 Paulo C. Rodriguez1
Paulo C. Rodriguez1 Amir A. Al-Khami
Amir A. Al-Khami- 1Augusta University, Georgia Cancer Center, Augusta, GA, USA
- 2Stanley S. Scott Cancer Center, Louisiana State University Health Sciences Center, New Orleans, LA, USA
- 3Department of Pediatrics, Louisiana State University Health Sciences Center, New Orleans, LA, USA
- 4Department of Genetics, Louisiana State University Health Sciences Center, New Orleans, LA, USA
Arginine metabolism has been a key catabolic and anabolic process throughout the evolution of the immune response. Accruing evidence indicates that arginine-catabolizing enzymes, mainly nitric oxide synthases and arginases, are closely integrated with the control of immune response under physiological and pathological conditions. Myeloid cells are major players that exploit the regulators of arginine metabolism to mediate diverse, although often opposing, immunological and functional consequences. In this article, we focus on the importance of arginine catabolism by myeloid cells in regulating innate and adaptive immunity. Revisiting this matter could result in novel therapeutic approaches by which the immunoregulatory nodes instructed by arginine metabolism can be targeted.
Introduction
Arginine (R) is considered a non-essential amino acid for healthy adult humans since it is endogenously synthesized from the amino acid citrulline as an immediate precursor in virtually all cell types. The small intestine is the major source of citrulline for arginine synthesis by the proximal tubules of the kidneys, known as the intestinal–renal axis for arginine synthesis (1, 2). The normal range of arginine in serum fluctuates between 50 and 150 µM (3, 4). However, arginine is generally classified as a semi or conditionally essential amino acid owing to the fact that arginine must be supplied in the diet in some pathological conditions, including sepsis, trauma, and cancer (5, 6). Arginine metabolism is regulated both through the expression of the y+ system of cationic amino acid transporters (7) and through the enzymes responsible for its catabolism. Arginine is metabolized intracellularly by nitric oxide synthase (NOS), arginase, arginine:glycine amidinotransferase (AGAT), and arginine decarboxylase (ADC). These enzymes are expressed in a tissue-specific manner, and some of them are induced under particular inflammatory settings.
Arginine metabolism has emerged as a critical regulator of innate and adaptive immune responses. The major arginine-catabolizing enzymes involved in inflammatory immune responses are the isoforms of NOS (NOS1–3) and arginase (arginase 1 and 2). It is becoming increasingly clear that cells of the myeloid lineage can augment or diminish the immune response via the differential regulation of these enzymes. These processes are fundamentally driven by a multitude of inflammatory cues within tissue microenvironments. Importantly, targeting arginine metabolism can modulate key aspects of these critical cells, resulting in a better disease control. As such, this review will discuss how arginine catabolic pathways can lead to heterogeneous, but often opposing, functional consequences and how these mechanisms can be harnessed for the treatment of multiple pathological conditions.
Arginine Metabolism: NOS
Three NOS isozymes, encoded by distinct genes, have been identified: NOS1 (known as neuronal NOS, nNOS), NOS2 (known as inducible NOS, iNOS, found in several myeloid cell populations and some T cell subsets), and NOS3 (known as endothelial NOS, eNOS) (2, 8, 9). All NOS enzymes metabolize arginine to produce nitric oxide, which crucially participates in processes associated with vasodilatation and cytotoxic mechanisms (9–11), in addition to citrulline generated as a byproduct. Both NOS1 and NOS3 are constitutively expressed in various types of cells, with their activities being dependent on calcium-calmodulin. On the contrary, NOS2 is controlled through inducible transcription in response to pro-inflammatory cytokines such as interferon γ (IFNγ), tumor necrosis factor α (TNFα), and IL-1β and bacterial lipopolysaccharide (Figure 1). Once stimulated, NOS2 is constantly activated and not controlled by calcium levels (8, 12, 13). Induction of NOS2 has been described primarily in macrophages (14) but also in other cells, including colon (15) and lung (16) epithelial cells and CD4+ T cells (17). NOS-derived nitric oxide can stimulate multiple enzymes and proteins inside the target cell. Among these pathways, activation of soluble guanylyl cyclase by nitric oxide to generate cyclic guanosine monophosphate is thought to be the most important (12). NOS is inhibited endogenously by asymmetric dimethylarginine (aDMA), an arginine analog and naturally occurring product of metabolism, or pharmacologically by arginine analogs such as l-NG-monomethylarginine (l-NMMA) among several others (18).
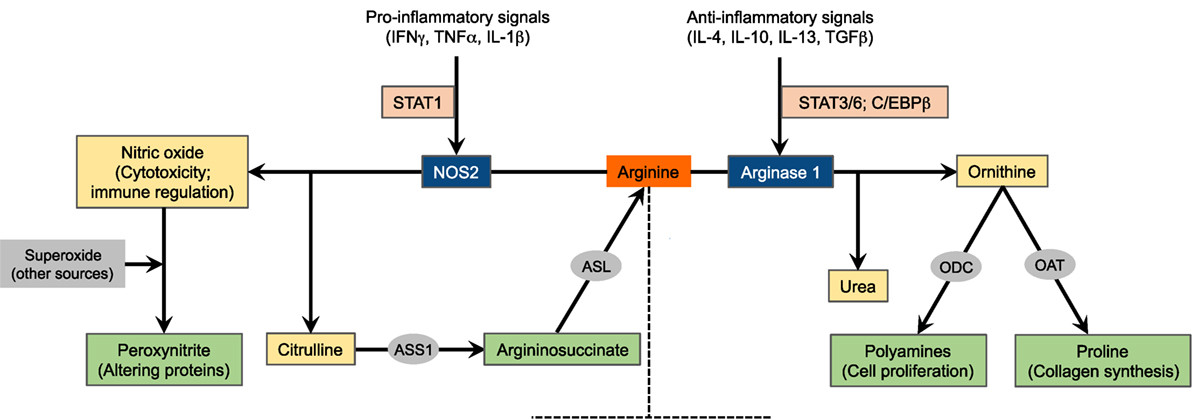
Figure 1. Schematic of arginine metabolism. For the sake of simplicity, the major arginine-catabolizing enzymes involved in inflammatory immune responses, NOS2 and arginase 1, are depicted. The expression of these enzymes is tightly regulated by microenvironmental inflammatory signals. This diagram, however, does not suggest that these enzymes are concurrently induced in a given cell type. NOS2, nitric oxide synthase 2; ASS1, argininosuccinate synthase 1; ASL, argininosuccinate lyase; ODC, ornithine decarboxylase; OAT, ornithine aminotransferase.
Arginine Metabolism: Arginase
Arginine is alternatively metabolized by arginases to produce ornithine and urea. Ornithine is the precursor for the production of polyamines via the ornithine decarboxylase (ODC) pathway as well as for the production of proline via the enzyme ornithine aminotransferase (OAT). While polyamines essentially regulate cell proliferation and differentiation, proline is critical for the synthesis of collagen, a primary protein in wound healing (2, 19, 20). Additionally, urea represents an important mechanism for detoxification of protein degradation. Arginase exists in two isoforms, arginase 1 and arginase 2, that induce the same reaction but are encoded by separate genes and differ in tissue distribution and intracellular localization. Arginase 1 is found in the cytosol of hepatocytes, macrophages, and other myeloid cells and in the granular compartment of human granulocytes. Recently, arginase 1 has been shown to be expressed by mouse and human innate lymphoid cells group 2 (21). Arginase 2, on the other hand, is a mitochondrial enzyme that is expressed in tissues such as kidneys, small intestine, and brain, in addition to most cells in the body (22, 23). The expression of arginase 1 is induced in myeloid cells by the T helper 2 (Th2) cytokines IL-4 and IL-13 (Figure 1). These cytokines activate the signal transducer and activator of transcription 6 (STAT6) that, with other transcription factors such as STAT3 and CCAAT/enhancer binding protein β (C/EBPβ), binds to an enhancer in the arginase 1 locus (24–26). Multiple other factors also induce the expression of arginase 1, including IL-10 (27), granulocyte-macrophage colony-stimulating factor (GM-CSF) (28), transforming growth factor β (TGFβ) (29), prostaglandin E2 (PGE2) (30), cyclic adenosine monophosphate (cAMP) (31), and toll-like receptor (TLR) agonists (32). Arginase 1 expression is also controlled by peroxisome proliferator-activated receptor transcription factors (33, 34). Conversely, arginase 2 is constitutively expressed. Given that the role of myeloid cell arginase 2 in shaping immune responses is not as clearly defined as arginase 1, we will only discuss the latter herein. However, recent evidence indicates that arginase 2 induced in other cell types like asthmatic airway epithelium (35) and activated T cells (36) regulates arginine flux, thereby redirecting the immune response and disease manifestation. This suggests that further investigation of arginase 2 in myeloid cells is warranted.
NOS and Arginase: Competitors for Arginine
Before we discuss the functional consequences of the regulated arginine metabolism in myeloid cells (Figure 2), it is important to emphasize the competition between NOS and arginase for the available intracellular arginine as a major mechanism that dictates the ultimate immune response outcome, as detailed below. The intracellular levels of arginine are in the range of 100–800 µM. The arginine Km of NOS is 3 µM, whereas that of arginase is close to 2 mM (2). Therefore, under physiological conditions, NOS should have a higher access to arginine than arginase. However, the Vmax of NOS is almost 1,000 times less than that of arginase, which equilibrates their capabilities to metabolize arginine (2, 37). As a proof of the balanced access of NOS and arginase to arginine is the fact that, despite the higher affinity of NOS to arginine, the production of nitric oxide depends of the extracellular levels of arginine (termed as arginine paradox) (38, 39). A potential explanation for this effect is the subcellular compartmentalization of arginine. However, studies that have regulated the localization of NOS and arginase have failed to confirm this concept (40). An additional level of complexity in the interaction between NOS and arginase is the uncoupling of NOS by arginase (41, 42). The uncoupled NOS produces less nitric oxide and uses more molecular oxygen to generate superoxide, thereby leading to the formation of peroxynitrite (PNT). Thus, the coexpression of arginase and NOS and the subsequent production of PNT in subsets of myeloid cells like myeloid-derived suppressor cells (MDSCs) could be the result of an uncoupled NOS. Moreover, increased arginase expression can limit NOS2 expression in immune cells by decreasing the arginine needed for NOS2 translation (43).
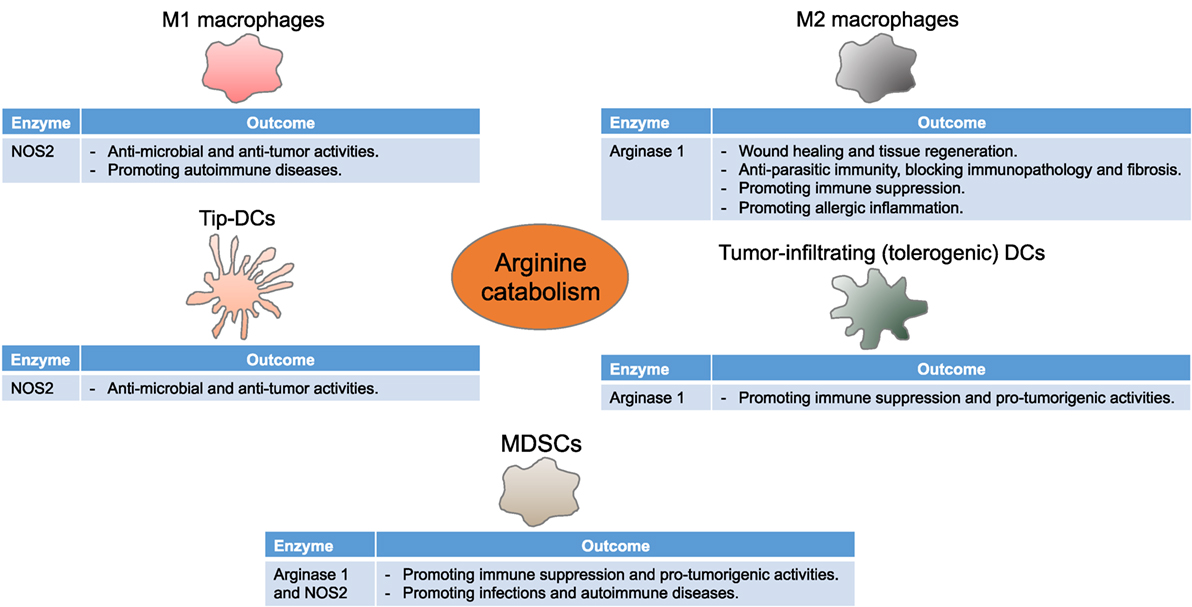
Figure 2. Arginine metabolism instructs myeloid cells to control immune responses. Myeloid cells differentially express NOS2 and arginase 1, thereby driving diverse, although seeming contradictory, immune and functional consequences in multiple disease settings. Tip-DCs, tumor necrosis factor α and inducible NOS-producing dendritic cells; MDSCs, myeloid-derived suppressor cells.
Arginine Metabolism in Macrophages
The concept of basing macrophage activation into M1 and M2 subsets with distinct functional consequences on the usage of arginine via NOS or arginase has been described several decades ago. M1 and M2 macrophages induce Th1 and Th2-like inflammatory responses that further intensify M1- and M2-type responses, respectively (44, 45). Stimulation of bone marrow-derived or peritoneal inflammatory macrophages with TLR agonists activates transcription factors such as nuclear factor kappa-light-chain-enhancer (NF-κB) that induce pro-inflammatory cytokines like IFNγ resulting in NOS2-expressing M1 macrophages, while cytokines like IL-4 and IL-13 activate STAT6 and lead to arginase 1-expressing M2 macrophages (46–49). Arginase 2, however, is not significantly modulated by Th1 or Th2 cytokines (4). Due to the fact that the in vitro culture systems do not precisely represent the multiple potential factors that affect macrophages in vivo as well as the discrepancies in the mouse and human results, the phenotypic and functional aspects of macrophage polarization remain to be tackled (50–52).
Arginine Metabolism in Classically Activated Macrophages
M1 macrophages, via NOS2, metabolize arginine to nitric oxide and citrulline (8, 14). Besides its multiple signaling pathways (9, 12), the cytotoxic properties of nitric oxide make M1 macrophages well suited to function as key effector cells for the elimination of intracellular pathogens and tumor cells. When induced, M1 macrophages likely use all imported arginine, and NOS2 generates nitric oxide in large quantities, also indicating the importance of arginine availability and uptake as a rate-limiting step for nitric oxide synthesis (53). To more efficiently produce nitric oxide, or when arginine is limited, citrulline is reused to synthesize nitric oxide via the so-called citrulline–nitric oxide cycle. These reactions involve two enzymes: argininosuccinate synthase (ASS1) and argininosuccinate lyase (ASL) (Figure 1). While ASS1 can be induced by TLR agonists and IFNγ, M1 macrophages constitutively express ASL (54, 55). In fact, mice lacking ASS1 fail to control mycobacteria infection, therefore confirming the importance of citrulline recycling via ASS1 and ASL in nitric oxide production by M1 macrophages (56).
Excessive nitric oxide synthesis can lead to unwanted host cytotoxicities and imbalanced immune responses. Upregulation of arginase 1, however, is a means by which macrophages limit the availability of arginine and regulate nitric oxide production (57). Besides the fact that the availability of arginine may control the translation of NOS2 mRNA (43), polyamines, which restrict the immune effector function of macrophages in response to TLR agonists (58, 59), also inhibit the cationic amino acid transporter 2B (CAT-2B) arginine transporter and nitric oxide synthesis in macrophages (60). Consistent with this, polyamines diminish Helicobacter pylori-induced NOS2 protein levels and nitric oxide production in macrophages through a post-transcriptional effect on NOS2 translation, whereas ODC inhibition enhances NOS2 protein expression and macrophage nitric oxide-dependent killing of bacteria (61). Another mechanism underlying the upregulation of arginase 1 in M1 macrophages involves a TLR-myeloid differentiation primary response 88 (MyD88)-dependent pathway. TLR signaling in mycobacteria-infected macrophages stimulates the production of cytokines like IL-6, IL-10, and granulocyte colony-stimulating factor (G-CSF) that provoke arginase 1 expression in an autocrine–paracrine fashion, involving the transcription factors STAT3 and C/EBPβ (62).
Given the complexity of inflammatory cues within diseased tissue microenvironments, it is essential to consider the interrelated and dichotomous regulation of macrophage arginine metabolism in determining the type and outcome of immune response against pathogens. For instance, in a model of Leishmania major, TNFα mediates protection by restraining the development of arginase 1-expressing M2 macrophages and dendritic cells (DCs), while maintaining the production of nitric oxide in situ (63). Other disease models, however, show different interactions between NOS2 and arginase 1. In tuberculosis granulomas, arginase 1-expressing M2 macrophages localize to the outer regions of granulomas, while NOS2-expressing M1 macrophages can be found in the inner regions. This provides an organized microenvironment within granulomas that separates anti-microbial (M1, NOS2-mediated) and anti-inflammatory (M2, arginase 1-mediated) responses to constraint lung pathology (64). In addition to its role in regulating NOS2 activity through arginine competition, arginase 1 also regulates T cell proliferation, thereby playing a significant role in the control of Mycobacterium tuberculosis growth and pathology independently of NOS2 suppression (65). Conversely, when M. tuberculosis-infected mice are coinfected with Schistosoma mansoni, arginase 1-expressing M2 macrophages expand and limit the microbicidal immune response, thus increasing the disease progression and severity (66). Such findings clearly point out to the significance of the signals within in vivo immune milieus that can instruct macrophages and can often not be uncovered through in vitro evaluation.
Arginine Metabolism in Alternatively Activated Macrophages
Arginase 1-expressing M2 macrophages play pivotal roles in multiple immunopathological settings. M2 macrophages regulate immune responses mostly through redirecting arginine away from NOS with arginase 1 or via ornithine production. Indeed, macrophages producing arginase 1 and ornithine have widely been considered critical for wound healing (67, 68). These results were recently confirmed using pharmacologic inhibition of arginase and, more importantly, macrophage-specific arginase 1 knockout mice (69). The decrease in arginase 1 expression results in a heightened infiltration of NOS2-expressing cells, reduced matrix deposition, and delayed healing (69).
M2 macrophage-specific arginase 1 blocks inflammation and fibrosis post infection with S. mansoni. For instance, conditional deletion of arginase 1 in macrophages is associated with accelerated mortality due to uncontrolled Th2 cytokine-induced pathology in the livers of infected mice (70). Although early studies of schistosomiasis pathology indicate that M2 macrophage arginase 1 might contribute to liver fibrosis via production of proline as a precursor of collagen (46), mice lacking arginase 1 in macrophages display increased liver fibrosis and collagen deposition (70). Likewise, macrophage-derived arginase 1 is protective against excessive injury of the intestinal tissue of infected mice (71). In addition to suppressing T cell proliferation, arginase 1 enhances regulatory T (Treg) cell and limits Th17 cell phenotype; furthermore, arginase 1 deficiency in macrophages results in IL-12/IL-23p40-dependent neutrophil-linked gut pathology (71). On the contrary, macrophage-specific arginase 1 is not essential in multiple murine models of Th2 lung inflammation and asthma (72), suggesting that the regulatory outcomes of macrophage-derived arginase 1 are organ specific. Although not technically supported, it is hypothesized that the function of arginase 1 varies according to the relative rate of perfusion, and hence ultimately arginine availability, in different organs (72). Nevertheless, other studies indicate that subsets of arginase 1-producing M2 macrophages may serve a role in allergic immune responses (73), indicating that this still is an exciting area for future investigation.
Macrophage arginase 1 also directly controls parasite growth. In a murine model of Heligmosomoides polygyrus, memory CD4+ T cells produce IL-4, therefore recruiting M2 macrophages that block larval parasite health and mobility via an arginase 1-dependent mechanism (74). Additionally, H. polygyrus-specific antibodies and helminth larvae induce arginase 1 expression in macrophages independent of IL-4Rα signaling, and the arginase 1 product ornithine directly inhibits larval motility (74). With this result, antibodies represent a novel pathway of macrophage alternative activation throughout Th2 immune responses.
Arginine Metabolism in DCs
Dendritic cells are the most professional antigen-presenting cells known as key mediators between innate and adaptive immune responses. They present pathogen-derived antigenic peptides and provide costimulatory molecules and cytokines crucial for T cell activation and differentiation (75, 76). As such, how properly DC function is regulated largely shapes T cell antigen-specific immunity in different disease scenarios. DCs are classified into several subpopulations with distinct phenotypes, functions, and locations. These include but are not limited to conventional DCs, monocyte-derived DCs, and plasmacytoid DCs (77, 78). The enzymes involved in arginine metabolism, NOS2 and arginase 1, are implicated in the function of subsets of DCs that evolve in response to local environmental stimuli. Similar to macrophages, the differential expression of these enzymes results in dichotomous functions within these critical immune cells.
A population of TNFα and iNOS-producing DCs (Tip-DCs) has recently been described (79, 80). These cells are characterized as CD11b+CD11c+Ly6C+MHC-II+. The initial reports indicate that Tip-DCs can mediate the resistance to pathogens such as Listeria (79), Brucella (81), and Leishmania (82). However, Tip-DCs can also contribute to the liver pathogenicity in Trypanosoma brucei-infected mice; in this context, TNFα and nitric oxide production is IFNγ and MyD88 signaling dependent (83). Moreover, Tip-DCs can interact with tumor-infiltrating antigen-specific CD8+ T cells to mediate tumor growth rejection (84). In this model, tumor antigen-reactive CD8+ T cells activate Tip-DCs that in turn present tumor-associated antigens, thereby enhancing T cell expansion and tumor killing via TNFα and nitric oxide production. While colony-stimulating factor 1 receptor (CSF-1R) signaling is not required, CD40–CD40L signaling is a key pathway for nitric oxide production and the antitumor response (84). Thus, activated T cells carry the possibility to modulate the inflammatory tumor microenvironment despite the fact that they are also targets of the suppressive elements of the same milieu, suggesting the tight balance occurring in tumors.
Tumors, however, can educate DCs to acquire an immunosuppressive phenotype, represented by low costimulatory molecule expression, poor antigen presentation, and high expression of regulatory receptors (85). Murine lung cancer-derived TGFβ and PGE2 favor the generation of tolerogenic DCs (CD11bhighCD11clowMHC-IIlow) that inhibit the proliferation of CD4+ T cells in vitro and in vivo (86). Arginase 1, induced mainly by PGE2, plays a significant role in this effect (86). IL-6 also promotes the expression and activity of arginase 1 that subsequently downregulates MHC-II in DCs and suppresses CD4+ T cell-mediated antitumor immunity (87). Although murine spontaneous mammary tumors are infiltrated with phenotypically mature DCs (CD11b+CD11chighMHC-IIhigh), these DCs suppress CD8+ T cell function via arginase 1 production, thus leading to impaired T cell antitumor immunity (88).
Arginine Metabolism in MDSCs
Myeloid-derived suppressor cells are associated with several immune regulatory aspects in conditions involving chronic inflammation, such as cancer (89, 90), infections (91–93), trauma (94), obesity (95, 96), graft versus host disease (97), and autoimmune diseases (98, 99). With a sustained status of abnormal myelopoiesis, MDSCs represent a heterogeneous population of myeloid progenitor cells distinct from mature myeloid cells like macrophages, DCs, and neutrophils. MDSCs potently suppress innate and adaptive immunity and comprise two major subsets, namely, monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs). In mice, M-MDSCs are CD11b+Ly6ChighLy6G−, while PMN-MDSCs are CD11b+Ly6ClowLy6G+. In humans, M-MDSCs are defined as CD33+CD14+CD15−HLA-DRlow, while PMN-MDSCs are CD33+CD14−CD15+HLA-DR−/low or CD33+CD14−CD66b+HLA-DR−/low (100).
Multiple tumor-associated factors drive MDSC accumulation and acquisition of immunosuppressive function. For instance, vascular endothelial growth factor is associated with an arrest in DC maturation, while concomitantly expanding MDSCs, through the inhibition of NF-κB signaling (101, 102). Whereas G-CSF plays a critical role in mobilizing and differentiating bone marrow granulocytic precursors within tumors (103), GM-CSF, depending on the stimulation magnitude and context, promotes MDSC accumulation in vitro and in vivo (104, 105). IL-1β is also a potent driver of MDSCs either directly or indirectly through stimulating other mediators such as IL-6 (106, 107). IL-4 and IL-13 evoke MDSC suppressive mechanisms via IL-4Rα-dependent STAT6 activation (108, 109). Other pro-inflammatory danger signals secreted mostly by myeloid cells such as S100A8/A9 proteins and high-mobility group box 1 can also enhance MDSC trafficking and function by signaling through cell membrane receptors like TLRs and the receptor for advanced glycation end-products (110, 111). Similarly, several transcription factors are critical for MDSCs. Among those, STAT1 activated by type 1 and 2 IFNs and IL-1β drives MDSC accumulation and regulatory mechanisms (NOS2 and arginase 1) (112, 113). The induction of STAT3 and STAT5 via G-CSF and GM-CSF, respectively, downregulates IFN regulatory factor 8 that essentially drives MDSC accumulation (114). STAT3 can also induce genes important for MDSC differentiation and proliferation (c-myc, cyclin D1, and S100A8/A9) and suppressive function (NADPH oxidase [NOX] subunits p47phox and gp91phox and C/EBPβ) (115–118). C/EBPβ-homologous protein (Chop) induced by tumor-derived reactive oxygen species (ROS) and reactive nitrogen species (RNS) and regulated by activating-transcription factor 4 promotes IL-6 production and activates C/EBPβ as well as STAT3, thereby mediating MDSC regulatory function (119).
Myeloid-derived suppressor cells promote immune dysfunction using different mechanisms, either directly via depriving T cells of essential metabolites such as arginine, tryptophan, and cysteine or interfering with T cell viability, migration, or activation or indirectly via inducing other immune regulatory cells such as Treg cells and tumor-associated macrophages (TAMs) (89, 90, 120). Depletion of arginine through arginase 1 is one of the first T cell suppressive mechanisms described in MDSCs. PMN-MDSCs, the major source of arginase 1 in tumor-bearing hosts, reduce extracellular arginine by arginine incorporation via CAT-2B or arginase 1 production (3, 121). Arginase 1 inhibitors such as Nω-hydroxy-nor-arginine (nor-NOHA) or Nω-hydroxy-arginine (NOHA) block MDSC suppressive activity and result in an immune-mediated, dose-dependent T cell antitumor immunity (3, 90). Starving T cells of arginine downregulates the expression of CD3ζ, a hallmark of T cell dysfunction in cancer patients (122). However, arginine-starved T cells produce IL-2 and upregulate the early activation markers CD25, CD69, and CD122, indicating that the effect induced by arginine depletion is not due to a defect in T cell receptor (TCR) signaling (123). On the other hand, arginine-starved T cells are arrested in the G0–G1 phase of the cell cycle as a result of an impaired expression of cyclin D3 and cyclin-dependent kinase 4 (cdk4) in T cells through a decreased mRNA stability and diminished translational rate (124, 125). Interestingly, in vivo deprivation of arginine impairs T cell responses due to a general control non-derepressible 2 (GCN2) kinase-dependent accumulation of MDSCs (126). More recently, arginine has been found to be critical for T cell metabolic fitness and survival, and therefore, increasing the intracellular arginine abundance in T cells prior to adoptive cellular therapy (ACT) enhances their persistence and antitumor responses (36). In addition to the high susceptibility to low extracellular arginine, T cells also fail to respond in environments that lack cysteine or in those having indoleamine 2,3-dioxgenase (IDO)-mediated tryptophan deprivation (127, 128). A potential role of the integrated stress responses has been suggested as a common mediator of the effects induced by amino acid deprivation (124, 129). Briefly, accumulation of empty aminoacyl-tRNAs caused by low amino acid content activates GCN2, which phosphorylates the eukaryotic translation initiation factor 2α (eIF2α). The phosphorylated form of eIF2α binds with higher affinity to eIF2β, blocking its ability to exchange guanosine diphosphate (GDP) for guanosine triphosphate (GTP), which then inhibits the binding of the eIF2 complex to methionine aminoacyl-tRNA. This results in a decreased initiation of global protein synthesis. Accordingly, culture of cells in the absence of arginine induces a significant phosphorylation of eIF2α and global decrease in protein synthesis (130). In addition, T cells from GCN2 knockout mice display a lower susceptibility to amino acid availability (129). Recent studies also point out to the essential role of rapamycin-insensitive companion of mammalian target of rapamycin (Rictor)/mTOR complex 2 in regulating the responses induced by limiting amino acids (131). As such, T cells lacking Rictor/mTOR are resistant to amino acid starvation-induced immunosuppression (132, 133), suggesting the relevance of this pathway in the suppression of T cell responses by amino acid depletion.
Myeloid-derived suppressor cells also exert their immunosuppressive effect through nitric oxide production by NOS2 in M-MDSCs and NOS3 in PMN-MDSCs (134). In addition to its direct apoptotic effects, nitric oxide negatively regulates T cells by impairing the IL-2R signaling pathways Jak-3, STAT5, ERK, and AKT (135, 136). Furthermore, MDSCs via the NOX subunits p22phox, p47phox, and gp91phox produce ROS such as superoxide and hydrogen peroxide that inhibit T cell CD3ζ expression and cytokine production (116). Nitric oxide can then react with superoxide to produce more detrimental RNS such as PNT generated by PMN-MDSCs depending on the expression of gp91phox and NOS3 (134). PNT can induce T cell apoptosis via the nitration of tyrosine residues, thereby blocking protein tyrosine phosphorylation (137). PNT also disrupts the conformational flexibility of the TCR-MHC/peptide binding by nitrating/nitrosylating the TCR and MHC, thus limiting T cell antitumor immunity (138, 139). Moreover, PNT hinders the infiltration of T cells, while facilitating the trafficking of MDSCs, into tumors mostly through the nitration of chemokines such as CCL2 and CCL5 or chemokine receptors such as CXCR4 (140, 141).
Arginine Methylation in Inflammation
Posttranslational methylation of arginine residues in proteins through the protein arginine methyltransferases (PRMTs) regulates multiple cellular signaling pathways related to cell differentiation, proliferation, and function. Arginine methylation by PRMTs activates or inhibits multiple transcription factors and other proteins, thereby regulating chromatin remodeling, RNA splicing, DNA damage repair, and protein–protein interactions. These events occur through the formation of aDMA, made by type I PRMTs, symmetric dimethylarginine (sDMA), made by type II PRMTs, or monomethylarginine (MMA), made by type III PRMTs. The major PRMTs associated with the regulation of immunity include the coactivator associated arginine methyltransferase 1 (CARM1 or PRMT4), PRMT1, PRMT5, and PRMT6. CARM1 targets proteins regulating chromatin remodeling and RNA-binding proteins (142). In addition, CARM1 is a major coactivator of NF-κB (143). Similarly, PRMT1 has been recognized as a major regulator of inflammation through its ability to methylate multiple proteins, including NF-κB (144), CITED2, STAT5 (145), and NFAT (146). Furthermore, PRMT5 and PRMT6 have been shown to increase the activity of NF-κB, thereby regulating the expression of IL-1α and IL-6 (147, 148). Although the capacity of arginine methylation to modulate multiple inflammatory signaling pathways has been described, its role in several pathologies and especially in patient populations remains to be investigated.
Therapeutic Implications and Concluding Remarks
Arginine deprivation is a novel therapeutic modality for several ASS1-deficient, arginine-auxotrophic solid and hematological malignancies. To this end, arginine-metabolizing enzymes, mycoplasma-derived arginine deiminase (ADI) and recombinant human arginase 1, have been pegylated for enhanced in vivo pharmacokinetics and pharmacodynamics (149, 150). Pegylated ADI (peg-ADI) controls tumor growth in multiple xenograft models, including melanoma, hepatocellular carcinoma, and lung cancer (151, 152). Promoting apoptosis and blocking angiogenesis and de novo protein synthesis are endorsed antitumor mechanisms for peg-ADI treatment (149, 150). Clinical investigations of peg-ADI have followed in melanoma and hepatocellular carcinoma, with response rates of 25 and 47%, respectively (153, 154). Several other phase II and phase III clinical trials are underway testing peg-ADI in patients with metastatic melanoma, advanced hepatocellular carcinoma, and small-cell lung cancer. With the clinical evidence for developing anti-peg-ADI neutralizing antibodies (155, 156), recombinant human arginase 1 represents an alternative arginine deprivation therapy. Pegylation extends the half-life of arginase 1 without altering its activity (157). Pegylated arginase 1 (peg-arginase 1) is effective against several cancers, such as melanoma, hepatocellular carcinoma, and leukemia (130, 157, 158). The antitumor effect of peg-arginase 1 is mediated via the induction of autophagy, apoptosis, and cell cycle arrest in malignant cells (130, 150, 159). Peg-arginase 1 is currently undergoing clinical investigation in patients with advanced hepatocellular carcinoma. The initial clinical evidence indicates that peg-arginase 1 is safe and results in prolonged arginine depletion (160). Interestingly, peg-arginase 1 also induces immune suppression by reducing the availability of arginine to primary T cells and through the induction of MDSCs (126). As such, peg-arginase 1 extends the survival of mice undergoing bone marrow transplantation and delays the appearance of graft versus host diseases, whereas enhancing the growth of Listeria (97, 161). Additionally, peg-arginase 1 exerts a potent anti-herpetic activity, blocking herpes simplex virus replication and virus-derived cytopathic effects in vitro (162). Arginine-catabolizing enzymes have a preclinical additive and/or synergistic effects with other treatments such as chemotherapy, radiotherapy, PI3K inhibitors, and autophagy regulators (149), and the clinical efficacy of these combinatorial approaches remains to be determined.
As outlined above, the arginine-metabolizing enzymes arginase 1 and NOS2 are key suppressive mechanisms by which immunoregulatory myeloid cells restrain T cell antitumor immunity (Figure 2), thus indeed paving the way for developing strategies to target these pathways. The arginase 1 inhibitor nor-NOHA and the ODC inhibitor α-difluoromethylornithine (DFMO) downregulate arginase 1 expression in tumor-associated MDSC and restore T cell antitumor immunity (3, 163). Since arginase 1 expression can be driven by cyclooxygenase 2 (COX2)/PGE2 axis, celecoxib, a selective COX2 inhibitor, also blocks arginase 1 expression, reduces MDSC accumulation, and elicits CD4+ and CD8+ antitumor immune responses (30). Moreover, dietary celecoxib synergizes with DC-based vaccination to extend the survival of mesothelioma-bearing mice (164). While these studies have examined the role of MDSCs, it is conceivable that these agents could also modulate other arginase 1-expressing myeloid cells like TAMs and tolerogenic DCs (85, 165). On the other hand, N(6)-(1-iminoethyl)-l-lysine-dihydrochloride (l-nil), a NOS2 selective inhibitor, constraints melanoma growth and improves the survival of tumor-bearing mice, and a combination of l-nil and cisplatin is better than either agent alone (166). Reports, however, recommend targeting both arginase 1 and NOS2 to augment the therapeutic effect. In a model of human prostatic adenocarcinomas, only concomitant inhibition of arginase 1 and NOS2 reduces PNT production and recovers tumor-infiltrating lymphocyte antitumor responsiveness (167). Phosphodiesterase-5 (PDE5) inhibitors (sildenafil, tadalafil, and vardenafil) decrease the expression of arginase 1 and NOS2, thereby blocking MDSC regulatory activity (168). Accordingly, PDE5 inhibitors promote intratumoral infiltration of activated T cells, control tumor growth, and enhance the efficacy of ACT (168). Likewise, nitric oxide-releasing aspirin (a typical aspirin linked to a nitric oxide donor) reduces arginase 1, NOS2, and PNT, while increasing the frequency and function of tumor-specific T cells, thus boosting the antitumor effect of cancer vaccination (169). Another small molecule that prevents PNT production in vivo, namely, 3-[(aminocarbonyl)furoxan-4-yl]methyl salicylate (AT38), also drives the infiltration of tumor antigen-specific T cells into tumors and synergizes with ACT (141). The fact that agents like PDE5 inhibitors and nitric oxide-releasing aspirin have been proven safe in patients provides a rationale to use these treatments in combination with other immunotherapeutic approaches such as ACT and checkpoint blockade. Additionally, as discussed above, NOS2-expressing Tip-DCs are critical for tumor rejection in the context of ACT in mice (84). Of interest, this antitumor response does not require lymphodepletion preconditioning prior to ACT. Therefore, this development could reprogram the immunosuppressive tumor microenvironment and, more importantly, obviate the need for other potentially toxic regimens.
Overall, arginine metabolism has evolved as a key player in the center of our immune system. At this point, it is very clear that the regulators of arginine metabolism can elicit dichotomous innate and adaptive immune responses for instance in controlled versus uncontrolled infection, autoimmunity versus self-tolerance, and antitumor immunity versus tumor-induced immune suppression. As such, a better understanding of arginine metabolic pathways within the complicated inflammatory microenvironments in vivo and in the human as opposed to the mouse system will facilitate the development of targeted therapeutic interventions in different diseases.
Author Contributions
PR wrote and critically revised the manuscript. AO discussed the manuscript. AAA wrote and critically revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was partially supported by CA18485 to PR, P30GM114732 to AAA (AO, Program Director), and U54GM104940 to AO and AAA.
References
2. Wu G, Morris SM Jr. Arginine metabolism: nitric oxide and beyond. Biochem J (1998) 336(Pt 1):1–17. doi: 10.1042/bj3360001
3. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64:5839–49. doi:10.1158/0008-5472.CAN-04-0465
4. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. l-Arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol (2003) 171:1232–9. doi:10.4049/jimmunol.171.3.1232
5. Nieves C Jr, Langkamp-Henken B. Arginine and immunity: a unique perspective. Biomed Pharmacother (2002) 56:471–82. doi:10.1016/S0753-3322(02)00291-3
6. Luiking YC, Poeze M, Dejong CH, Ramsay G, Deutz NE. Sepsis: an arginine deficiency state? Crit Care Med (2004) 32:2135–45. doi:10.1097/01.CCM.0000142939.81045.A0
7. Closs EI, Simon A, Vekony N, Rotmann A. Plasma membrane transporters for arginine. J Nutr (2004) 134:2752S–9S.
8. Bogdan C. Nitric oxide and the immune response. Nat Immunol (2001) 2:907–16. doi:10.1038/ni1001-907
9. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J (2012) 33:829d–37d. doi:10.1093/eurheartj/ehr304
10. Amber IJ, Hibbs JB Jr, Parker CJ, Johnson BB, Taintor RR, Vavrin Z. Activated macrophage conditioned medium: identification of the soluble factors inducing cytotoxicity and the l-arginine dependent effector mechanism. J Leukoc Biol (1991) 49:610–20.
11. Hibbs JB Jr, Taintor RR, Vavrin Z. Macrophage cytotoxicity: role for l-arginine deiminase and imino nitrogen oxidation to nitrite. Science (1987) 235:473–6. doi:10.1126/science.2432665
12. Bogdan C. Nitric oxide and the regulation of gene expression. Trends Cell Biol (2001) 11:66–75. doi:10.1016/S0962-8924(00)01900-0
13. Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol (2015) 36:161–78. doi:10.1016/j.it.2015.01.003
14. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol (1997) 15:323–50. doi:10.1146/annurev.immunol.15.1.323
15. Kolios G, Rooney N, Murphy CT, Robertson DA, Westwick J. Expression of inducible nitric oxide synthase activity in human colon epithelial cells: modulation by T lymphocyte derived cytokines. Gut (1998) 43:56–63. doi:10.1136/gut.43.1.56
16. Robbins RA, Barnes PJ, Springall DR, Warren JB, Kwon OJ, Buttery LD, et al. Expression of inducible nitric oxide in human lung epithelial cells. Biochem Biophys Res Commun (1994) 203:209–18. doi:10.1006/bbrc.1994.2169
17. Jianjun Y, Zhang R, Lu G, Shen Y, Peng L, Zhu C, et al. T cell-derived inducible nitric oxide synthase switches off Th17 cell differentiation. J Exp Med (2013) 210:1447–62. doi:10.1084/jem.20122494
18. Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet (1992) 339:572–5. doi:10.1016/0140-6736(92)90865-Z
19. Morris SM Jr. Regulation of enzymes of the urea cycle and arginine metabolism. Annu Rev Nutr (2002) 22:87–105. doi:10.1146/annurev.nutr.22.110801.140547
20. Cederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, Iyer RK. Arginases I and II: do their functions overlap? Mol Genet Metab (2004) 81(Suppl 1):S38–44. doi:10.1016/j.ymgme.2003.10.012
21. Monticelli LA, Buck MD, Flamar AL, Saenz SA, Tait Wojno ED, Yudanin NA, et al. Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat Immunol (2016) 17:656–65. doi:10.1038/ni.3421
23. Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol (2009) 158:638–51. doi:10.1111/j.1476-5381.2009.00291.x
24. Gray MJ, Poljakovic M, Kepka-Lenhart D, Morris SM Jr. Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPbeta. Gene (2005) 353:98–106. doi:10.1016/j.gene.2005.04.004
25. Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol (2004) 172:7565–73. doi:10.4049/jimmunol.172.12.7565
26. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest (2013) 123:1580–9. doi:10.1172/JCI60083
27. Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol (2002) 169:2253–63. doi:10.4049/jimmunol.169.5.2253
28. Jost MM, Ninci E, Meder B, Kempf C, Van RN, Hua J, et al. Divergent effects of GM-CSF and TGFbeta1 on bone marrow-derived macrophage arginase-1 activity, MCP-1 expression, and matrix metalloproteinase-12: a potential role during arteriogenesis. FASEB J (2003) 17:2281–3. doi:10.1096/fj.03-0071fje
29. Boutard V, Havouis R, Fouqueray B, Philippe C, Moulinoux JP, Baud L. Transforming growth factor-beta stimulates arginase activity in macrophages. Implications for the regulation of macrophage cytotoxicity. J Immunol (1995) 155:2077–84.
30. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202:931–9. doi:10.1084/jem.20050715
31. Corraliza IM, Modolell M, Ferber E, Soler G. Involvement of protein kinase A in the induction of arginase in murine bone marrow-derived macrophages. Biochim Biophys Acta (1997) 1334:123–8. doi:10.1016/S0304-4165(96)00081-5
32. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol (2008) 9:1399–406. doi:10.1038/ni.1671
33. Chawla A. Control of macrophage activation and function by PPARs. Circ Res (2010) 106:1559–69. doi:10.1161/CIRCRESAHA.110.216523
34. Gallardo-Soler A, Gomez-Nieto C, Campo ML, Marathe C, Tontonoz P, Castrillo A, et al. Arginase I induction by modified lipoproteins in macrophages: a peroxisome proliferator-activated receptor-gamma/delta-mediated effect that links lipid metabolism and immunity. Mol Endocrinol (2008) 22:1394–402. doi:10.1210/me.2007-0525
35. Xu W, Ghosh S, Comhair SA, Asosingh K, Janocha AJ, Mavrakis DA, et al. Increased mitochondrial arginine metabolism supports bioenergetics in asthma. J Clin Invest (2016) 126:2465–81. doi:10.1172/JCI82925
36. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. l-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167:829–42. doi:10.1016/j.cell.2016.09.031
37. Forstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, et al. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension (1994) 23:1121–31. doi:10.1161/01.HYP.23.6.1121
38. Shin S, Mohan S, Fung HL. Intracellular l-arginine concentration does not determine NO production in endothelial cells: implications on the “l-arginine paradox”. Biochem Biophys Res Commun (2011) 414:660–3. doi:10.1016/j.bbrc.2011.09.112
39. Zani BG, Bohlen HG. Transport of extracellular l-arginine via cationic amino acid transporter is required during in vivo endothelial nitric oxide production. Am J Physiol Heart Circ Physiol (2005) 289:H1381–90. doi:10.1152/ajpheart.01231.2004
40. Elms S, Chen F, Wang Y, Qian J, Askari B, Yu Y, et al. Insights into the arginine paradox: evidence against the importance of subcellular location of arginase and eNOS. Am J Physiol Heart Circ Physiol (2013) 305:H651–66. doi:10.1152/ajpheart.00755.2012
41. Romero MJ, Platt DH, Tawfik HE, Labazi M, El-Remessy AB, Bartoli M, et al. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res (2008) 102:95–102. doi:10.1161/CIRCRESAHA.107.155028
42. White AR, Ryoo S, Li D, Champion HC, Steppan J, Wang D, et al. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension (2006) 47:245–51. doi:10.1161/01.HYP.0000198543.34502.d7
43. Lee J, Ryu H, Ferrante RJ, Morris SM Jr, Ratan RR. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc Natl Acad Sci U S A (2003) 100:4843–8. doi:10.1073/pnas.0735876100
44. Mills CD. Macrophage arginine metabolism to ornithine/urea or nitric oxide/citrulline: a life or death issue. Crit Rev Immunol (2001) 21:399–425. doi:10.1615/CritRevImmunol.v21.i5.10
45. Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol (2012) 32:463–88. doi:10.1615/CritRevImmunol.v32.i6.10
46. Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, et al. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of l-arginine metabolism. J Immunol (2001) 167:6533–44. doi:10.4049/jimmunol.167.11.6533
47. Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol (1998) 160:5347–54.
48. Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol (1999) 163:3771–7.
49. Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol (2001) 166:2173–7. doi:10.4049/jimmunol.166.4.2173
50. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
51. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi:10.1172/JCI59643
52. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
53. Yeramian A, Martin L, Serrat N, Arpa L, Soler C, Bertran J, et al. Arginine transport via cationic amino acid transporter 2 plays a critical regulatory role in classical or alternative activation of macrophages. J Immunol (2006) 176:5918–24. doi:10.4049/jimmunol.176.10.5918
54. Wu GY, Brosnan JT. Macrophages can convert citrulline into arginine. Biochem J (1992) 281(Pt 1):45–8. doi:10.1042/bj2810045
55. Nussler AK, Billiar TR, Liu ZZ, Morris SM Jr. Coinduction of nitric oxide synthase and argininosuccinate synthetase in a murine macrophage cell line. Implications for regulation of nitric oxide production. J Biol Chem (1994) 269:1257–61.
56. Qualls JE, Subramanian C, Rafi W, Smith AM, Balouzian L, DeFreitas AA, et al. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe (2012) 12:313–23. doi:10.1016/j.chom.2012.07.012
57. Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol (1995) 25:1101–4. doi:10.1002/eji.1830250436
58. Szabo C, Southan GJ, Thiemermann C, Vane JR. The mechanism of the inhibitory effect of polyamines on the induction of nitric oxide synthase: role of aldehyde metabolites. Br J Pharmacol (1994) 113:757–66. doi:10.1111/j.1476-5381.1994.tb17058.x
59. Zhang M, Caragine T, Wang H, Cohen PS, Botchkina G, Soda K, et al. Spermine inhibits proinflammatory cytokine synthesis in human mononuclear cells: a counterregulatory mechanism that restrains the immune response. J Exp Med (1997) 185:1759–68. doi:10.1084/jem.185.10.1759
60. Mossner J, Hammermann R, Racke K. Concomitant down-regulation of l-arginine transport and nitric oxide (NO) synthesis in rat alveolar macrophages by the polyamine spermine. Pulm Pharmacol Ther (2001) 14:297–305. doi:10.1006/pupt.2001.0297
61. Bussiere FI, Chaturvedi R, Cheng Y, Gobert AP, Asim M, Blumberg DR, et al. Spermine causes loss of innate immune response to Helicobacter pylori by inhibition of inducible nitric-oxide synthase translation. J Biol Chem (2005) 280:2409–12. doi:10.1074/jbc.C400498200
62. Qualls JE, Neale G, Smith AM, Koo MS, DeFreitas AA, Zhang H, et al. Arginine usage in mycobacteria-infected macrophages depends on autocrine-paracrine cytokine signaling. Sci Signal (2010) 3:ra62. doi:10.1126/scisignal.2000955
63. Schleicher U, Paduch K, Debus A, Obermeyer S, Konig T, Kling JC, et al. TNF-mediated restriction of arginase 1 expression in myeloid cells triggers type 2 NO synthase activity at the site of infection. Cell Rep (2016) 15:1062–75. doi:10.1016/j.celrep.2016.04.001
64. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol (2013) 191:773–84. doi:10.4049/jimmunol.1300113
65. Duque-Correa MA, Kuhl AA, Rodriguez PC, Zedler U, Schommer-Leitner S, Rao M, et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc Natl Acad Sci U S A (2014) 111:E4024–32. doi:10.1073/pnas.1408839111
66. Monin L, Griffiths KL, Lam WY, Gopal R, Kang DD, Ahmed M, et al. Helminth-induced arginase-1 exacerbates lung inflammation and disease severity in tuberculosis. J Clin Invest (2015) 125:4699–713. doi:10.1172/JCI77378
67. Albina JE, Mills CD, Barbul A, Thirkill CE, Henry WL Jr, Mastrofrancesco B, et al. Arginine metabolism in wounds. Am J Physiol (1988) 254:E459–67.
68. Albina JE, Mills CD, Henry WL Jr, Caldwell MD. Temporal expression of different pathways of 1-arginine metabolism in healing wounds. J Immunol (1990) 144:3877–80.
69. Campbell L, Saville CR, Murray PJ, Cruickshank SM, Hardman MJ. Local arginase 1 activity is required for cutaneous wound healing. J Invest Dermatol (2013) 133:2461–70. doi:10.1038/jid.2013.164
70. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog (2009) 5:e1000371. doi:10.1371/journal.ppat.1000371
71. Herbert DR, Orekov T, Roloson A, Ilies M, Perkins C, O’Brien W, et al. Arginase I suppresses IL-12/IL-23p40-driven intestinal inflammation during acute schistosomiasis. J Immunol (2010) 184:6438–46. doi:10.4049/jimmunol.0902009
72. Barron L, Smith AM, El Kasmi KC, Qualls JE, Huang X, Cheever A, et al. Role of arginase 1 from myeloid cells in th2-dominated lung inflammation. PLoS One (2013) 8:e61961. doi:10.1371/journal.pone.0061961
73. Jiang Z, Zhu L. Update on the role of alternatively activated macrophages in asthma. J Asthma Allergy (2016) 9:101–7. doi:10.2147/JAA.S104508
74. Esser-von BJ, Mosconi I, Guiet R, Piersgilli A, Volpe B, Chen F, et al. Antibodies trap tissue migrating helminth larvae and prevent tissue damage by driving IL-4Ralpha-independent alternative differentiation of macrophages. PLoS Pathog (2013) 9:e1003771. doi:10.1371/journal.ppat.1003771
75. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med (1973) 137:1142–62. doi:10.1084/jem.137.5.1142
76. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol (2006) 311:17–58. doi:10.1007/3-540-32636-7_2
77. Ueno H, Schmitt N, Klechevsky E, Pedroza-Gonzalez A, Matsui T, Zurawski G, et al. Harnessing human dendritic cell subsets for medicine. Immunol Rev (2010) 234:199–212. doi:10.1111/j.0105-2896.2009.00884.x
78. Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity (2014) 40:642–56. doi:10.1016/j.immuni.2014.04.016
79. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity (2003) 19:59–70. doi:10.1016/S1074-7613(03)00171-7
80. Geissmann F, Auffray C, Palframan R, Wirrig C, Ciocca A, Campisi L, et al. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol Cell Biol (2008) 86:398–408. doi:10.1038/icb.2008.19
81. Copin R, De BP, Carlier Y, Letesson JJ, Muraille E. MyD88-dependent activation of B220-CD11b+LY-6C+ dendritic cells during Brucella melitensis infection. J Immunol (2007) 178:5182–91. doi:10.4049/jimmunol.178.8.5182
82. De TC, Magez S, Akira S, Ryffel B, Carlier Y, Muraille E. iNOS-producing inflammatory dendritic cells constitute the major infected cell type during the chronic Leishmania major infection phase of C57BL/6 resistant mice. PLoS Pathog (2009) 5:e1000494. doi:10.1371/journal.ppat.1000494
83. Bosschaerts T, Guilliams M, Stijlemans B, Morias Y, Engel D, Tacke F, et al. Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-gamma and MyD88 signaling. PLoS Pathog (2010) 6:e1001045. doi:10.1371/journal.ppat.1001045
84. Marigo I, Zilio S, Desantis G, Mlecnik B, Agnellini AH, Ugel S, et al. T cell cancer therapy requires CD40-CD40L activation of tumor necrosis factor and inducible nitric-oxide-synthase-producing dendritic cells. Cancer Cell (2016) 30:651. doi:10.1016/j.ccell.2016.08.004
85. Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol (2015) 194:2985–91. doi:10.4049/jimmunol.1403134
86. Liu Q, Zhang C, Sun A, Zheng Y, Wang L, Cao X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J Immunol (2009) 182:6207–16. doi:10.4049/jimmunol.0803926
87. Narita Y, Kitamura H, Wakita D, Sumida K, Masuko K, Terada S, et al. The key role of IL-6-arginase cascade for inducing dendritic cell-dependent CD4(+) T cell dysfunction in tumor-bearing mice. J Immunol (2013) 190:812–20. doi:10.4049/jimmunol.1103797
88. Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, et al. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via l-arginine metabolism. Cancer Res (2009) 69:3086–94. doi:10.1158/0008-5472.CAN-08-2826
89. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi:10.1038/nri3175
90. Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev (2008) 222:180–91. doi:10.1111/j.1600-065X.2008.00608.x
91. Rieber N, Singh A, Oz H, Carevic M, Bouzani M, Amich J, et al. Pathogenic fungi regulate immunity by inducing neutrophilic myeloid-derived suppressor cells. Cell Host Microbe (2015) 17:507–14. doi:10.1016/j.chom.2015.02.007
92. Macatangay BJ, Landay AL, Rinaldo CR. MDSC: a new player in HIV immunopathogenesis. AIDS (2012) 26:1567–9. doi:10.1097/QAD.0b013e328355e682
93. Goh CC, Roggerson KM, Lee HC, Golden-Mason L, Rosen HR, Hahn YS. Hepatitis C virus-induced myeloid-derived suppressor cells suppress NK cell IFN-gamma production by altering cellular metabolism via arginase-1. J Immunol (2016) 196:2283–92. doi:10.4049/jimmunol.1501881
94. Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol (2006) 176:2085–94. doi:10.4049/jimmunol.176.4.2085
95. Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov (2016) 6:852–69. doi:10.1158/2159-8290.CD-15-1177
96. Xia S, Sha H, Yang L, Ji Y, Ostrand-Rosenberg S, Qi L. Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J Biol Chem (2011) 286:23591–9. doi:10.1074/jbc.M111.237123
97. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood (2010) 116:5738–47. doi:10.1182/blood-2010-06-287839
98. Zhu B, Bando Y, Xiao S, Yang K, Anderson AC, Kuchroo VK, et al. CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol (2007) 179:5228–37. doi:10.4049/jimmunol.179.8.5228
99. Wu H, Zhen Y, Ma Z, Li H, Yu J, Xu ZG, et al. Arginase-1-dependent promotion of TH17 differentiation and disease progression by MDSCs in systemic lupus erythematosus. Sci Transl Med (2016) 8:331ra40. doi:10.1126/scitranslmed.aae0482
100. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi:10.1038/ncomms12150
101. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood (1998) 92:4150–66.
102. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol (1998) 160:1224–32.
103. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A (2010) 107:21248–55. doi:10.1073/pnas.1015855107
104. Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat (2010) 123:39–49. doi:10.1007/s10549-009-0622-8
105. Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, et al. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood (2000) 96:3838–46.
106. Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol (2006) 176:284–90. doi:10.4049/jimmunol.176.1.284
107. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res (2007) 67:10019–26. doi:10.1158/0008-5472.CAN-07-2354
108. Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol (2003) 170:270–8. doi:10.4049/jimmunol.170.1.270
109. Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest (2006) 116:2777–90. doi:10.1172/JCI28828
110. Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res (2014) 74:5723–33. doi:10.1158/0008-5472.CAN-13-2347
111. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol (2008) 181:4666–75. doi:10.4049/jimmunol.181.7.4666
112. Hix LM, Karavitis J, Khan MW, Shi YH, Khazaie K, Zhang M. Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J Biol Chem (2013) 288:11676–88. doi:10.1074/jbc.M112.441402
113. Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol (2005) 174:4880–91. doi:10.4049/jimmunol.174.8.4880
114. Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest (2013) 123:4464–78. doi:10.1172/JCI68189
115. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med (2008) 205:2235–49. doi:10.1084/jem.20080132
116. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182:5693–701. doi:10.4049/jimmunol.0900092
117. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity (2010) 32:790–802. doi:10.1016/j.immuni.2010.05.010
118. Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res (2009) 69:2506–13. doi:10.1158/0008-5472.CAN-08-4323
119. Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity (2014) 41:389–401. doi:10.1016/j.immuni.2014.08.015
120. Ugel S, De SF, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest (2015) 125:3365–76. doi:10.1172/JCI80006
121. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res (2009) 69:1553–60. doi:10.1158/0008-5472.CAN-08-1921
122. Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, et al. l-Arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell Immunol (2004) 232:21–31. doi:10.1016/j.cellimm.2005.01.004
123. Taheri F, Ochoa JB, Faghiri Z, Culotta K, Park HJ, Lan MS, et al. l-Arginine regulates the expression of the T-cell receptor zeta chain (CD3zeta) in Jurkat cells. Clin Cancer Res (2001) 7:958s–65s.
124. Rodriguez PC, Quiceno DG, Ochoa AC. l-Arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109:1568–73. doi:10.1182/blood-2006-06-031856
125. Rodriguez PC, Hernandez CP, Morrow K, Sierra R, Zabaleta J, Wyczechowska DD, et al. l-Arginine deprivation regulates cyclin D3 mRNA stability in human T cells by controlling HuR expression. J Immunol (2010) 185:5198–204. doi:10.4049/jimmunol.1001224
126. Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res (2015) 75:275–83. doi:10.1158/0008-5472.CAN-14-1491
127. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281:1191–3. doi:10.1126/science.281.5380.1191
128. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70:68–77. doi:10.1158/0008-5472.CAN-09-2587
129. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity (2005) 22:633–42. doi:10.1016/j.immuni.2005.03.013
130. Hernandez CP, Morrow K, Lopez-Barcons LA, Zabaleta J, Sierra R, Velasco C, et al. Pegylated arginase I: a potential therapeutic approach in T-ALL. Blood (2010) 115:5214–21. doi:10.1182/blood-2009-12-258822
131. Van de Velde LA, Murray PJ. Proliferating helper T cells require Rictor/mTORC2 complex to integrate signals from limiting environmental amino acids. J Biol Chem (2016) 291:25815–22. doi:10.1074/jbc.C116.763623
132. Van de Velde LA, Subramanian C, Smith AM, Barron L, Qualls JE, Neale G, et al. T cells encountering myeloid cells programmed for amino acid-dependent immunosuppression use Rictor/mTORC2 for proliferative checkpoint decisions. J Biol Chem (2017) 292:15–30. doi:10.1074/jbc.M116.766238
133. Van de Velde LA, Guo XJ, Barbaric L, Smith AM, Oguin TH III, Thomas PG, et al. Stress kinase GCN2 controls the proliferative fitness and trafficking of cytotoxic T cells independent of environmental amino acid sensing. Cell Rep (2016) 17:2247–58. doi:10.1016/j.celrep.2016.10.079
134. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer (2014) 134:2853–64. doi:10.1002/ijc.28622
135. Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol (2002) 168:689–95. doi:10.4049/jimmunol.168.2.689
136. Saio M, Radoja S, Marino M, Frey AB. Tumor-infiltrating macrophages induce apoptosis in activated CD8(+) T cells by a mechanism requiring cell contact and mediated by both the cell-associated form of TNF and nitric oxide. J Immunol (2001) 167:5583–93. doi:10.4049/jimmunol.167.10.5583
137. Brito C, Naviliat M, Tiscornia AC, Vuillier F, Gualco G, Dighiero G, et al. Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol (1999) 162:3356–66.
138. Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest (2011) 121:4015–29. doi:10.1172/JCI45862
139. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13:828–35. doi:10.1038/nm1609
140. De Sanctis F, Sandri S, Ferrarini G, Pagliarello I, Sartoris S, Ugel S, et al. The emerging immunological role of post-translational modifications by reactive nitrogen species in cancer microenvironment. Front Immunol (2014) 5:69. doi:10.3389/fimmu.2014.00069
141. Molon B, Ugel S, Del PF, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208:1949–62. doi:10.1084/jem.20101956
142. Cheng D, Cote J, Shaaban S, Bedford MT. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell (2007) 25:71–83. doi:10.1016/j.molcel.2006.11.019
143. Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, et al. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J (2005) 24:85–96. doi:10.1038/sj.emboj.7600500
144. Hassa PO, Covic M, Bedford MT, Hottiger MO. Protein arginine methyltransferase 1 coactivates NF-kappaB-dependent gene expression synergistically with CARM1 and PARP1. J Mol Biol (2008) 377:668–78. doi:10.1016/j.jmb.2008.01.044
145. Kleinschmidt MA, Streubel G, Samans B, Krause M, Bauer UM. The protein arginine methyltransferases CARM1 and PRMT1 cooperate in gene regulation. Nucleic Acids Res (2008) 36:3202–13. doi:10.1093/nar/gkn166
146. Mowen KA, Schurter BT, Fathman JW, David M, Glimcher LH. Arginine methylation of NIP45 modulates cytokine gene expression in effector T lymphocytes. Mol Cell (2004) 15:559–71. doi:10.1016/j.molcel.2004.06.042
147. Di LA, Yang Y, Macaluso M, Bedford MT. A gain-of-function mouse model identifies PRMT6 as a NF-kappaB coactivator. Nucleic Acids Res (2014) 42:8297–309. doi:10.1093/nar/gku530
148. Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang DB, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proc Natl Acad Sci U S A (2013) 110:13516–21. doi:10.1073/pnas.1311784110
149. Phillips MM, Sheaff MT, Szlosarek PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res Treat (2013) 45:251–62. doi:10.4143/crt.2013.45.4.251
150. Patil MD, Bhaumik J, Babykutty S, Banerjee UC, Fukumura D. Arginine dependence of tumor cells: targeting a chink in cancer’s armor. Oncogene (2016) 35:4957–72. doi:10.1038/onc.2016.37
151. Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Pegylated arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res (2002) 62:5443–50.
152. Kelly MP, Jungbluth AA, Wu BW, Bomalaski J, Old LJ, Ritter G. Arginine deiminase PEG20 inhibits growth of small cell lung cancers lacking expression of argininosuccinate synthetase. Br J Cancer (2012) 106:324–32. doi:10.1038/bjc.2011.524
153. Ascierto PA, Scala S, Castello G, Daponte A, Simeone E, Ottaiano A, et al. Pegylated arginine deiminase treatment of patients with metastatic melanoma: results from phase I and II studies. J Clin Oncol (2005) 23:7660–8. doi:10.1200/JCO.2005.02.0933
154. Izzo F, Marra P, Beneduce G, Castello G, Vallone P, De Rosa V, et al. Pegylated arginine deiminase treatment of patients with unresectable hepatocellular carcinoma: results from phase I/II studies. J Clin Oncol (2004) 22:1815–22. doi:10.1200/JCO.2004.11.120
155. Glazer ES, Piccirillo M, Albino V, Di GR, Palaia R, Mastro AA, et al. Phase II study of pegylated arginine deiminase for nonresectable and metastatic hepatocellular carcinoma. J Clin Oncol (2010) 28:2220–6. doi:10.1200/JCO.2009.26.7765
156. Szlosarek PW, Luong P, Phillips MM, Baccarini M, Stephen E, Szyszko T, et al. Metabolic response to pegylated arginine deiminase in mesothelioma with promoter methylation of argininosuccinate synthetase. J Clin Oncol (2013) 31:e111–3. doi:10.1200/JCO.2012.42.1784
157. Cheng PN, Lam TL, Lam WM, Tsui SM, Cheng AW, Lo WH, et al. Pegylated recombinant human arginase (rhArg-peg5,000mw) inhibits the in vitro and in vivo proliferation of human hepatocellular carcinoma through arginine depletion. Cancer Res (2007) 67:309–17. doi:10.1158/0008-5472.CAN-06-1945
158. Lam TL, Wong GK, Chow HY, Chong HC, Chow TL, Kwok SY, et al. Recombinant human arginase inhibits the in vitro and in vivo proliferation of human melanoma by inducing cell cycle arrest and apoptosis. Pigment Cell Melanoma Res (2011) 24:366–76. doi:10.1111/j.1755-148X.2010.00798.x
159. Morrow K, Hernandez CP, Raber P, Del VL, Wilk AM, Majumdar S, et al. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia (2013) 27:569–77. doi:10.1038/leu.2012.247
160. Yau T, Cheng PN, Chan P, Chan W, Chen L, Yuen J, et al. A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Invest New Drugs (2013) 31:99–107. doi:10.1007/s10637-012-9807-9
161. Zhu X, Pribis JP, Rodriguez PC, Morris SM Jr, Vodovotz Y, Billiar TR, et al. The central role of arginine catabolism in T-cell dysfunction and increased susceptibility to infection after physical injury. Ann Surg (2014) 259:171–8. doi:10.1097/SLA.0b013e31828611f8
162. Sanchez MD, Ochoa AC, Foster TP. Development and evaluation of a host-targeted antiviral that abrogates herpes simplex virus replication through modulation of arginine-associated metabolic pathways. Antiviral Res (2016) 132:13–25. doi:10.1016/j.antiviral.2016.05.009
163. Ye C, Geng Z, Dominguez D, Chen S, Fan J, Qin L, et al. Targeting ornithine decarboxylase by alpha-difluoromethylornithine inhibits tumor growth by impairing myeloid-derived suppressor cells. J Immunol (2016) 196:915–23. doi:10.4049/jimmunol.1500729
164. Veltman JD, Lambers ME, van NM, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer (2010) 10:464. doi:10.1186/1471-2407-10-464
165. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41:49–61. doi:10.1016/j.immuni.2014.06.010
166. Sikora AG, Gelbard A, Davies MA, Sano D, Ekmekcioglu S, Kwon J, et al. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin Cancer Res (2010) 16:1834–44. doi:10.1158/1078-0432.CCR-09-3123
167. Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med (2005) 201:1257–68. doi:10.1084/jem.20042028
168. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med (2006) 203:2691–702. doi:10.1084/jem.20061104
Keywords: arginine, nitric oxide synthase, arginase, immune response, M1 and M2, macrophage, dendritic cell, MDSC
Citation: Rodriguez PC, Ochoa AC and Al-Khami AA (2017) Arginine Metabolism in Myeloid Cells Shapes Innate and Adaptive Immunity. Front. Immunol. 8:93. doi: 10.3389/fimmu.2017.00093
Received: 29 October 2016; Accepted: 19 January 2017;
Published: 07 February 2017
Edited by:
Philip Calder, University of Southampton, UKReviewed by:
Mourad Aribi, University of Tlemcen, AlgeriaMarkus Munder, University of Mainz, Germany
Copyright: © 2017 Rodriguez, Ochoa and Al-Khami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amir A. Al-Khami, YWFsa2hhQGxzdWhzYy5lZHU=