
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol., 27 March 2017
Sec. T Cell Biology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00316
This article is part of the Research TopicOn the Origin and Function of Human NK-like CD8+ T Cells: Charting New TerritoriesView all 14 articles
 Alice Barbarin1,2†
Alice Barbarin1,2† Emilie Cayssials1,2,3,4†Florence Jacomet1,2,4,5Nicolas Gonzalo Nunez6,7,8Sara Basbous1,4Lucie Lefèvre1
Emilie Cayssials1,2,3,4†Florence Jacomet1,2,4,5Nicolas Gonzalo Nunez6,7,8Sara Basbous1,4Lucie Lefèvre1 Myriam Abdallah1,2Nathalie Piccirilli1,2Benjamin Morin1Vincent Lavoue9,10Véronique Catros10,11,12Eliane Piaggio6,7,8
Myriam Abdallah1,2Nathalie Piccirilli1,2Benjamin Morin1Vincent Lavoue9,10Véronique Catros10,11,12Eliane Piaggio6,7,8 André Herbelin1,2,4*‡
André Herbelin1,2,4*‡ Jean-Marc Gombert1,2,4,5*‡
Jean-Marc Gombert1,2,4,5*‡
Unconventional T cells are defined by their capacity to respond to signals other than the well-known complex of peptides and major histocompatibility complex proteins. Among the burgeoning family of unconventional T cells, innate-like CD8(+) T cells in the mouse were discovered in the early 2000s. This subset of CD8(+) T cells bears a memory phenotype without having encountered a foreign antigen and can respond to innate-like IL-12 + IL-18 stimulation. Although the concept of innate memory CD8(+) T cells is now well established in mice, whether an equivalent memory NK-like T-cell population exists in humans remains under debate. We recently reported that CD8(+) T cells responding to innate-like IL-12 + IL-18 stimulation and co-expressing the transcription factor Eomesodermin (Eomes) and KIR/NKG2A membrane receptors with a memory/EMRA phenotype may represent a new, functionally distinct innate T cell subset in humans. In this review, after a summary on the known innate CD8(+) T-cell features in the mouse, we propose Eomes together with KIR/NKG2A and CD49d as a signature to standardize the identification of this innate CD8(+) T-cell subset in humans. Next, we discuss IL-4 and IL-15 involvement in the generation of innate CD8(+) T cells and particularly its possible dependency on the promyelocytic leukemia zinc-finger factor expressing iNKT cells, an innate T cell subset well documented for its susceptibility to tumor immune subversion. After that, focusing on cancer diseases, we provide new insights into the potential role of these innate CD8(+) T cells in a physiopathological context in humans. Based on empirical data obtained in cases of chronic myeloid leukemia, a myeloproliferative syndrome controlled by the immune system, and in solid tumors, we observe both the possible contribution of innate CD8(+) T cells to cancer disease control and their susceptibility to tumor immune subversion. Finally, we note that during tumor progression, innate CD8(+) T lymphocytes could be controlled by immune checkpoints. This study significantly contributes to understanding of the role of NK-like CD8(+) T cells and raises the question of the possible involvement of an iNKT/innate CD8(+) T cell axis in cancer.
The traditional view of the immune system distinguishes innate immunity from adaptive or acquired immunity. Innate immunity is derived from cells expressing the receptors specific for molecules from microbial pathogens called pathogen-associated molecular patterns or self-molecules from the healthy or unhealthy individual called damage-associated molecular patterns. One of the key characteristics of innate immunity effector mechanisms is their capacity for very rapid response to pro-inflammatory cytokines such as IL-12, IL-18, and IL-33.
The effectors of adaptive immunity possess receptors characterized by highly diverse and specific antigens. A major feature of adaptive immunity consists in its serving as an essential support for the immunologic memory, which means that it can remember and quite effectively respond to an antigen long after having encountered it for the first time.
However, numerous works over the past 20 years have shown the distinction between innate immunity and adaptive immunity to be less clear-cut and more tenuous than it first appeared. This revised perception is based, in particular, on description of non-conventional T cells responding to stimuli that had previously been considered as being recognized solely by innate cells. These populations of T lymphocytes include not only certain T-cell receptor (TCR)-γδ cells but also TCR-αβ cells such as natural killer T (iNKT) cells and innate mucosal-associated invariant T (MAIT) cells [for a list of the different cells, see Ref. (1, 2)].
A new contingent of innate T cells was described in the early 2000s in the mouse, partially in the thymus, where they were termed «innate memory» (IM) CD8(+) T cells, and partially in the spleen, where they were termed «virtual memory» (VM) CD8(+) T cells (3, 4). Aside from possessing a phenotype of activated memory cells, one characteristic of these cells consists in their differentiating into memory cells independently of a foreign antigen. In parallel, CD8(+) T cells in humans were described as cells possessing innate characteristics including NK markers. They were found in human cord blood, a finding consistent with the hypothesis that their development does not depend on foreign antigens. These cells hence were termed NK-like CD8(+) T cells.
At the outset of this review, we shall compare the human NK-like CD8(+) T cells with IM/VM CD8(+) T cells in mice. On the basis of this comparison and with regard to humans, we shall focus first on expression of the transcription factor Eomesodermin (Eomes) as a lineage marker of that population of cells, and then on their innate functions (cytotoxicity and TCR-independent IFN-γ expression), along with their memory phenotype, and on the roles of IL-4- and promyelocytic leukemia zinc-finger factor (PLZF)-expressing T cells in differentiation of these cells, hereafter referred to as innate CD8(+) T cells. We shall discuss the use of membrane markers, particularly the α4-integrin CD49d, in order to obtain a more well-defined phenotype correlating with their functions and/or explaining their possible physiological role. Finally, we shall discuss the implication of innate CD8(+) T cells in anticancer immunity in humans.
Studies conducted shortly after 2000 by Forman et al. (5, 6) demonstrated the existence of CD8(+) T cells producing IFN-γ in response to innate signals occurring independently from the TCR. These CD8(+) T cells possessed a CD44(+) CD62L(−) CD122(+) memory phenotype and, in vivo, provided protection against Listeria monocytogenes (LM) infection (5–7). Their mobilization depended on the production of IL-12 and IL-18. Interestingly, the Forman team subsequently showed that this cell population was present in the thymus of C57BL/6 wild-type mice and that it was enriched in C57BL/6 H-2 Kb−/−Db−/− mice not having undergone stimulation by foreign antigens (7).
A second series of studies having to do with Itk−/− (inducing T cell kinase), Rlk−/− (resting lymphocyte kinase), or Itk−/−Rlk−/− mice led to identification of a population of thymic CD8(+) T cells expressing an activated memory [CD44(+) CD62L(−) CD122(+)] phenotype and called IM CD8(+) T cells (8–12). Interestingly, these cells developed in the thymus and are exported to the spleen and the lymph nodes (LNs) where they could fulfill an anti-infective function against LM, particularly through production of IFN-γ following TCR-independent stimulation by IL-12 and IL-18. An important point in the studies dedicated to this population has been the demonstration that its differentiation depended on expression of the Eomes transcription factor and IL-15 (13–15). Eomes expression initiates the differentiation program of these cells and induces the expression of CD122 (the β chain of the IL-2 and IL-15 receptor). IL-15 has a critical role in the expansion of IM CD8(+) T cells (8, 16). It should also be noted that this population is present in Itk−/−Kb−/−Db−/− mice, a finding suggesting that at least some IM CD8(+) T cells are selected by non-classical major histocompatibility complex (MHC) class I molecules (9, 17–19).
A final series of studies has described in mouse spleens and peripheral lymphoid organs a population of activated [CD122(+)] and memory [CD44(+) CD62L(−)] CD8(+) T lymphocytes of which the differentiation into memory cells occurs independently of any recognition of a foreign antigen. This population consists in the so-called VM CD8(+) T cells. As is the case with IM T CD8(+) cells, Eomes and IL-15 are the two key factors in their differentiation. This population is capable of producing IFN-γ in response to innate stimulation by IL-12 + IL-18 (11, 20–22).
The expression by mouse thymic IM CD8(+) T cells of some integrins, such as CD49d [an α4-integrin, or VLA-α4, which is most often matched with a β7-integrin (which is bound to Madcam and VCAM-1 or CD106), or a β1-integrin], has been described (14, 21, 23, 24). Hence, CD49d is used to discriminate between IM and VM T cells (Figure 1A) arising from the thymus or the spleen, respectively. In this model, a possible filiation link between IM CD8(+) and VM CD8(+) T-cell populations remains to be investigated. A gating strategy to identify IM/VM CD8(+) T cells in mice, taking Eomes, CD44, and CD122 together with CD49d as delineating markers is depicted in Figure 1B and Figure S1 in Supplementary Material. Based on this gating strategy, in the C57BL/6 mouse strain, the vast majority of memory [CD44(+) Eomes(+) CD122(+)] CD8(+) T cells are IM CD8(+) T cells in the thymus vs. VM CD8(+) T cells in the spleen, as attested by their differential CD49d expression. Interestingly, in the BALB/c strain, thymic IM CD8(+) T cells are minority cells among memory CD44(+) CD122(+) Eomes(+) cells, raising the question of a possible link between IM and VM CD8(+) T cells, as well as the association of CD49d with the innate functions of IM/VM CD8(+) T cells (see CD49d, a Functional Marker of Innate CD8(+) T Cells in Humans).
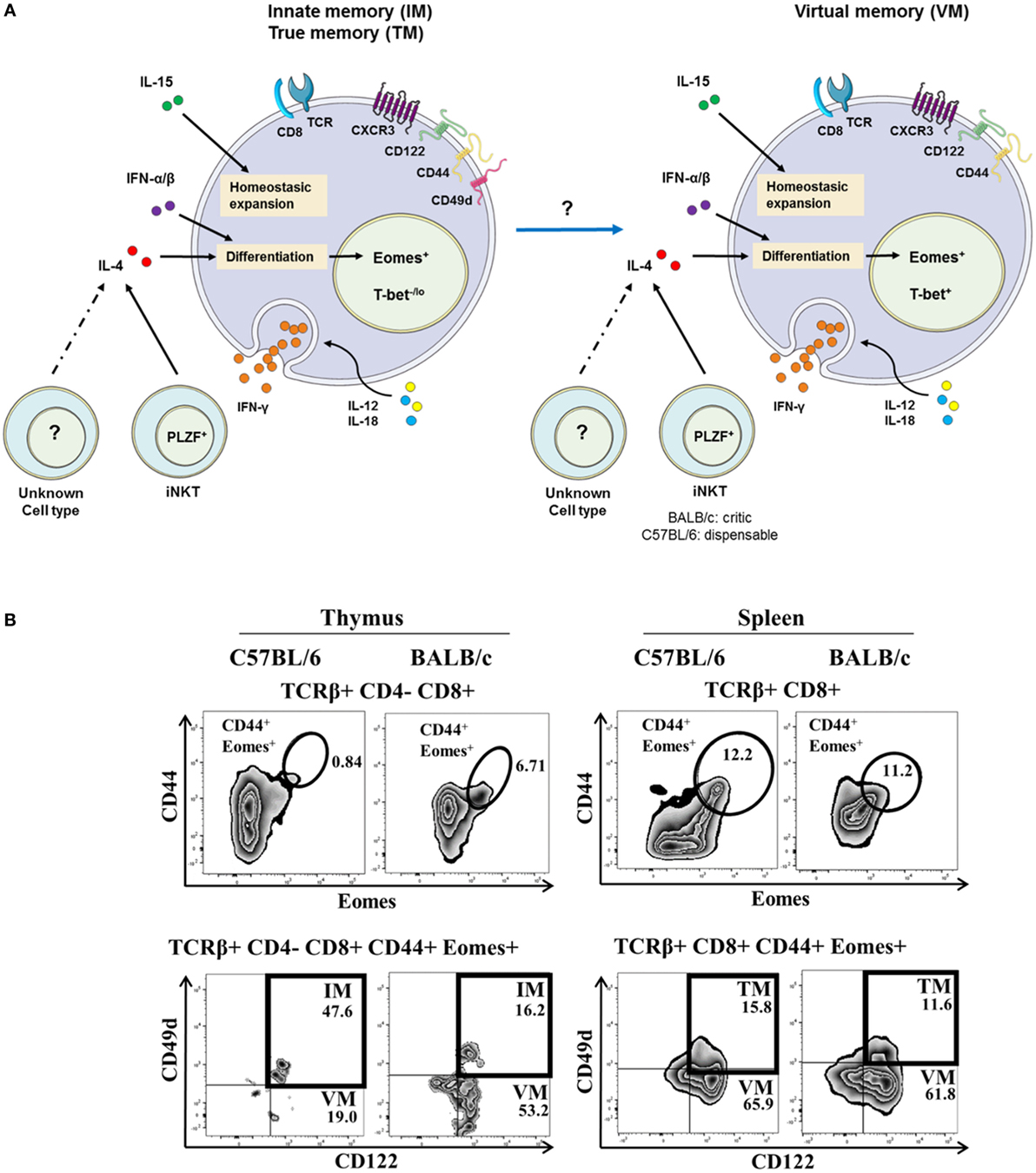
Figure 1. The innate/true memory (IM/TM) and virtual memory (VM) CD8(+) T cells in mice. (A) Representative view of IM/TM and VM CD8(+) T cells in mice. CXCR3(+) CD122(+) CD44(+) CD49d(+) Eomes(+) and CXCR3(+) CD122(+) CD44(+) Eomes(+) T-bet(+) expression defined the IM/TM CD8(+) and the VM CD8(+) T-cell subsets, respectively. Both subsets have the capacity to respond to the IL-12 + IL-18 innate-like stimulation, and their development and homeostasis are regulated by IL-4 [mainly produced by PLZF(+) iNKT cells], IL-15, and type I interferons. Note that possible existence of a filiation link in mice between IM CD8(+) and VM CD8(+) T-cell populations remains to be investigated. (B) Gating strategy in C57BL/6 and BALB/c mouse strains for IM and VM CD8(+) T cells in the thymus (left panel) and for TM and VM CD8(+) T cells in the spleen (right panel). IM/TM and VM population frequencies are displayed in the upper and lower right quadrants, respectively.
IL-15 plays a key role in the homeostatic expansion of CD8(+) T cells, and it has been reported in several studies that this cytokine is implicated in differentiation into VM CD8(+) T cells after homeostatic proliferation (4, 25–28). However, August and his team have shown that the size of the IM/VM CD8(+) T population is not modified by T-cell depletion prior to bone marrow transplantation, a finding suggesting that lymphoid precursors are differentiated into IM/VM CD8(+) T cells following “tuning” by cells expressing MHC class I molecules. Moreover, in the same study, IM/VM CD8(+) T cells are distinguished from homeostatic proliferation CD8(+) T cells by a different transcriptional profile (29). Another study suggests that contrary to naive CD8(+) T cells and homeostatic proliferation CD8(+) T cells, acquisition of the phenotype of IM CD8(+) T cells necessitates engagement of their TCR (30).
In parallel with the previously described work on mice, several studies conducted in the early 2000s demonstrated the existence in humans of CD8(+) T cell-expressing markers and receptors of NK cells including CD56, KIR, NKG2A and NKG2C (CD159a and c), and CD94 (31–36). Several studies precisely characterized the phenotype of the KIR(+) CD8(+) T cells and showed that they possess an EMRA memory phenotype [CD45RA(+) CCR7(−) CD57(+)] (33, 34). Finally, Björkström et al. (33), showed that EMRA(+) KIR(+) CD8(+) T cells have a skewed repertoire using fewer different Vβ than their EMRA(+) KIR(−) CD8(+) T cell counterpart, a finding suggesting the role of antigenic pressure in the acquisition of this phenotype. An equivalent to this population of KIR/NKG2(+) CD8(+) T cells is present in human cord blood, where they possess an EMRA memory phenotype and rapidly express IFN-γ following TCR stimulation (36). In fact, this is a population of T cells that has been educated and has differentiated in the absence of foreign antigenic stimulation, into terminal effector memory T cells.
These KIR(+) CD8(+) T cells have a weaker response to TCR stimulation than their KIR(−) CD8(+) counterparts with regard to the expression of IFN-γ and TNF-α or to the degranulation evaluated by CD107a staining. The KIR(+) CD8(+) T cells expressing two different KIRs have a weaker response to TCR stimulation than cells expressing a single KIR or without KIR (33, 34). Remarkably, this CD56(+) [or KIR(+)] CD8(+) T cell subset responds quite effectively to innate stimuli, one example being the association of IL-12 with IL-18 (37). The same team reported that loss of this function in IL-12R−/− patients is associated with a risk of severe infections from intracellular germs, particularly mycobacteria and Salmonella (37).
The study by Jacomet et al. (38) showed that KIR(+) and/or NKG2A(+) CD8(+) T cells preferentially express Eomes. These KIR/NKG2A(+) Eomes(+) CD8(+) T cells have a memory phenotype and share functional and phenotypic features (4, 38) with IM CD8(+) T cells in mice (8–11). Moreover, with respect to KIR/NKG2A(−) CD8(+) T cells, they possess an EMRA phenotype [CD45RA(+) CCR7(−)] and preferentially express the surface molecule CD57, which is a terminal differentiation marker (Figure 2A). Figure 2B shows a gating strategy designed to analyze these cells in human blood. Remarkably, the majority (around 60–70%) of KIR/NKG2A(+) Eomes(+) CD8(+) T cells express the T-box transcription factor T-bet, a phenotype comparable to the phenotype described for mouse VM CD8(+) T cells (22). These cells are characterized by increased frequency of CD16(+) cells in comparison to conventional memory [KIR/NKG2A(−) Eomes(+)] CD8(+) T cells. Moreover, CD16 expression is substantially increased in KIR/NKG2A(+) Eomes(+) CD8(+) T cells following 48 h of IL-15 in vitro treatment (Figure 3A).
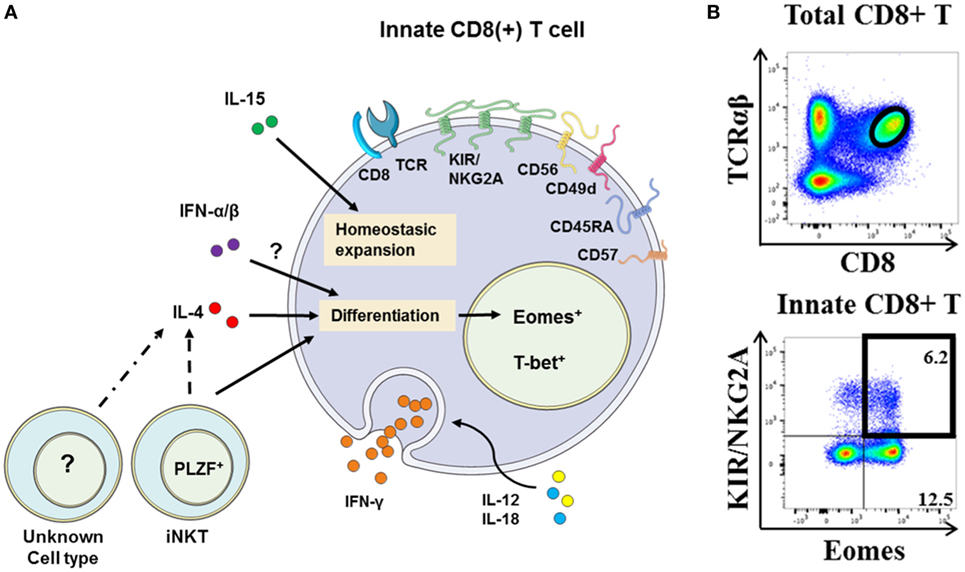
Figure 2. The innate CD8(+) T cells in humans. (A) Representative view of human innate CD8(+) T cells. The membrane markers KIR/NKG2A, CD56, CD57, CD45RA, and CD49d and nuclear transcription factors Eomes and T-bet define this new unique innate CD8(+) T-cell population [for details, see Ref. (4, 38)]. Innate CD8(+) T cells in humans have the capacity to respond to IL-12 + IL-18 innate-like stimulation. Their development and homeostasis are regulated by IL-15, IL-4 [presumably by PLZF(+) iNKT cells], and expectedly type I interferons. Note that the precise origin (central vs. peripheral) of innate CD8(+) T-cell populations in humans remains under debate. (B) Gating strategy for innate CD8(+) T cells among peripheral blood mononuclear cells from healthy donors. After gating on T-cell receptor (TCR)-αβ(+) CD8(+) cells, the innate CD8(+) T cells are defined as KIR/NKG2A(+) Eomes(+). Frequency of innate CD8(+) T cells is displayed in the upper right corner.
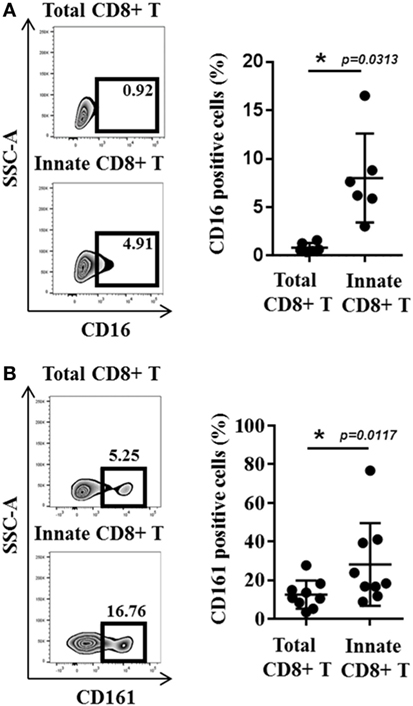
Figure 3. CD16 and CD161 are slightly enriched in innate CD8(+) T cells. (A) Peripheral blood mononuclear cells (PBMCs) from healthy donors (HDs) (n = 6) were cultured for 48 h in the presence of IL-15 and then analyzed by flow cytometry. One representative sample is shown for CD16 expression among total (upper panel) and innate (lower panel) CD8(+) T-cell subsets. Frequencies are displayed in the gate and the histogram (right panel) presents the full cohort of CD16-positive cells (mean ± SD) in both T-cell subsets. (B) PBMCs from HD (n = 9) were analyzed ex vivo by flow cytometry. One representative sample is shown for CD161 expression among total (upper panel) and innate (lower panel) CD8(+) T-cell subsets. Frequencies are displayed in the gates (left panel). The histogram (right panel) represents the full cohort of CD161-positive cells (mean ± SD) in both T-cell subsets.
Together with their marked NK-like phenotype, a hallmark of KIR/NKG2A(+) Eomes(+) CD8(+) T cells is their capacity to rapidly produce large amounts of IFN-γ in response to innate-like stimulation by IL-12 + IL-18 (38). So it is that among the CD8(+) T cells, 60–70% of those possessing a capacity for innate response to IL-12 + IL-18 are KIR/NKG2A(+) Eomes(+) CD8(+) T cells. Moreover, the frequency of the cells producing IFN-γ in response to IL-12 + IL-18 stimulation is four times greater than that of the same cells stimulated with anti-CD3 and anti-CD28 agonistic monoclonal antibodies (mAbs) (38). In the same way, these cells possess a cytotoxic arsenal: perforin and granzyme B. They possess an innate cytotoxic potential induced by an anti-CD16 antibody and revealed by the CD107a test, which assesses degranulation (38). In the final analysis, these data demonstrate that KIR/NKG2A(+) Eomes(+) CD8(+) T cells display innate activity by responding to innate stimuli with response efficacy greater than that of their response to adaptive stimuli (i.e., via their TCR), as has also been shown for innate T-cell populations such as dendritic epidermal T cells (39).
A final element appearing to favor the innate character of KIR/NKG2A(+) Eomes(+) CD8(+) T cells is their differentiation without any exogenous antigenic stimulation, as it is possible to show the existence of these cells in the fetal thymus (40), and as our team has shown them to be present in cord blood. Interestingly, in cord blood, they express an EMRA phenotype with a lower CD57 cell frequency than in the adult, suggesting that there exist supplementary steps in the terminal maturation of these peripheral cells (38). Functionally, as is the case in adult cells, these KIR/NKG2A(+) Eomes(+) CD8(+) T cord blood cells express IFN-γ after innate-like stimulation by IL-12 + IL-18.
All in all, we described for the first time a CD8(+) T cells population with innate features by associating Eomes and KIR/NKG2A markers with the capacity to respond to IL-12 + IL-18 stimulation. These features validate the KIR/NKG2A(+) Eomes(+) CD8(+) T-cell compartment as a new member of the innate T cell family in humans, and we have termed them innate CD8(+) T cells (41).
There exist a number of hypotheses on the mechanisms involved in the reprogramming of conventional T lymphocytes into innate CD8(+) T lymphocytes. One of these assumptions is based on a cross talk between IL-12R and TCR signalosome, in which IL-12 recruits Tyk2 and Fyn tyrosine kinases to activate CD3ζ–TCR signal transduction pathways (42).
Some studies conducted in mice suggest that IM and/or VM CD8(+) T cells could be selected by non-classical MHC class I molecules (9, 17–19). In humans, our results (Figure S2 in Supplementary Material) show that a relatively small fraction of KIR/NKG2A(+) Eomes(+) CD8(+) cells are MAIT cells. This finding suggests that at least some human innate CD8(+) T cells could be selected by non-classical class I MHC molecules, and it raises the possibility of the presence in this population of cells being restricted by non-classical MHC class I HLA-E molecules (18).
We have sought out other markers of innate CD8(+) T cells in humans. Among them, we tested CD56, a marker in humans associated with NK cells, but our results showed that this marker is no more effective in distinguishing the innate CD8(+) T-cell population than the KIR/NKG2A markers. More precisely, this marker delineate only 20–30% of the CD8(+) T cells expressing IFN-γ after innate stimulation by IL-12 + IL-18 (as opposed to the approximately 70% exhibited by the KIR/NKG2A markers) [data not shown; (38)]. We also tested CD161, of which the expression is a common feature of human innate T cell subsets including iNKT cells, TCR-γδ T cells, and MAIT cells (43). However, as for CD56, CD161 is expressed by only approximately 20% of the Eomes(+) KIR/NKG2A(+) CD8(+) T cells (Figure 3B).
Reasoning by analogy with the mouse model, we tested CD49d expression by innate CD8(+) T cells in humans (Figures 4A,B). Interestingly, the CD8(+) T cells with the KIR/NKG2A(+) Eomes(+) phenotype strongly expressed CD49d, as compared to the CD8(+) T-cell population taken as a whole (Figure 4A). Moreover, the majority (almost 70%) of the cells with a more pronounced expression of CD49d consisted in those expressing IFN-γ after innate-like stimulation by IL-12 + IL-18 (Figure 4B). These results show CD49d to be closely associated with the innate effector functions of the innate CD8(+) T cells. On the other hand, CD49d cannot substitute for Eomes and KIR/NKG2A, given the fact that only 20–40% of CD49d(+) cells are actually innate CD8(+) T cells (Figure S3 in Supplementary Material). However, as innate CD8(+) T cells arise from the thymus, the functional link between higher CD49d expression and IFN-γ secretion in response to IL-12 + IL-18 stimulation should be tested in cord blood, which is an accessible source of cells providing an approximate reflection of thymic cells in humans.
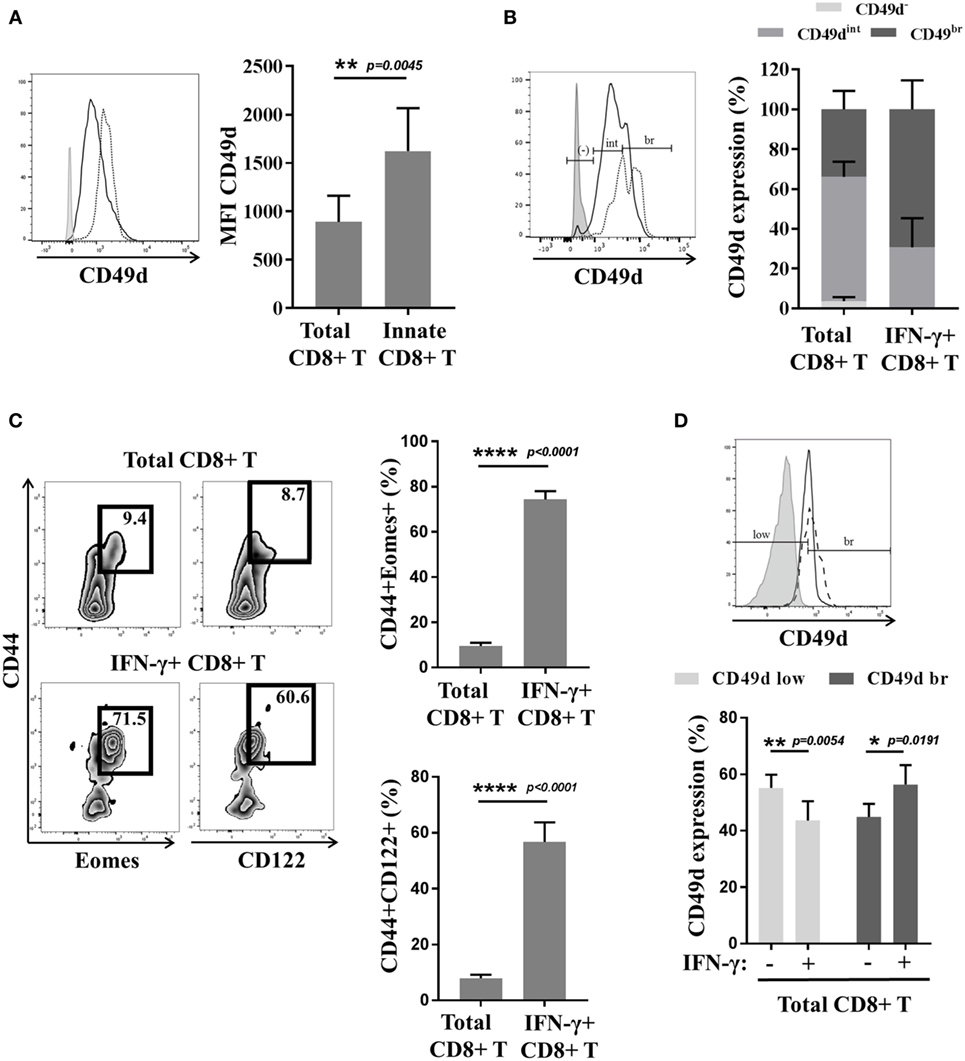
Figure 4. The CD49d molecule correlates with the innate function of innate CD8(+) T cells in mice and humans. (A) Peripheral blood mononuclear cells (PBMCs) from healthy donors (HDs) (n = 6) were analyzed ex vivo by flow cytometry for CD49d expression in total and innate CD8(+) T cells. One representative sample is shown (left): total CD8(+) T cells (solid dark line), innate CD8(+) T cells (dotted black line), and isotype control (gray). Full cohort (MFI ± SD) is shown on the histogram (right). (B) PBMCs from HD (n = 5) were cultured 48 h in the presence of IL-12 + IL-18 and then analyzed ex vivo by flow cytometry for CD49d expression. Distribution of CD49d (percentage ± SD) in total CD8(+) T cells and in IFN-γ(+) CD8(+) T cells is shown on the histogram (right). One representative sample is shown (left): total CD8(+) T cells (solid dark line), IFN-γ(+) CD8(+) T cells (dotted black line), and isotype control (gray). (C,D) Splenocytes from Eomes-GFP mice were isolated, cultured for 16 h in the presence of IL-12 + IL-18, and analyzed by flow cytometry. (C) One representative sample is shown for the presence of CD44(+) Eomes(+) and CD44(+) CD122(+) cells among total CD8(+) (upper left panel) or IFN-γ(+) CD8(+) T cells (lower left panel). Frequency of each subset is displayed in the gate. Full cohort frequencies for CD44(+) Eomes(+) (upper histogram) and CD44(+) CD122(+) (lower histogram) cells among total CD8(+) and IFN-γ(+) CD8(+) T cells are shown. (D) CD49d expression was analyzed in total and IFN-γ(+) CD8(+) T cells. One representative sample is shown (upper panel): total CD8(+) T cells (solid dark line), IFN-γ(+) CD8(+) T cells (dotted black line), and isotype control (gray). Full cohort histograms for frequencies of CD49d(low) and CD49d(bright) are shown (lower panel) in total CD8(+) and IFN-γ(+) CD8(+) T cells.
These results have led us to inquire about the biological meaning of CD49d expression by thymic vs. splenic (or IM vs. VM) CD8(+) T lymphocytes in mice. The results of several teams (14, 21) suggest that contrary to thymic IM CD8(+) cells, splenic (so-called VM) innate CD8(+) T cells do not express CD49d (14, 21) (Figure 1). We have observed, as in humans, that a large majority of splenic CD8(+) T cells expressing IFN-γ after innate-like stimulation by IL-12 + IL-18 were those harboring the IM [Eomes(+) CD44(+) CD122(+)] phenotype (Figure 4C). Moreover, these IFN-γ-producing cells were mostly those that more pronouncedly expressed CD49d as compared to the total CD8(+) T-cell population (Figure 4D).
Taken as a whole, these different results suggest that CD49d is associated with the innate-like functions of NK-like CD8(+) T cells as much in humans as in mice. According to the β chain with which CD49d is matched, the cells are variably liable to migrate toward different territories. While the α4β7-integrin is associated with the migration of T lymphocytes toward the digestive mucosa, the α4β1-integrin is associated with migration toward the oral mucosa, the salivary glands, the vaginal mucosa, and the central nervous system (44, 45). Indeed, therapeutic targeting of CD49d is used in treatment of multiple sclerosis (MS). Several studies have documented the implication of CD49d in the penetration of pathogenic T lymphocytes during MS or experimental allergic encephalomyelitis (EAE) (46, 47). Taken together, these different observations raise the possibility of the implication of the innate CD8(+) T cells in the pathogenesis of MS/EAE.
In mice, differentiation of IM CD8(+) T lymphocytes depends on soluble factors such as type I IFN and the IL-4 and IL-15 cytokines.
Several studies have shown that IL-4 favors the arising of VM CD8(+) T cells (48–51). More recent studies confirm the involvement of IL-4 in IM/VM CD8(+) T cell generation with a more critical role being assumed in the BALB/c (22, 30) than in the C57BL/6 mouse strain (14). IL-4-producing PLZF(+) T cells, including at least partially iNKT lymphocytes (52, 53), elicit the generation of IM CD8(+) T cells. Other results from the Hogquist group confirm the link between iNKT PLZF(+) cells, IL-4, and IM CD8(+) T cell generation by comparing different mouse strains (22). Figure 1B shows the higher frequency of T CD8(+) IM in the thymus of BALB/c in comparison with C57BL/6 mice. However, the August group shows results suggesting that IL-4 produced by differentiating CD4(+) CD8(+) double-positive thymocytes controls the generation of IM CD8(+) thymocytes in the Itk−/− C57BL/6 background (16, 29). As concerns VM CD8(+) T lymphocytes, IL-4 is likewise at least partially implicated. IL-4 is likely to act by eliciting Eomes expression, of which the action would entail a heightened level of expression of CD122, and thereby sensitize the cells to IL-15 (14, 16).
Type I IFNs such as IFN-β and/or IFN-α (24) could favor the differentiation of lymphocytes to VM and/or IM CD8(+) T lymphocytes. The underlying mechanism described by the authors is the induction of Eomes expression by naive CD8(+) T cells.
Finally, IL-15 is a determining factor in the maintenance and/or homeostatic expansion of IM and VM CD8(+) T cells (8, 14, 16).
Few studies have addressed the role of these factors in the development of innate CD8(+) T cells in humans (38, 40). Interestingly, our own results (38) have shown a relation of proportionality between PLZF expression in T and iNKT lymphocytes and Eomes expression by innate CD8(+) T cells in cord blood, suggesting that iNKT cells control the differentiation of human innate CD8(+) T cells. Our results in vitro (41) suggest that IL-4 favors Eomes expression and the generation and/or expansion of human innate CD8(+) T cells.
There exist only sparse data in either humans or mice describing the functions of innate CD8(+) T lymphocytes. An initial set of results consisted in a demonstration in mice of a protective role of IM/VM CD8(+) T lymphocytes against viral (30, 54) and bacterial (11) infections. Our studies have been focused on the numeric/functional status of innate CD8(+) T lymphocytes during the multistep development of human tumors.
Similarly to iNKT cells (55–58), innate CD8(+) T cells fulfill functions providing them with anticancer potential. Hence, a deficiency of these cells in CML (Box 1) on diagnosis is likely to constitute, as with iNKT cells (Box 2), an immune subversion signature. If this is indeed the case, parallel study of iNKT cells and innate CD8(+) T cells in CML both at diagnosis and following molecular remission by tyrosine kinase inhibitor (TKI) therapy could perhaps answer questions concerning a dynamic process of generation of innate CD8(+) T cells in humans that would depend on iNKT cells.
Box 1. Chronic myeloid leukemia (CML), a myeloproliferative syndrome controlled by the immune system.
Chronic myeloid leukemia is the first malignant disorder with a specific genetic abnormality in the background. It is due to the formation of the chimeric oncogene BCR–ABL. This oncogene is responsible for the transformation of hematopoietic stem cells (HSC) into leukemic stem cells, which results in a leukemic syndrome of mature myeloid cells characterizing the chronic phase (CP) and ineluctably evolving, without treatment, to acute leukemia (59). The ABL domain of the chimeric oncogene BCR–ABL presents deregulated tyrosine kinase activity, which is responsible for the transformation of the HSC. Since the outset of the 2000s, new CML treatment has consisted in the tyrosine kinase inhibitors of BCR–ABL. Some arguments suggest that CML is a disease in which the immune system has a key role [for review, see Ref. (60)]. In addition, during the chronic phase of CML, numerous innate anomalies in the innate immune system have been evidenced. Indeed, defective differentiation of the plasmacytoid dendritic cells, defective IFN-α production by mononuclear cells, and defective functions of NK cells have been observed (61, 62).
Box 2. Functional deficiency of iNKT cells in chronic phase of CML (CML-CP) patients.
Our team has shown in CML-CP patients at diagnosis a major defect in the iNKT lymphocyte functions, particularly as concerns their proliferative response to T-cell receptor (TCR) stimulation (63) and their cytotoxic arsenal, with a loss in the expression of perforin and FasL, two elements implicated in cancer immunosurveillance by iNKT cells in mice (57, 64). It must also be emphasized that the iNKT cells of CML-CP patients have lost their expression of the transcription factor promyelocytic leukemia zinc-finger factor and no longer produce IL-4 during TCR engagement; on the other hand, they show normal expression of IFN-γ in comparison with the iNKT lymphocytes of healthy donors or patients in complete remission following treatment by tyrosine kinase inhibitors such as imatinib mesylate (63).
There exists a major defect in the innate CD8(+) T cells of chronic phase of CML (CML-CP) patients compared to those of healthy subjects or patients in complete remission following TKI treatment (41). This numerical defect is associated with a loss of IFN-γ expression after innate-like stimulation by IL-12 + IL-18 cytokines and with a loss of degranulation after stimulation via CD16. On the contrary, IFN-γ expression after TCR stimulation (instead of IL-12 + IL-18 stimulation) by the same innate CD8(+) T cells during CP is conserved, thereby showing that the functional defect affecting our population of interest is innate rather than adaptive. Finally, there exists a partial reconstitution in CML remission patients of the frequency and functions of innate CD8(+) T lymphocytes in terms of IFN-γ expression in response to IL-12 + IL-18 and as regards the displaying of cytotoxic functions after CD16 stimulation.
In CML patients, the numerical and functional status of innate CD8(+) T cells seems closely linked to that of the iNKT cells. In analysis of a cohort of CP patients and those in complete remission, we have observed a correlation between Eomes expression by innate CD8(+) T cells and PLZF expression by iNKT cells (41). This finding underscores the possible pathophysiological significance of IL-4 expression by iNKT cells in CML patients; while deficient during the CP, IL-4 expression is restored in remission (63) and could determine the status of innate CD8(+) T cells during the disease or its treatment.
More generally, a scenario can now be outlined as a possible explanation, during CML, for immune subversion of innate CD8(+) T cells by leukemic cells. Immune subversion could result from dysfunction of the antigen-presenting cells (APCs), particularly leukemia myeloid dendritic cells (DCs) and their environment; they might be considered as responsible for the loss of function of the innate immune cells, including NK and innate CD8(+) T cells. As regards iNKT cells, their loss of function could be due to a loss of CD1d expression by the leukemic APCs expressing the BCR–ABL oncogene [Figure 5; (65)]. The loss of CD1d expression could reprogram iNKT cells by favoring the loss of PLZF expression and, consequently, of IL-4, thereby decreasing their capacity to orient a differentiation of CD8(+) T cells into innate CD8(+) T cells (Figure 5).
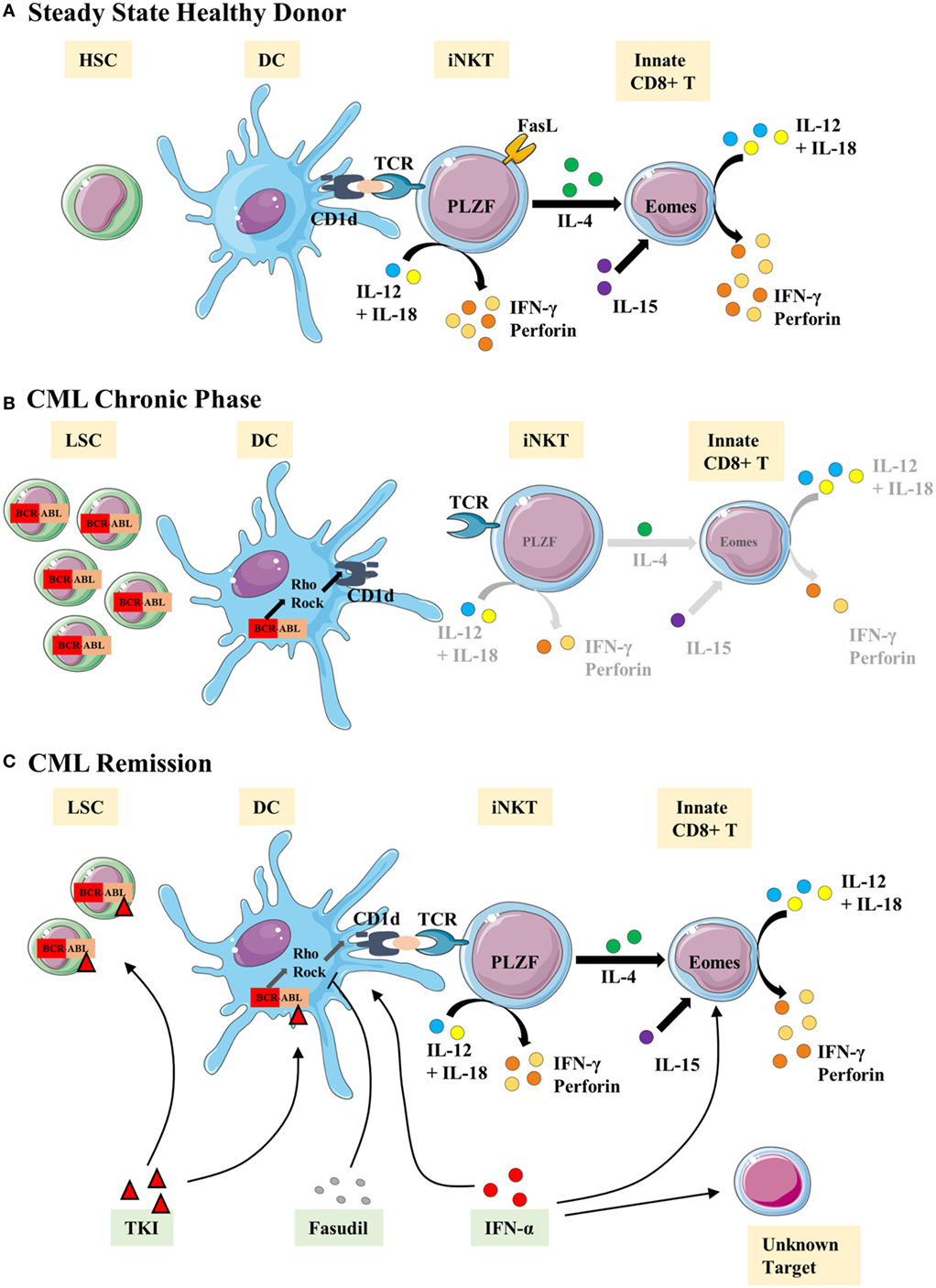
Figure 5. Representative view of the iNKT/innate CD8(+) T-cell axis hypothesis in chronic myeloid leukemia (CML). We propose the following scenario in CML: (A) steady state/healthy situation. Normal hematopoietic stem cells (HSC) generate normal immune cells. Antigens are presented via the CD1d molecule by dendritic cells (DCs) to iNKT cells. We propose that activated iNKT cells produce IL-4 but the possibility of a T-cell receptor (TCR)-independent mechanism for IL-4 secretion cannot be ruled out. IL-4 is thought to take part with IL-15 in the development/homeostasis of innate CD8(+) T cells. iNKT and innate CD8(+) T cells produce IFN-γ and perforin in response to the innate-like IL-12 + IL-18 stimulation. (B) Chronic phase of CML. Leukemic stem cells (LSC) produce modified immune cells bearing BCR–ABL translocation, including DCs. Impaired CD1d antigen presentation by DCs results from activation of the Rho/Rock pathway via the DH-PH domain of the ABL part of BCR–ABL. iNKT cell development/stimulation is thereby impaired, especially in terms of promyelocytic leukemia zinc-finger factor (PLZF) expression and IL-4 production. Consequently, we surmise that the innate CD8(+) T subset is defective in number and function. (C) Restoration of the iNKT/innate CD8(+) T-cell axis by therapies. IFN-α therapy is thought to help restoring DCs and innate CD8(+) T cells as well as other unidentified cells. Tyrosine kinase inhibitor (TKI) therapies targeting the ABL tyrosine kinase domain clear/control the generation of LSC and abnormal immune cells, including DCs. Fasudil therapy, combined with TKI, restores the CD1d presentation by DCs to iNKT cells and is one possible mechanism to restore the iNKT/innate CD8(+) T-cell axis.
Another, non-exclusive hypothesis is that of a loss of IL-15 expression by leukemic cells or of the leukemic milieu, as has been shown in colon cancer (66). A different study leads to the suggestion that during CML, TKI might favor the capacity of the DCs to trans-present IL-15 to T cells/NK cells (67). The scope of these works should be broadened, leading to a search for a defect in the expression and trans-presentation of IL-15 during CML-CP. Another relevant element is the loss in sensitivity to IL-15 or signalization of the latter by the innate CD8(+) T cells. Our preliminary results suggest a loss in response to IL-15 by total and innate CD8(+) T lymphocytes (Figure S4 in Supplementary Material). The possible implication of this loss of sensitivity to IL-15 in the dysregulation of the innate CD8(+) T lymphocytes during CML-CP remains to be investigated.
To conclude, CML can be considered as a model in study of the loss of innate effectors (NK cells) and innate T effectors [iNKT cells and innate CD8(+) T cells] and their relevance in the leukemic process. The escape of leukemic cells from the immune system could depend on loss of the coordinated functions of the effectors of classical innate immunity and innate T-cell immunity.
Since we cannot rule out the possibility that the anticancer role attributed to innate CD8(+) T lymphocytes is specific to leukemia, we have sought to extend the scope of our hypothesis on the antitumor role of the innate CD8(+) T lymphocytes by assessing the presence of these cells in solid tumors or metastasized tissues. The characteristics of tumor micro-environments differ according to cancers, and it is for that reason that we have assessed the presence of innate CD8(+) T lymphocytes in two types of solid tumors: breast cancer and ovarian cancer.
Study of lymph nodes (LN) invaded by tumor cells in breast cancer has highlighted the significantly frequent presence of innate CD8(+) T lymphocytes (Figure 6). The results of this preliminary study seem to justify organization of a large-scale study on breast cancer patients aimed at determining a possible link between, on the one hand, the numerical and functional status of the innate CD8(+) T cells present in the invaded LN draining the tumor and, on the other hand, disease prognosis.
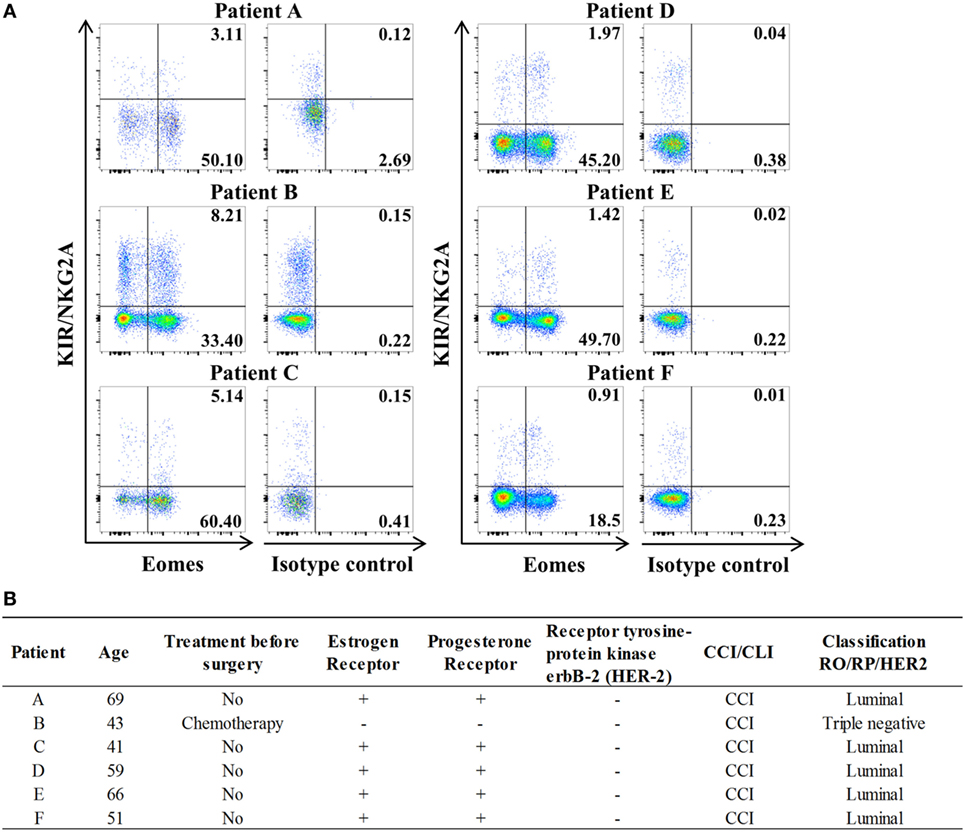
Figure 6. Innate CD8(+) T cells are present in tumor-draining lymph nodes (TDLNs) from breast cancer patients. (A) Frequencies of innate CD8(+) T cells among total CD8(+) T cells in axillary TDLNs were analyzed ex vivo by flow cytometry. The full cohort of five luminal breast cancer patients and one triple negative patient with chemotherapy is shown. Quadrant frequencies are displayed in upper right corners. (B) Clinical patient characteristics. CCI: invasive ductal carcinoma. CLI: invasive lobular carcinoma.
Study of intra-tumoral lymphocytes in ovarian cancer has shown the significantly frequent presence of innate CD8(+) T lymphocytes, which are also present in the peritoneal carcinosis of ovarian cancers and in ascite fluids (Figure 7). Interestingly, there was a significant higher frequency of innate CD8(+) T lymphocytes in primitive tumors than in carcinosis (a proximity metastasis), indicating that these cells not only penetrate tumors in ovarian cancer but also might undergo immune subversion in the peritoneal environment. Moreover, like in CML, our data have shown a positive correlation between Eomes expression in innate CD8(+) T lymphocytes and PLZF expression in iNKT cells both in peripheral blood mononuclear cells (PBMCs) and tumor material (but not in carcinosis and ascite) from ovarian cancer patients (Figure 7C).
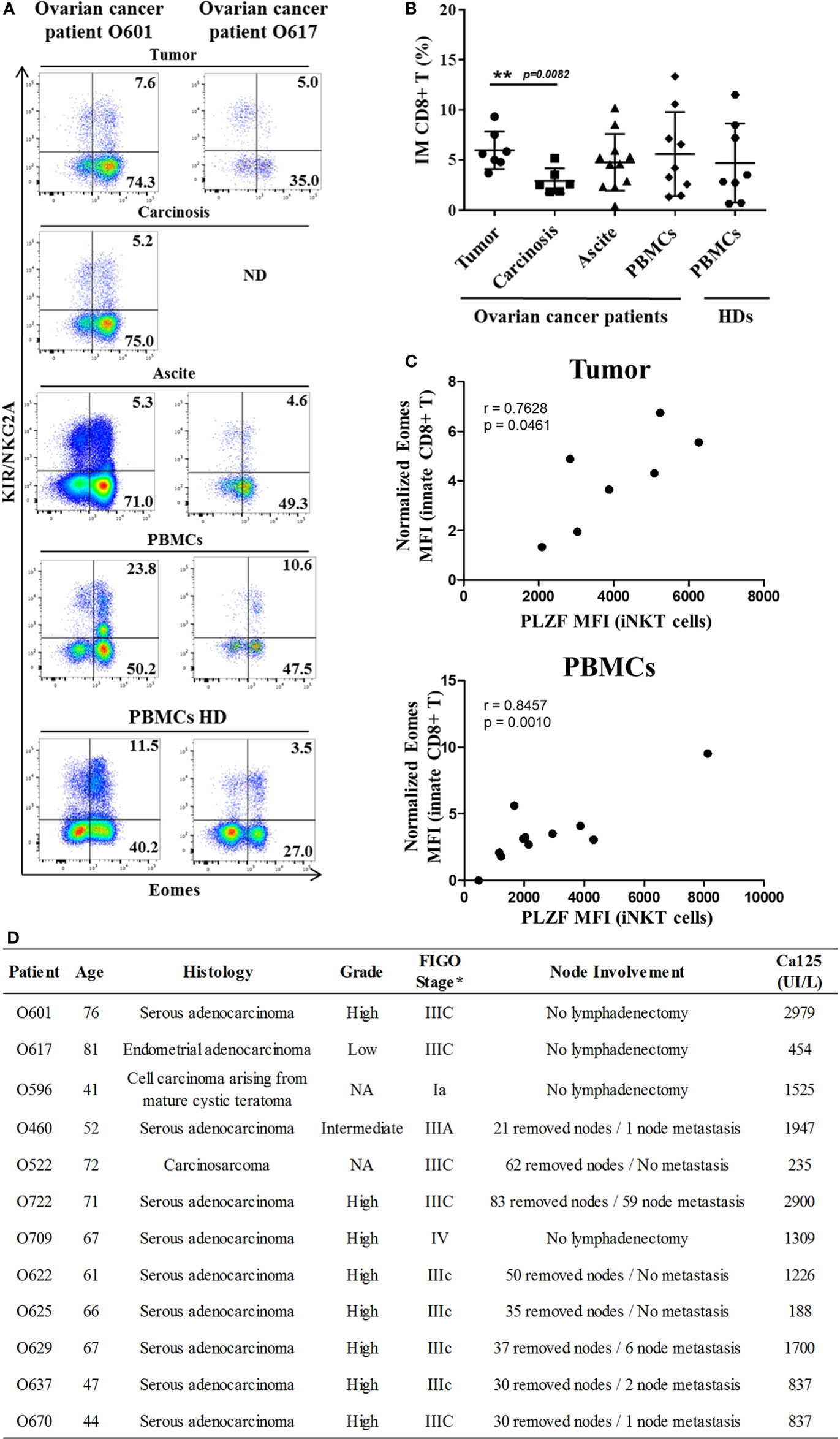
Figure 7. Innate CD8(+) T cells are present in tumors and tumor fluids from ovarian cancer patients. Cells from tumors, carcinoma, peritoneal ascites, and peripheral blood mononuclear cells (PBMCs) from ovarian cancer patients and PBMCs from healthy individuals [healthy donors (HDs)] were analyzed ex vivo by flow cytometry. (A) Two representative samples reflecting the presence of innate CD8(+) T cells in tumor, carcinoma, peritoneal ascites, and PBMCs from ovarian cancer patients and PBMCs from two HDs. ND, not determined. (B) Cohort study of innate CD8(+) T-cell frequency [expressed as percentage ± SD of T cells among total CD8(+) T cells] in tumors (n = 7), carcinoma (n = 7), peritoneal fluids (n = 12), and PBMCs from ovarian cancer patients (n = 10) and PBMCs from HDs (n = 8). (C) Eomes and promyelocytic leukemia zinc-finger factor (PLZF) expression were analyzed in innate CD8(+) T cells [CD3(+) CD8(+) KIR/NKG2A(+) Eomes(+) cells] and iNKT cells [CD3(+) 6B11(+) cells], respectively. Eomes MFI values are expressed relative to that of CD45(+) CD3(−) KIR/NKG2A(−) cells. The MFI of PLZF-expressing iNKT cells correlate positively with Eomes MFI in innate CD8(+) T both in tumor and PBMCs but not in carcinosis (r = −0.04673, p = 0.8853) and ascites (r = 0.5424, p = 0.2084) from ovarian cancer patients (correlation Spearman test). (D) Clinical patient characteristics. NA, not available.
Taken as a whole, these different results show that innate CD8(+) T lymphocytes are present in tumors and probably integrated in the dynamics of anticancer responses. However, at this time, it is not possible to determine their intra-tumoral functional status and, more specifically, their prognostic usefulness.
Numerous studies have dealt with the role of Eomes in the exhaustion of CD8(+) T lymphocytes during chronic infections or cancers (68–72). Our results in healthy donors (HDs) attesting a significant proliferative response to IL-15 (Figure S4 in Supplementary Material) do not lend support to an exhaustion phenotype of innate CD8(+) T cells in normal conditions. During cancer progression, on the other hand, the elevated frequency of innate CD8(+) T lymphocytes in contact with the tumor present in severely ill patients raises the question of their possible exhausted immune-exhaustion status. Furthermore, during CML, we have observed a functional defect in the innate CD8(+) T-cell population, a finding suggesting not only that CML innate CD8(+) T cells are in the process of being exhausted but also that they could be a target of immune blocking checkpoints. Monitoring multifunctionality of immune-exhausted CD8(+) T cells co-expressing Eomes and KIR/NKG2A in patients with solid tumors and CML will help to determine whether the evasion/subversion mechanisms used by tumor cells also apply to innate CD8(+) T cells in humans. At this stage, it would be interesting to determine whether the innate CD8(+) T cells express CXCR3 and CXCR5, two chemokine receptors that were recently described as being associated with CD8(+) T lymphocytes during cancers or chronic viral infection and of which the proliferation is restored following anti-programmed cell death 1 treatment (73).
Taken together, having availed ourselves of different types of empirical data, we propose that in humans, innate CD8(+) T lymphocytes constitute a new cellular component that could have a role in antitumor immunity. Our results during CML and ovarian cancer are in favor of the existence of an axis composed of innate T cells with an antitumoral potential, which consist of iNKT cells and innate CD8(+) T lymphocytes. Differentiation of these unconventional CD8(+) T cells in humans is associated with Eomes expression and could depend on IL-4 and PLZF(+) iNKT cells. Our results suggest that during CML, these innate CD8(+) T lymphocytes could be controlled by immune checkpoints. While the results of our studies on solid cancers corroborate our hypothesis concerning the role of innate CD8(+) T lymphocytes in antitumor immunity, as of now we are unable to determine whether this role is protective, permissive, and/or detrimental in these other types of cancer.
Healthy donors were volunteers from the Pôle Biologie Santé (Poitiers, France). PBMCs were isolated from blood samples by density gradient centrifugation (Histopaque®-1077, Sigma-Aldrich), resuspended in 90% fetal calf serum with 10% DMSO, and placed in a controlled rate freezer for cryopreservation at −80°C until use. For this series of data, age range was between 21 and 65 with a sex ratio of 0.6.
Invaded tumor-draining lymph nodes were collected from six untreated luminal breast cancer patients undergoing standard surgery at Institute Curie Hospital (Paris, France), in accordance with institutional ethical guidelines. Precisely, all patients gave informed consent in a written form in accordance with the Declaration of Helsinki for participation in this study, which was approved by the scientific committee of the Institute Curie Hospital.
Cells from tumors, carcinomatosis, peritoneal ascites, and PBMCs were collected from eight untreated patients with ovarian carcinoma undergoing standard surgery at CHU of Rennes. Human samples were obtained from the processing of biological samples through the Centre de Resources Biologiques Santé of Rennes (BB-0033-00056). The research protocol was conducted under French legal guidelines and fulfilled the requirements of the local institutional ethics committee.
Tissue samples were cut into small fragments, digested with 0.1 mg/ml Liberase TL (Roche) in the presence of 0.1 mg/ml DNase (Roche) for 15–30 min before the addition of 20% FCS. Cells were filtered on a 40-μm cell strainer (BD), washed, and cryopreserved for further study. Ascite cells were obtained after centrifugation (400 g, 10 min).
The 8-to-12-week-old female C57BL/6JRj Eomes-GFP transgenic mice (74) and BALB/c wild-type mice (Janvier) were used and bred in our animal facility (PREBIOS, Platform of Research and Experimentation in Health Biology of the University of Poitiers) under specific pathogen-free conditions. Spleen and thymus were collected immediately after cervical dislocation. Splenocytes and thymocytes were isolated and analyzed ex vivo by flow cytometry. In some experiments, splenocytes were cultured for 16 h in the presence of 20 ng/ml of each cytokine (IL-12: R&D Systems; IL-18: MBL International) prior to analysis by flow cytometry. All procedures were performed in accordance with the recommendations of the European Accreditation of Laboratory Animal Care and French institutional committee of Poitou-Charentes (COMETHEA, C2EA-84, no. 2016072216352833).
Peripheral blood mononuclear cells (1 × 106 cells/ml) were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FCS and antibiotics. For IL-12 + IL-18 or IL-15 stimulation, PBMCs from HD were seeded at 1.106 cells/ml into 24-well plates and incubated for 48 h with 20 ng/ml of each cytokine (IL-12: R&D Systems; IL-18: MBL International; IL-15: Miltenyi). Golgistop (BD Biosciences) was added for the last 5 h of culture for IL-12 + IL-18 stimulation.
In humans, phenotypic analysis of cells from HD or breast/ovarian cancer patients was performed by flow cytometry either ex vivo or after culture. Expression of different markers was assessed by staining with appropriate combinations of the following antibodies (mAbs): anti-TCR-αβ BV421 (clone: IP26, BioLegend), anti-CD8 PE-Cy7 (clone: RPA-T8, Biolegend), anti-IFN-γ FITC (clone: B27, BioLegend), anti-TCR-Vα7.2 BV421 (clone: 3C10, BioLegend), anti-CD161 PerCP-Cy5.5 (clone: HP-3G10, Biolegend), and anti-Eomes eFluor® 660 (clone: WD1928, eBiosciences). KIR/NKG2A referred to staining with the mix of the three following antibodies from Miltenyi Biotech: anti-KIR2D PE (clone: NKVFS1), anti-KIR3DL1/KIR3DL2 (CD158e/k) PE (clone: 5.133), and anti-NKG2A (CD159a) PE (clone: REA110). For nuclear Eomes staining and intracytoplasmic IFN-γ staining, cells were permeabilized with an anti-human FoxP3 staining kit (eBioscience) and a Cytofix/Cytoperm kit (BD Biosciences), respectively.
In mice, splenocytes and thymocytes were stained with appropriate combinations of the following antibodies: anti-TCR-β PerCP-Cy5.5 (clone: H57-597, BD Biosciences), anti-CD8 BV510 (clone: 53-6.7, BD Biosciences), anti-CD44 PE-Cy7 (clone: IM7, BD Biosciences), anti-CD49d Vioblue (clone: R1-2, Miltenyi Biotec), anti-CD122 APC (clone: TM-B1, Biolegend), anti-CD4 PE (clone: RM4-5, BD Biosciences), anti-CD24 PE (clone: M1/69, BD Biosciences), and anti-Eomes AF488 (clone: Dan11mag, eBioscience). For nuclear Eomes staining and intracytoplasmic IFN-γ staining, cells were permeabilized with an anti-human FoxP3 staining kit (eBioscience).
Dead cells were excluded using the Live/Dead® Fixable NearIR Dead Cell Stain kit (Life Technologies). Cells were analyzed on a Fortessa flow cytometer (BD Biosciences) or a FACSVerse™ cytometer with FACSuite™ software (BD Biosciences) using FlowJo v10 (TreeStar, Inc.).
Statistical analyses were performed using GraphPad Prism version 7.0 (GraphPad Software). The statistical significance of differences and of mean values was analyzed by the two-tailed Wilcoxon test in Figure 3, paired t-test in Figure 4, and Mann–Whitney non-parametric test in Figure 7. Results were considered to be statistically significant when p < 0.05.
All patients gave informed consent in a written form in accordance with the recommendations of “the Declaration of Helsinki” for participation in the study, which was approved by the scientific committees of the Clinic Investigator Center Inserm CIC-1402 (Poitiers, France), the Biological Resource Center of CHU of Poitiers (NF S96-900 certification since February 2014), the Biological Resource Center of CHU of Rennes (NF S96-900, certification since May 2009), and the Institute Curie Hospital (Paris, France).
AB, EC, and FJ designed the experiments, performed the experiments, analyzed and interpreted the data, and wrote the manuscript. LL, MA, NN, NP, BM, and SB contributed to sample preparation from patients and healthy controls, designed the experiments, performed the experiments, and analyzed and interpreted the data. EP, VC, and VL provided clinical samples and contributed to the interpretation of data. AH and J-MG together were responsible for the overall study design, supervised the project, and took primary responsibility for writing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are especially indebted to Jeffrey Arsham for editing the English of their manuscript. They acknowledge Thierry Walzer (Centre International de Recherche en Infectiologie, INSERM U1111-CNRS UMR 5308, Lyon, France) for the kind gift of Eomes-GFP mice. They thank Christine Sedlik (Institut Curie, Paris, France) for fruitful discussions and help with samples from breast cancer patients. They thank Image UP (Université de Poitiers) flow cytometry core facilities. This study was supported by INSERM, CHU de Poitiers, Université de Poitiers, Ligue contre le Cancer du Grand Ouest (Comités départementaux de la Vienne, de la Charente, de la Charente Maritime et des Deux-Sèvres), Association pour la Recherche en Immunologie-Poitou-Charentes (ARIM-PC), les Régions Aquitaine Limousin Poitou-Charentes, Bretagne, Centre et Pays de la Loire (PlasTICO project), Association Laurette Fugain, Ministère de la Recherche, Sport and Collection, and INCa-DGOS 8658 (PRT-K 2015-052).
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00316/full#supplementary-material.
1. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol (2015) 16:1114–23. doi:10.1038/ni.3298
2. Lanier LL. Shades of grey – the blurring view of innate and adaptive immunity. Nat Rev Immunol (2013) 13:73–4. doi:10.1038/nri3389
3. Jameson SC, Lee YJ, Hogquist KA. Innate memory T cells. Adv Immunol (2015) 126:173–213. doi:10.1016/bs.ai.2014.12.001
4. Van Kaer L. Innate and virtual memory T cells in man. Eur J Immunol (2015) 45:1916–20. doi:10.1002/eji.201545761
5. Berg RE, Cordes CJ, Forman J. Contribution of CD8+ T cells to innate immunity: IFN-gamma secretion induced by IL-12 and IL-18. Eur J Immunol (2002) 32:2807–16. doi:10.1002/1521-4141(2002010)32:10<2807::AID-IMMU2807>3.0.CO;2-0
6. Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J Exp Med (2003) 198:1583–93. doi:10.1084/jem.20031051
7. Su J, Berg RE, Murray S, Forman J. Thymus-dependent memory phenotype CD8 T cells in naive B6.H-2Kb-/-Db-/- animals mediate an antigen-specific response against Listeria monocytogenes. J Immunol (2005) 175:6450–7. doi:10.4049/jimmunol.175.10.6450
8. Atherly LO, Lucas JA, Felices M, Yin CC, Reiner SL, Berg LJ. The Tec family tyrosine kinases Itk and Rlk regulate the development of conventional CD8+ T cells. Immunity (2006) 25:79–91. doi:10.1016/j.immuni.2006.05.012
9. Broussard C, Fleischacker C, Horai R, Chetana M, Venegas AM, Sharp LL, et al. Altered development of CD8+ T cell lineages in mice deficient for the Tec kinases Itk and Rlk. Immunity (2006) 25:93–104. doi:10.1016/j.immuni.2006.05.011
10. Horai R, Mueller KL, Handon RA, Cannons JL, Anderson SM, Kirby MR, et al. Requirements for selection of conventional and innate T lymphocyte lineages. Immunity (2007) 27:775–85. doi:10.1016/j.immuni.2007.09.012
11. Hu J, Sahu N, Walsh E, August A. Memory phenotype CD8+ T cells with innate function selectively develop in the absence of active Itk. Eur J Immunol (2007) 37:2892–9. doi:10.1002/eji.200737311
12. Schaeffer EM, Broussard C, Debnath J, Anderson S, McVicar DW, Schwartzberg PL. Tec family kinases modulate thresholds for thymocyte development and selection. J Exp Med (2000) 192:987–1000. doi:10.1084/jem.192.7.987
13. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and Eomesodermin. Nat Immunol (2005) 6:1236–44. doi:10.1038/ni1268
14. Sosinowski T, White JT, Cross EW, Haluszczak C, Marrack P, Gapin L, et al. CD8α+ dendritic cell trans presentation of IL-15 to naive CD8+ T cells produces antigen-inexperienced T cells in the periphery with memory phenotype and function. J Immunol (2013) 190:1936–47. doi:10.4049/jimmunol.1203149
15. Zhou X, Yu S, Zhao D-M, Harty JT, Badovinac VP, Xue H-H. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity (2010) 33:229–40. doi:10.1016/j.immuni.2010.08.002
16. Prince AL, Kraus Z, Carty SA, Ng C, Yin CC, Jordan MS, et al. Development of innate CD4+ and CD8+ T cells in Itk-deficient mice is regulated by distinct pathways. J Immunol (2014) 193:688–99. doi:10.4049/jimmunol.1302059
17. Cho H, Bediako Y, Xu H, Choi H-J, Wang C-R. Positive selecting cell type determines the phenotype of MHC class Ib-restricted CD8+ T cells. Proc Natl Acad Sci U S A (2011) 108:13241–6. doi:10.1073/pnas.1105118108
18. Kim H-J, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature (2010) 467:328–32. doi:10.1038/nature09370
19. Urdahl KB, Sun JC, Bevan MJ. Positive selection of MHC class Ib-restricted CD8(+) T cells on hematopoietic cells. Nat Immunol (2002) 3:772–9. doi:10.1038/ni814
20. Akue AD, Lee J-Y, Jameson SC. Derivation and maintenance of virtual memory CD8 T cells. J Immunol (2012) 188:2516–23. doi:10.4049/jimmunol.1102213
21. Haluszczak C, Akue AD, Hamilton SE, Johnson LDS, Pujanauski L, Teodorovic L, et al. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med (2009) 206:435–48. doi:10.1084/jem.20081829
22. Lee J-Y, Hamilton SE, Akue AD, Hogquist KA, Jameson SC. Virtual memory CD8 T cells display unique functional properties. Proc Natl Acad Sci U S A (2013) 110:13498–503. doi:10.1073/pnas.1307572110
23. Marshall HD, Prince AL, Berg LJ, Welsh RM. IFN-alpha beta and self-MHC divert CD8 T cells into a distinct differentiation pathway characterized by rapid acquisition of effector functions. J Immunol (2010) 185:1419–28. doi:10.4049/jimmunol.1001140
24. Martinet V, Tonon S, Torres D, Azouz A, Nguyen M, Kohler A, et al. Type I interferons regulate Eomesodermin expression and the development of unconventional memory CD8(+) T cells. Nat Commun (2015) 6:7089. doi:10.1038/ncomms8089
25. Becker TC, Coley SM, Wherry EJ, Ahmed R. Bone marrow is a preferred site for homeostatic proliferation of memory CD8 T cells. J Immunol (2005) 174:1269–73. doi:10.4049/jimmunol.174.3.1269
26. Cho BK, Rao VP, Ge Q, Eisen HN, Chen J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med (2000) 192:549–56. doi:10.1084/jem.192.4.549
27. Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med (2000) 192:557–64. doi:10.1084/jem.192.4.557
28. Murali-Krishna K, Ahmed R. Cutting edge: naive T cells masquerading as memory cells. J Immunol (2000) 165:1733–7. doi:10.4049/jimmunol.165.4.1733
29. Huang W, Hu J, August A. Cutting edge: innate memory CD8+ T cells are distinct from homeostatic expanded CD8+ T cells and rapidly respond to primary antigenic stimuli. J Immunol (2013) 190:2490–4. doi:10.4049/jimmunol.1202988
30. Renkema KR, Li G, Wu A, Smithey MJ, Nikolich-Žugich J. Two separate defects affecting true naive or virtual memory T cell precursors combine to reduce naive T cell responses with aging. J Immunol (2014) 192:151–9. doi:10.4049/jimmunol.1301453
31. Arlettaz L, Degermann S, De Rham C, Roosnek E, Huard B. Expression of inhibitory KIR is confined to CD8+ effector T cells and limits their proliferative capacity. Eur J Immunol (2004) 34:3413–22. doi:10.1002/eji.200324756
32. Arlettaz L, Villard J, de Rham C, Degermann S, Chapuis B, Huard B, et al. Activating CD94:NKG2C and inhibitory CD94:NKG2A receptors are expressed by distinct subsets of committed CD8+ TCR alphabeta lymphocytes. Eur J Immunol (2004) 34:3456–64. doi:10.1002/eji.200425210
33. Björkström NK, Béziat V, Cichocki F, Liu LL, Levine J, Larsson S, et al. CD8 T cells express randomly selected KIRs with distinct specificities compared with NK cells. Blood (2012) 120:3455–65. doi:10.1182/blood-2012-03-416867
34. Huard B, Karlsson L. A subpopulation of CD8+ T cells specific for melanocyte differentiation antigens expresses killer inhibitory receptors (KIR) in healthy donors: evidence for a role of KIR in the control of peripheral tolerance. Eur J Immunol (2000) 30:1665–75. doi:10.1002/1521-4141(200006)30:6<1665::AID-IMMU1665>3.0.CO;2-2
35. van Veken LT, Diez Campelo M, van der Hoorn MA, Hagedoorn RS, van Egmond HM, van Bergen J, et al. Functional analysis of killer Ig-like receptor-expressing cytomegalovirus-specific CD8+ T cells. J Immunol (2009) 182:92–101. doi:10.4049/jimmunol.182.1.92
36. Warren HS, Rana PM, Rieger DT, Hewitt KA, Dahlstrom JE, Kent AL. CD8 T cells expressing killer Ig-like receptors and NKG2A are present in cord blood and express a more naïve phenotype than their counterparts in adult blood. J Leukoc Biol (2006) 79:1252–9. doi:10.1189/jlb.0905536
37. Guia S, Cognet C, de Beaucoudrey L, Tessmer MS, Jouanguy E, Berger C, et al. A role for interleukin-12/23 in the maturation of human natural killer and CD56+ T cells in vivo. Blood (2008) 111:5008–16. doi:10.1182/blood-2007-11-122259
38. Jacomet F, Cayssials E, Basbous S, Levescot A, Piccirilli N, Desmier D, et al. Evidence for Eomesodermin-expressing innate-like CD8(+) KIR/NKG2A(+) T cells in human adults and cord blood samples. Eur J Immunol (2015) 45:1926–33. doi:10.1002/eji.201545539
39. Wencker M, Turchinovich G, Di Marco Barros R, Deban L, Jandke A, Cope A, et al. Innate-like T cells straddle innate and adaptive immunity by altering antigen-receptor responsiveness. Nat Immunol (2014) 15:80–7. doi:10.1038/ni.2773
40. Min HS, Lee YJ, Jeon YK, Kim EJ, Kang BH, Jung KC, et al. MHC class II-restricted interaction between thymocytes plays an essential role in the production of innate CD8+ T cells. J Immunol (2011) 186:5749–57. doi:10.4049/jimmunol.1002825
41. Jacomet F, Cayssials E, Barbarin A, Desmier D, Basbous S, Lefèvre L, et al. The hypothesis of the human iNKT/innate CD8(+) T-cell axis applied to cancer: evidence for a deficiency in chronic myeloid leukemia. Front Immunol (2016) 7:688. doi:10.3389/fimmu.2016.00688
42. Goplen NP, Saxena V, Knudson KM, Schrum AG, Gil D, Daniels MA, et al. IL-12 signals through the TCR to support CD8 innate immune responses. J Immunol (2016) 197:2434–43. doi:10.4049/jimmunol.1600037
43. Gao Y, Williams AP. Role of innate T cells in anti-bacterial immunity. Front Immunol (2015) 6:302. doi:10.3389/fimmu.2015.00302
44. Denucci CC, Mitchell JS, Shimizu Y. Integrin function in T-cell homing to lymphoid and nonlymphoid sites: getting there and staying there. Crit Rev Immunol (2009) 29:87–109. doi:10.1615/CritRevImmunol.v29.i2.10
45. Martin-Blondel G, Pignolet B, Tietz S, Yshii L, Gebauer C, Perinat T, et al. Migration of encephalitogenic CD8 T cells into the central nervous system is dependent on the α4β1-integrin. Eur J Immunol (2015) 45:3302–12. doi:10.1002/eji.201545632
46. Polman C, Schellekens H, Killestein J. Neutralizing antibodies to interferon-beta may persist after cessation of therapy: what impact could they have? Mult Scler Houndmills Basingstoke Engl (2006) 12:245–6. doi:10.1191/135248506ms1284ed
47. Chen Y, Bord E, Tompkins T, Miller J, Tan CS, Kinkel RP, et al. Asymptomatic reactivation of JC virus in patients treated with natalizumab. N Engl J Med (2009) 361:1067–74. doi:10.1056/NEJMoa0904267
48. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science (2006) 311:1924–7. doi:10.1126/science.1122927
49. Ueda N, Kuki H, Kamimura D, Sawa S, Seino K, Tashiro T, et al. CD1d-restricted NKT cell activation enhanced homeostatic proliferation of CD8+ T cells in a manner dependent on IL-4. Int Immunol (2006) 18:1397–404. doi:10.1093/intimm/dxl073
50. Morris SC, Heidorn SM, Herbert DR, Perkins C, Hildeman DA, Khodoun MV, et al. Endogenously produced IL-4 nonredundantly stimulates CD8+ T cell proliferation. J Immunol (2009) 182:1429–38. doi:10.4049/jimmunol.182.3.1429
51. Mbitikon-Kobo F-M, Vocanson M, Michallet M-C, Tomkowiak M, Cottalorda A, Angelov GS, et al. Characterization of a CD44/CD122int memory CD8 T cell subset generated under sterile inflammatory conditions. J Immunol (2009) 182:3846–54. doi:10.4049/jimmunol.0802438
52. Verykokakis M, Boos MD, Bendelac A, Kee BL. SAP protein-dependent natural killer T-like cells regulate the development of CD8(+) T cells with innate lymphocyte characteristics. Immunity (2010) 33:203–15. doi:10.1016/j.immuni.2010.07.013
53. Weinreich MA, Odumade OA, Jameson SC, Hogquist KA. T cells expressing the transcription factor PLZF regulate the development of memory-like CD8+ T cells. Nat Immunol (2010) 11:709–16. doi:10.1038/ni.1898
54. Lee A, Park SP, Park CH, Kang BH, Park SH, Ha S-J, et al. IL-4 induced innate CD8+ T cells control persistent viral infection. PLoS Pathog (2015) 11:e1005193. doi:10.1371/journal.ppat.1005193
55. Fujii S-I, Shimizu K, Okamoto Y, Kunii N, Nakayama T, Motohashi S, et al. NKT cells as an ideal anti-tumor immunotherapeutic. Front Immunol (2013) 4:409. doi:10.3389/fimmu.2013.00409
56. Crowe NY, Smyth MJ, Godfrey DI. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J Exp Med (2002) 196:119–27. doi:10.1084/jem.20020092
57. Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med (2000) 191:661–8. doi:10.1084/jem.191.4.661
58. Street SE, Cretney E, Smyth MJ. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood (2001) 97:192–7. doi:10.1182/blood.V97.1.192
59. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am J Hematol (2014) 89:547–56. doi:10.1002/ajh.23691
60. Guilhot F, Roy L, Saulnier P-J, Guilhot J, Barra A, Gombert J-M, et al. Immunotherapeutic approaches in chronic myelogenous leukemia. Leuk Lymphoma (2008) 49:629–34. doi:10.1080/10428190801927510
61. Mohty M, Jourdan E, Mami NB, Vey N, Damaj G, Blaise D, et al. Imatinib and plasmacytoid dendritic cell function in patients with chronic myeloid leukemia. Blood (2004) 103:4666–8. doi:10.1182/blood-2003-09-3220
62. Verfaillie C, Kay N, Miller W, McGlave P. Diminished A-LAK cytotoxicity and proliferation accompany disease progression in chronic myelogenous leukemia. Blood (1990) 76:401–8.
63. Rossignol A, Levescot A, Jacomet F, Robin A, Basbous S, Giraud C, et al. Evidence for BCR-ABL-dependent dysfunctions of iNKT cells from chronic myeloid leukemia patients. Eur J Immunol (2012) 42:1870–5. doi:10.1002/eji.201142043
64. Wingender G, Krebs P, Beutler B, Kronenberg M. Antigen-specific cytotoxicity by invariant NKT cells in vivo is CD95/CD178-dependent and is correlated with antigenic potency. J Immunol (2010) 185:2721–9. doi:10.4049/jimmunol.1001018
65. Basbous S, Levescot A, Piccirilli N, Brizard F, Guilhot F, Roy L, et al. The Rho-ROCK pathway as a new pathological mechanism of innate immune subversion in chronic myeloid leukaemia. J Pathol (2016) 240:262–8. doi:10.1002/path.4779
66. Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci Transl Med (2014) 6:228ra37. doi:10.1126/scitranslmed.3007240
67. Powers JJ, Dubovsky JA, Epling-Burnette PK, Moscinski L, Zhang L, Mustjoki S, et al. A molecular and functional analysis of large granular lymphocyte expansions in patients with chronic myelogenous leukemia treated with tyrosine kinase inhibitors. Leuk Lymphoma (2011) 52:668–79. doi:10.3109/10428194.2010.550074
68. Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27:670–84. doi:10.1016/j.immuni.2007.09.006
69. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science (2012) 338:1220–5. doi:10.1126/science.1229620
70. Gill S, Vasey AE, De Souza A, Baker J, Smith AT, Kohrt HE, et al. Rapid development of exhaustion and down-regulation of Eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood (2012) 119:5758–68. doi:10.1182/blood-2012-03-415364
71. Lu B, Chen L, Liu L, Zhu Y, Wu C, Jiang J, et al. T-cell-mediated tumor immune surveillance and expression of B7 co-inhibitory molecules in cancers of the upper gastrointestinal tract. Immunol Res (2011) 50:269–75. doi:10.1007/s12026-011-8227-9
72. Dielmann A, Letsch A, Nonnenmacher A, Miller K, Keilholz U, Busse A. Favorable prognostic influence of T-box transcription factor Eomesodermin in metastatic renal cell cancer patients. Cancer Immunol Immunother (2016) 65:181–92. doi:10.1007/s00262-015-1786-1
73. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8(+) T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537:417–21. doi:10.1038/nature19330
Keywords: innate memory CD8(+) T cells, NK-like T cells, iNKT cells, natural killer receptors, Eomesodermin, CD49d, chronic myeloid leukemia, solid cancers
Citation: Barbarin A, Cayssials E, Jacomet F, Nunez NG, Basbous S, Lefèvre L, Abdallah M, Piccirilli N, Morin B, Lavoue V, Catros V, Piaggio E, Herbelin A and Gombert J-M (2017) Phenotype of NK-Like CD8(+) T Cells with Innate Features in Humans and Their Relevance in Cancer Diseases. Front. Immunol. 8:316. doi: 10.3389/fimmu.2017.00316
Received: 15 December 2016; Accepted: 06 March 2017;
Published: 27 March 2017
Edited by:
Fernando A. Arosa, Universidade da Beira Interior, PortugalReviewed by:
Avery August, Cornell University, USACopyright: © 2017 Barbarin, Cayssials, Jacomet, Nunez, Basbous, Lefèvre, Abdallah, Piccirilli, Morin, Lavoue, Catros, Piaggio, Herbelin and Gombert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: André Herbelin, YW5kcmUuaGVyYmVsaW5AaW5zZXJtLmZy;
Jean-Marc Gombert, am0uZ29tYmVydEBjaHUtcG9pdGllcnMuZnI=
†These authors have contributed equally to this work.
‡These authors have contributed equally to this work as senior authors.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.