 Shouxiong Huang
Shouxiong Huang- Department of Environmental Health, University of Cincinnati College of Medicine, Cincinnati, OH, USA
Peptide-specific conventional T cells have been major targets for designing most antimycobacterial vaccines. Immune responses mediated by conventional T cells exhibit a delayed onset upon primary infection and are highly variable in different human populations. In contrast, innate-like T cells quickly respond to pathogens and display effector functions without undergoing extensive clonal expansion. Specifically, the activation of innate-like T cells depends on the promiscuous interaction of highly conserved antigen-presenting molecules, non-peptidic antigens, and likely semi-invariant T cell receptors. In antimicrobial immune responses, mucosal-associated invariant T cells are activated by riboflavin precursor metabolites presented by major histocompatibility complex-related protein I, while lipid-specific T cells including natural killer T cells are activated by lipid metabolites presented by CD1 proteins. Multiple innate-like T cell subsets have been shown to be protective or responsive in mycobacterial infections. Through rapid cytokine secretion, innate-like T cells function in early defense and memory response, offering novel advantages over conventional T cells in the design of anti-tuberculosis strategies.
Host immune responses are critical in controlling pandemic and life-threatening Mycobacterium tuberculosis (M. tuberculosis) infection in humans through the development of protective immunity in a T cell-dependent manner (1–3). An effective T cell response leads to a low lifetime risk of developing active tubercular diseases. In mice, the activation of CD4+ or CD8+ T cells is important for maintaining a low bacterial load in tissues, as shown using antibody depletion and T cell adoptive transfer in various T cell-deficient mice (4–9). In humans, the importance of CD4+ T cell response is supported by the dramatically increased risk of active tuberculosis with the co-infection of human immunodeficiency virus (HIV), which reduces the number of CD4+ T cells in patients (10). Abundant circumstantial evidence also supports that the effector functions of human CD8+ T cells are able to suppress mycobacterial growth (3), although the activating elements for different CD8+ T cell populations remain elusive (3, 11, 12). Thus, therapeutic and vaccine strategies aimed at the development of protective T cell responses would be beneficial for minimizing the treatment course of antibiotics, preventing the spread of drug-resistant M. tuberculosis, and reducing lung inflammatory responses and injury (13).
The licensed vaccine Bacillus Calmette–Guérin (BCG) has saved many children’s lives, despite offering insufficient protection against pulmonary tuberculosis in adults and no evidenced efficacy in controlling the prevalence of tuberculosis (14). Upon discovery of virulence-associated genetic complexes from M. tuberculosis, virulent factors encoded by these pathogenicity islands have been shown to induce protective T cell responses (15, 16). Many vaccination approaches in the pipeline of development are based on immune response mediated by these virulent proteins through antigen presentation by major histocompatibility complex (MHC) class I or MHC class II molecules (15, 17, 18). Immune responses mediated by conventional T cells feature highly specific and heterogeneous immune responses that are determined by tri-molecular interactions among highly polymorphic antigen-presenting molecules, diversified antigenic peptides, and variable T cell receptors in human populations (19). Meanwhile, conventional T cells require a relatively long priming stage allowing naïve T cells to differentiate into effector cells with antimycobacterial functions. As discussed below, innate-like T cells display conserved receptor interaction and fast-responding kinetics in antigen presentation and effector responses, which contribute to unique antimycobacterial immune defenses.
Discoveries of Unconventional T Cells in M. Tuberculosis Infections
Until recently, CD8+ mucosal-associated invariant T (MAIT) cells in antimycobacterial responses were difficult to distinguish from conventional CD8+ T cells. High-frequency CD8+ T cells in both infected and uninfected individuals are reactive to M. tuberculosis, potentially challenging the notion that CD8+ T cells are conventional T cells activated by mycobacterial peptides in infected humans. Limited dilution analysis showed that a high percentage of these M. tuberculosis-reactive CD8+ T cells were not restricted by classical MHC class I molecules (12, 20). Surprisingly, these non-conventional T cells, which covered 80% of the total CD8+ T cell clones generated from individual healthy donors, 70% from latently infected donors, and about 35% from donors with active tuberculosis, were identified as MAIT cells in a blocking assay using an antibody against the MHC-related protein I (MR1) (12). This finding indicates that a high percentage of CD8+ T cells are not restricted by MHC class I proteins and do not respond to peptide antigens. In fact, they are restricted by MHC class I-like MR1 protein, as confirmed with functional blockage using an anti-MR1 antibody (12, 20). The existence of a high percentage of mycobacterial-reactive CD8+ T cells in uninfected healthy humans is also striking, as the stimulants for these CD8+ T cells must be shared between non-mycobacterial and mycobacterial organisms, and peptide antigens are least likely to explain this result. To date, we know MAIT cells respond to riboflavin precursor metabolites produced by a variety of bacterial species, including M. tuberculosis (21). Concurrently, MAIT cells have been shown to be protective against mycobacterial infection using infected mouse models deficient of MR1 protein or with overexpression of the MAIT cell TCR (20).
The presence of antimycobacterial T cells restricted by the cluster of differentiation I (CD1) proteins has been reported along with the initial discovery of a CD1 antigen presentation function (22–26). The expression of an invariant TCR sequence likely supports a unique activation mechanism diverted from conventional T cells (27). Indeed, CD1-restricted T cells from peripheral blood can be stimulated by autologous immature CD1+ dendritic cells and respond at a significant magnitude and frequency in asymptomatic M. tuberculosis-infected donors (23). It appears that lipid-stimulated T cell proliferation is minimally detectable or absent in the blood samples from active tuberculosis patients and become detectable 2 weeks after the start of antibiotic treatment. This interesting finding suggests that M. tuberculosis-reactive CD4+ T cells respond to M. tuberculosis lipid antigens presented by CD1 proteins and exist abundantly in healthy individuals with previous exposure to M. tuberculosis (23).
A Glance at Unconventional T Cells
Unlike conventional T cells, which are restricted by the antigen-presenting molecules encoded by the MHC genetic complexes, unconventional T cells are activated by MHC class I-like molecules that are encoded by genes outside the MHC complexes. As shown in Table 1, unconventional T cells are mostly restricted by CD1 and MR1 proteins. Specifically for two major invariant T cell populations, MAIT cells are activated by riboflavin precursor metabolites presented by the MR1 protein, and natural killer T (NKT) cells are activated by various lipid metabolites presented by the CD1d protein (Table 1). CD1- and MR1-restricted T cell subsets are in fact abundant in human peripheral blood or tissues. In particular, MR1-restricted MAIT cells and CD1a- and CD1c-restricted T cells are highly frequent in human blood (12, 28, 59); MAIT cells and iNKT cells are also abundant in human liver tissues (29, 30). The functional uniqueness of MAIT and iNKT cells is mostly attributable to their invariant TCRα sequences, which were initially characterized in the early 1990s (27). The expression of invariant TCRα chains with biased usage of TCRβ chains is now known as a major feature in MAIT, iNKT, and other unconventional T cell populations (Table 1), contributing to the quick-responding kinetics described below.
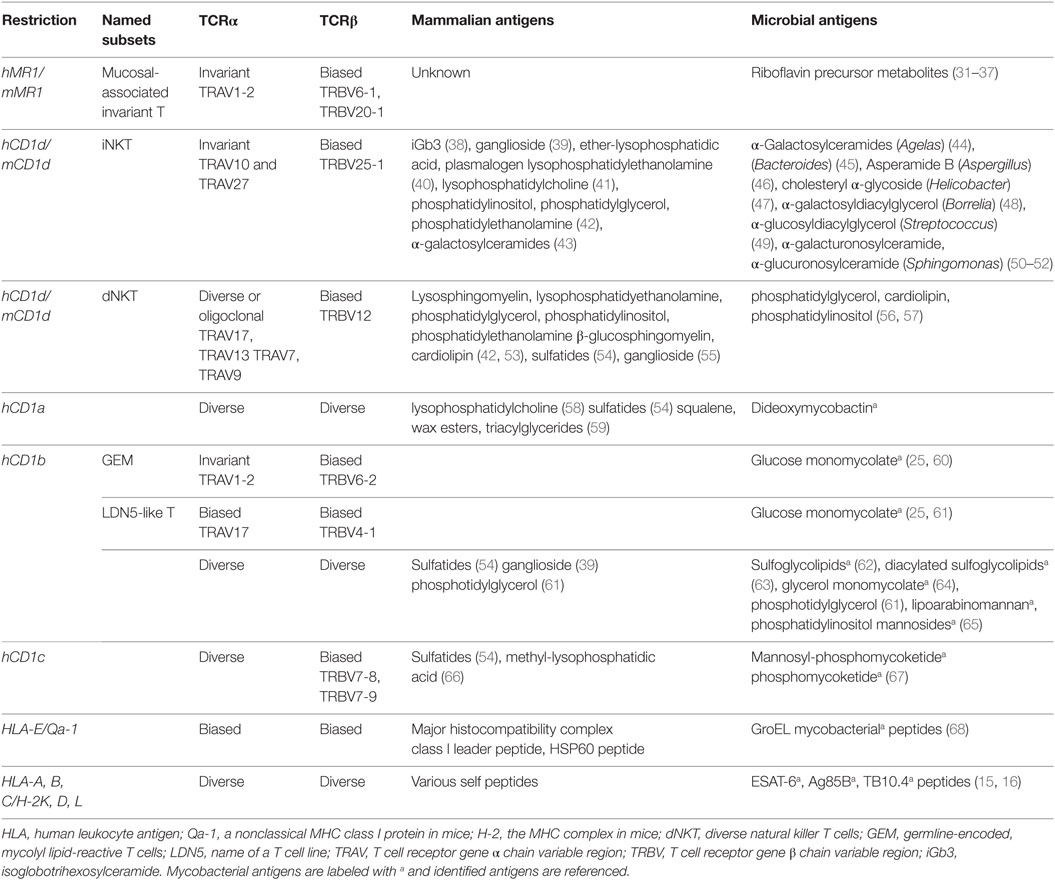
Table 1. Antigen-presenting molecules, antigens, and TCRs for unconventional T cells.
The identification of mycobacterial antigens for unconventional T cell activation has focused on the lipid antigens presented by group I CD1 proteins (CD1a, CD1b, and CD1c). Unconventional T cells against M. tuberculosis were in fact initially discovered to respond to CD1b-restricted mycobacterial lipid antigen (22). Thereafter, more antimycobacterial lipid-specific T cells were discovered to detect mycobacterial lipid antigens presented by group I CD1 proteins (Table 1). Subsets of CD1a-restricted T cells, represented by the cell line CD8-2, are reactive to dideoxymycobactin (DDM) (24). CD1b-restricted T cells are able to recognize more complex mycobacterial lipids, including glycerol monomycolate (64), glucose monomycolate (25), free mycolic acid (69), diacylated sulfoglycolipids (63), and phosphatidylinositol mannosides (70). Several lines of CD1c-restricted T cells have also been derived in response to a different class of mycobacterial lipid, mycoketides, including the T cell lines CD8-1, which responds to mycobacterial β-mannosyl phosphomycoketide from mycobacterial lipid extracts, and DN-6, which recognizes phosphomycoketide (26, 67). As summarized, these unconventional T cells exhibit different features from conventional T cells in antigen presentation (Table 1).
Innate-Like Postulate and Fast-Responding Kinetics
To consider T cell populations as being innate-like requires comparison of the biological features of T cells with those of cells from the innate and adaptive immune systems (Table 2). One measurable characteristic of an innate-like postulate is the quick activation kinetics from pathogen-unexposed precursors or naïve cells to effector cells in an antigen-specific manner. The activation of conventional naïve T cells requires prolonged antigenic priming for days and weeks following a primary infection to stimulate clonal expansion and effector function (71). Conventional CD8+ T cells have been shown with antimycobacterial responses, as supported by the M. tuberculosis-infected β2m−/− and TAP1−/− mice, which are unable to control M. tuberculosis replication in the lung and cause premature death (2, 72, 73). These mycobacterial peptide-specific CD8+ T cells occur in the draining lymph node, become detectable in lung tissues within 2 weeks, and peak around 5–8 weeks in lung tissues after a primary infection in mice (Figure 1A) (3). Using a skin test, immune responses can be detected 5–6 weeks after M. tuberculosis infection in humans (74). In a drastic contrast to fast T cell responses to other intracellular bacteria, such as Listeria infection in mouse spleen (75), a much slower kinetics of T cell responses in M. tuberculosis infection may attribute to a slower growing curve of M. tuberculosis, bacterial inhibition of migratory activity, bacterial modulation of host antigen-presenting cells, and relatively immunoprivileged nature of the healthy alveolar tissues (2, 76). The antigen stimulation for conventional T cell activation occurs faster in vitro than in vivo because sufficient antigen presentation is usually warranted when setting up cell culture. In cell culture, mouse naïve CD8+ T cells require antigen stimulation for at least a week in vitro to observe the cytotoxic activity to specific antigens (77). In fact, several rounds of stimulation with peptide-pulsed macrophages for multiple weeks are required to achieve stronger responses of naïve CD8+ T cells, such as the generation of anti-HIV cytolytic T cells (78). Distinctively, the response of macrophages and other innate immune cells to uptake bacterial materials for priming adaptive immune responses is believed in a very early stage, generally within hours, after infection (79).
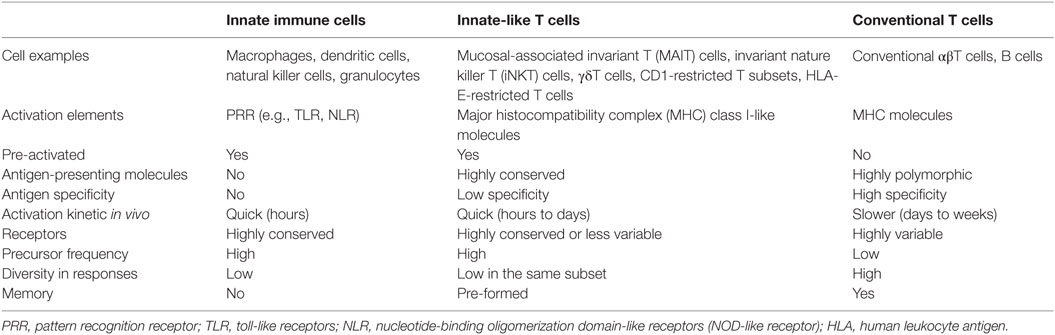
Table 2. Characteristics of innate-like T cells.
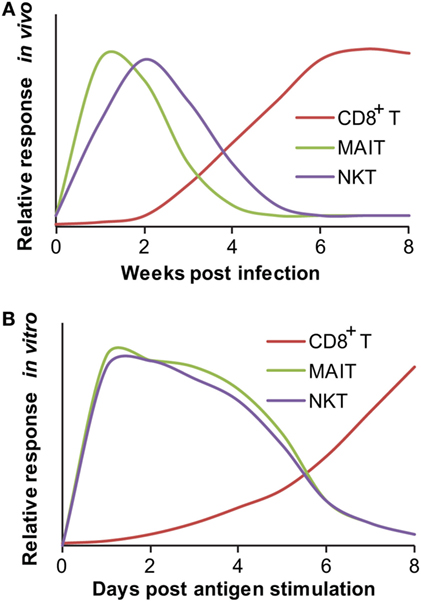
Figure 1. Responding kinetics of mucosal-associated invariant T (MAIT) and natural killer T (NKT) cells. (A) In vivo responding kinetics were hypothesized based on the presence of mycobacterial antigen-specific CD8+ T cells in lung tissues (3), the ability of MAIT cells to inhibit Bacillus Calmette–Guérin (BCG) growth in lung tissues (80), and the ability of transferred iNKT to inhibit M. tuberculosis growth in lung tissues (81). (B) In vitro-responding kinetics were estimated according to the acquisition of cytolytic function by CD8+ T cells upon in vitro peptide stimulation (77), cytokine production by polyclonal MAIT cells upon stimulation with BCG-infected macrophages (80), and cytokine production by tetramer-isolated human polyclonal NKT cells upon antigen-specific activation (82).
Unlike conventional T cells, innate-like T cells, such as MAIT cells, are ready to respond to mycobacterial or other antigens within hours to days (83–85). When pathogen-unexposed mouse MAIT cells were cocultured with BCG-infected macrophages, the production of a large amount of IFN-γ and IL-17a was detectable from MAIT cells within 4 days of coculture (80), although the time course needs to be further determined and the actual responding time is likely shorter. The early in vitro response has also been supported by the upregulation of CD69 expression on human and mouse MAIT cells following overnight incubation with E. coli- or Mycobacterium abscessus-infected monocytes (12, 20), or M. tuberculosis-infected lung epithelial cells (12). The secretion of IFN-γ (12, 20) and tumor necrosis factor-α (TNF-α) (12, 86) upon MAIT cell activation can be observed as well after incubating bacterial-infected antigen-presenting cells and pathogen-unexposed MAIT cells for 16–48 h (Figure 1B). This in vitro responding kinetics of MAIT cells is much faster than that of naïve conventional T cells stimulated with peptide antigens, which usually need 7 days or more to exhibit proliferation or effector function (78, 87). In vivo, MAIT cell accumulation in the primary infected lung tissue starts less than 10 days post BCG infection, as detected by an MR1-tetramer for measuring the frequency of MAIT cells isolated from bronchoalveolar lavage fluid (BALF) and lung tissue (33). Taking into account the slow growing curve and immune-escaping mechanism mediated by M. tuberculosis as discussed (2, 76), the kinetics of MAIT cell responses is essentially faster than the response of conventional CD8+ T cells in the lung infection of M. tuberculosis (Figure 1A). It appears that both the in vitro and in vivo kinetics of MAIT cells upon mycobacterial infection can be considered to resemble that of conventional memory T cells (33, 88).
In addition to MAIT cells, other unconventional T cells also exhibit quick-responding kinetics in their “primary” responses, including various CD1-restricted T cell subsets and HLA-E-restricted T cells (Table 1). NKT cells were identified in response to mycobacterial infection by the fact that the adoptive transfer of naïve splenic iNKT cells from uninfected mice resulted in a significantly reduced burden of M. tuberculosis in the lungs of infected mice (81). This adoptive transfer assay provided evidence that CD1d-restricted NKT cells mediate protection against M. tuberculosis in vivo using innate-like responding kinetics, which allows visualization of the inhibition curve of M. tuberculosis within 3 weeks after the transfer of naïve iNKT cells, supporting the quick-responding kinetics of naïve iNKT cells (81). As characterized, the iNKT antigen α-galactosylceramide (α-GalCer) also ameliorated the course of tuberculosis in mice in the early stage of M. tuberculosis infection but provided no additional survival benefit for an extended application (89). Although the lack of long-term protection of α-GalCer in the mycobacterial-infected mouse model is difficult to explain, it is perhaps consistent with the observation that CD1d−/− mice are not more sensitive to M. tuberculosis infection (8). Moreover, the protection provided by iNKT cells appears similar to the protection offered by MAIT cells for decreasing BCG growth in lung tissues at 10 days of infection rather than at 30 days. These initial results support that innate-like MAIT and iNKT cells provide protection against mycobacterial infection in the early stage of infection. Although MAIT cells produce more cytokines at 30 days, whether innate-like T cells are able to further control the bacterial growth in later stages, cross-talk with innate immune cells, bridge the activation of conventional T cells, and regulate inflammatory responses in infected tissues are interesting topics that require further investigations (80).
The kinetics for group I CD1-restricted T cells in responding to mycobacterial and other antigens remain poorly understood, mainly due to the lack of expression of human CD1a, CD1b, and CD1c homologs in mouse models. Interestingly, a recent study using humanized mice showed that the kinetics of primary activation and memory response of group I CD1-restricted T cells were somewhat delayed (90). The antimycobacterial immune responses of group I CD1-restricted T cells peaked at 7 days following the immunization of M. tuberculosis lipids and showed a bit more rapid secondary responses at 5 days (90). Whether naïve group I CD1-restricted T cells are able to quickly respond to mycobacterial infections similarly to iNKT cells and have an innate-like response in vitro or in vivo remains unknown.
The innate-like kinetics of activation for unconventional T cells are correlated to highly conserved structures and interacting modes of three types of key molecules, antigen-presenting molecules, antigens, and TCRs, which are variable for conventional T cell activation. As detailed below, the conserved genetic feature of each type of molecules is not restricted by the individual donor and will contribute to promiscuous tri-molecular interaction in a donor-unrestricted manner, providing an immunogenetic basis for explaining innate-like responses in unconventional T cells (91).
Highly Conserved Antigen-Presenting Molecules
In 1989, Janeway hypothesized that the innate immune system is able to regulate adaptive immune responses through innate immune recognition, such as pattern recognition (92). This prediction has been validated and has contributed to the understanding of the regulatory role of the innate immune system in adaptive immune responses (93). The innate immune system displays a remarkable homology of various innate immune molecules, including pattern recognition receptors (PRR) such as toll-like receptors (TLRs), which either recognize pathogen-associated microbial patterns (PAMP) or endogenous damage-associated molecular patterns (DAMP) (94–96). The common feature of these innate immune receptors is that they are germline-encoded and are considered highly conserved, with limited single-nucleotide polymorphisms (SNPs) in humans (97, 98). Innate cells with PRR expression are capable of mounting rapid effector responses independently of clonal expansion. This strategy of early pathogen detection in the innate immune system contrasts with that in the adaptive immune system. The HLA gene complex is highly diverse, with more than 3,000 variant alleles discovered in the HLA-A, B, or C loci, to date, in human populations, and it can also form 15,000–70,000 possible α and β chain combinations for HLA-DQ, DR, and DP molecules (from the IMGT/HLA website: http://www.ebi.ac.uk/ipd/imgt/hla/stats.html, September 13, 2016). This profound heterogeneity confers hypervariable interaction in antigen presentation for conventional T cell activation in human populations and is also functionally reflected in the difficulty of finding non-rejected graft donors for organ transplantation except for identical twins (99).
Similar to PRR molecules and drastically different from conventional HLA proteins, HLA class I-like proteins, such as MR1, CD1, and HLA-E, are considered to be highly conserved in humans (85, 91, 100, 101). Few SNPs have been identified by gene sequencing for HLA class I-like proteins, although more will likely be identified when human genome sequences are further available. The MR1 gene is generally considered invariant in humans, except for two silent mutations, one of which generates a STOP codon in the α2 domain for an MR1 pseudogene (102). Different splicing variants of the MR1 gene have also been identified in other mammals (100, 103). The sequences of the ligand-binding and TCR-interacting domains (α1 and α2 domains) of the MR1 protein are identical between humans and chimpanzees, and they are highly homologous in mammals (100). It is difficult to imagine that human populations have such sequence variation, but humans and chimpanzees share the same sequence in the α1 and α2 domains of the MR1 protein. In regard to CD1 genes, SNPs in the α1 domain (104, 105) and the non-coding regions (106) of the human CD1a and CD1d genes have been identified, and non-synonymous mutations have also been deduced from exon 2 of both genes (107). Few identified SNPs support that MR1 and CD1 genes are still very conserved and appear similar to innate immune receptors such as TLRs in terms of sequence stability in human populations. Although these rare MR1 or CD1 variants may exist in some individuals, nearly monomorphic sequence of MHC class I-like molecules in humans provides an almost identical platform for antigen presentation and T cell activation in M. tuberculosis infections. Thus, a vaccination strategy designed based on these highly conserved structures of antigen-presenting molecules would be highly applicable to a broad range of the human population, in contrast to vaccination strategies based on the classical HLA system (108, 109).
Cross-Species Conservation of Non-Peptidic Antigens
Since the discovery of T cells using neonatally thymectomized mice in 1961 (110, 111), synthetic polypeptides have been used as model compounds for the elucidation of the molecular basis of immune responses (112), and peptide antigens have been shown to interact with MHC proteins for T cell activation (113). Peptides are probably better signatures for distinguishing non-self from self and fit nicely to the immunological paradigm in which high specificity for recognition and clearance of non-self is a major theme. In contrast to the chemical specificity of peptide compounds, lipids, and other small metabolites with the same or similar structures at least partially shared by humans, mammals, and even microbes are perhaps less consistent with the immunological theory of “self–non-self discrimination.” Interestingly, these highly conserved and structurally similar metabolites cross organisms, similar to PAMP or DAMP, which bind to innate receptors, are products of the fundamental metabolic pathways that are critical for cellular structures and functions in organisms. For the development and expansion of MAIT cells, the molecular factors provided by commensal bacteria are critical because MAIT cells are less detectable in germ-free mice (114). The result that germ-free mice do not demonstrate expanded MAIT cells suggests that microbial factors are critical for the activation and development of MAIT cells. Recently, bacterial metabolites in vitamin B2 and B9 metabolism were identified as MAIT cell antigens or ligands associated with the MR1 protein (31, 34). The first category of MR1 ligands, 6-formylpterin (6-FP) and acetyl-6-formylpterin (Ac-6-FP) as intermediate metabolites from vitamin B9 (folic acid) metabolism, has been identified from culture media. These two ligands function as antagonist molecules for blocking known MAIT cell activation (31, 35). The second category of MR1 ligands derives from the precursor metabolites in vitamin B2 (riboflavin) metabolism and includes three ribityllumazine species, 7-hydroxy-6-methyl-8-d-ribityllumazine (RL-6-Me-7-OH), 6,7-dimethyl-8-d-ribityllumazine (RL-6,7-diMe), and reduced 6-hydroxymethyl-8-d-ribityllumazine (rRL-6-CH2OH). These ligands exist in bacterial culture supernatant (31, 115) and have a bicyclic core similar to the formyl-pterins but are functional as agonists for stimulating MAIT cells when presented by the MR1 protein. The presence of a ribityl side chain from ribityllumazine species is critical for interacting with TCR for MAIT cell activation (31, 34, 35). A recently added third category of MR1 ligands includes two pyrimidine metabolites in riboflavin metabolism, 5-(2-oxopropylideneamino)-6-d-ribitylaminouracil (5-OP-RU) and 5-(2-oxoethylideneamino)-6-d-ribitylaminouracil (5-OE-RU) (36), which are considered to be the most potent known MAIT cell activators. Interesting research from Australia and France used bacterial strains defective in enzymes for riboflavin synthesis to show that mutations in the upstream genes involved in the synthesis of 5-amino-6-d-ribitylaminouracil (5-A-RU), the precursor of 5-OP-RU and 5-OE-RU, ablate MAIT cell activation by bacterial supernatants, whereas mutated genes involved in reactions downstream of 5-A-RU do not (36, 116, 117).
These precursor metabolites in riboflavin metabolism for MAIT cell activation provide several unique features that are different from those of peptide antigens for conventional T cells. First, the same or a similar group of riboflavin precursor metabolites is produced in a wide variety of bacteria and fungi, such as Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae, Staphylococcus aureus, Salmonella enterica serovar Typhimurium, M. tuberculosis, and Candida albicans, but not in Listeria monocytogenes and certain strains of Enterobacter or Streptococcus (20, 31, 32). It appears that riboflavin biosynthesis is a canonical pathway for these stimulatory bacterial species and involves a number of enzymes essential for producing MAIT cell antigens (37). Second, the riboflavin pathway is essential for the survival and biological function of many bacteria (118–120). These metabolite antigens, which are generated by commensal bacteria or pathogenic microorganisms, are presumably pre-existing in humans unexposed to other pathogens, such as M. tuberculosis. If this hypothesis is true, MAIT cells likely have encountered the shared bacterial riboflavin metabolites in individuals uninfected by M. tuberculosis. Thus, these metabolite antigens derived from non-mycobacterial species contribute to the pre-activated state of precursor MAIT cells. Upon the primary infection of M. tuberculosis, these pre-activated host MAIT cells are able to quickly respond to the same or similar antigens derived from M. tuberculosis. Third, the presentation of small metabolites does not require the processing of proteasome or endocytic peptide digestion which is needed for peptide antigen presentation mediated by MHC class I and class II molecules (121). However, the production of bacterial and host small molecules connects antigen presentation to the metabolism of various lipid and vitamin metabolites, which are involved in broad biological functions. Finally, metabolite antigens will result in a chemically conserved interaction with antigen-presenting molecules and TCRs, in which TCR contact will be less dependent on antigen structures, as described below.
Unlike conventional peptide antigens, lipid antigens for CD1-restricted T cells also show some cross-species similarities that contribute to the innate-like responses of CD1-restricted T cells. Recently, lipodomic analyses have identified a broad range of lipid metabolites that associate with CD1a, CD1b, CD1c, and CD1d proteins. These lipid metabolites broadly include various species of sphingolipids, glycosphingolipids, glycerophospholipids, lysophospohlipids, ether-linked phospholipids, acylglycerols, wax esters, and fatty acids, as reported and reviewed (41, 59, 122–125). Similar interesting features are usually shared by lipid metabolites that are identified from different bacteria or between mammals and bacteria (Table 1). For example, NKT cells respond to an exogenous α-galactosylceramide that was originally identified in the marine sponge Agelas mauritianus (44). Although most glycosyl sphingosines in mammals show a β-anomeric linkage, α-glycosylceramides were also recently detected in mammalian cells (43). Similar lipid antigens, glycosphingolipids, have been identified in various bacterial species, including non-pathogenic Sphingomonas (19, 50, 51) and Bacteroides species that are part of the gut microbiota (45, 126–128). Although bacterial and mammalian lipid antigens presented by CD1 proteins display some structural similarities, Mycobacterium spp. are considered highly unique in lipid metabolites. Several identified CD1-presented lipid antigens, including CD1a-presented mycobactin metabolites (24), CD1b-presented monomycolate, mycolic acid, sulfoglycolipids (25, 63, 64, 69), and CD1c-presented phosphatidylinositol mannosides (70), and mannosyl phosphomycoketide (26), have not yet been fully investigated in terms of their cross-reactivity to a high percentage of lipid-specific T cells.
To bind to MHC class I proteins, nonamer peptides optimally fit to the length of the groove in MHC class I proteins and longer peptides (~14–20mers) extend beyond the whole length of the ligand-binding groove in MHC class II proteins (109, 121, 129, 130). Dependent on the ionic and hydrogen bonding interactions between peptides and MHC proteins, the anchor residues from antigenic peptides and the corresponding binding pockets of MHC proteins can be defined (131–133). As a result, classical MHC proteins discriminate specific residues of antigenic peptides and their positions in the peptide sequence. This recognition is determined by the products of genetic codes, which are subject to mutation, genetic inheritance, and functional selection. However, antigen binding to an MHC class I-like protein uses a manner of interaction different from that of MHC–peptide interaction.
A Cyclic Anchor and Central Portal for MR1
The MR1 ligands identified to date share one or two cyclic structures, which are embedded within the MR1 ligand-binding cleft through interactions with aromatic or basic residues of MR1 proteins (Figure 2). Different from the ligand-binding grooves of MHC class I or class II proteins, which are shallow and open to access by antigenic ligands, the MR1 ligand-binding cleft has a small open portal in the middle portion of the MR1 protein (Figure 2). The MR1 ligand is located centrally within the MR1 ligand-binding cleft in a very small region, with the cyclic structure toward the base of the β-sheet and the ribityl side chain positioned upward, as shown in MR1 structure with ligand RL-6-Me-7-OH (Figures 2A,B). Interestingly, similar to ligand binding in CD1 proteins (19, 91), antigen binding to MR1 proteins exhibits very limited solvent accessibility and is largely buried within the MR1 protein. The interaction between RL-6-Me-7-OH and MR1 protein is dominated by hydrophobic interactions, with Tyr 7, Tyr 62, Trp 69, and Trp 156 forming an “aromatic cradle” that sequesters the ligand (Figures 2A,B) (31, 34). The physical interaction between the MR1 ligand-binding cleft and the ligand is consistent with the functional result in a mutagenesis study that shows the mutation of Tyr 7, Arg 9, and Arg 94 reduces MR1 expression and MAIT cell activation (Figure 2A) (84). By taking a similar mode of interaction with the MR1 protein, both bicyclic and monocyclic moieties of MR1 ligands occupy a small area of the MR1 ligand-binding cleft (Figures 2 and 3A) (36), although RL-6-Me-7-OH occupies a large region of the cavity, making numerous contacts within the cleft to correctly orient for T cell recognition in the absence of Schiff base bond formation (34).
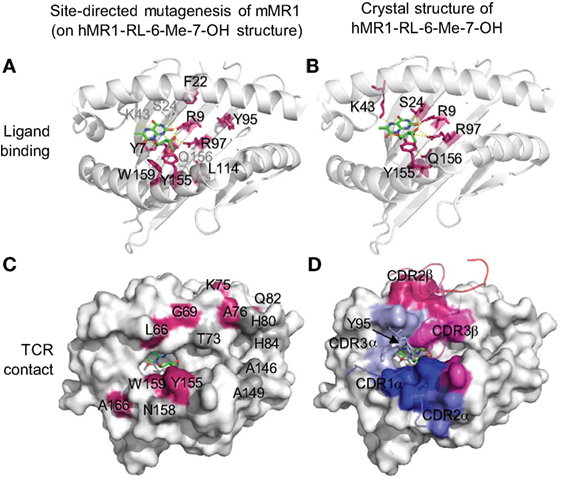
Figure 2. Ligand-binding and TCR contact of MR1 protein inferred from mutagenesis (84) and structural (34) studies. (A) Mutated mouse MR1 residues (84) are annotated on a human MR1 crystal structure (hMR1-RL-6-Me-7-OH) (34) to show the resulted functional impact on MR1 surface expression and mucosal-associated invariant T (MAIT) cell activation through predicted ligand interactions (green). Red: mutated residues on the β-sheet or proximal regions showing low MR1 expression and/or inhibited MAIT cell activation; gray: ligand-interacting residues not mutated. (B) Structural interactions between MR1 protein and the ligand RL-6-Me-7-OH (green) (34). (C) Mutated mouse MR1 residues (84) are annotated on hMR1 crystal structure (34) to show the resulted functional impact on T cell activation through predicted interaction with MAIT cell TCR. Red: mutated residues on helical regions showing inhibited MAIT cell activation; gray: mutated helical residues not showing functional impact. (D) Surface MR1 residues that structurally interact with corresponding complementarity-determining regions (CDRs) are shown with the same color (34).
Hydrophobic Anchor and Right-Sided Portal for CD1
CD1-lipid binding also shows a very different mechanism from the peptide antigen loading in classical MHC proteins. Lipid metabolites usually consist of aliphatic hydrocarbon chains present in the alkyl and polyketide tails with repeating methylene unites and hydrophilic head group (101, 136, 137). A lipid metabolite usually contacts with the non-polar residues on the inner surface of the CD1 ligand-binding cavity through non-specific hydrophobic interactions (138–140). With major aliphatic chains buried in the CD1 proteins, head groups of lipid antigens protrude out from the small portals to the right side of the CD1 ligand-binding cleft (Figure 3). Unlike the importance of the position of anchor residues for determining the orientation of peptide antigen binding to MHC molecules (141), the hydrophobic inner surfaces in CD1 proteins interact with proximal, center, and distal ends of the aliphatic chains of lipids biochemically independently of the position of the methylene unites. Although the binding of lipid ligands with CD1 proteins uses a more promiscuous, position-independent mode, the size of the clefts and individual pockets within the CD1 proteins places an upper limit on lipid antigen binding, as previously reviewed (142). However, this promiscuous interacting mode allows CD1 proteins to bind to a very broad spectrum of cellular lipids, leaving less restriction on lipid metabolite binding than on peptide binding to MHC molecules.
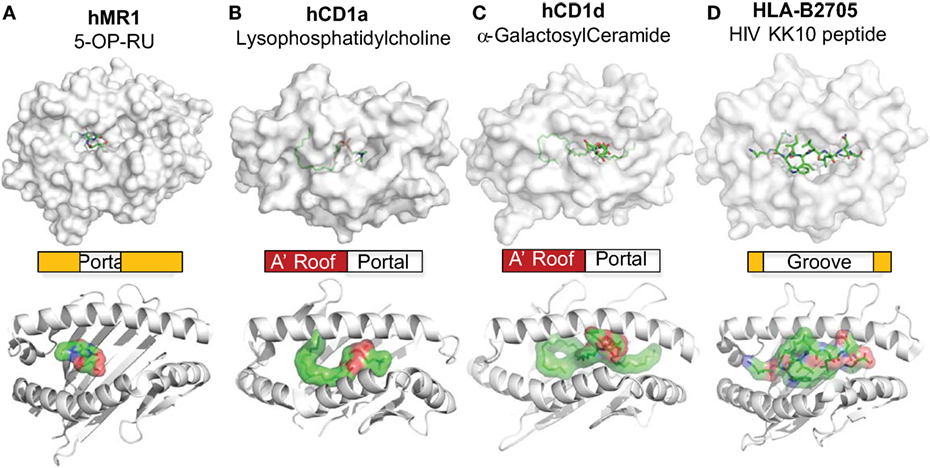
Figure 3. MR1 and CD1 ligand-binding clefts. The crystal structures of MR1 (A) (34), CD1a (B) (58), and CD1d (C) (134) molecules are shown with associated ligands (green) in comparison to the HLA-B2705 protein (D) (135). The positions of the portals for the MR1 and CD1 ligand-binding clefts are annotated. The shapes and relative sizes of the MR1 and CD1 ligand-binding clefts are also shown with ribbon view.
Dual Presentation of Self and Non-Self Ligands
Whereas most models of T-cell recognition emphasize TCR discrimination of self and foreign antigens, the innate immune system recognizes both self and foreign ligands. Similar to PRRs, which have evolved to bind pathogen-derived and stress-induced specific molecular patterns (94, 95), MHC class I-like molecules also bind both self and non-self ligands (Table 1). For example, the CD1a protein binds both mycobacterial DDM (24) and endogenous lysophosphatidylcholine (LPC) (58). CD1b autoreactive T cells have been shown to recognize phosphatidylglycerol (PG) lipids derived from mammalian cells, Salmonella, Staphylococcus, and other bacteria (61). Alternatively speaking, CD1b did not discriminate the structural differences that distinguish mammalian PG from bacterial PG or distinguish Salmonella PG from other bacterial PG, supporting the dual recognition of self and bacterial lipids by CD1b-restricted autoreactive T cells. The CD1c- and CD1d-mediated presentation of both self and non-self lipids has also been observed (Table 1) (51, 67, 143). Moreover, Qa1 protein, a mouse homolog of human HLA-E protein, binds signal peptides derived from self class I molecules as NK sensors for viral infection as well as GroEL peptides from Salmonella typhimurium or mycobacterial peptides to stimulate a cytotoxic T-lymphocyte response against infection (Table 1) (144, 145). Interestingly, these GroEL-specific cytotoxic T-lymphocytes cross-react with the stress-induced HSP60 peptide as a potential danger signal (144, 145). Moreover, mouse MAIT cells have been demonstrated to respond to both endogenous and exogenous antigens (84, 100, 146–148). Human MAIT cells are reactive to bacterial antigens in bacterial infection, which likely also induce endogenous antigens, similar to the activation of CD1d-restricted NKT cells (51). It appears very common for innate-like T cells to be reactive to both self and foreign antigens, supporting an innate-like feature of immune response.
Semi-Invariant TCR Beyond the Public TCR
TCRs of conventional T cells are expressed through rearrangement of the V, D, J, and C gene fragments of the β chain and the V, J, and C gene fragments of the α chain upon antigen stimulation. TCR gene rearrangement also accompanies T cell clonal expansion to proliferate a cluster of new T cells that express a rearranged TCR with specificity to bacterial priming antigens. While undergoing TCR rearrangement and clonal expansion in the priming stage, the primary response of conventional T cells is delayed, as in M. tuberculosis infection (Figure 1). However, public TCRs of conventional T cells can occur in small populations of donors sharing the same MHC allele in an infection of the same pathogen that expresses an immunodominant antigen (149). In this case, the same microbe must infect multiple individuals and prime T cell responses with the same antigens. These individuals should share similar HLA gene sequences to be able to present the same set of peptides. The generation of a public TCR in this setting is a highly coincident scenario and a rare event, as HLA alleles are highly variable in general human populations. Even though the public TCR is expressed, it yet exists in a very small human population. In contrast, the V and J rearrangements that define the MAIT and NKT TCRα chains are highly conserved in humans and mice. The invariant rearrangement of V and J gene fragments possibly occurs upon the stimulation of structurally conserved metabolites available prior to the infection of microbial pathogens. These semi-invariant TCRs are much more “popular” during the development and common in human populations than are the public TCRs (27, 150, 151). In addition to the public TCRs from conventional T cells, MAIT, NKT, GEM, and LDN5-like T cells express semi-invariant TCRs (Table 1). Unlike the fortuitously rearranged conventional TCRs, MAIT TCR is assembled by a canonical TRAV1-2-TRAJ33 (hVα7.2-Jα33 and mVα19-Jα33) chain paired with limitedly variable β chains TRBV6 and TRBV20 (114). The iNKT cell TCR is assembled by the TRAV10-TRAJ18 (hVα24-Jα18; mVα14-Jα18) chain mainly paired with TRBV25. Together with other invariant or biased TCR rearrangement (Table 1), MAIT and iNKT cells offer semi-invariant T cell receptors for the recognition of antigen-presenting molecules and antigens.
Limited Interference of Metabolite Antigens
The docking mode of classical MHC molecules and TCRs utilizes various degrees of diagonal orientation through recognition of both peptide antigens and MHC proteins, as defined by the disulfide bond between TCRα and β chains with respect to the α1 and α2 helixes (19, 152, 153). In the MHC–peptide–TCR tri-molecular interaction for the activation of conventional T cells, multiple complementarity-determining regions (CDRs) of TCR usually come into contact with residues of antigenic peptides (19, 153). However, most contacting sites from the TCRs of MAIT cells and lipid-specific T cells interact with the surface of MR1 or CD1 proteins rather than the antigens (Figures 2 and 4). Thus, in a very different manner, the TCRs of lipid-specific T cells use an asymmetric mode with little interference from associated lipid metabolites to dock on the surface of the CD1-lipid antigen complex as reviewed (91). Briefly, the lipid metabolites associated with CD1 proteins are either amphipathic or purely hydrophobic. The amphipathic lipids, such as phospholipids and glycosphingolipids, typically use the hydrophobic aliphatic chain to crawl into the ligand-binding cleft of CD1 proteins, with the hydrophilic head group protruding outside of the portal of the ligand-binding cleft for TCR interactions (Figures 3 and 4). The non-amphipathic lipid metabolites, such as wax ester and squalene, are likely fully embedded within the ligand-binding cleft of CD1a protein from direct interactions with TCRs (59). Interestingly, upon lipid association with CD1 proteins, the portals of the ligand-binding cleft usually open to the right side and form a laterally asymmetric displaying platform. On a large portion of the CD1 protein surface, an “A’ roof” forms to the left side to cover the A’ pocket of CD1 proteins (91). Therefore, the TCRs of lipid-specific T cells are able to utilize an interacting mode called “limited interference” to contact CD1 surfaces without an exposed lipid motif (Figure 4B). For example, the BK6 TCR binds on the left side of the CD1a platform and exclusively contacts the “A’ roof” of the CD1a protein, without contact with the associated LPC or fatty acids within the CD1a ligand-binding cleft (Figure 4B) (58). Alternatively, TCR interacts with the right side of the CD1 antigen-displaying platform TCR recognition of CD1d–α-galacosyldiacylglycerol (Figure 4C) (134) or CD1d–α-galacosylceramide (154). Both manners of interaction provide the major portion of TCR contacting surface with “limited interference” from the embedded lipid antigen underneath (91) (Figure 3).
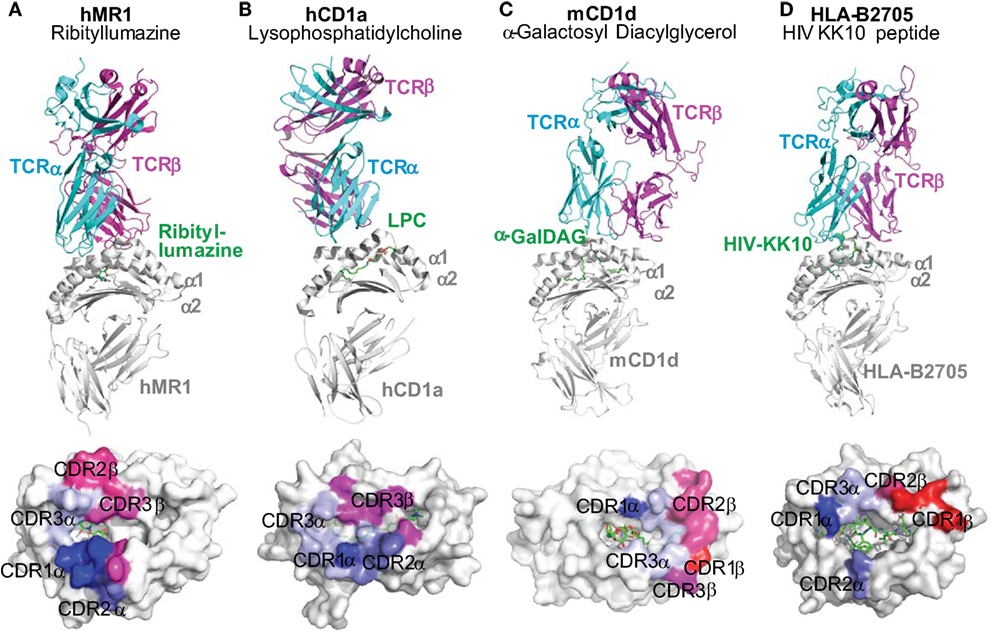
Figure 4. Docking mode of T cell receptors to MR1, CD1, and HLA-B2705 proteins. Tri-molecular interaction among an antigen-presenting molecule, an antigen, and a TCR in the activation of unconventional T cells is represented with the tri-molecular complexes of (A) human MR1-ribityllumazine–mucosal-associated invariant T cell TCR (34), (B) human CD1a–lysophosphatidylcholine–TCR of BK6 T cell (58), (C) mouse CD1d–α-galactosyl diacylglycerol (α-GalDAG)–iNKT TCR (134), and (D) HLA-B2705-KK10 peptide from human immunodeficiency virus Gag protein-TCR (135). The color-coated residues annotate the regions on the surface of antigen-presenting molecules to interact with CDR regions of TCRs (bluish colors for TCRα interactions and reddish colors for TCRβ interactions). TCRs from unconventional T cells more dominantly contact the surfaces of MR1 and CD1 proteins than the metabolite antigens. However, TCRs from conventional T cells recognize both HLA proteins and peptide antigens.
Similarly, the MAIT cell TCR mainly resides on the MR1 protein surface, although the direct interaction of MAIT TCR with the antigen appears critical. Human MR1/RL-6-Me-7-OH/MAIT TCR complex shows a hydrogen bond interaction between the Y95 residue at the TCR CDR3α region and the ribityl side chain of the antigen RL-6-Me-7-OH (Figure 2D and Figure 4A). The other stimulatory MAIT cell antigens with a ribityl side chain, such as 5-OP-RU and 5-OE-RU, also form a hydrogen bond with the TCR CDR3α region (34, 36, 155). The tri-molecular interaction has been further recapitulated with two additional xenoreactive human MR1/antigen/bovine MAIT TCR complexes (155, 156). Both ternary structures and functional studies suggest that the ribityl side chain is critical for TCR recognition and T cell activation (Figure 2D), as the ribityl side chain is unavailable to interact with MAIT cell TCR, 6-FP and Ac-6-FP are unable to directly contact TCRs and activate MAIT cells (37). It also appears that a Schiff base bond formation strengthens the binding of ribitylaminouracil (5-OE-RU and 5-OP-RU) to the MR1 protein and the tri-molecular interaction in TCR-5-OP-RU-MR1 and TCR-5-OE-RU-MR1 complexes, supporting a strong potency for MAIT cell activation (37).
Compared to the interaction between conventional TCR and peptidic antigens, TCR interaction with mostly embedded metabolite antigens is limited in terms of the number of contacting sites between TCR and antigens (Figure 4). Therefore, a “limited interference model” can be proposed to describe the degree of TCR–antigen interaction required for the activation of innate-like T cells. This model will not understate the importance of the interactions between TCR and some antigenic motifs, which are likely determinative for the activation of MAIT and iNKT cells by some antigens, such as the ribityl side chain for MAIT cell TCRs (37) and the galactose moiety for iNKT cell TCR (154). Functionally, a “limited interference model” better explains the quick activation kinetics in innate-like T cells, which are less dependent on the priming with structurally identical antigenic metabolites.
Low Antigen Discrimination in Tetramer Detection
It is known that the tetramers formulated with classical MHC proteins and bacterial peptide antigens usually detect antigen-specific conventional T cells upon the infection of the corresponding bacterial pathogens (3). In contrast to this highly specific reactivity of tetramer staining using classical MHC proteins and bacterial peptides, the tetramers of MHC class I-like proteins for detecting innate-like T cells are highly conserved and appear to act in an antigen non-discriminative manner. Conservation of the semi-invariant TCR of innate-like T cells is reflected by the positive detection of MAIT or iNKT cells in uninfected human individuals using MR1 or CD1d tetramers with bacterial metabolite antigens (44, 45). Recently, hMR1-rRL-6-CH2OH (32), MR1-5-OPRU, and MR1-5-OE-RU tetramers (36) were used to efficiently detect human MAIT cells in peripheral blood mononuclear cells (PBMCs). The detected MAIT cells were considered to cover most MAIT cells in humans or mice with or without exposure to antigen-producing bacterial pathogens (33, 157, 158). It has also been claimed that MR1-5-OP-RU and MR1-5-OE-RU tetramers stain all human MAIT cells in peripheral blood, possibly because of the high affinity of these pyrimidine metabolites to interact with both MR1 protein and MAIT TCRs (36). Similarly, CD1d–α-GalCer tetramer also detects iNKT cells in healthy mice from “naïve” T cell populations (82, 159, 160). The tetramer recognition of innate-like T cells in both healthy donors and tuberculosis patients raises an interesting question regarding whether the tetramer-positive T cells in healthy and infected individuals function differently. Since the stimulatory antigens for eliciting MAIT cell and NKT cell responses can be derived from different bacterial species (32, 33, 157), the ability of mycobacterial metabolites to induce protective antimycobacterial immune responses could feasibly be compared with that of other bacterial metabolites.
Xenoreactivity Beyond Alloreactivity
Cross-species activation confers an interesting innate-like manner of reactivity that is unlikely to exist in the adaptive immune system. In addition to broad alloreactivity in human populations, MHC class I-like molecules also generate very interesting cross-species reactivity in mammalian systems, as called xenoreactivity. Xenoreactivity is usually mediated by innate receptors such as NK cell receptors (161). These germline-encoded receptors are able to recognize conserved molecular motifs on targeted cells from different species; for example, human NK cells adhere to and lyse porcine cells (161). However, the activation of conventional T cells is determined by the recognition of both highly variable MHC molecules and heterogeneous peptide antigens (Figures 3D and 4D), which are unlikely to activate conventional T cells of a different species. By taking advantage of promiscuous and conserved tri-molecular interactions, MR1- and CD1-restricted T cells exhibit the interesting ability of cross-species activation. The xenoreactivity of T cells supports conserved structures and functions for antigen presentation (100). Indeed, the MR1 protein confers a high percentage of sequence homology among mammalian species, which is higher than 80% for rodents, bovines, and humans for the α1 and α2 domains of MR1 (100). Mouse MR1 is able to activate both mouse and human MAIT cells. Human MR1 with a single residue mutation to the corresponding residue in mouse MR1 (L151Q) is able to activate human MAIT cells (100). Moreover, the iNKTs in mice recognize the lipid antigens presented by human CD1d proteins. This is also true vice versa (162, 163), although CD1d sequences are not as homologous as MR1 proteins between mice and humans. Structural analyses of NKT TCRs responding to mouse and human CD1d–α-GalCer complex reveal that a contiguous CDR3β sequence is conserved between humans and mice to provide structural plasticity to accommodate a variety of glycolipid antigens presented by CD1d (164). Similarly, in comparison to conventional peptide-presented T cells, peptides presented by human HLA-E bind to the mouse homolog Qa-1b molecule (165).
Proliferation and Exhaustion
The available number of effector cells and the effectiveness of each effector are important criteria for estimating the final efficiency of immune cells in antimycobacterial immune defense. Conventional T cells specifically respond to bacterial peptide antigens and undergo clonal expansion to achieve multiple sets of functional capacity in immune defense, which include: (i) effector functions for cytokine production or cytolysis; (ii) memory functions for long-term antimicrobial immune responses; (iii) ability to amplify their cell number from an undetectable or absent state to high frequency; and (iv) ability to migrate from antigen-priming tissues to lesion tissues, for example, from lymph nodes to mycobacterial-infected alveolar tissues. A clonal expansion is usually required for conventional T cells to achieve a sufficient number of effector T cells upon antigen stimulation. In the case of aerosol infection with M. tuberculosis, mouse CD8+ T cells positive for the tetramer H-2Kb-TB10.3/10.4 will proliferate from undetectable to 1 million cells/lung, peak around 4 weeks after primary infection, and have an 8- to 12-fold increase in secondary challenge with the aerosol infection in comparison to the unchallenged (166). Thus, the clonal expansion of conventional T cells is vast upon antigen stimulation.
However, the cell frequency change of MAIT cells in M. tuberculosis-infected individuals is distinct from that of conventional CD8+ T cells. MAIT cells dramatically decrease in peripheral blood and are likely increased in lung tissues, as demonstrated in active M. tuberculosis-infected patients, suggesting the migration of activated MAIT cells to infected tissues (12). In Vα19iCα–/–MR1+/+ mice, MAIT cells accumulate three-fold more in BALF in comparison to that found in Vα19iCα–/–MR1–/– mice, as detected with MR1-rRL-6-Me-7-OH and MR1-5-OP-RU tetramers (33). With a high basal frequency of precursor MAIT cells prior to primary infection, the clonal expansion of MAIT cells upon mycobacterial infection appears much weaker than the clonal expansion of conventional T cells. Without undergoing potent clonal expansion, MAIT cells are ready to exhibit effector functions upon activation, although further expansion, migration, or reduction of precursor cells can occur.
Whether the lower frequency of MAIT cells in tuberculosis patients is attributable to MAIT cell exhaustion is an interesting question. When MAIT cells are analyzed in patients with pulmonary tuberculosis, tuberculous pleurisy, and tuberculous peritonitis by flow cytometry, a dramatically reduced MAIT cell number is usually observed. On the one hand, patients with active tuberculosis have a significantly higher production of cytokines IFN-γ and TNF-α from MAIT cells in responding to ex vivo BCG stimulation but not to E. coli stimulation, as compared to healthy control subjects (167). On the other hand, it is interesting that MAIT cells in patients with active tuberculosis exhibit an elevated expression of programed cell death protein-1 (PD-1), and the blockade of PD-1 signaling results in a significantly higher frequency of BCG-stimulated IFN-γ production from MAIT cells (167, 168). Whether the expression of PD-1 protein in MAIT cells indicates an exhaustion phenotype is controversial in regard to tuberculosis, particularly considering that PD-1–/– mice are sensitive to M. tuberculosis infection (169). However, the decreased frequency of MAIT cells may also be due to a decreased expression of CD161 in the existing MAIT cells rather than to an actual reduction in the MAIT cell number. Exhaustion or apoptosis in NKT cells has also been observed. The poor response to α-GalCer in M. tuberculosis-infected patients has been found to be due to increased NKT cell apoptosis, reduced CD1d expression, and a defect in NKT cells (170). Similarly, M. tuberculosis infection is associated with an elevated expression of the inhibitory PD-1 receptor on NKT cells, and the blockade of PD-1 signaling has been shown to enhance the response to α-GalCer.
Innate-Like T Cells in Bacterial Killing and Cytokine Production
Effector functions of MAIT cells contributing to protectivity against mycobacterial infections have been recently demonstrated upon MAIT cell activation (Figures 5A,B). MAIT cells isolated from the thymus and peripheral blood have shown the ability to kill mycobacterial-infected antigen-presenting cells in vitro. Pathogen-unexposed MAIT cells respond to lung epithelial cells infected with M. tuberculosis and produce tumor necrosis factor-α (Figure 5) (12, 86). As a dominant effector cytokine secreted by MAIT cells in most in vitro assays, TNF-α is an inflammatory cytokine and plays a critical protective role against mycobacterial infection, at least in the early stage of infection. The protectivity of TNF-α is supported by the fact that chemical blockers of TNF-α used for treating rheumatoid arthritis have caused the reactivation of tuberculosis (171). However, TNF-α also has the potential to worsen the inflammatory pathology at a later stage as a “double-edge sword.” Thus, maintaining an advantageous balance between TNF-α-mediated protective functions and pathogenic outcomes is critical in M. tuberculosis infections (172, 173). MAIT cells also express other pro-inflammatory cytokines, including IFN-γ and IL-17 (Figure 5B) (20, 30, 83). When pathogen-unexposed mouse MAIT cells are cocultured with BCG-infected macrophages, MAIT cells are quickly activated, as reflected by the production of large amounts of IFN-γ and IL-17a. Cytokines IFN-γ and IL-17a contribute to controlling BCG growth in macrophages, as supported by the effect of anti-IFN-γ and anti-IL-17a antibodies on impairing macrophage immunity (Figure 5B) (80). IFN-γ is also a critical antimycobacterial cytokine produced by conventional CD8+ T and CD4+ T cells (2) that has demonstrated a protective function in most stages of mycobacterial infections. While it is produced by MAIT cells, IL-17a is able to inhibit BCG growth in infected macrophages (83). However, the long-term impact of IL-17 in M. tuberculosis infections remains unclear (2). Moreover, cytotoxic reactivity allows MAIT cells to lyse Shigella flexneri-infected epithelial cell line HeLa cells (174) and M. semegmatis-infected lung epithelial A549 cells (175). MAIT cells upregulate the expression of perforin, granzyme B, and surface CD107a (LAMP1) upon the stimulation with anti-CD3 and CD28-coated beads, supporting their cytolytic function (Figure 5B) (174). Similar to NK and NKT cells, a high percentage of MAIT cells express the CD161 molecule, which is believed to modulate the cytokine response, such as IFN-γ, TNF-α, and IL-17 cytokine secretion (174, 176). But CD161 may not regulate the cytotoxic activity of MAIT cells (174). This is unlike the inhibitory effect of the CD161 molecule previously shown on NK and CD8+ T cells (177, 178). Overall, MAIT cells have a mixed Th1/Th17-associated pattern of cytokine production, including TNF-α, IFN-γ, and IL-17 at a striking level similar to conventional memory T cells, to inhibit mycobacterial growth in infected macrophages (30, 83). For in vivo mycobacterial infections in MR1 knockout or MAIT cell over-expressing mice, MAIT cells have shown the capacity to decrease bacterial loads (20, 33, 83, 158, 174), upregulate the Th1-, Th2-, or Th17-like cytokines, and enhance their cytotoxicity and frequency.
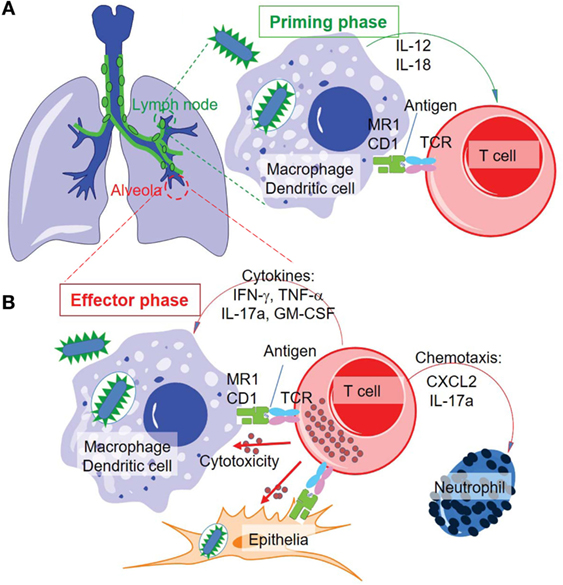
Figure 5. Proposed priming and effector phases of innate-like T cell responses in tuberculosis. (A) The priming phase may occur in the lymph nodes of lung tissues. Naïve or precursor innate-like T cells are activated through interaction with MR1 and CD1 proteins and/or stimulation of cytokines. (B) Activated innate-like T cells migrate to infected tissues, such as alveolar regions, to perform cytotoxic function and secrete cytokines and chemokines in anti-M. tuberculosis effector response.
Similarly, overall observations show that NKT cells have a decreased frequency in peripheral blood and a weaker response in human M. tuberculosis infection (170) and HIV infection (179). When cocultured with splenocytes isolated from uninfected mice, M. tuberculosis-infected macrophages suppress bacterial replication in macrophages dependent on the function of iNKT cells (81), which is similar to the suppression of BCG replication in macrophages by MAIT cells (80). Cytotoxicity, IFN-γ, and GM-CSF production have been shown upon activation of iNKT cells (81, 180). The adoptive transfer of naïve splenic iNKT cells from uninfected mice significantly reduces the bacterial burden in the lungs of infected mice (81). This effector function and protectivity does not require in vitro or in vivo stimulation of iNKT cells with a strong agonist of α-GalCer as shown previously (89). Thus, in therapeutic development, it would be interesting to further understand whether endogenous or mycobacterial lipid antigens are critical for CD1d-mediated iNKT cell effector function. In addition to the direct effect of bacterial inhibition and killing, the stimulation of HLA-E-restricted T cells and CD1d-restricted T cells is able to enhance the secretion of Th2 cytokines (IL-4, -5, -10, and -13), regulating B-cell activation and antibody production in antimycobacterial immune defense (181–183).
However, the role of lipid-specific T cells restricted by group I CD1 proteins in antimycobacterial effector responses is less understood. Several early studies showed that the frequency of group I CD1-restricted T cells increases in M. tuberculosis-infected individuals, suggesting the activation and expansion of M. tuberculosis-specific CD1-restricted T cells (23, 64, 184). The overall effector responses of lipid-specific T cells restricted by group I CD1 proteins are similar to those of iNKT and MAIT cells and are characterized by the production of cytokines IFN-γ and TNF-α upon stimulation with mycobacterial lipid antigens. Since mice do not express group I CD1 proteins, CD1a, CD1b, or CD1c, a study using a guinea pig model expressing human CD1b and CD1c homologs (185, 186) showed that immunization with mycobacterial lipids elicits the antigen-specific proliferation and cytotoxicity of group I CD1-restricted T cells (187) and minimizes the lung tissue damage induced by M. tuberculosis challenge (188). However, group I CD1 protein responses to M. tuberculosis infection exhibit relatively delayed kinetics in responding to mycobacterial lipid-pulsed mouse DCs, as shown using a humanized mouse model expressing human CD1a, CD1b, and CD1c proteins (90). This response peaks 7 days after immunization in unexposed mice, but it is difficult to compare this kinetics with those of conventional T cells because of the different immunization approaches used in various experiments. For example, the memory responses of DO11.10 T cells in mouse draining lymph nodes to the OVA peptide expressed in infected Salmonella typhimurium take about 5 days (189). Thus, whether group I CD1-restricted T cells utilize fast or slow kinetics in primary immune response remains unclear. Further, tetramer staining of group I CD1-restricted T cells seems to be a promising approach to determine the frequency and antigen specificity of T cells. Current reports of tetramer staining in infected and healthy human blood samples mostly show a minimal frequency of CD1a and CD1b tetramer-positive T cells (67, 190–192). The underlying mechanism may be multi-dimensional, for example, more immunodominant or conserved antigens may exist to detect a higher frequency of group I CD1-restricted T cells, or an antigen re-stimulation may be needed to expand the number of group I CD1-restricted T cells. However, CD1c loaded with phosphomycoketide lacking a carbohydrate head group is able to stain a high percentage of polyclonal T cells in the blood samples of donors with latent M. tuberculosis infection (67), suggesting the potential to detect a high frequency of some group I CD1-restricted T cells in patients.
Memory Phenotype
Memory T cells are T cells that can be quickly reactivated and become responsive to bacteria immediately after infections (193). The described relative quick kinetics for activated innate-like T cells in lung tissues is consistent with the memory phenotype of innate-like T cells. Although the kinetics of lung T cell activation can be impacted by the delayed or detrimental growth of M. tuberculosis in infection (194, 195), the memory phenotype of innate-like T cells can be still reflected by the expression of surface memory markers. It has been shown that these semi-invariant MAIT cells express memory markers (mainly CD45RAlowCD45ROhigh) in humans without additional in vitro stimulation (150). Further detailed phenotyping of MAIT cells using a MAIT cell over-expressing mouse model, Vα19+Ca−/−MR1+/+, has indicated that a high percentage of MAIT cells are CD44highCD45RBlowCD62LlowCD25high (196) upon TCR ligation with anti-CD3 and anti-CD28 antibodies. Recently, multiple reports further confirmed the expression of memory markers CD44highCD62Llow in mice (158) and the expression of CD45RA−CD45RO+CD95highCD62Llow, an effector-memory phenotype, in humans (30, 197, 198). Understanding the acquisition of this memory phenotype over the lifetime of humans is critical for determining the immune therapeutic strategy upon mycobacterial infection. In adults, MAIT cells have an effector-memory phenotype (largely CD45RA−CD45RO+CCR7lowCD62Llow) (199). In cord blood, MAIT cells express the markers of naïve cells (CD45RA+CD45RO−) (200, 201). These cells are also CCR7lowCD62Llow, suggesting that CD45RO positive expression is obtained after birth, which is consistent with the stepwise MAIT cell development facilitated by the maturation of gut microbiota (200). These critical observations of the acquisition of an effector-memory phenotype during the stepwise development of MAIT cells support the notion that antigen stimulation in the early lifetime facilitates the formation of the memory phenotype. It is likely that the microbial cyclic small molecules, such as riboflavin precursor metabolites defined at later time (31, 36), facilitate the process of memory formation. Similarly, NKT cells from the pleural fluid mononuclear cells of M. tuberculosis-infected patients also express CD45ROhighCD62LlowCCR7low that supports an effector memory phenotype for NKT cells (202).
The determination of whether microbial antigens are involved in the acquisition of the memory phenotype of MAIT cells helps in answering the question of whether MAIT cells are pre-activated by commonly distributed cross-species conserved antigens. This pre-activation is likely a major mechanism through which MAIT cells acquire an activated or memory phenotype and allows MAIT cells to be ready to respond to stimulation by different and unexposed pathogens. Similar or identical conserved antigens shared between gut microbiota and pathogens, such as riboflavin precursor metabolites, may be driving elements for the formation of memory or activated phenotypes of MAIT cells. Thus, the activated and memory phenotypes of MAIT cells in adults may have been shaped by the stimulation of microbial antigens during the early stage of individual development. The term “innate-like T cells” broadly defines the pre-activated memory phenotype of T cells and a quick activating kinetics upon encountering previously unexposed pathogens that potentially express metabolite antigens with the same or similar chemical structures to the “pre-activating” metabolites. The quick activation kinetics and memory phenotype of innate-like T cells suggest that the effector and regulatory functions of innate-like T cells may occur prior to the activation of the adaptive immune system (Figure 1).
Tissue Tropism in Early Defense
In M. tuberculosis infection, a rapid and regional immune response is important for containing the bacteria and infected cells in the initial stage of infection. Upon aerosol infection with M. tuberculosis, the acquired cellular immune responses are slow to be induced and take effect within the lung (2). This lagged period allows the invading slow-growing M. tuberculosis to grow and initiate conventional T cell activation. The stimulation of conventional T cells against M. tuberculosis requires the interaction of antigen-presenting cells, bacteria, and T cells in lung tissue or draining lymph nodes. Failure in this battle in lung tissue will lead to bacterial outgrowth, host pathology, and bacterial dissemination to distal tissues. In a beneficial response, around 8–9 days post mycobacterial infection, dendritic cells and macrophages will sample M. tuberculosis and migrate to draining lymph nodes (Figure 5A), and naïve T cells will be primed to proliferate and become effector cells. The activated effector cells will reversely migrate to the lung tissue at around 18–20 days post infection as a consequence of inflammatory responses to kill infected phagocytes and produce cytokines (2). MAIT cells have been shown to accumulate in lung tissues but decrease in peripheral blood in infected humans, indicating a mechanism allowing MAIT cell migration to lung tissues (12). Although the in vivo kinetics of MAIT cell stimulation upon mycobacterial infection are not clear, an initial assay using BCG-infected mice suggests that MAIT cells are critical for protecting mice from the high bacterial burden at day 10 but not at day 30 following infection (80), which is consistent with the protection conferred by the innate-like responding kinetics. Further in vivo studies are needed to determine the protection and early activation kinetics of MAIT cell responses to mycobacterial infections. In parallel, the role of group I CD1-restricted T cells will likely be further understood using animal models expressing group I CD1 proteins and having similar pathologies to mycobacterial infection in humans. Thus, the gap between the dissemination of bacteria and the onset of conventional T cell responses (2, 203, 204) is ideally filled by the responses of innate-like T cells. It seems promising that innate-like T cells could contain M. tuberculosis in the early stage of infection and contribute to a reduced rate of active disease in humans.
Therapeutic Value and Remaining Questions
Targeting innate-like T cell activation will provide novel therapeutics applicable to various human populations for controlling the early stage of mycobacterial infection, in a manner complementary to conventional T cell-based therapies. Multiple lines of evidence support the unique therapeutic values of innate-like T cells in tuberculosis. First, an effective early T cell response likely leads to a low lifetime risk of developing active tuberculosis (3, 10). Second, innate-like T cell populations, especially MAIT and iNKT cells are ideal candidates to offer a protective effect in the early stage of mycobacterial infections (83, 180, 205). Third, previous application of α-GalCer in the early stage of M. tuberculosis infection was shown to protect mycobacterial-infected mice against a high bacterial burden and severe pathology (89). This protective effect is encouraging for further investigations of other vaccine strategies based on innate-like T cells. For MAIT cells, the mouse study supports the protection of MAIT cells in M. abscessus-infected mice (20) and a newly published study shows the upregulation of cytotoxic MAIT cells in BCG-vaccinated macaque (206). In humans, high frequency of MAIT cells in latent infections of tuberculosis or healthy donors and low frequency in active tuberculosis support the association of high MAIT cell frequency with healthy conditions (12). Fourth, current vaccine candidates in the clinical trial pipeline mostly target mycobacterial secretory proteins and induce dominant responses by conventional CD4+ and CD8+ T cells (14). To overcome the variation in generating conventional T cell responses in large human populations, as demonstrated in a recent clinical trial in South Africa (207–209), innate-like T cells with donor-unrestricted features are expected to be applicable in most or all human populations if showing effectiveness in small groups of donors. Moreover, in comparison to these gene-based or microorganism-based vaccination strategies (14), the application of small metabolite antigens for activating innate-like T cells is expected to be safer, as naturally existing small molecules are not genetic materials that potentially induce inheritable complications. Metabolite-activated innate-like T cell are abundant T cell populations and the protein-based vaccines in clinical trial pipeline (16) will miss these abundant targets, demanding the need of new strategies for antimycobacterial vaccine design (89).
However, it is critical to further understand the in vivo responding kinetics and protectivity of innate-like T cells or donor-unrestricted T cells in controlling M. tuberculosis infections, especially chronic infections. Approaches are needed to improve the efficiency of the antimycobacterial T cell responses mediated by innate-like T cells, for example, reducing the frequency of exhausted MAIT cells in active tuberculosis. Moreover, it is important to understand whether M. tuberculosis expresses unique small molecule antigens for inducing different MAIT and iNKT cell responses, similar to those induced by other bacterial antigens. What other conserved or unique antigens are able to induce the protective function of these MR1- and CD1-restricted T cells? Answering these questions will facilitate targeting candidate metabolite antigens and innate-like T cells for developing novel vaccination and therapeutic strategies against tuberculosis.
Author Contribution
The author confirms being the sole contributor who analyzed the literature and wrote the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AK and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Funding
The author thanks National Institute of Allergy and Infectious Diseases (AI115358) and National Institute of Environmental Health Sciences (ES006096) Center for Environmental Genetics (CEG) for grant support.
References
1. Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol (2001) 19:93–129. doi:10.1146/annurev.immunol.19.1.93
2. Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol (2009) 27:393–422. doi:10.1146/annurev.immunol.021908.132703
3. Behar SM. Antigen-specific CD8(+) T cells and protective immunity to tuberculosis. Adv Exp Med Biol (2013) 783:141–63. doi:10.1007/978-1-4614-6111-1_8
4. Orme IM, Collins FM. Adoptive protection of the Mycobacterium tuberculosis-infected lung. Dissociation between cells that passively transfer protective immunity and those that transfer delayed-type hypersensitivity to tuberculin. Cell Immunol (1984) 84(1):113–20. doi:10.1016/0008-8749(84)90082-0
5. Tascon RE, Stavropoulos E, Lukacs KV, Colston MJ. Protection against Mycobacterium tuberculosis infection by CD8+ T cells requires the production of gamma interferon. Infect Immun (1998) 66(2):830–4.
6. Caruso AM, Serbina N, Klein E, Triebold K, Bloom BR, Flynn JL. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J Immunol (1999) 162(9):5407–16.
7. Flynn JL, Goldstein MM, Triebold KJ, Koller B, Bloom BR. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A (1992) 89(24):12013–7. doi:10.1073/pnas.89.24.12013
8. Behar SM, Dascher CC, Grusby MJ, Wang CR, Brenner MB. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. J Exp Med (1999) 189(12):1973–80. doi:10.1084/jem.189.12.1973
9. Urdahl KB, Liggitt D, Bevan MJ. CD8+ T cells accumulate in the lungs of Mycobacterium tuberculosis-infected Kb-/-Db-/- mice, but provide minimal protection. J Immunol (2003) 170(4):1987–94. doi:10.4049/jimmunol.170.4.1987
10. Selwyn PA, Hartel D, Lewis VA, Schoenbaum EE, Vermund SH, Klein RS, et al. A prospective study of the risk of tuberculosis among intravenous drug users with human immunodeficiency virus infection. N Engl J Med (1989) 320(9):545–50. doi:10.1056/NEJM198903023200901
11. Grotzke JE, Lewinsohn DM. Role of CD8+ T lymphocytes in control of Mycobacterium tuberculosis infection. Microbes Infect (2005) 7(4):776–88. doi:10.1016/j.micinf.2005.03.001
12. Gold MC, Cerri S, Smyk-Pearson S, Cansler ME, Vogt TM, Delepine J, et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol (2010) 8(6):e1000407. doi:10.1371/journal.pbio.1000407
13. Wallis RS, Hafner R. Advancing host-directed therapy for tuberculosis. Nat Rev Immunol (2015) 15(4):255–63. doi:10.1038/nri3813
14. Kaufmann SH. Tuberculosis vaccines: time to think about the next generation. Semin Immunol (2013) 25(2):172–81. doi:10.1016/j.smim.2013.04.006
15. Kaufmann SH, Hussey G, Lambert PH. New vaccines for tuberculosis. Lancet (2010) 375(9731):2110–9. doi:10.1016/S0140-6736(10)60393-5
16. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet (2013) 381(9871):1021–8. doi:10.1016/S0140-6736(13)60177-4
17. Nunes-Alves C, Booty MG, Carpenter SM, Jayaraman P, Rothchild AC, Behar SM. In search of a new paradigm for protective immunity to TB. Nat Rev Microbiol (2014) 12(4):289–99. doi:10.1038/nrmicro3230
18. Lin PL, Flynn JL. CD8 T cells and Mycobacterium tuberculosis infection. Semin Immunopathol (2015) 37(3):239–49. doi:10.1007/s00281-015-0490-8
19. Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. T cell antigen receptor recognition of antigen-presenting molecules. Annu Rev Immunol (2015) 33:169–200. doi:10.1146/annurev-immunol-032414-112334
20. Le Bourhis L, Martin E, Peguillet I, Guihot A, Froux N, Core M, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol (2010) 11(8):701–8. doi:10.1038/ni.1890
21. Napier RJ, Adams EJ, Gold MC, Lewinsohn DM. The role of mucosal associated invariant T Cells in antimicrobial immunity. Front Immunol (2015) 6:344. doi:10.3389/fimmu.2015.00344
22. Porcelli S, Morita CT, Brenner MB. CD1b restricts the response of human CD4-8- T lymphocytes to a microbial antigen. Nature (1992) 360(6404):593–7. doi:10.1038/360593a0
23. Ulrichs T, Moody DB, Grant E, Kaufmann SH, Porcelli SA. T-cell responses to CD1-presented lipid antigens in humans with Mycobacterium tuberculosis infection. Infect Immun (2003) 71(6):3076–87. doi:10.1128/IAI.71.6.3076-3087.2003
24. Moody DB, Young DC, Cheng TY, Rosat JP, Roura-Mir C, O’Connor PB, et al. T cell activation by lipopeptide antigens. Science (2004) 303(5657):527–31. doi:10.1126/science.1089353
25. Moody DB, Reinhold BB, Guy MR, Beckman EM, Frederique DE, Furlong ST, et al. Structural requirements for glycolipid antigen recognition by CD1b-restricted T cells. Science (1997) 278(5336):283–6. doi:10.1126/science.278.5336.283
26. Moody DB, Ulrichs T, Muhlecker W, Young DC, Gurcha SS, Grant E, et al. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature (2000) 404(6780):884–8. doi:10.1038/35009119
27. Porcelli S, Yockey CE, Brenner MB, Balk SP. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med (1993) 178(1):1–16. doi:10.1084/jem.178.1.1
28. de Lalla C, Lepore M, Piccolo FM, Rinaldi A, Scelfo A, Garavaglia C, et al. High-frequency and adaptive-like dynamics of human CD1 self-reactive T cells. Eur J Immunol (2011) 41(3):602–10. doi:10.1002/eji.201041211
29. Jeffery HC, van Wilgenburg B, Kurioka A, Parekh K, Stirling K, Roberts S, et al. Biliary epithelium and liver B cells exposed to bacteria activate intrahepatic MAIT cells through MR1. J Hepatol (2016) 64(5):1118–27. doi:10.1016/j.jhep.2015.12.017
30. Dusseaux M, Martin E, Serriari N, Peguillet I, Premel V, Louis D, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood (2011) 117(4):1250–9. doi:10.1182/blood-2010-08-303339
31. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature (2012) 491(7426):717–23. doi:10.1038/nature11605
32. Gold MC, McLaren JE, Reistetter JA, Smyk-Pearson S, Ladell K, Swarbrick GM, et al. MR1-restricted MAIT cells display ligand discrimination and pathogen selectivity through distinct T cell receptor usage. J Exp Med (2014) 211(8):1601–10. doi:10.1084/jem.20140507
33. Sakala IG, Kjer-Nielsen L, Eickhoff CS, Wang X, Blazevic A, Liu L, et al. Functional heterogeneity and antimycobacterial effects of mouse mucosal-associated invariant T cells specific for riboflavin metabolites. J Immunol (2015) 195(2):587–601. doi:10.4049/jimmunol.1402545
34. Patel O, Kjer-Nielsen L, Le Nours J, Eckle SB, Birkinshaw R, Beddoe T, et al. Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat Commun (2013) 4:2142. doi:10.1038/ncomms3142
35. Eckle SB, Birkinshaw RW, Kostenko L, Corbett AJ, McWilliam HE, Reantragoon R, et al. A molecular basis underpinning the T cell receptor heterogeneity of mucosal-associated invariant T cells. J Exp Med (2014) 211(8):1585–600. doi:10.1084/jem.20140484
36. Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature (2014) 509(7500):361–5. doi:10.1038/nature13160
37. Eckle SB, Corbett AJ, Keller AN, Chen Z, Godfrey DI, Liu L, et al. Recognition of vitamin B precursors and byproducts by mucosal associated invariant T cells. J Biol Chem (2015) 290(51):30204–11. doi:10.1074/jbc.R115.685990
38. Zhou D, Mattner J, Cantu C III, Schrantz N, Yin N, Gao Y, et al. Lysosomal glycosphingolipid recognition by NKT cells. Science (2004) 306(5702):1786–9. doi:10.1126/science.1103440
39. Shamshiev A, Donda A, Prigozy TI, Mori L, Chigorno V, Benedict CA, et al. The alphabeta T cell response to self-glycolipids shows a novel mechanism of CD1b loading and a requirement for complex oligosaccharides. Immunity (2000) 13(2):255–64. doi:10.1016/S1074-7613(00)00025-X
40. Facciotti F, Ramanjaneyulu GS, Lepore M, Sansano S, Cavallari M, Kistowska M, et al. Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat Immunol (2012) 13(5):474–80. doi:10.1038/ni.2245
41. Fox LM, Cox DG, Lockridge JL, Wang X, Chen X, Scharf L, et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol (2009) 7(10):e1000228. doi:10.1371/journal.pbio.1000228
42. Gumperz JE, Roy C, Makowska A, Lum D, Sugita M, Podrebarac T, et al. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity (2000) 12(2):211–21. doi:10.1016/S1074-7613(00)80174-0
43. Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, et al. The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian alpha-linked glycosylceramides. Immunity (2014) 41(4):543–54. doi:10.1016/j.immuni.2014.08.017
44. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science (1997) 278(5343):1626–9. doi:10.1126/science.278.5343.1626
45. Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, Kronenberg M, et al. Production of alpha-galactosylceramide by a prominent member of the human gut microbiota. PLoS Biol (2013) 11(7):e1001610. doi:10.1371/journal.pbio.1001610
46. Albacker LA, Chaudhary V, Chang YJ, Kim HY, Chuang YT, Pichavant M, et al. Invariant natural killer T cells recognize a fungal glycosphingolipid that can induce airway hyperreactivity. Nat Med (2013) 19(10):1297–304. doi:10.1038/nm.3321
47. Ito Y, Vela JL, Matsumura F, Hoshino H, Tyznik A, Lee H, et al. Helicobacter pylori cholesteryl alpha-glucosides contribute to its pathogenicity and immune response by natural killer T cells. PLoS One (2013) 8(12):e78191. doi:10.1371/journal.pone.0078191
48. Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol (2006) 7(9):978–86. doi:10.1038/ni1380
49. Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol (2011) 12(10):966–74. doi:10.1038/ni.2096
50. Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature (2005) 434(7032):520–5. doi:10.1038/nature03407
51. Mattner J, Debord KL, Ismail N, Goff RD, Cantu C III, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature (2005) 434(7032):525–9. doi:10.1038/nature03408
52. Sriram V, Du W, Gervay-Hague J, Brutkiewicz RR. Cell wall glycosphingolipids of Sphingomonas paucimobilis are CD1d-specific ligands for NKT cells. Eur J Immunol (2005) 35(6):1692–701. doi:10.1002/eji.200526157
53. Macho-Fernandez E, Brigl M. The extended family of CD1d-restricted NKT cells: sifting through a mixed bag of TCRs, antigens, and functions. Front Immunol (2015) 6:362. doi:10.3389/fimmu.2015.00362
54. Shamshiev A, Gober HJ, Donda A, Mazorra Z, Mori L, De Libero G. Presentation of the same glycolipid by different CD1 molecules. J Exp Med (2002) 195(8):1013–21. doi:10.1084/jem.20011963
55. Wu DY, Segal NH, Sidobre S, Kronenberg M, Chapman PB. Cross-presentation of disialoganglioside GD3 to natural killer T cells. J Exp Med (2003) 198(1):173–81. doi:10.1084/jem.20030446
56. Wolf BJ, Tatituri RV, Almeida CF, Le Nours J, Bhowruth V, Johnson D, et al. Identification of a potent microbial lipid antigen for diverse NKT cells. J Immunol (2015) 195(6):2540–51. doi:10.4049/jimmunol.1501019
57. Tatituri RV, Watts GF, Bhowruth V, Barton N, Rothchild A, Hsu FF, et al. Recognition of microbial and mammalian phospholipid antigens by NKT cells with diverse TCRs. Proc Natl Acad Sci U S A (2013) 110(5):1827–32. doi:10.1073/pnas.1220601110
58. Birkinshaw RW, Pellicci DG, Cheng TY, Keller AN, Sandoval-Romero M, Gras S, et al. αβ T cell antigen receptor recognition of CD1a presenting self lipid ligands. Nat Immunol (2015) 16(3):258–66. doi:10.1038/ni.3098
59. de Jong A, Cheng TY, Huang S, Gras S, Birkinshaw RW, Kasmar AG, et al. CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nat Immunol (2014) 15(2):177–85. doi:10.1038/ni.2790
60. Van Rhijn I, Kasmar A, de Jong A, Gras S, Bhati M, Doorenspleet ME, et al. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat Immunol (2013) 14(7):706–13. doi:10.1038/ni.2630
61. Van Rhijn I, van Berlo T, Hilmenyuk T, Cheng TY, Wolf BJ, Tatituri RV, et al. Human autoreactive T cells recognize CD1b and phospholipids. Proc Natl Acad Sci U S A (2016) 113(2):380–5. doi:10.1073/pnas.1520947112
62. Garcia-Alles LF, Collmann A, Versluis C, Lindner B, Guiard J, Maveyraud L, et al. Structural reorganization of the antigen-binding groove of human CD1b for presentation of mycobacterial sulfoglycolipids. Proc Natl Acad Sci U S A (2011) 108(43):17755–60. doi:10.1073/pnas.1110118108
63. Gilleron M, Stenger S, Mazorra Z, Wittke F, Mariotti S, Bohmer G, et al. Diacylated sulfoglycolipids are novel mycobacterial antigens stimulating CD1-restricted T cells during infection with Mycobacterium tuberculosis. J Exp Med (2004) 199(5):649–59. doi:10.1084/jem.20031097
64. Layre E, Collmann A, Bastian M, Mariotti S, Czaplicki J, Prandi J, et al. Mycolic acids constitute a scaffold for mycobacterial lipid antigens stimulating CD1-restricted T cells. Chem Biol (2009) 16(1):82–92. doi:10.1016/j.chembiol.2008.11.008
65. Sieling PA, Chatterjee D, Porcelli SA, Prigozy TI, Mazzaccaro RJ, Soriano T, et al. CD1-restricted T cell recognition of microbial lipoglycan antigens. Science (1995) 269(5221):227–30. doi:10.1126/science.7542404
66. Lepore M, de Lalla C, Gundimeda SR, Gsellinger H, Consonni M, Garavaglia C, et al. A novel self-lipid antigen targets human T cells against CD1c(+) leukemias. J Exp Med (2014) 211(7):1363–77. doi:10.1084/jem.20140410
67. Ly D, Kasmar AG, Cheng TY, de Jong A, Huang S, Roy S, et al. CD1c tetramers detect ex vivo T cell responses to processed phosphomycoketide antigens. J Exp Med (2013) 210(4):729–41. doi:10.1084/jem.20120624
68. Caccamo N, Pietra G, Sullivan LC, Brooks AG, Prezzemolo T, La Manna MP, et al. Human CD8 T lymphocytes recognize Mycobacterium tuberculosis antigens presented by HLA-E during active tuberculosis and express type 2 cytokines. Eur J Immunol (2015) 45(4):1069–81. doi:10.1002/eji.201445193
69. Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature (1994) 372(6507):691–4. doi:10.1038/372691a0
70. Ernst WA, Maher J, Cho S, Niazi KR, Chatterjee D, Moody DB, et al. Molecular interaction of CD1b with lipoglycan antigens. Immunity (1998) 8(3):331–40. doi:10.1016/S1074-7613(00)80538-5
71. Winslow GM, Cooper A, Reiley W, Chatterjee M, Woodland DL. Early T-cell responses in tuberculosis immunity. Immunol Rev (2008) 225:284–99. doi:10.1111/j.1600-065X.2008.00693.x
72. Sperber SJ, Gornish N. Reactivation of tuberculosis during therapy with corticosteroids. Clin Infect Dis (1992) 15(6):1073–4. doi:10.1093/clind/15.6.1073
73. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med (2001) 345(15):1098–104. doi:10.1056/NEJMoa011110
74. Wallgren A. The time-table of tuberculosis. Tubercle (1948) 29(11):245–51. doi:10.1016/S0041-3879(48)80033-4
75. Lara-Tejero M, Pamer EG. T cell responses to Listeria monocytogenes. Curr Opin Microbiol (2004) 7(1):45–50. doi:10.1016/j.mib.2003.12.002
76. Goldberg MF, Saini NK, Porcelli SA. Evasion of innate and adaptive immunity by Mycobacterium tuberculosis. Microbiol Spectr (2014) 2(5):1–24. doi:10.1128/microbiolspec.MGM2-0005-2013
77. Wang B, Primeau TM, Myers N, Rohrs HW, Gross ML, Lybarger L, et al. A single peptide-MHC complex positively selects a diverse and specific CD8 T cell repertoire. Science (2009) 326(5954):871–4. doi:10.1126/science.1177627
78. Zarling AL, Johnson JG, Hoffman RW, Lee DR. Induction of primary human CD8+ T lymphocyte responses in vitro using dendritic cells. J Immunol (1999) 162(9):5197–204.
79. Geijtenbeek TB, Van Vliet SJ, Koppel EA, Sanchez-Hernandez M, Vandenbroucke-Grauls CM, Appelmelk B, et al. Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med (2003) 197(1):7–17. doi:10.1084/jem.20021229
80. Chua WJ, Truscott SM, Eickhoff CS, Blazevic A, Hoft DF, Hansen TH. Polyclonal MAIT cells have unique innate functions in bacterial infection. Infect Immun (2012) 80(9):3256–67. doi:10.1128/IAI.00279-12
81. Sada-Ovalle I, Chiba A, Gonzales A, Brenner MB, Behar SM. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog (2008) 4(12):e1000239. doi:10.1371/journal.ppat.1000239
82. Metelitsa LS, Naidenko OV, Kant A, Wu HW, Loza MJ, Perussia B, et al. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J Immunol (2001) 167(6):3114–22. doi:10.4049/jimmunol.167.6.3114
83. Chua WJ, Truscott SM, Eickhoff CS, Blazevic A, Hoft DF, Hansen TH. Polyclonal mucosa-associated invariant T cells have unique innate functions in bacterial infection. Infect Immun (2012) 80(9):3256–67. doi:10.1128/IAI.00279-12
84. Huang S, Gilfillan S, Cella M, Miley MJ, Lantz O, Lybarger L, et al. Evidence for MR1 antigen presentation to mucosal-associated invariant T cells. J Biol Chem (2005) 280(22):21183–93. doi:10.1074/jbc.M501087200
85. Hansen TH, Huang S, Arnold PL, Fremont DH. Patterns of nonclassical MHC antigen presentation. Nat Immunol (2007) 8(6):563–8. doi:10.1038/ni1475
86. Gold MC, Eid T, Smyk-Pearson S, Eberling Y, Swarbrick GM, Langley SM, et al. Human thymic MR1-restricted MAIT cells are innate pathogen-reactive effectors that adapt following thymic egress. Mucosal Immunol (2013) 6(1):35–44. doi:10.1038/mi.2012.45
87. Alexander MA, Damico CA, Wieties KM, Hansen TH, Connolly JM. Correlation between CD8 dependency and determinant density using peptide-induced, Ld-restricted cytotoxic T lymphocytes. J Exp Med (1991) 173(4):849–58.
88. Serbina NV, Flynn JL. CD8(+) T cells participate in the memory immune response to Mycobacterium tuberculosis. Infect Immun (2001) 69(7):4320–8. doi:10.1128/IAI.69.7.4320-4328.2001
89. Chackerian A, Alt J, Perera V, Behar SM. Activation of NKT cells protects mice from tuberculosis. Infect Immun (2002) 70(11):6302–9. doi:10.1128/IAI.70.11.6302-6309.2002
90. Felio K, Nguyen H, Dascher CC, Choi HJ, Li S, Zimmer MI, et al. CD1-restricted adaptive immune responses to mycobacteria in human group 1 CD1 transgenic mice. J Exp Med (2009) 206(11):2497–509. doi:10.1084/jem.20090898
91. Huang S, Moody DB. Donor-unrestricted T cells in the human CD1 system. Immunogenetics (2016) 68(8):577–96. doi:10.1007/s00251-016-0942-x
92. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol (1989) 54(Pt 1):1–13. doi:10.1101/SQB.1989.054.01.003
93. Medzhitov R. Approaching the asymptote: 20 years later. Immunity (2009) 30(6):766–75. doi:10.1016/j.immuni.2009.06.004
94. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol (2001) 1(2):135–45. doi:10.1038/35100529
95. Matzinger P. The danger model: a renewed sense of self. Science (2002) 296(5566):301–5. doi:10.1126/science.1071059
96. Trinchieri G, Sher A. Cooperation of toll-like receptor signals in innate immune defence. Nat Rev Immunol (2007) 7(3):179–90. doi:10.1038/nri2038
97. Jaeger M, Stappers MH, Joosten LA, Gyssens IC, Netea MG. Genetic variation in pattern recognition receptors: functional consequences and susceptibility to infectious disease. Future Microbiol (2015) 10(6):989–1008. doi:10.2217/fmb.15.37
98. Azad AK, Sadee W, Schlesinger LS. Innate immune gene polymorphisms in tuberculosis. Infect Immun (2012) 80(10):3343–59. doi:10.1128/IAI.00443-12
99. Tiercy JM, Claas F. Impact of HLA diversity on donor selection in organ and stem cell transplantation. Hum Hered (2013) 76(3–4):178–86. doi:10.1159/000358798
100. Huang S, Martin E, Kim S, Yu L, Soudais C, Fremont DH, et al. MR1 antigen presentation to mucosal-associated invariant T cells was highly conserved in evolution. Proc Natl Acad Sci U S A (2009) 106(20):8290–5. doi:10.1073/pnas.0903196106
101. Van Rhijn I, Godfrey DI, Rossjohn J, Moody DB. Lipid and small-molecule display by CD1 and MR1. Nat Rev Immunol (2015) 15(10):643–54. doi:10.1038/nri3889
102. Parra-Cuadrado JF, Navarro P, Mirones I, Setien F, Oteo M, Martinez-Naves E. A study on the polymorphism of human MHC class I-related MR1 gene and identification of an MR1-like pseudogene. Tissue Antigens (2000) 56(2):170–2. doi:10.1034/j.1399-0039.2000.560211.x
103. Goldfinch N, Reinink P, Connelley T, Koets A, Morrison I, Van Rhijn I. Conservation of mucosal associated invariant T (MAIT) cells and the MR1 restriction element in ruminants, and abundance of MAIT cells in spleen. Vet Res (2010) 41(5):62. doi:10.1051/vetres/2010034
104. Aruffo A, Seed B. Expression of cDNA clones encoding the thymocyte antigens CD1a, b, c demonstrates a hierarchy of exclusion in fibroblasts. J Immunol (1989) 143(5):1723–30.
105. Zajonc DM, Crispin MD, Bowden TA, Young DC, Cheng TY, Hu J, et al. Molecular mechanism of lipopeptide presentation by CD1a. Immunity (2005) 22(2):209–19. doi:10.1016/j.immuni.2004.12.009
106. Seshadri C, Thuong NT, Yen NT, Bang ND, Chau TT, Thwaites GE, et al. A polymorphism in human CD1A is associated with susceptibility to tuberculosis. Genes Immun (2014) 15(3):195–8. doi:10.1038/gene.2014.5
107. Han M, Hannick LI, DiBrino M, Robinson MA. Polymorphism of human CD1 genes. Tissue Antigens (1999) 54(2):122–7. doi:10.1034/j.1399-0039.1999.540202.x
108. Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol (2009) 9(7):503–13. doi:10.1038/nri2575
109. Hansen TH, Connolly JM, Gould KG, Fremont DH. Basic and translational applications of engineered MHC class I proteins. Trends Immunol (2010) 31(10):363–9. doi:10.1016/j.it.2010.07.003
110. Miller JF. Immunological function of the thymus. Lancet (1961) 2(7205):748–9. doi:10.1016/S0140-6736(61)90693-6
111. Miller JF. Revisiting thymus function. Front Immunol (2014) 5:411. doi:10.3389/fimmu.2014.00411
112. Ben-Efraim S, Fuchs S, Sela M. Differences in immune response to synthetic antigens in two inbred strains of guinea-pigs. Immunology (1967) 12(5):573–81.
113. Bevan MJ. Interaction antigens detected by cytotoxic T cells with the major histocompatibility complex as modifier. Nature (1975) 256(5516):419–21. doi:10.1038/256419a0
114. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature (2003) 422(6928):164–9. doi:10.1038/nature01433
115. Reantragoon R, Kjer-Nielsen L, Patel O, Chen Z, Illing PT, Bhati M, et al. Structural insight into MR1-mediated recognition of the mucosal associated invariant T cell receptor. J Exp Med (2012) 209(4):761–74. doi:10.1084/jem.20112095
116. Le Bourhis L, Mburu YK, Lantz O. MAIT cells, surveyors of a new class of antigen: development and functions. Curr Opin Immunol (2013) 25(2):174–80. doi:10.1016/j.coi.2013.01.005
117. Soudais C, Samassa F, Sarkis M, Le Bourhis L, Bessoles S, Blanot D, et al. In vitro and in vivo analysis of the Gram-negative bacteria-derived riboflavin precursor derivatives activating mouse MAIT cells. J Immunol (2015) 194(10):4641–9. doi:10.4049/jimmunol.1403224
118. Bonomi HR, Marchesini MI, Klinke S, Ugalde JE, Zylberman V, Ugalde RA, et al. An atypical riboflavin pathway is essential for Brucella abortus virulence. PLoS One (2010) 5(2):e9435. doi:10.1371/journal.pone.0009435
119. Gutierrez-Preciado A, Torres AG, Merino E, Bonomi HR, Goldbaum FA, Garcia-Angulo VA. Extensive identification of bacterial riboflavin transporters and their distribution across bacterial species. PLoS One (2015) 10(5):e0126124. doi:10.1371/journal.pone.0126124
120. Mack M, van Loon AP, Hohmann HP. Regulation of riboflavin biosynthesis in Bacillus subtilis is affected by the activity of the flavokinase/flavin adenine dinucleotide synthetase encoded by ribC. J Bacteriol (1998) 180(4):950–5.
121. Roche PA, Cresswell P. Antigen processing and presentation mechanisms in myeloid cells. Microbiol Spectr (2016) 4(3):1–14. doi:10.1128/microbiolspec.MCHD-0008-2015
122. Huang S, Cheng TY, Young DC, Layre E, Madigan CA, Shires J, et al. Discovery of deoxyceramides and diacylglycerols as CD1b scaffold lipids among diverse groove-blocking lipids of the human CD1 system. Proc Natl Acad Sci U S A (2011) 108(48):19335–40. doi:10.1073/pnas.1112969108
123. Yuan W, Kang SJ, Evans JE, Cresswell P. Natural lipid ligands associated with human CD1d targeted to different subcellular compartments. J Immunol (2009) 182(8):4784–91. doi:10.4049/jimmunol.0803981
124. Cox D, Fox L, Tian R, Bardet W, Skaley M, Mojsilovic D, et al. Determination of cellular lipids bound to human CD1d molecules. PLoS One (2009) 4(5):e5325. doi:10.1371/journal.pone.0005325
125. De Libero G, Mori L. Novel insights into lipid antigen presentation. Trends Immunol (2012) 33(3):103–11. doi:10.1016/j.it.2012.01.005
126. An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell (2014) 156(1–2):123–33. doi:10.1016/j.cell.2013.11.042
127. Olszak T, Neves JF, Dowds CM, Baker K, Glickman J, Davidson NO, et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature (2014) 509(7501):497–502. doi:10.1038/nature13150
128. Telesford KM, Yan W, Ochoa-Reparaz J, Pant A, Kircher C, Christy MA, et al. A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and Treg function. Gut Microbes (2015) 6(4):234–42. doi:10.1080/19490976.2015.1056973
129. Mohan JF, Unanue ER. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat Rev Immunol (2012) 12(10):721–8. doi:10.1038/nri3294
130. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol (2013) 31:443–73. doi:10.1146/annurev-immunol-032712-095910
131. Madden DR, Gorga JC, Strominger JL, Wiley DC. The structure of HLA-B27 reveals nonamer self-peptides bound in an extended conformation. Nature (1991) 353(6342):321–5. doi:10.1038/353321a0
132. Miley MJ, Messaoudi I, Metzner BM, Wu Y, Nikolich-Zugich J, Fremont DH. Structural basis for the restoration of TCR recognition of an MHC allelic variant by peptide secondary anchor substitution. J Exp Med (2004) 200(11):1445–54. doi:10.1084/jem.20040217
133. Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol (2006) 6(4):271–82. doi:10.1038/nri1805
134. Li Y, Girardi E, Wang J, Yu ED, Painter GF, Kronenberg M, et al. The Valpha14 invariant natural killer T cell TCR forces microbial glycolipids and CD1d into a conserved binding mode. J Exp Med (2010) 207(11):2383–93. doi:10.1084/jem.20101335
135. Ladell K, Hashimoto M, Iglesias MC, Wilmann PG, McLaren JE, Gras S, et al. A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity (2013) 38(3):425–36. doi:10.1016/j.immuni.2012.11.021
136. Moody DB, Zajonc DM, Wilson IA. Anatomy of CD1-lipid antigen complexes. Nat Rev Immunol (2005) 5(5):387–99. doi:10.1038/nri1605
137. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13(2):101–17. doi:10.1038/nri3369
138. Zeng Z, Castano AR, Segelke BW, Stura EA, Peterson PA, Wilson IA. Crystal structure of mouse CD1: an MHC-like fold with a large hydrophobic binding groove. Science (1997) 277(5324):339–45. doi:10.1126/science.277.5324.339
139. Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI. Recognition of CD1d-restricted antigens by natural killer T cells. Nat Rev Immunol (2012) 12(12):845–57. doi:10.1038/nri3328
140. Zajonc DM, Girardi E. Recognition of microbial glycolipids by natural killer T cells. Front Immunol (2015) 6:400. doi:10.3389/fimmu.2015.00400
141. Stern LJ, Wiley DC. Antigenic peptide binding by class I and class II histocompatibility proteins. Behring Inst Mitt (1994) 94:1–10.
142. Ly D, Moody DB. The CD1 size problem: lipid antigens, ligands, and scaffolds. Cell Mol Life Sci (2014) 71(16):3069–79. doi:10.1007/s00018-014-1603-6
143. Mansour S, Tocheva AS, Cave-Ayland C, Machelett MM, Sander B, Lissin NM, et al. Cholesteryl esters stabilize human CD1c conformations for recognition by self-reactive T cells. Proc Natl Acad Sci U S A (2016) 113(9):E1266–75. doi:10.1073/pnas.1519246113
144. Soloski MJ, Metcalf ES. The involvement of class Ib molecules in the host response to infection with Salmonella and its relevance to autoimmunity. Microbes Infect (2001) 3(14–15):1249–59. doi:10.1016/S1286-4579(01)01485-X
145. Rodgers JR, Cook RG. MHC class Ib molecules bridge innate and acquired immunity. Nat Rev Immunol (2005) 5(6):459–71. doi:10.1038/nri1635
146. Young MH, U’Ren L, Huang S, Mallevaey T, Scott-Browne J, Crawford F, et al. MAIT cell recognition of MR1 on bacterially infected and uninfected cells. PLoS One (2013) 8(1):e53789. doi:10.1371/journal.pone.0053789
147. Chua WJ, Kim S, Myers N, Huang S, Yu L, Fremont DH, et al. Endogenous MHC-related protein 1 is transiently expressed on the plasma membrane in a conformation that activates mucosal-associated invariant T cells. J Immunol (2011) 186(8):4744–50. doi:10.4049/jimmunol.1003254
148. Huang S, Gilfillan S, Kim S, Thompson B, Wang X, Sant AJ, et al. MR1 uses an endocytic pathway to activate mucosal-associated invariant T cells. J Exp Med (2008) 205(5):1201–11. doi:10.1084/jem.20072579
149. Venturi V, Price DA, Douek DC, Davenport MP. The molecular basis for public T-cell responses? Nat Rev Immunol (2008) 8(3):231–8. doi:10.1038/nri2260
150. Tilloy F, Treiner E, Park SH, Garcia C, Lemonnier F, de la Salle H, et al. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med (1999) 189(12):1907–21. doi:10.1084/jem.189.12.1907
151. Boudinot P, Mondot S, Jouneau L, Teyton L, Lefranc MP, Lantz O. Restricting nonclassical MHC genes coevolve with TRAV genes used by innate-like T cells in mammals. Proc Natl Acad Sci U S A (2016) 113(21):E2983–92. doi:10.1073/pnas.1600674113
152. Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol (2006) 24:419–66. doi:10.1146/annurev.immunol.23.021704.115658
153. Castro CD, Luoma AM, Adams EJ. Coevolution of T-cell receptors with MHC and non-MHC ligands. Immunol Rev (2015) 267(1):30–55. doi:10.1111/imr.12327
154. Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature (2007) 448(7149):44–9. doi:10.1038/nature05907
155. Lopez-Sagaseta J, Dulberger CL, Crooks JE, Parks CD, Luoma AM, McFedries A, et al. The molecular basis for mucosal-associated invariant T cell recognition of MR1 proteins. Proc Natl Acad Sci U S A (2013) 110(19):E1771–8. doi:10.1073/pnas.1222678110
156. Lopez-Sagaseta J, Dulberger CL, McFedries A, Cushman M, Saghatelian A, Adams EJ. MAIT recognition of a stimulatory bacterial antigen bound to MR1. J Immunol (2013) 191(10):5268–77. doi:10.4049/jimmunol.1301958
157. Reantragoon R, Corbett AJ, Sakala IG, Gherardin NA, Furness JB, Chen Z, et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J Exp Med (2013) 210(11):2305–20. doi:10.1084/jem.20130958
158. Rahimpour A, Koay HF, Enders A, Clanchy R, Eckle SB, Meehan B, et al. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J Exp Med (2015) 212(7):1095–108. doi:10.1084/jem.20142110
159. Benlagha K, Weiss A, Beavis A, Teyton L, Bendelac A. In vivo identification of glycolipid antigen-specific T cells using fluorescent CD1d tetramers. J Exp Med (2000) 191(11):1895–903. doi:10.1084/jem.191.11.1895
160. Gumperz JE, Miyake S, Yamamura T, Brenner MB. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J Exp Med (2002) 195(5):625–36. doi:10.1084/jem.20011786
161. Forte P, Pazmany L, Matter-Reissmann UB, Stussi G, Schneider MK, Seebach JD. HLA-G inhibits rolling adhesion of activated human NK cells on porcine endothelial cells. J Immunol (2001) 167(10):6002–8. doi:10.4049/jimmunol.167.10.6002
162. Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med (1998) 188(8):1521–8. doi:10.1084/jem.188.8.1521
163. Brossay L, Kronenberg M. Highly conserved antigen-presenting function of CD1d molecules. Immunogenetics (1999) 50(3–4):146–51. doi:10.1007/s002510050590
164. Kjer-Nielsen L, Borg NA, Pellicci DG, Beddoe T, Kostenko L, Clements CS, et al. A structural basis for selection and cross-species reactivity of the semi-invariant NKT cell receptor in CD1d/glycolipid recognition. J Exp Med (2006) 203(3):661–73. doi:10.1084/jem.20051777
165. Kurepa Z, Hasemann CA, Forman J. Qa-1b binds conserved class I leader peptides derived from several mammalian species. J Exp Med (1998) 188(5):973–8. doi:10.1084/jem.188.5.973
166. Kamath A, Woodworth JS, Behar SM. Antigen-specific CD8+ T cells and the development of central memory during Mycobacterium tuberculosis infection. J Immunol (2006) 177(9):6361–9. doi:10.4049/jimmunol.177.9.6361
167. Jiang J, Wang X, An H, Yang B, Cao Z, Liu Y, et al. Mucosal-associated invariant T-cell function is modulated by programmed death-1 signaling in patients with active tuberculosis. Am J Respir Crit Care Med (2014) 190(3):329–39. doi:10.1164/rccm.201401-0106OC
168. Leeansyah E, Ganesh A, Quigley MF, Sonnerborg A, Andersson J, Hunt PW, et al. Activation, exhaustion, and persistent decline of the antimicrobial MR1-restricted MAIT-cell population in chronic HIV-1 infection. Blood (2013) 121(7):1124–35. doi:10.1182/blood-2012-07-445429
169. Lazar-Molnar E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, et al. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci U S A (2010) 107(30):13402–7. doi:10.1073/pnas.1007394107
170. Kee SJ, Kwon YS, Park YW, Cho YN, Lee SJ, Kim TJ, et al. Dysfunction of natural killer T cells in patients with active Mycobacterium tuberculosis infection. Infect Immun (2012) 80(6):2100–8. doi:10.1128/IAI.06018-11
171. Harris J, Keane J. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin Exp Immunol (2010) 161(1):1–9. doi:10.1111/j.1365-2249.2010.04146.x
172. Mootoo A, Stylianou E, Arias MA, Reljic R. TNF-alpha in tuberculosis: a cytokine with a split personality. Inflamm Allergy Drug Targets (2009) 8(1):53–62. doi:10.2174/187152809787582543
174. Le Bourhis L, Dusseaux M, Bohineust A, Bessoles S, Martin E, Premel V, et al. MAIT cells detect and efficiently lyse bacterially-infected epithelial cells. PLoS Pathog (2013) 9(10):e1003681. doi:10.1371/journal.ppat.1003681
175. Sharma PK, Wong EB, Napier RJ, Bishai WR, Ndung’u T, Kasprowicz VO, et al. High expression of CD26 accurately identifies human bacteria-reactive MR1-restricted MAIT cells. Immunology (2015) 145(3):443–53. doi:10.1111/imm.12461
176. Fergusson JR, Smith KE, Fleming VM, Rajoriya N, Newell EW, Simmons R, et al. CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell Rep (2014) 9(3):1075–88. doi:10.1016/j.celrep.2014.09.045
177. Germain C, Meier A, Jensen T, Knapnougel P, Poupon G, Lazzari A, et al. Induction of lectin-like transcript 1 (LLT1) protein cell surface expression by pathogens and interferon-gamma contributes to modulate immune responses. J Biol Chem (2011) 286(44):37964–75. doi:10.1074/jbc.M111.285312
178. Rosen DB, Cao W, Avery DT, Tangye SG, Liu YJ, Houchins JP, et al. Functional consequences of interactions between human NKR-P1A and its ligand LLT1 expressed on activated dendritic cells and B cells. J Immunol (2008) 180(10):6508–17. doi:10.4049/jimmunol.180.10.6508
179. Sandberg JK, Fast NM, Palacios EH, Fennelly G, Dobroszycki J, Palumbo P, et al. Selective loss of innate CD4(+) V alpha 24 natural killer T cells in human immunodeficiency virus infection. J Virol (2002) 76(15):7528–34. doi:10.1128/JVI.76.15.7528-7534.2002
180. Rothchild AC, Jayaraman P, Nunes-Alves C, Behar SM. iNKT cell production of GM-CSF controls Mycobacterium tuberculosis. PLoS Pathog (2014) 10(1):e1003805. doi:10.1371/journal.ppat.1003805
181. van Meijgaarden KE, Haks MC, Caccamo N, Dieli F, Ottenhoff TH, Joosten SA. Human CD8+ T-cells recognizing peptides from Mycobacterium tuberculosis (Mtb) presented by HLA-E have an unorthodox Th2-like, multifunctional, Mtb inhibitory phenotype and represent a novel human T-cell subset. PLoS Pathog (2015) 11(3):e1004671. doi:10.1371/journal.ppat.1004671
182. Wu C, Li Z, Fu X, Yu S, Lao S, Yang B. Antigen-specific human NKT cells from tuberculosis patients produce IL-21 to help B cells for the production of immunoglobulins. Oncotarget (2015) 6(30):28633–45. doi:10.18632/oncotarget.5764
183. Joosten SA, van Meijgaarden KE, van Weeren PC, Kazi F, Geluk A, Savage ND, et al. Mycobacterium tuberculosis peptides presented by HLA-E molecules are targets for human CD8 T-cells with cytotoxic as well as regulatory activity. PLoS Pathog (2010) 6(2):e1000782. doi:10.1371/journal.ppat.1000782
184. Sieling PA, Jullien D, Dahlem M, Tedder TF, Rea TH, Modlin RL, et al. CD1 expression by dendritic cells in human leprosy lesions: correlation with effective host immunity. J Immunol (1999) 162(3):1851–8.
186. Dascher CC, Hiromatsu K, Naylor JW, Brauer PP, Brown KA, Storey JR, et al. Conservation of a CD1 multigene family in the guinea pig. J Immunol (1999) 163(10):5478–88.
187. Hiromatsu K, Dascher CC, LeClair KP, Sugita M, Furlong ST, Brenner MB, et al. Induction of CD1-restricted immune responses in guinea pigs by immunization with mycobacterial lipid antigens. J Immunol (2002) 169(1):330–9. doi:10.4049/jimmunol.169.1.330
188. Dascher CC, Hiromatsu K, Xiong X, Morehouse C, Watts G, Liu G, et al. Immunization with a mycobacterial lipid vaccine improves pulmonary pathology in the guinea pig model of tuberculosis. Int Immunol (2003) 15(8):915–25. doi:10.1093/intimm/dxg091
189. Chen ZM, Jenkins MK. Clonal expansion of antigen-specific CD4 T cells following infection with Salmonella typhimurium is similar in susceptible (Itys) and resistant (Ityr) BALB/c mice. Infect Immun (1999) 67(4):2025–9.
190. Kasmar AG, van Rhijn I, Cheng TY, Turner M, Seshadri C, Schiefner A, et al. CD1b tetramers bind {alpha}{beta} T cell receptors to identify a mycobacterial glycolipid-reactive T cell repertoire in humans. J Exp Med (2011) 208(9):1741–7. doi:10.1084/jem.20110665
191. Kasmar AG, Van Rhijn I, Magalhaes KG, Young DC, Cheng TY, Turner MT, et al. Cutting Edge: CD1a tetramers and dextramers identify human lipopeptide-specific T cells ex vivo. J Immunol (2013) 191(9):4499–503. doi:10.4049/jimmunol.1301660
192. Van Rhijn I, Gherardin NA, Kasmar A, de Jager W, Pellicci DG, Kostenko L, et al. TCR bias and affinity define two compartments of the CD1b-glycolipid-specific T cell repertoire. J Immunol (2014) 192(9):4054–60. doi:10.4049/jimmunol.1400158
193. Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol (2016) 16(2):102–11. doi:10.1038/nri.2015.10
194. Jeong YH, Jeon BY, Gu SH, Cho SN, Shin SJ, Chang J, et al. Differentiation of antigen-specific T cells with limited functional capacity during Mycobacterium tuberculosis infection. Infect Immun (2014) 82(1):132–9. doi:10.1128/IAI.00480-13
195. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity (2007) 27(2):281–95. doi:10.1016/j.immuni.2007.07.010
196. Kawachi I, Maldonado J, Strader C, Gilfillan S. MR1-restricted V alpha 19i mucosal-associated invariant T cells are innate T cells in the gut lamina propria that provide a rapid and diverse cytokine response. J Immunol (2006) 176(3):1618–27. doi:10.4049/jimmunol.176.3.1618
197. Slichter CK, McDavid A, Miller HW, Finak G, Seymour BJ, McNevin JP, et al. Distinct activation thresholds of human conventional and innate-like memory T cells. JCI Insight (2016) 1(8):e86292. doi:10.1172/jci.insight.86292
198. Gherardin NA, Keller AN, Woolley RE, Le Nours J, Ritchie DS, Neeson PJ, et al. Diversity of T cells restricted by the MHC class I-related molecule MR1 facilitates differential antigen recognition. Immunity (2016) 44(1):32–45. doi:10.1016/j.immuni.2015.12.005
199. Billerbeck E, Kang YH, Walker L, Lockstone H, Grafmueller S, Fleming V, et al. Analysis of CD161 expression on human CD8+ T cells defines a distinct functional subset with tissue-homing properties. Proc Natl Acad Sci U S A (2010) 107(7):3006–11. doi:10.1073/pnas.0914839107
200. Martin E, Treiner E, Duban L, Guerri L, Laude H, Toly C, et al. Stepwise development of MAIT cells in mouse and human. PLoS Biol (2009) 7(3):e54. doi:10.1371/journal.pbio.1000054
201. Walker LJ, Kang YH, Smith MO, Tharmalingham H, Ramamurthy N, Fleming VM, et al. Human MAIT and CD8αα cells develop from a pool of type-17 precommitted CD8+ T cells. Blood (2012) 119(2):422–33. doi:10.1182/blood-2011-05-353789
202. Li Z, Yang B, Zhang Y, Ma J, Chen X, Lao S, et al. Mycobacterium tuberculosis-specific memory NKT cells in patients with tuberculous pleurisy. J Clin Immunol (2014) 34(8):979–90. doi:10.1007/s10875-014-0090-8
203. Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, et al. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med (2008) 205(1):105–15. doi:10.1084/jem.20071367
204. Reiley WW, Calayag MD, Wittmer ST, Huntington JL, Pearl JE, Fountain JJ, et al. ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proc Natl Acad Sci U S A (2008) 105(31):10961–6. doi:10.1073/pnas.0801496105
205. Behar SM, Carpenter SM, Booty MG, Barber DL, Jayaraman P. Orchestration of pulmonary T cell immunity during Mycobacterium tuberculosis infection: immunity interruptus. Semin Immunol (2014) 26(6):559–77. doi:10.1016/j.smim.2014.09.003
206. Greene JM, Dash P, Roy S, McMurtrey C, Awad W, Reed JS, et al. MR1-restricted mucosal-associated invariant T (MAIT) cells respond to mycobacterial vaccination and infection in nonhuman primates. Mucosal Immunol (2016). doi:10.1038/mi.2016.91
207. Pollock KM, Whitworth HS, Montamat-Sicotte DJ, Grass L, Cooke GS, Kapembwa MS, et al. T-cell immunophenotyping distinguishes active from latent tuberculosis. J Infect Dis (2013) 208(6):952–68. doi:10.1093/infdis/jit265
208. Lindestam Arlehamn CS, Gerasimova A, Mele F, Henderson R, Swann J, Greenbaum JA, et al. Memory T cells in latent Mycobacterium tuberculosis infection are directed against three antigenic islands and largely contained in a CXCR3+CCR6+ Th1 subset. PLoS Pathog (2013) 9(1):e1003130. doi:10.1371/journal.ppat.1003130
209. Rahman S, Gudetta B, Fink J, Granath A, Ashenafi S, Aseffa A, et al. Compartmentalization of immune responses in human tuberculosis: few CD8+ effector T cells but elevated levels of FoxP3+ regulatory t cells in the granulomatous lesions. Am J Pathol (2009) 174(6):2211–24. doi:10.2353/ajpath.2009.080941
Keywords: vaccine, innate-like T cells, Mycobacterium tuberculosis, lipid, riboflavin metabolites, CD1, MR1, antigen presentation
Citation: Huang S (2016) Targeting Innate-Like T Cells in Tuberculosis. Front. Immunol. 7:594. doi: 10.3389/fimmu.2016.00594
Received: 30 September 2016; Accepted: 29 November 2016;
Published: 21 December 2016
Edited by:
Jeffrey K. Actor, University of Texas Health Science Center at Houston, USAReviewed by:
Arshad Khan, University of Texas Health Science Center at Houston, USABuka Samten, University of Texas Health Science Center at Tyler, USA
Copyright: © 2016 Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shouxiong Huang, c2hvdXhpb25nLmh1YW5nQHVjLmVkdQ==