 Miguel López-Botet1,2,3*
Miguel López-Botet1,2,3* Carlos Vilches4
Carlos Vilches4 Dolores Redondo-Pachón1,5
Dolores Redondo-Pachón1,5 Aura Muntasell1Aldi Pupuleku3
Aura Muntasell1Aldi Pupuleku3 José Yélamos1,2Julio Pascual1,5Marta Crespo1,5
José Yélamos1,2Julio Pascual1,5Marta Crespo1,5
- 1Hospital del Mar Medical Research Institute (IMIM), Barcelona, Spain
- 2Department of Immunology, Hospital del Mar, Barcelona, Spain
- 3Univ. Pompeu Fabra, Barcelona, Spain
- 4Immunogenetics-Histocompatibility, Instituto de Investigación Sanitaria Puerta de Hierro, Majadahonda, Spain
- 5Department of Nephrology, Hospital del Mar, Barcelona, Spain
Allograft rejection constitutes a major complication of solid organ transplantation requiring prophylactic/therapeutic immunosuppression, which increases susceptibility of patients to infections and cancer. Beyond the pivotal role of alloantigen-specific T cells and antibodies in the pathogenesis of rejection, natural killer (NK) cells may display alloreactive potential in case of mismatch between recipient inhibitory killer-cell immunoglobulin-like receptors (KIRs) and graft HLA class I molecules. Several studies have addressed the impact of this variable in kidney transplant with conflicting conclusions; yet, increasing evidence supports that alloantibody-mediated NK cell activation via FcγRIIIA (CD16) contributes to rejection. On the other hand, human cytomegalovirus (HCMV) infection constitutes a risk factor directly associated with the rate of graft loss and reduced host survival. The levels of HCMV-specific CD8+ T cells have been reported to predict the risk of posttransplant infection, and KIR-B haplotypes containing activating KIR genes have been related with protection. HCMV infection promotes to a variable extent an adaptive differentiation and expansion of a subset of mature NK cells, which display the CD94/NKG2C-activating receptor. Evidence supporting that adaptive NKG2C+ NK cells may contribute to control the viral infection in kidney transplant recipients has been recently obtained. The dual role of NK cells in the interrelation of HCMV infection with rejection deserves attention. Further phenotypic, functional, and genetic analyses of NK cells may provide additional insights on the pathogenesis of solid organ transplant complications, leading to the development of biomarkers with potential clinical value.
Introduction
Kidney transplantation is a widely used therapeutic intervention for chronic renal failure. Graft rejection remains a major complication, requiring prophylactic/therapeutic administration of immunosuppressive drugs. Consequently, kidney transplant recipients (KTR) are exposed to an increased susceptibility to infections, particularly by herpesviruses (e.g., cytomegalovirus and Epstein–Barr virus). Besides the pivotal role played by alloantigen-specific T cells and antibodies in the pathogenesis of graft rejection, natural killer (NK) cells alloreactivity and their contribution to antiviral defense receive increasing attention.
Diversity of the Human NK Cell Receptor Repertoire and NK Cell Subsets Distribution
Natural killer cells constitute an innate lymphoid lineage involved in early defense against certain intracellular pathogens and tumors, which mediate cytotoxicity and pro-inflammatory cytokine production upon interaction with pathological cells (1–3). NK cells are controlled by an array of germ line-encoded inhibitory and activating/co-stimulatory receptors (NKR), as well as by different cytokines (e.g., IL-2, IL-12, IL-15, IL-18, and type I interferons), which regulate their differentiation, proliferation, and effector functions. Inhibitory killer-cell immunoglobulin-like receptors (KIRs) and CD94/NKG2A complement each other, scanning potential target cells for altered surface expression of HLA class I (HLA-I) molecules.
The combinatorial distribution of these NKR along differentiation determines the existence of a variety of NK cell subsets capable of responding against pathological cells, which have downregulated HLA-I expression, as predicted by the “missing-self” hypothesis (4). In the context of transplantation, NK cell subsets may also react against normal allogeneic cells lacking specific HLA-I ligands for their inhibitory KIR (iKIR).
Killer-cell immunoglobulin-like receptor and NKG2 NK cell receptor families include other members with activating function whose physiological role is being investigated. At late differentiation stages, cytolytic T lymphocytes (TCRαβ CD8+, CD4+, and TCRγδ) may also display HLA-specific NKR (i.e., KIR, CD94/NKG2A, CD94/NKG2C, and LILRB1) (5, 6).
KIRs for HLA-A, -B, and -C
The human KIR family comprises (i) six receptors (four KIR2DL and two KIR3DL) with cytoplasmic “immunoreceptor tyrosine-based inhibition motifs” (ITIMs), which recruit the SHP-1/2 tyrosine phosphatases preventing NK cell activation; (ii) six KIR with short cytoplasmic tails lacking ITIMs (i.e., KIR2DS and KIR3DS), which interact with DAP12; this adaptor molecule contains “immunoreceptor tyrosine-based activation motifs” (ITAM) linked to protein tyrosine kinase (PTK) activation pathways; and (iii) two KIR (2DL4 and 3DL3) displaying ambiguous signaling motifs (7, 8).
Most iKIRs specifically recognize sets of HLA class Ia (i.e., HLA-A, -B, and -C) allotypes sharing structural polymorphisms at the α1 domain; yet, the ligands for some of them (e.g., KIR2DL5) and most activating KIR (aKIR) remain elusive. In an example of convergent evolution, the physiological role of KIR is undertaken in mice by members of the Ly49 lectin-like family; the Ly49H receptor triggers NK cell functions upon interaction with the m157 viral protein, contributing to defense against murine CMV (9–11). The low affinity interaction of some aKIR with HLA-I molecules suggests that they might specifically recognize pathogen-derived HLA–peptide complexes or other as yet unknown molecules.
At the population level, KIR repertoires are quite diverse due to the fact that not all KIR loci are found in the genome of every individual, and to the existence of a variety of alleles. Each KIR is encoded by a different gene in chromosome 19q13.4, and multiple KIR haplotypes/genotypes have been described worldwide (8). Moreover, iKIR–ligand interactions modulate functional NK cell maturation through an education process termed “licensing,” ill-defined at the molecular level, which dictates that most mature NK cells display at least an inhibitory NKR specific for self HLA-I molecules (12, 13).
CD94/NKG2 Killer Lectin-Like Receptors for HLA-E
CD94 and members of the NKG2 family are lectin-like membrane glycoproteins encoded at the NK gene complex on human chromosome 12. Similar to KIRs, the CD94/NKG2A heterodimer constitutes an inhibitory receptor linked to the SHP-1 tyrosine phosphatase, and CD94/NKG2C is coupled through DAP12 to a PTK activation pathway (14). The specific ligand for both CD94/NKG2 receptors is constituted by the HLA-E class Ib molecule, which binds to leader sequence peptides from other HLA-I molecules, including alleles not recognized by iKIRs (15–17). Thus, CD94/NKG2A prevents the response against cells with a normal expression of HLA-I molecules, complementing the function of KIRs. HLA-E may present pathogen-derived peptides [e.g., human cytomegalovirus (HCMV), HIV-1, and HCV] altering CD94/NKG2A recognition (18–20). On the other hand, CD94/NKG2C binds to HLA-E with lower affinity than its inhibitory counterpart (21, 22) and has been reported to be involved in the response to human HCMV (see Adaptive NK Cell Response to HCMV).
Additional Activating and Inhibitory NKR
The CD16A (FcγRIIIA) receptor is coupled through CD3ζ or FcεRIγ chain adapters to a PTK activation pathway, triggering cytotoxicity and cytokine production upon interaction with IgG-opsonized cells (23). A CD16A allelic dimorphism (158V or F) influences the affinity of its interaction with IgG, modulating receptor-mediated signaling and activation of effector functions (24). Surface CD16 expression is downregulated in activated NK cells through a shedding process mediated by ADAM-17 metalloprotease (25, 26).
The human NKG2D C-type lectin triggers phosphatidyl inositol-3 kinase signaling through the DAP10 adaptor (27). NKG2D functions as an activating/co-stimulatory receptor specific for a set of ligands (MICA, MICB, and “UL16-binding proteins”) displayed by pathological cells, which are also inducible by cellular stress in normal tissues (6). Several immune evasion mechanisms that prevent NKG2D ligand (NKG2D-L) expression in HCMV-infected cells have been identified (28).
Natural cytotoxicity receptors, i.e., NCR1 (NKp46), NCR2 (NKp44), and NCR3 (NKp30), are connected to PTK signaling pathways through different ITAM-bearing adapters (29). In addition to their putative role in recognition of pathogen-derived molecules, there is evidence supporting the expression of ligands in normal cells that may trigger NK cell functions when control by inhibitory receptors is reduced (30). NKp46 is coupled to the CD3ζ or FcεRIγ chain, triggering cytotoxicity and cytokine production upon recognition of an ill-defined cellular ligand(s). NKp46 has been shown to be involved in the NK cell response to HCMV-infected dendritic cells and macrophages (31, 32). The nature of cellular ligands for NKp44 also remains open, and several ligands have been reported for the CD3ζ-linked NKp30 (30, 33).
In addition to the pivotal role played by adhesion molecules (i.e., LFA-1 and CD2) in the NK cell interaction with target cells, engagement of DNAM1, a co-stimulatory receptor specific for Nectin-2 (CD112) and PVR (CD155), contributes to the response against tumor and virus-infected cells (32, 34). NK cells may acquire additional inhibitory NKR upon activation or at late differentiation stages. Among these checkpoints, LILRB1 (ILT2, LIR-1, or CD85j) interacts with a wide spectrum of HLA-I molecules and binds with a higher affinity to the UL18 HCMV glycoprotein (35, 36); similarly, TIGIT (T cell Ig and ITIM domain) binds to CD155 competing with DNAM1 (37).
Peripheral Blood NK Cell Subsets
The human peripheral blood NK cell compartment includes a variety of cell subsets, which represent distinct maturation stages and display different combinations of HLA-I-specific NKR. Similar to T and B lymphocytes, NK cells may undergo clonal expansion and late differentiation events, skewing the NKR repertoire and further diversifying their phenotypic/functional profile (Figure 1).
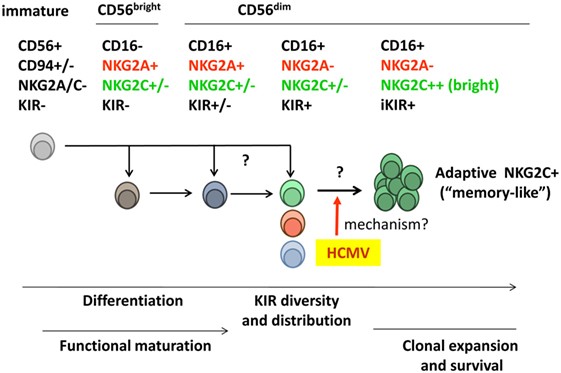
Figure 1. Diversity of the human peripheral blood natural killer (NK) cell compartment. Two main NK cell populations are identified according to expression levels of the CD56 marker. CD56bright NK cells secrete pro-inflammatory cytokines but display a low cytotoxic potential and are often considered to represent an early maturation stage. According to such view, this subset is believed to differentiate into the major cytotoxic CD56dim NK cell population, which includes a variety of subsets differing in NKR expression [e.g., killer-cell immunoglobulin-like receptors (KIRs), CD94/NKG2A, and CD94/NKG2C]. Whether some CD56dim subsets (e.g., NKG2A− KIR+) might directly derive from immature NK cell precursors rather than from NKG2A+ KIR− CD56bright NK cells is not formally ruled out. Human cytomegalovirus (HCMV) infection promotes the differentiation and stable expansion of an NK cell subset, which displays high levels of the CD94/NKG2C receptor and an oligoclonal inhibitory KIR (iKIR) expression pattern, associated with other phenotypic and functional characteristics (see details in Section “Adaptive NK Cell Response to HCMV”). The nature of their precursors and the mechanism(s) underlying such adaptive NK cell response to HCMV are investigated.
Two NK cell populations are identified in peripheral blood according to their surface expression levels of the CD56 neural-cell adhesion molecule isoform (i.e., CD56bright and CD56dim) (38). CD56bright NK cells constitute a minor fraction (~10%) of the normal circulating NK cell compartment. They display a low cytotoxic potential but secrete pro-inflammatory cytokines and are conventionally considered to represent an early maturation stage (39). Most CD56bright NK cells express CD94/NKG2A, NKG2D, and NCR, but lack KIR and CD16. The predominant (~90%) CD16+NKG2D+CD56dim NK cell population comprises distinct subsets, defined according to KIR, NKG2A, and NKG2C expression (e.g., NKG2A+KIR+NKG2C+/− and NKG2A−KIR+NKG2C+/−). Evidences have been obtained indirectly supporting a linear differentiation model in which CD56bright NK cells sequentially give rise to the other NK cell subsets (38, 40). Yet, the possibility that alternative differentiation pathways branching from NK cell precursors may independently generate CD56bright and CD56dim subsets cannot be formally ruled out.
Further levels of NK cell phenotypic/functional heterogeneity are determined by (i) the diversity of human NKR repertoires, conditioned by the existence of hundreds of different KIR haplotypes diverging in gene and allotype content; (ii) the clonal distribution of KIR combinations among CD56dim NK cells, modulated by the influence of KIR–ligand interactions on NK cell maturation; (iii) the oligoclonal adaptive expansion of NK cell subsets in response to HCMV infection (see Adaptive NK Cell Response to HCMV); and (iv) the incidence of late differentiation events, which determine additional phenotypic and functional changes (e.g., expression of CD57 and LILRB1) (Figure 1).
NK Cells and HCMV Infection in KTR
Human cytomegalovirus is a member of the herpesviridae family which causes highly prevalent lifelong infections in all human populations, generally asymptomatic in immunocompetent hosts. The virus establishes latency, undergoing occasional reactivation which allows its efficient transmission through secretions (41, 42). HCMV may cause severe congenital disorders (43) and increases the morbidity/mortality rate in immunocompromised individuals (44, 45), being associated with some chronic inflammatory disorders (i.e., atherosclerosis) and immune senescence (46). As a consequence of immunosuppression to prevent graft rejection, KTR are exposed to HCMV reactivation/reinfection, leading to potentially severe complications (47, 48).
Together with specific T lymphocytes and antibodies, commonly analyzed to assess the adaptive immune response to HCMV, NK cells contribute to defense against this pathogen (49, 50). To escape from CD8+ T cells, HCMV downregulates surface expression of HLA-I molecules in infected cells, interfering with antigen presentation (51, 52). Consequently, engagement of inhibitory NKR is impaired promoting NK cell activation, which is counteracted by a variety of viral immune evasion strategies (53–55).
Adaptive NK Cell Response to HCMV
In 2004, we discovered that healthy HCMV-seropositive (HCMV+) individuals display increased proportions of NK and T cells hallmarked by high surface levels of CD94/NKG2C (NKG2Cbright) (56). The imprint of HCMV in the NK cell compartment is perceived to a variable extent only in some HCMV+ subjects, persisting under steady state conditions. A number of reports have extended these observations in different settings, and the terms “adaptive” or “memory-like” are currently employed to designate the human differentiated NKG2Cbright NK cell population (55). For the sake of precision, we have strictly used this original definition along the text. Yet, it is of note that these terms have been used by some authors to define other NK cell populations (e.g., in vitro cytokine-differentiated NK cells) (57).
Expansions of NKG2Cbright cells are not induced by other herpesviruses (i.e., EBV and HSV-1) but have been reported in the course of different viral infections, yet associated with HCMV coinfection (58–61). As compared to other NK cell subsets, including the low proportions of NKG2Cdim cells detected in HCMV(−) and some HCMV(+) individuals, adaptive NKG2C+ NK cells display a phenotype characterized by an oligoclonal pattern of iKIR specific for self HLA-I molecules (preferentially HLA-C). Moreover, they express reduced levels of NCR (i.e., NKp30 and NKp46), Siglec7, and CD161 (56, 62–64), acquire late differentiation markers (e.g., CD57 and LILRB1) (65, 66), maintain surface expression of NKG2D and CD16, and display increased levels of CD2 involved in their activation (67, 68). Epigenetic downregulation of signaling molecules (e.g., FcεRIγ chain and Syk) and certain transcription factors have been associated with adaptive NK cell differentiation (69, 70). From a functional standpoint, they contain greater levels of Granzyme B and efficiently secrete TNF-α and IFN-γ (62, 63), mediating antibody-dependent cytotoxicity (ADCC) and cytokine production against HCMV-infected cells (71–73).
Expansions of NKG2C+ cells following HCMV infection were reported in immunosuppressed transplant recipients (65, 66, 74), in a severe T cell primary immunodeficiency (75), as well as in children and newborns with congenital or postnatal HCMV infection (76, 77), independently of aging (78–80). Altogether, these observations suggest that the magnitude of the HCMV imprint on the NK cell compartment in healthy individuals is likely fixed at the time of primary infection, presumably depending on host/virus genetics and other circumstantial factors (e.g., age at infection, viral load, etc.) (81).
By analogy with the role of Ly49H+ cells in the response to murine CMV (82), we hypothesized that CD94/NKG2C-mediated specific recognition of virus-infected cells drives the adaptive differentiation, proliferation, and survival of this lymphocyte subset (55). Indirectly supporting this view, in vitro stimulation of PBMC from HCMV+ donors with virus-infected cells elicited a preferential expansion of CD94/NKG2C+ NK cells (83, 84). Yet, at variance with Ly49H, the nature of a hypothetical viral ligand remains uncertain, and there is no experimental evidence supporting that the CD94/NKG2C receptor may trigger NK cell effector functions against HCMV-infected cells (32, 55, 83, 85). By contrast, NKG2C+ adaptive NK cells have been shown to efficiently mediate antibody-dependent effector functions, particularly pro-inflammatory cytokine production, against HCMV and HSV-1 infected cells (24, 71). It is of note that CD16 remains functionally coupled to the CD3ζ adapter (73) following downregulation of FcεRIγ. The molecular mechanisms driving this pattern of response to HCMV and the existence of a putative CD94/NKG2C viral ligand are investigated (Figure 2).
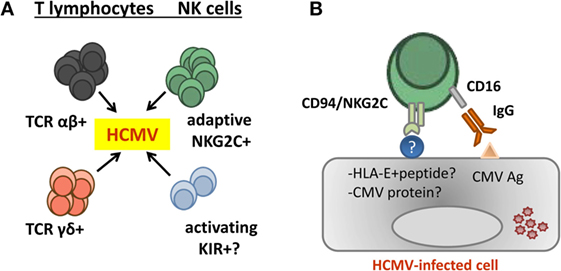
Figure 2. Contribution of adaptive natural killer (NK) cells to human cytomegalovirus (HCMV) control. (A) Evidences supporting a contribution of different T and NK cell subsets in the control of HCMV infection in kidney transplant recipients have been reported. (B) Adaptive NKG2Cbright NK cells generated in response to HCMV infection efficiently mediate antibody-dependent cytotoxicity and cytokine production (e.g., TNF-α and IFN-γ) in response to HCMV-infected cells. Yet, there is no consistent evidence supporting an involvement of CD94/NKG2C in triggering NK cell effector functions against infected cells, and the nature of a hypothetical viral ligand remains elusive.
A deletion of the NKG2C gene (officially designated KLRC2) is frequently detected in different human populations, with some variation depending on their ethnic/geographic origin (86–89). NKG2C gene copy number is directly related with surface expression levels and the activating function of CD94/NKG2C (62). Moreover, the NKG2C genotype is as well associated with steady state numbers of circulating NKG2C+ NK cells, which appear reduced in NKG2C+/del as compared to NKG2C+/+ individuals, further supporting a role of the NKR in driving the generation of adaptive NK cells (62, 76, 88). The identification of ~5% HCMV(+) healthy NKG2Cdel/del blood donors illustrates that the receptor is dispensable for controlling the viral infection under normal conditions, being redundant with other cell types (i.e., T lymphocytes). Moreover, NKG2C− NK cell subsets sharing some phenotypic features with canonical adaptive NKG2C+ NK cells have been reported in HCMV(+) NKG2Cdel/del blood donors (68, 90) and HCMV-infected hematopoietic stem cell transplantation (HSCT) recipients (91). On the other hand, the lack of NKG2C+ NK cells has been suggested to alter the control of primary HCMV infection in childhood (88); a putative relevance of the NKG2C deletion in immunosuppressed patients is discussed in the next section.
NK Cell Response to HCMV Infection in KTR
Posttransplant HCMV infection constitutes a risk factor for cardiac and renal allograft vasculopathy associated with chronic graft dysfunction and is directly associated with the rate of graft loss and reduced host survival (47, 48, 92). Antiviral prophylaxis is commonly administered to HCMV(−) KTR transplanted from an HCMV(+) donor or treated with intensive immunosuppression; patients developing HCMV viremia receive antiviral therapy, not free of adverse effects. Identification of biomarkers predicting the risk of posttransplant HCMV infection is warranted to improve its clinical management. Regular immunosuppressive therapy in KTR is aimed to prevent rejection, impairing the development of alloreactive T cells and production of alloantibodies, but has been proposed to be less effective on differentiated CTL and mature NK cells (93). Yet, alterations of the phenotypic and functional profile of circulating NK cells following immunosuppression were detected in other studies (94, 95). After low-dose therapy with anti-thymocyte globulin (ATG) NK cells recovered faster than T cells (96). In this regard, following induction with ATG functionally competent NK cells were reported to display for several months an NKG2A+ KIR− phenotype (97). Thus, it is plausible that NK cells may contribute to antiviral defense in KTR, partially compensating their impaired T cell response.
The putative influence of KIR and HLA-I genotypes in the control of HCMV infection in KTR has been addressed. A relation of the KIR repertoire with viral load was reported in primary HCMV infection (98), even though the risk of HCMV disease was not influenced by KIR–ligand matching (99). De Rham et al. detected increased numbers of KIR3DL1+ NK cells in KTR during the acute phase of HCMV reactivation (100). In both KTR and healthy blood donors, this NK cell subset efficiently killed in vitro infected fibroblasts; different interpretations for this observation were proposed. On the other hand, KIR-B haplotypes encoding aKIR were related with a lower rate of HCMV infection (101). In cases receiving thymoglobulin and intensive immunosuppression, KIR-associated control of HCMV was limited to seropositive KTR (102). A role of activating NKR in the control of other viral infections (e.g., BK and varicella zoster) has been also proposed (103, 104).
We recently explored the relationship of adaptive NKG2C+ NK cells with the outcome of HCMV infection in KTR, monitoring pre- and posttransplant the NK cell immunophenotype and the incidence of viremia (105). NKG2C+ NK cell expansions did not systematically follow detection of HCMV viremia in KTR, thus suggesting that a prompt control of the infection by antiviral therapy and preexisting differentiated CTL may hamper the adaptive NK cell response development. Conversely, late NKG2C+ NK cell expansions might reflect clinically unnoticed HCMV replication after withdrawal of antiviral therapy. In this regard, symptom-free HCMV reactivations in KTR have been associated with altered phenotypic and functional profiles of NK cells, which expressed LILRB1 and downregulated FcϵRIγ (106). In the same line, increased proportions of LILRB1+ (LIR-1+) NK cells were originally associated with HCMV infection in lung transplant recipients (107).
Regular immunosuppressive protocols did not modify the levels of adaptive NK cells in KTR without detectable viremia along the follow-up, nor did they impair their expansion in some cases undergoing HCMV infection (105). Nevertheless, the possibility that immunosuppression may interfere with de novo adaptive NK cell differentiation, as it does with alloreactive T cell development, is not ruled out. Further studies are warranted to precisely assess the impact of different drugs on the development and effector functions of adaptive NK cells.
Of note, high pretransplant levels of NKG2C+ NK cells were associated with a reduced incidence of posttransplant HCMV viremia, independently of other related variables (e.g., thymoglobulin induction, antiviral prophylaxis, and age), suggesting that adaptive NK cells might confer some protection against viral reactivation/reinfection (105). In this regard, a low NK cell count post-liver transplantation has been reported to be an independent risk factor for HCMV disease (108). Despite their limited direct in vitro response against HCMV-infected cells, adaptive NKG2C+ NK cells may contribute to antiviral defense. In particular, they efficiently mediate antibody-dependent effector functions and likely participate in the response to HCMV reactivation in KTR, in combination with specific IgG (70, 71, 73) (Figure 2B). In this context, the influence of CD16A dimorphism and IgG allotypes on the magnitude of ADCC deserves attention (24). The possibility that aKIR may be involved in the putative antiviral effect of adaptive NKG2C+ NK cells appears unlikely, considering that they do express iKIR (63, 64, 90) and that their expansion is independent of KIR-A/B haplotypes (56). Nevertheless, NK cell subsets expressing CD94/NKG2C or aKIR might play complementary roles in the response to HCMV.
The frequencies of TcRαβ T cells specific for HCMV antigens (e.g., IE-1 and pp65) have been reported to predict the risk of posttransplant infection (109, 110); moreover, TcRγδ T cells were associated with control of posttransplant HCMV viremia (111). Adaptive NKG2C+ NK cells and CTL have been proposed to be independent (78–80). Thus the possibility that the association of adaptive NKG2C+ NK cells with a lower risk of HCMV infection might indirectly reflect a central role of HCMV-specific TcRαβ T cells (Figure 3) appears unlikely; further studies are warranted to precisely address this issue.
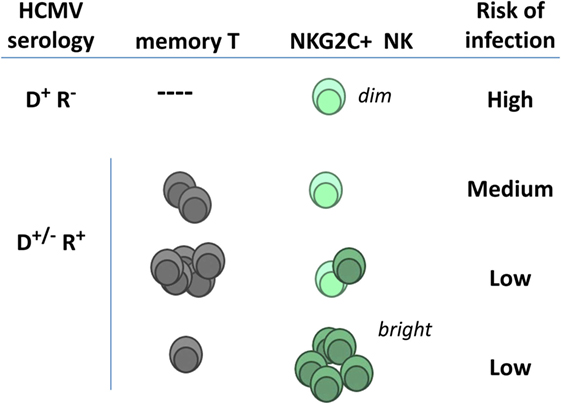
Figure 3. Hypothetical relation of adaptive natural killer (NK) cells and specific T lymphocytes with posttransplant infection risk in kidney transplant recipients. NKG2Cdim NK cells detected in human cytomegalovirus (HCMV)(−) individuals express lower surface levels of the receptor and differ phenotypically and functionally from adaptive NK cells expanding in response to HCMV infection (NKG2Cbright) (see details in Section “Adaptive NK Cell Response to HCMV”). Pretransplant frequencies of virus-specific T lymphocytes and of NKG2C+ NK cells in seropositive recipients have been independently related with a reduced incidence of HCMV infection (D, donor; R, recipient).
The distributions of the NKG2C genotypes in two different KTR cohorts, studied pre- and posttransplant, appeared comparable to the frequencies detected in blood donors; as reported, the magnitude of the NKG2C+ NK cell expansion was greater in NKG2C+/+ than in NKG2C+/del subjects (105). Remarkably, somewhat increased frequencies of the NKG2C+/del genotype and a reciprocal reduction of NKG2C+/+ cases were detected among KTR suffering symptomatic HCMV infection; unexpectedly, an opposite reduction of the NKG2Cdel/del frequency was observed among this KTR group. Despite that differences did not reach statistical significance, the coincident trends in both cohorts suggested a relation of NKG2C copy number with the outcome of HCMV infection and its impact in KTR; larger studies are warranted to confirm these observations.
Altogether these results indirectly support that adaptive NKG2C+ NK cells may play an active role in defense against HCMV, partially compensating in KTR the effect of immunosuppression on T cells. High pretransplant levels of NKG2C+ cells may predict a lower risk of posttransplant HCMV replication/disease in KTR receiving regular immunosuppression, particularly in NKG2C+/+ HCMV(+) patients (Figure 3). On the other hand, posttransplant expansions of differentiated adaptive NKG2C+ NK cells reflect the incidence of viral replication and, once established, might contribute to its control. It is plausible that antibody-mediated response to other viral infections may as well contribute to the expansion of adaptive NKG2C+ cells (70, 112). It is uncertain whether adaptive NK cells may comparably respond to HCMV reactivation or reinfection, reported to have a different clinical impact (113). From a practical standpoint, monitoring basal and posttransplant levels of adaptive NK cells may provide biomarkers to evaluate the control of HCMV, with practical implications in the clinical management of the viral infection. Assessing the relation with other phenotypic features displayed at late stages of adaptive NKG2C+ NK cells differentiation (e.g., CD57 expression and FcεRIγ chain loss) deserves attention. Furthermore, studies in larger cohorts are required to assess the relation of the adaptive NK cell response in KTR with the incidence of other viral infections, as well as with the risk of chronic graft rejection, cardiovascular disease, and cancer (48, 114). In this regard, the possibility that antibody-dependent activation of adaptive NK cells may participate in donor-specific alloantibodies (DSA)-mediated rejection is addressed in the next section.
NK Cells and Allograft Rejection
NK Cell Alloreactivity
Mature NK cells whose iKIR fail to recognize HLA-I alleles on an allograft are predicted to mediate cytotoxicity and pro-inflammatory cytokine production, as long as activating NKR are engaged by ligands displayed on target cells. Some degree of KIR–ligand mismatching between donors and recipients is estimated to occur in 50–75% of HLA non-identical transplants, and several studies have addressed the impact of this variable in kidney transplant outcome (Figure 4A). On one hand, KIR–ligand mismatches were suggested to influence short-term outcome in KTR (115) and were associated with a reduced long-term graft survival in HLA-incompatible KTR (116), proposing a beneficial effect of NK cell-targeted immunosuppression. Conversely, KIR–ligand mismatch was reported by others to be irrelevant for predicting long-term allograft survival (117) and, in the same line, no effect on the risk of rejection was perceived after reduction of immunosuppressive therapy (118).
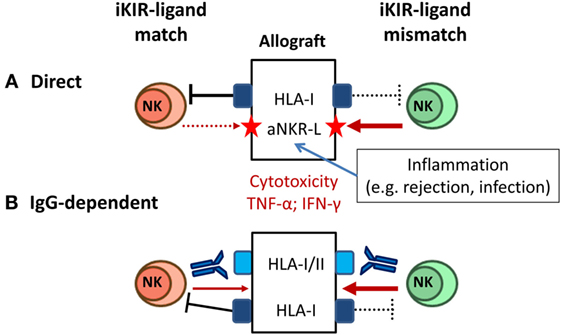
Figure 4. Natural killer (NK) cell-mediated alloreactivity in solid organ transplantation. (A) NK cells lacking inhibitory KIR (iKIR) specific for donor HLA class I ligands (“mismatched,” right) may potentially mediate cytotoxicity and cytokine production against the allograft. NK cell alloreactivity is favored by cellular stress conditions (e.g., pro-inflammatory stimuli) inducing graft expression of ligands for activating NKR (e.g., NKG2D ligand). (B) Donor-specific alloantibodies (DSA) trigger CD16+ NK cell-mediated cytotoxicity and cytokine production against the allograft, overcoming the control by inhibitory NKR (e.g., iKIR) (left); iKIR–ligand mismatch might synergize with a DSA-CD16-mediated response (right).
These apparently conflicting observations might be reconciled considering the implications of inhibitory NKR–MHC class I mismatch in other experimental and clinical transplant settings. Classical animal models of “F1-hybrid resistance” revealed a role of NK cells in rejection of allogeneic hematopoietic transplants, but not of other tissues grafts (119). In HSCT, donor NK cell-mediated alloreactivity has been shown to potentially exert an antileukemic effect without promoting graft-versus-host disease (120). In the same line, adoptive immunotherapy with allogeneic NK cells in HSCT recipients has been proven a safe procedure (121, 122). Altogether these observations support that the NK cell alloreactive potential, determined by KIR–ligand mismatch, may have a negligible pathogenic impact in solid organ transplantation, unless engagement of activating NKR triggers NK cell effector functions. Accordingly, NK cell alloreactivity would be favored by stimuli promoting graft expression of activating NKR ligands (e.g., NKG2D-L). This situation may take place in the context of infections (e.g., HCMV) or T cell/DSA-mediated rejection reactions, enhancing the pathogenic impact of these adverse events (Figure 4A). From a methodological standpoint, the genotypic prediction of KIR–ligand mismatching should be complemented by a direct assessment of the frequencies of potential alloreactive NK cells, using specific mAbs to discriminate homologous activating and iKIR as reported for HSCT (123).
Alloantibody-Dependent NK Cell Activation
Posttransplant donor-specific anti-HLA antibodies (DSA) are a major risk factor in kidney transplant, causing microvascular damage associated with humoral rejection. In addition to complement activation, HLA-specific alloantibodies may trigger NK cells through CD16 to mediate ADCC and cytokine production (Figure 4B). Indications that NK cells contribute to chronic antibody-mediated rejection (ABMR) have been obtained in experimental models and analyzing kidney biopsies (124, 125). Consistent with a pathogenic role of NK cells, increased CD56+ cells have been observed in graft lesions from patients suffering ABMR. NK cell-associated gene expression has been associated with microvascular inflammation (126, 127), providing biomarkers with potential diagnostic/prognostic value (128–130). CD16A is also expressed by TCRγδ and some TCRαβ T lymphocyte subsets (131, 132). CD16+ TCRγδ T cells have been related with the response to posttransplant HCMV infection in KTR, and evidences supporting their involvement in ABMR have been reported (111, 133).
CD16 downregulation and expression of activation markers have been observed in circulating NK cells from KTR, likely reflecting IgG-dependent NK cell activation triggered by infectious pathogens (e.g., HCMV) or DSA (134). In the same line, altered distributions of circulating NK cells have been associated with the presence of alloantibodies in KTR. DSA+ patients were reported to display lower proportions of the major CD56dim NK cell subset as compared with cases without anti-HLA antibodies (95). Increased proportions of CD56bright and CD56dim NKG2A+ cells, but not their absolute numbers, were observed in DSA+ KTR (135). The data suggest that alloantibody-mediated activation of NK cells via CD16 may promote their turnover, accounting for the imbalanced NK cell subset distribution.
This hypothesis predicts that CD56bright NKG2A+ CD16− NK cells should be spared from the effect of alloantibodies, consistent with their increased proportions in DSA+ KTR. On the other hand, the association of DSA with increased proportions of CD56dimNKG2A+ NK cells suggests that engagement of CD94/NKG2A by HLA-E, conserved in all individuals, might also dampen the alloantibody effect on this subpopulation. Conversely, KIR–ligand mismatch would add to alloantibody activation of CD56dimNKG2A− KIR+ NK cells, synergizing with the pathogenic effects of DSA and accelerating their turnover. Given the oligoclonal expression by adaptive NKG2C+ NK cells of self-reactive KIR, preferentially specific for HLA-C molecules (63, 64), and their ability to mediate antibody-dependent effector functions (71, 73), it is likely that they may play a relevant pathogenic role in DSA-mediated graft rejection of KIR–ligand-I mismatched transplants.
In summary, consistent evidence has been obtained supporting a functional duality of NK cells in the context of kidney transplantation, reflected by their positive involvement in the response to HCMV infection as opposed to their participation in graft rejection. Further studies integrating phenotypic, functional, and genetic analysis of NK cells should provide valuable insights on the pathogenesis of solid organ transplant complications, leading to the potential development of clinically useful biomarkers.
Author Contributions
All authors have actively contributed to build up the conceptual framework developed in this review and revised the draft written by ML-B.
Conflict of Interest Statement
The authors individually declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the technical help of Dulce Soto, Judit Rigol, Andrea Vera, Sara Alvarez, Anna Faura, and Rosa Causadias.
Funding
The authors are supported by a coordinated research project from Fundació La Marató de TV3 (137/C/2012) and by grants from: Plan Estatal I+D Retos (SAF2013-49063-C2-1-R), Spanish Ministry of Economy and Competitiveness (MINECO, FEDER); EU FP7-MINECO Infect-ERA program (PCIN-2015-191-C02-01); FIS-FEDER PI13/00598 and 16/00617, Intensification Programs (Spanish Ministry of Health ISCIII); and RedinRen RD16/0009/0013. AM is supported by Asociación Española Contra el Cáncer (AECC). AP is supported by EU-FP7 Marie Curie Training Network (NATURIMMUN FP7-PEOPLE-2012-ITN-317013).
References
1. Moretta A, Bottino C, Mingari MC, Biassoni R, Moretta L. What is a natural killer cell? Nat Immunol (2002) 3(1):6–8.
3. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi: 10.1038/ni1582
4. Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today (1990) 11:237–44.
5. Vivier E, Anfossi N. Inhibitory NK-cell receptors on T cells: witness of the past, actors of the future. Nat Rev Immunol (2004) 4(3):190–8.
6. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi:10.1146/annurev-immunol-032712-095951
7. Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol (2002) 20:217–51.
8. Parham P, Moffett A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol (2013) 13(2):133–44. doi:10.1038/nri3370
9. Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science (2002) 296(5571):1323–6.
10. Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A (2002) 99(13):8826–31.
11. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature (2009) 457(7229):557–61. doi:10.1038/nature07665
12. Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell (2010) 142(6):847–56. doi:10.1016/j.cell.2010.08.031
13. Horowitz A, Djaoud Z, Nemat-Gorgani N, Blokhuis J, Hilton HG, Béziat V, et al. Class I HLA haplotypes form two schools that educate NK cells in different ways. Sci Immunol (2016) 1(3):eaag1672.
14. López-Botet M, Bellón T. Natural killer cell activation and inhibition by receptors for MHC class I. Curr Opin Immunol (1999) 11(3):301–7.
15. Braud VM, Allan DS, O’Callaghan CA, Soderstrom K, D’Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature (1998) 391(6669):795–9.
16. Lee N, Llano M, Carretero M, Ishitani A, Navarro F, López-Botet M, et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A (1998) 95:5199–204.
17. Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med (1998) 187(5):813–8.
18. Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, et al. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science (2000) 287(5455):1031–33.
19. Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity (1999) 10(6):661–71.
20. Nattermann J, Nischalke HD, Hofmeister V, Ahlenstiel G, Zimmermann H, Leifeld L, et al. The HLA-A2 restricted T cell epitope HCV core 35-44 stabilizes HLA-E expression and inhibits cytolysis mediated by natural killer cells. Am J Pathol (2005) 166(2):443–53.
21. Valés-Gómez M, Reyburn HT, Erskine RA, López-Botet M, Strominger JL. Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. EMBO J (1999) 18(15):4250–60.
22. Heatley SL, Pietra G, Lin J, Widjaja JM, Harpur CM, Lester S, et al. Polymorphism in human cytomegalovirus UL40 impacts on recognition of human leukocyte antigen-E (HLA-E) by natural killer cells. J Biol Chem (2013) 288(12):8679–90. doi:10.1074/jbc.M112.409672
23. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood (2006) 107(1):159–66.
24. Moraru M, Black LE, Muntasell A, Portero F, López-Botet M, Reyburn HT, et al. NK cell and Ig interplay in defense against herpes simplex virus type 1: epistatic interaction of CD16A and IgG1 allotypes of variable affinities modulates antibody-dependent cellular cytotoxicity and susceptibility to clinical reactivation. J Immunol (2015) 195(4):1676–84. doi:10.4049/jimmunol.1500872
25. Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine activation induces human memory-like NK cells. Blood (2012) 120(24):4751–60. doi:10.1182/blood-2012-04-419283
26. Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8(+) T cells. Nat Rev Immunol (2011) 11(10):645–57. doi:10.1038/nri3044
27. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol (2003) 3(10):781–90.
28. Jonjic S, Babic M, Polic B, Krmpotic A. Immune evasion of natural killer cells by viruses. Curr Opin Immunol (2008) 20(1):30–8. doi:10.1016/j.coi.2007.11.002
29. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol (2001) 19:197–223.
30. Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol (2013) 34(4):182–91. doi:10.1016/j.it.2013.01.003
31. Romo N, Magri G, Muntasell A, Heredia G, Baia D, Angulo A, et al. Natural killer cell-mediated response to human cytomegalovirus-infected macrophages is modulated by their functional polarization. J Leukoc Biol (2011) 90(4):717–26. doi:10.1189/jlb.0311171
32. Magri G, Muntasell A, Romo N, Sáez-Borderias A, Pende D, Geraghty DE, et al. NKp46 and DNAM-1 NK-cell receptors drive the response to human cytomegalovirus-infected myeloid dendritic cells overcoming viral immune evasion strategies. Blood (2011) 117(3):848–56. doi:10.1182/blood-2010-08-301374
33. Kruse PH, Matta J, Ugolini S, Vivier E. Natural cytotoxicity receptors and their ligands. Immunol Cell Biol (2014) 92(3):221–9. doi:10.1038/icb.2013.98
34. Pende D, Spaggiari GM, Marcenaro S, Martini S, Rivera P, Capobianco A, et al. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: evidence for the involvement of the poliovirus receptor (CD155) and Nectin-2 (CD112). Blood (2005) 105(5):2066–73.
35. Colonna M, Navarro F, Bellón T, Llano M, García P, Samaridis J, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med (1997) 186(11):1809–18.
36. Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity (1997) 7(2):273–82.
37. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A (2009) 106(42):17858–63. doi:10.1073/pnas.0903474106
38. Caligiuri MA. Human natural killer cells. Blood (2008) 112(3):461–9. doi:10.1182/blood-2007-09-077438
39. Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, et al. Human CD56bright NK cells: an update. J Immunol (2016) 196(7):2923–31. doi:10.4049/jimmunol.1502570
40. Bjorkström NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood (2010) 116(19):3853–64. doi:10.1182/blood-2010-04-281675
41. Mocarski ES, Shenk T, Griffiths PD, Pass RF. Cytomegaloviruses. 6th ed. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins (2013). p. 1960–2015.
42. Crumpacker CS. Cytomegalovirus. 5th ed. In: Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. Philadelphia: Churchill Livingston (2000). p. 1586–99.
43. Pass RF, Fowler KB, Boppana SB, Britt WJ, Stagno S. Congenital cytomegalovirus infection following first trimester maternal infection: symptoms at birth and outcome. J Clin Virol (2006) 35(2):216–20.
44. Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med (2007) 357(25):2601–14.
45. Boeckh M, Ljungman P. How we treat cytomegalovirus in hematopoietic cell transplant recipients. Blood (2009) 113(23):5711–9. doi:10.1182/blood-2008-10-143560
46. Pawelec G, Goldeck D, Derhovanessian E. Inflammation, ageing and chronic disease. Curr Opin Immunol (2014) 29:23–8. doi:10.1016/j.coi.2014.03.007
47. Sagedal S, Hartmann A, Nordal KP, Osnes K, Leivestad T, Foss A, et al. Impact of early cytomegalovirus infection and disease on long-term recipient and kidney graft survival. Kidney Int (2004) 66(1):329–37.
48. Tong CY, Bakran A, Peiris JS, Muir P, Herrington CS. The association of viral infection and chronic allograft nephropathy with graft dysfunction after renal transplantation. Transplantation (2002) 74(4):576–8.
49. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol (2013) 132(3):515–25. doi:10.1016/j.jaci.2013.07.020
50. Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol (1999) 17:189–220.
51. Halenius A, Gerke C, Hengel H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: so many targets-but how many arrows in the quiver? Cell Mol Immunol (2015) 12(2):139–53. doi:10.1038/cmi.2014.105
52. Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. Viral subversion of the immune system. Annu Rev Immunol (2000) 18:861–926.
53. Lisnic B, Lisnic VJ, Jonjic S. NK cell interplay with cytomegaloviruses. Curr Opin Virol (2015) 15:9–18. doi:10.1016/j.coviro.2015.07.001
54. Wilkinson GW, Tomasec P, Stanton RJ, Armstrong M, Prod’homme V, Aicheler R, et al. Modulation of natural killer cells by human cytomegalovirus. J Clin Virol (2008) 41(3):206–12.
55. López-Botet M, Muntasell A, Vilches C. The CD94/NKG2C+ NK-cell subset on the edge of innate and adaptive immunity to human cytomegalovirus infection. Semin Immunol (2014) 26(2):145–51. doi:10.1016/j.smim.2014.03.002
56. Gumá M, Angulo A, Vilches C, Gómez-Lozano N, Malats N, López-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104(12):3664–71.
57. Wagner JA, Fehniger TA. Human adaptive natural killer cells: beyond NKG2C. Trends Immunol (2016) 37(6):351–3. doi:10.1016/j.it.2016.05.001
58. Gumá M, Cabrera C, Erkizia I, Bofill M, Clotet B, Ruiz L, et al. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J Infect Dis (2006) 194(1):38–41.
59. Petitdemange C, Becquart P, Wauquier N, Béziat V, Debré P, Leroy EM, et al. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog (2011) 7(9):e1002268. doi:10.1371/journal.ppat.1002268
60. Béziat V, Dalgard O, Asselah T, Halfon P, Bedossa P, Boudifa A, et al. CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur J Immunol (2012) 42(2):447–57. doi:10.1002/eji.201141826
61. Bjorkström NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med (2011) 208(1):13–21. doi:10.1084/jem.20100762
62. Muntasell A, López-Montañés M, Vera A, Heredia G, Romo N, Peñafiel J, et al. NKG2C zygosity influences CD94/NKG2C receptor function and the NK-cell compartment redistribution in response to human cytomegalovirus. Eur J Immunol (2013) 43:3268–78. doi:10.1002/eji.201343773
63. Béziat V, Liu L, Malmberg JA, Ivarsson MA, Sohlberg E, Bjorklund AT, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood (2013) 121:2678–88. doi:10.1182/blood-2012-10-459545
64. Djaoud Z, David G, Bressollette C, Willem C, Rettman P, Gagne K, et al. Amplified NKG2C+ NK cells in cytomegalovirus (CMV) infection preferentially express killer cell Ig-like receptor 2DL: functional impact in controlling CMV-infected dendritic cells. J Immunol (2013) 191(5):2708–16. doi:10.4049/jimmunol.1301138
65. López-Vergès S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, et al. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci U S A (2011) 108(36):14725–32. doi:10.1073/pnas.1110900108
66. Della Chiesa M, Falco M, Podesta M, Locatelli F, Moretta L, Frassoni F, et al. Phenotypic and functional heterogeneity of human NK cells developing after umbilical cord blood transplantation: a role for human cytomegalovirus? Blood (2012) 119(2):399–410. doi:10.1182/blood-2011-08-372003
67. Rölle A, Halenius A, Ewen EM, Cerwenka A, Hengel H, Momburg F. CD2-CD58 interactions are pivotal for the activation and function of adaptive natural killer cells in human cytomegalovirus infection. Eur J Immunol (2016) 46(10):2420–5. doi:10.1002/eji.201646492
68. Liu LL, Landskron J, Ask EH, Enqvist M, Sohlberg E, Traherne JA, et al. Critical role of CD2 co-stimulation in adaptive natural killer cell responses revealed in NKG2C-deficient humans. Cell Rep (2016) 15(5):1088–99. doi:10.1016/j.celrep.2016.04.005
69. Schlums H, Cichocki F, Tesi B, Theorell J, Béziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity (2015) 42(3):443–56. doi:10.1016/j.immuni.2015.02.008
70. Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity (2015) 42(3):431–42. doi:10.1016/j.immuni.2015.02.013
71. Costa-García M, Vera A, Moraru M, Vilches C, López-Botet M, Muntasell A. Antibody-mediated response of NKG2Cbright NK cells against human cytomegalovirus. J Immunol (2015) 194(6):2715–24. doi:10.4049/jimmunol.1402281
72. Wu Z, Sinzger C, Frascaroli G, Reichel J, Bayer C, Wang L, et al. Human cytomegalovirus-induced NKG2C(hi) CD57(hi) natural killer cells are effectors dependent on humoral antiviral immunity. J Virol (2013) 87(13):7717–25. doi:10.1128/JVI.01096-13
73. Zhang T, Scott JM, Hwang I, Kim S. Cutting edge: antibody-dependent memory-like NK cells distinguished by FcRγ deficiency. J Immunol (2013) 190(4):1402–6. doi:10.4049/jimmunol.1203034
74. Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood (2012) 119(11):2665–74. doi:10.1182/blood-2011-10-386995
75. Kuijpers TW, Baars PA, Dantin C, van den Burg M, van Lier RA, Roosnek E. Human NK cells can control CMV infection in the absence of T cells. Blood (2008) 112(3):914–5. doi:10.1182/blood-2008-05-157354
76. Noyola DE, Fortuny C, Muntasell A, Noguera-Julián A, Muñoz-Almagro C, Alarcón A, et al. Influence of congenital human cytomegalovirus infection and the NKG2C genotype on NK-cell subset distribution in children. Eur J Immunol (2012) 42:3256–66. doi:10.1002/eji.201242752
77. Noyola DE, Alarcón A, Noguera-Julian A, Muntasell A, Muñoz-Almagro C, García J, et al. Dynamics of the NK-cell subset redistribution induced by cytomegalovirus infection in preterm infants. Hum Immunol (2015) 76(2–3):118–23. doi:10.1016/j.humimm.2015.01.017
78. Bengner M, Béziat V, Ernerudh J, Nilsson BO, Lofgren S, Wikby A, et al. Independent skewing of the T cell and NK cell compartments associated with cytomegalovirus infection suggests division of labor between innate and adaptive immunity. Age (Dordr) (2014) 336(2):571–82. doi:10.1007/s11357-013-9587-y
79. Bigley AB, Spielmann G, Agha N, O’Connor DP, Simpson RJ. Dichotomous effects of latent CMV infection on the phenotype and functional properties of CD8+ T-cells and NK-cells. Cell Immunol (2016) 300:26–32. doi:10.1016/j.cellimm.2015.11.005
80. López-Botet M, Muntasell A, Martínez-Rodríguez JE, López-Montañés M, Costa-García M, Pupuleku A. Development of the adaptive NK cell response to human cytomegalovirus in the context of aging. Mech Ageing Dev (2016) 158:23–6. doi:10.1016/j.mad.2016.06.010
81. Muntasell A, Vilches C, Angulo A, López-Botet M. Adaptive reconfiguration of the human NK-cell compartment in response to cytomegalovirus: a different perspective of the host-pathogen interaction. Eur J Immunol (2013) 43(5):1133–41. doi:10.1002/eji.201243117
82. O’Sullivan TE, Sun JC, Lanier LL. Natural killer cell memory. Immunity (2015) 43(4):634–45. doi:10.1016/j.immuni.2015.09.013
83. Gumá M, Budt M, Sáez A, Brckalo T, Hengel H, Angulo A, et al. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood (2006) 107(9):3624–31.
84. Rölle A, Pollmann J, Ewen EM, Le VT, Halenius A, Hengel H, et al. IL-12-producing monocytes and HLA-E control HCMV-driven NKG2C+ NK cell expansion. J Clin Invest (2014) 124(12):5305–16. doi:10.1172/JCI77440
85. Djaoud Z, Riou R, Gavlovsky PJ, Mehlal S, Bressollette C, Gérard N, et al. Cytomegalovirus-infected primary endothelial cells trigger NKG2C+ natural killer cells. J Innate Immun (2016) 8(4):374–85. doi:10.1159/000445320
86. Miyashita R, Tsuchiya N, Hikami K, Kuroki K, Fukazawa T, Bijl M, et al. Molecular genetic analyses of human NKG2C (KLRC2) gene deletion. Int Immunol (2004) 16(1):163–8.
87. Moraru M, Cañiizares M, Muntasell A, de Pablo R, López-Botet M, Vilches C. Assessment of copy-number variation in the NKG2C receptor gene in a single-tube and characterization of a reference cell panel, using standard polymerase chain reaction. Tissue Antigens (2012) 80(2):184–7. doi:10.1111/j.1399-0039.2012.01911.x
88. Goodier MR, White MJ, Darboe A, Nielsen CM, Goncalves A, Bottomley C, et al. Rapid NK cell differentiation in a population with near-universal human cytomegalovirus infection is attenuated by NKG2C deletions. Blood (2014) 124(14):2213–22. doi:10.1182/blood-2014-05-576124
89. Rangel-Ramírez VV, García-Sepúlveda CA, Escalante-Padrón F, Pérez-González LF, Rangel-Castilla A, Aranda-Romo S, et al. NKG2C gene deletion in the Mexican population and lack of association to respiratory viral infections. Int J Immunogenet (2014) 41(2):126–30. doi:10.1111/iji.12104
90. Muntasell A, Pupuleku A, Cisneros E, Vera A, Moraru M, Vilches C, et al. Relationship of NKG2C copy number with the distribution of distinct cytomegalovirus-induced adaptive NK cell subsets. J Immunol (2016) 196:3818–27. doi:10.4049/jimmunol.1502438
91. Della Chiesa M, Falco M, Bertaina A, Muccio L, Alicata C, Frassoni F, et al. Human cytomegalovirus infection promotes rapid maturation of NK cells expressing activating killer Ig-like receptor in patients transplanted with NKG2C-/- umbilical cord blood. J Immunol (2014) 192:1471–9. doi:10.4049/jimmunol.1302053
92. Razonable RR, Humar A. Cytomegalovirus in solid organ transplantation. Am J Transplant (2013) 13(Suppl 4):93–106. doi:10.1111/ajt.12103
93. Demmers MW, Korevaar SS, Betjes MG, Weimar W, Rowshani AT, Baan CC. Limited efficacy of immunosuppressive drugs on CD8+ T cell-mediated and natural killer cell-mediated lysis of human renal tubular epithelial cells. Transplantation (2014) 97(11):1110–8. doi:10.1097/TP.0000000000000108
94. Morteau O, Blundell S, Chakera A, Bennett S, Christou CM, Mason PD, et al. Renal transplant immunosuppression impairs natural killer cell function in vitro and in vivo. PLoS One (2010) 5(10):e13294. doi:10.1371/journal.pone.0013294
95. Neudoerfl C, Mueller BJ, Blume C, Daemen K, Stevanovic-Meyer M, Keil J, et al. The peripheral NK cell repertoire after kidney transplantation is modulated by different immunosuppressive drugs. Front Immunol (2013) 4:46. doi:10.3389/fimmu
96. Kho MM, Bouvy AP, Cadogan M, Kraaijeveld R, Baan CC, Weimar W. The effect of low and ultra-low dosages thymoglobulin on peripheral T, B and NK cells in kidney transplant recipients. Transpl Immunol (2012) 26(4):186–90. doi:10.1016/j.trim.2012.02.003
97. Hadaya K, Avila Y, Valloton L, de Rham, Bandelier C, Ferrari-Lacraz S, et al. Natural killer cell receptor – repertoire and functions after induction therapy by polyclonal rabbit anti-thymocyte globulin in unsensitized kidney transplant recipients. Clin Immunol (2010) 137(2):250–60. doi:10.1016/j.clim.2010.07.004
98. Jones DC, Peacock S, Hughes D, Traherne JA, Allen RL, Barnardo MC, et al. Killer immunoglobulin-like receptor gene repertoire influences viral load of primary human cytomegalovirus infection in renal transplant patients. Genes Immun (2014) 15(8):562–8. doi:10.1038/gene.2014.53
99. Michelo CM, van der Meer A, Tijssen HJ, Zomer R, Stelma F, Hilbrands LB, et al. KIR and human leukocyte antigen genotype associated risk of cytomegalovirus disease in renal transplant patients. Transplantation (2015) 99(7):1506–13. doi:10.1097/TP.0000000000000497
100. De Rham C, Hadaya K, Bandelier C, Ferrari-Lacraz S, Villard J. Expression of killer cell immunoglobulin-like receptors (KIRs) by natural killer cells during acute CMV infection after kidney transplantation. Transpl Immunol (2014) 31(3):157–64. doi:10.1016/j.trim.2014.08.002
101. Stern M, Hadaya K, Honger G, Martin PY, Steiger J, Hess C, et al. Telomeric rather than centromeric activating KIR genes protect from cytomegalovirus infection after kidney transplantation. Am J Transplant (2011) 11(6):1302–7. doi:10.1111/j.1600-6143.2011.03516.x
102. González A, Schmitter K, Hirsch HH, Garzoni C, van Delden C, Boggian K, et al. KIR-associated protection from CMV replication requires pre-existing immunity: a prospective study in solid organ transplant recipients. Genes Immun (2014) 15(7):495–9. doi:10.1038/gene.2014.39
103. Trydzenskaya H, Juerchott K, Lachmann N, Kotsch K, Kunert K, Weist B, et al. The genetic predisposition of natural killer cell to BK virus-associated nephropathy in renal transplant patients. Kidney Int (2013) 84(2):359–65. doi:10.1038/ki.2013.59
104. Schmied L, Terszowski G, González A, Schmitter K, Hirsch HH, Garzoni C, et al. Protection from varicella zoster in solid organ transplant recipients carrying killer cell immunoglobulin-like receptor B haplotypes. Transplantation (2015) 99(12):2651–5. doi:10.1097/TP.0000000000000778
105. Redondo-Pachón D, Crespo M, Yélamos J, Muntasell A, Pérez-Sáez MJ, Pérez-Fernández S, et al. Adaptive NKG2C+ NK cell response and the risk of cytomegalovirus infection in kidney transplant recipients. J Immunol (2017) 198(1):94–101.
106. Makwana NB, Foley B, Lee S, Fernandez S, Irish AB, Price P. Asymptomatic CMV infections in long-term renal transplant recipients are associated with the loss of FcRgamma from LIR-1+ NK cells. Eur J Immunol (2016) 46(11):2597–608. doi:10.1002/eji.201646422
107. Berg L, Riise GC, Cosman D, Bergström T, Olofsson S, Kärre K, et al. LIR-1 expression on lymphocytes, and cytomegalovirus disease in lung-transplant recipients. Lancet (2003) 361(9363):1099–101.
108. Fernández-Ruiz M, Silva JT, López-Medrano F, Allende LM, San Juan R, Cambra F, et al. Post-transplant monitoring of NK cell counts as a simple approach to predict the occurrence of opportunistic infection in liver transplant recipients. Transpl Infect Dis (2016) 18(4):552–65. doi:10.1111/tid.12564
109. Bestard O, Lucia M, Crespo E, Van LB, Palacio D, Melilli E, et al. Pretransplant immediately early-1-specific T cell responses provide protection for CMV infection after kidney transplantation. Am J Transplant (2013) 13(7):1793–805. doi:10.1111/ajt.12256
110. Lucia M, Crespo E, Cruzado JM, Grinyó JM, Bestard O. Human CMV-specific T-cell responses in kidney transplantation; toward changing current risk-stratification paradigm. Transpl Int (2014) 27(7):643–56. doi:10.1111/tri.12318
111. Kaminski H, Garrigue I, Couzi L, Taton B, Bachelet T, Moreau JF, et al. Surveillance of gammadelta T cells predicts cytomegalovirus infection resolution in kidney transplants. J Am Soc Nephrol (2016) 27(2):637–45. doi:10.1681/ASN.2014100985
112. Saghafian-Hedengren S, Sohlberg E, Theorell J, Carvalho-Queiroz C, Nagy N, Persson JO, et al. Epstein-Barr virus coinfection in children boosts cytomegalovirus-induced differentiation of natural killer cells. J Virol (2013) 87(24):13446–55. doi:10.1128/JVI.02382-13
113. Ishibashi K, Yamaguchi O, Suzutani T. Reinfection of cytomegalovirus in renal transplantation. Fukushima J Med Sci (2011) 57(1):1–10.
114. Achour A, Baychelier F, Besson C, Arnoux A, Marty M, Hannoun L, et al. Expansion of CMV-mediated NKG2C+ NK cells associates with the development of specific de novo malignancies in liver-transplanted patients. J Immunol (2014) 192(1):503–11. doi:10.4049/jimmunol.1301951
115. Kunert K, Seiler M, Mashreghi MF, Klippert K, Schonemann C, Neumann K, et al. KIR/HLA ligand incompatibility in kidney transplantation. Transplantation (2007) 84(11):1527–33.
116. Van Bergen J, Thompson A, Haasnoot GW, Roodnat JI, de Fijter JW, Claas FH, et al. KIR-ligand mismatches are associated with reduced long-term graft survival in HLA-compatible kidney transplantation. Am J Transplant (2011) 11(9):1959–64. doi:10.1111/j.1600-6143.2011.03621.x
117. Tran TH, Unterrainer C, Fiedler G, Dohler B, Scherer S, Ruhenstroth A, et al. No impact of KIR-ligand mismatch on allograft outcome in HLA-compatible kidney transplantation. Am J Transplant (2013) 13(4):1063–8. doi:10.1111/ajt.12134
118. Kreijveld E, van der Meer A, Tijssen HJ, Hilbrands LB, Joosten I. KIR gene and KIR ligand analysis to predict graft rejection after renal transplantation. Transplantation (2007) 84(8):1045–51.
119. Kumar V, George T, Yu YYL, Liu J, Bennett M. Role of murine NK cells and their receptors in hybrid resistance. Curr Opin Immunol (1997) 9:52–6.
120. Velardi A, Ruggeri L, Mancusi A. Killer-cell immunoglobulin-like receptors reactivity and outcome of stem cell transplant. Curr Opin Hematol (2012) 19(4):319–23. doi:10.1097/MOH.0b013e32835423c3
121. Locatelli F, Moretta F, Brescia L, Merli P. Natural killer cells in the treatment of high-risk acute leukaemia. Semin Immunol (2014) 26(2):173–9. doi:10.1016/j.smim.2014.02.004
122. Knorr DA, Bachanova V, Verneris MR, Miller JS. Clinical utility of natural killer cells in cancer therapy and transplantation. Semin Immunol (2014) 26(2):161–72. doi:10.1016/j.smim.2014.02.002
123. Locatelli F, Pende D, Mingari MC, Bertaina A, Falco M, Moretta A, et al. Cellular and molecular basis of haploidentical hematopoietic stem cell transplantation in the successful treatment of high-risk leukemias: role of alloreactive NK cells. Front Immunol (2013) 4:15. doi:10.3389/fimmu.2013.00015
124. Zhang ZX, Huang X, Jiang J, Lau A, Yin Z, Liu W, et al. Natural killer cells mediate long-term kidney allograft injury. Transplantation (2015) 99(5):916–24. doi:10.1097/TP.0000000000000665
125. Baldwin WM III, Valujskikh A, Fairchild RL. Mechanisms of antibody-mediated acute and chronic rejection of kidney allografts. Curr Opin Organ Transplant (2016) 21(1):7–14. doi:10.1097/MOT.0000000000000262
126. Hidalgo LG, Sellares J, Sis B, Mengel M, Chang J, Halloran PF. Interpreting NK cell transcripts versus T cell transcripts in renal transplant biopsies. Am J Transplant (2012) 12(5):1180–91. doi:10.1111/j.1600-6143.2011.03970.x
127. Venner JM, Hidalgo LG, Famulski KS, Chang J, Halloran PF. The molecular landscape of antibody-mediated kidney transplant rejection: evidence for NK involvement through CD16a Fc receptors. Am J Transplant (2015) 15(5):1336–48. doi:10.1111/ajt.13115
128. Sellares J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant (2012) 12(2):388–99. doi:10.1111/j.1600-6143.2011.03840.x
129. Gupta A, Broin PO, Bao Y, Pullman J, Kamal L, Ajaimy M, et al. Clinical and molecular significance of microvascular inflammation in transplant kidney biopsies. Kidney Int (2016) 89(1):217–25. doi:10.1038/ki.2015.276
130. Halloran PF, Famulski K, Reeve J. The molecular phenotypes of rejection in kidney transplant biopsies. Curr Opin Organ Transplant (2015) 20(3):359–67. doi:10.1097/MOT.0000000000000193
131. Clemenceau B, Vivien R, Berthome M, Robillard N, Garand R, Gallot G, et al. Effector memory alphabeta T lymphocytes can express FcgammaRIIIa and mediate antibody-dependent cellular cytotoxicity. J Immunol (2008) 180(8):5327–34.
132. Couzi L, Pitard V, Sicard X, Garrigue I, Hawchar O, Merville P, et al. Antibody-dependent anti-cytomegalovirus activity of human gammadelta T cells expressing CD16 (FcgammaRIIIa). Blood (2012) 119(6):1418–27. doi:10.1182/blood-2011-06-363655
133. Bachelet T, Couzi L, Pitard V, Sicard X, Rigothier C, Lepreux S, et al. Cytomegalovirus-responsive gammadelta T cells: novel effector cells in antibody-mediated kidney allograft microcirculation lesions. J Am Soc Nephrol (2014) 25(11):2471–82. doi:10.1681/ASN.2013101052
134. Hoffmann U, Neudorfl C, Daemen K, Keil J, Stevanovic-Meyer M, Lehner F, et al. NK cells of kidney transplant recipients display an activated phenotype that is influenced by immunosuppression and pathological staging. PLoS One (2015) 10(7):e0132484. doi:10.1371/journal.pone.0132484
Keywords: human, natural killer, cytomegalovirus, renal, transplantation, rejection
Citation: López-Botet M, Vilches C, Redondo-Pachón D, Muntasell A, Pupuleku A, Yélamos J, Pascual J and Crespo M (2017) Dual Role of Natural Killer Cells on Graft Rejection and Control of Cytomegalovirus Infection in Renal Transplantation. Front. Immunol. 8:166. doi: 10.3389/fimmu.2017.00166
Received: 02 December 2016; Accepted: 02 February 2017;
Published: 16 February 2017
Edited by:
Emanuela Marcenaro, University of Genoa, ItalyReviewed by:
Michael G. Brown, University of Virginia, USAEnnio Carbone, Magna Græcia University, Italy
Copyright: © 2017 López-Botet, Vilches, Redondo-Pachón, Muntasell, Pupuleku, Yélamos, Pascual and Crespo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel López-Botet, bGJvdGV0QGltaW0uZXM=, bWlndWVsLmxvcGV6LWJvdGV0QHVwZi5lZHU=