 Emily M. Mace
Emily M. Mace Jordan S. Orange
Jordan S. Orange- Center for Human Immunobiology, Baylor College of Medicine and Texas Children’s Hospital, Houston, TX, USA
Human NK cells play critical roles in human host defense, particularly the control of viral infection and malignancy, and patients with congenital immunodeficiency affecting NK cell function or number can suffer from severe illness. The importance of NK cell function is particularly underscored in patients with primary immunodeficiency in which NK cells are the primary or sole affected population (NK cell deficiency, NKD). While NKD may lead to the absence of NK cells, we are also gaining an increasing appreciation of the effect that NKD may have on the generation of specific NK cell subsets. In turn, this leads to improved insights into the requirements for human NK cell subset generation, as well as their importance in immune homeostasis. The presence of inherently abnormally developed or functionally impaired NK cells, in particular, appears to be problematic in the way of interfering with normal human host defense and may be more impactful than low numbers of NK cells alone. Here, we review the known genetic causes of NKD and the insight that is derived by these into the requirements for human subset generation and, by extension, for NK cell-mediated immunity.
Introduction
While human NK cells and NK cell subsets have been well described, understanding the true physiologic role that they serve in humans has been difficult. The heterogeneity of NK cells as a result of both genetic and environmental diversity, as well as the divergence in phenotypic markers between mouse and man has made advances in the field incremental. An important tool in the understanding of human NK cells and NK cell subsets has been the discovery of human inborn genetic immunodeficiency diseases (primary immunodeficiencies) that affect the generation or homeostasis of NK cells or specific NK cell subsets as well as those that affect NK cell function. There are over 300 genetic deficiencies of human immunity, and nearly 50 are known to have at least some impact on NK cells. This latter category has been reviewed by a number of authors in recent years (1–4). Five of these primary immunodeficiencies, however, may have their primary impact upon NK cells and are known as NK cell deficiency (NKD) (these are summarized in Table 1). These have provided insight into the requirements for human NK cell maturation, the importance of the successful execution of NK cell development, and the overall value of certain NK cell functions. Here, we will describe recent advances in our understanding of the importance of human NK cell subsets and NK cell function as informed by NKD.
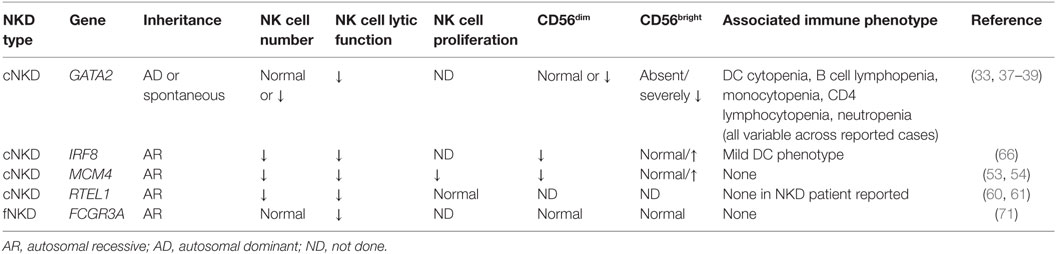
Table 1. Human NK cell deficiencies and their effect on NK cell subsets, function, and proliferation.
Human NK Cell Subsets
Human NK cells in peripheral blood are broadly classified as being CD56+CD3− and generally comprise 1–17% of mononuclear cells within peripheral blood of healthy adults (5). They are found primarily in two broadly defined phenotypic subsets, namely the CD56bright and CD56dim subsets, with the CD56dim subset being the predominant one in peripheral blood. While the basis for this stratification is the surface density of CD56 itself, each is a distinct functional subset with its own repertoire of markers that represent its capabilities. CD56bright NK cells are broadly defined as being the potent producers of cytokines, whereas the CD56dim subset is the classical mediator of contact-dependent cytotoxicity, both natural and antibody-dependent. However, exceptions to these rules exist, and there is evidence that both subsets can perform the tasks commonly attributed to the other (6, 7). In addition, there are several well-defined functional and phenotypic intermediaries between the two, as well as several subsets that are specifically expanded after infection or other immune stimulation. These include the CD56neg subset, which is commonly found expanded in patients with HIV and HCV (8, 9). It should be noted that these cells are not truly CD56 negative, although the expression of CD56 on them is significantly lower than that of the CD56dim subset. They retain expression of CD16, killer cell immunoglobulin receptors (KIR, CD158 family), and CD57 and are generally thought to be terminally differentiated or exhausted cells (10). An additional important subset expanded in disease is the adaptive NKG2C+ population expanded following certain viral infections, particularly CMV (11–13), but also in cases of reinfection of CMV seropositive individuals by hantavirus (14), HIV (15, 16), Epstein–Barr Virus (17), Hepatitis C Virus (18), and Chikungunya virus (19). Further studies have revealed distinct subsets associated with the generation of adaptive NK cells following CMV infection. These cells are high affinity IgE receptor (FcɛRIγ)-deficient and express low amounts of Syk and/or EWS/FLI1-activated transcript 2 (EAT2) (20, 21). Further adaptation of the NK cell repertoire occurs with epigenetic silencing of promyelocytic leukemia zinc finger (PLZF) (22). There is also remarkable diversity generated by both environmental and genetic factors within the human population as a whole with regards to NK cell receptor expression and functional potential (23, 24).
The relationship between CD56bright and CD56dim subsets is not entirely clear, although it is thought that they represent the two terminally mature populations of NK cells derived from less mature NK cell precursors. There are oft-cited lines of evidence suggesting that CD56bright NK cells are the direct precursors of the CD56dim subset, namely the longer telomeres in CD56bright NK cells (25), their earlier regeneration after HSCT (26), and the generation of CD56dim NK cells from CD56bright cells in certain experimental systems (27–29). The expression of CD56 on NK cells is unique to human and non-human primate systems, which has made the study of this specific aspect of NK cell development difficult to model prior to the advent of humanized mouse models. The use of such humanized mice, in which human NK cells develop in a rodent system, showed that CD56bright NK cells give rise to CD56dim cells (29); however, a recent study using genetic bar coding of NK cell lineages in rhesus macaques was indicative of the two subsets having distinct ontologies (30). Ultimately, answering this important question for NK cell biology will require a dissection of the mechanism that drives NK cell development using a satisfying human model. The study of primary human NKDs that affect NK cell subset generation are an informative tool in this endeavor, specifically for identifying novel requirements for the generation and homeostasis of human NK cell subsets (discussed further below). While complex, these disorders also provide insight into the functions of particular NK cell subsets in humans, as well as the overall value of NK cells in human host defense.
Classical NKD
Classical (c)NKD refers to NKD as a result of absent or profoundly decreased CD56+ CD3− NK cell number, specifically defined as ≤1% of peripheral lymphocytes (3, 31). As a result, these are disorders in which overall NK cell development and/or homeostasis is impaired. Recent studies enabled by next generation sequencing techniques have identified genetic causes for previously described but unidentified cNKD. In combination with these genetic advances, we have also seen an increase in our understanding of NK cell subsets and the cell biological tools used to evaluate them. As a result, we can now appreciate that cNKD also includes cases in which human peripheral blood NK cell subsets are selectively underrepresented or absent. Thus, while cNKD has been historically used to define individuals with very low percentages of NK cells among peripheral blood lymphocytes, we would propose that this definition be expanded to include any example of impaired development or survival leading to the gross underrepresentation of a major NK cell subset. We would also argue that abnormally developed NK cells or the absence of particular NK cell subsets in the presence of others is far more relevant to host defense than simply having low overall numbers of NK cells.
To date, there are four genetically defined reported causes of cNKD, and of these at least three (MCM4, GATA2, IRF8) contain an aspect of subset aberration, in addition to NK cell numbers ranging from the low end of normal to undetectable. The fourth (RTEL1) presents information too limited to make a definitive statement other than there being underrepresented NK cells seemingly in isolation from other immune defects. In some cases, namely GATA2 deficiency, a single gene defect may account for a widely variable clinical phenotype with regards to NK cell number. This likely is related to the effect of specific mutations but may also be reflective of innate plasticity of NK cell numbers and subsets over time, in even healthy individuals, and the infectious history of the patient (5, 23, 24).
GATA2
GATA2 deficiency is the genetic lesion behind the most frequently cited case of classic NKD in the literature. A 13-year-old girl, originally presenting with varicella pneumonia, was found to have decreased NK cell number and function in the presence of T cell and B cell function (32). In addition to VZV, CMV and HSV infections followed, and she ultimately developed aplastic anemia and died during the course of hematopoietic stem cell transplantation (32, 33). Posthumous sequencing from a cryopreserved T cell line identified a pathogenic frameshift GATA mutation (c.1025_1026insGCCG; p.A342GfsX41), confirming GATA2 haploinsufficiency as the cause of her disease (33). GATA2 haploinsufficiency was also described as a cause of DC and monocyte deficiency (MonoMAC, DCML) by independent groups in 2009, and NK cell cytopenia was noted as a feature of disease (34–36). More detailed phenotyping has revealed that there is variation in the NK cell number in these patients, with many of them having less than 1% NK cells within their lymphocyte population. However, there is a substantive range in NK cell number, with some patients having NK cells within the normal range of healthy donors. Despite the presence of NK cells in some patients, a consistent and notable feature of GATA2 deficiency is the absolute loss of the CD56bright NK cell subset (33). Originally described in a cohort of eight patients, this has since been reported by other groups (37, 38) and is a consistent feature of the >30 patients that we have studied with GATA2 deficiency and accompanying infectious history or hematologic disease.
While it is difficult to estimate the prevalence of GATA2 mutation within the general population, the report of a cohort of 57 patients, of which 82% have clinically detected NK cell lymphopenia and 70% have severe and early onset viral infection suggestive of NK cell dysfunction, is the largest group of NKD patients that we are aware of (39). This suggests it is perhaps the most common NKD, as this cohort alone significantly outnumbers those reported with RTEL1, MCM4, IRF8, and FCGR3A mutations. Furthermore, GATA2 is also the only gene associated with NKD that is autosomal dominant (via haploinsufficiency) and the only for which spontaneous cases have been reported (35, 36), thus supporting the statement of commonality from a genetic standpoint as well.
GATA2 deficiency is complex as it can be a multi-syndromic disease affecting multiple organs and presenting in multiple different ways. Patients can be susceptible to atypical mycobacterial infections, fungal infections, and severe and recalcitrant viral infections. Interestingly, there is a seemingly progressive nature of the disease, with many patients presenting in young adulthood and some even later in life. The range of clinical presentations and natural history has been well described elsewhere (39). Interestingly, one of the earliest clinical features to appear is a susceptibility to HPV disease, which could point to a role for NK cell-mediated defenses. In addition to immune deficiency, GATA2 deficiency is a cause of familial bone marrow failure (40), and a recent study of over 400 children and adolescents revealed GATA2 mutation to be the most common germline mutation leading to myelodysplastic syndrome in children and young adults (41). While this group did not specifically examine NKD in their cohort, they report half of their patients to have immunodeficiency, suggesting that the high rates of NK cell cytopenias reported by Spinner et al. (39) are likely present in this group.
However, there are some notable aspects of GATA2 deficiency with regards to NK cell biology. While other NK cell deficiencies have been reported as affecting the frequency of subset distribution (particularly MCM4, described below), the loss of the CD56bright NK cell subset is as near to absolute as has been described. The mechanism by which GATA2 regulates the development or maintenance of the CD56bright NK cell pool is not understood. GATA2 is a zinc finger transcription factor that is required for embryonic hematopoiesis, and maintenance of the stem cell pool in adults (42, 43) and GATA2 haploinsufficiency can also lead to loss of dendritic cell subsets and B cell cytopenias. Therefore, given its important role in stem cell maintenance and hematopoiesis, GATA2 deficiency may affect multiple immune cell lineages, which are again highly variable from patient to patient. In vitro differentiation of NK cells from patient hematopoietic stem cells leads to aberrant NK cell development, suggesting that NKD is cell intrinsic in these patients (33). However, given the interdependence on particularly NK and DC cross talk, it is likely that loss of other subsets affects NK cell numbers and functions in these patients, although this has not been explicitly studied. Immune manifestations include susceptibility to mycobacterial disease, frequently M. avium and M. kansasii, susceptibility to fungal infection, and severe herpesviral infections (39). An increased rate of malignancy may be in part due to viral infection as this includes increased rates of cervical cancer in young women potentially attributable to human papillomavirus infection. Less common clinical symptoms include miscarriage, solid organ tumors, and lymphedema (Emberger’s syndrome) (39).
Due to the requirement for GATA2 in renewal of the adult stem cells, it is thought that depletion of the stem cell pool leads to subsequent lymphopenia, and in vitro differentiation experiments demonstrate an NK cell intrinsic role for GATA2 through the phenocopying of the CD56bright NK cell subset loss in NK cells derived from patient CD34+ HSC (33). Whether GATA2 is required for the generation or homeostasis of CD56bright NK cells, or whether the defect arises earlier in lineage commitment, remains to be determined. GATA2 is highly expressed not only in CD34+ HSC, but also across a range of lineages including monocytes, monocyte-derived dendritic cells, B cells, and mature NK cells, as well as the common myeloid precursor (44–46). Conditional deletion of Gata2 in mice has recently demonstrated its requirement for DC differentiation from lineage negative precursors (47). Interestingly, this is at least partially through repression of genes that control T cell and ILC lineages, including Tcf7, Eomes, and Gata3. Direct binding of the Gata3 promoter by Gata2 in common myeloid progenitor cells suggests that these two transcription factors may play a role in tuning fate decisions similar to the well described GATA switch that occurs between GATA1 and GATA2 and is independent of transcription factor expression (47, 48).
This particular deficiency also allows for one to hypothesize regarding potential contribution of NK cells to human host defense. Given that some GATA2 deficiency patients lack only the CD56bright subset, it could speak to an important role for human NK cell-derived cytokine production specifically in the defense against HPV and/or herpesviruses. As there is strong animal model support for this role (49–51), the connection is at least plausible. It is not possible, however, to draw a direct connection as there is the variable impact upon other elements of innate immunity in these patients as discussed above. Furthermore, there are also those GATA2 deficiency patients that essentially lack NK cells altogether as well as those who have CD56dim NK cells. As deficient natural cytotoxicity is a conserved clinical hallmark of GATA2 deficiency (32, 33), this leaves open the contribution of contact-dependent cytotoxicity as the critical NK cell-mediated component of viral control. Thus, while GATA2 deficiency provides both insights and compelling leads, further work needs be done to formally attach a specific role to NK cell defenses through this disorder.
MCM4
Mutations in minichromosomal maintenance complex member 4 (MCM4) were described in 2012 as a cause of NKD accompanied by adrenal insufficiency, developmental delay, and short stature in a population of endogamous Irish travelers (52–55). Notably, patients in this cohort had increased susceptibility to viral infection, including cytomegalovirus, and malignancy (52–54). An initial description selective NKD (52) was followed by a genetic diagnosis of splice-site mutations in MCM4 and more detailed analysis of the NK cell phenotype (53, 54). MCM4 patients were the first to have demonstrated heritability to NKD and were the first listed in the Online Mendelian Inheritance in Man database as such (OMIM# 609981). Patients with MCM4 mutation have decreased frequency of CD56dim NK cells and, as a result, a relative overrepresentation of the CD56bright subset with an overall decrease in absolute numbers of NK cells (53). Careful analysis of both NK cells and fibroblasts from six patients in the Irish traveler cohort yielded novel insight into both NK cell biology and the role of the MCM complex in NK cell homeostasis and human disease. Specifically, CD56bright NK cells from patients had decreased rates of proliferation and impaired terminal maturation. This likely accounts for the severe viral susceptibility in these patients as they are lacking the more mature subset of NK cells and have at least some instability within the CD56bright population (53).
Why does MCM4 mutation exert such a specific, profound effect on NK cell maturation and function? MCM4, along with MCM2–7, MCM10, and GINS1 form a highly conserved complex that plays a critical role in the initiation and elongation of eukaryotic DNA replication (56, 57). Embryos from the Mcm4 knockout mouse do not survive implantation, and mice carrying the hypomorphic Chaos3 mutation affecting Mcm4 have genomic instability reminiscent of that found in MCM4 patients (58). Careful analysis of the patients’ fibroblasts showed normal MCM2–7 complex formation and DNA-binding but impaired DNA replication and, subsequently, cell cycle arrest (53). Genomic instability frequently accompanies impaired replication, and this is also the case in MCM4 patient fibroblasts, which had increased rates of chromosomal breakage (53). Interestingly, accompanying the decrease in NK cell number and specific decrease in frequency of CD56dim NK cells, patient NK cells had impaired proliferation in response to cytokine stimulation and increased rates of apoptosis (53).
The apparent role of MCM4 in the generation of CD56dim NK cells is not fully understood; however, it may speak to a previously hypothesized requirement for homeostatic proliferation, specifically in the CD56bright subset, leading to generation of the CD56dim subset. This model was originally proposed based on differential rates of apoptosis and proliferation detected in CD56bright and CD56dim NK cells in peripheral blood (59). The rapid proliferation of peripheral blood CD56bright cells with little apoptosis suggests that this subset either becomes recruited to tissue or generates the CD56dim subset, as substantiated by mouse xenograft studies (29). Should the latter be the case, the loss of CD56dim NK cells in MCM4 patients could be explained by the inability of the CD56bright NK cells to successfully undergo this proliferative burst and effectively generate CD56dim NK cells. Clearly, MCM4 deficiency raises many important new questions in NK cell biology, and answers will undoubtedly provide leads into both how NK cells develop as well as how developing NK cells figure into human host defense.
RTEL1
The third case of classic NKD, similarly to GATA2, is a case of a young girl who died of varicella infection at the age of 2 years. Originally reported in 2005 (60), her genetic lesion was recently solved by whole exome sequencing and found to be due to homozygous mutation in RTEL1 (61). Consistent with RTEL1 being a cause of specific NKD, the girl had seemingly normal immunoglobulin levels and specific antibody titers, normal T and B lymphocyte counts and subsets, and absence of bone marrow failure. Notably, however, she had severely decreased NK cell number and function that was not rescued by IL-2. As common γ chain and JAK3 mutations lead to SCID, her IL-15 levels and signaling pathways were tested, yet were found to be intact (61). RTEL1 deficiency is a reported cause of Hoyeraal–Hreidarsson Syndrome, an X-linked telomere deficiency prevalent among the Ashkenazi Jewish population that causes dyskeratosis congenita, bone marrow failure syndrome, and immunodeficiency (62). The mutation reported in this (2-year-old female) patient is a founder mutation causing Hoyeraal–Hreidarsson syndrome and lymphopenia, including progressive NK cell immunodeficiency (63, 64). Interestingly, the patient had no clinical features of Hoyeraal–Hreidarsson syndrome, and her growth and neurological development were normal for her age (personal communication, A. Etzioni, 2016). She did have an inverted CD4/CD8 T cell ratio but was virally infected (61). The patient also did not have recognizable platelet abnormalities or anemia (personal communication, A. Etzioni, 2016). Since the discovery of this patient, there have not been other reports of biallelic RTEL1 mutations causing selective NKD. However, given the molecular mechanism of MCM4 deficiency, the link between RTEL1, which also plays a role in DNA repair, and NK cell development is interesting. As the patient was deceased at the time of genetic study, unfortunately in-depth studies of NK cell subsets or developmental processes were not tenable. The effect of RTEL1 deficiency on NK cell development underscores the specific sensitivity of human NK cells to DNA damage and the requirement for the DNA damage response in normal human NK cell development and homeostasis.
IRF8
The most recent description of cNKD identifies the cause of NKD in a family first described in 1982 with severe EBV susceptibility and absent NK cell function in affected individuals (65). The longitudinal study of a surviving affected sibling shows a distinctive, stable NK cell phenotype, with decreased NK cell function and increased CD56bright NK cells relative to the CD56dim subset (66). This phenotype is conserved in other patients with biallelic IRF8 mutation and is NK cell intrinsic. Gene expression analysis of NK cells from the proband shows deregulation of genes involved in NK cell maturation and effector function, suggesting that IRF8 is playing a role in the regulation of genes that include NFIL3, PRDM1, TBX21, GRZB, STAT5a, STAT5b, and PRF. Strikingly, a similar block in terminal maturation is also observed in mice with homozygous, but not heterozygous Irf8 mutations. IRF8 deficiency as a result of both homozygous and heterozygous mutation can lead to DC deficiency (67, 68), although DC subsets in the proband reported here were only minimally affected (66). While mutation and gene dosage effects may be at play, the NK cell phenotype was conserved between all patients with biallelic IRF8 mutations studied but was not identified in any individuals with heterozygous IRF8 mutation. These findings demonstrate a requirement for IRF8 in terminal NK cell maturation and human antiviral defense and identify biallelic mutations in IRF8 as a newly described cause of cNKD.
Functional NKD
In contrast to cNKD, functional NKD (fNKD) represents a scenario in which NK cells are present in normal numbers but have some impaired functional capacity. In fNKD, a patient’s NK cells appear to have gone through appropriate developmental progression and are not unduly susceptible to cell death (i.e., impaired survival). This distinction makes this category particularly difficult to diagnose clinically as the most common screening tests of quantitative flow cytometry will most likely be normal. Thus, a suspicion given a patient’s particular clinical susceptibility is necessary to prompt a more substantive assessment of NK cell function. While presently fewer in number, it is possible that the fNKD category may ultimately outweigh the cNKD category in number of mechanistically defined diagnoses. Presently, there is only one known fNKD that is caused by biallelic specific mutations in FCGR3A.
FCGR3A
The only cause of fNKD reported to date is a rare mutation in FCGR3A (OMIM #615705), the gene encoding the low affinity IgG Fc receptor found on NK cells and macrophages (FcγRIIIA, CD16). Originally reported as the first monogenic cause of isolated NKD (69, 70), the c.230T-A transversion leads to L66H substitution in the first extracellular Ig domain of CD16 and is disease-causing when homozygous (69–71). Notably, while protein is expressed, the L66H substitution leads to loss of recognition of CD16 by mAb B73.1 while retaining recognition by the more commonly used 3G8 mAb (71). Therefore, dual detection with these antibodies is a rapid screen for this rare mutation as in certain individuals the B73.1 epitope will not be detected, although genotyping is required to confirm this molecular diagnosis (as individuals have been identified lacking the epitope for other reasons). Aside from the apparent loss of CD16+ NK cells if using mAb B73.1, NK cells from these patients appear to have appropriate development and subset generation (71).
This cause of fNKD was first identified in two patients with recurrent frequent upper respiratory infections and recurrent HSV (as well as herpes whitlow in one patient and recurrent varicella zoster in the second) (69, 70). Our group subsequently identified homozygous L66H substitution in a 14-year-old male with recurrent EBV-driven Castleman’s disease and recalcitrant cutaneous warts (71). While CD16 function is required to mediate ADCC, strikingly the patients with CD16 L66H mutation have normal ADCC-mediated cellular cytotoxicity, yet impaired natural cytotoxicity. This seemingly paradoxical effect is explained by the location of the mutation in the membrane distal Ig-like domain, which does not affect binding to IgG Fc mediated by the membrane proximal domain. Instead, molecular studies of the effect of L66H mutation revealed a role for the distal Ig-like domain of CD16 in binding to and stabilizing the NK cell coactivating receptor CD2, thus delineating a crucial role for CD16 in the co-stimulation of natural cytotoxicity in addition to its well-defined role in ADCC (71). Recently, the biology of this interaction has been further advanced through the study of individuals who do not possess the NKG2C locus in which a compensatory co-stimulatory relationship between CD2 and CD16 were demonstrated to be of value (72). Thus while, only one example of fNKD, this disorder has provided valuable insights into how NK cells react to challenges and focus upon patients with abnormal NK cell function, but grossly normal appearing NK cells should lead to important new leads both biologically and clinically.
Putting it all in Context
While originally classified as classical or functional, the lines between the types of NK cell deficiencies has been blurred by increased resolution of NK cell subsets as well as the normative ranges and diversity of human NK cells (5, 23, 24). This is exemplified by the example of GATA2 mutation, which was initially reported as leading to absence of circulating NK cells but which, upon careful analysis was shown to lead to decreased NK cell number, specifically loss of the CD56bright NK cell subset, accompanied by decreased function (33). Thus, it is possible to have a scenario in which NK cells may be present by clinical enumeration in a patient with classical NKD. The use of multiparametric flow cytometry to evaluate even the most rudimentary of NK cell subsets provides invaluable insight into the potential source of NKD. This is particularly true of GATA2 deficiency, which has a distinctive and seemingly immutable phenotype with regards to the absence of CD56bright NK cells (33, 37, 38).
A fascinating biological question has emerged from these discoveries of NKD, namely that of the interdependence of subsets upon each other for function, as well as the relative importance of NK cell number versus functional potential. Particularly in the case of GATA2 and MCM4, the skewing of CD56bright and CD56dim subset generation has a seemingly profound effect on the function of the other subset (33, 53). Namely, in cases where CD56dim NK cells are present in GATA2 patients, these cells remain unable to mediate the contact-dependent target cell lysis that is a functional hallmark of the CD56dim subset. In the case of MCM4 deficiency, although it seems that the CD56bright subset is impacted more so than the CD56dim subset, profoundly decreased NK cell function by the CD56dim subset is a likely a contributing cause of the patients’ disease (54).
These observations add to an interesting but complicated literature on the ontology of human NK cells and ultimately speak to the need for a better understanding of the mechanism of, and requirement for human NK cell development. Finally, the question of the relationship between NK cell number, subset distribution, function, and disease state remains incompletely understood. Whether it is more advantageous to have fewer NK cells that are appropriately developed, as opposed to a greater number of inappropriately mature cells, remains to be seen. The evidence points to the former, begging the analogy that it is more important to have a small, functional team than a large one with badly behaved players. By extension, a small team may be a result of a disruptive individual, so determining the relative contribution of the deregulation of function and/or phenotype will be increasingly insightful into the pathogenesis of NKD. Emerging evidence in transplantation for primary immunodeficiency emphasizes this point, as having potentially very low numbers of NK cells that have the potential to work effectively may suffice at the right time and under the right circumstance. Along these lines, it is even possible that having a few defective NK cells could be more damaging than having none at all. Answering these questions will require further studies that carefully and thoughtfully dissect human NK cell development in health and disease. Furthermore, there are likely to be context-, time-, and exposure-specific components of NKD as there are likely to be critical windows in which an absence of NK cell subsets or functions may be particularly relevant for human host defense.
It should also be noted that all these studies in NKD will benefit from a greater understanding of NK cells within unique microenvironments. By necessity, most of this work has used peripheral blood as the source of NK cells, yet the tissue-specific NK cell phenotypes in liver, gravid uterus, and secondary lymphoid tissue speak to their shaping by these sites. Understanding how these seldom-visualized cells are affected by disease will be an important component of a greater understanding of the non-redundant function of human NK cells in health and disease. The present state of knowledge, however, suggests that there are patients with deficiencies of immunity reflected primarily within the NK cell compartment who are in need of mechanistic and therapeutic answers. Further study into individuals with suggestive phenotypes will likely produce new causative genes for NKD and insights into NK cell biology that can ultimately lead to new and improved therapeutic directions.
Author Contributions
Both the authors contributed equally to the writing and editing of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank their clinical and research collaborators, as well as patients and their families. They also acknowledge funding support to JO by NIH-NIAID Grants R01AI067946 and R01AI120989 and the Jeffrey Modell Foundation.
References
1. Ham H, Billadeau DD. Human immunodeficiency syndromes affecting human natural killer cell cytolytic activity. Front Immunol (2014) 5:2. doi:10.3389/fimmu.2014.00002
2. Orange JS. Human natural killer cell deficiencies. Curr Opin Allergy Clin Immunol (2006) 6(6):399–409. doi:10.1097/ACI.0b013e3280106b65
3. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol (2013) 132(3):515–25; quiz 526. doi:10.1016/j.jaci.2013.07.020
4. Voss M, Bryceson YT. Natural killer cell biology illuminated by primary immunodeficiency syndromes in humans. Clin Immunol (2015). doi:10.1016/j.clim.2015.11.004
5. Angelo LS, Banerjee PP, Monaco-Shawver L, Rosen JB, Makedonas G, Forbes LR, et al. Practical NK cell phenotyping and variability in healthy adults. Immunol Res (2015) 62(3):341–56. doi:10.1007/s12026-015-8664-y
6. De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci U S A (2011) 108(2):728–32. doi:10.1073/pnas.1012356108
7. Nielsen N, Ødum N, Ursø B, Lanier LL, Spee P. Cytotoxicity of CD56(bright) NK cells towards autologous activated CD4+ T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A. PLoS One (2012) 7(2):e31959. doi:10.1371/journal.pone.0031959
8. Gonzalez VD, Falconer K, Björkström NK, Blom KG, Weiland O, Ljunggren HG, et al. Expansion of functionally skewed CD56-negative NK cells in chronic hepatitis C virus infection: correlation with outcome of pegylated IFN-alpha and ribavirin treatment. J Immunol (2009) 183(10):6612–8. doi:10.4049/jimmunol.0901437
9. Hu PF, Hultin LE, Hultin P, Hausner MA, Hirji K, Jewett A, et al. Natural killer cell immunodeficiency in HIV disease is manifest by profoundly decreased numbers of CD16+CD56+ cells and expansion of a population of CD16dimCD56- cells with low lytic activity. J Acquir Immune Defic Syndr Hum Retrovirol (1995) 10(3):331–40. doi:10.1097/00042560-199511000-00005
10. Bjorkstrom NK, Ljunggren HG, Sandberg JK. CD56 negative NK cells: origin, function, and role in chronic viral disease. Trends Immunol (2010) 31(11):401–6. doi:10.1016/j.it.2010.08.003
11. Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood (2012) 119(11):2665–74. doi:10.1182/blood-2011-10-386995
12. Gumá M, Angulo A, Vilches C, Gómez-Lozano N, Malats N, López-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104(12):3664–71. doi:10.1182/blood-2004-05-2058
13. Gumá M, Budt M, Sáez A, Brckalo T, Hengel H, Angulo A, et al. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood (2006) 107(9):3624–31. doi:10.1182/blood-2005-09-3682
14. Björkström NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med (2011) 208(1):13–21. doi:10.1084/jem.20100762
15. Goodier MR, Mela CM, Steel A, Gazzard B, Bower M, Gotch F. NKG2C+ NK cells are enriched in AIDS patients with advanced-stage Kaposi’s sarcoma. J Virol (2007) 81(1):430–3. doi:10.1128/JVI.01567-06
16. Brunetta E, Fogli M, Varchetta S, Bozzo L, Hudspeth KL, Marcenaro E, et al. Chronic HIV-1 viremia reverses NKG2A/NKG2C ratio on natural killer cells in patients with human cytomegalovirus co-infection. AIDS (2010) 24(1):27–34. doi:10.1097/QAD.0b013e3283328d1f
17. Sohlberg E, Saghafian-Hedengren S, Rasul E, Marchini G, Nilsson C, Klein E, et al. Cytomegalovirus-seropositive children show inhibition of in vitro EBV infection that is associated with CD8+CD57+ T cell enrichment and IFN-gamma. J Immunol (2013) 191(11):5669–76. doi:10.4049/jimmunol.1301343
18. Béziat V, Dalgard O, Asselah T, Halfon P, Bedossa P, Boudifa A, et al. CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur J Immunol (2012) 42(2):447–57. doi:10.1002/eji.201141826
19. Petitdemange C, Becquart P, Wauquier N, Béziat V, Debré P, Leroy EM, et al. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog (2011) 7(9):e1002268. doi:10.1371/journal.ppat.1002268
20. Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity (2015) 42(3):431–42. doi:10.1016/j.immuni.2015.02.013
21. Zhang T, Scott JM, Hwang I, Kim S. Cutting edge: antibody-dependent memory-like NK cells distinguished by FcRγ deficiency. J Immunol (2013) 190(4):1402–6. doi:10.4049/jimmunol.1203034
22. Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity (2015) 42(3):443–56. doi:10.1016/j.immuni.2015.02.008
23. Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med (2013) 5(208):208ra145. doi:10.1126/scitranslmed.3006702
24. Strauss-Albee DM, Fukuyama J, Liang EC, Yao Y, Jarrell JA, Drake AL, et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med (2015) 7(297):297ra115. doi:10.1126/scitranslmed.aac5722
25. Romagnani C, Juelke K, Falco M, Morandi B, D’Agostino A, Costa R, et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol (2007) 178(8):4947–55. doi:10.4049/jimmunol.178.8.4947
26. Dulphy N, Haas P, Busson M, Belhadj S, Peffault de Latour R, Robin M, et al. An unusual CD56(bright) CD16(low) NK cell subset dominates the early posttransplant period following HLA-matched hematopoietic stem cell transplantation. J Immunol (2008) 181(3):2227–37. doi:10.4049/jimmunol.181.3.2227
27. Chan A, Hong DL, Atzberger A, Kollnberger S, Filer AD, Buckley CD, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol (2007) 179(1):89–94. doi:10.4049/jimmunol.179.1.89
28. Cooley S, Xiao F, Pitt M, Gleason M, McCullar V, Bergemann TL, et al. A subpopulation of human peripheral blood NK cells that lacks inhibitory receptors for self-MHC is developmentally immature. Blood (2007) 110(2):578–86. doi:10.1182/blood-2006-07-036228
29. Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med (2009) 206(1):25–34. doi:10.1084/jem.20082013
30. Wu C, Li B, Lu R, Koelle SJ, Yang Y, Jares A, et al. Clonal tracking of rhesus macaque hematopoiesis highlights a distinct lineage origin for natural killer cells. Cell Stem Cell (2014) 14(4):486–99. doi:10.1016/j.stem.2014.01.020
31. Orange JS. Unraveling human natural killer cell deficiency. J Clin Invest (2012) 122(3):798–801. doi:10.1172/JCI62620
32. Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med (1989) 320(26):1731–5. doi:10.1056/NEJM198906293202605
33. Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood (2013) 121(14):2669–77. doi:10.1182/blood-2012-09-453969
34. Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood (2011) 118(10):2656–8. doi:10.1182/blood-2011-06-360313
35. Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood (2011) 118(10):2653–5. doi:10.1182/blood-2011-05-356352
36. Vinh DC, Patel SY, Uzel G, Anderson VL, Freeman AF, Olivier KN, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood (2010) 115(8):1519–29. doi:10.1182/blood-2009-03-208629
37. Maciejewski-Duval A, Meuris F, Bignon A, Aknin ML, Balabanian K, Faivre L, et al. Altered chemotactic response to CXCL12 in patients carrying GATA2 mutations. J Leukoc Biol (2016) 99(6):1065–76. doi:10.1189/jlb.5MA0815-388R
38. Dickinson RE, Milne P, Jardine L, Zandi S, Swierczek SI, McGovern N, et al. The evolution of cellular deficiency in GATA2 mutation. Blood (2014) 123(6):863–74. doi:10.1182/blood-2013-07-517151
39. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood (2014) 123(6):809–21. doi:10.1182/blood-2013-07-515528
40. Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet (2011) 43(10):1012–7. doi:10.1038/ng.913
41. Wlodarski MW, Hirabayashi S, Pastor V, Starý J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood (2016) 127(11):1387–97. doi:10.1182/blood-2015-09-669937
42. Rodrigues NP, Janzen V, Forkert R, Dombkowski DM, Boyd AS, Orkin SH, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood (2005) 106(2):477–84. doi:10.1182/blood-2004-08-2989
43. Tipping AJ, Pina C, Castor A, Hong D, Rodrigues NP, Lazzari L, et al. High GATA-2 expression inhibits human hematopoietic stem and progenitor cell function by effects on cell cycle. Blood (2009) 113(12):2661–72. doi:10.1182/blood-2008-06-161117
44. Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A (2004) 101(16):6062–7. doi:10.1073/pnas.0400782101
45. Mabbott NA, Baillie JK, Brown H, Freeman TC, Hume DA. An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genomics (2013) 14:632. doi:10.1186/1471-2164-14-632
46. Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol (2009) 10(11):R130. doi:10.1186/gb-2009-10-11-r130
47. Onodera K, Fujiwara T, Onishi Y, Itoh-Nakadai A, Okitsu Y, Fukuhara N, et al. GATA2 regulates dendritic cell differentiation. Blood (2016) 128(4):508–18. doi:10.1182/blood-2016-02-698118
48. Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S. GATA switches as developmental drivers. J Biol Chem (2010) 285(41):31087–93. doi:10.1074/jbc.R110.159079
49. Bukowski JF, Woda BA, Welsh RM. Pathogenesis of murine cytomegalovirus infection in natural killer cell-depleted mice. J Virol (1984) 52(1):119–28.
50. Welsh RM, Brubaker JO, Vargas-Cortes M, O’Donnell CL. Natural killer (NK) cell response to virus infections in mice with severe combined immunodeficiency. The stimulation of NK cells and the NK cell-dependent control of virus infections occur independently of T and B cell function. J Exp Med (1991) 173(5):1053–63. doi:10.1084/jem.173.5.1053
51. Welsh RM, O’Donnell CL, Shultz LD. Antiviral activity of NK 1.1+ natural killer cells in C57BL/6 scid mice infected with murine cytomegalovirus. Nat Immun (1994) 13(5):239–45.
52. Eidenschenk C, Dunne J, Jouanguy E, Fourlinnie C, Gineau L, Bacq D, et al. A novel primary immunodeficiency with specific natural-killer cell deficiency maps to the centromeric region of chromosome 8. Am J Hum Genet (2006) 78(4):721–7. doi:10.1086/503269
53. Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest (2012) 122(3):821–32. doi:10.1172/JCI61014
54. Hughes CR, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest (2012) 122(3):814–20. doi:10.1172/JCI60224
55. O’Riordan SM, Lynch SA, Hindmarsh PC, Chan LF, Clark AJ, Costigan C. A novel variant of familial glucocorticoid deficiency prevalent among the Irish Traveler population. J Clin Endocrinol Metab (2008) 93(7):2896–9. doi:10.1210/jc.2008-0034
56. Aparicio T, Guillou E, Coloma J, Montoya G, Méndez J. The human GINS complex associates with Cdc45 and MCM and is essential for DNA replication. Nucleic Acids Res (2009) 37(7):2087–95. doi:10.1093/nar/gkp065
57. Costa A, Onesti S. Structural biology of MCM helicases. Crit Rev Biochem Mol Biol (2009) 44(5):326–42. doi:10.1080/10409230903186012
58. Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, et al. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat Genet (2007) 39(1):93–8. doi:10.1038/ng1936
59. Lutz CT, Karapetyan A, Al-Attar A, Shelton BJ, Holt KJ, Tucker JH, et al. Human NK cells proliferate and die in vivo more rapidly than T cells in healthy young and elderly adults. J Immunol (2011) 186(8):4590–8. doi:10.4049/jimmunol.1002732
60. Etzioni A, Eidenschenk C, Katz R, Beck R, Casanova JL, Pollack S. Fatal varicella associated with selective natural killer cell deficiency. J Pediatr (2005) 146(3):423–5. doi:10.1016/j.jpeds.2004.11.022
61. Hanna S, Béziat V, Jouanguy E, Casanova JL, Etzioni A. A homozygous mutation of RTEL1 in a child presenting with an apparently isolated natural killer cell deficiency. J Allergy Clin Immunol (2015) 136(4):1113–4. doi:10.1016/j.jaci.2015.04.021
62. Fedick AM, Shi L, Jalas C, Treff NR, Ekstein J, Kornreich R, et al. Carrier screening of RTEL1 mutations in the Ashkenazi Jewish population. Clin Genet (2015) 88(2):177–81. doi:10.1111/cge.12459
63. Ballew BJ, Joseph V, De S, Sarek G, Vannier JB, Stracker T, et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet (2013) 9(8):e1003695. doi:10.1371/journal.pgen.1003695
64. Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet (2013) 132(4):473–80. doi:10.1007/s00439-013-1265-8
65. Fleisher G, Starr S, Koven N, Kamiya H, Douglas SD, Henle W. A non-x-linked syndrome with susceptibility to severe Epstein-Barr virus infections. J Pediatr (1982) 100(5):727–30. doi:10.1016/S0022-3476(82)80572-6
66. Mace EM, Bigley V, Gunesch JT, Chinn IK, Cao TN, Keller MD, et al. Biallelic Mutations in IRF8 Cause Human NK Cell Deficiency. Barcelona, Spain: 17th Biennial Meeting of the European Society for Immunodeficiencies (2016).
67. Salem S, Langlais D, Lefebvre F, Bourque G, Bigley V, Haniffa M, et al. Functional characterization of the human dendritic cell immunodeficiency associated with the IRF8(K108E) mutation. Blood (2014) 124(12):1894–904. doi:10.1182/blood-2014-04-570879
68. Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med (2011) 365(2):127–38. doi:10.1056/NEJMoa1100066
69. de Vries E, Koene HR, Vossen JM, Gratama JW, von dem Borne AE, Waaijer JL, et al. Identification of an unusual Fc gamma receptor IIIa (CD16) on natural killer cells in a patient with recurrent infections. Blood (1996) 88(8):3022–7.
70. Jawahar S, Moody C, Chan M, Finberg R, Geha R, Chatila T. Natural killer (NK) cell deficiency associated with an epitope-deficient Fc receptor type IIIA (CD16-II). Clin Exp Immunol (1996) 103(3):408–13. doi:10.1111/j.1365-2249.1996.tb08295.x
71. Grier JT, Forbes LR, Monaco-Shawver L, Oshinsky J, Atkinson TP, Moody C, et al. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest (2012) 122(10):3769–80. doi:10.1172/JCI64837
Keywords: NK cell deficiency, NK cell development, NK cells, primary immunodeficiency, immune homeostasis
Citation: Mace EM and Orange JS (2016) Genetic Causes of Human NK Cell Deficiency and Their Effect on NK Cell Subsets. Front. Immunol. 7:545. doi: 10.3389/fimmu.2016.00545
Received: 23 September 2016; Accepted: 16 November 2016;
Published: 02 December 2016
Edited by:
Laurent Brossay, Brown University, USAReviewed by:
Michael G. Brown, University of Virginia, USAStephen Noel Waggoner, Cincinnati Children’s Hospital Medical Center, USA
Aimee Beaulieu, Rutgers New Jersey Medical School, USA
Copyright: © 2016 Mace and Orange. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emily M. Mace, bWFjZUBiY20uZWR1;
Jordan S. Orange, b3JhbmdlQGJjbS5lZHU=