 Tomomi Toubai
Tomomi Toubai Nathan D. Mathewson
Nathan D. Mathewson John Magenau
John Magenau Pavan Reddy
Pavan Reddy- 1Division of Hematology and Oncology, Department of Internal Medicine, University of Michigan Comprehensive Cancer Center, Ann Arbor, MI, USA
- 2Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA, USA
Graft-versus-host response after allogeneic hematopoietic stem cell transplantation (allo-HCT) represents one of the most intense inflammatory responses observed in humans. Host conditioning facilitates engraftment of donor cells, but the tissue injury caused from it primes the critical first steps in the development of acute graft-versus-host disease (GVHD). Tissue injuries release pro-inflammatory cytokines (such as TNF-α, IL-1β, and IL-6) through widespread stimulation of pattern recognition receptors (PRRs) by the release of danger stimuli, such as damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). DAMPs and PAMPs function as potent stimulators for host and donor-derived antigen presenting cells (APCs) that in turn activate and amplify the responses of alloreactive donor T cells. Emerging data also point towards a role for suppression of DAMP induced inflammation by the APCs and donor T cells in mitigating GVHD severity. In this review, we summarize the current understanding on the role of danger stimuli, such as the DAMPs and PAMPs, in GVHD.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HCT) has become widely used as a curative therapy for a variety of life-threatening hematological malignancies and congenital immune deficiencies (1). However, graft-versus-host disease (GVHD) remains as significant obstacle to improving the success of this treatment (2). The cause of GVHD reflects a complex process involving immune dysregulation in the context of recovering immunocompetent donor cells in recipients of allo-HCT. Donor T cells play a central role in the pathogenesis of acute GVHD. However, emerging data in the past 15 years have demonstrated a key role for donor, or recipient antigen presenting cells (APCs), derived from both hematopoietic and non-hematopoietic cells. Although current strategies of the prevention and treatment of acute GVHD mainly target T cells, modulating APC function represents a promising additional strategy for reducing acute GVHD. Therefore, a greater understanding of how APCs are activated and regulated is of significant interest. Myeloablative or reduced intensity conditioning regimens are a prerequisite for facilitating engraftment of donor hematopoietic cells, and for eliminating residual tumor cells, but they also cause significant host tissue damage. The impact of damage responses on APCs has become an active area of research. Host tissue injuries by conditioning regimens release “danger signals” including pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharides (LPS), and damage-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB-1) as well as pro-inflammatory cytokines, such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α from the inflamed tissues. These danger signals activate host or donor APCs that in turn present allo-antigens via major histocompatibility complex (MHC) class I or class II to donor T cells. In addition, activated APCs produce an abundance of T-cell stimulating cytokines, such as IL-12, which further escalate the inflammatory response. In this review, we describe several encouraging investigations that have been conducted in both experimental bone marrow transplantation (BMT) models and humans over the last two decades. We further summarize the updated findings of how DAMPs and PAMPs amplify or mitigate GVHD and explore potential new strategies for the regulation of these “danger signals” in the regulation of GVHD.
Danger Signals in GVHD Development
PAMPs are non-host derive molecules derived from microbes and are recognized by pattern recognition receptors (PRRs) that initiate and sustain the innate immune responses for protecting host from foreign pathogens (3). DAMPs are host-derived molecules released by host tissue damages and binds to PRRs that initiate and sustain non-infectious immune responses (4). These DAMPs and PAMPs are released as a consequence of conditioning-related tissue damage after allo-HCT. They activate APCs that in turn stimulate donor T cell proliferation and differentiation into effector T cells that migrate to target organs and cause GVHD. Upon target tissue destruction, additional PAMPs and DAMPs are released that perpetuate and amplify GVHD (Figure 1). Therefore, our understanding of the release of PAMPs/DAMPs and ways to limit this potentially lethal immunologic cascade by ameliorating tissue damages by inhibiting danger signaling with specific inhibitors may be important for mitigating the intensity of GVHD.
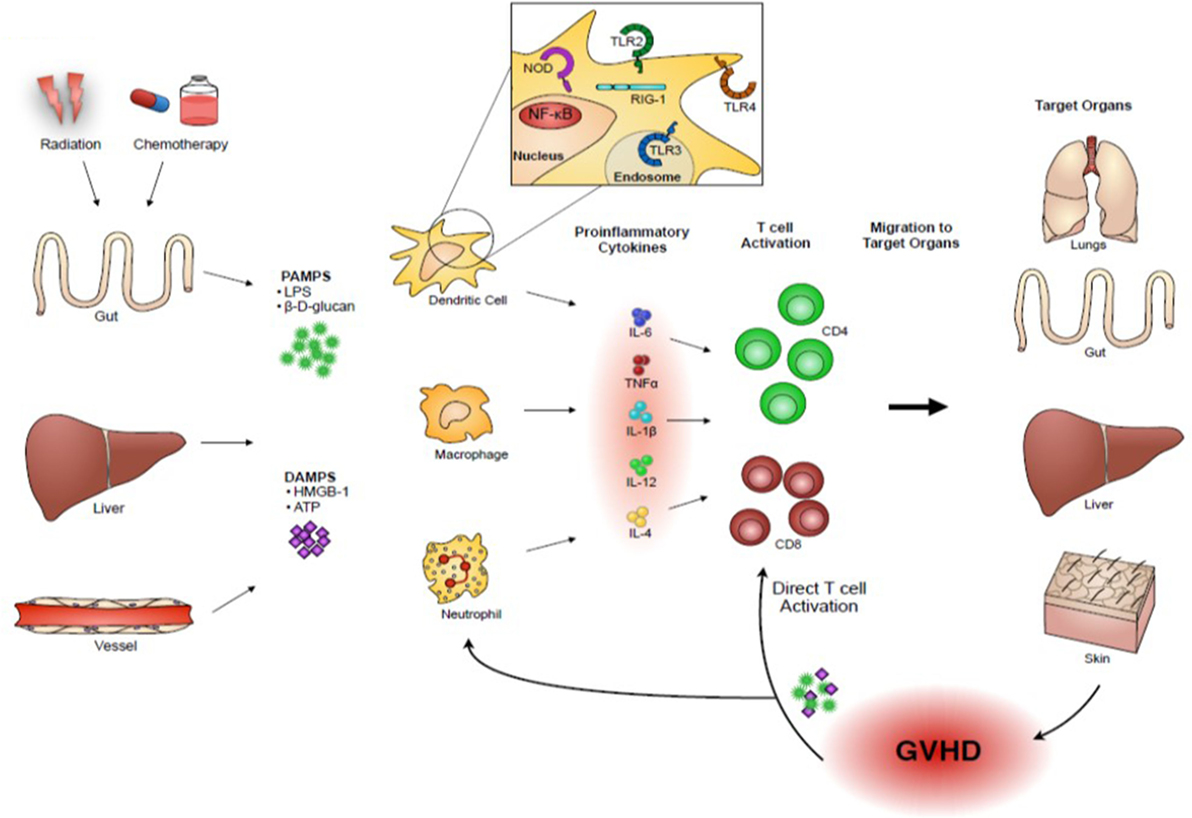
Figure 1. Danger signals play an important role in acute GVHD pathogenesis. Host tissue injuries by conditioning regimens release “danger signals” including pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharides (LPS) and β-D-glucans, and damage-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB-1) and adenosine triphosphate (ATP). These danger signals activate host or donor antigen-presenting cells (APCs), such as dendritic cells and macrophages, which in turn present alloantigens via major histocompatibility complex (MHC) class I or class II to donor T cells. In addition, activated APCs produce an abundance of pro-inflammatory cytokines, such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α, and T-cell stimulating cytokines, such as IL-12, which further escalate the inflammatory response. Activated donor T cells proliferate and differentiate into effector T cells that migrate to target organs and cause GVHD. Upon target tissue destruction, additional PAMPs and DAMPs are released and they might perpetuate GVHD responses.
Role of Specific PRRs in GVHD
Danger signaling is transmitted through PRRs when they bind PAPMs and DAMPs. Several signaling pathways, such as toll-like receptor (TLR), Nucleotide-binding oligomerization domain (NOD)-like receptor (NLR), and retinoic acid-inducible gene 1 (RIG-I) signaling, are recognized. The detailed mechanisms are recently reviewed in several articles (5–7). In this review, we focus on some of these receptors that have been implicated in GVHD.
Toll-Like Receptors
Toll-like receptors are one of the PRRs and play a key role in innate immune responses by recognizing PAMPs as well as DAMPs (8). TLRs are expressed on a variety of cells derived from both hematopoietic and non-hematopoietic lineages (8). We discuss below the experimental studies of TLRs in the pathogenesis of acute GVHD. The studies are also summarized in Table 1.
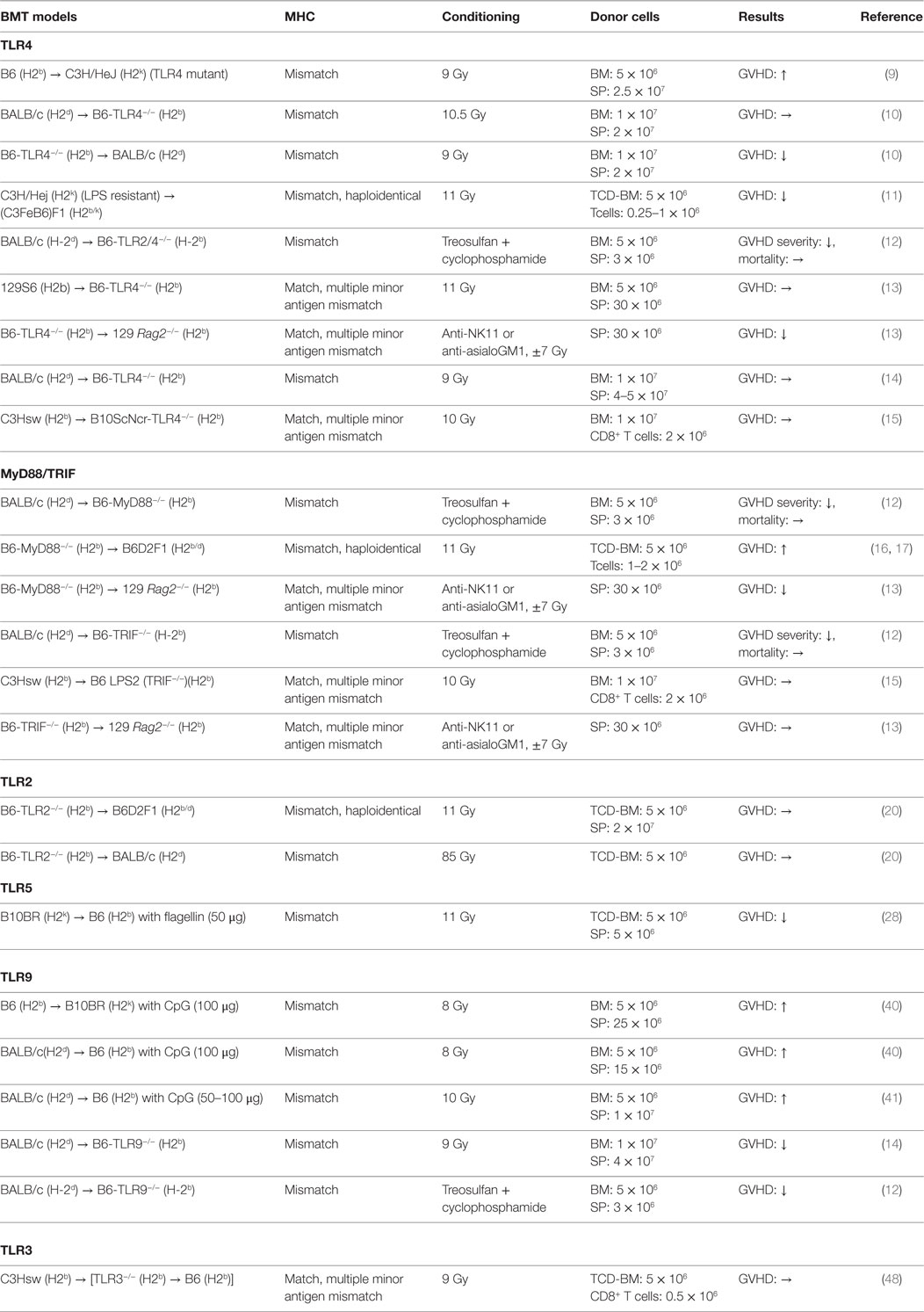
Table 1. The role of TLRs in the pathogenesis of acute GVHD.
TLR4
TLR4 is a cell-surface receptor for PAMPs such as LPS and also for DAMPs. TLR4 is broadly expressed on many immune cells, such as dendritic cells (DCs). TLR4 signaling is transmitted through intracellular adaptor molecules myeloid differentiation primary response gene 88 (MyD88) and Toll/IL-1receptor (TIR)-domain-containing adaptor-inducing interferon β (TRIF) that activate NF-κB signaling that potently enhances expression of pro-inflammatory cytokines. The role of TLR4 in APCs for mediating acute GVHD remains controversial. Mutations in TLR4 are involved in the reduction of GVHD responses by hyporesponsiveness of APCs to LPS stimulation while over activation of TLR4 signaling results in exacerbation of GVHD (9). When TLR4−/− animals were used as either donor or recipient, acute GVHD severity and mortality were significantly ameliorated in MHC-mismatched B6 into BALB/c model by altering DC functions in TLR4−/− APCs (10). This finding was consistent with a previous report in which recipients receiving LPS resistant donor cells demonstrated less GVHD and prolonged survival in MHC-mismatched haploidentical BMT (11). TLR2/4−/− animals receiving MHC-mismatched BMT (BALB/c into B6) also showed significantly less intestinal GVHD, but reduction was appeared dependent on conditioning intensity (12). In minor mismatched BMT context, MyD88-mediated TLR4 signaling on donor, but not recipient cells, was required for mediating acute GVHD (13). In addition, when TLR4 signaling was impaired in host APCs, that is, in the case of the absence of MyD88, TRIF, or both MyD88 and TRIF expression, acute GVHD severity and mortality were equivalent to WT animals (14, 15). While there is an increasing understanding of the key role of TLR4 signaling in contributing to the initiating event of GVHD, such disparate findings indicate that the role of TLR4 signaling for mediating GVHD may differ depending on the strain, the cell type where TLR4 is mutated, and the conditioning.
MyD88/TRIF
As MyD88 is required for the signaling of many TLRs, when MyD88−/− animals were used as the host, acute GVHD was significantly improved (12). In contrast, the recipients that received MyD88−/− T cell depleted BM (TCD-BM) cells showed greater intestinal GVHD (16) but reduced hepatic GVHD. This was found to be dependent on myeloid-derived suppressor cells (MDSCs) (17). In addition, MyD88−/− donor T cells reduced graft-versus-tumor (GVT) activity through the expansion of Foxp3- and IL-4-producing T cells in MHC-mismatched haploidentical B6 into B6D2F1 model (18). However, donor MyD88 was shown to be required in minor mismatch GVHD (13). TRIF is also required to transmit TLR signaling, but its role seems to be negligible in the development of GVHD (12, 13, 15). Collectively, these studies suggest that MyD88 may have pleiotropic functions that are cell intrinsic during allo-HCT.
TLR2
TLR2 is a cell-surface receptor expressed on APCs as well as T cells. TLR2 recognizes cell-wall components such as peptidoglycan (PGN) from gram-positive bacteria as well as zymosan from yeast. Intriguingly, granulocyte-colony stimulating factor (G-CSF) mobilized donor grafts showed the increase level of TLR2 expression on myeloid cell populations (19), but upregulated TLR2 expression did not correlate with enhanced allogeneic responses (20). The studies utilizing TLR2−/− animals as either donor or host demonstrated that TLR2 has little effect on acute GVHD (12, 20).
TLR5
TLR5 recognizes flagellin that is an essential component of bacterial flagella from both gram-negative and -positive bacteria and regulates immunity (21–26). CBLB502, a TLR5 agonist and a polypeptide drug derived from Salmonella flagellin, protects intestinal and hematopoietic cells from total body irradiation (TBI) in mice and primates (27). Consistent with this observation, when TLR5 agonist was administered before conditioning, acute GVHD was reduced with enhanced anti-CMV immunity in both MHC-mismatched and haploidentical murine models (28, 29). By contrast, TLR5 mRNA expression on peripheral blood, especially in the Lin(−)HLADR(−)CD33(+) CD16(+) and CD14(++)CD16(−) monocytes, was increased in the patients with GVHD after receiving adaptive Treg infusion for prevention of GVHD in human (30).
TLR7
TLR7 recognizes endosomal single strand ribonucleic acids (RNAs) leading to production of type I interferons (IFNs), pro-inflammatory cytokines, as well as regulatory cytokines (31, 32). TLR7 is critical for antiviral immunity and the development of autoimmune diseases (33–36). The contribution of TLR7 to acute GVHD is not well characterized. TLR7 ligand imiquimod was shown to increase alloreactivity of host-derived DCs and Langerhans cells (LCs) in the skin and to enhance donor lymphocyte infusions (DLIs)-mediated GVHD in MHC-matched multiple minor antigen-mismatched model of BMT (37). Because Type I IFNs are indispensable in the antitumor responses, whether TLR7 agonists increase GVT activities without enhancing GVHD after allo-HCT is of significant interest.
TLR9
TLR9 recognizes unmethylated cytosine-phophorothionate-guanine (CpG) dinucleatides in the bacterial DNA and triggers a Th1-mediated inflammatory response (38). CpG-mediated immune responses through TLR9 distinguish bacterial DNA from self-DNAs. TLR9 is expressed intracellularly in both immune and non-hematopoietic derived cells, such as endothelial and epithelial cells (39). Administration of CpG oligonucleotides (CpG ODNs), a synthetic TLR9 ligand, exacerbates acute GVHD in a host APC and IFN-γ dependent manner (40, 41). In addition, CpG ODNs enhances rejection donor HSCs in donor-derived APC dependent manner (40). When lethally or sub-lethally conditioned TLR9−/− animals were used as hosts, acute GVHD severity and mortality was ameliorated. This was dependent on the expression of TLR9 expression on the non-hematopoietic cells (12, 14). However, clinical studies exploring TLR9 polymorphisms in allo-HCT hosts suggested that those with homozygous CC gene variant of TLR9 (which correlates with lower expression of TLR9 mRNA) showed significantly improved overall survival (OS) and reduced relapse rate with no difference in acute GVHD when compared with patients having TC/TT gene variants (42, 43). Donor TLR9 gene tag single nucleotide polymorphisms (SNPs), +1174A/G (rs352139) and +1635 C/T (rs 352140), respectively, correlated increased severity of acute GVHD and CMV reactivation (44).
TLR3
TLR3 recognizes double-stranded RNA (dsRNA), which is produced by most of viruses. It signals through interferon regulatory factor (IRF)3 and activates NF-κB and enhances production of type I IFNs (45, 46). TLR3 also plays an important role in enhancing antigen presentation in APCs (47). Using chimeric recipients with TLR3−/− hematopoietic cells, we have already demonstrated that TLR3 deficiency in host APCs showed equivalent GVHD severity and mortality to WT animals but impaired GVT activity in MHC-matched multiple minor mismatched BMT model. Activation of TLR3 by polyinosine-polycytidylic acid (Poly I:C) improved GVT activity without enhancing GVHD (48). Because type I IFNs are essential role in antitumor immune responses (49, 50), TLR3 may have greater influence on GVT activity.
NLR Signaling
Nucleotide-binding oligomerization domain-like receptors are subtype of PPRs that function as cytoplasmic sensors of PAMPs and DAMPs. NLRs are expressed by majority of immune cells and some non-immune cells. NLRs have been extensively studies for their role in innate immunity. NOD1 and NOD2 are the most widely investigated NLRs in GVHD. NOD1 and NOD2 recognize different kinds of PGN fragments from bacterial cell wall. NOD1 binds to diaminopimelate-containing N-acetyl glucosamine-N-acetylmuramic acid (GluNAc-MurNac) tripeptide from gram-negative bacterial PGN (51, 52), while NOD2 binds to muramyl dipeptide (MDP) that is produced by all bacteria (53). Once NOD1 and NOD2 are ligated, NF-κB and mitogen-activated protein kinase (MAPK) pathways are activated through the caspase recruitment domain (CARD)-containing serine/threonine kinase, receptor-interacting protein 2 (Rip2) (54), and induce pro-inflammatory cytokines. NOD1 and NOD2 signaling is also involved in endoplasmic reticulum (ER) stress induced inflammation through the IRE1α/TRAF2 signaling pathway (55) and production of antimicrobial peptides in the intestinal tract (56). The role of NOD2 signaling pathway in allo-HCT is somewhat controversial. NOD2 polymorphism in both donor and recipient was associated with increased transplant-related mortality in humans after HLA-identical sibling HCT or T cell depleted HCT as well as increased GVHD severity (43, 57–64). However, other studies demonstrated that NOD2 had no impact on outcome including GVHD severity and mortality (44, 65–69). In addition, intriguingly, NOD2 polymorphism was associated with increased relapse of leukemia after unrelated HCT (70). In experimental models, NOD2−/− recipient animals showed exacerbated GVHD severity and morality, particularly intestinal GVHD. This was due to host APC activation in experimental BMT (71). By contrast, donor NOD−/− BM cells reduced GVHD-related mortality in MHC-mismatched haploidentical BMT model (72).
Inflammasomes
Inflammasomes are multiprotein molecules that are in the cytoplasm of immune cells, such as APCs, as well as non-hematopoietic cells. They consist of an adaptor protein, apoptosis associated speck-like protein containing a caspase recruit domain (CARD) (ASC), which has pyrin domain (PYD) and CARD, pro-caspase 1, and certain receptor proteins, such as NLR family members (NLRP1, NLRP3, NLRC4, NLRP6, NLRP7, and NLRP12), the protein absent in melanoma 2 (AIM2) (73, 74). Once inflammasomes are activated, they produce inflammatory cytokines, specifically IL-1β and IL-18, and induce pyroptosis, a highly inflammatory form of programed cell death (73, 74). This is called canonical inflammasome pathway, which in contrast to the non-canonical pathway activates caspase 11 in mouse and caspases 4 and 5 in humans (75). The detailed molecular and activation pathways of inflammasomes have recently been summarized in excellent reviews (73, 74). Studies exploring the role of inflammasomes in acute GVHD have recently been published. NLRP3 activation by the intestinal commensal bacteria and uric acid released after conditioning, enhanced GVHD severity and mortality by increasing levels of caspase-1, IL-1β, and TH17 cells (76). Another mechanism of enhanced GVHD by NLRP3 was shown to be associated with microRNA-155 dependent host DC migration toward sites of ATP release (77). In addition, inflammasome activation in the inflammatory milieu ameliorated the immune suppressive function of MDSCs and exacerbated GVHD (78). Relevant to clinical translation, the addition of TBI or busulfan and cyclophosphamide (BU/CY) conditioning is capable of mediating NLRP3 activation in the liver and enhancing inflammation (79). As therapeutic strategy, inhibiting NLRP3 activation by nucleotide reverse transcriptase inhibitors decreased GVHD severity and mortality (80). Further, NLRP3 inflammasome in human CD4+ T cells promotes IFN-γ production and Th1 differentiation by enhancing caspase 1-dependent IL-1β secretion mediated through intracellular C5 activation (81). These data suggest that NLRP3 inflammasome contribute to the functional important mediators of GVHD: donor T cells, APCs, and non-hematopoietic cells in target tissue. Consistent with experimental models, donor polymorphisms in the NLRP3 inflammasome have been associated with outcomes after allo-HCT. TT genotype at rs10925027 in NLRP3 was associated with disease relapse and donor GG genotype at rs1043684 in NLRP2 was associated with non-relapse mortality (NRM) and OS. Also, patient AA genotype at rs5862 in NLRP1 was associated with NRM and OS after HLA-matched sibling HCT (82). We recently found that a related but distinct inflammasome, NLRP6, expressed in intestinal epithelial cells regulates innate immune responses and intestine homeostasis though the regulating normal commensal bacteria (83, 84). Absence of NLRP6 improved GVHD contrary to models of inflammatory bowel disease (IBD). Intriguingly, NLRP6−/− animals showed enhanced mucin family protein MUC2 expression in epithelial cells after allo-BMT (85). These results suggest that depending on the context, NLRP6 may exert opposite effects in various inflammatory disorders.
RIG-I Signaling
RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) are known as RIG-I-like receptors (RLRs). RIG-I and MDA5 contain a DExD/H box RNA helicase domain and CARD, but LGP2 has no CARD-like domain. These receptors bind to intracellular dsRNA or ssRNA and trigger innate antiviral responses by producing type I IFNs (86–91). Therefore, RIG-I pathways also play an important role in PAMPs and DAMPs-mediated inflammatory responses. However, whether RIG-I pathways facilitate GVHD development is presently unknown. Preliminary study suggests RIG-I-induced type I IFNs promote the regeneration of intestinal stem cells during acute tissue damage may ameliorate GVHD severity with preserving GVL activities in mouse model (92, 93).
C-Type Lectin Receptors
C-type lectin receptors (CLRs) are expressed on myeloid-derived APCs as soluble or transmembrane embedded proteins. They directly activate NF-κB through spleen tyrosine kinase (SYK) (94) or indirectly, by cooperating with other PRRs such as TLRs (95–97). Stimulation of CLRs promotes the production of pro-inflammatory cytokines, effector T cell differentiation into Th1 and Th17 (98). CLRs are divided into two groups; group 1 CLRs belong to the mannose receptor family and group 2 CLRs belong to the asialoglycoprotein receptor family which has subfamilies, the DC-associated C-type lectin1 (Dectin-1) and DC immunoreceptor (DCIR) subfamily including Dectin-2 (95). These recognize mannose, fucose, and glucan carbo-hydrate structures of bacteria, fungi. Both Dectin1 and Dectin2 activate NF-κB by enhancing SYK signaling through either the cytoplasmic immunoreceptor tyrosine-based activation motif (ITAM) in Dectin1 (94) or the ITAM-containing adaptor molecules, such as Fc receptor γ-chain (Fcr γ) or DAP12 in Dectin2 (99). Clinical studies have suggested that the incidence of acute GVHD increases with candida colonization in dectin1 gene dependent manner (100, 101). In murine model, α-mannan, which is a major component of fungal cell wall, mediated Th17 dependent pulmonary GVHD in a host dectin2 dependent manner (102).
Role of Specific PAMPs and DAMPs Proteins in GVHD
Both exogenous and endogenous danger signal proteins are released from damaged tissues and abnormal intestinal microbial colonies after conditioning. In addition, blood stream infection (BSI) caused by gut translocation of colonized bacteria is another critical source of PAMPs after allo-HCT (103). Experimental data showed that individual PAMPs and DAMPs proteins can function either independently or cooperatively to initiate GVHD.
Lipopolysaccharides
Lipopolysaccharides (endotoxin) are membrane component of many gram-negative bacteria representing one of the earliest and most investigated PAMPs in GVHD. The role of LPS in GVHD is complex and controversial. LPS translocation due to gastrointestinal (GI) tract damage is correlated with conditioning intensity (104) and shown to contribute to GVHD in select model systems. LPS activates APCs including DCs and macrophages (MFs) triggering production of pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6 (105, 106). These events contribute idiopathic lung injuries after allo-BMT (107). Persistent exposure of LPS precipitates pulmonary GVHD pathogenesis because recipient mice directly exposed to repeated inhaled LPS after allo-BMT showed pulmonary GVHD in hematopoietic donor-derived C-C motif ligand 2 (CCL2) and C-C motif receptor (CCR2) dependent manner (108, 109). In addition to host-derived cells, sensitivity to LPS in donor non-T cells has been suggested to be involved in GVHD severity (11). LPS is one of the ligands of TLR4, which plays a key role in innate immune responses (110), and its signaling is transmitted through the common MyD88 and TRIF pathway that can nuclear translocation of NF-kB to induce expression of inflammatory cytokine genes (111–113). TLR4 mutations lead to LPS hyporesponsiveness (110). As noted above, role of TLR4 and MyD88 in GVHD seems to depend on the model system.
Flagellin
The recipient animals treated with flagellin before allo-HCT demonstrated reduced GVHD mortality and enhanced immune reconstitution with preservation of antiviral and GVT effects after allo-HCT (28). Modulating TLR5 functions with flagellin enhanced GVT without exacerbating GVHD in CD8+ T cell and NK cell dependent model (114). Tumor reactive T cells engineered to produce flagellin along with expression of a melanoma-specific antigen-augmented antitumor responses. Contrary to enhanced T cell-mediated antitumor responses, TLR5-dependent commensal bacteria promote tumor development by expanding MDSCs and dampen antitumor immunity in TLR5- and IL-6-dependent manner (115). In addition, mesenchymal stem cells (MSCs), pretreated by flagellin showed increased Foxp3 expression, enhanced IL-10 production, and suppressed GVHD (116). Interestingly, TLR5 stimulation with flagellin protects gut mucosal tissue from damages caused by irradiation (27, 117).
Damage-Associated Molecular Patterns
HMGB1
HMGB1 is a ubiquitous DNA-binding nuclear protein of all eukaryotic cells, binds to nucleosome and regulates gene transcriptions (118). By contrast, HMGB1 plays an important role in initiating innate immune responses because endogenous HMGB-1 that is located in nucleus in resting cells is acetylated and is released from damaged tissues. HMGB-1 binds to TLRs (TLR2 or TLR4) or receptor for advanced glycation endproducts (RAGE) and activates NF-κB or MAPK signaling to produce pro-inflammatory cytokines in especially DCs or MFs (119–123). Therefore, extracellular HMGB1 functions as a DAMP. In addition, the inflammatory milieu with abundant IFN-γ, TNF-α, as well as LPS may promote further HMGB1 release from DCs and MFs (124). Inflammasome and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) 1 pathways are involved in molecular mechanisms of HMGB-1 release, which requires its acetylation and translocation from nucleus to cytoplasm and released to extracellular space through unique protein releasing pathway, such as pyroptosis. Recent reports suggests that HMGB1 promotes not only immune suppressive function through the facilitating MDSCs proliferation in cancer (125) but also protection from tissue injury in IBD by regulating cellular autophagy and apoptosis (126). Patients with HMGB1 polymorphism, the 2351insT minor allele, showed reduced grade II to IV acute GVHD following myeloablative allo-HCT (127). Increased serum levels of HMGB1 were observed in acute GVHD patients and donors treated with granulocyte-colony-stimulating factor (G-CSF) (128, 129). Myeloablative conditioning such as TBI or cyclophosphamide + TBI also increased serum HMGB1 levels consistent with its function as a DAMP (130).
Adenosine Triphosphate
All cells generate adenosine triphosphate (ATP) as the primary energy source via glycolysis and oxidative phosphorylation (OXPHOS) that is stored within cytoplasm and mitochondria (131). Once cells are exposed to stress or injury, ATP is released from damaged cells and the concentration of ATP in extracellular space is increased. Released ATP binds to purinergic receptor families, such as P2X expressed on the hematopoietic and non-hematopoietic cells, and can function as a potent DAMP (132, 133). In GVHD, extracellular ATP is dramatically increased after TBI and binds to P2X7R on host APCs. After its ligation, host APCs expressed greater co-stimulatory molecules, such as CD80 and CD86, and enhanced stimulation of donor CD4+ T cells and production of IFN-γ, and decreased Tregs. These results were associated with reduction of STAT5 phosphorylation and enhanced GVHD (134). Another purinergic receptor, P2Y2, in host hematopoietic derived APCs was shown to enhance GVHD (135). Increased extracellular ATP is regulated by ecto-nucleotidases, such as CD39, which phosphohydrolyzes ATP to adenosine diphosphate (ADP) and adenosine monophosphate (AMP) and then dephosphorylate into adenosine by CD73, also known as ecto-5′-nucleotidase (136). In line with this, agonists of the adenosine receptors (AR) decreased GVHD (137). Loss of this regulatory mechanism by CD73−/− T cells or in APCs exacerbated GVHD (138, 139). The recent study showed that inhibiting Notch 1 signaling by inducing expression of A2A receptor in CD73 dependent manner is a critical mechanism of Treg-induced GVHD suppression (140). The immunosuppression of BM-derived MSCs in GVHD was also shown to be partially dependent on CD73 activity (141).
Uric Acid
Uric acid is a metabolite of purine nucleotide and hyperuricemia is known to lead gout (142). Uric acid is also released from injured cells, stimulates DC maturation, activates CD8+ T cell cytotoxic functions, and is recognized as an endogenous DAMP (143). Recent data show that uric acid contributes to GVHD severity by stimulating with NLRP3 inflammasome (76). Patients with acute GVHD show a high level of serum uric acid during the pretransplantation period and the patients received a recombinant urate oxidase appeared to show significantly reduced GVHD in phase I study. These results are consistent with a study that blood uric acid homeostasis may be altered after allo-HCT by conditioning and using cyclosporine A (144). However, a recent study showed low serum level of uric acid was associated with GVHD severity (145).
Heat Shock Proteins
Heat shock proteins (HSPs) work as molecular chaperones that enhance protein folding and intracellular transportation (146). HSPs have been demonstrated the association with chronic inflammatory diseases as well as autoimmune disease (147, 148). HSPs bind to TLR2/4 and mediate inflammatory responses. In GVHD, expression of HSP70 in lymphoid and target organs is increased and correlated with severity in both human and experimental GVHD (149–151). HSP70 homogene polymorphism (+2763 A/A) was associated with the development of GVHD (152). Another study demonstrated that antibodies to HSP70 and HSP90 increased in the patients with GVHD after allogeneic peripheral blood stem cell transplantation (allo-PBMCT) (153). HSP90 expression is increased in activated T cells and facilitates effector function and survival in activated T cells (154). HSP90-specific inhibitor decreased allogeneic T cell responses in vitro (155), but the in vivo effects on GVHD were not studied.
Heparan Sulfate Proteoglycans
Heparan sulfate proteoglycans (HSPGs) are component of extracellular matrix and play fundamental role in cell development, metabolism, and immunity (156, 157). HSPGs are crucial role in enhancing innate immune responses by stimulating DCs to enhance production of pro-inflammatory cytokines through TLR4 pathway (158). HSPGs promote neutrophils recruitment into the site of inflammation (159, 160) while enhance neutrophil infiltration exacerbates GVHD (161). The serum level of syndecan-1, which is one of the HSPGs, and heparin sulfate are increased in patients with GVHD (162, 163). In experimental models, heparin sulfate activates TLR4 signaling on DCs and leads to enhanced DC maturation and allogeneic T cell proliferation and increased GVHD severity (163). On the other hand, the absence of syndecan-4, which is one of the HSPGs and a ligand of DC-HIL that functions as a co-inhibitory pathway of donor T cell immune responses worsened GVHD (164).
Alpha-Mannan
The alpha-mannan (α-mannan) is a component of fungal cell wall as a known DAMP. The α-mannan is recognized by dectin1 and dectin2, which is one of the CLRs and activates NF-κB signaling through SYK and produces pro-inflammatory cytokines and effector T cell differentiation (96). α-mannan stimulated macrophages through dectin2, enhanced Th17 differentiation, and worsened lung GVHD (102). Colonization of candida species exacerbated GVHD in clinical studies (100, 101).
Therapeutic Strategies through Modulating Danger Signaling
“Danger signals” are indispensable role in initiating and developing acute GVHD. Regulating danger signal in an efficient manner in early phase of allo-SCT would ameliorate GVHD and have a great therapeutic strategy. Herein, we summarize potential therapeutic strategies for prevention and treatment of GVHD through modulating this signaling pathway. The studies are also summarized in Table 2.
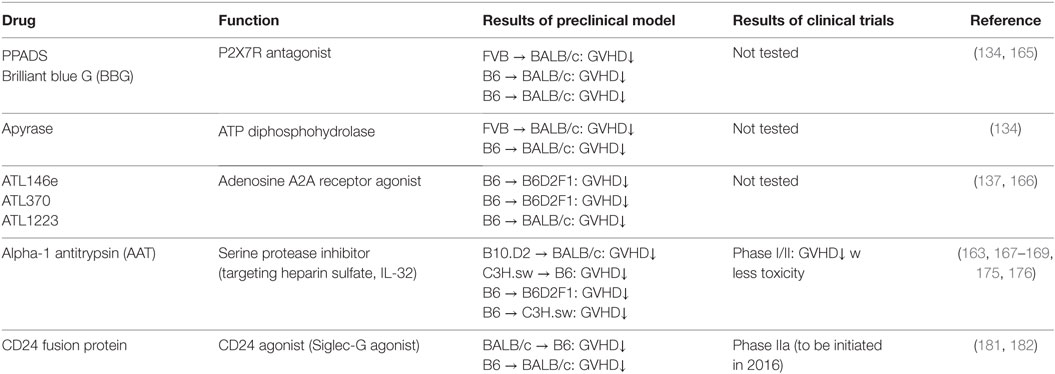
Table 2. Targeting danger signals to ameliorate GVHD.
Purinergic Receptor Antagonist
P2X7 receptor on APCs binds to extracellular ATPs, which released from damaged tissues by conditioning, and activates APCs to produce pro-inflammatory cytokines. Therefore, P2X7R plays a key role in DAMPs-mediated inflammatory responses. Systemic administration of the broad-spectrum P2X7R antagonist, pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS), or an ATP diphosphohydrolase, apyrase attenuated GVHD by suppressing APC activation (134). Additionally, another P2X7R receptor antagonist, brilliant blue G (BBG) improved liver function by regulating the infiltration of donor MFs and neutrophils in liver and attenuated GVHD (165). Beside P2X7R antagonist, adenosine A2A receptor agonist, ATL146e, decreased GVHD severity by modulating T cell activation and Treg function in experimental model (137, 166). Whether modulating other purinergic receptors ameliorates GVHD in the context allo-HCT needs to be studied.
Alpha-1 Antitrypsin
Serine protease inhibitor alpha-1 antitrypsin (AAT) attenuates GVHD by inhibiting HS, one of the DAMPs, and reducing HS mediated allogenic T cell responses in murine model (163). Clinical investigation demonstrates that the patients who have GVHD increased the serum level of HS after allo-HCT (163). In addition, we and others found that AAT attenuates GVHD with reducing serum levels of pro-inflammatory cytokines but increasing IL-10 levels by modulating function of donor and host APCs as well as altering the ratio of donor effector T cells to Tregs (167, 168). AAT also inhibits IL-32 activation mediated by proteinase-3, which is a neutrophil granule serine proteinase (169, 170). AAT homeostasis after allo-HCT may be important for regulating allogeneic responses because elevated AAT clearance in stool was correlated with the severity of GI-GVHD and steroid resistant GVHD (SR-GVHD) in some studies (171–173), but not others (174). Consistent with this, we and others have demonstrated that AAT treatment for SR-GVHD-improved GVHD manifestations without significant adverse effects or increased rates of infection in a multicenter prospective or single institution phase I/II study (175, 176). This data indicates that AAT may be a rational first-line therapy for SR-GVHD or other high risk GVHD, which is associated with high mortality. Although the mechanism how AAT suppresses SR-GVHD has not been clearly elucidated, these encouraging findings warrant further prospective, randomized, and multi-centered study.
Siglec-G: A Potential Negative Signaling for DAMPs-Mediated Inflammation
Sialic acid-binding immunoglobulin like lectins (Siglecs) have an immunoreceptor tyrosine-based inhibitory motifs (ITIM) or ITIM-like regions in their intracellular domains and negatively regulate DAMPs-mediated innate inflammatory responses (177, 178) or induce B-cell tolerance by suppressing NF-κB pathways (179, 180). We observed that Siglec-G expression in host APCs plays an important role in protecting DAMPs-mediated GVHD following conditioning mediated tissue damage. Interaction of Siglec-G with CD24, a small glycosyl-phosphatidyl-inositol (GPI)-anchored glycoprotein on T cells that is recognized as a ligand of Siglec-G (181), was critical for protection from GVHD. Enhancing Siglec-G-CD24 axis by a novel CD24 fusion protein (CD24Fc), consisting of the extracellular domain of mature human CD24 linked to the human immunoglobulin G1 (IgG1) Fc domain, mitigated GVHD in multiple experimental BMT models (181). We also found that enhancing the interactions between Siglec-G on T cells and CD24 on APCs with CD24Fc mitigated GVHD while preserving GVT effects in experimental models as well as human PBMCs (182). The summarized mechanism of Siglec-G-CD24 axis for controlling GVHD is shown in Figure 2. Based on these preclinical studies, a prospective, randomized multi-centered phase IIa study is currently investigating whether the addition of CD24Fc to standard immune-prophylaxis can limit the incidence and severity of acute GVHD following myeloablative allo-HCT.
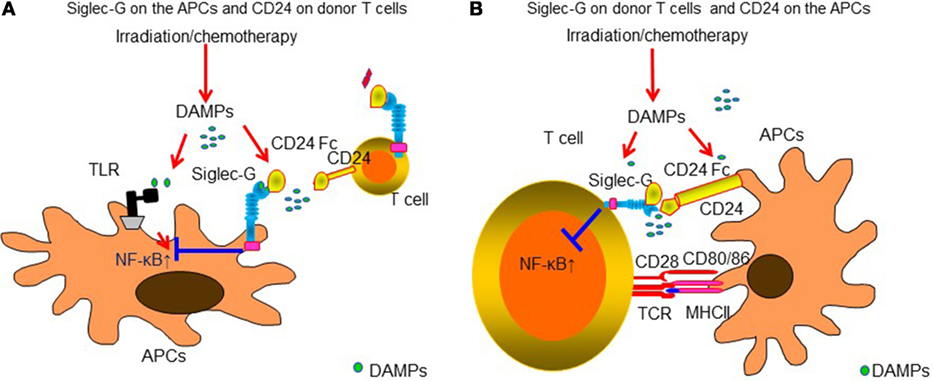
Figure 2. Siglec-G-CD24 axis is critical for regulating acute GVHD. Sialic acid-binding immunoglobulin-like lectins (Siglecs) have an immunoreceptor tyrosine-based inhibitory motifs (ITIM) or ITIM-like regions in their intracellular domains and negatively regulate DAMPs-mediated innate inflammatory responses. Siglec-G expression in host APCs plays an important role in protecting from DAMPs-mediated GVHD following conditioning-mediated tissue damage. Interaction of Siglec-G with CD24, a small glycosyl-phosphatidyl-inositol (GPI)-anchored glycoprotein on T cells that is recognized as a ligand of Siglec-G was critical for protection from GVHD. Enhancing Siglec-G-CD24 axis by a novel CD24 fusion protein (CD24Fc) mitigated GVHD (A). In addition, enhancing the interactions between Siglec-G on T cells and CD24 on APCs with CD24Fc mitigated GVHD (B).
Closing Remark
Danger signals mediate inflammatory responses through a multitude of PRRs that play a key role in the pathogenesis of GVHD. Once danger signals are released after conditioning, multiple innate immune signaling pathways are activated and amplified. Therefore, regulating danger signaling pathways in an effective manner is complex. Preclinical data suggest that targeting one specific signaling pathway or molecule may have only limited effects in reducing GVHD. In addition, the specific timing of regulation by blockade using antagonists may be a critical factor to consider. However, it is plausible that stimulating the negative regulating pathway that is commonly employed by several DAMPs may be a more rational way to mitigate GVHD. Thus exploring novel mechanisms of negative regulation of danger DAMP signaling that mediate lethal inflammatory responses should be carefully examined as new strategy of the prevention and treatment of GVHD. One potential benefit of regulating danger signaling is that GVT responses may be preserved due to selective attenuation of APCs with presumably limited effects on donor tumor-specific T cells necessary for mediating GVT responses. Clinical trials that investigate critical mediators of the danger response hold promise in the prevention of GVHD without affecting GVT responses.
Author Contributions
The review article was designed and written by TT and PR. Both NM and JM helped with designing figures. together with the help of co-authors NM and JM. All of them performed literature review and critically discussed the published literature.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health grants HL-128046 (PR), HL-090775 (PR), CA-173878(PR), and CA-203542 (PR), and American Society of Blood and Marrow Transplantation New Investigator Award (TT).
References
1. Jenq RR, van den Brink MR. Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer. Nat Rev Cancer (2010) 10(3):213–21. doi:10.1038/nrc2804
2. Choi SW, Reddy P. Current and emerging strategies for the prevention of graft-versus-host disease. Nat Rev Clin oncol. (2014) 11(9):536–47. doi:10.1038/nrclinonc.2014.102
3. Medzhitov R, Janeway CA Jr. Innate immunity: the virtues of a nonclonal system of recognition. Cell (1997) 91(3):295–8. doi:10.1016/S0092-8674(00)80412-2
4. Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. (2004) 4(6):469–78. doi:10.1038/nri1372
5. Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. (2016) 16(1):35–50. doi:10.1038/nri.2015.8
6. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. (2014) 14(8):546–58. doi:10.1038/nri3713
7. Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol. (2012) 12(3):168–79. doi:10.1038/nri3151
8. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi:10.1146/annurev.immunol.20.083001.084359
9. Imado T, Iwasaki T, Kitano S, Satake A, Kuroiwa T, Tsunemi S, et al. The protective role of host Toll-like receptor-4 in acute graft-versus-host disease. Transplantation (2010) 90(10):1063–70. doi:10.1097/TP.0b013e3181f86947
10. Zhao Y, Liu Q, Yang L, He D, Wang L, Tian J, et al. TLR4 inactivation protects from graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Cell Mol Immunol (2013) 10(2):165–75. doi:10.1038/cmi.2012.58
11. Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J Jr, et al. Tumor necrosis factor-alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest (1998) 102(10):1882–91. doi:10.1172/JCI4285
12. Heimesaat MM, Nogai A, Bereswill S, Plickert R, Fischer A, Loddenkemper C, et al. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut (2010) 59(8):1079–87. doi:10.1136/gut.2009.197434
13. Nalle SC, Kwak HA, Edelblum KL, Joseph NE, Singh G, Khramtsova GF, et al. Recipient NK cell inactivation and intestinal barrier loss are required for MHC-matched graft-versus-host disease. Sci Transl Med (2014) 6(243):243ra87. doi:10.1126/scitranslmed.3008941
14. Calcaterra C, Sfondrini L, Rossini A, Sommariva M, Rumio C, Menard S, et al. Critical role of TLR9 in acute graft-versus-host disease. J Immunol (2008) 181(9):6132–9. doi:10.4049/jimmunol.181.9.6132
15. Li H, Matte-Martone C, Tan HS, Venkatesan S, McNiff J, Demetris AJ, et al. Graft-versus-host disease is independent of innate signaling pathways triggered by pathogens in host hematopoietic cells. J Immunol (2011) 186(1):230–41. doi:10.4049/jimmunol.1002965
16. Lim JY, Lee YK, Lee SE, Ju JM, Eom KS, Kim YJ, et al. MyD88 in donor bone marrow cells is critical for protection from acute intestinal graft-vs.-host disease. Mucosal Immunol (2016) 9(3):730–43. doi:10.1038/mi.2015.96
17. Lim JY, Lee YK, Lee SE, Ju JM, Park G, Choi EY, et al. Attenuation of hepatic graft-versus-host disease in allogeneic recipients of MyD88-deficient donor bone marrow. Immune Netw. (2015) 15(3):125–34. doi:10.4110/in.2015.15.3.125
18. Lim JY, Ryu DB, Lee SE, Park G, Choi EY, Min CK. Differential effect of MyD88 signal in donor T cells on graft-versus-leukemia effect and graft-versus-host disease after experimental allogeneic stem cell transplantation. Mol Cells (2015) 38(11):966–74. doi:10.14348/molcells.2015.0158
19. Joo YD, Lee WS, Won HJ, Lee SM, Choi JH, Lee SM, et al. Upregulation of TLR2 expression on G-CSF-mobilized peripheral blood stem cells is responsible for their rapid engraftment after allogeneic hematopoietic stem cell transplantation. Cytokine (2011) 54(1):36–42. doi:10.1016/j.cyto.2010.12.020
20. Lee WS, Kim JY, Won HJ, Lee SM, Suh YS, Joo YD, et al. Effect of upregulated TLR2 expression from G-CSF-mobilized donor grafts on acute graft-versus-host disease. Int Immunopharmacol (2015) 29(2):488–93. doi:10.1016/j.intimp.2015.10.007
21. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature (2001) 410(6832):1099–103. doi:10.1038/35074106
22. Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol (2005) 175(12):8051–9. doi:10.4049/jimmunol.175.12.8051
23. Wilson RH, Maruoka S, Whitehead GS, Foley JF, Flake GP, Sever ML, et al. The Toll-like receptor 5 ligand flagellin promotes asthma by priming allergic responses to indoor allergens. Nat Med (2012) 18(11):1705–10. doi:10.1038/nm.2920
24. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity (2012) 36(2):276–87. doi:10.1016/j.immuni.2011.12.011
25. Uematsu S, Fujimoto K, Jang MH, Yang BG, Jung YJ, Nishiyama M, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol (2008) 9(7):769–76. doi:10.1038/ni.1622
26. Uematsu S, Jang MH, Chevrier N, Guo Z, Kumagai Y, Yamamoto M, et al. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat Immunol (2006) 7(8):868–74. doi:10.1038/ni1362
27. Burdelya LG, Krivokrysenko VI, Tallant TC, Strom E, Gleiberman AS, Gupta D, et al. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science (2008) 320(5873):226–30. doi:10.1126/science.1154986
28. Hossain MS, Jaye DL, Pollack BP, Farris AB, Tselanyane ML, David E, et al. Flagellin, a TLR5 agonist, reduces graft-versus-host disease in allogeneic hematopoietic stem cell transplantation recipients while enhancing antiviral immunity. J Immunol (2011) 187(10):5130–40. doi:10.4049/jimmunol.1101334
29. Hossain MS, Ramachandiran S, Gewirtz AT, Waller EK. Recombinant TLR5 agonist CBLB502 promotes NK cell-mediated anti-CMV immunity in mice. PLoS One (2014) 9(5):e96165. doi:10.1371/journal.pone.0096165
30. Sawitzki B, Brunstein C, Meisel C, Schumann J, Vogt K, Appelt C, et al. Prevention of graft-versus-host disease by adoptive T regulatory therapy is associated with active repression of peripheral blood Toll-like receptor 5 mRNA expression. Biol Blood Marrow Transplant (2014) 20(2):173–82. doi:10.1016/j.bbmt.2013.10.022
31. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303(5663):1529–31. doi:10.1126/science.1093616
32. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (2004) 303(5663):1526–9. doi:10.1126/science.1093620
33. Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med (2005) 202(11):1575–85. doi:10.1084/jem.20051696
34. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science (2006) 312(5780):1669–72. doi:10.1126/science.1124978
35. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity (2006) 25(3):417–28. doi:10.1016/j.immuni.2006.07.013
36. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity (2007) 27(5):801–10. doi:10.1016/j.immuni.2007.09.009
37. Durakovic N, Bezak KB, Skarica M, Radojcic V, Fuchs EJ, Murphy GF, et al. Host-derived Langerhans cells persist after MHC-matched allografting independent of donor T cells and critically influence the alloresponses mediated by donor lymphocyte infusions. J Immunol (2006) 177(7):4414–25. doi:10.4049/jimmunol.177.7.4414
38. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature (2000) 408(6813):740–5. doi:10.1038/35047123
39. Saitoh S, Miyake K. Regulatory molecules required for nucleotide-sensing Toll-like receptors. Immunol Rev (2009) 227(1):32–43. doi:10.1111/j.1600-065X.2008.00729.x
40. Taylor PA, Ehrhardt MJ, Lees CJ, Panoskaltsis-Mortari A, Krieg AM, Sharpe AH, et al. TLR agonists regulate alloresponses and uncover a critical role for donor APCs in allogeneic bone marrow rejection. Blood (2008) 112(8):3508–16. doi:10.1182/blood-2007-09-113670
41. Liu J, Guo YM, Onai N, Ohyagi H, Hirokawa M, Takahashi N, et al. Cytosine-phosphorothionate-guanine oligodeoxynucleotides exacerbates hemophagocytosis by inducing tumor necrosis factor-alpha production in mice after bone marrow transplantation. Biol Blood Marrow Transplant (2016) 22(4):627–36. doi:10.1016/j.bbmt.2015.12.018
42. Elmaagacli AH, Koldehoff M, Beelen DW. Improved outcome of hematopoietic SCT in patients with homozygous gene variant of Toll-like receptor 9. Bone Marrow Transplant (2009) 44(5):295–302. doi:10.1038/bmt.2009.32
43. Elmaagacli AH, Steckel N, Ditschkowski M, Hegerfeldt Y, Ottinger H, Trenschel R, et al. Toll-like receptor 9, NOD2 and IL23R gene polymorphisms influenced outcome in AML patients transplanted from HLA-identical sibling donors. Bone Marrow Transplant (2011) 46(5):702–8. doi:10.1038/bmt.2010.166
44. Xiao HW, Luo Y, Lai XY, Shi JM, Tan YM, He JS, et al. Donor TLR9 gene tagSNPs influence susceptibility to a GVHD and CMV reactivation in the allo-HSCT setting without polymorphisms in the TLR4 and NOD2 genes. Bone Marrow Transplant (2014) 49(2):241–7. doi:10.1038/bmt.2013.160
45. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature (2001) 413(6857):732–8. doi:10.1038/35099560
46. Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity (2002) 17(3):251–63. doi:10.1016/S1074-7613(02)00390-4
47. Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature (2005) 433(7028):887–92. doi:10.1038/nature03326
48. Toubai T, Sun Y, Luker G, Liu J, Luker KE, Tawara I, et al. Host-derived CD8+ dendritic cells are required for induction of optimal graft-versus-tumor responses after experimental allogeneic bone marrow transplantation. Blood (2013) 121(20):4231–41. doi:10.1182/blood-2012-05-432872
49. Avery RJ, Norton JD, Jones JS, Burke DC, Morris AG. Interferon inhibits transformation by murine sarcoma viruses before integration of provirus. Nature (1980) 288(5786):93–5. doi:10.1038/288093a0
50. Lacour J, Lacour F, Spira A, Michelson M, Petit JY, Delage G, et al. Adjuvant treatment with polyadenylic-polyuridylic acid (Polya.Polyu) in operable breast cancer. Lancet (1980) 2(8187):161–4. doi:10.1016/S0140-6736(80)90057-4
51. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science (2003) 300(5625):1584–7. doi:10.1126/science.1084677
52. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol (2003) 4(7):702–7. doi:10.1038/ni945
53. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278(11):8869–72. doi:10.1074/jbc.C200651200
54. Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature (2002) 416(6877):194–9. doi:10.1038/416194a
55. Keestra-Gounder AM, Byndloss MX, Seyffert N, Young BM, Chavez-Arroyo A, Tsai AY, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature (2016) 532(7599):394–7. doi:10.1038/nature17631
56. Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science (2005) 307(5710):731–4. doi:10.1126/science.1104911
57. Elmaagacli AH, Koldehoff M, Hindahl H, Steckel NK, Trenschel R, Peceny R, et al. Mutations in innate immune system NOD2/CARD 15 and TLR-4 (Thr399Ile) genes influence the risk for severe acute graft-versus-host disease in patients who underwent an allogeneic transplantation. Transplantation (2006) 81(2):247–54. doi:10.1097/01.tp.0000188671.94646.16
58. Holler E, Rogler G, Brenmoehl J, Hahn J, Herfarth H, Greinix H, et al. Prognostic significance of NOD2/CARD15 variants in HLA-identical sibling hematopoietic stem cell transplantation: effect on long-term outcome is confirmed in 2 independent cohorts and may be modulated by the type of gastrointestinal decontamination. Blood (2006) 107(10):4189–93. doi:10.1182/blood-2005-09-3741
59. Hildebrandt GC, Granell M, Urbano-Ispizua A, Wolff D, Hertenstein B, Greinix HT, et al. Recipient NOD2/CARD15 variants: a novel independent risk factor for the development of bronchiolitis obliterans after allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2008) 14(1):67–74. doi:10.1016/j.bbmt.2007.09.009
60. Holler E, Rogler G, Brenmoehl J, Hahn J, Greinix H, Dickinson AM, et al. The role of genetic variants of NOD2/CARD15, a receptor of the innate immune system, in GvHD and complications following related and unrelated donor haematopoietic stem cell transplantation. Int J Immunogenet (2008) 35(4–5):381–4. doi:10.1111/j.1744-313X.2008.00795.x
61. Granell M, Urbano-Ispizua A, Arostegui JI, Fernandez-Aviles F, Martinez C, Rovira M, et al. Effect of NOD2/CARD15 variants in T-cell depleted allogeneic stem cell transplantation. Haematologica (2006) 91(10):1372–6.
62. Landfried K, Bataille F, Rogler G, Brenmoehl J, Kosovac K, Wolff D, et al. Recipient NOD2/CARD15 status affects cellular infiltrates in human intestinal graft-versus-host disease. Clin Exp Immunol (2010) 159(1):87–92. doi:10.1111/j.1365-2249.2009.04049.x
63. Jaskula E, Lange A, Kyrcz-Krzemien S, Markiewicz M, Dzierzak-Mietla M, Jedrzejczak WW, et al. NOD2/CARD15 single nucleotide polymorphism 13 (3020insC) is associated with risk of sepsis and single nucleotide polymorphism 8 (2104C>T) with herpes viruses reactivation in patients after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2014) 20(3):409–14. doi:10.1016/j.bbmt.2013.12.558
64. van der Velden WJ, Blijlevens NM, Maas FM, Schaap NP, Jansen JH, van der Reijden BA, et al. NOD2 polymorphisms predict severe acute graft-versus-host and treatment-related mortality in T-cell-depleted haematopoietic stem cell transplantation. Bone Marrow Transplant (2009) 44(4):243–8. doi:10.1038/bmt.2009.21
65. Sairafi D, Uzunel M, Remberger M, Ringden O, Mattsson J. No impact of NOD2/CARD15 on outcome after SCT. Bone Marrow Transplant (2008) 41(11):961–4. doi:10.1038/bmt.2008.9
66. Gruhn B, Intek J, Pfaffendorf N, Zell R, Corbacioglu S, Zintl F, et al. Polymorphism of interleukin-23 receptor gene but not of NOD2/CARD15 is associated with graft-versus-host disease after hematopoietic stem cell transplantation in children. Biol Blood Marrow Transplant (2009) 15(12):1571–7. doi:10.1016/j.bbmt.2009.08.001
67. Nguyen Y, Al-Lehibi A, Gorbe E, Li E, Haagenson M, Wang T, et al. Insufficient evidence for association of NOD2/CARD15 or other inflammatory bowel disease-associated markers on GVHD incidence or other adverse outcomes in T-replete, unrelated donor transplantation. Blood (2010) 115(17):3625–31. doi:10.1182/blood-2009-09-243840
68. van der Straaten HM, Paquay MM, Tilanus MG, van Geloven N, Verdonck LF, Huisman C. NOD2/CARD15 variants are not a risk factor for clinical outcome after nonmyeloablative allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2011) 17(8):1231–6. doi:10.1016/j.bbmt.2010.12.709
69. Tanabe T, Yamaguchi N, Matsuda K, Yamazaki K, Takahashi S, Tojo A, et al. Association analysis of the NOD2 gene with susceptibility to graft-versus-host disease in a Japanese population. Int J Hematol (2011) 93(6):771–8. doi:10.1007/s12185-011-0860-5
70. Mayor NP, Shaw BE, Hughes DA, Maldonado-Torres H, Madrigal JA, Keshav S, et al. Single nucleotide polymorphisms in the NOD2/CARD15 gene are associated with an increased risk of relapse and death for patients with acute leukemia after hematopoietic stem-cell transplantation with unrelated donors. J Clin Oncol (2007) 25(27):4262–9. doi:10.1200/JCO.2007.12.1897
71. Penack O, Smith OM, Cunningham-Bussel A, Liu X, Rao U, Yim N, et al. NOD2 regulates hematopoietic cell function during graft-versus-host disease. J Exp Med (2009) 206(10):2101–10. doi:10.1084/jem.20090623
72. Galvani RG, Lemos R, Areal RB, Salvador PA, Zamboni DS, Wanderley JL, et al. Disease severity and mortality can be independently regulated in a mouse model of experimental graft versus host disease. PLoS One (2015) 10(2):e0118079. doi:10.1371/journal.pone.0118079
73. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16(7):407–20. doi:10.1038/nri.2016.58
74. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481(7381):278–86. doi:10.1038/nature10759
75. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi:10.1038/nature10558
76. Jankovic D, Ganesan J, Bscheider M, Stickel N, Weber FC, Guarda G, et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J Exp Med (2013) 210(10):1899–910. doi:10.1084/jem.20130084
77. Chen S, Smith BA, Iype J, Prestipino A, Pfeifer D, Grundmann S, et al. MicroRNA-155-deficient dendritic cells cause less severe GVHD through reduced migration and defective inflammasome activation. Blood (2015) 126(1):103–12. doi:10.1182/blood-2014-12-617258
78. Koehn BH, Apostolova P, Haverkamp JM, Miller JS, McCullar V, Tolar J, et al. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood (2015) 126(13):1621–8. doi:10.1182/blood-2015-03-634691
79. Qiao J, Huang Y, Xia Y, Chu P, Yao H, Xu L, et al. Busulfan and cyclosphamide induce liver inflammation through NLRP3 activation in mice after hematopoietic stem cell transplantation. Sci Rep (2015) 5:17828. doi:10.1038/srep17828
80. Fowler BJ, Gelfand BD, Kim Y, Kerur N, Tarallo V, Hirano Y, et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science (2014) 346(6212):1000–3. doi:10.1126/science.1261754
81. Arbore G, West EE, Spolski R, Robertson AA, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science (2016) 352(6292):aad1210. doi:10.1126/science.aad1210
82. Granell M, Urbano-Ispizua A, Pons A, Arostegui JI, Gel B, Navarro A, et al. Common variants in NLRP2 and NLRP3 genes are strong prognostic factors for the outcome of HLA-identical sibling allogeneic stem cell transplantation. Blood (2008) 112(10):4337–42. doi:10.1182/blood-2007-12-129247
83. Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature (2012) 488(7411):389–93. doi:10.1038/nature11250
84. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145(5):745–57. doi:10.1016/j.cell.2011.04.022
85. Toubai T, Rossi C, Tamaki H, Mathewson N, Oravecz-Wilson K, Zajac C, et al. NLRP6 in host target tissues exacerbates graft-versus-host disease. Blood (2015) 126(23):148.
86. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol (2004) 5(7):730–7. doi:10.1038/ni1087
87. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol (2005) 6(10):981–8. doi:10.1038/ni1243
88. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature (2005) 437(7062):1167–72. doi:10.1038/nature04193
89. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature (2006) 441(7089):101–5. doi:10.1038/nature04734
90. Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science (2006) 314(5801):997–1001. doi:10.1126/science.1132998
91. Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A (2007) 104(2):582–7. doi:10.1073/pnas.0606699104
92. Fischer JC, Bscheider M, Eisenkolb G, Wintges A, Lindemans CA, Heidegger S, et al. RIG-I-induced type I IFNs promote regeneration of the intestinal stem cell compartment during acute tissue damage. Blood (2015) 126(23):3072.
93. Fischer JC, Lindemans CA, Lin CC, Wintges A, Bscheider M, Wertheimer T, et al. The role of the cytoplasmic RNA sensor retinoic acid inducible gene-I (RIG-I) in allogeneic stem cell transplantation. Blood (2014) 124(21):3827.
94. Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity (2005) 22(4):507–17. doi:10.1016/j.immuni.2005.06.005
95. Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. (2009) 9(7):465–79. doi:10.1038/nri2569
96. Geijtenbeek TB, Gringhuis SI. C-type lectin receptors in the control of T helper cell differentiation. Nat Rev Immunol. (2016) 16(7):433–48. doi:10.1038/nri.2016.55
97. Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Wevers B, Bruijns SC, et al. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol (2009) 10(2):203–13. doi:10.1038/ni.1692
98. LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol (2007) 8(6):630–8. doi:10.1038/ni1460
99. Sato K, Yang XL, Yudate T, Chung JS, Wu J, Luby-Phelps K, et al. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J Biol Chem (2006) 281(50):38854–66. doi:10.1074/jbc.M606542200
100. van der Velden WJ, Netea MG, de Haan AF, Huls GA, Donnelly JP, Blijlevens NM. Role of the mycobiome in human acute graft-versus-host disease. Biol Blood Marrow Transplant (2013) 19(2):329–32. doi:10.1016/j.bbmt.2012.11.008
101. van der Velden WJ, Plantinga TS, Feuth T, Donnelly JP, Netea MG, Blijlevens NM. The incidence of acute graft-versus-host disease increases with Candida colonization depending the dectin-1 gene status. Clin Immunol (2010) 136(2):302–6. doi:10.1016/j.clim.2010.04.007
102. Uryu H, Hashimoto D, Kato K, Hayase E, Matsuoka S, Ogasawara R, et al. Alpha-Mannan induces Th17-mediated pulmonary graft-versus-host disease in mice. Blood (2015) 125(19):3014–23. doi:10.1182/blood-2014-12-615781
103. Poutsiaka DD, Munson D, Price LL, Chan GW, Snydman DR. Blood stream infection (BSI) and acute GVHD after hematopoietic SCT (HSCT) are associated. Bone Marrow Transplant (2011) 46(2):300–7. doi:10.1038/bmt.2010.112
104. Hill GR, Teshima T, Gerbitz A, Pan L, Cooke KR, Brinson YS, et al. Differential roles of IL-1 and TNF-alpha on graft-versus-host disease and graft versus leukemia. J Clin Invest (1999) 104(4):459–67. doi:10.1172/JCI6896
105. Nestel FP, Price KS, Seemayer TA, Lapp WS. Macrophage priming and lipopolysaccharide-triggered release of tumor necrosis factor alpha during graft-versus-host disease. J Exp Med (1992) 175(2):405–13. doi:10.1084/jem.175.2.405
106. Eissner G, Lindner H, Behrends U, Kolch W, Hieke A, Klauke I, et al. Influence of bacterial endotoxin on radiation-induced activation of human endothelial cells in vitro and in vivo: protective role of IL-10. Transplantation (1996) 62(6):819–27. doi:10.1097/00007890-199609270-00020
107. Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr, Crawford JM, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. (1996) 88(8):3230–9.
108. Martinu T, Gowdy KM, Nugent JL, Sun J, Kinnier CV, Nelson ME, et al. Role of C-C motif ligand 2 and C-C motif receptor 2 in murine pulmonary graft-versus-host disease after lipopolysaccharide inhalations. Am J Respir Cell Mol Biol (2014) 51(6):810–21. doi:10.1165/rcmb.2013-0451OC
109. Martinu T, Kinnier CV, Sun J, Kelly FL, Nelson ME, Garantziotis S, et al. Allogeneic splenocyte transfer and lipopolysaccharide inhalations induce differential T cell expansion and lung injury: a novel model of pulmonary graft-versus-host disease. PLoS One (2014) 9(5):e97951. doi:10.1371/journal.pone.0097951
110. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282(5396):2085–8. doi:10.1126/science.282.5396.2085
111. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature (1997) 388(6640):394–7. doi:10.1038/41131
112. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity (1999) 11(1):115–22. doi:10.1016/S1074-7613(00)80086-2
113. Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature (2003) 424(6950):743–8. doi:10.1038/nature01889
114. Ding X, Bian G, Leigh ND, Qiu J, McCarthy PL, Liu H, et al. A TLR5 agonist enhances CD8(+) T cell-mediated graft-versus-tumor effect without exacerbating graft-versus-host disease. J Immunol (2012) 189(10):4719–27. doi:10.4049/jimmunol.1201206
115. Rutkowski MR, Stephen TL, Svoronos N, Allegrezza MJ, Tesone AJ, Perales-Puchalt A, et al. Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell (2015) 27(1):27–40. doi:10.1016/j.ccell.2014.11.009
116. Linard C, Strup-Perrot C, Lacave-Lapalun JV, Benderitter M. Flagellin preconditioning enhances the efficacy of mesenchymal stem cells in an irradiation-induced proctitis model. J Leukoc Biol (2016) 100(3):569–80. doi:10.1189/jlb.3A0915-393R
117. Jones RM, Sloane VM, Wu H, Luo L, Kumar A, Kumar MV, et al. Flagellin administration protects gut mucosal tissue from irradiation-induced apoptosis via MKP-7 activity. Gut (2011) 60(5):648–57. doi:10.1136/gut.2010.223891
118. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. (2005) 5(4):331–42. doi:10.1038/nri1594
119. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285(5425):248–51. doi:10.1126/science.285.5425.248
120. Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem (1995) 270(43):25752–61. doi:10.1074/jbc.270.43.25752
121. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem (2004) 279(9):7370–7. doi:10.1074/jbc.M306793200
122. Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med (2000) 192(4):565–70. doi:10.1084/jem.192.4.565
123. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418(6894):191–5. doi:10.1038/nature00858
124. Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol (2003) 170(7):3890–7. doi:10.4049/jimmunol.170.7.3890
125. Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res (2014) 74(20):5723–33. doi:10.1158/0008-5472.CAN-13-2347
126. Zhu X, Messer JS, Wang Y, Lin F, Cham CM, Chang J, et al. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest (2015) 125(3):1098–110. doi:10.1172/JCI76344
127. Kornblit B, Masmas T, Petersen SL, Madsen HO, Heilmann C, Schejbel L, et al. Association of HMGB1 polymorphisms with outcome after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant (2010) 16(2):239–52. doi:10.1016/j.bbmt.2009.10.002
128. Tagami K, Yujiri T, Tanimura A, Mitani N, Nakamura Y, Ariyoshi K, et al. Elevation of serum high-mobility group box 1 protein during granulocyte colony-stimulating factor-induced peripheral blood stem cell mobilisation. Br J Haematol (2006) 135(4):567–9. doi:10.1111/j.1365-2141.2006.06335.x
129. Yujiri T, Tagami K, Tanaka Y, Mitani N, Nakamura Y, Ariyoshi K, et al. Increased serum levels of high-mobility group box 1 protein in patients who developed acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Eur J Haematol (2010) 85(4):366–7. doi:10.1111/j.1600-0609.2010.01507.x
130. Nomura S, Maeda Y, Ishii K, Katayama Y, Yagi H, Fujishima N, et al. Relationship between HMGB1 and PAI-1 after allogeneic hematopoietic stem cell transplantation. J Blood Med. (2016) 7:1–4. doi:10.2147/JBM.S93008
131. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi:10.1126/science.1160809
132. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature (2014) 509(7500):310–7. doi:10.1038/nature13085
133. Khakh BS, North RA. P2X receptors as cell-surface ATP sensors in health and disease. Nature (2006) 442(7102):527–32. doi:10.1038/nature04886
134. Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med (2010) 16(12):1434–8. doi:10.1038/nm.2242
135. Klambt V, Wohlfeil SA, Schwab L, Hulsdunker J, Ayata K, Apostolova P, et al. A novel function for P2Y2 in myeloid recipient-derived cells during graft-versus-host disease. J Immunol (2015) 195(12):5795–804. doi:10.4049/jimmunol.1501357
136. Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev (1998) 161:95–109. doi:10.1111/j.1600-065X.1998.tb01574.x
137. Lappas CM, Liu PC, Linden J, Kang EM, Malech HL. Adenosine A2A receptor activation limits graft-versus-host disease after allogenic hematopoietic stem cell transplantation. J Leukoc Biol (2010) 87(2):345–54. doi:10.1189/jlb.0609388
138. Tsukamoto H, Chernogorova P, Ayata K, Gerlach UV, Rughani A, Ritchey JW, et al. Deficiency of CD73/ecto-5′-nucleotidase in mice enhances acute graft-versus-host disease. Blood (2012) 119(19):4554–64. doi:10.1182/blood-2011-09-375899
139. Wang L, Fan J, Chen S, Zhang Y, Curiel TJ, Zhang B. Graft-versus-host disease is enhanced by selective CD73 blockade in mice. PLoS One (2013) 8(3):e58397. doi:10.1371/journal.pone.0058397
140. Del Papa B, Pierini A, Sportoletti P, Baldoni S, Cecchini D, Rosati E, et al. The NOTCH1/CD39 axis: a Treg trip-switch for GVHD. Leukemia (2016) 30(9):1931–4. doi:10.1038/leu.2016.87
141. Amarnath S, Foley JE, Farthing DE, Gress RE, Laurence A, Eckhaus MA, et al. Bone marrow-derived mesenchymal stromal cells harness purinergenic signaling to tolerize human Th1 cells in vivo. Stem Cells (2015) 33(4):1200–12. doi:10.1002/stem.1934
142. Macdonald AD. Nitrite of amyl an eliminator of uric acid; its employment in the treatment of gout. Br Med J (1885) 1(1273):1039–40. doi:10.1136/bmj.1.1273.1039-a
143. Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature (2003) 425(6957):516–21. doi:10.1038/nature01991
144. Deliliers GL, Annaloro C. Hyperuricemia and bone marrow transplantation. Contrib Nephrol (2005) 147:105–14. doi:10.1159/000082548
145. Ostendorf BN, Blau O, Uharek L, Blau IW, Penack O. Association between low uric acid levels and acute graft-versus-host disease. Ann Hematol (2015) 94(1):139–44. doi:10.1007/s00277-014-2180-3
146. Gething MJ, Sambrook J. Protein folding in the cell. Nature (1992) 355(6355):33–45. doi:10.1038/355033a0
147. De Graeff-Meeder ER, van der Zee R, Rijkers GT, Schuurman HJ, Kuis W, Bijlsma JW, et al. Recognition of human 60 kD heat shock protein by mononuclear cells from patients with juvenile chronic arthritis. Lancet (1991) 337(8754):1368–72. doi:10.1016/0140-6736(91)93057-G
148. Selmaj K, Brosnan CF, Raine CS. Colocalization of lymphocytes bearing gamma delta T-cell receptor and heat shock protein hsp65+ oligodendrocytes in multiple sclerosis. Proc Natl Acad Sci U S A (1991) 88(15):6452–6. doi:10.1073/pnas.88.15.6452
149. Goral J, Mathews HL, Clancy J Jr. Expression of 70-kDa heat-shock protein during acute graft-versus-host disease. Clin Immunol Immunopathol (1998) 86(3):252–8. doi:10.1006/clin.1997.4473
150. Jarvis M, Marzolini M, Wang XN, Jackson G, Sviland L, Dickinson AM. Heat shock protein 70: correlation of expression with degree of graft-versus-host response and clinical graft-versus-host disease. Transplantation (2003) 76(5):849–53. doi:10.1097/01.TP.0000083556.73555.78
151. Novota P, Sviland L, Zinocker S, Stocki P, Balavarca Y, Bickeboller H, et al. Correlation of Hsp70-1 and Hsp70-2 gene expression with the degree of graft-versus-host reaction in a rat skin explant model. Transplantation (2008) 85(12):1809–16. doi:10.1097/TP.0b013e31817753f7
152. Bogunia-Kubik K, Lange A. HSP70-hom gene polymorphism in allogeneic hematopoietic stem-cell transplant recipients correlates with the development of acute graft-versus-host disease. Transplantation (2005) 79(7):815–20. doi:10.1097/01.TP.0000153157.97736.2C
153. Goral J, Shenoy S, Mohanakumar T, Clancy J Jr. Antibodies to 70 kD and 90 kD heat shock proteins are associated with graft-versus-host disease in peripheral blood stem cell transplant recipients. Clin Exp Immunol (2002) 127(3):553–9. doi:10.1046/j.1365-2249.2002.01770.x
154. Ferris DK, Harel-Bellan A, Morimoto RI, Welch WJ, Farrar WL. Mitogen and lymphokine stimulation of heat shock proteins in T lymphocytes. Proc Natl Acad Sci U S A (1988) 85(11):3850–4. doi:10.1073/pnas.85.11.3850
155. Stuehler C, Mielke S, Chatterjee M, Duell J, Lurati S, Rueckert F, et al. Selective depletion of alloreactive T cells by targeted therapy of heat shock protein 90: a novel strategy for control of graft-versus-host disease. Blood (2009) 114(13):2829–36. doi:10.1182/blood-2009-06-224600
156. Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature (2007) 446(7139):1030–7. doi:10.1038/nature05817
157. Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. (2006) 6(9):633–43. doi:10.1038/nri1918
158. Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol (2002) 168(10):5233–9. doi:10.4049/jimmunol.168.10.5233
159. Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell (2002) 111(5):635–46. doi:10.1016/S0092-8674(02)01079-6
160. Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs l-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol (2005) 6(9):902–10. doi:10.1038/ni1233
161. Schwab L, Goroncy L, Palaniyandi S, Gautam S, Triantafyllopoulou A, Mocsai A, et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat Med (2014) 20(6):648–54. doi:10.1038/nm.3517
162. Seidel C, Ringden O, Remberger M. Increased levels of syndecan-1 in serum during acute graft-versus-host disease. Transplantation (2003) 76(2):423–6. doi:10.1097/01.TP.0000074316.76104.A5
163. Brennan TV, Lin L, Huang X, Cardona DM, Li Z, Dredge K, et al. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood (2012) 120(14):2899–908. doi:10.1182/blood-2011-07-368720
164. Chung JS, Tomihari M, Tamura K, Kojima T, Cruz PD Jr, Ariizumi K. The DC-HIL ligand syndecan-4 is a negative regulator of T-cell allo-reactivity responsible for graft-versus-host disease. Immunology (2013) 138(2):173–82. doi:10.1111/imm.12027
165. Zhong X, Zhu F, Qiao J, Zhao K, Zhu S, Zeng L, et al. The impact of P2X7 receptor antagonist, brilliant blue G on graft-versus-host disease in mice after allogeneic hematopoietic stem cell transplantation. Cell Immunol (2016). doi:10.1016/j.cellimm.2016.07.014
166. Han KL, Thomas SV, Koontz SM, Changpriroa CM, Ha SK, Malech HL, et al. Adenosine A(2)A receptor agonist-mediated increase in donor-derived regulatory T cells suppresses development of graft-versus-host disease. J Immunol (2013) 190(1):458–68. doi:10.4049/jimmunol.1201325
167. Tawara I, Sun Y, Lewis EC, Toubai T, Evers R, Nieves E, et al. Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci U S A (2012) 109(2):564–9. doi:10.1073/pnas.1117665109
168. Marcondes AM, Karoopongse E, Lesnikova M, Margineantu D, Welte T, Dinarello CA, et al. Alpha-1-Antitrypsin (AAT)-modified donor cells suppress GVHD but enhance the GVL effect: a role for mitochondrial bioenergetics. Blood (2014) 124(18):2881–91. doi:10.1182/blood-2014-04-570440
169. Marcondes AM, Li X, Tabellini L, Bartenstein M, Kabacka J, Sale GE, et al. Inhibition of IL-32 activation by alpha-1 antitrypsin suppresses alloreactivity and increases survival in an allogeneic murine marrow transplantation model. Blood (2011) 118(18):5031–9. doi:10.1182/blood-2011-07-365247
170. Novick D, Rubinstein M, Azam T, Rabinkov A, Dinarello CA, Kim SH. Proteinase 3 is an IL-32 binding protein. Proc Natl Acad Sci U S A (2006) 103(9):3316–21. doi:10.1073/pnas.0511206103
171. Hagen LE, Schechter T, Luk Y, Brodovitch A, Gassas A, Doyle JJ. High alpha-1 antitrypsin clearance predicts severity of gut graft-versus-host disease (GVHD) in children. Pediatr Transplant (2011) 15(6):659–63. doi:10.1111/j.1399-3046.2011.01553.x
172. Rodriguez-Otero P, Porcher R, Peffault de Latour R, Contreras M, Bouhnik Y, Xhaard A, et al. Fecal calprotectin and alpha-1 antitrypsin predict severity and response to corticosteroids in gastrointestinal graft-versus-host disease. Blood (2012) 119(24):5909–17. doi:10.1182/blood-2011-12-397968
173. O’Meara A, Kapel N, Xhaard A, Sicre de Fontbrune F, Manene D, Dhedin N, et al. Fecal calprotectin and alpha1-antitrypsin dynamics in gastrointestinal GVHD. Bone Marrow Transplant (2015) 50(8):1105–9. doi:10.1038/bmt.2015.109
174. Lorenz F, Marklund S, Werner M, Palmqvist R, Wahlin BE, Wahlin A. Fecal calprotectin as a biomarker of intestinal graft versus host disease after allogeneic hematopoietic stem cell transplantation. Sci Rep (2015) 5:7920. doi:10.1038/srep07920
175. Goldstein SC, Koreth J, Magenau JM, Soiffer RJ, Braun T, Kennel M, et al. Alpha 1 anti-trypsin (AAT): novel strategy to treat steroid refractory acute graft versus host disease. Biol Blood Marrow Transplant (2016) 22(3):S57–S. doi:10.1016/j.bbmt.2015.11.346
176. Marcondes AM, Hockenbery D, Lesnikova M, Dinarello CA, Woolfrey A, Gernsheimer T, et al. Response of steroid-refractory acute GVHD to alpha1-antitrypsin. Biol Blood Marrow Transplant (2016) 22(9):1596–601. doi:10.1016/j.bbmt.2016.05.011
177. Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science (2009) 323(5922):1722–5. doi:10.1126/science.1168988
178. Chen GY, Chen X, King S, Cavassani KA, Cheng J, Zheng X, et al. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat Biotechnol (2011) 29(5):428–35. doi:10.1038/nbt.1846
179. Hoffmann A, Kerr S, Jellusova J, Zhang J, Weisel F, Wellmann U, et al. Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat Immunol (2007) 8(7):695–704. doi:10.1038/ni1480
180. Duong BH, Tian H, Ota T, Completo G, Han S, Vela JL, et al. Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. J Exp Med (2010) 207(1):173–87. doi:10.1084/jem.20091873
181. Toubai T, Hou G, Mathewson N, Liu C, Wang Y, Oravecz-Wilson K, et al. Siglec-G-CD24 axis controls the severity of graft-versus-host disease in mice. Blood (2014) 123(22):3512–23. doi:10.1182/blood-2013-12-545335
Keywords: allogeneic hematopoietic stem cell transplantation, graft-versus-host-disease, danger signals, pathogen-associated molecular patterns, damage-associated molecular patterns
Citation: Toubai T, Mathewson ND, Magenau J and Reddy P (2016) Danger Signals and Graft-versus-host Disease: Current Understanding and Future Perspectives. Front. Immunol. 7:539. doi: 10.3389/fimmu.2016.00539
Received: 29 October 2016; Accepted: 15 November 2016;
Published: 29 November 2016
Edited by:
Abdulraouf Ramadan, Indiana University – Purdue University Indianapolis, USAReviewed by:
Robert Zeiser, University of Freiburg, GermanyFrances T. Hakim, National Cancer Institute, USA
Copyright: © 2016 Toubai, Mathewson, Magenau and Reddy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomomi Toubai, dG9tb21pdEB1bWljaC5lZHU=;
Pavan Reddy, cmVkZHlwckBtZWQudW1pY2guZWR1