might subsequently alter T cell immunometabolism and immune function due to nutrient competition and by generating metabolic products with detrimental effects on T cell immune function (208).
Coinhibitory receptors have a major impact on the metabolic programs that drive the differentiation and functional fate of naïve T cells. These coinhibitory receptors have a major role in reprogramming the metabolic preferences of TEFF cells leading to metabolic and functional switches. Because coinhibitory receptors mediate distinct effects on the activation of signaling pathways, the role of coinhibitory receptors on altering T cell immunometabolism is also anticipated to be distinct. Identifying and targeting the specific immunometabolic determinants relayed by different coinhibitory receptors might have significant clinical implications as selective receptor targeting might be employed in order to achieve specific desired modifications in the metabolic programs and functional fates of T cells.
Metabolic determinants of different microenvironments provide important cues to drive differentiation and function of T cells by metabolism-based mechanisms. Expression and engagement of coinhibitory receptors after T cell activation can provide the necessary signals to terminate immune responses by altering the metabolic programs of antigen-specific TEFF cells. Such metabolic reprogramming might lead to conversion of TEFF to TREG and TM for termination of immune activation and maintenance of the T cell repertoire, respectively. Understanding and recapitulating the metabolic programs mediated by coinhibitory receptors will have implications for the induction of self-tolerance and the treatment of autoimmune diseases. Conversely, blocking and reversing metabolic programs mediated by coinhibitory receptors will promote TEFF differentiation and function and will improve immune-based therapies mediated by checkpoint inhibitors and by adoptive immunotherapy of ex vivo engineered T cells for the treatment of cancer and chronic infections. Exploiting the role of the coinhibitory receptors in T cells, innate immune cells and cancer will be a major challenge.
NP and JW wrote the main body of the manuscript and generated figures; LS wrote several sections of the manuscript; CH participated in the preparation of the manuscript and generated the Supplementary Material; PS participated in the writing of various sections of the manuscript; and VB had the overall supervision in the preparation of the manuscript and figures.
Vassiliki A. Boussiotis has patents on the PD-1 pathway licensed by Bristol-Myers Squibb, Roche, Merck, EMD-Serono, Boehringer Ingelheim, AstraZeneca, Novartis and Dako. The authors declare no additional potential conflicts of interest.
This work was supported by NIH grants CA183605, CA183605S1, and AI098129-01 and by the DoD grant PC140571.
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00330/full#supplementary-material.
1. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol (2009) 27:591–619. doi:10.1146/annurev.immunol.021908.132706
2. Marko AJ, Miller RA, Kelman A, Frauwirth KA. Induction of glucose metabolism in stimulated T lymphocytes is regulated by mitogen-activated protein kinase signaling. PLoS One (2010) 5:e15425. doi:10.1371/journal.pone.0015425
3. Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol (2003) 23:7315–28. doi:10.1128/MCB.23.20.7315-7328.2003
4. Schultze SM, Hemmings BA, Niessen M, Tschopp O. PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Rev Mol Med (2012) 14:e1. doi:10.1017/S1462399411002109
5. Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol (2004) 172:4661–5. doi:10.4049/jimmunol.172.8.4661
6. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16:769–77. doi:10.1016/S1074-7613(02)00323-0
7. Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol (2003) 33:2223–32. doi:10.1002/eji.200324048
8. Lafferty KJ, Warren HS, Woolnough JA, Talmage DW. Immunological induction of T lymphocytes: role of antigen and the lymphocyte costimulator. Blood Cells (1978) 4:395–406.
9. Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A (1999) 96:185–90. doi:10.1073/pnas.96.1.185
10. Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, et al. The interaction properties of costimulatory molecules revisited. Immunity (2002) 17:201–10. doi:10.1016/S1074-7613(02)00362-X
11. Appleman LJ, Boussiotis VA. T cell anergy and costimulation. Immunol Rev (2003) 192:161–80. doi:10.1034/j.1600-065X.2003.00009.x
12. Chambers CA, Allison JP. Co-stimulation in T cell responses. Curr Opin Immunol (1997) 9:396–404. doi:10.1016/S0952-7915(97)80087-8
13. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol (2016) 34:539–73. doi:10.1146/annurev-immunol-032414-112049
14. Kamphorst AO, Ahmed R. Manipulating the PD-1 pathway to improve immunity. Curr Opin Immunol (2013) 25:381–8. doi:10.1016/j.coi.2013.03.003
15. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi:10.1111/j.1600-065X.2010.00923.x
16. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in CTLA-4. Science (1995) 270:985–8. doi:10.1126/science.270.5238.985
17. Tivol EA, Borriello F, Schweitzer NA, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA4. Immunity (1995) 3:541–7. doi:10.1016/1074-7613(95)90125-6
18. Ward-Kavanagh LK, Lin WW, Šedý JR, Ware CF. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity (2016) 44:1005–19. doi:10.1016/j.immuni.2016.04.019
19. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi:10.1016/j.immuni.2016.05.001
20. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity (2016) 44:955–72. doi:10.1016/j.immuni.2016.05.002
21. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi:10.1084/jem.20090847
22. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi:10.1126/science.1160062
23. Attanasio J, Wherry EJ. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity (2016) 44:1052–68. doi:10.1016/j.immuni.2016.04.022
24. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342:1242454. doi:10.1126/science.1242454
25. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
26. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38:633–43. doi:10.1016/j.immuni.2013.04.005
27. Wu Q, Liu Y, Chen C, Ikenoue T, Qiao Y, Li CS, et al. The tuberous sclerosis complex-mammalian target of rapamycin pathway maintains the quiescence and survival of naive T cells. J Immunol (2011) 187:1106–12. doi:10.4049/jimmunol.1003968
28. Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol (2011) 12:888–97. doi:10.1038/ni.2068
29. Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol (2001) 167:6869–76. doi:10.4049/jimmunol.167.12.6869
30. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi:10.1016/j.immuni.2011.09.021
31. Colombetti S, Basso V, Mueller DL, Mondino A. Prolonged TCR/CD28 engagement drives IL-2-independent T cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J Immunol (2006) 176:2730–8. doi:10.4049/jimmunol.176.5.2730
32. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180:4476–86. doi:10.4049/jimmunol.180.7.4476
33. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol (2012) 30:39–68. doi:10.1146/annurev-immunol-020711-075024
34. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol (2011) 12:21–35. doi:10.1038/nrm3025
35. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36:68–78. doi:10.1016/j.immuni.2011.12.007
36. Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell (2016) 166:63–76. doi:10.1016/j.cell.2016.05.035
37. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41:75–88. doi:10.1016/j.immuni.2014.06.005
38. Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science (2007) 315:1687–91. doi:10.1126/science.1139393
39. Verbist KC, Guy CS, Milasta S, Liedmann S, Kamiński MM, Wang R, et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature (2016) 532:389–93. doi:10.1038/nature17442
40. Rohr JC, Gerlach C, Kok L, Schumacher TN. Single cell behavior in T cell differentiation. Trends Immunol (2014) 35:170–7. doi:10.1016/j.it.2014.02.006
41. Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature (2008) 452:356–60. doi:10.1038/nature06672
42. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460:108–12. doi:10.1038/nature08155
43. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi:10.1038/nature08097
44. Zhang L, Tschumi BO, Lopez-Mejia IC, Oberle SG, Meyer M, Samson G, et al. Mammalian target of rapamycin complex 2 controls CD8 T cell memory differentiation in a foxo1-dependent manner. Cell Rep (2016) 14:1206–17. doi:10.1016/j.celrep.2015.12.095
45. Kopf H, de la Rosa GM, Howard OM, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol (2007) 7:1819–24. doi:10.1016/j.intimp.2007.08.027
46. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44. doi:10.1016/j.immuni.2009.04.014
47. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186:3299–303. doi:10.4049/jimmunol.1003613
48. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
49. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
50. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12:295–303. doi:10.1038/ni.2005
51. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi:10.1016/j.cell.2013.05.016
52. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol (2013) 14:1064–72. doi:10.1038/ni.2687
53. Cao Y, Rathmell JC, Macintyre AN. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS One (2014) 9:e104104. doi:10.1371/journal.pone.0104104
54. Marjanovic S, Eriksson I, Nelson BD. Expression of a new set of glycolytic isozymes in activated human peripheral lymphocytes. Biochim Biophys Acta (1990) 1087:1–6. doi:10.1016/0167-4781(90)90113-G
55. Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol (2012) 8:839–47. doi:10.1038/nchembio1212-1008b
56. Appleman LJ, van Puijenbroek AA, Shu KM, Nadler LM, Boussiotis VA. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J Immunol (2002) 168:2729–36. doi:10.4049/jimmunol.168.6.2729
57. Cornish GH, Sinclair LV, Cantrell DA. Differential regulation of T-cell growth by IL-2 and IL-15. Blood (2006) 108:600–8. doi:10.1182/blood-2005-12-4827
58. Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell (2007) 18:1437–46. doi:10.1091/mbc.E06-07-0593
59. Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem (1997) 272:17269–75. doi:10.1074/jbc.272.28.17269
60. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129:1261–74. doi:10.1016/j.cell.2007.06.009
61. Xu X, Ye L, Araki K, Ahmed R. mTOR, linking metabolism and immunity. Semin Immunol (2012) 24:429–35. doi:10.1016/j.smim.2012.12.005
62. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185:1037–44. doi:10.4049/jimmunol.0903586
63. Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, et al. Transcription factor HIF-1 is a necessary mediator of the Pasteur effect in mammalian cells. Mol Cell Biol (2001) 21:3436–44. doi:10.1128/MCB.21.10.3436-3444.2001
64. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol (2013) 14:1173–82. doi:10.1038/ni.2714
65. Boussiotis VA, Barber DL, Nakarai T, Freeman GJ, Gribben JG, Bernstein GM, et al. Prevention of T cell anergy by signaling through the γc chain of the IL-2 receptor. Science (1994) 266:1039–42. doi:10.1126/science.7973657
66. Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood (2008) 111:2101–11. doi:10.1182/blood-2007-06-096297
67. McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev (1990) 70:391–425.
68. Tamás P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med (2006) 203:1665–70. doi:10.1084/jem.20052469
69. Blagih J, Krawczyk CM, Jones RG. LKB1 and AMPK: central regulators of lymphocyte metabolism and function. Immunol Rev (2012) 249:59–71. doi:10.1111/j.1600-065X.2012.01157.x
70. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42:41–54. doi:10.1016/j.immuni.2014.12.030
71. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol (2012) 13:251–62. doi:10.1038/nrm3311
72. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell (2003) 115:577–90. doi:10.1016/S0092-8674(03)00929-2
73. Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev (2004) 18:1533–8. doi:10.1101/gad.1199104
74. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev (2003) 17:1829–34. doi:10.1101/gad.1110003
75. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30:214–26. doi:10.1016/j.molcel.2008.03.003
76. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174:561–9. doi:10.1084/jem.174.3.561
77. Chambers CA, Kuhns MS, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates primary and secondary peptide-specific CD4(+) T cell responses. Proc Natl Acad Sci U S A (1999) 96:8603–8. doi:10.1073/pnas.96.15.8603
78. Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity (2001) 14:145–55. doi:10.1016/S1074-7613(01)00097-8
79. Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med (1996) 183:2541–50. doi:10.1084/jem.183.6.2541
80. Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med (1996) 183:2533–40. doi:10.1084/jem.183.6.2533
81. Gattinoni L, Ranganathan A, Surman DR, Palmer DC, Antony PA, Theoret MR, et al. CTLA-4 dysregulation of self/tumor-reactive CD8+ T-cell function is CD4+ T-cell dependent. Blood (2006) 108:3818–23. doi:10.1182/blood-2006-07-034066
82. Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4–deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity (1997) 7:885–95. doi:10.1016/S1074-7613(00)80406-9
83. Chambers CA, Sullivan TJ, Truong T, Allison JP. Secondary but not primary T cell responses are enhanced in CTLA-4-deficient CD8+ T cells. Eur J Immunol (1998) 28:3137–43. doi:10.1002/(SICI)1521-4141(199810)28:10<3137::AID-IMMU3137>3.3.CO;2-O
84. Luhder F, Chambers C, Allison JP, Benoist C, Mathis D. Pinpointing when T cell costimulatory receptor CTLA-4 must be engaged to dampen diabetogenic T cells. Proc Natl Acad Sci U S A (2000) 97:12204–9. doi:10.1073/pnas.200348397
85. Tai X, Van Laethem F, Sharpe AH, Singer A. Induction of autoimmune disease in CTLA-4-/- mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc Natl Acad Sci U S A (2007) 104:13756–61. doi:10.1073/pnas.0706509104
86. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi:10.1126/science.1202947
87. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol (2002) 3:1097–101. doi:10.1038/ni846
88. Carreno BM, Bennett F, Chau TA, Ling V, Luxenberg D, Jussif J, et al. CTLA-4 (CD152) can inhibit T cell activation by two different mechanisms depending on its level of cell surface expression. J Immunol (2000) 165:1352–6. doi:10.4049/jimmunol.165.3.1352
89. Chikuma S, Imboden JB, Bluestone JA. Negative regulation of T cell receptor-lipid raft interaction by cytotoxic T lymphocyte-associated antigen 4. J Exp Med (2003) 197:129–35. doi:10.1084/jem.20021646
90. Chuang E, Lee KM, Robbins MD, Duerr JM, Alegre ML, Hambor JE, et al. Regulation of cytotoxic T lymphocyte-associated molecule-4 by Src kinases. J Immunol (1999) 162:1270–7.
91. Schneider H, Schwartzberg PL, Rudd CE. Resting lymphocyte kinase (Rlk/Txk) phosphorylates the YVKM motif and regulates PI 3-kinase binding to T-cell antigen CTLA-4. Biochem Biophys Res Commun (1998) 252:14–9. doi:10.1006/bbrc.1998.9559
92. Miyatake S, Nakaseko C, Umemori H, Yamamoto T, Saito T. Src family tyrosine kinases associate with and phosphorylate CTLA-4 (CD152). Biochem Biophys Res Commun (1998) 249:444–8. doi:10.1006/bbrc.1998.9191
93. Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, et al. Molecular basis of T cell inactivation by CTLA-4. Science (1998) 282:2263–6. doi:10.1126/science.282.5397.2263
94. Marengère LE, Waterhouse P, Duncan GS, Mittrücker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science (1996) 272:1170–3. doi:10.1126/science.272.5265.1170
95. Schneider H, Smith X, Liu H, Bismuth G, Rudd CE. CTLA-4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur J Immunol (2008) 38:40–7. doi:10.1002/eji.200737423
96. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313:1972–5. doi:10.1126/science.1131078
97. Baroja ML, Vijayakrishnan L, Bettelli E, Darlington PJ, Chau TA, Ling V, et al. Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J Immunol (2002) 168:5070–8. doi:10.4049/jimmunol.168.10.5070
98. Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, et al. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity (2000) 13:313–22. doi:10.1016/S1074-7613(00)00031-5
99. Olsson C, Riesbeck K, Dohlsten M, Michaëlsson E. CTLA-4 ligation suppresses CD28-induced NF-kappaB and AP-1 activity in mouse T cell blasts. J Biol Chem (1999) 274:14400–5. doi:10.1074/jbc.274.20.14400
100. Fraser JH, Rincón M, McCoy KD, Le Gros G. CTLA4 ligation attenuates AP-1, NFAT and NF-kappaB activity in activated T cells. Eur J Immunol (1999) 29:838–44. doi:10.1002/(SICI)1521-4141(199903)29:03<838::AID-IMMU838>3.0.CO;2-P
101. Leung HT, Bradshaw J, Cleaveland JS, Linsley PS. Cytotoxic T lymphocyte-associated molecule-4, a high-avidity receptor for CD80 and CD86, contains an intracellular localization motif in its cytoplasmic tail. J Biol Chem (1995) 270:25107–14. doi:10.1074/jbc.270.42.25107
102. Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity (1996) 4:535–43. doi:10.1016/S1074-7613(00)80480-X
103. Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol (1996) 157:4762–70.
104. Chuang E, Alegre ML, Duckett CS, Noel PJ, Vander Heiden MG, Thompson CB. Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J Immunol (1997) 159:144–51.
105. Zhang Y, Allison JP. Interaction of CTLA-4 with AP50, a clathrin-coated pit adaptor protein. Proc Natl Acad Sci U S A (1997) 94:9273–8. doi:10.1073/pnas.94.17.9273
106. Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS, et al. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity (1997) 6:583–9. doi:10.1016/S1074-7613(00)80346-5
107. Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity (2002) 16:23–35. doi:10.1016/S1074-7613(01)00259-X
108. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515:577–81. doi:10.1038/nature13988
109. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11:3887–95.
110. Nishimura H, Agata Y, Kawasaki A, Sato M, Imamura S, Minato N, et al. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int Immunol (1996) 8:773–80. doi:10.1093/intimm/8.5.773
111. Mathieu M, Cotta-Grand N, Daudelin JF, Thébault P, Labrecque N. Notch signaling regulates PD-1 expression during CD8(+) T-cell activation. Immunol Cell Biol (2013) 91:82–8. doi:10.1038/icb.2012.53
112. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity (2014) 41:802–14. doi:10.1016/j.immuni.2014.10.013
113. Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol (2008) 181:4832–9. doi:10.4049/jimmunol.181.7.4832
115. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A (2008) 105:15016–21. doi:10.1073/pnas.0801497105
116. Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol (2013) 14:152–61. doi:10.1038/ni.2496
117. Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med (2006) 203:2281–92. doi:10.1084/jem.20061496
118. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol (2007) 19:813–24. doi:10.1093/intimm/dxm057
119. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol (2004) 173:945–54. doi:10.4049/jimmunol.173.2.945
120. Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol (2013) 33:3091–8. doi:10.1128/MCB.00319-13
121. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
122. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem (1998) 273:13375–8. doi:10.1074/jbc.273.22.13375
123. Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A (1999) 96:2110–5. doi:10.1073/pnas.96.5.2110
124. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal (2012) 5:ra46. doi:10.1126/scisignal.2002796
125. Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science (1998) 280:1082–6. doi:10.1126/science.280.5366.1082
126. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi:10.1038/ncomms7692
127. Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med (2008) 205:565–74. doi:10.1084/jem.20071477
128. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med (1992) 176:327–37. doi:10.1084/jem.176.2.327
129. Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol (1995) 25:2718–21. doi:10.1002/eji.1830250949
130. Goldberg MV, Drake CG. LAG-3 in cancer immunotherapy. Curr Top Microbiol Immunol (2011) 344:269–78. doi:10.1007/82_2010_114
131. Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol (2004) 172:5450–5. doi:10.4049/jimmunol.172.9.5450
132. Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol (2002) 169:5392–5. doi:10.4049/jimmunol.169.10.5392
133. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest (2007) 117:3383–92. doi:10.1172/JCI31184
134. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21:503–13. doi:10.1016/j.immuni.2004.08.010
135. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol (2008) 180:5916–26. doi:10.4049/jimmunol.180.9.5916
136. Durham NM, Nirschl CJ, Jackson CM, Elias J, Kochel CM, Anders RA, et al. Lymphocyte activation gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS One (2014) 9:e109080. doi:10.1371/journal.pone.0109080
137. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2011) 72:917–27. doi:10.1158/0008-5472.CAN-11-1620
138. Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J (2007) 26:494–504. doi:10.1038/sj.emboj.7601520
139. Delmastro MM, et al. Modulation of redox balance leaves murine diabetogenic TH1 T cells “LAG-3-ing” behind. Diabetes (2012) 61:1760–8. doi:10.2337/db11-1591
140. Previte D, Delmastro-Greenwood M, Piganelli J. Lymphocyte activation gene 3 (LAG-3) regulates activation-induced CD4+ T cell metabolic transition (IRM6P. 725) [abstract]. J Immunol (2014) 192(Suppl 1):63.17.
141. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415:536–41. doi:10.1038/415536a
142. Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The TIM gene family regulates autoimmune and allergic diseases. Trends Mol Med (2005) 11:362–9. doi:10.1016/j.molmed.2005.06.008
143. Sánchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol (2003) 4:1093–101. doi:10.1038/ni987
144. Sabatos CA, Chakravarti S, Cha E, Schubart A, Sánchez-Fueyo A, Zheng XX, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol (2003) 4:1102–10. doi:10.1038/ni988
145. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6:1245–52. doi:10.1038/ni1271
146. Cao E, Zang X, Ramagopal UA, Mukhopadhaya A, Fedorov A, Fedorov E, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity (2007) 26:311–21. doi:10.1016/j.immuni.2007.01.016
147. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517:386–90. doi:10.1038/nature13848
148. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol (2010) 184:1918–30. doi:10.4049/jimmunol.0903059
149. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol (2011) 31:3963–74. doi:10.1128/MCB.05297-11
150. van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun (2006) 351:571–6. doi:10.1016/j.bbrc.2006.10.079
151. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med (2012) 18:1394–400. doi:10.1038/nm.2871
152. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med (2008) 205:2763–79. doi:10.1084/jem.20081398
153. Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol (2009) 83:9122–30. doi:10.1128/JVI.00639-09
154. McMahan RH, Golden-Mason L, Nishimura MI, McMahon BJ, Kemper M, Allen TM, et al. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J Clin Invest (2010) 120:4546–57. doi:10.1172/JCI43127
155. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A (2010) 107:14733–8. doi:10.1073/pnas.1009731107
156. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207:2187–94. doi:10.1084/jem.20100643
157. Ferris RL, Lu B, Kane LP. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J Immunol (2014) 193:1525–30. doi:10.4049/jimmunol.1400557
158. Nebbia G, Peppa D, Schurich A, Khanna P, Singh HD, Cheng Y, et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS One (2012) 7:e47648. doi:10.1371/journal.pone.0047648
159. Yang ZZ, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, et al. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest (2012) 122:1271–82. doi:10.1172/JCI59806
160. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One (2012) 7:e30676. doi:10.1371/journal.pone.0030676
161. Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207:2175–86. doi:10.1084/jem.20100637
162. Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L, et al. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc Natl Acad Sci U S A (2012) 109:5399–404. doi:10.1073/pnas.1120606109
163. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol (2011) 41:902–15. doi:10.1002/eji.201041136
164. Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol (2015) 15:243–54. doi:10.1038/nri3799
165. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10:48–57. doi:10.1038/ni.1674
166. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol (2011) 186:1338–42. doi:10.4049/jimmunol.1003081
167. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40:569–81. doi:10.1016/j.immuni.2014.02.012
168. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest (2015) 125:2046–58. doi:10.1172/JCI80445
169. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell (2014) 26:923–37. doi:10.1016/j.ccell.2014.10.018
170. Maïza H, Leca G, Mansur IG, Schiavon V, Boumsell L, Bensussan A. A novel 80-kD cell surface structure identifies human circulating lymphocytes with natural killer activity. J Exp Med (1993) 178:1121–6. doi:10.1084/jem.178.3.1121
171. Anumanthan A, Bensussan A, Boumsell L, Christ AD, Blumberg RS, Voss SD, et al. Cloning of BY55, a novel Ig superfamily member expressed on NK cells, CTL, and intestinal intraepithelial lymphocytes. J Immunol (1998) 161:2780–90.
172. Agrawal S, Marquet J, Freeman GJ, Tawab A, Bouteiller PL, Roth P, et al. Cutting edge: MHC class I triggering by a novel cell surface ligand costimulates proliferation of activated human T cells. J Immunol (1999) 162:1223–6.
173. Bensussan A, Rabian C, Schiavon V, Bengoufa D, Leca G, Boumsell L. Significant enlargement of a specific subset of CD3+CD8+ peripheral blood leukocytes mediating cytotoxic T-lymphocyte activity during human immunodeficiency virus infection. Proc Natl Acad Sci U S A (1993) 90:9427–30. doi:10.1073/pnas.90.20.9427
174. Han P, Goularte OD, Rufner K, Wilkinson B, Kaye J. An inhibitory Ig superfamily protein expressed by lymphocytes and APCs is also an early marker of thymocyte positive selection. J Immunol (2004) 172:5931–9. doi:10.4049/jimmunol.172.10.5931
175. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol (2003) 4:670–9. doi:10.1038/ni944
176. Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory receptor expression depends more dominantly on differentiation and activation than “exhaustion” of human CD8 T cells. Front Immunol (2013) 4:455. doi:10.3389/fimmu.2013.00455
177. Hurchla MA, Sedy JR, Gavrieli M, Drake CG, Murphy TL, Murphy KM. B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly Induced in anergic CD4+ T cells. J Immunol (2005) 174:3377–85. doi:10.4049/jimmunol.174.9.5884a
178. Oya Y, Watanabe N, Owada T, Oki M, Hirose K, Suto A, et al. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheum (2008) 58:2498–510. doi:10.1002/art.23674
179. Liu X, Alexiou M, Martin-Orozco N, Chung Y, Nurieva RI, Ma L, et al. Cutting edge: a critical role of B and T lymphocyte attenuator in peripheral T cell tolerance induction. J Immunol (2009) 182:4516–20. doi:10.4049/jimmunol.0803161
180. Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol (2005) 6:90–8. doi:10.1038/ni1144
181. Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol (2008) 9:176–85. doi:10.1038/ni1554
182. Ware CF. Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol Rev (2008) 223:186–201. doi:10.1111/j.1600-065X.2008.00629.x
183. Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell (1996) 87:427–36. doi:10.1016/S0092-8674(00)81363-X
184. Morel Y, Truneh A, Sweet RW, Olive D, Costello RT. The TNF superfamily members LIGHT and CD154 (CD40 ligand) costimulate induction of dendritic cell maturation and elicit specific CTL activity. J Immunol (2001) 167:2479–86. doi:10.4049/jimmunol.167.5.2479
185. Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem Biophys Res Commun (2003) 312:1236–43. doi:10.1016/j.bbrc.2003.11.070
186. Nikolova M, Marie-Cardine A, Boumsell L, Bensussan A. BY55/CD160 acts as a co-receptor in TCR signal transduction of a human circulating cytotoxic effector T lymphocyte subset lacking CD28 expression. Int Immunol (2002) 14:445–51. doi:10.1093/intimm/14.5.445
187. Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol (2008) 38:2438–50. doi:10.1002/eji.200838289
188. Riley JL. PD-1 signaling in primary T cells. Immunol Rev (2009) 229:114–25. doi:10.1111/j.1600-065X.2009.00767.x
189. Wang MC, O’Rourke EJ, Ruvkun G. Fat metabolism links germline stem cells and longevity in C. elegans. Science (2008) 322:957–60. doi:10.1126/science.1162011
190. Saha A, Aoyama K, Taylor PA, Koehn BH, Veenstra RG, Panoskaltsis-Mortari A, et al. Host programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood (2013) 122:3062–73. doi:10.1182/blood-2013-05-500801
191. Kaminski MM, Sauer SW, Klemke CD, Süss D, Okun JG, Krammer PH, et al. Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J Immunol (2010) 184:4827–41. doi:10.4049/jimmunol.0901662
192. Kamiński MM, Sauer SW, Kamiński M, Opp S, Ruppert T, Grigaravičius P, et al. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep (2012) 2:1300–15. doi:10.1016/j.celrep.2012.10.009
193. Tkachev V, Goodell S, Opipari AW, Hao LY, Franchi L, Glick GD, et al. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J Immunol (2015) 194:5789–800. doi:10.4049/jimmunol.1402180
194. St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell (2006) 127:397–408. doi:10.1016/j.cell.2006.09.024
195. Bhalla K, Hwang BJ, Dewi RE, Ou L, Twaddel W, Fang HB, et al. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res (2011) 71:6888–98. doi:10.1158/0008-5472.CAN-11-1011
196. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature (2007) 450:736–40. doi:10.1038/nature06322
197. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity (2016) 45:358–73. doi:10.1016/j.immuni.2016.07.008
198. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey R, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity (2016) 45:701–3. doi:10.1016/j.immuni.2016.08.009
199. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med (2015) 212:1125–37. doi:10.1084/jem.20142237
200. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2:261–8. doi:10.1038/85330
201. Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci U S A (2013) 110:E2480–9. doi:10.1073/pnas.1305394110
202. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity (2015) 43:435–49. doi:10.1016/j.immuni.2015.09.001
203. Qorraj M, Bruns H, Böttcher M, Weigand L, Saul D, Mackensen A, et al. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia (2017) 31(2):470–8. doi:10.1038/leu.2016.214
204. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi:10.1016/j.cell.2015.08.016
205. Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood (2008) 111:3635–43. doi:10.1182/blood-2007-11-123141
206. Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell (2015) 162:1242–56. doi:10.1016/j.cell.2015.08.052
207. Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature (2008) 451:345–9. doi:10.1038/nature06489
Keywords: T cells, costimulation, coinhibitory molecules, metabolism and bioenergetics, T cell differentiation
Citation: Patsoukis N, Weaver JD, Strauss L, Herbel C, Seth P and Boussiotis VA (2017) Immunometabolic Regulations Mediated by Coinhibitory Receptors and Their Impact on T Cell Immune Responses. Front. Immunol. 8:330. doi: 10.3389/fimmu.2017.00330
Received: 23 January 2017; Accepted: 08 March 2017;
Published: 11 April 2017
Edited by:
Christoph Hess, University of Basel, SwitzerlandReviewed by:
Bernd Lepenies, University of Veterinary Medicine, GermanyCopyright: © 2017 Patsoukis, Weaver, Strauss, Herbel, Seth and Boussiotis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vassiliki A. Boussiotis, dmJvdXNzaW9AYmlkbWMuaGFydmFyZC5lZHU=
†First coauthors listed alphabetically.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.