 Md Zahidul Alam
Md Zahidul Alam Samir Devalaraja
Samir Devalaraja Malay Haldar
Malay Haldar- Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Erythroid function and development is intimately linked to macrophages. The primary function of erythrocytes is oxygen delivery, which is mediated by iron-containing hemoglobin. The major source of this iron is a recycling pathway where macrophages scavenge old and damaged erythrocytes to release iron contained within the heme moiety. Macrophages also promote erythropoiesis by providing a supportive niche in the bone marrow as an integral component of “erythorblastic islands.” Importantly, inflammation leads to alterations in iron handling by macrophages with significant impact on iron homeostasis and erythropoiesis. The importance of macrophages in erythropoiesis and iron homeostasis is well established and has been extensively reviewed. However, this developmental relationship is not one way, and erythrocytes can also regulate macrophage development and function. Erythrocyte-derived heme can induce the development of iron-recycling macrophages from monocytes, engage pattern recognition receptors to activate macrophages, and act as ligand for specific nuclear receptors to modulate macrophage function. Here, we discuss the role of heme as a signaling molecule impacting macrophage homeostasis. We will review these actions of heme within the framework of our current understanding of the role of micro-environmental factors in macrophage development and function.
Macrophage Diversity
Macrophages are prominent cells of the innate immune system characterized by their high phagocytic capacity and the ability to process antigens. While best known for their roles in controlling immune responses, they are functionally very versatile with roles in wound repair, tissue morphogenesis, and tissue homeostasis (1, 2). Almost every tissue in the body harbors resident macrophage population that performs tissue-specific homeostatic function (2). Examples include surfactant recycling by alveolar macrophages in the lung and iron recycling by splenic red pulp macrophages (RPM). The functional distinction between tissue-resident macrophages is reflected in their distinct gene-expression profile and dependence on specific transcription factors. As examples, RPM development is dependent on the transcription factor SpiC, Kuffer cells on transcription factor Id3, peritoneal macrophages on transcription factor Gata6, alveolar macrophages on transcription factor PPAR-γ, and splenic marginal zone macrophages on transcription factor Lxrα (3–7). An important question is how divergent tissue-resident macrophages are generated from a common precursor. Recent work suggests a prominent role of the tissue microenvironment in this process. For instance, high levels of heme in the red pulp area of spleen were found to induce the expression of the transcription factor SpiC, which promoted the development of monocytes into RPM (8). Likewise, local retinoic acid levels were found to control the development and localization of peritoneal macrophages (6). An additional line of evidence comes from the finding that tissue macrophages can be reprogramed when transplanted into a new tissue microenvironment (9). Therefore, local microenvironment-associated factors play key role in generating phenotypic and functional diversity of tissue-resident macrophages.
Monocytes were considered to be the primary source of tissue-resident macrophages. However, definitive studies in recent years have changed this paradigm, and we now recognize two distinct precursors of tissue macrophages: circulating monocytes and embryonic precursors that seed various tissue before birth and are maintained by local proliferation (10). Embryonic precursors of tissue macrophages develop independent of hematopoietic stem cells, either from the yolk sac during early stages of embryogenesis or from fetal monocyte-like cells in later stages. In adults, circulating monocytes can differentiate into tissue macrophages, especially during inflammation or tissue injury. Monocytes have a limited lifespan of a few days in the circulation and are continuously replenished from hematopoietic stem cells in the bone marrow (11, 12). The implication of the dual origin (embryonic vs. adult monocyte) on macrophage function is currently not clear. Recent studies suggest that the relative distribution of embryonic and monocyte-derived macrophages in tissue undergoes significant changes with age and pathological conditions. In both instances, the contribution of monocyte-derived macrophages appears to increase (13). Tissue inflammation lead to recruitment of circulating monocytes that differentiate locally into macrophages or closely related antigen-presenting cells known as dendritic cells (DCs) depending on local micro-environmental factors that are not well understood (14).
Macrophage function can also be categorized into pro-inflammatory as embodied by the “M1-polarized” macrophage and anti-inflammatory as represented by the “M2-polarized” macrophage (15). These polarization states represent the two extremes of a functional spectrum from an anti-inflammatory phenotype that support tissue repair and homeostasis to an inflammatory phenotype that induces and support immune responses (16, 17). Tissue macrophages generally have features of M2, while M1 macrophages are generated in inflammatory and infectious settings. Functional polarization of macrophages is driven by various activation signals including cytokines, which has been extensively reviewed elsewhere (15). Notably, the activation state of a macrophage can impact how it will respond to tissue-associated factors. As an example, high levels of heme induce the iron exporter ferroportin (SLC40A1) in macrophages to help recycle iron (18, 19). However, macrophages can strongly downregulate ferroportin if they sense the presence of bacterial pathogens in the environment in an attempt to sequester iron from extracellular pathogens (18, 20, 21). Therefore, macrophages integrate diverse cues from its environment to formulate a context-appropriate response.
In summary, local microenvironment-associated factors have significant impact on macrophage differentiation and function. Additionally, the activation state of the macrophage influences how it will respond to such microenvironment-associated factors.
Iron-Recycling Macrophages
Heme comprises of a protoporphyrin IX ring with an iron atom in the center (22). The capacity of the iron to undergo reversible change in oxidation status is central to heme’s ability to catalyze diverse reactions such as delivery of oxygen to tissue, electron transfer, and oxidation reactions (23). The specific function of heme depends on the protein to which it is attached as a prosthetic group. There are many distinct types of hemoproteins performing various functions. However, the vast majority of the heme in vertebrates is sequestered within hemoglobin and myoglobin in erythrocytes and muscle cells, respectively, which can be released upon damage to these cells (24). Macrophages are the primary cells responsible for the uptake and disposal of heme, which is important for three main reasons: (1) preventing heme and iron-mediated cellular toxicity, (2) recycling iron to sustain erythropoiesis, and (3) preventing pathogen’s access to iron during infection (Figure 1) (25, 26). Each erythrocyte contains approximately 1.2 × 109 heme moieties associated with hemoglobin, and approximately 200 billion erythrocytes reach senescence each day (26). Hemoglobin released from senescent or damaged erythrocytes can be readily oxidized releasing the prosthetic heme group. Free heme can catalyze oxidation of proteins, generate lipid peroxides, and damage DNA through oxidative stress (27). Iron released from heme can also generate harmful free radicals via Fenton chemistry (28). Therefore, without a proper disposal mechanism there is a considerable threat from the large amount of heme that can be released from erythrocytes. Alterations in membranes of senescent and damaged erythrocytes are detected by specialized erythrophagocytic macrophages in the spleen, liver, and bone marrow, which remove these erythrocytes before they undergo hemolysis (Figure 1) (29–31). Cell-free hemoglobin and heme binds to carrier proteins haptoglobin and hemopexin, respectively (31). Hemoglobin–haptoglobin and heme–hemopexin complexes are taken up by macrophages via cognate receptors CD163 and CD91, respectively (Figure 1) (32, 33). Therefore, macrophages prevent heme-mediated toxicity by phagocytosing old erythrocytes before they rupture as well as by taking up heme and hemoglobin that is already released from erythrocytes.
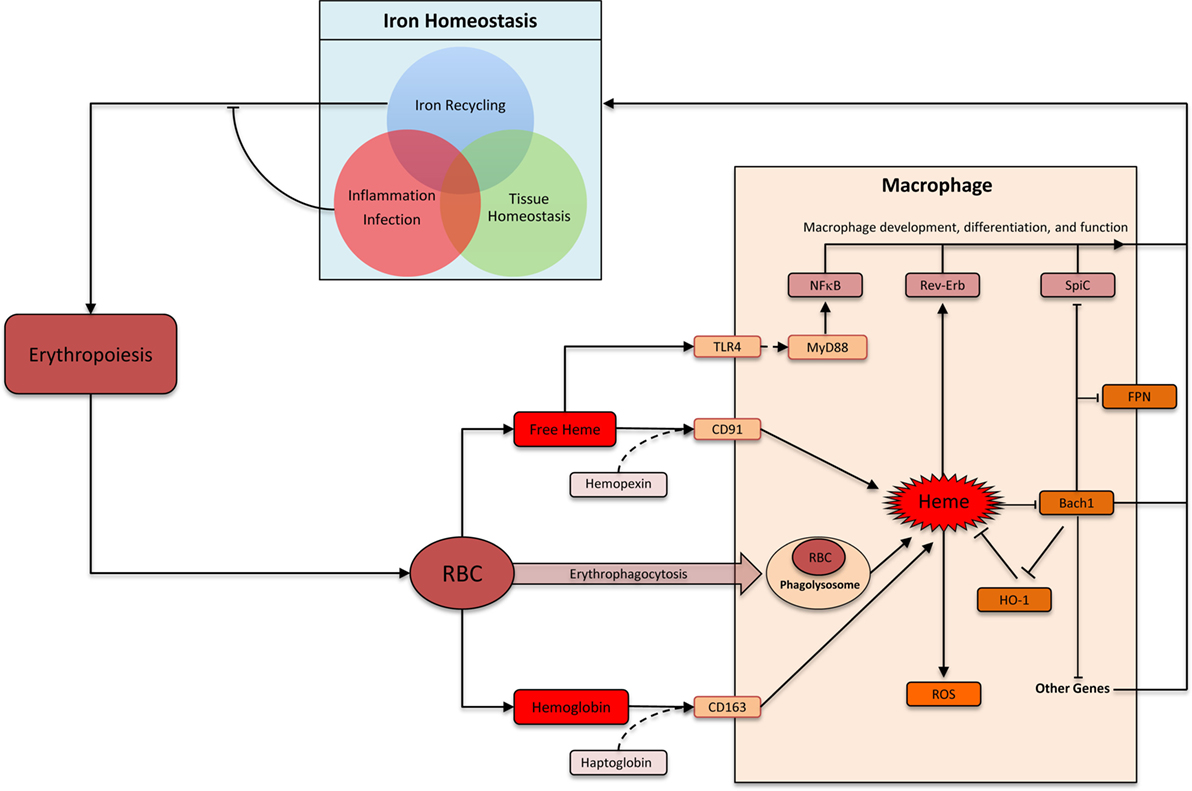
Figure 1. Heme: linking erythropoiesis and macrophage function. Abbreviations: RBC, red blood cells; ROS, reactive oxygen species; FPN, ferroportin (Slc40A1); HO-1, heme oxygenase-1. Heme is taken up by macrophages by several distinct pathways. Heme-induced HO-1 leads to heme degradation. Inside macrophages, heme triggers multiple distinct pathways that impacts macrophage function and differentiation. In the context of iron homeostasis, heme-induced functional changes can be divided into three groups: iron recycling at the steady state, iron-sequestration during infection and inflammation, and preventing heme toxicity to maintain tissue homeostasis.
Heme is metabolized inside macrophages by the sequential activity of Heme oxygenase (HO) and biliverdin reductase (BVR). HO has two isoforms: the inducible HO-1 and the constitutively expressed HO-2 (34). HO-1 levels increase upon heme accumulation in macrophage and catalyze the breakdown of heme into biliverdin. BVR then converts biliverdin to bilirubin, which is exported out of the cells to be incorporated into bile acid in the liver (26, 31, 34). Iron is released upon degradation of heme to bilirubin. This iron can be either stored inside the macrophages in combination with ferritin or exported out of the cells via the iron exporter ferroportin (26, 31). The decision to store or release iron depends upon various factors such as iron requirements of the host and the presence or absence of inflammation and infection. At the steady state, the vast majority (>90%) of the body’s iron requirement is provided by this macrophage-based recycling machinery (Figure 1). Inflammation and infection with extracellular pathogens leads to a much greater propensity to store, rather than release, iron inside macrophages (35). One underlying mechanism is the release of hepcidin from hepatocytes in response to inflammation, which leads to internalization and degradation of ferroportin (36). Indeed, such infectious settings promote iron accumulation by macrophages leading to a systemic reduction of circulating iron. This is a form of evolutionarily conserved immune response aimed at limiting pathogen’s access to critical nutrients (35). Therefore, iron recycling by macrophages is carefully regulated to strike a balance between iron requirements of the host, avoiding deleterious effects of heme/iron, and preventing pathogen access to iron.
Heme-Mediated Regulation of Iron-Recycling Macrophages
Iron recycling by macrophages is a highly specialized function, which involves several closely coordinated steps from heme acquisition and heme degradation to iron storage and release. As the first element in this metabolic cascade, heme regulates many of the subsequent steps in this pathway. Heme can physically bind the transcriptional factor Bach1 (BTB and CNC homology 1) via a dipeptide motif of cysteine and proline, which inhibits its functions (37). Bach1 and the closely related Bach2 belong to the basic leucine zipper family of transcription factors, which control gene expression by forming heterodimers with small Maf proteins (38). Bach–Maf heterodimers bind to Maf recognition elements (MARE) in the genome to suppress target genes such as HO1, ferroportin, and ferritin (38). Direct binding of heme to Bach1 inhibits Bach1 activity by (1) altering its DNA binding, (2) promoting its nuclear export, and (3) inducing polyubiquitination and degradation of Bach1 protein (39–42). In the absence of Bach1, Maf binds to transcription factor Nrf2. Nrf2–Maf heterodimers also bind MARE sequences, but leads to target gene activation (38, 43). Therefore, heme levels inside a cell can control the relative abundance of “activating” Nrf2-Maf and “repressive” Bach1–Maf heterodimers. This is a key mechanism by which high levels of environmental heme induce the expression of HO1 (44). Induced HO1 reduces heme levels, which then decreases the rate of Bach1 degradation. This “feedback loop” ensures that Bach1, HO1, and Fpn levels are carefully titrated against heme level.
Bach1 appears to serve as an important transcriptional sensor of heme. Its targets include genes involved in heme degradation (for example: HO1, ferritin, and ferroportin), redox homeostasis (for example: GCLC and GCLM) as well as cell-cycle and apoptosis related (for example: CALM1, BCL2L11, and SQSTM1) (38). One notable target is the transcription factor SpiC, which is highly expressed by iron-recycling macrophages in the spleen (RPM) and is required for their development (3). SpiC is a member of the Spi subfamily of Ets transcription factor, which is also expressed in B cells (45, 46). High levels of heme in splenic red pulp were found to induce degradation of Bach1 protein in monocytes. This occurred via direct interaction of heme and Bach1 protein leading to proteasome-dependent degradation of Bach1, which de-repressed SpiC to promote the development of RPM from monocytes (8). This pathway is particularly important during hemolysis and conditions leading to erythrocyte damage, which requires greater number of RPM (8). Hemolysis may be associated with sterile conditions (such as sickle cell anemia) or infectious diseases (such as a traumatic wound or malaria), which are associated with distinct iron-recycling requirements. While in the sterile setting it is safe to “turn on” the RPM transcriptional program aimed at releasing the heme-associated iron back into the circulation, in infectious setting iron is usually sequestered inside the macrophages. Currently, it is unclear whether and how pathogenic stimuli with or without heme influences SpiC expression in macrophages. Finally, SpiC induction is one of the many consequences of Bach1 inhibition, and Bach1 inhibition itself is one of the many consequences of increased heme levels. The full repertoire heme’s impact on macrophage development remains to be elucidated.
Heme-Mediated Regulation of Macrophage Activation
Heme can influence macrophage function directly or indirectly in a number of ways. Direct mechanisms include heme’s activity as a prosthetic group in hemoproteins, inhibition of Bach transcription factors, binding to nuclear receptors such as RevErb, interactions with toll-like receptor 4 (TLR4), and direct binding to DNA in the nucleus. Indirect actions are more diverse and include heme’s ability to induce ROS and generate metabolites such as iron, carbon monoxide, bilirubin, and biliverdin. The source of heme for the aforementioned activities can be cell intrinsic (all cells can produce heme) or exogenous. Here, we will restrict our discussion to the direct actions of cell-exogenous heme on macrophages.
The role of heme in inducing iron-recycling macrophages via Bach1 degradation is discussed above. This pathway ensures safe disposal and recycling of heme. Excess heme in circulation or tissue may also indicate damage to erythrocytes or muscle, which contains the majority of heme in the body. In this context, heme may act as an alarmin, which are endogenous factors released upon tissue damage to activate immune system (24). Heme has been shown to activate TLR4 to induce TNF production in macrophages (47). TLRs are evolutionarily conserved transmembrane receptors that recognize structurally conserved molecular motifs associated with pathogens (48). TLR4 engagement by prototypical agonist LPS activates two distinct downstream pathways: MyD88 dependent and TRIF dependent. Heme appears to selectively activate MyD88-dependent pathway and induces a different repertoire of cytokines when compared to LPS, underscoring their distinct mode of action through TLR4 (47). Notably, heme showed synergistic effects with lower doses of LPS on the production of inflammatory cytokines such as TNF and IL6 (49). This synergism required the activity of spleen tyrosine kinase and was also observed with activation of TLR2, TLR3, and TLR9 (49). This synergy may be particularly relevant to infectious diseases associated with hemolysis, such as malaria, where heme may help control infection by “boosting” TLR signaling when pathogen levels (hence TLR agonist levels) are low. Heme has also been reported to promote IL1-β processing by NLRP3 inflammasome component further supporting its role as an alarmin (50).
Macrophage activation via heme is also relevant in certain non-infectious diseases. Recent work has shown that heme released upon hemolysis in sickle cell disease leads to the induction of a pro-inflammatory “M1” polarization in macrophages, which was found to be dependent on TLR4 activity and ROS production (51). Importantly, exogenous administration of the heme scavenger hemopexin was found to counteract the induction of this inflammatory phenotype in macrophages of a mouse model of sickle cell disease. Heme is also a natural ligand for nuclear receptors RevErbα and RevErbβ (52–54). Heme binding to RevErbα has been shown to repress IL10 transcription in human monocytes and macrophages (55). IL10 has anti-inflammatory role and its suppression by heme RevErbα was suggested to maintain a pro-inflammatory “M1” phenotype in macrophages. Notably, genetic cis regulatory elements near IL10 locus promoting this action of RevErbα were present in humans but not mice (55). More global transcriptional analysis suggested that RevErbα and RevErbβ generally suppress enhancer-directed transcription in macrophages but the relevance of this in the context of heme activity in macrophage remains to be fully elucidated (56).
Concluding Remarks
Nature has delegated macrophages the task of handling heme, which is aimed at avoiding tissue toxicity, defending against pathogens, and recycling iron to support ongoing erythropoiesis. These diverse physiological requirements involve distinct but overlapping functional modules in macrophages (Figure 1). The core functional module is the ability to “sense” and “degrade” heme. Other functional modules are context dependent. As an example, iron released upon heme degradation is exported outside the cell via Fpn at the steady state. During inflammation, however, Fpn is downregulated leading to an accumulation of iron inside macrophages. Indeed, macrophages excel at homeostatic functions by integrating myriad extracellular signals to formulate an appropriate physiological response.
An exciting and relatively new finding is the ability of “free” heme to act as a signaling molecule for monocyte differentiation via activation of the transcription factor SpiC. During inflammation or injury circulating monocytes can enter tissue to differentiate into wound-healing and anti-inflammatory macrophages or pro-inflammatory and immune-stimulatory DCs (57). Therefore, the ability of heme to influence the differentiation of a monocyte into DCs vs. macrophage has important implications on immunity and tissue homeostasis, which remains to be fully explored. In this context, it will also be important to uncover factors and pathways that control the movement of heme within distinct cellular compartments after its uptake. Several heme transporters have been described: Flvcr regulating heme export from cytoplasm, HRG-1 regulating heme transport from lysosome into cytoplasm, and Mrp5 regulating heme export from cytoplasm (58–61). Future research will likely provide new insights into pathways controlling such chaperoned movement of heme between and within cells.
Recent work has led to a gradual shift in our perception of free heme: from a mere product of erythrocyte degradation to an active signaling molecule. As future work reveals additional facets of heme activity, the number of ways in which erythrocyte-derived heme can influence macrophage development and function will grow. This may also open up new opportunities to therapeutically modulate macrophage function in various diseases.
Author Contributions
MA and MH wrote, organized, and edited the manuscript. SD prepared the figure and helped edit the manuscript.
Conflict of Interest Statement
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be a potential conflict of interest.
Funding
This work was supported by National Institutes of Health (K08AI106953), Burroughs Wellcome Fund (CAMS award), and American Society of Hematology (scholar award).
References
1. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
2. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14:986–95. doi:10.1038/ni.2705
3. Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature (2009) 457:318–21. doi:10.1038/nature07472
4. Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science (2016) 353:1–12. doi:10.1126/science.aaf4238
5. A-Gonzalez N, Guillen JA, Gallardo G, Diaz M, de la Rosa JV, Hernandez IH, et al. The nuclear receptor LXRα controls the functional specialization of splenic macrophages. Nat Immunol (2013) 14:831–9. doi:10.1038/ni.2622
6. Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell (2014) 157:832–44. doi:10.1016/j.cell.2014.04.016
7. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol (2014) 15:1026–37. doi:10.1038/ni.3005
8. Haldar M, Kohyama M, So AY-L, Kc W, Wu X, Briseño CG, et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell (2014) 156:1223–34. doi:10.1016/j.cell.2014.01.069
9. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159:1312–26. doi:10.1016/j.cell.2014.11.018
10. Hoeffel G, Ginhoux F. Ontogeny of tissue-resident macrophages. Front Immunol (2015) 6:486. doi:10.3389/fimmu.2015.00486
11. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol (2014) 14:392–404. doi:10.1038/nri3671
12. Bain CC, Bravo-Blas A, Scott CL, Gomez Perdiguero E, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol (2014) 15:929–37. doi:10.1038/ni.2967
13. Haldar M, Murphy KM. Origin, development, and homeostasis of tissue-resident macrophages. Immunol Rev (2014) 262:25–35. doi:10.1111/imr.12215
14. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14:571–8. doi:10.1038/nri3712
15. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol (2014) 5:514. doi:10.3389/fimmu.2014.00514
16. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi:10.1038/nri2448
17. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40:274–88. doi:10.1016/j.immuni.2014.01.006
18. Drakesmith H, Nemeth E, Ganz T. Ironing out ferroportin. Cell Metab (2015) 22:777–87. doi:10.1016/j.cmet.2015.09.006
19. Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab (2005) 1:191–200. doi:10.1016/j.cmet.2005.01.003
20. Guida C, Altamura S, Klein FA, Galy B, Boutros M, Ulmer AJ, et al. A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood (2015) 125:2265–75. doi:10.1182/blood-2014-08-595256
21. Liu X-B, Nguyen N-BH, Marquess KD, Yang F, Haile DJ. Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells Mol Dis (2005) 35:47–56. doi:10.1016/j.bcmd.2005.04.006
22. Chung J, Chen C, Paw BH. Heme metabolism and erythropoiesis. Curr Opin Hematol (2012) 19:156–62. doi:10.1097/MOH.0b013e328351c48b
23. Kosman DJ. Redox cycling in iron uptake, efflux, and trafficking. J Biol Chem (2010) 285:26729–35. doi:10.1074/jbc.R110.113217
24. Soares MP, Bozza MT. Red alert: labile heme is an alarmin. Curr Opin Immunol (2016) 38:94–100. doi:10.1016/j.coi.2015.11.006
25. Korolnek T, Hamza I. Macrophages and iron trafficking at the birth and death of red cells. Blood (2015) 125:2893–7. doi:10.1182/blood-2014-12-567776
26. Soares MP, Hamza I. Macrophages and iron metabolism. Immunity (2016) 44:492–504. doi:10.1016/j.immuni.2016.02.016
27. Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicol Lett (2005) 157:175–88. doi:10.1016/j.toxlet.2005.03.004
28. Gozzelino R, Soares MP. Coupling heme and iron metabolism via ferritin H chain. Antioxid Redox Signal (2014) 20:1754–69. doi:10.1089/ars.2013.5666
29. Bratosin D, Mazurier J, Tissier JP, Estaquier J, Huart JJ, Ameisen JC, et al. Cellular and molecular mechanisms of senescent erythrocyte phagocytosis by macrophages. A review. Biochimie (1998) 80:173–95. doi:10.1016/S0300-9084(98)80024-2
30. de Back DZ, Kostova EB, van Kraaij M, van den Berg TK, van Bruggen R. Of macrophages and red blood cells; a complex love story. Front Physiol (2014) 5:9. doi:10.3389/fphys.2014.00009
31. Ganz T. Macrophages and systemic iron homeostasis. J Innate Immun (2012) 4:446–53. doi:10.1159/000336423
32. Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature (2001) 409:198–201. doi:10.1038/35051594
33. Hvidberg V, Maniecki MB, Jacobsen C, Højrup P, Møller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood (2005) 106:2572–9. doi:10.1182/blood-2005-03-1185
34. Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol (1997) 37:517–54. doi:10.1146/annurev.pharmtox.37.1.517
35. Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol (2015) 15:500–10. doi:10.1038/nri3863
36. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science (2004) 306:2090–3. doi:10.1126/science.1104742
37. Igarashi K, Watanabe-Matsui M. Wearing red for signaling: the heme-bach axis in heme metabolism, oxidative stress response and iron immunology. Tohoku J Exp Med (2014) 232:229–53. doi:10.1620/tjem.232.229
38. Zhou Y, Wu H, Zhao M, Chang C, Lu Q. The bach family of transcription factors: a comprehensive review. Clin Rev Allergy Immunol (2016) 50:345–56. doi:10.1007/s12016-016-8538-7
39. Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J (2001) 20:2835–43. doi:10.1093/emboj/20.11.2835
40. Suzuki H, Tashiro S, Hira S, Sun J, Yamazaki C, Zenke Y, et al. Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J (2004) 23:2544–53. doi:10.1038/sj.emboj.7600248
41. Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, et al. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol (2007) 27:6962–71. doi:10.1128/MCB.02415-06
42. Tan M-KM, Lim H-J, Bennett EJ, Shi Y, Harper JW. Parallel SCF adaptor capture proteomics reveals a role for SCFFBXL17 in NRF2 activation via BACH1 repressor turnover. Mol Cell (2013) 52:9–24. doi:10.1016/j.molcel.2013.08.018
43. Oyake T, Itoh K, Motohashi H, Hayashi N, Hoshino H, Nishizawa M, et al. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol Cell Biol (1996) 16:6083–95. doi:10.1128/MCB.16.11.6083
44. Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci U S A (2004) 101:1461–6. doi:10.1073/pnas.0308083100
45. Bednarski JJ, Pandey R, Schulte E, White LS, Chen B-R, Sandoval GJ, et al. RAG-mediated DNA double-strand breaks activate a cell type-specific checkpoint to inhibit pre-B cell receptor signals. J Exp Med (2016) 213:209–23. doi:10.1084/jem.20151048
46. Bemark M, Mårtensson A, Liberg D, Leanderson T. Spi-C, a novel Ets protein that is temporally regulated during B lymphocyte development. J Biol Chem (1999) 274:10259–67. doi:10.1074/jbc.274.15.10259
47. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of toll-like receptor 4. J Biol Chem (2007) 282:20221–9. doi:10.1074/jbc.M610737200
48. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
49. Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourão-Sa D, Fortes GB, et al. Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J Biol Chem (2010) 285:32844–51. doi:10.1074/jbc.M110.146076
50. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A (2014) 111:E4110–8. doi:10.1073/pnas.1405023111
51. Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood (2016) 127:473–86. doi:10.1182/blood-2015-08-663245
52. Carter EL, Gupta N, Ragsdale SW. High affinity heme binding to a heme regulatory motif on the nuclear receptor Rev-erbβ leads to its degradation and indirectly regulates its interaction with nuclear receptor corepressor. J Biol Chem (2016) 291:2196–222. doi:10.1074/jbc.M115.670281
53. Raghuram S, Stayrook KR, Huang P, Rogers PM, Nosie AK, McClure DB, et al. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REV-ERBbeta. Nat Struct Mol Biol (2007) 14:1207–13. doi:10.1038/nsmb1344
54. Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, et al. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science (2007) 318:1786–9. doi:10.1126/science.1150179
55. Chandra V, Mahajan S, Saini A, Dkhar HK, Nanduri R, Raj EB, et al. Human IL10 gene repression by Rev-erbα ameliorates Mycobacterium tuberculosis clearance. J Biol Chem (2013) 288:10692–702. doi:10.1074/jbc.M113.455915
56. Lam MTY, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature (2013) 498:511–5. doi:10.1038/nature12209
57. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (2010) 327:656–61. doi:10.1126/science.1178331
58. Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, et al. Identification of a human heme exporter that is essential for erythropoiesis. Cell (2004) 118:757–66. doi:10.1016/j.cell.2004.08.014
59. Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science (2008) 319:825–8. doi:10.1126/science.1151133
60. Korolnek T, Zhang J, Beardsley S, Scheffer GL, Hamza I. Control of metazoan heme homeostasis by a conserved multidrug resistance protein. Cell Metab (2014) 19:1008–19. doi:10.1016/j.cmet.2014.03.030
Keywords: heme, iron-recycling macrophage, erythrophagocytosis, SpiC, Bach1
Citation: Alam MZ, Devalaraja S and Haldar M (2017) The Heme Connection: Linking Erythrocytes and Macrophage Biology. Front. Immunol. 8:33. doi: 10.3389/fimmu.2017.00033
Received: 30 October 2016; Accepted: 09 January 2017;
Published: 24 January 2017
Edited by:
Robert F. Paulson, Pennsylvania State University, USACopyright: © 2017 Alam, Devalaraja and Haldar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Malay Haldar, bWhhbGRhckBtYWlsLm1lZC51cGVubi5lZHU=