
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 08 February 2017
Sec. Primary Immunodeficiencies
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00075
This article is part of the Research Topic Etiopathogenesis of Systemic Sclerosis: An Update View all 11 articles
 Silvia Svegliati1†Donatella Amico1†Tatiana Spadoni1†
Silvia Svegliati1†Donatella Amico1†Tatiana Spadoni1† Colomba Fischetti1Doreen Finke1
Colomba Fischetti1Doreen Finke1 Gianluca Moroncini1
Gianluca Moroncini1 Chiara Paolini1Cecilia Tonnini1Antonella Grieco1Marina Rovinelli1
Chiara Paolini1Cecilia Tonnini1Antonella Grieco1Marina Rovinelli1 Ada Funaro2
Ada Funaro2 Armando Gabrielli1*
Armando Gabrielli1*
One of the earliest events in the pathogenesis of systemic sclerosis (SSc) is microvasculature damage with intimal hyperplasia and accumulation of cells expressing PDGF receptor. Stimulatory autoantibodies targeting PDGF receptor have been detected in SSc patients and demonstrated to induce fibrosis in vivo and convert in vitro normal fibroblasts into SSc-like cells. Since there is no evidence of the role of anti-PDGF receptor autoantibodies in the pathogenesis of SSc vascular lesions, we investigated the biologic effect of agonistic anti-PDGF receptor autoantibodies from SSc patients on human pulmonary artery smooth muscle cells and the signaling pathways involved. The synthetic (proliferation, migration, and type I collagen gene α1 chain expression) and contractile (smooth muscle-myosin heavy chain and smooth muscle-calponin expression) profiles of human pulmonary artery smooth muscle cells were assessed in vitro after incubation with SSc anti-PDGF receptors stimulatory autoantibodies. The role of reactive oxygen species, NOX isoforms, and mammalian target of rapamycin (mTOR) was investigated. Human pulmonary artery smooth muscle cells acquired a synthetic phenotype characterized by higher growth rate, migratory activity, gene expression of type I collagen α1 chain, and less expression of markers characteristic of the contractile phenotype such as smooth muscle-myosin heavy chain and smooth muscle-calponin when stimulated with PDGF and autoantibodies against PDGF receptor, but not with normal IgG. This phenotypic profile is mediated by increased generation of reactive oxygen species and expression of NOX4 and mTORC1. Our data indicate that agonistic anti-PDGF receptor autoantibodies may contribute to the pathogenesis of SSc intimal hyperplasia.
Systemic sclerosis (SSc; scleroderma) is a multiorgan disorder characterized by microvasculature damage, circulating autoantibodies, and fibroblast activation leading to fibrosis of the skin and visceral organs (1–3).
Vascular involvement is an early and very likely primary event in the pathogenesis of scleroderma, precedes fibrosis, and is characterized by endothelial cell (EC) injury and dysfunction, altered capillary permeability, increased expression of adhesion molecules, abnormal secretion of vasoactive mediators, and activation of platelets and fibrinolytic pathways (4–7). Microvascular injury and damage lead to vascular remodeling with striking intimal hyperplasia (also called neointima) due to increase in cell number and extracellular matrix and marked luminal narrowing (7, 8). The process culminates in rarefaction of blood vessels on nailfold capillaroscopy of SSc patients with late-stage disease (9). Loss of microvasculature is associated with tissue hypoxia, which induces strong expression of VEGF and its receptors (10–12).
The source of the intimal cells in scleroderma vasculopathy is a matter of debate. Medial smooth muscle cells, pericytes, circulating fibrocytes, ECs, adventitial fibroblasts, and adventitial stem cells have all been held responsible (13). Furthermore, the exact cause of vascular injury in SSc is unknown, and it may include reactive oxygen species (ROS)-mediated damage, viral agents, anti-EC antibodies, ischemia–reperfusion events, cytotoxic T-lymphocytes, and antibody-dependent cellular cytotoxicity [see for review Ref. (14)]. A further mechanisms has been reported by Riemekasten et al. who detected serum autoantibodies against angiotensin II type I receptor and endothelin-1 type 1 receptor, which induced extracellular signal-regulated kinase 1/2 (ERK 1/2) phosphorylation and increased TGF-β gene expression in ECs and were associated with severe disease manifestations (15).
While over the years much attention has been devoted to EC injury in scleroderma, oddly enough no in vitro studies focused on smooth muscle cells that are rich in PDGF receptors (PDGFR) (16), a key signaling molecule in the pathogenesis of SSc fibrosis. High levels of PDGF and PDGF receptor β (PDGFR β) have been found in skin lesions from patients with scleroderma (17, 18) and may contribute to the differentiation of perivascular pericytes into vascular smooth muscle cells, fibroblasts, and myofibroblasts (19). The beneficial effects of selective inhibitors of PDGF signaling on dermal fibrosis (20, 21) and lung fibrosis (22) further indicate the importance of PDGF in scleroderma. Finally, the relevance of PDGFR has been further emphasized by the high prevalence of anti-PDGFRα autoantibodies in SSc sera (23, 24).
Anti-PDGFRα autoantibodies play a role in the pathogenesis of scleroderma since they convert in vitro normal fibroblasts into SSc-like cells via the ROS, RAS, and ERK 1/2 pathway (23–26) and are capable to induce fibrosis in vivo (27). No report has, however, described their effect on human smooth muscle cells, and since a better understanding of the molecular mechanisms involved in scleroderma vascular events could help to prevent severe complications such as digital ulcers, pulmonary hypertension, and renal crisis, which are responsible for a substantially reduced survival and impaired quality of life (28–30), we decided to investigate the biological effects of SSc agonistic anti-PDGFR autoantibodies on human pulmonary artery smooth muscle cells (HPASMC) in vitro.
Serum samples were obtained from 11 SSc patients (2 were male and 9 were female) with median age of 56 years (range, 43–73 years), and median disease duration (defined as the time from the onset of the first non-Raynaud’s phenomenon clinical manifestation) was 7 years (2–21). Diagnosis was made following the ACR/EULAR preliminary criteria for the classification of SSc (31). Five patients had limited cutaneous SSc and six diffuse cutaneous SSc according to LeRoy et al. (32). Six patients were antitopoisomerase I positive; two patients were anticentromere positive, and three were ANA positive but lacked specific SSc autoantibodies. In the whole group, median PAPs detected by echocardiography was 30 mmHg (range, 28.5–36.5 mmHg) (Table 1). The study was approved by the local ethic committee. A written informed consent was obtained from all patients. At the time of the investigation, patients who had never been on immunosuppressive therapy had not received any treatment for the previous 6 weeks. Control sera were obtained from 10 age-, sex-, and race-matched normal, non-smoking, healthy volunteers. All subjects provided informed consent and the study protocol was approved by the Institutional Ethics Committee of Università Politecnica delle Marche.

Table 1. Clinical characteristics of systemic sclerosis patients (n = 11).
Immunoglobulins were purified from serum using gravity flow column packed with protein A/G agarose following manufacturer instruction (Pierce). Concentrations of all samples were calculated based on absorbance at 280 nm. The eluted IgG fractions that contain the highest absorbance were subjected to buffer exchange with PBS using desalting column, a size-exclusion chromatography with an average molecular weight exclusion limit of 5 KDa (Pierce). Purity of IgG preparations was checked by Coomassie blue staining, and PDGF and TGF-β contaminations were ruled out by immunoblotting with biotinylated anti-human PDGF-BB and anti-human TGF-β antibodies (Abcam), respectively, with detection limit of 0.1 ng cytokine/200 μg IgG.
Agonistic (VHPAM-Vκ16F4) and non-agonistic (VHPAM-Vκ13B8) human monoclonal autoantibodies targeting PDGFRα were generated from SSc memory B cells as already described (24).
Human pulmonary artery smooth muscle cells (HPASMC) were purchased from Lonza and maintained in Medium 231 supplemented with smooth muscle growth medium (SMGM) and 100 U/ml penicillin/streptomycin (all from Life Technologies). Cells between passages 3 and 9 were used in all experiments performed after 24 h of starvation (0.1% SMGM).
Total IgGs were isolated from each patient and control and were singly tested in triplicate. The same conditions were applied to human monoclonal antibodies. Each experiment was performed at least three times. Means of all experiments are shown.
Relative changes in intracellular ROS were monitored using the fluorescent probe 2′, 7′-dichlorfluorescein-diacetate (DCFH-DA; Life Technologies). Cells were plated at 2.5 × 105/ml and then incubated with scleroderma (SSc IgG) or normal subjects IgG (N IgG) (200 µg/ml), PDGF (15 ng/ml), or human monoclonal anti-PDGFRα autoantibodies (10 µg/ml) for 15 min and then stained with 10 µM DCFH-DA (Life Technologies) for 10 min at 37°C. Fluorescence was read using a plate reader fluorimeter (Victor 2, Perkin Elmer).
For confocal microscopy experiments, cells were seeded on coverslides and treated as described earlier. Cells were loaded with 10 µM DCFH-DA for 30 min, washed with PBS, and then visualized on a confocal microscope Eclipse C1 (Nikon). Data were analyzed by ImageJ software.
The intracellular content of ROS was also measured at a single-cell level by flow cytometry (FACS, Becton Dickinson). Briefly, cells were detached by trypsin, washed with PBS, and incubated with 5 µM DCFH-DA for 20 min at 37°C. Cells were then washed and resuspended in 1 ml PBS. For each sample, 10,000 events were collected, and intracellular ROS formation was detected as a result of oxidation of DCFH-DA at a wavelength of 520 nm. Data analysis was carried out using the WinMDI software.
Where indicated, the inhibitor of flavoprotein-dependent oxidases diphenyleneiodonium (DPI; 10 µM, Calbiochem), a general ROS inhibitor N-acetyl-cysteine (NAC; 10 mM, Sigma), and the PDGFR chemical inhibitor AG1296 (2 µM, Calbiochem) were added 1 h before stimulation.
Cell proliferation was determined by 5-bromo-2′-deoxyuridine (BrdU) incorporation assay (Roche). Briefly, HPASMC were seeded in 96-well pates at a density of 2.5 × 105 cells/ml and then treated with SSc IgG or N IgG (200 µg/ml), PDGF (15 ng/ml), or human monoclonal anti-PDGFRα autoantibodies (10 µg/ml) for 48 h. Cells were pulsed for 6 h with BrdU, and plates were analyzed in a plate reader at 450 nm (Victor 2, PerkinElmer).
Migration was studied by the wound scratch assay (33). Briefly, the cell monolayer was scraped by a straight line to create a “scratch” with a pipette tip. Cells were incubated for 24 h with SSc IgG, N IgG (200 µg/ml), PDGF (15 ng/ml), or human monoclonal anti-PDGFRα autoantibodies (10 µg/ml) in the presence or absence of inhibitors. Mitomycin was added to inhibit cell proliferation. Unstimulated cells were used as negative controls. The percentage of cells repopulating the scratch wound was assessed after 24 h.
Total RNA was isolated from HPASMC with Pure Link RNA Minikit (Life Technologies) and reverse transcribed using IScript cDNA Synthesis Kit (Bio-Rad Laboratories) according to the instructions of the manufacturer. Gene expression was quantified by SYBR Green real-time PCR in a iCycler iQ™ real-time PCR Detection System (Bio-Rad). Specific primer pairs for each gene were designed with the Universal ProbeLibrary Assay Design Centre by Roche Applied Science and were as follows: NOX1 5′-CTGTTTGTGGATGCCTTCCT-3′ (forward), 5′-TGTGGAAGGTGAGG-TTGTGA-3′ (reverse); NOX2 5′-TCACTTCCTCCACCAAAACC-3′ (forward), 5′GGGATTGGGCATTCCTTTAT-3′ (reverse); NOX4 5′-CTTCCGTTGGTTTGCAGATT-3′ (forward), 5′-GAATTGGGCCACAACAGA-3′ (reverse); type I collagen α1 chain (COL1A1) 5′-AGGGCCAAGACGAAGACATC-3′ (forward), 5′-AGATCACGTCATCGCACAACA-3′ (reverse); smooth muscle-myosin heavy chain (SM-MHC) 5′-CAGGCGTTCCGCCAACGCTA-3′ (forward), 5′-TCCCGTCCATGAAGCCTTTGG-3′ (reverse); smooth muscle-calponin (sm-calponin) 5′-TTTTGAGGCCAACGACCTGT-3′ (forward), 5′-TCCTTTCGTCTTCGCCATG-3′ (reverse); and GAPDH 5′-TGCACCACCAACTGCTTAGC-3′ (forward), 5′-TGGGATTTCCATTGATGA-CAAGC-3′ (reverse).
GAPDH was used to test the quality of cDNA and as a house keeping gene in real-time PCR. The threshold cycle (Ct) was used to detect the increase in the signal associated with an exponential growth of PCR product during the log-linear phase. The relative expression was calculated using the following formula: 2−ΔΔCt. The ΔCt validation experiments showed similar amplification efficiency for all templates used (difference between linear slopes for all templates less than 0.1).
Cells were lysed in RIPA buffer [1× PBS, 1% non-idet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 2 mM sodium orthovanadate] supplemented with a cocktail of protease inhibitors (Sigma). Proteins were separated on 4–12% gradient SDS–polyacrylamide gels, transferred onto nitrocellulose membranes, and immunoblotted with antibodies against NOX4, β-actin (Santa Cruz Biotechnology), and phospho mTOR and mTOR (Cell Signalling). Signals were detected by chemiluminescence (Pierce). Densitometric analysis was performed with Quantity One software (BioRad). Where indicated rapamycin (10 nM) was added 48 h before stimulation.
For intracellular staining assay, HPASMC were treated with SSc IgG or N IgG (200 µg/ml) and PDGF (15 ng/ml) for 24 h and then permeabilized with Cyto Fix/Perm (Becton Dickinson). Cells were detached by trypsin, washed with PBS, and incubated with antibody against NOX4 (AbCam) for 24 h at 37°C. Cells were then washed and incubated with FITC-conjugated secondary antibody and resuspended in 1 ml PBS. For each sample, 10,000 events were collected. Data analysis was carried out using the WinMDI software.
Nox4 was silenced using siRNA oligoribonucleotides (Qiagen) using Lipofectamine™ 2000 reagent (Life Technologies) following the manufacturer’s instructions. Briefly, 10 µM NOX4 siRNA or scrambled oligos and Lipofectamine 2000 were mixed in serum-free Medium 231 without antibiotics and then added to the cells. Medium was changed after 4 h. Gene silencing and ROS production were monitored 72 h after transfection.
Statistical analysis was performed using GraphPad Prism version 4.0 by two-tailed Student’s t-test. P values less than 0.05 were considered significant.
Since the pathogenesis of scleroderma is characterized by an abnormal generation of ROS [for review, see Ref. (34)] and several lines of evidence implicate oxidative stress in the pathogenesis of PAH (35), we exploited our previous demonstration that agonistic anti-PDGFRα autoantibodies isolated from SSc sera induce an abnormal generation of ROS in normal fibroblasts via NOX (23, 24, 36). Hence, HPASMC were stimulated in vitro with IgG isolated from serum of distinct scleroderma patients (SSc IgG; n = 11) or distinct normal controls (N IgG; n = 10) (200 µg/ml, 15 min) and then incubated with the peroxide-sensitive fluorophore DCHF-DA. Confocal microscopy (Figure 1A), plate reader fluorimeter (Figure 1B), or FACS analysis (Figure 1C) showed that the levels of ROS, induced by PDGF and SSc IgG, were higher than the levels obtained in unstimulated cells (p < 0.05) and were inhibited by preincubating cells with PDGFR tyrosine kinase inhibitor AG 1296 (2 µM, 1 h) (p < 0.05), indicating that ROS production occurred through PDGFRα. The amount of ROS generated by N IgG was not significantly different from that obtained in unstimulated cells (Figures 1A–C).
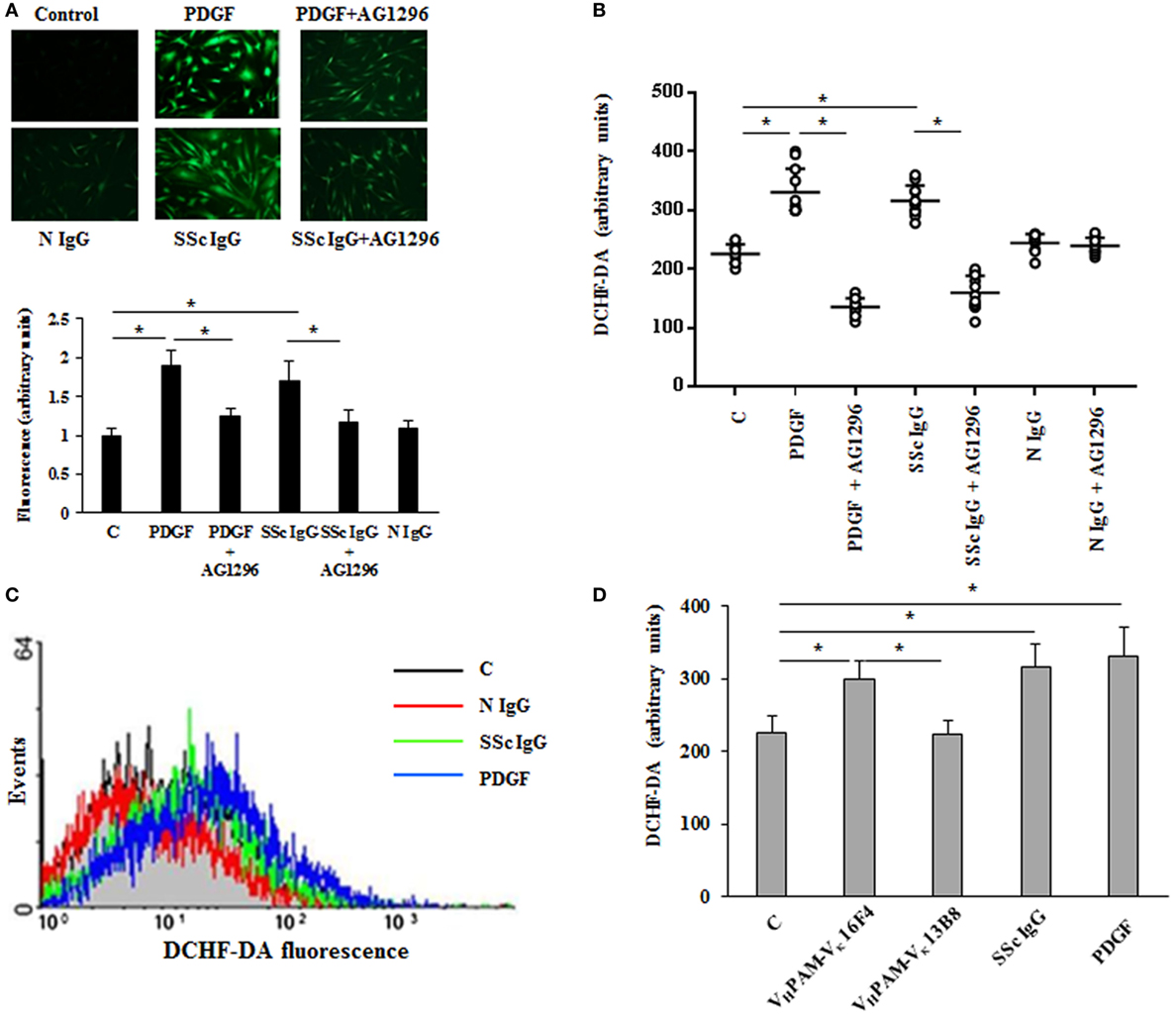
Figure 1. Agonistic anti-PDGF receptor autoantibodies from systemic sclerosis (SSc) patients induce increased reactive oxygen species (ROS) generation in HPASMC. HPASMC were exposed to PDGF (15 ng/ml) or IgG isolated from each SSc patients (SSc IgG; n = 11; 200 µg/ml) or normal controls (N IgG; n = 10; 200 µg/ml) for 15 min in the presence or absence of AG1296 (2 µM) that was added 1 h before stimulation with Ig. Unstimulated cells were used as a control (C). (A) Generation of intracellular ROS was evaluated by fluorescence microscopy using DCHF-DA as a oxidative probe. Representative images of three independent experiments are shown (upper panel). Scale bar = 100 µm. The fluorescence was quantified by ImageJ software (lower panel). The IgG from each subject was tested. Data are presented as mean ± SD of three independent experiments. *p < 0.05. (B) Intracellular ROS production measured by plate reader fluorimeter. The IgG from each subject was tested in triplicate. Data are presented as mean ± SD of three independent experiments. *p < 0.05. (C) ROS production of untreated cells (C) (black line), SSc IgG-treated cells (green line), N IgG-treated cells (red line), and PDGF-treated cells (blue line) by FACS analysis. A representative histogram of three independent experiments is shown. (D) HPASMC were stimulated for 15 min with PDGF (15 ng/ml), SSc IgG (200 µg/ml), and the agonistic (VHPAM-Vκ16F4) and non-agonistic (VHPAM-Vκ13B8) human monoclonal anti-PDGFRα autoantibody (10 µg/ml each). Unstimulated cells were used as a control (C). ROS generation was evaluated by fluorometry. Data are presented as mean ± SD of three independent experiments. *p < 0.05.
Systemic sclerosis IgGs contain a multitude of SSc disease-specific and non-specific antibodies, including agonistic autoantibodies against angiotensin II receptor type 1 and the endothelin receptor type A (37). Thus, to demonstrate that the findings reported above are related to stimulatory antibodies targeting PDGFRα, HPASMC were exposed to combinatorial human monoclonal anti-PDGFRα autoantibody VHPAM-Vκ16F4 (10 µg/ml), which binds and stimulate the PDGFRα in vitro, and to antibody VHPAM-Vκ13B8 (10 µg/ml), which binds but does not stimulate the receptor in vitro (24). Figure 1D shows that the agonistic antibody VHPAM-VK16F4-stimulated cells produced significantly larger amount of ROS compared to unstimulated cells and VHPAM-VK13B8-treated HPASMC. No difference was observed between VHPAM-VK13B8-treated and unstimulated cells.
The in vitro scratch assay was used to study the effect of SSc IgG on HPASMC migration (33). Incubation with PDGF (15 ng/ml) or SSc IgG (200 µg/ml) for 24 h enhanced migratory ability of HPASMC compared to cells not stimulated used as controls (50 and 45%, respectively, over control cells, p < 0.05) (Figure 2A). Migration after N IgG was not statistically different from that of unstimulated HPSMC. The addition of AG1296 (2 µM, 1 h) significantly reduced PDGF- or SSc IgG-induced migration (p < 0.05). AG1296-treated cells did not differ from unstimulated or N IgG-treated cells (p = n.s.) (Figure 2A).
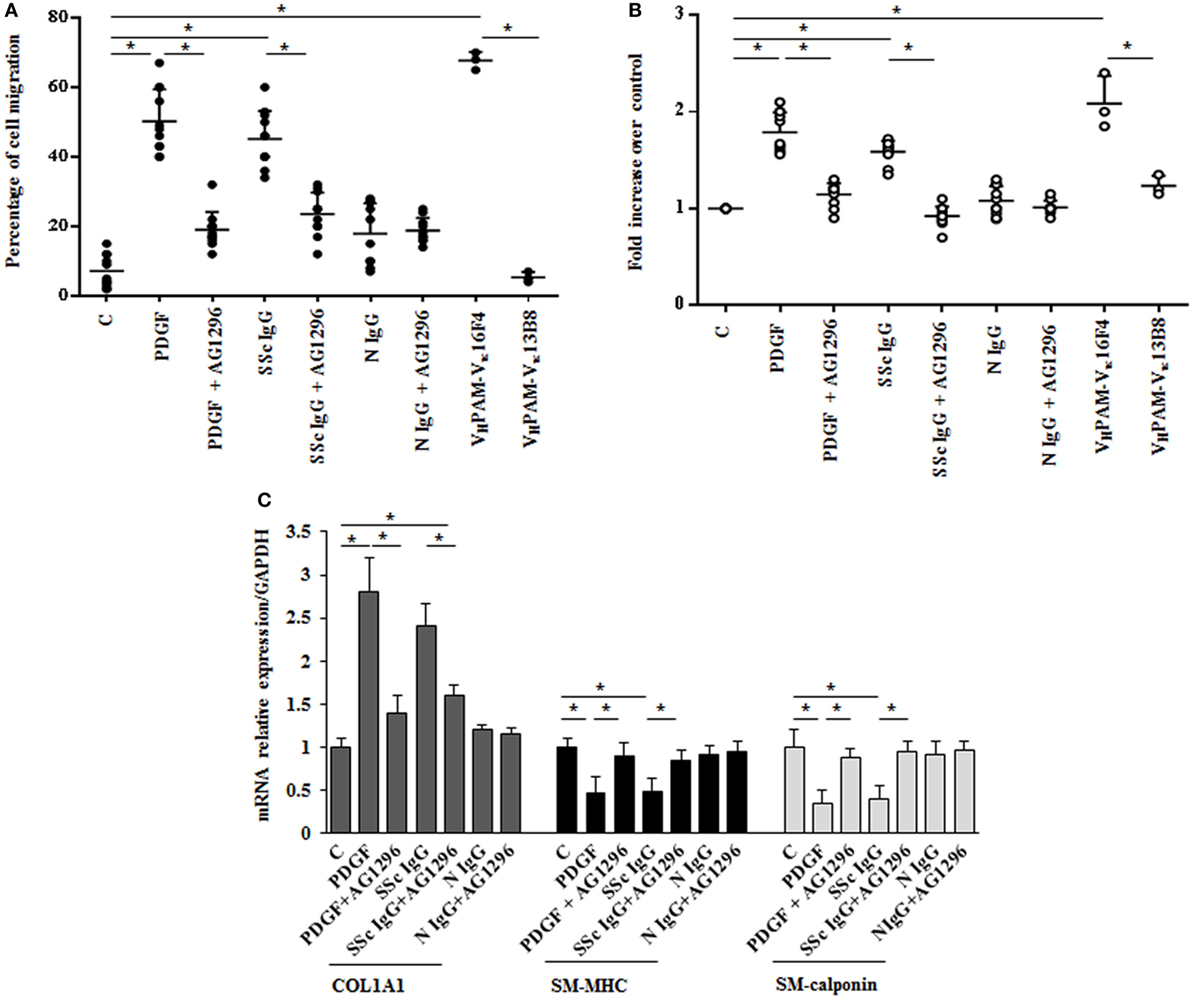
Figure 2. Biological effects induced by systemic sclerosis (SSc) IgGs are mediated by PDGF receptor α (PDGFRα). (A) Migration was analyzed by the wound healing scratch test. Cells were scraped and stimulated with PDGF (15 ng/ml), distinct SSc IgG (200 µg/ml, n = 11), or distinct N IgG (200 µg/ml, n = 10) in the presence or absence of AG1296 or with agonistic (VHPAM-Vκ16F4) and non-agonistic (VHPAM-Vκ13B8) human monoclonal anti-PDGFRα autoantibody. After 24 h, the gap size was measured and expressed as a percentage of the original wound. The IgG from each subject was tested in triplicate. Data are presented as mean ± SD of three independent experiments. *p < 0.05. (B) HPASMC treated with PDGF (15 ng/ml), single-tested SSc IgG (200 µg/ml; n = 11), or N IgG (200 µg/ml; n = 10) or with the human monoclonal anti-PDGFRα autoantibody VHPAM-Vk16F4 and VHPAM-Vκ13B8 for 48 h in the presence or the absence of AG1296 (2 µM). Proliferation was determined by 5-bromo-2′-deoxyuridine assay and expressed as fold increase over controls. The IgG from each subject were tested in triplicate. Data are presented as mean ± SD of five independent experiments. *p < 0.05. (C) HPASMC were treated with AG1296 (2 µM) for 1 h and then exposed to PDGF (15 ng/ml), SSc IgG (200 µg/ml; n = 11), or N IgG (200 µg/ml; n = 10) for 24 h. COL1A1, smooth muscle-myosin heavy chain, and SM-calponin mRNA levels were measured by real-time PCR. Data are presented as mean ± SD of three independent experiments. *p < 0.05. The IgG from each subject was single tested.
Agonistic VHPAM-VK16F4 (10 µg/ml) significantly stimulated HPSMC migration when compared to unstimulated cells and VHPAM-VK13B8-treated cells (60 and 62%, respectively; p < 0.05) (Figure 2A). No difference was detected between unstimulated and VHPAM-VK13B8-treated cells (p = n.s.) (Figure 2A).
HPASMC were incubated with PDGF (15 ng/ml) or IgG isolated from serum of distinct scleroderma patients (SSc IgG; n = 11) or normal controls (N IgG; n = 10) (200 µg/ml) for 48 h, and cell proliferation was determined by monitoring BrdU incorporation for 6 h. SSc IgG and PDGF significantly increased the number of BrdU-positive cells compared to unstimulated cells (1.8- and 1.58-fold, respectively; p < 0.05). This effect was reverted by incubating HPSMC with AG1296 (2 µM, 1 h) (Figure 2B). No difference was demonstrated between unstimulated cells and N IgG-treated cells. These findings were confirmed when cells were exposed to VHPAM-VK16F4 and proliferation compared to that obtained with VHPAM-VK13B8 (Figure 2B).
To determine whether SSc IgG contributed to establish a phenotypic modulation of HPASMC cells favoring a transition from contractile to synthetic state, we detected the expression of transcripts of COL1A1 and smooth muscle cell-specific contractile markers, SM-MHC, and SM-calponin mRNA (38). PDGF and SSc IgG increased COL1A1 gene expression (2.8 ± 0.7- and 2.4 ± 0.2-fold, respectively, when compared to unstimulated cells; p < 0.05) and decreased SM-MHC expression (0.46 ± 0.2- and 0.5 ± 0.1-fold, respectively, when compared to unstimulated cells; p < 0.05) and SM-calponin transcription (0.35 ± 016- and 0.4 ± 0.16-fold, respectively, when compared to unstimulated cells, respectively; p < 0.05) consistent with a synthetic phenotype (Figure 2C). Cells incubated with N IgG showed the same levels of transcripts of unstimulated cells. Experiments performed in the presence of AG1296 confirmed the role of PDGFR as a primary target of SSc IgG. All these findings are consistent with the induction of a synthetic phenotype in HPSMC by anti-PDGFRα autoantibodies.
The stimulation of HPASMC by single SSc IgG occurred through enhanced ROS generation, since the addition of the generic antioxidant NAC (10 mM) and the selective NADPH oxidase inhibitor DPI (10 µM) for 1 h before stimulation significantly prevented cell migration and proliferation induced by PDGF (15 ng/ml) and SSc IgG (200 µg/ml; n = 11) (p < 0.05) (Figures 3A,B). Moreover, the upregulation of COL1A1 and the reduction of mRNA transcript for SM-MHC and SM-calponin induced by PDGF and SSc IgG were reverted by the addition of NAC and DPI (Figure 3C).
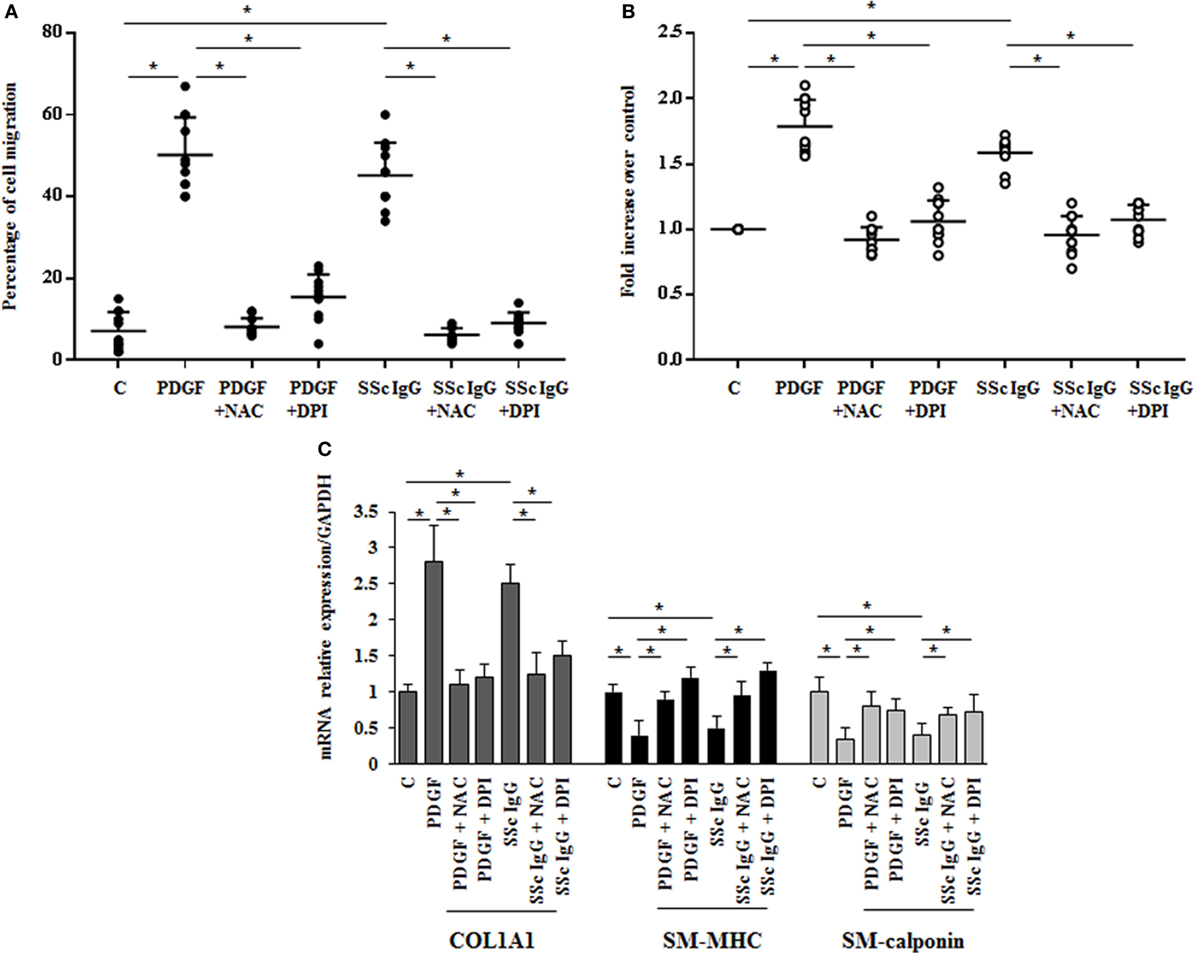
Figure 3. Biological effects induced by systemic sclerosis (SSc) IgG are mediated by reactive oxygen species. (A) HPASMC were scraped and stimulated with PDGF (15 ng/ml) or IgG isolated from each SSc patient (200 µg/ml; n = 11) and single tested in the presence or absence of N-acetyl-cysteine (NAC) (10 mM) or diphenyleneiodonium (DPI) (10 µM). After 24 h, the gap size was measured and expressed as a percentage of the original wound. Data are presented as mean ± SD of five independent experiments. *p < 0.05. (B) HPASMC treated with PDGF (15 ng/ml) and SSc IgG (200 µg/ml) for 48 h in the presence or absence of NAC (10 mM) or DPI (10 µM) for 1 h. Proliferation was determined by 5-bromo-2′-deoxyuridine assay and expressed as fold increase over controls. Data are presented as mean ± SD of five independent experiments. *p < 0.05. The IgG from each subject was single tested. (C) HPASMC were treated with NAC (10 mM) or DPI (10 µM) for 1 h and then exposed to PDGF (15 ng/ml) and distinct SSc IgG (200 µg/ml, n = 11) for 24 h. COL1A1, smooth muscle-myosin heavy chain, and SM-calponin mRNA levels were measured by real-time PCR. Data are presented as mean ± SD of three independent experiments. *p < 0.05.
These results indicate that SSc IgG induce phenotypic modulation and affect the rate of proliferation and migration of HPASMC through ROS produced by NOX activation.
To identify which NOX isoforms were upregulated in stimulated HPASMC, we treated HPASMC with PDGF (15 ng/ml) or single SSc IgG (200 µg/ml; n = 11) or IgG from normal subjects (N IgG; n = 10) (200 µg/ml) for 24 h, and the mRNA levels of the different NOX isoforms were assessed by real-time PCR. After stimulation, NOX4 mRNA levels were significantly upregulated compared to unstimulated cells (8.7 ± 1.5 and 8 ± 1.6 fold, respectively, p < 0.05), whereas NOX2, DUOX1, and DUOX2 were unaffected by the treatment (Figure 4A). PDGF, but not SSc IgG, increased NOX1 mRNA relative to control (2.9 ± 0.2-fold) but, as demonstrated elsewhere (36), the silencing of NOX1 did not influence the ROS generation in these cells (data not shown).
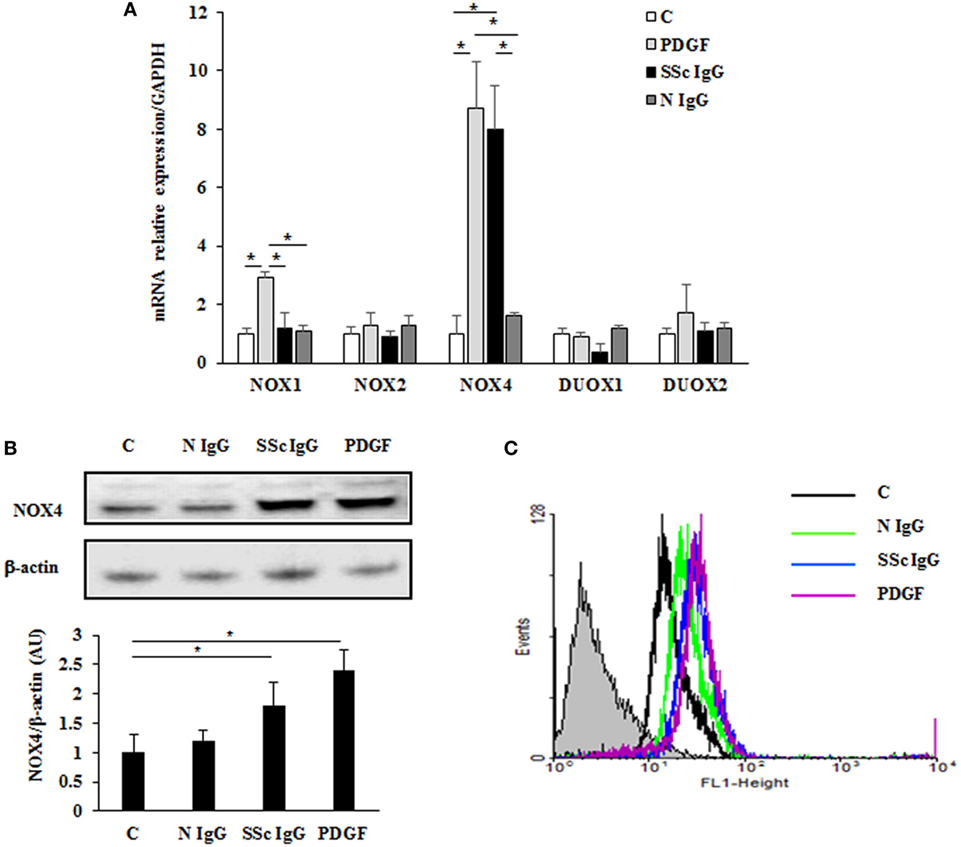
Figure 4. Induction of NOX4 levels by systemic sclerosis (SSc) IgG. (A) Real-time PCR analysis of NOX isoforms in HPASMC treated with PDGF (15 ng/ml) or IgG isolated from distinct SSc patients (200 µg/ml; n = 11) or distinct N IgG (200 µg/ml; n = 10) and single tested for 24 h. Data are presented as mean ± SD of three independent experiments. *p < 0.05 compared to controls. (B) Total cell lysates from HPASMC stimulated with PDGF (15 ng/ml), single SSc IgG (200 µg/ml; n = 5), or single N IgG (200 µg/ml; n = 5) for 24 h were analyzed by immunoblotting with a specific antibody against NOX4. β-actin was used as a control for normalization. One representative experiment is shown in the upper panel. Densitometric analysis from three independent experiments is reported in the lower panel. Data are presented as mean ± SD from three independent experiments. *p < 0.05. (C) One representative FACS analysis of NOX4 expression is shown. Cells were stimulated with PDGF (15 ng/ml), distinct SSc IgG (200 µg/ml; n = 11), or N IgG (200 µg/ml; n = 10); permealized; and then labeled with anti-NOX4 and secondary FITC-conjugated antibody.
A significant increase of NOX4 protein expression compared to basal conditions was observed by immunoblotting (Figure 4B; p < 0.05) and FACS analysis (Figure 4C; p < 0.05) after a 24-hour exposure to PDGF or SSc IgG, whereas no effect was observed after N IgG addition (200 µg/ml; n = 10).
To dissect the role of NOX4 in ROS generation after PDGF or SSc IgG stimulation, we silenced NOX4 using small interfering RNA (siRNA) (p < 0.05) (Figure 5A). The specific siRNA selectively prevented ROS generation in HPASMC stimulated with either 15 ng/ml PDGF or 200 µg/ml SSc IgG for 15 min (Figure 5B) and reverted the effects of PDGF or SSc IgG on migration and proliferation (Figures 5C,D) compared to unstimulated cells. Silencing of NOX4 inhibited PDGF, SSc IgG induction of COL1A1, and downregulation of SM-MHC and SM-calponin, indicating a regulatory role of NOX4 in the phenotypic modulation of HPASMC (Figure 6).
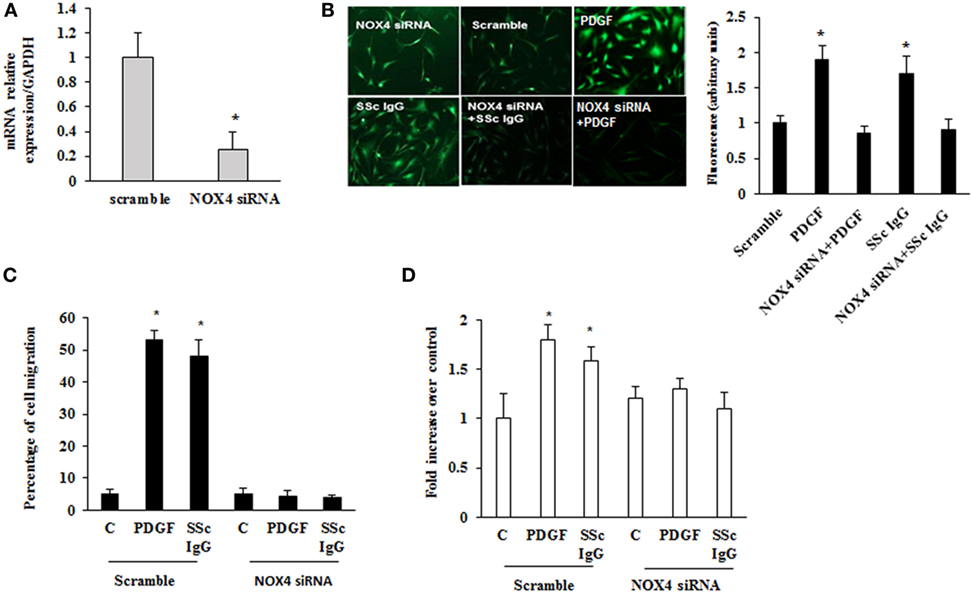
Figure 5. Inhibition of systemic sclerosis (SSc) IgG biological effects by silencing NOX4. (A) HPASMC were transfected with NOX4 or scramble siRNA for 24 h, and NOX4 mRNA levels were analyzed by real-time PCR analysis. Data are presented as mean ± SD of three independent experiments. *p < 0.05 compared to cells transfected with scramble siRNA. (B) HPASMC were transfected with NOX4 siRNA or scramble siRNA for 24 h and then were stimulated with PDGF (15 ng/ml) or distinct SSc IgG (200 µg/ml; n = 11) for 15 min. Cells were incubated with DCHF-DA, and free radical production was evaluated by fluorescence microscopy. Representative images of three independent experiments are shown. The fluorescence was quantified using ImageJ software (right panel). (C) HPASMC were transfected with NOX4 or scramble siRNA for 24 h, scraped, and stimulated with PDGF (15 ng/ml) or single SSc IgG (200 µg/ml). After 24 h, the gap size was measured and expressed as a percentage of the original wound. Data are presented as mean ± SD of five independent experiments. *p < 0.05. (D) Transfected cells with NOX4 or scramble siRNA for 24 h were treated with PDGF (15 ng/ml) or single SSc IgG (200 µg/ml; n = 11) for 48 h. Proliferation was determined by 5-bromo-2′-deoxyuridine assay and expressed as fold increase over controls. Data are presented as mean ± SD of five independent experiments. *p < 0.05 compared to unstimulated cells.
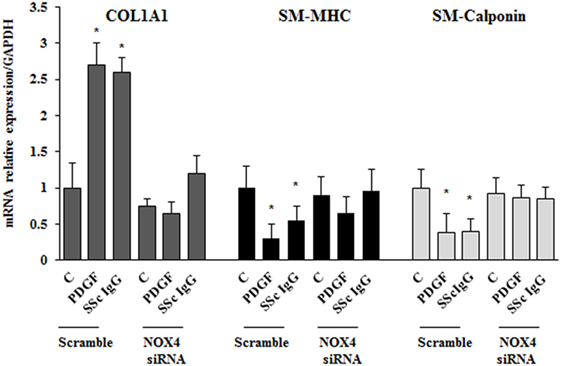
Figure 6. NOX4 silencing drives a synthetic phenotype in HPASMC. Transfected cells with NOX4 siRNA or scramble siRNA for 24 h were treated with PDGF (15 ng/ml) or systemic sclerosis IgG (200 µg/ml; n = 11) for 24 h. COL1A1, smooth muscle-myosin heavy chain, and SM-calponin mRNA levels were measured by real-time PCR. Data are presented as mean ± SD of three independent experiments. *p < 0.05 compared to unstimulated cells.
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase that acts as a major activator of cell growth and proliferation. mTOR has also been shown to play a role in proliferation of vascular smooth muscle cells and when dysregulated has been implicated in vascular remodeling (39). To evaluate the role of mTOR as a possible downstream effector in stimulated HPASMC, we treated the cells with PDGF (15 ng/ml) or distinct SSc IgG (200 µg/ml; n = 11) for 24 h in the presence or absence of rapamycin (10 nM), a specific inhibitor of mTOR added 48 h before. Phosphorylation levels of mTOR were higher in samples incubated with PDGF or SSc IgG compared to unstimulated cells and were reverted by rapamycin (Figure 7A). Moreover, the treatment with rapamycin significantly decreased the migration rate (Figure 7B), proliferation (Figure 7C); and COL1A1 expression (Figure 7D) and restored the levels of SM-MHC, and SM-calponin (Figure 7D) in HPASMC incubated with PDGF or SSc IgG. These results demonstrated that mTOR is a crucial downstream signaling of SSc IgG effects via PDGFR.
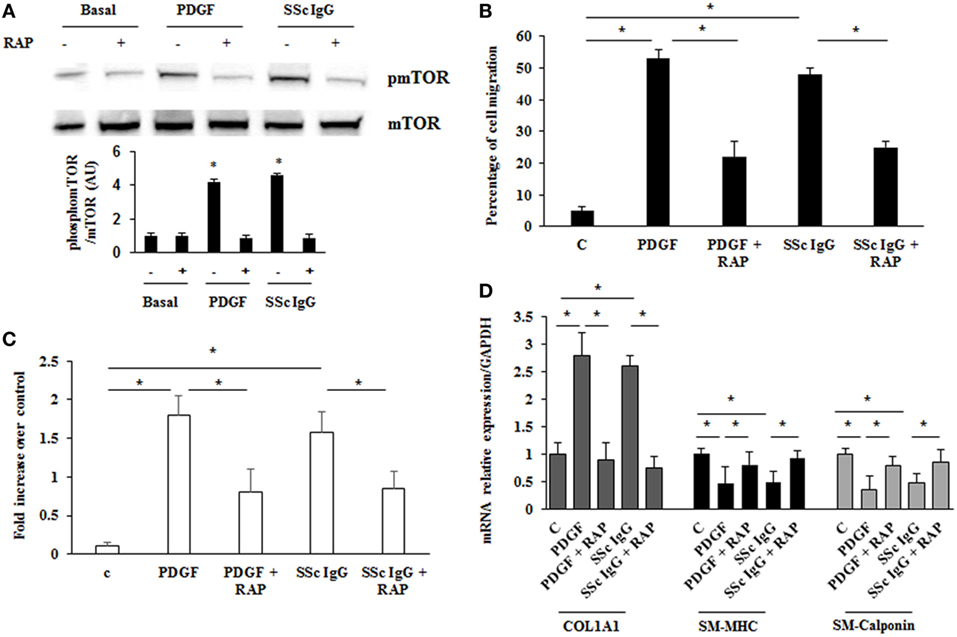
Figure 7. Modulation of systemic sclerosis (SSc) IgG effects by rapamycin. (A) HPASMC were stimulated with PDGF (15 ng/ml) or SSc IgG (200 µg/ml; n = 11) for 24 h in the presence or absence of rapamycin (10 nM) added 48 h before. Phosphorylation of mTOR was detected by immunoblotting, and anti-total mTOR antibody was used as a control for normalization. One representative experiment is shown in the upper panel. Densitometric analysis from three independent experiments is reported in the lower panel. Data are presented as mean ± SD. *p < 0.05. (B) Migration was analyzed through wound healing scratch test. Cells were scraped and stimulated with PDGF (15 ng/ml) or single SSc IgG (200 µg/ml; n = 11) in the presence or absence of rapamycin (10 nM) added 48 h before. After 24 h, the gap size was measured and expressed as a percentage of the original wound. Data are presented as mean ± SD of five independent experiments. *p < 0.05. (C) HPASMC cells treated with PDGF (15 ng/ml) or single SSc IgG (200 µg/ml; n = 11) for 48 h in the presence or the absence of rapamycin (10 nM) added 48 h before. Proliferation was determined by 5-bromo-2′-deoxyuridine assay and expressed as fold increase over controls. Data are presented as mean ± SD of five independent experiments. *p < 0.05. (D) HPASMC were treated with rapamycin and then exposed to PDGF (15 ng/ml) or SSc IgG (200 µg/ml; n = 11) for 24 h. COL1A1, smooth muscle-myosin heavy chain, and SM-calponin mRNA levels were measured by real-time PCR. Data are presented as mean ± SD of three independent experiments. *p < 0.05. The IgG from each subject was single tested.
Vascular smooth muscle cells are highly specialized cells whose principal functions are contraction and regulation of blood vessel tone. In the mature arterial wall, they exhibit a contractile phenotype characterized by low rate of proliferation and expression of specific markers such as α-SMA, SM-MHC, and SM-calponin. In response to vascular injury, they can dedifferentiate into a proliferative and synthetic phenotype. This phenotypic switching contributes to intimal hyperplasia and has been linked to the development and progression of several vascular disease (38). The functional state of smooth muscle cells is controlled by a complex combination of local environmental cues and epigenetic programs that influences synthetic or contractile, as well as intermediate phenotypes (40–44).
In this study, we analyzed the expression of distinct functional markers of smooth muscle cells from human pulmonary arteries exposed in vitro to anti-PDGFR autoantibodies from SSc patients. In our experimental conditions, the data show that HPASMC acquire a synthetic phenotype characterized by higher growth rate, migratory activity, type I collagen gene expression, and minimal expression of markers characteristic of the contractile phenotype such as SM-MHC and smooth muscle-calponin. Thus, our findings indicate that anti-PDGFR autoantibodies may contribute not only to the development of SSc fibrotic lesions (23, 26) but also to the development of the vascular features. However, it is important to point out that our data do not allow to establish whether the new phenotype is due to the conversion of normal contractile vascular smooth muscle cells to a less differentiate state or to the expansion of medial-derived multipotent vascular stem cells (45).
Furthermore, since none of the SSc IgGs were from patients with pulmonary arterial hypertension, it seems that the impact of anti-PDGFRα autoantibodies on vascular smooth muscle cells reflects a general phenomenon, that is, scleroderma vasculopathy, and the association of their serum levels to specific clinical features (pulmonary arterial hypertension, digital ulcers, and scleroderma renal crisis) must be addressed in a larger cohort of SSc patients.
The activation of PDGFR by SSc IgG was both selective and ROS dependent since the presence of AG 1296 or NAC reduced the proliferation and migration of HPASMC and increased the expression of the differentiation markers. Our data are at variance with the findings reported by Arts et al. (46) who demonstrated that in smooth muscle cells SSc IgG engaged PDGFR leading to the activation of the epidermal growth factor receptor through a PDGFR-independent pathway. Furthermore, even if SSc IgG induced a profibrotic change in smooth muscle cells, collagen gene expression and cell proliferation were not influenced. Finally, some crucial experimental variables such as the source of the cells (aortic rat versus human pulmonary smooth muscle cells) and the timing of stimulation may explain the difference between their and our data.
Reactive oxygen species are known to mediate a variety of intracellular process, and the identification of the sources of these ROS has facilitated our understanding of a number of biological functions. Here, we demonstrated that the SSc IgG-induced abnormal oxidative stress through NOX4 activation facilitated the synthetic phenotype of human pulmonary artery smooth muscle cells. Interestingly and similar to what occurs in SSc fibroblasts (36), SSc IgG increased the expression of NOX4, while NOX1 was unaffected. These findings are the exact opposite of what reported by Lassègue et al. (47). If this discrepancy is to be ascribed to the different cell line and methodology used or indicates a different pathophysiological role of smooth muscle cells, NOX isoforms in response to distinct environmental cues remain to be established. Furthermore, at variance with what we found in normal fibroblasts (36), in HPSMC, SSc IgG did not induce NOX2, suggesting that individual NOX enzymes may play distinct roles between cell types. Thus, the precise interplay of NOX2 and NOX4 in HPSMC following SSc IgG stimulation needs further studies.
Cell proliferation and migration are thought to be a critical step in fibrogenesis, and since there is a strict interplay between mTOR and redox-based signaling (48, 49), we addressed the contribution of mTOR signaling to these phenotypes in cells exposed to ROS inducing stimuli.
mTOR is a serine/threonine kinase that consists of two distinct complexes mTORC1 and mTORC2. Growth factors, nutrients, DNA damage, and hypoxia among others regulate mTOR, which, once activated, phosphorylates multiple downstream signals playing a key role in cellular protein synthesis, control of eukaryotic cell growth, and proliferation, including cell cycle progression, differentiation, protein degradation, apoptosis, and angiogenesis (50). The role of mTOR in the vasculature is subject to intense investigation, particularly in the field of pulmonary arterial hypertension, where evidence has been provided of its importance in the events leading to proliferation and survival of pulmonary vascular cells and its modulation by growth factors, vasoactive agents, vascular Ca2+ channels, and chronic hypoxia (50).
Interestingly, Eid et al. (51), in a different experimental system, have shown that the pharmacological inhibition of mTOR with rapamycin decreases NOX4 activity, ROS production, and podocytes apoptosis induced by high glucose, suggesting that rapamycin may represent a therapeutic modality of diabetic kidney disease (51). In line with their observations, we show that ROS-inducing SSc IgG activated mTOR, and its pharmacological inhibition with the antifungal macrolide rapamycin blocked PDGF and SSc IgG ability to promote crucial functions of vascular smooth muscle cells such as proliferation, migration, and collagen production, implying that mTORC1 mediated the phenotypic conversion.
These findings underscore the potential importance of mTOR in the pathogenesis of SSc, also in light of studies conducted on fibroblasts in vitro and in experimental models of fibrosis. mTOR activation contributes to type I collagen production by dermal fibroblasts (52, 53), and rapamycin improves skin fibrosis in two mouse models of fibrosis (54). Treatment of a small cohort of SSc patients with rapamycin was safe, but the efficacy was not impressive (55). However, the clinical benefit of a more efficient mTOR blockade should be explored exploiting the novel information regarding mTOR signaling targets.
In conclusion, our study demonstrates that stimulatory autoantibodies targeting PDGFR activate smooth muscle cells and may, thus, contribute to the development of SSc vascular lesions.
However, to better define their role in the pathogenesis of the individual SSc vascular manifestations, such as pulmonary arterial hypertension, digital ulcers, and SSc renal disease, it is necessary to study vascular smooth muscle cells of distinct organs, those isolated from tissues of SSc patients, and to establish whether there is indeed an association between the serum levels of anti-PDGFRα autoantibodies and particular vascular features. Nevertheless, the present findings and those already published (23, 24, 27) highlight the importance of an early treatment of scleroderma aimed at downregulating B cell immune response.
SS designed the experimental work analyzed results and wrote the first draft of the manuscript. DA, TS, and MR performed experimental work and analyzed results. CF and DF selected and provided samples and analyzed clinical data. GM, CP, CT, and AGR provided the monoclonal autoantibodies. AF contributed to write the final revised version of the manuscript. AGA designed, supervised, evaluated the experiments, and wrote the final version of the manuscript. All the authors read and approved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Fondazione di Medicina Molecolare e Terapia Cellulare- Università Politecnica delle Marche and by grants from Ministero Italiano per l’ Università e la Ricerca Scientifica and AILS (Associazione Italiana lotta alla Sclerodermia).
1. Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med (2009) 360:1989–2003. doi: 10.1056/NEJMra0806188
2. Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest (2007) 117:557–67. doi:10.1172/JCI31139
3. Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Primers (2015) 1:15002. doi:10.1038/nrdp.2015.2
4. Prescott RJ, Freemont AJ, Jones CJ, Hoyland J, Fielding P. Sequential dermal microvascular and perivascular changes in the development of scleroderma. J Pathol (1992) 166:255–63. doi:10.1002/path.1711660307
5. Kahaleh B. Vascular disease in scleroderma: mechanisms of vascular injury. Rheum Dis Clin North Am (2008) 34:57–71. doi:10.1016/j.rdc.2007.12.004
6. Trojanowska M. Cellular and molecular aspects of vascular dysfunction in systemic sclerosis. Nat Rev Rheumatol (2010) 6:453–60. doi:10.1038/nrrheum.2010.102
7. Pattanaik D, Brown M, Postlethwaite AE. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res (2011) 1:105–25. doi:10.2147/JIR.S18145
8. Rodnan GP, Myerowitz RL, Justh GO. Morphologic changes in the digital arteries of patients with progressive systemic sclerosis (scleroderma) and Raynaud phenomenon. Medicine (1980) 59:393–408. doi:10.1097/00005792-198011000-00001
9. Maricq HR, Downey JA, LeRoy EC. Standstill of nailfold capillary blood flow during cooling in scleroderma and Raynaud’s syndrome. Blood Vessels (1976) 13:338–49.
10. Davies CA, Jeziorska M, Freemont AJ, Herrick AL. The differential expression of VEGF, VEGFR-2, and GLUT-1 proteins in disease subtypes of systemic sclerosis. Hum Pathol (2006) 37:190–7. doi:10.1016/j.humpath.2005.10.007
11. Distler O, Distler JH, Scheid A, Acker T, Hirth A, Rethage J, et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res (2004) 95:109–16. doi:10.1161/01.RES.0000134644.89917.96
12. Mackiewicz Z, Sukura A, Povilenaité D, Ceponis A, Virtanen I, Hukkanen M, et al. Increased but imbalanced expression of VEGF and its receptors has no positive effect on angiogenesis in systemic sclerosis skin. Clin Exp Rheumatol (2002) 20:641–6.
13. Matucci-Cerinic M, Kahaleh B, Wigley FM. Evidence that systemic sclerosis is a vascular disease. Arthritis Rheum (2013) 65:1953–62. doi:10.1002/art.37988
14. Wigley FM. Vascular disease in scleroderma. Clin Rev Allergy Immunol (2009) 36:150–75. doi:10.1007/s12016-008-8106-x
15. Riemekasten G, Philippe A, Näther M, Slowinski T, Müller DN, Heidecke H, et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann Rheum Dis (2011) 70:530–6. doi:10.1136/ard.2010.135772
16. Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev (1999) 79:1283–316.
17. Gay S, Jones RE Jr, Huang GQ, Gar RE. Immunohistologic demonstration of platelet-derived growth factor (PDGF) and sis-oncogene expression in scleroderma. J Invest Dermatol (1989) 92:301–3. doi:10.1111/1523-1747.ep12276895
18. Klareskog L, Gustafsson R, Scheynius A, Hallgren R. Increased expression of platelet-derived growth factor type B receptors in the skin of patients with systemic sclerosis. Arthritis Rheum (1990) 33:1534–41. doi:10.1002/art.1780331011
19. Rajkumar VS, Howell K, Csiszar K, Denton CP, Black CM, Abraham DJ. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res Ther (2005) 7:R1113–23. doi:10.1186/ar1790
20. Akhmetshina A, Dees C, Pileckyte M, Maurer B, Axmann R, Jüngel A, et al. Dual inhibition of c-abl and PDGF receptor signaling by dasatinib and nilotinib for the treatment of dermal fibrosis. FASEB J (2008) 22:2214–22. doi:10.1096/fj.07-105627
21. Olivieri A, Locatelli F, Zecca M, Sanna A, Cimminiello M, Raimondi R, et al. Imatinib for refractory chronic graft-versus-host disease with fibrotic features. Blood (2009) 114:700–18. doi:10.1182/blood-2009-02-204156
22. Fraticelli P, Gabrielli B, Pomponio G, Valentini G, Bosello S, Riboldi P, et al. Low-dose oral imatinib in the treatment of systemic sclerosis interstitial lung disease unresponsive to cyclophosphamide: a phase II pilot study. Arthritis Res Ther (2014) 16:R144. doi:10.1186/ar4606
23. Svegliati Baroni S, Santillo M, Bevilacqua F, Luchetti M, Spadoni T, Mancini M, et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med (2006) 354:2667–76. doi:10.1056/NEJMoa052955
24. Moroncini G, Grieco A, Nacci G, Paolini C, Tonnini C, Pozniak C, et al. Epitope specificity determines pathogenicity and detectability of anti-PDGFRα autoantibodies in systemic sclerosis. Arthritis Rheumatol (2015) 67:1891–903. doi:10.1002/art.39125
25. Svegliati S, Marrone G, Pezone A, Spadoni T, Grieco A, Moroncini M, et al. Oxidative DNA damage induces the ATM-mediated transcriptional suppression of the WNT inhibitor WIF-1 in systemic sclerosis and fibrosis. Sci Signal (2014) 7:ra84. doi:10.1126/scisignal.2004592
26. Svegliati S, Cancello R, Sambo P, Luchetti M, Paroncini P, Orlandini G, et al. Platelet-derived growth factor and reactive oxygen species (ROS) regulate Ras protein levels in primary human fibroblasts via ERK1/2. Amplification of ROS and Ras in systemic sclerosis fibroblasts. J Biol Chem (2005) 280:36474–82. doi:10.1074/jbc.M502851200
27. Luchetti MM, Moroncini G, Escamez MJ, Svegliati Baroni S, Spadoni T, Grieco A, et al. Induction of scleroderma fibrosis in skin-humanized mice by anti-platelet-derived growth factor receptor agonistic autoantibodies. Arthritis Rheumatol (2016) 68:2263–73. doi:10.1002/art.39728
28. Gelber AC, Manno RL, Shah AA, Woods A, Le EN, Boin F, et al. Race and association with disease manifestations and mortality in scleroderma: a 20-year experience at the Johns Hopkins Scleroderma Center and review of the literature. Medicine (2013) 92:191–205. doi:10.1097/MD.0b013e31829be125
29. Launay D, Sitbon O, Hachulla E, Mouthon L, Gressin V, Rottat L, et al. Survival in systemic sclerosis-associated pulmonary arterial hypertension in the modern management era. Ann Rheum Dis (2013) 72:1940–6. doi:10.1136/annrheumdis-2012-202489
30. Guillevin L, Hunsche E, Denton CP, Krieg T, Schwierin B, Rosenberg D, et al. Functional impairment of systemic scleroderma patients with digital ulcerations: results from the DUO Registry. Clin Exp Rheumatol (2013) 31(2 Supplement 76):71–80.
31. van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheum (2013) 65:2737–47. doi:10.1002/art.38098
32. LeRoy EC, Black CM, Fleischmajer R, Jablonska S, Krieg T, Medsger TA Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol (1988) 12:217–23.
33. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc (2007) 2:329–33. doi:10.1038/nprot.2007.30
34. Gabrielli A, Svegliati S, Moroncini G, Amico D. New insights into the role of oxidative stress in scleroderma fibrosis. Open J Rheumatol (2012) 6:87–95. doi:10.2174/1874312901206010087
35. Fessel JP, West JD. Redox biology in pulmonary arterial hypertension (2013 Grover Conference series). Pulm Circ (2015) 5:599–609. doi:10.1086/683814
36. Spadoni T, Svegliati Baroni S, Amico D, Albani L, Moroncini G, Avvedimento EV, et al. A reactive oxygen species–mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol (2015) 67:1611–22. doi:10.1002/art.39084
37. Riemekasten G, Cabral-Marques O. Vascular hypothesis revisited: role of stimulating antibodies against angiotensin and endothelin receptors in the pathogenesis of systemic sclerosis. Autoimmun Rev (2016) 15(7):690–4. doi:10.1016/j.autrev.2016.03.005
38. Rensen SS, Doevendans PA, Van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J (2007) 15:100–8. doi:10.1007/BF03085963
39. Montezano A, Touyz M. Mammalian target of rapamycin: a novel pathway in vascular calcification. Can J Cardiol (2014) 30:482–4. doi:10.1016/j.cjca.2014.03.001
40. Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW, Gabbiani G, et al. Smooth muscle cell plasticity: fact or fiction? Circ Res (2013) 112:17–22. doi:10.1161/CIRCRESAHA.112.281048
41. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev (2004) 84:767–801. doi:10.1152/physrev.00041.2003
42. Frid MG, Moiseeva EP, Stenmark KR. Multiple phenotypically distinct smooth muscle cell populations exist in the adult and developing bovine pulmonary arterial media in vivo. Circ Res (1994) 75:669–81. doi:10.1161/01.RES.75.4.669
43. Christen T, Verin V, Bochaton-Piallat M, Popowski Y, Ramaekers F, Debruyne P, et al. Mechanisms of neointima formation and remodeling in the porcine coronary artery. Circulation (2001) 103:882–8. doi:10.1161/01.CIR.103.6.882
44. Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest (2000) 106:1139–47. doi:10.1172/JCI10522
45. Majesky MW. Development basis of vascular smooth muscle diversity. Arterioscler Tromb Vasc Biol (2007) 27:1248–58. doi:10.1161/ATVBAHA.107.141069
46. Arts MR, Baron M, Chokr N, Fritzler MJ, Canadian Scleroderma Research Group (CSRG), Servant MJ. Systemic sclerosis immunoglobulin induces growth and pro-fibrotic state in vascular smooth muscle cells through the epidermal growth factor receptor. PLoS One (2014) 9:e100035. doi:10.1371/journal.pone.0100035
47. Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, et al. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res (2001) 88:888–94. doi:10.1161/hh0901.090299
48. Sarbassov DD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem (2005) 280(47):39505–9. doi:10.1074/jbc.M506096200
50. Goncharova EA. mTOR and vascular remodeling in lung diseases: current challenges and therapeutic prospects. FASEB J (2013) 27:1796–807. doi:10.1096/fj.12-222224
51. Eid A, Ford B, Bhandary B, De Cassia Cavaglieri R, Block K, Barnes J, et al. Mammalian target of rapamycin regulates NOX4-mediated podocyte depletion in diabetic renal injury. Diabetes (2013) 62:2935–47. doi:10.2337/db12-1504
52. Ponticos M, Papaioannou I, Xu S, Holmes AM, Khan K, Denton CP, et al. Failed degradation of JunB contributes to overproduction of type I collagen and development of dermal fibrosis in patients with systemic sclerosis. Arthritis Rheumatol (2015) 67:243–53. doi:10.1002/art.38897
53. Tamaki Z, Asano Y, Kubo M, Ihn H, Tada Y, Sugaya M, et al. Effects of the immunosuppressant rapamycin on the expression of human α2(I) collagen and matrix metalloproteinase 1 genes in scleroderma dermal fibroblasts. J Dermatol Sci (2014) 74:251–9. doi:10.1016/j.jdermsci.2014.02.002
54. Yoshizaki A, Yanaba K, Yoshizaki A, Iwata Y, Komura K, Ogawa F, et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum (2010) 62:2476–87. doi:10.1002/art.27498
Keywords: systemic sclerosis, autoantibodies, vascular smooth muscle cells, platelet-derived growth factor, synthetic phenotype
Citation: Svegliati S, Amico D, Spadoni T, Fischetti C, Finke D, Moroncini G, Paolini C, Tonnini C, Grieco A, Rovinelli M, Funaro A and Gabrielli A (2017) Agonistic Anti-PDGF Receptor Autoantibodies from Patients with Systemic Sclerosis Impact Human Pulmonary Artery Smooth Muscle Cells Function In Vitro. Front. Immunol. 8:75. doi: 10.3389/fimmu.2017.00075
Received: 28 October 2016; Accepted: 17 January 2017;
Published: 08 February 2017
Edited by:
Raffaele De Palma, Seconda Università degli Studi di Napoli, ItalyReviewed by:
Marta Rizzi, University Medical Center Freiburg, GermanyCopyright: © 2017 Svegliati, Amico, Spadoni, Fischetti, Finke, Moroncini, Paolini, Tonnini, Grieco, Rovinelli, Funaro and Gabrielli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Armando Gabrielli, YS5nYWJyaWVsbGlAdW5pdnBtLml0
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.