 Thomas R. L. Klei
Thomas R. L. Klei Sanne M. Meinderts
Sanne M. Meinderts Timo K. van den Berg
Timo K. van den Berg Robin van Bruggen
Robin van Bruggen- Department of Blood Cell Research, Sanquin Research and Landsteiner Laboratory, University of Amsterdam, Amsterdam, Netherlands
Erythropoiesis is a highly regulated process where sequential events ensure the proper differentiation of hematopoietic stem cells into, ultimately, red blood cells (RBCs). Macrophages in the bone marrow play an important role in hematopoiesis by providing signals that induce differentiation and proliferation of the earliest committed erythroid progenitors. Subsequent differentiation toward the erythroblast stage is accompanied by the formation of so-called erythroblastic islands where a central macrophage provides further cues to induce erythroblast differentiation, expansion, and hemoglobinization. Finally, erythroblasts extrude their nuclei that are phagocytosed by macrophages whereas the reticulocytes are released into the circulation. While in circulation, RBCs slowly accumulate damage that is repaired by macrophages of the spleen. Finally, after 120 days of circulation, senescent RBCs are removed from the circulation by splenic and liver macrophages. Macrophages are thus important for RBCs throughout their lifespan. Finally, in a range of diseases, the delicate interplay between macrophages and both developing and mature RBCs is disturbed. Here, we review the current knowledge on the contribution of macrophages to erythropoiesis and erythrophagocytosis in health and disease.
Introduction
Red blood cell (RBC) turnover is a complex process that is regulated at different levels by many factors, particularly by macrophages. Macrophages shape and direct the developing RBC throughout erythropoiesis and ultimately phagocytose senescent RBCs. To perform these tasks, macrophages rely on adhesion molecules, a range of soluble and mechanical factors, and it requires cross talk with the developing or senescent RBC. Here, we review the role macrophages in erythropoiesis and erythrophagocytosis in health and disease.
Macrophage Function in Erythropoiesis
Erythropoiesis is a highly coordinated and regulated process that consists of defined developmental stages, which are regulated by macrophages in a variety of ways. First, macrophages retain hematopoietic stem cells (HSCs) in the hematopoietic niche. Growth factors and cytokines including interleukin-3 (IL-3) and granulocyte-macrophage colony-stimulating factor (GM-CSF) facilitate proliferation and differentiation of the earliest primitive erythroid progenitor cells, so-called burst-forming unit-erythroid cells (BFU-e) (1). These slowly proliferating erythroid progenitors can then, under the influence of kidney-derived erythropoietin (Epo), differentiate into rapidly dividing colony-forming unit-erythroid cells (CFU-e) (2). Subsequent differentiation into the erythroblast stage is accompanied by the formation of so-called erythroblastic islands. Here, a central macrophage interacts with up to 30 erythroblasts through various adhesion molecules both on the macrophage as well as on the erythroblasts (3). In these erythroblastic islands, macrophages facilitate erythroblast proliferation, differentiation, and are involved in iron supply. Finally, macrophages phagocytose and digest the nuclei extruded by erythroblasts during their transition to the reticulocyte stage (4).
Macrophages and Erythroblasts Form Erythroblastic Islands through Intercellular Adhesion Molecules
Erythroblastic islands can be found in all tissue compartments that support erythropoiesis including the yolk sac, fetal liver, bone marrow, and in certain cases the splenic red pulp (5). They are important for proper erythropoiesis as depletion of bone marrow macrophages, or interference with the adhesion molecules that contribute to the formation of erythroblastic islands, leads to pro-erythroblast differentiation arrest, a reduction in proliferation, increased apoptosis, and ultimately anemia (6–8). Several adhesion molecules have been identified that are important for formation and stabilization of erythroblastic islands and thus for erythropoiesis (Figure 1).
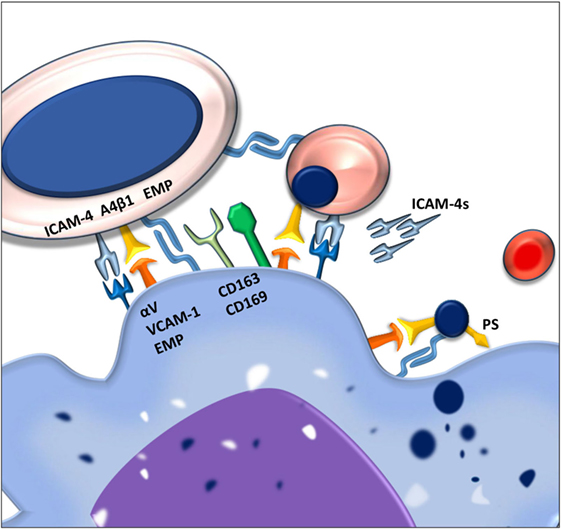
Figure 1. Macrophages and erythroblasts form erythroblastic islands through adhesion molecules. Macrophages express several adhesion molecules that facilitate interactions with erythroid precursors of various stages. The intercellular adhesion molecule 4 (ICAM-4) and the α4β1 integrin on erythroblasts interact with the αVβ1 integrin and vascular cell adhesion molecule-1 (VCAM-1) on the macrophage. Gradual loss of the α4β1 integrin and EMP on the erythroblast facilitates the release of reticulocytes from the erythroblastic island. Persistence of these adhesion molecules on the pyrenocytes is contributing to its phagocytosis by the central macrophage.
One of the first adhesion molecules described to be important for erythroblastic island formation is erythroblast macrophage protein (Emp). Emp is expressed both on the central macrophage as well as on the erythroblast (9). The homotypic interaction between Emp on the macrophage and Emp on the erythroblast is one of the prerequisites for efficient erythropoiesis since interfering with this interaction, using blocking antibodies, decreases proliferation and results in increased erythroblast apoptosis (10). Even though macrophages have been shown to be able to interact with Emp-null erythroblasts, these erythroblasts do not enucleate, underlining the importance of this adhesion molecule in erythropoiesis.
Next to Emp, several other adhesion molecules are important in the formation of stable erythroblastic islands. These include the α4β1 integrin and intercellular adhesion molecule 4 (ICAM-4) expressed on erythroid precursors. These molecules interact with vascular cell adhesion molecule-1 (VCAM-1) and αV integrins, respectively, such as αVβ1 and αVβ5 (11). By interfering with the interactions between these receptor pairs by use of blocking antibodies, it was found that these adhesion molecules are important for erythroblastic island integrity. Interfering with the ICAM-4–αV integrin interaction was shown to lead to a reduction in erythroblastic island number as well as the total number of erythroblasts per island (6). Similarly, loss of α4β1-VCAM-1 interaction led to erythroblastic island disruption (8). Since abrogation of the ICAM-4–αV integrin binding was found to lead to reduced interaction between erythroblasts and macrophages, and since ICAM-4 expression is decreasing during erythropoiesis, the discovery of a secreted form of ICAM-4, ICAM-4S, prompted the hypothesis that ICAM-4S is involved in the release of terminally differentiated erythroblasts from the erythroblastic island (12).
Apart from promoting erythroblastic island stability and formation, several adhesion molecules found on the central macrophages, such as CD163 and CD169, have been shown to promote proliferation. CD163 consists of nine so-called scavenger receptor cysteine rich (SRCR) domains. The third SRCR domain within CD163 functions to scavenge hemoglobin–haptoglobin complexes from the circulation (13). A 13 amino acid motif within the second scavenger domain of CD163 was found to directly interact with erythroblasts (14). It was furthermore established that CD163 interaction with erythroblasts promoted expansion but not differentiation. This was shown to be dependent on the maturation stage of the erythroblast as the interaction between macrophage CD163 and the erythroblast is quickly lost past the pro-erythroblast stage, indicating that its function is to regulate early pro-survival signals.
The counter receptor for CD163 on the erythroblast has not yet been identified. What is known is that upon interaction of CD163 with the hemoglobin–haptoglobin complex, a cascade of intracellular signaling is induced within the macrophage, including calcium mobilization and inositol triphosphate production. Subsequently, through an autocrine feedback loop, the secretion of interleukin-6 and macrophage colony-stimulating factor (CSF-1) leads to macrophage survival (15). It is of interest to assess whether these, or similar downstream signaling events, also occur after binding of erythroblasts and thus stimulate macrophage survival through CSF-1, indirectly aiding erythroblast expansion. Another macrophage adhesion molecule that is important for erythropoiesis is CD169 (16), a surface molecule originally identified as the “sheep erythrocyte receptor” or sialoadhesin, based on its capacity to recognize sialic acid residues (17). An interesting study by the group of Jacobsen showed the importance of CD169+ macrophages in erythropoiesis. In this study, treatment of mice with granulocyte colony-stimulating factor (G-CSF) was found to impair bone marrow erythropoiesis. Searching for the underlying cause they established that bone marrow macrophage numbers were greatly reduced. Characterization of bone marrow macrophages showed that CD169+ but not CD169− macrophages were lost upon G-CSF treatment (7). Currently, it is unclear why CD169+ macrophages in the bone marrow are mainly affected, especially since G-CSF treatment did not ablate CD169+ macrophages in the spleen. It was furthermore established that splenic CD169+ macrophages, upon CD169+ macrophage depletion from the bone marrow, could facilitate extramedullary hematopoiesis (EMH) in the spleen which is speculated to compensate for the G-CSF induced dyserythropoiesis in the bone marrow. In polycythemia vera (PV), where the Janus Kinase V617F mutation JAK2(V617F) is hyper sensitizing erythroid precursors to EPO, the depletion of CD169+ macrophages was found to reduce splenomegaly and reticulocytosis resulting in normalization of the erythroid compartment in mice (16). This finding indicates that erythropoiesis in PV is still sensitive to the stimulating activity of macrophages and identifies these macrophages as a potential therapeutic target to counteract the exaggerated erythropoiesis induced by the JAK2(V617F) mutation. Another chronic stress condition resulting in anemia is beta-thalassemia (18). In this disease, the absence or reduced expression of β-globin chains causes excess α-globin chains to precipitate in RBCs and precursors, leading to premature RBC destruction and ultimately anemia. High Epo levels here result in increasing erythroblast numbers. To facilitate the high rate of erythropoiesis, iron absorption is increased which in chronic situations leads to toxic iron overload. Ablation of CD169+ macrophages in a PV mouse model using clodronate liposomes broke this cycle of continuous-accelerated inefficient erythropoiesis. Already 40 h after clodronate treatment, an improvement of the anemia was observed (19). Within the bone marrow and spleen, the depletion of macrophages led to a reduction of erythroid progenitors and a proportional increase in differentiated erythroid cells. This clearly shows that although Epo/EpoR/JAK2 are master regulators in erythrocytosis in PV, CD169+ macrophages play an important role in the exaggerated levels of erythropoiesis during chronic stress conditions, which is ameliorated upon macrophage depletion. How exactly CD169+ macrophages exert this proliferative potential upon erythropoiesis is still not clear. What is known though is that CD169+ macrophages are pivotal in retaining HSCs in the bone marrow mesenchymal stem cell niche (20). Furthermore, also during later stages of hematopoietic development, CD169+ macrophages are important for erythroid proliferation. This was addressed in a study where through electron microscopy it was found that CD169+ macrophages induced loose interactions with erythroblasts leading to the formation of erythroblastic islands, allowing rapid proliferation and inducing pro-erythroblast cytokinesis (21). In contrast, CD169− macrophages engaged tight interactions with erythroblasts leading to erythroblast phagocytosis and impaired proliferation (21). How, mechanistically, CD169+ macrophages induce cytokinesis is still subject to investigation. However, an interesting candidate to direct future research toward is Mucin-1 as it was found to act as a ligand for CD169 (22). This heavily O-glycosylated protein is gradually expressed during erythropoiesis and found to be absent in RBCs. Similar to other known SIGLECs, the cytoplasmic tail of CD169 contains tyrosine motifs that induce intracellular signaling upon ligation. Upon ligation of CD169 by antibody crosslinking, clathrin-mediated endocytosis was observed (23). Current focus regarding CD169-mediated endocytosis involves toxins and lipid antigen uptake, as a means of antigen internalization and ultimately presentation. It would be of interest to determine how CD169 on macrophages contributes to proliferation of erythroblasts.
Secretion of Soluble Factors Important in Erythroblastic Islands
Various soluble factors have been identified that are important in defined stages of erythropoiesis. Within erythroblastic islands, both macrophages and erythroblasts secrete, or are dependent on, soluble factors. Some of these factors have been found to be important for erythroblastic island integrity, but some also for proliferation, differentiation, and hemoglobinization of RBC precursors.
Macrophages and erythroblasts cross-stimulate each other to promote proliferation. Macrophages have, for example, been shown to secrete insulin-like growth factor-1 and bone morphogenetic protein 4 (BMP4) to stimulate BFU-e and CFU-e proliferation (24, 25). On the other hand, erythroblasts secrete vascular endothelial growth factor A placental growth factor and growth-arrest specific 6 that are thought to aid macrophage proliferation (26–29). Another factor that is important for erythropoiesis is ferritin. In splenic and bone marrow macrophages but also hepatocytes and Kupffer cells, excess iron is stored in this globular protein (30). It was found that bone marrow and spleen derived macrophages, in absence of transferrin, could synthesize ferritin and excrete it. Subsequently, erythroid precursors were found to internalize ferritin through endocytosis, surprisingly, through transferrin receptor-1 (31–33). Some findings suggest that macrophages directly supply ferritin to surrounding developing erythroblasts. Ferritin molecules were, for example, found to be localized between the membranes of the central erythroblastic island macrophage and the erythroblast (34). However, in mice, serum ferritin was found to contain low levels of iron. It is thus questioned whether serum-derived ferritin contributes, in any significant way, to hemoglobin synthesis in erythroid precursors (32).
Aside from factors positively influencing erythroid and macrophage proliferation, several secreted factors negatively regulate erythropoiesis. These include the cytokines transforming growth factor-β (TGF-β), tumor necrosis factor-α (TNF-α), and interferon-γ (INF-γ) (35–38). INF-γ and TNF-α negatively regulate erythropoiesis mainly through induction of apoptosis (37, 39). Production of TNF-α by macrophages, for example, induces caspase-mediated cleavage of transcription factor GATA-1 (37). GATA-1 is critical for pro-erythroblast differentiation. This is not only illustrated by the observation that pro-erythroblasts from chimeric GATA-1 knockout mice fail to mature beyond this stage and go into apoptosis (40) but also by individuals carrying mutations in the GATA1 gene who suffer from dyserythropoietic anemia (41–43). Production of TGF-β by macrophages negatively regulates erythropoiesis quite differently. It was found that TGF-β markedly accelerated and increased erythroid differentiation but at the same time induces loss of proliferative potential in RBC precursors thereby causing erythropoietic arrest (36).
Besides positive and negative regulators of normal erythropoiesis, several secreted factors have been identified that are important during stress erythropoiesis, as is found during anemia due to trauma or sepsis. In stress erythropoiesis, RBC output by CFU-e cells is insufficient due to the limited amount of divisions these cells undergo. BFU-e progenitor cells in such conditions are therefore induced to form new CFU-e cells. Several factors have been identified that play a role during stress erythropoiesis. These factors include glucocorticoids (GC), BMP4, and stem cell factor (44–46). Mice lacking the GC receptor, for example, display normal erythropoiesis but are incapable of inducing stress erythropoiesis as they fail to induce CFU-e proliferation in the spleen as seen in wild-type mice (44). Similarly, BMP4 expression in the spleen, as a response to acute anemia, was found to drive differentiation of immature progenitors into so-called stress BFU-e cells, allowing rapid expansion and erythropoiesis (47).
Macrophages Support Terminal Differentiation in Erythropoiesis
The last stages of erythroid development include the expulsion of the nucleus and membrane remodeling of the nascent reticulocyte in the circulation (48–50). Macrophages phagocytose expelled nuclei in the bone marrow and facilitate the maturation process of circulating reticulocytes in the spleen.
One of the key elements in the clearance of the expelled nuclei, the so-called pyrenocytes, by macrophages, is the sorting process of membrane proteins. During nuclear extrusion, proteins are selectively being directed toward either the reticulocyte or pyrenocyte membrane. Cytoskeletal proteins and integral membrane proteins important for RBC shape and deformability such as ankyrin, spectrin, and the glycophorins are sorted toward the reticulocyte membrane. On the other hand, proteins associated with cellular adhesion such as Emp1 and β1 integrins are sorted toward the pyrenocyte (48). Although it is currently unclear how this sorting process works, it is hypothesized that cytoskeletal interactions play an important role since protein sorting is defective in disorders such as elliptocytosis, spherocytosis, and sickle cell disease (51, 52). It is clear, however, that during terminal maturation, proteins in the RBC membrane undergo dramatic reorganization. The sorting of adhesive molecules Emp and β1 integrins toward the pyrenocyte allows for the interaction with macrophage Emp and VCAM-1, respectively. In addition, it is thought that a combination of ATP depletion and concomitant phosphatidylserine (PS) exposure signals the macrophage through one of its PS receptors to phagocytose the pyrenocyte (53).
Finally, reticulocytes are released into the circulation where they mature into RBCs. During this stage, reticulocytes lose volume, become biconcave, and acquire the typical flexibility of an RBC (54, 55). Furthermore, expression of the transferrin receptor is quickly downregulated through exosome release to prevent excessive iron import and toxicity (56). It is thought that red pulp macrophages (RPMs) of the spleen play an important role in this maturation process, as depletion of macrophages by clodronate treatment inhibits the maturation of reticulocytes in the circulation (57). How splenic macrophages come into contact with early reticulocytes and promote their maturation into RBCs is still not clear. One hypothesis involves a mechanism similar to recognition of aged RBCs, a process that is described more extensively below, and is based on the capacity of the red pulp of the spleen to filter out rigid RBCs. A characteristic that old RBCs and reticulocytes share is a relative lack of deformability. Treatment of healthy RBCs with agents that alter their deformability, such as chloroquine, leads to RBC accumulation in the spleen (58). Thus, reticulocytes may be trapped in the spleen by their lack of deformability, thereby coming into close proximity to the RPMs. A protein that seems to be involved in reticulocyte maturation and differentially regulates membrane stiffness of reticulocytes and RBCs is glycophorin-A (GpA). It was found that in reticulocytes GpA is strongly interacting with the underlying cytoskeleton and thereby largely contributes to membrane rigidity. In terminally differentiated RBCs, however, the interaction with the cytoskeleton is less prominent, resulting in a more deformable membrane (59). It is therefore postulated that the spleen acts as sieve where non-deformable cells are captured and that, here, macrophages await to facilitate reticulocyte remodeling.
Role of Macrophages in EMH
Hematopoiesis occurring outside the bone marrow is referred to as EMH. In healthy mice, roughly 10% of erythropoietic output is facilitated by the spleen (60). In human spleen, although some hematopoietic elements are usually present, it is generally accepted that, during steady state, its contribution to hematopoiesis is negligible (61, 62). There are certain circumstances during which EMH occurs. During fetal development, for example, EMH occurs in the yolk sac, fetal liver, and spleen (63–65). In adulthood, bone marrow abnormalities including overproduction of marrow elements, damage to the bone marrow, or alterations in cellular bone marrow constituents can cause EMH. Also, prolonged hypoxic conditions, chronic inflammation and infection, and genetic disorders can contribute to EMH (66).
A commonly used agent to induce and study EMH is G-CSF. G-CSF treatment in mice leads to the depletion of macrophages from the bone marrow, by a mechanism that is still obscure. In these mice, erythropoiesis is arrested at the pro-erythroblast stage. It is well recognized that this causes EMH (67). It is believed that mobilization of HSC to the spleen, especially, when G-CSF treatment is combined with cyclophosphamide treatment is contributing to EMH (68). Macrophages play a key role in capturing the mobilized HSCs through vascular VCAM-1 (69).
Additional to retaining HSCs in the spleen, macrophages were found to play a central role in ongoing EMH. Hepatic sinusoidal macrophages were found to form erythroblastic islands upon induction of anemia in mice by injection of phenyl hydrazine (70). To assess the importance of CD169+ macrophages in EMH of the spleen and bone marrow, a mouse model was used where the diphtheria toxin receptor (DTR) was knocked-in downstream of the Siglec 1 promotor, a protein found specifically in macrophages. Treatment of these CD169DTR/+ mice with diphtheria toxin leads to ablation of these macrophages. Short exposure to DT resulted in CD169+ macrophage ablation from bone marrow but not from spleen. Using phenylhydrazine it was found that splenic VCAM-1, an adhesion molecule facilitating erythroblastic island interactions, on CD169+ bone marrow macrophages, was critical to recover from hemolytic anemia (16).
EMH can also be observed during infection. Systemic infection by E. coli in mice was found to lead to a 744-fold increase in G-CSF serum levels, directly inducing HSC mobilization to the spleen (71). Similarly, S. aureus was shown to induce production of IL-3 and GM-CSF in spleen cell cultures of mice (72). In hemaglobinopathies such as β-thalassemia and sickle cell disease, the premature destruction of RBCs and erythroid precursors causes anemia, overproduction of Epo and ultimately EMH in spleen and liver (73). Since in these diseases the erythroid progenitors are susceptible to apoptosis, increased Epo levels here do not compensate the anemia.
Taken together, disease, infection, and genetic disorders all can lead to EMH of which the underlying mechanism is not always clear. During anemia, the body attempts to increase erythropoiesis by excretion of hematopoietic factors and mobilization of HSCs to other tissues such as the spleen. Here, macrophages can trap HSCs through, amongst others, VCAM-1 and in the presence of the right factors initiate erythroblastic island formation and EMH. The relationship between anemia and EMH is intimate but still not well understood at the molecular level. Future studies may shed further light on the factors that drive EMH.
Repair and Clearance of RBC by Macrophages in the Spleen
After their development in the bone marrow, RBCs have a life span of approximately 120 days (74), during which they have numerous interactions with macrophages of the spleen and liver. With time the plasma membrane of the RBC undergoes deleterious changes that make the cell susceptible to clearance by macrophages (75). The spleen is the primary organ to filter effete RBC out of the blood. It is anatomically divided in red pulp, which is responsible for the filtering function, and white pulp, which is dedicated to adaptive immunity. The red and white pulp are divided by the marginal zone (76). With its specialized structure of the venous system, the red pulp of the spleen has the unique capacity to retain old RBC that has become less deformable (74, 75). The membrane of RBCs is extremely elastic. This deformability is of great importance since it allows RBCs to pass through capillaries that are narrower than their own diameter (77, 78). Over time, RBCs lose their elasticity. RPMs located in the cords of the red pulp phagocytize the RBCs that are too rigid to pass through the inter-endothelial slits of the red pulp (74). The high percentage of macrophages in the red pulp and the unique structure of the spleen give it its specialized ability to scrutinize the integrity of RBCs (75). Decreased RBC deformability is not only a characteristic of senescent RBCs (79). In many pathological conditions such as sickle cell disease, hereditary spherocytosis, or sepsis RBC clearance is enhanced due to rigidification of the RBC membrane (80–84). Stiffening of the RBC membrane is also observed in malaria which is discussed below. On the opposite side, hyperactivation of macrophages may also lead to enhanced clearance. This can occur for instance in the hyperinflammatory syndromes, hemophagocytic lymphohistiocytosis (HLH) (85). HLH is a rare disorder that is characterized by spiking fevers and hemophagocytosis by activated macrophages (86). It can be caused by various disorders which can be hereditary or secondary to several pathologies such as infection, malignancy, or autoimmune disease (87). Depending on the background of the disease, numerous mechanisms have been proposed to explain hemophagocytosis in HLH. It is suggested that in most if not all forms of HLH, the hyperactivation of macrophages is caused by a cytokine storm induced by uncontrolled activated NK cells and cytotoxic T-lymphocytes (88).
In addition to inspecting RBC membrane deformability, macrophages also remove inclusion bodies from circulating RBCs (89). Already in the late 1950s, it was shown that this process is exclusively executed in the spleen. Crosby et al. showed that injected-labeled siderocytes (RBCs containing iron filled granules) in subjects with a spleen decreased over time while in splenectomized subjects the amount of siderocytes remained unchanged (89). Moreover, splenectomized patients show a high amount of RBCs with Howell-Jolly bodies (nuclear chromatin inclusions), Heinz bodies (inclusions of denatured hemoglobin), siderocytes, and Pappenheimer bodies (inclusions formed by phagosomes that have taken up excessive amounts of iron) (90). Hence, it seems that the spleen and its residing RPMs not only clear senescent RBCs but are also responsible for keeping RBC “healthy.” RBCs contain a variety of enzymes to protect the cell from oxidative damage. Yet, RBCs are not capable of synthesizing new proteins and in time oxidative damage will accumulate resulting in the formation of inclusion bodies (91). The exact mechanism underlying the removal of these inclusion bodies is not known, but it has been proposed that RBC vesiculation is involved in the disposal of RBC damage (92).
Red Pulp Macrophages
The RPMs are extremely potent in neutralizing the toxic effects of hemoglobin. They express high levels of the hemoglobin scavenger receptor CD163 and the enzyme heme-oxygenase 1 (HO-1), which plays a crucial role in heme degradation (13, 76, 93). Recycling of iron through erythrophagocytosis by macrophages is the largest contributor to iron homeostasis. After phagocytosis of senescent RBCs, hemoglobin is degraded into heme and iron after which it is exported through ferroportin into the plasma. Here, transferrin traffics iron to cells that require iron including developing erythroid cells in particular [reviewed in Ref. (94)]. HO-1 is one of the critical enzymes in this cascade. Its absence leads to splenic and liver macrophage apoptosis and concomitant release of non-metabolized heme. In the spleen, this leads to red pulp fibrosis, atrophy, and ultimately functional aspleny (95).
The origin of RPMs is a current point of discussion. Recent studies by Geissmann et al. suggest that primitive macrophages originate from the yolk sac (96). However, subsequent studies propose that fetal HSCs give rise to RPMs (76, 97–99). The general view is that in the homeostatic situation macrophage populations are maintained by local proliferation. Nonetheless, in pathological conditions, such as infection and inflammation, circulating monocytes may enter the spleen and differentiate into RPMs (99–101). It seems that in inflammatory situations, the “steady-state” macrophage populations are substituted for an inflammatory monocyte derived pool of macrophages. Besides macrophages also other phagocytes may contribute to RBC clearance in pathological situations. Stijlemans et al. show that in mice trypanosomiasis, a parasitic disease, increases erythrophagocytosis by neutrophils and monocytes (102). This indicates that besides macrophages other phagocytes can play a role in RBC clearance during infection.
Clearance of Blood-Borne Infections in the Spleen
The primary function of RPMs is believed to be the scavenging of senescent RBCs. Nevertheless, besides their homeostatic role, RPMs are important to control blood-borne diseases through their ability to control iron availability for infecting pathogens. Several diseases are associated with loss of RPMs due to for instance auto-splenectomy as observed in SCD. A combination of factors causes functional asplenism in these patients. First, slow RBC circulation speeds and high oxygen extraction rates cause the RBCs to sickle. Mechanical obstruction by sickled RBCs but also adhesion of RBCs to endothelium obstructs red pulp sinuses of the spleen and can contribute to infarction and in the long-term functional aspleny (103, 104). Since macrophages in the spleen play a key role in removing both senescent RBCs as well as blood-borne pathogens from the circulation, the functional aspleny causes these patients to become more susceptible to blood-borne infections. Moreover, in SCD and thalassemia patients, or in patients suffering from iron overload in general, infection by pathogens such as Y. enterocolitica but also to streptococci is often observed (105, 106). Besides taking up whole RBC, macrophages can also take up immune complexes or pathogens bound to complement receptor 1 on RBCs while leaving the RBC intact, a process termed immune adherence clearance (107).
Red pulp macrophages are suggested to be specifically important in the control of Plasmodium-infected RBCs. Malaria is the most frequently acquired infection affecting RBC and has a major impact on RBC deformability (75). Plasmodium parasites have a complex life cycle that starts off in hepatocytes and is followed by asexual stages in the RBC. This involves major restructuring of the RBC membrane and cytoskeleton (108). While the young asexual stages do not impact RBC deformability to a great extent, the mature asexual stages of the parasite cause stiffening of the RBC membrane, which makes them susceptible for removal in the spleen (75). Splenectomized mice show increased susceptibility toward Plasmodium chabaudi infections (109). Moreover, Couper et al. show that macrophage depletion in Plasmodium yoelii-infected mice results in exacerbated parasite growth and more profound anemia (110). These findings indicate a vital role of macrophages in the spleen in malaria infections. Although it is generally believed that RPMs play a central role in control of malaria infections, the extent of their importance is debatable. Studies show that parasitized RBCs are found in the red pulp (109), RPMs can remove parasites internalized by the RBC while leaving the RBC intact, a process called pitting (111), and RPMs expand during malaria infections (112). However, Kim et al. show that while RPMs produce large amounts of type I interferons during P. chabaudi infections Spic−/− mice, which lack RPMs completely, eliminate the parasite as efficiently as Wt mice (112). It was concluded that RPMs contribute to early immune infection recognition and activation, while they may be dispensable for control of the infection due to compensation by infiltrating monocytes.
RBC Clearance in the Liver
Besides the spleen also the liver is an important RBC depot and has an important role in RBC clearance and iron recycling (102, 113, 114). Current research suggests that under pathological conditions the liver and not the spleen is the primary site of RBC clearance. Theurl et al. show in a mouse model that infused-stressed RBCs predominantly end up in the liver (114). The presence of stressed or damaged RBCs led to a rapid recruitment of monocytes. These monocytes give rise to “transient macrophages” that are well equipped to phagocytose RBCs and recycle the iron. This specific macrophage population was only found in the liver, and the researchers suggest that therefore the liver is uniquely adapted to respond to an increased demand for RBC phagocytosis. This hypothesis is supported by the work of Stijlmans et al. in which it is shown in mice that LPS injection or T. brucei infection leads to enhanced erythrophagocytosis primarily in the liver (102).
Band 3-Mediated Clearance of RBC by Macrophages
Red blood cell clearance can be viewed as a two component process. On the one hand, mechanical retention and RBC deformability determine if an RBC gets cleared from the circulation; on the other hand, cell biological factors are involved in RBC removal through erythrophagocytosis in the spleen. So-called “eat me” signals accumulate on the cell membrane of the RBC over time, and these signals can trigger RBC clearance by macrophages (Figure 2) (115–117). There is no clear consensus about what these “eat me” signals exactly are (113). Yet, there have been several proposed mechanisms of RBC clearance. Firstly, it has been suggested that band 3, a highly abundant transmembrane protein, is the main target of natural occurring antibodies (Nabs) (116, 118, 119). Nabs are antibodies present in the serum of healthy individuals that have been formed without the apparent antigen exposure, for instance, anti-A antibodies in B-positive individuals (120). These immunoglobins are preferentially of the IgM isotype, and their production already starts before birth by B cells of the B1 type in the marginal zones of the spleen (121). Nabs recognize self-, altered-self, and foreign antigens. Besides protecting from invading pathogens they are important regulators for clearance of necrotic or apoptotic cells (122). Over time, band 3 is believed to be modified mostly due to oxidative damage, which leads to the formation of epitopes for Nabs binding (118, 123). The exact nature of epitope formation allowing Nab binding is still under debate. It is hypothesized that oxidative damage to hemoglobin leads to the formation of hemichromes (hemoglobin denaturation products), which bind to band 3 and cause band 3 clustering. This in turn results in Nab binding (124). An alternative hypothesis for band 3 epitope recognition by Nabs is proteolytic degradation of band 3 (119). Kay et al. showed that IgG eluted from scenescent RBCs rebound to proteolytic fragments of band 3. This led to the hypothesis that a cell-age-specific antigen is exposed by a proteolytic modification (125). Due to their low affinity and low numbers in the circulation, Nabs are not efficient opsonins. It has been proposed that erythrophagocytosis can be enhanced by complement activation through the classical pathway after opsonization of RBCs with Nabs (116). Complex formation between Nabs and complement C3 fragments is potent opsonins that are readily recognized by complement receptors on macrophages (and other phagocytes) (123, 126, 127).
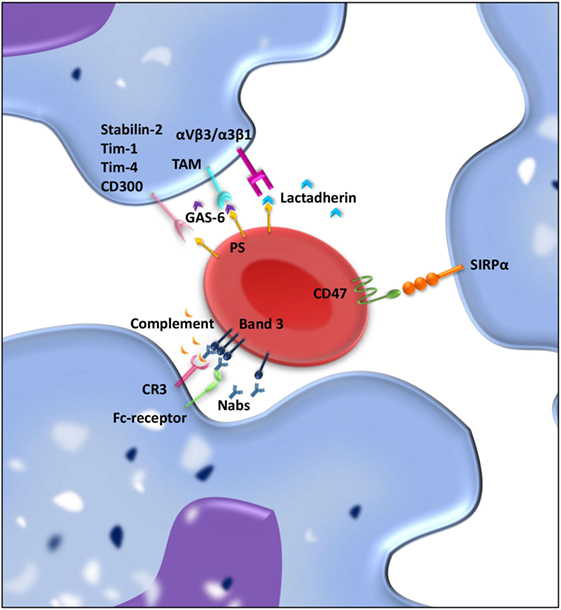
Figure 2. “Eat me” and “don’t eat me” signals involved in interaction between macrophages and red blood cell (RBC) regulating clearance. RBCs and macrophages interact with each other through ligand–ligand interactions. Over time, “eat me” signals accumulate on the RBC membrane. Phosphatidylserine (PS) exposed on the RBC membrane can directly bind Stabilin-2, Tim-1, Tim-4, or CD300 on the macrophages and is presumed to give a pro-phagocytic signal. Moreover, bridging molecules such as GAS-6 and lactadherin can facilitate RBC-macrophage interaction by binding PS on RBCs and TAM receptors or αvβ3/β5 integrins on the macrophage. Band 3 clustering and opsonization with Nabs and complement on the RBC enables binding to the macrophage via Fc receptors and CR-1 and thereby facilitates phagocytosis. As a counterbalance, CD47–SIRPα interactions inhibit phagocytosis of RBCs by the macrophage.
Oxidative Stress/Band 3-Mediated Clearance in Disease
In the homeostatic situation, RBC removal and erythropoiesis is balanced. In pathological situations, the oxidative insults modifying band 3 can be rapid and massive leading to accelerated RBC removal. Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an example of a genetic RBC defect that exhibits insufficient antioxidant defense and hence anemia due to enhanced RBC destruction (118). G6PD is an enzyme involved in the production of NADPH, a cofactor used in anabolic reactions. NADPH is crucial for the protection of the cell against oxidative stress (128). Due to its role as oxygen carrier, RBCs are particularly exposed to oxidative damage. G6PD patients therefore have an increased risk for RBC destruction due to oxidative stress which can be triggered by medication, illness, or certain foods such as fava beans. In addition to RBC defects affecting antioxidant defense hereditary, RBC disorders such as thalassemias and hemoglobinopathies also show increased oxidative stress (129, 130). Besides inadequate ROS detoxification free heme and iron may as well give rise to enhanced oxidative stress since partially oxygenated hemoglobin is susceptible to redox reactions (118, 131). In all these diseases, high levels of anti-band 3 antibodies and C3b are found bound to RBCs (132, 133). Increased RBC phagocytosis is observed which can be ameliorated in many patients by a splenectomy (124).
Besides their role in genetic RBC defects, anti-band 3 antibodies are also said to be important in RBC clearance during malarial infections (115, 134, 135). Infected RBCs demonstrate hemichrome binding to band 3 followed by band 3 clustering and opsonization with Nabs and complement (135, 136). Sickle cell trait, thalassemia trait, and G6PD-deficient individuals are known to have a selective advantage with respect to malarial infections. RBCs of these patients are more sensitive to oxidative stress exerted by the malaria parasite (124). Already in early stages of the infection, RBCs are opsonized with Nabs, which may be one of the many reasons that malaria infections are more efficiently cleared in these patients.
PS-Mediated RBC Clearance
A second mechanism that has been proposed to be important for RBC removal is PS exposure at the cell surface of the RBC. In healthy RBCs, PS is located on the inner leaflet of the RBC membrane (137). On the outer leaflet of the RBC, PS exposure is suggested to be an “eat me” signal that triggers phagocytosis by macrophages. PS exposure can be induced by an increase in cytosolic Ca2+ concentration, which can be a result of oxidative stress, osmotic shock, or other forms of cell stress (138). PS exposure as a signal for clearance is not restricted to RBCs but is seen as a general removal signal of senescent or damaged cells. In nucleated cells, PS exposure is part of the apoptotic process. Since RBCs are non-nucleated cells, apoptosis does not occur, but the regulated process of cell death in RBC has been given the term eryptosis (139). PS-dependent apoptotic cell phagocytosis is evolutionary conserved and not only found in mammals but also in lower organisms such as C. elegans and Drosophila (140). Macrophages display an array of receptors for PS including Tim-1, Tim-4, Stabilin-2, and CD300 (141–144). Furthermore, there are various bridging molecules, which can bind to PS on the cell surface, such as lactadherin, GAS-6, and protein S. These proteins than facilitate binding to αvβ3/5 integrins or TAM receptors, respectively, on the macrophage and thereby induce clearance of PS-exposing RBCs (145).
PS-Mediated RBC Clearance in Disease
There is a multitude of diseases associated with PS-mediated RBC clearance. Some examples of pathologies where increased oxidative stress activates Ca2+ cation channels and thereby induces PS exposure and eryptosis are sickle cell anemia, thalassemia, G6PD, iron deficiency, chronic kidney disease, and diabetes (138). Other diseases that are postulated to result in eryptosis are malignancies, cytostatic drug-induced anemia, hepatic failure, heart failure, and dehydration (138). PS exposure induced by the malaria parasite has been widely investigated. To ensure pathogen survival, Plasmodium falciparum activates oxidant-sensitive ion channels in the RBC membrane by inducing oxidative stress. These channels facilitate cellular uptake of nutrients, Na+ and Ca2+, and waste removal, which are crucial for parasite survival, yet a secondary concequence is PS exposure (146, 147). It has been suggested that P. falciparum prolongs RBC lifespan by sequestrating Ca2+ and digesting hemoglobin to diminish RBC swelling and preserve osmotic stability in the RBC (138, 148). The genetic resistance toward malaria infections found in sickle cell trait, thalassemia trait, and G6PD-deficient individuals has partially been attributed to the sensitivity of their RBC to induce PS exposure and erypotosis upon infection with as a result accelerated clearance of infected RBCs (149).
CD47-Mediated RBC Clearance
Although there is still debate on the exact nature of “eat me” signals on RBC membrane, it has been generally accepted that CD47 is a “don’t eat me” signal that plays a crucial role in RBC homeostasis (150, 151). CD47 is a ubiquitously expressed protein that binds the inhibitory receptor signal-regulatory protein alpha (SIRPα) present on macrophages but also other myeloid cells. The CD47–SIRPα interaction inhibits immune responses such as phagocytosis (152). The strong negative signal for phagocytosis brands CD47 a marker of “self” (150). Besides functioning as a “don’t eat me” signal, our group has shown that a conformational change in the CD47 protein can switch the molecule from a “don’t eat me” to an “eat me” signal (117). This conformational change in CD47 can be induced by oxidative stress and promotes TSP-1 binding to CD47, which creates a novel binding site for SIRPα. This alternative binding site for SIRPα induces a pro/phagocytic signal for the phagocyte. The dual role of CD47–SIRPα in regulating RBC uptake demonstrates the complexity of RBC clearance (117).
CD47-Mediated RBC Clearance in Disease
The role of CD47 as a “don’t eat me” or as an “eat me” signal in RBC clearance in pathological conditions is still rather unclear. It has been suggested that anemia in untreated Gaucher disease can be partly explained by reduced expression of CD47 on RBCs (153, 154). In mice, it has been shown that CD47–SIRPα interactions have a profound effect on disease severity of autoimmune hemolytic anemia (AIHA) (155). However, in human, AIHA patients’ CD47 expression is normal (156, 157). In sickle cell disease, CD47 expression is not significantly altered in patients compared to healthy controls. Nonetheless, hydroxyurea, a commonly used therapy, induces overexpression of CD47 on RBCs (158). How CD47 expression is influenced in human infection models is mostly unknown. Yi et al. showed that the absence of CD47 on RBCs may be important for the process of detecting and mounting an immune response against infected RBCs. They showed that the lack of CD47 on RBCs activates dendritic cells which in turn stimulate CD4+ T cells and antibody responses (159). Chambers et al. show that in vitro exposure of RBC to P. falciparum has no effect on CD47 expression (160). Yet, interfering with the CD47–SIRPα interaction might be a promising therapeutic strategy in the control of malaria infections. Ayi et al. show that disrupting the CD47–SIRPα interaction enhances phagocytosis of infected RBCs (161). Moverover, Banerjee et al. demonstrated that Plasmodium yoelii preferentially infects RBCs expressing high levels of CD47 and that CD47−/− is highly resistant to P. yoelii infections (162). In contrast to Chambers et al., these authors do find reduced CD47 expression on human P. falciparum-infected RBCs. All in all, the role of CD47 in pathological situation is still largely unknown. It is apparent that RBC clearance is a complex and multifactorial process that can be influenced by numerous factors such as, inflammation, infection, chemical substances, oxidative stress, or osmotic imbalance. Eventually, the balance between “eat me” and “don’t eat me” signals will determine if an RBC gets cleared from the circulation or not.
Concluding Remarks
The interplay between macrophages and RBC shapes RBC formation, repair, and clearance. The intricate molecular mechanisms driving these processes are only partially understood. These mechanisms are plastic and differ greatly between steady-state and pathological conditions. In SCD, thalassemia, or hemolytic anemia, the body cannot always cope with the adverse effects observed. Modulating the interaction between macrophages and developing erythroblasts has already been shown to lead to favorable results. An example of this is the study by Chow et al. where depletion of CD169+ macrophages was found to normalize erythropoiesis in PV (16). Therefore, studying alternative mechanisms to fine-tune erythropoiesis and erythrophagocytosis in disease is important in reducing the clinical symptoms of several RBC-related diseases.
Author Contributions
SM and TK wrote the review and designed the figures. TB and RB supervised and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
SM was supported by a grant from the Dutch Ministry of Health awarded to TB. TK was supported by a grant from the Landsteiner Foundation for Blood Transfusion Research awarded to RB.
Abbreviations
AIHA, autoimmune hemolytic anemia; BFU-e, burst-forming unit-erythroid cells; BMP4, bone morphogenetic protein 4; CFU-e, colony-forming unit-erythroid cells; CSF-1, macrophage colony-stimulating factor; DT, diphtheria toxin; EMH, extramedullary hematopoietis; Emp, erythroblast macrophage protein; Epo, erythropoietin; EpoR, erythropoietin receptor; G6PD, glucose-6-phosphate dehydrogenase; GAS-6, growth-arrest specific 6; GC, glucocorticoids; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; GpA, glycophorin-A; HO-1, heme-oxygenase 1; HSC, hematopoietic stem cell; ICAM-4, intercellular adhesion molecule 4; INF-γ, interferon-γ; IGF-1, insulin-like growth factor-1; IL-3, interleukin-3; Nabs, natural occurring antibodies; PGF, placental growth factor; PS, phosphatidylserine; PV, polycythemia vera; RBC, red blood cell; SIRPα, signal-regulatory protein alpha; SRCR, scavenger receptor cysteine rich; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α; TRF-1, transferrin receptor-1; VCAM-1, vascular cell adhesion molecule-1; VEGF-A, vascular endothelial growth factor A.
References
1. Bot FJ, Van Eijk L, Schipper P, Lowenberg B. Human granulocyte-macrophage colony-stimulating factor (GM-CSF) stimulates immature marrow precursors but no CFU-GM, CFU-G, or CFU-M. Exp Hematol (1989) 17:292–5.
2. Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood (2011) 118:6258–68. doi:10.1182/blood-2011-07-356006
3. Manwani D, Bieker JJ. The erythroblastic island. Curr Top Dev Biol (2008) 82:23–53. doi:10.1016/S0070-2153(07)00002-6
4. Chasis JA. Erythroblastic islands: specialized microenvironmental niches for erythropoiesis. Curr Opin Hematol (2006) 13:137–41. doi:10.1097/01.moh.0000219657.57915.30
5. Allen TD, Dexter TM. Ultrastructural aspects of erythropoietic differentiation in long-term bone marrow culture. Differentiation (1982) 21:86–94. doi:10.1111/j.1432-0436.1982.tb01201.x
6. Lee G, Lo A, Short SA, Mankelow TJ, Spring F, Parsons SF, et al. Targeted gene deletion demonstrates that the cell adhesion molecule ICAM-4 is critical for erythroblastic island formation. Blood (2006) 108:2064–71. doi:10.1182/blood-2006-03-006759
7. Jacobsen RN, Forristal CE, Raggatt LJ, Nowlan B, Barbier V, Kaur S, et al. Mobilization with granulocyte colony-stimulating factor blocks medullar erythropoiesis by depleting F4/80(+)VCAM1(+)CD169(+)ER-HR3(+)Ly6G(+) erythroid island macrophages in the mouse. Exp Hematol (2014) 42:547–61.e4. doi:10.1016/j.exphem.2014.03.009
8. Sadahira Y, Yoshino T, Monobe Y. Very late activation antigen 4-vascular cell adhesion molecule 1 interaction is involved in the formation of erythroblastic islands. J Exp Med (1995) 181:411–5. doi:10.1084/jem.181.1.411
9. Hanspal M, Hanspal JS. The association of erythroblasts with macrophages promotes erythroid proliferation and maturation: a 30-kD heparin-binding protein is involved in this contact. Blood (1994) 84:3494–504.
10. Soni S, Bala S, Gwynn B, Sahr KE, Peters LL, Hanspal M. Absence of erythroblast macrophage protein (Emp) leads to failure of erythroblast nuclear extrusion. J Biol Chem (2006) 281:20181–9. doi:10.1074/jbc.M603226200
11. Spring FA, Parsons SF, Ortlepp S, Olsson ML, Sessions R, Brady RL, et al. Intercellular adhesion molecule-4 binds alpha(4)beta(1) and alpha(V)-family integrins through novel integrin-binding mechanisms. Blood (2001) 98:458–66. doi:10.1182/blood.V98.2.458
12. Lee G, Spring FA, Parsons SF, Mankelow TJ, Peters LL, Koury MJ, et al. Novel secreted isoform of adhesion molecule ICAM-4: potential regulator of membrane-associated ICAM-4 interactions. Blood (2003) 101:1790–7. doi:10.1182/blood-2002-08-2529
13. Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature (2001) 409:198–201. doi:10.1038/35051594
14. Fabriek BO, Polfliet MM, Vloet RP, van der Schors RC, Ligtenberg AJ, Weaver LK, et al. The macrophage CD163 surface glycoprotein is an erythroblast adhesion receptor. Blood (2007) 109:5223–9. doi:10.1182/blood-2006-08-036467
15. Van den Heuvel MM, Tensen CP, van As JH, den Berg TK, Fluitsma DM, Dijkstra CD, et al. Regulation of CD 163 on human macrophages: cross-linking of CD163 induces signaling and activation. J Leukoc Biol (1999) 66:858–66.
16. Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med (2013) 19:429–36. doi:10.1038/nm.3057
17. Crocker PR, Gordon S. Mouse macrophage hemagglutinin (sheep erythrocyte receptor) with specificity for sialylated glycoconjugates characterized by a monoclonal antibody. J Exp Med (1989) 169:1333–46. doi:10.1084/jem.169.4.1333
18. Wickramasinghe SN, Hughes M. Globin chain precipitation, deranged iron metabolism and dyserythropoiesis in some thalassaemia syndromes. Haematologia (Budap) (1984) 17:35–55.
19. Ramos P, Casu C, Gardenghi S, Breda L, Crielaard BJ, Guy E, et al. Macrophages support pathological erythropoiesis in polycythemia vera and beta-thalassemia. Nat Med (2013) 19:437–45. doi:10.1038/nm.3126
20. Chow A, Lucas D, Hidalgo A, Mendez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med (2011) 208:261–71. doi:10.1084/jem.20101688
21. Falchi M, Varricchio L, Martelli F, Masiello F, Federici G, Zingariello M, et al. Dexamethasone targeted directly to macrophages induces macrophage niches that promote erythroid expansion. Haematologica (2015) 100:178–87. doi:10.3324/haematol.2014.114405
22. Rughetti A, Biffoni M, Pierelli L, Rahimi H, Bonanno G, Barachini S, et al. Regulated expression of MUC1 epithelial antigen in erythropoiesis. Br J Haematol (2003) 120:344–52. doi:10.1046/j.1365-2141.2003.04038.x
23. Delputte PL, Van Gorp H, Favoreel HW, Hoebeke I, Delrue I, Dewerchin H, et al. Porcine sialoadhesin (CD169/Siglec-1) is an endocytic receptor that allows targeted delivery of toxins and antigens to macrophages. PLoS One (2011) 6:e16827. doi:10.1371/journal.pone.0016827
24. Sawada K, Krantz SB, Dessypris EN, Koury ST, Sawyer ST. Human colony-forming units-erythroid do not require accessory cells, but do require direct interaction with insulin-like growth factor I and/or insulin for erythroid development. J Clin Invest (1989) 83:1701–9. doi:10.1172/JCI114070
25. Millot S, Andrieu V, Letteron P, Lyoumi S, Hurtado-Nedelec M, Karim Z, et al. Erythropoietin stimulates spleen BMP4-dependent stress erythropoiesis and partially corrects anemia in a mouse model of generalized inflammation. Blood (2010) 116:6072–81. doi:10.1182/blood-2010-04-281840
26. Angelillo-Scherrer A, Burnier L, Lambrechts D, Fish RJ, Tjwa M, Plaisance S, et al. Role of Gas6 in erythropoiesis and anemia in mice. J Clin Invest (2008) 118:583–96. doi:10.1172/JCI30375
27. Tordjman R, Delaire S, Plouet J, Ting S, Gaulard P, Fichelson S, et al. Erythroblasts are a source of angiogenic factors. Blood (2001) 97:1968–74. doi:10.1182/blood.V97.7.1968
28. Akinosoglou KS, Solomou EE, Gogos CA. Malaria: a haematological disease. Hematology (2012) 17:106–14. doi:10.1179/102453312X13221316477336
29. Majka M, Janowska-Wieczorek A, Ratajczak J, Ehrenman K, Pietrzkowski Z, Kowalska MA, et al. Numerous growth factors, cytokines, and chemokines are secreted by human CD34(+) cells, myeloblasts, erythroblasts, and megakaryoblasts and regulate normal hematopoiesis in an autocrine/paracrine manner. Blood (2001) 97:3075–85. doi:10.1182/blood.V97.10.3075
30. Matsuno T, Mori M, Awai M. Distribution of ferritin and hemosiderin in the liver, spleen and bone marrow of normal, phlebotomized and iron overloaded rats. Acta Med Okayama (1985) 39:347–60.
31. Leimberg MJ, Prus E, Konijn AM, Fibach E. Macrophages function as a ferritin iron source for cultured human erythroid precursors. J Cell Biochem (2008) 103:1211–8. doi:10.1002/jcb.21499
32. Cohen LA, Gutierrez L, Weiss A, Leichtmann-Bardoogo Y, Zhang DL, Crooks DR, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood (2010) 116:1574–84. doi:10.1182/blood-2009-11-253815
33. Li L, Fang CJ, Ryan JC, Niemi EC, Lebron JA, Bjorkman PJ, et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc Natl Acad Sci U S A (2010) 107:3505–10. doi:10.1073/pnas.0913192107
34. Policard A, Bessis M. Micropinocytosis and rhopheocytosis. Nature (1962) 194:110–1. doi:10.1038/194110a0
35. Zamai L, Secchiero P, Pierpaoli S, Bassini A, Papa S, Alnemri ES, et al. TNF-related apoptosis-inducing ligand (TRAIL) as a negative regulator of normal human erythropoiesis. Blood (2000) 95:3716–24.
36. Zermati Y, Fichelson S, Valensi F, Freyssinier JM, Rouyer-Fessard P, Cramer E, et al. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp Hematol (2000) 28:885–94. doi:10.1016/S0301-472X(00)00488-4
37. De Maria R, Zeuner A, Eramo A, Domenichelli C, Bonci D, Grignani F, et al. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature (1999) 401:489–93. doi:10.1038/46809
38. Libregts SF, Gutierrez L, de Bruin AM, Wensveen FM, Papadopoulos P, van Ijcken W, et al. Chronic IFN-gamma production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood (2011) 118:2578–88. doi:10.1182/blood-2010-10-315218
39. Dai C, Krantz SB. Interferon gamma induces upregulation and activation of caspases 1, 3, and 8 to produce apoptosis in human erythroid progenitor cells. Blood (1999) 93:3309–16.
40. Pevny L, Lin CS, D’Agati V, Simon MC, Orkin SH, Costantini F. Development of hematopoietic cells lacking transcription factor GATA-1. Development (1995) 121:163–72.
41. Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio JA, Beggs AH, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest (2012) 122:2439–43. doi:10.1172/JCI63597
42. Del Vecchio GC, Giordani L, De Santis A, De Mattia D. Dyserythropoietic anemia and thrombocytopenia due to a novel mutation in GATA-1. Acta Haematol (2005) 114:113–6. doi:10.1159/000086586
43. Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet (2000) 24:266–70. doi:10.1038/73480
44. Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, et al. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev (1999) 13:2996–3002. doi:10.1101/gad.13.22.2996
45. Wu DC, Paulson RF. Hypoxia regulates BMP4 expression in the murine spleen during the recovery from acute anemia. PLoS One (2010) 5:e11303. doi:10.1371/journal.pone.0011303
46. Broudy VC, Lin NL, Priestley GV, Nocka K, Wolf NS. Interaction of stem cell factor and its receptor c-kit mediates lodgment and acute expansion of hematopoietic cells in the murine spleen. Blood (1996) 88:75–81.
47. Lenox LE, Perry JM, Paulson RF. BMP4 and Madh5 regulate the erythroid response to acute anemia. Blood (2005) 105:2741–8. doi:10.1182/blood-2004-02-0703
48. Bell AJ, Satchwell TJ, Heesom KJ, Hawley BR, Kupzig S, Hazell M, et al. Protein distribution during human erythroblast enucleation in vitro. PLoS One (2013) 8:e60300. doi:10.1371/journal.pone.0060300
49. Liu J, Guo X, Mohandas N, Chasis JA, An X. Membrane remodeling during reticulocyte maturation. Blood (2010) 115:2021–7. doi:10.1182/blood-2009-08-241182
50. Chasis JA, Prenant M, Leung A, Mohandas N. Membrane assembly and remodeling during reticulocyte maturation. Blood (1989) 74:1112–20.
51. Salomao M, Chen K, Villalobos J, Mohandas N, An X, Chasis JA. Hereditary spherocytosis and hereditary elliptocytosis: aberrant protein sorting during erythroblast enucleation. Blood (2010) 116:267–9. doi:10.1182/blood-2010-02-264127
52. Yuditskaya S, Suffredini AF, Kato GJ. The proteome of sickle cell disease: insights from exploratory proteomic profiling. Expert Rev Proteomics (2010) 7:833–48. doi:10.1586/epr.10.88
53. Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature (2005) 437:754–8. doi:10.1038/nature03964
54. Waugh RE, Mckenney JB, Bauserman RG, Brooks DM, Valeri CR, Snyder LM. Surface area and volume changes during maturation of reticulocytes in the circulation of the baboon. J Lab Clin Med (1997) 129:527–35. doi:10.1016/S0022-2143(97)90007-X
55. Waugh RE, Mantalaris A, Bauserman RG, Hwang WC, Wu JH. Membrane instability in late-stage erythropoiesis. Blood (2001) 97:1869–75. doi:10.1182/blood.V97.6.1869
56. Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol (1983) 97:329–39. doi:10.1083/jcb.97.2.329
57. Rhodes MM, Koury ST, Kopsombut P, Alford CE, Price JO, Koury MJ. Stress reticulocytes lose transferrin receptors by an extrinsic process involving spleen and macrophages. Am J Hematol (2016) 91:875–82. doi:10.1002/ajh.24421
58. Huang S, Amaladoss A, Liu M, Chen H, Zhang R, Preiser PR, et al. In vivo splenic clearance correlates with in vitro deformability of red blood cells from Plasmodium yoelii-infected mice. Infect Immun (2014) 82:2532–41. doi:10.1128/IAI.01525-13
59. Lee JC, Gimm JA, Lo AJ, Koury MJ, Krauss SW, Mohandas N, et al. Mechanism of protein sorting during erythroblast enucleation: role of cytoskeletal connectivity. Blood (2004) 103:1912–9. doi:10.1182/blood-2003-03-0928
60. Pantel K, Loeffler M, Bungart B, Wichmann HE. A mathematical model of erythropoiesis in mice and rats. Part 4: differences between bone marrow and spleen. Cell Tissue Kinet (1990) 23:283–97.
61. O’Malley DP, Kim YS, Perkins SL, Baldridge L, Juliar BE, Orazi A. Morphologic and immunohistochemical evaluation of splenic hematopoietic proliferations in neoplastic and benign disorders. Mod Pathol (2005) 18:1550–61.
62. Freedman MH, Saunders EF. Hematopoiesis in the human spleen. Am J Hematol (1981) 11:271–5. doi:10.1002/ajh.2830110307
63. Samokhvalov IM, Samokhvalova NI, Nishikawa S. Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature (2007) 446:1056–61. doi:10.1038/nature05725
64. Lux CT, Yoshimoto M, Mcgrath K, Conway SJ, Palis J, Yoder MC. All primitive and definitive hematopoietic progenitor cells emerging before E10 in the mouse embryo are products of the yolk sac. Blood (2008) 111:3435–8. doi:10.1182/blood-2007-08-107086
65. Wolber FM, Leonard E, Michael S, Orschell-Traycoff CM, Yoder MC, Srour EF. Roles of spleen and liver in development of the murine hematopoietic system. Exp Hematol (2002) 30:1010–9. doi:10.1016/S0301-472X(02)00881-0
66. Johns JL, Christopher MM. Extramedullary hematopoiesis: a new look at the underlying stem cell niche, theories of development, and occurrence in animals. Vet Pathol (2012) 49:508–23. doi:10.1177/0300985811432344
67. Platzbecker U, Prange-Krex G, Bornhauser M, Koch R, Soucek S, Aikele P, et al. Spleen enlargement in healthy donors during G-CSF mobilization of PBPCs. Transfusion (2001) 41:184–9. doi:10.1046/j.1537-2995.2001.41020184.x
68. Morrison SJ, Wright DE, Weissman IL. Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc Natl Acad Sci U S A (1997) 94:1908–13. doi:10.1073/pnas.94.5.1908
69. Dutta P, Hoyer FF, Grigoryeva LS, Sager HB, Leuschner F, Courties G, et al. Macrophages retain hematopoietic stem cells in the spleen via VCAM-1. J Exp Med (2015) 212:497–512. doi:10.1084/jem.20141642
70. Sonoda Y, Sasaki K. Hepatic extramedullary hematopoiesis and macrophages in the adult mouse: histometrical and immunohistochemical studies. Cells Tissues Organs (2012) 196:555–64. doi:10.1159/000338336
71. Burberry A, Zeng MY, Ding L, Wicks I, Inohara N, Morrison SJ, et al. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and Toll-like receptor signaling. Cell Host Microbe (2014) 15:779–91. doi:10.1016/j.chom.2014.05.004
72. Galelli A, Anderson S, Charlot B, Alouf JE. Induction of murine hemopoietic growth factors by toxic shock syndrome toxin-1. J Immunol (1989) 142:2855–63.
73. Ribeil JA, Arlet JB, Dussiot M, Moura IC, Courtois G, Hermine O. Ineffective erythropoiesis in beta-thalassemia. ScientificWorldJournal (2013) 2013:394295. doi:10.1155/2013/394295
74. Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol (2005) 5:606–16. doi:10.1038/nri1669
75. Duez J, Holleran JP, Ndour PA, Pionneau C, Diakite S, Roussel C, et al. Mechanical clearance of red blood cells by the human spleen: potential therapeutic applications of a biomimetic RBC filtration method. Transfus Clin Biol (2015) 22:151–7. doi:10.1016/j.tracli.2015.05.004
76. Borges da Silva H, Fonseca R, Pereira RM, Cassado Ados A, Alvarez JM, D’Império Lima MR. Splenic macrophage subsets and their function during blood-borne infections. Front Immunol (2015) 6:480. doi:10.3389/fimmu.2015.00480
77. Buffet PA, Milon G, Brousse V, Correas JM, Dousset B, Couvelard A, et al. Ex vivo perfusion of human spleens maintains clearing and processing functions. Blood (2006) 107:3745–52. doi:10.1182/blood-2005-10-4094
78. Groom AC, Schmidt EE, Macdonald IC. Microcirculatory pathways and blood flow in spleen: new insights from washout kinetics, corrosion casts, and quantitative intravital videomicroscopy. Scanning Microsc (1991) 5:159–73; discussion 173–4.
79. Mohanty JG, Nagababu E, Rifkind JM. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front Physiol (2014) 5:84. doi:10.3389/fphys.2014.00084
80. Dondorp AM, Angus BJ, Chotivanich K, Silamut K, Ruangveerayuth R, Hardeman MR, et al. Red blood cell deformability as a predictor of anemia in severe falciparum malaria. Am J Trop Med Hyg (1999) 60:733–7.
81. Deplaine G, Safeukui I, Jeddi F, Lacoste F, Brousse V, Perrot S, et al. The sensing of poorly deformable red blood cells by the human spleen can be mimicked in vitro. Blood (2011) 117:e88–95. doi:10.1182/blood-2010-10-312801
82. Barabino GA, Platt MO, Kaul DK. Sickle cell biomechanics. Annu Rev Biomed Eng (2010) 12:345–67. doi:10.1146/annurev-bioeng-070909-105339
83. Betticher DC, Keller H, Maly FE, Reinhart WH. The effect of endotoxin and tumour necrosis factor on erythrocyte and leucocyte deformability in vitro. Br J Haematol (1993) 83:130–7. doi:10.1111/j.1365-2141.1993.tb04643.x
84. Baskurt OK, Gelmont D, Meiselman HJ. Red blood cell deformability in sepsis. Am J Respir Crit Care Med (1998) 157:421–7. doi:10.1164/ajrccm.157.2.9611103
85. Hayden A, Park S, Giustini D, Lee AY, Chen LY. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: a systematic scoping review. Blood Rev (2016) 30:411–20. doi:10.1016/j.blre.2016.05.001
86. Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematology Am Soc Hematol Educ Program (2013) 2013:605–11. doi:10.1182/asheducation-2013.1.605
87. Morimoto A, Nakazawa Y, Ishii E. Hemophagocytic lymphohistiocytosis: pathogenesis, diagnosis, and management. Pediatr Int (2016) 58:817–25. doi:10.1111/ped.13064
88. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood (2004) 104:735–43. doi:10.1182/blood-2003-10-3413
90. Wright BS, Wright DH. The Illustrated Pathology of the Spleen. Cambridge, UK: Cambridge University Press (2000).
91. Harley JD. Role of reduced glutathione in human erythrocytes. Nature (1965) 206:1054–5. doi:10.1038/2061054a0
92. Willekens FL, Roerdinkholder-Stoelwinder B, Groenen-Döpp YA, Bos HJ, Bosman GJ, van den Bos AG, et al. Hemoglobin loss from erythrocytes in vivo results from spleen-facilitated vesiculation. Blood (2003) 101:747–51. doi:10.1182/blood-2002-02-0500
93. Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature (2009) 457:318–21. doi:10.1038/nature07472
94. Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell (2004) 117:285–97. doi:10.1016/S0092-8674(04)00343-5
95. Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood (2010) 116:6054–62. doi:10.1182/blood-2010-03-272138
96. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (2010) 327:656–61. doi:10.1126/science.1178331
97. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41:21–35. doi:10.1016/j.immuni.2014.06.013
98. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38:792–804. doi:10.1016/j.immuni.2013.04.004
99. Kurotaki D, Uede T, Tamura T. Functions and development of red pulp macrophages. Microbiol Immunol (2015) 59:55–62. doi:10.1111/1348-0421.12228
100. Bain CC, Hawley CA, Garner H, Scott CL, Schridde A, Steers NJ, et al. Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat Commun (2016) 7:ncomms11852. doi:10.1038/ncomms11852
101. Haldar M, Kohyama M, So AY, Kc W, Wu X, Briseno CG, et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell (2014) 156:1223–34. doi:10.1016/j.cell.2014.01.069
102. Stijlemans B, Cnops J, Naniima P, Vaast A, Bockstal V, De Baetselier P, et al. Development of a pHrodo-based assay for the assessment of in vitro and in vivo erythrophagocytosis during experimental trypanosomosis. PLoS Negl Trop Dis (2015) 9:e0003561. doi:10.1371/journal.pntd.0003561
103. El Nemer W, Wautier MP, Rahuel C, Gane P, Hermand P, Galacteros F, et al. Endothelial Lu/BCAM glycoproteins are novel ligands for red blood cell alpha4beta1 integrin: role in adhesion of sickle red blood cells to endothelial cells. Blood (2007) 109:3544–51. doi:10.1182/blood-2006-07-035139
104. El Nemer W, Gauthier E, Wautier MP, Rahuel C, Gane P, Galacteros F, et al. Role of Lu/BCAM in abnormal adhesion of sickle red blood cells to vascular endothelium. Transfus Clin Biol (2008) 15:29–33. doi:10.1016/j.tracli.2008.05.002
105. Abcarian PW, Demas BE. Systemic Yersinia enterocolitica infection associated with iron overload and deferoxamine therapy. AJR Am J Roentgenol (1991) 157:773–5. doi:10.2214/ajr.157.4.1892033
106. Chirio R, Collignon A, Sabbah L, Lestradet H, Torlotin JC. [Yersinia enterocolitica infections and thalassemia major in children]. Ann Pediatr (Paris) (1989) 36(308):311–4.
107. Wilson JG, Andriopoulos NA, Fearon DT. CR1 and the cell membrane proteins that bind C3 and C4. A basic and clinical review. Immunol Res (1987) 6:192–209. doi:10.1007/BF02918091
108. Maier AG, Cooke BM, Cowman AF, Tilley L. Malaria parasite proteins that remodel the host erythrocyte. Nat Rev Microbiol (2009) 7:341–54. doi:10.1038/nrmicro2110
109. Yadava A, Kumar S, Dvorak JA, Milon G, Miller LH. Trafficking of Plasmodium chabaudi adami-infected erythrocytes within the mouse spleen. Proc Natl Acad Sci U S A (1996) 93:4595–9. doi:10.1073/pnas.93.10.4595
110. Couper KN, Blount DG, Hafalla JC, van Rooijen N, de Souza JB, Riley EM. Macrophage-mediated but gamma interferon-independent innate immune responses control the primary wave of Plasmodium yoelii parasitemia. Infect Immun (2007) 75:5806–18. doi:10.1128/IAI.01005-07
111. Schnitzer B, Sodeman T, Mead ML, Contacos PG. Pitting function of the spleen in malaria: ultrastructural observations. Science (1972) 177:175–7. doi:10.1126/science.177.4044.175
112. Kim CC, Nelson CS, Wilson EB, Hou B, Defranco AL, Derisi JL. Splenic red pulp macrophages produce type I interferons as early sentinels of malaria infection but are dispensable for control. PLoS One (2012) 7:e48126. doi:10.1371/journal.pone.0048126
113. de Back DZ, Kostova EB, van Kraaij M, van den Berg TK, van Bruggen R. Of macrophages and red blood cells; a complex love story. Front Physiol (2014) 5:9. doi:10.3389/fphys.2014.00009
114. Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med (2016) 22:945–51. doi:10.1038/nm.4146
115. Turrini F, Ginsburg H, Bussolino F, Pescarmona GP, Serra MV, Arese P. Phagocytosis of Plasmodium falciparum-infected human red blood cells by human monocytes: involvement of immune and nonimmune determinants and dependence on parasite developmental stage. Blood (1992) 80:801–8.
116. Lutz HU. Innate immune and non-immune mediators of erythrocyte clearance. Cell Mol Biol (Noisy-le-grand) (2004) 50:107–16.
117. Burger P, Hilarius-Stokman P, de Korte D, van den Berg TK, van Bruggen R. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood (2012) 119:5512–21. doi:10.1182/blood-2011-10-386805
118. Arese P, Turrini F, Schwarzer E. Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes. Cell Physiol Biochem (2005) 16:133–46. doi:10.1159/000089839
119. Kay MM. Band 3 and its alterations in health and disease. Cell Mol Biol (Noisy-le-grand) (2004) 50:117–38.
120. Kidd JG, Friedewald WF. A natural antibody that reacts in vitro with a sedimentable constituent of normal tissue cells: II. Specificity of the phenomenon: general discussion. J Exp Med (1942) 76:557–78. doi:10.1084/jem.76.6.543
121. Lutz HU, Binder CJ, Kaveri S. Naturally occurring auto-antibodies in homeostasis and disease. Trends Immunol (2009) 30:43–51. doi:10.1016/j.it.2008.10.002
122. Simon HU, Spath PJ. IVIG – mechanisms of action. Allergy (2003) 58:543–52. doi:10.1034/j.1398-9995.2003.00239.x
123. Lutz HU, Bussolino F, Flepp R, Fasler S, Stammler P, Kazatchkine MD, et al. Naturally occurring anti-band-3 antibodies and complement together mediate phagocytosis of oxidatively stressed human erythrocytes. Proc Natl Acad Sci U S A (1987) 84:7368–72. doi:10.1073/pnas.84.21.7368
124. Pantaleo A, Giribaldi G, Mannu F, Arese P, Turrini F. Naturally occurring anti-band 3 antibodies and red blood cell removal under physiological and pathological conditions. Autoimmun Rev (2008) 7:457–62. doi:10.1016/j.autrev.2008.03.017
125. Kay MM. Isolation of the phagocytosis-inducing IgG-binding antigen on senescent somatic cells. Nature (1981) 289:491–4. doi:10.1038/289491a0
126. Gattegno L, Saffar L, Vaysse J. Inhibition by monoclonal anticomplement receptor type 1 on interactions between senescent human red blood cells and monocytic-macrophagic cells. J Leukoc Biol (1989) 45:422–8.
127. Lutz HU, Fasler S, Stammler P, Bussolino F, Arese P. Naturally occurring anti-band 3 antibodies and complement in phagocytosis of oxidatively-stressed and in clearance of senescent red cells. Blood Cells (1988) 14:175–203.
128. Beutler E. Red cell metabolism. A. Defects not causing hemolytic disease. B. Environmental modification. Biochimie (1972) 54:759–64. doi:10.1016/S0300-9084(72)80181-0
129. Aslan M, Freeman BA. Redox-dependent impairment of vascular function in sickle cell disease. Free Radic Biol Med (2007) 43:1469–83. doi:10.1016/j.freeradbiomed.2007.08.014
130. Origa R, Galanello R. Pathophysiology of beta thalassaemia. Pediatr Endocrinol Rev (2011) 8(Suppl 2):263–70.
131. van Zwieten R, Verhoeven AJ, Roos D. Inborn defects in the antioxidant systems of human red blood cells. Free Radic Biol Med (2014) 67:377–86. doi:10.1016/j.freeradbiomed.2013.11.022
132. Mannu F, Arese P, Cappellini MD, Fiorelli G, Cappadoro M, Giribaldi G, et al. Role of hemichrome binding to erythrocyte membrane in the generation of band-3 alterations in beta-thalassemia intermedia erythrocytes. Blood (1995) 86:2014–20.
133. Yuan J, Kannan R, Shinar E, Rachmilewitz EA, Low PS. Isolation, characterization, and immunoprecipitation studies of immune complexes from membranes of beta-thalassemic erythrocytes. Blood (1992) 79:3007–13.
134. Ayi K, Turrini F, Piga A, Arese P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: a common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood (2004) 104:3364–71. doi:10.1182/blood-2003-11-3820
135. Giribaldi G, Ulliers D, Mannu F, Arese P, Turrini F. Growth of Plasmodium falciparum induces stage-dependent haemichrome formation, oxidative aggregation of band 3, membrane deposition of complement and antibodies, and phagocytosis of parasitized erythrocytes. Br J Haematol (2001) 113:492–9. doi:10.1046/j.1365-2141.2001.02707.x
136. Turrini F, Giribaldi G, Carta F, Mannu F, Arese P. Mechanisms of band 3 oxidation and clustering in the phagocytosis of Plasmodium falciparum-infected erythrocytes. Redox Rep (2003) 8:300–3. doi:10.1179/135100003225002943
137. Fernandez-Boyanapalli RF, Frasch SC, Mcphillips K, Vandivier RW, Harry BL, Riches DW, et al. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood (2009) 113:2047–55. doi:10.1182/blood-2008-05-160564
138. Lang F, Jilani K, Lang E. Therapeutic potential of manipulating suicidal erythrocyte death. Expert Opin Ther Targets (2015) 19:1219–27. doi:10.1517/14728222.2015.1051306
139. Lang KS, Lang PA, Bauer C, Duranton C, Wieder T, Huber SM, et al. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem (2005) 15:195–202. doi:10.1159/000086406
140. Segawa K, Nagata S. An apoptotic ‘Eat Me’ signal: phosphatidylserine exposure. Trends Cell Biol (2015) 25:639–50. doi:10.1016/j.tcb.2015.08.003
141. Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity (2007) 27:927–40. doi:10.1016/j.immuni.2007.11.011
142. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ (2008) 15:192–201. doi:10.1038/sj.cdd.4402242
143. Murakami Y, Tian L, Voss OH, Margulies DH, Krzewski K, Coligan JE. CD300b regulates the phagocytosis of apoptotic cells via phosphatidylserine recognition. Cell Death Differ (2014) 21:1746–57. doi:10.1038/cdd.2014.86
144. Simhadri VR, Andersen JF, Calvo E, Choi SC, Coligan JE, Borrego F. Human CD300a binds to phosphatidylethanolamine and phosphatidylserine, and modulates the phagocytosis of dead cells. Blood (2012) 119:2799–809. doi:10.1182/blood-2011-08-372425
145. Raymond A, Ensslin MA, Shur BD. SED1/MFG-E8: a bi-motif protein that orchestrates diverse cellular interactions. J Cell Biochem (2009) 106:957–66. doi:10.1002/jcb.22076
146. Lang F, Abed M, Lang E, Foller M. Oxidative stress and suicidal erythrocyte death. Antioxid Redox Signal (2014) 21:138–53. doi:10.1089/ars.2013.5747
147. Foller M, Bobbala D, Koka S, Huber SM, Gulbins E, Lang F. Suicide for survival – death of infected erythrocytes as a host mechanism to survive malaria. Cell Physiol Biochem (2009) 24:133–40. doi:10.1159/000233238
148. Lew VL, Tiffert T, Ginsburg H. Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum-infected red blood cells. Blood (2003) 101:4189–94. doi:10.1182/blood-2002-08-2654
149. Taylor SM, Fairhurst RM. Malaria parasites and red cell variants: when a house is not a home. Curr Opin Hematol (2014) 21:193–200. doi:10.1097/MOH.0000000000000039
150. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science (2000) 288:2051–4. doi:10.1126/science.288.5473.2051
151. Oldenborg PA, Gresham HD, Lindberg FP. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J Exp Med (2001) 193:855–62. doi:10.1084/jem.193.7.855
152. Barclay AN, van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol (2014) 32:25–50. doi:10.1146/annurev-immunol-032713-120142
153. Bratosin D, Tissier JP, Lapillonne H, Hermine O, De Villemeur TB, Cotoraci C, et al. A cytometric study of the red blood cells in Gaucher disease reveals their abnormal shape that may be involved in increased erythrophagocytosis. Cytometry B Clin Cytom (2011) 80:28–37. doi:10.1002/cyto.b.20539
154. Oldenborg PA. CD47: a cell surface glycoprotein which regulates multiple functions of hematopoietic cells in health and disease. ISRN Hematol (2013) 2013:614619. doi:10.1155/2013/614619
155. Oldenborg PA, Gresham HD, Chen Y, Izui S, Lindberg FP. Lethal autoimmune hemolytic anemia in CD47-deficient nonobese diabetic (NOD) mice. Blood (2002) 99:3500–4. doi:10.1182/blood.V99.10.3500
156. Ahrens N, Pagenkopf C, Kiesewetter H, Salama A. CD47 is expressed at normal levels in patients with autoimmune haemolytic anaemia and/or immune thrombocytopenia. Transfus Med (2006) 16:397–402. doi:10.1111/j.1365-3148.2006.00688.x
157. Barros MM, Yamamoto M, Figueiredo MS, Cancado R, Kimura EY, Langhi DM Jr, et al. Expression levels of CD47, CD35, CD55, and CD59 on red blood cells and signal-regulatory protein-alpha,beta on monocytes from patients with warm autoimmune hemolytic anemia. Transfusion (2009) 49:154–60. doi:10.1111/j.1537-2995.2008.01936.x
158. Cartron JP, Elion J. Erythroid adhesion molecules in sickle cell disease: effect of hydroxyurea. Transfus Clin Biol (2008) 15:39–50. doi:10.1016/j.tracli.2008.05.001
159. Yi T, Li J, Chen H, Wu J, An J, Xu Y, et al. Splenic dendritic cells survey red blood cells for missing self-CD47 to trigger adaptive immune responses. Immunity (2015) 43:764–75. doi:10.1016/j.immuni.2015.08.021
160. Chambers DR, Procter J, Muratova O, Byrne K, Keister D, Shanks D, et al. In vitro RBC exposure to Plasmodium falciparum has no effect on RBC antigen expression. Transfus Med (2002) 12:213–9. doi:10.1046/j.1365-3148.2002.00375.x
161. Ayi K, Lu Z, Serghides L, Ho JM, Finney C, Wang JC, et al. CD47-SIRPalpha interactions regulate macrophage uptake of Plasmodium falciparum-infected erythrocytes and clearance of malaria in vivo. Infect Immun (2016) 84:2002–11. doi:10.1128/IAI.01426-15
Keywords: macrophages, red blood cells, erythropoiesis, adhesion molecules, RBC clearance, erythrophagocytosis
Citation: Klei TRL, Meinderts SM, van den Berg TK and van Bruggen R (2017) From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 8:73. doi: 10.3389/fimmu.2017.00073
Received: 25 October 2016; Accepted: 17 January 2017;
Published: 02 February 2017
Edited by:
Robert F. Paulson, Pennsylvania State University, USAReviewed by:
Xinjian Chen, University of Utah, USAReinhard Obst, Ludwig Maximilian University of Munich, Germany
Copyright: © 2017 Klei, Meinderts, van den Berg and van Bruggen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robin van Bruggen, ci52YW5icnVnZ2VuQHNhbnF1aW4ubmw=
†These authors have contributed equally to this work.