 Tao Zeng
Tao Zeng- Institute of Toxicology, School of Public Health, Shandong University, Jinan, China
Alcoholic liver disease (ALD) encompasses a spectrum of liver injury ranging from steatosis to steatohepatitis, fibrosis, and finally cirrhosis. Accumulating evidences have demonstrated that Kupffer cells (KCs) play critical roles in the pathogenesis of both chronic and acute ALD. It has become clear that alcohol exposure can result in increased hepatic translocation of gut-sourced endotoxin/lipopolysaccharide, which is a strong M1 polarization inducer of KCs. The activated KCs then produce a large amount of reactive oxygen species (ROS), pro-inflammatory cytokines, and chemokines, which finally lead to liver injury. The critical roles of KCs and related inflammatory cascade in the pathogenesis of ALD make it a promising target in pharmaceutical drug developments for ALD treatment. Several drugs (such as rifaximin, pentoxifylline, and infliximab) have been evaluated or are under evaluation for ALD treatment in randomized clinical trials. Furthermore, screening pharmacological regulators for KCs toward M2 polarization may provide additional therapeutic agents. The combination of these potentially therapeutic drugs with hepatoprotective agents (such as zinc, melatonin, and silymarin) may bring encouraging results.
Introduction
Alcoholic liver disease (ALD) remains one of the predominant causes of liver-related morbidity and mortality worldwide (1). ALD encompasses a spectrum of progressively aggregated liver diseases, from simple steatosis, to steatohepatitis, fibrosis, and finally cirrhosis (2, 3). It has been generally accepted that ALD is a multifactorial disease, and both parenchymal cells (hepatocytes) and non-parenchymal cells in the liver are involved in the pathogenesis of ALD. Accumulating evidence suggests that Kupffer cells (KCs), the resident macrophages in the liver, play crucial roles (4–6). KCs originate from bone narrow-derived monocytes and account for about 20–25% of non-parenchymal cells in the liver (7). KCs play key roles in host defense by removing foreign, toxic and infective substances from the portal blood and have been demonstrated to be involved in the pathogenesis of many kinds of liver diseases (8, 9). It has been demonstrated that KCs are activated by gut-derived endotoxin/lipopolysaccharide (LPS), and then release many hepatotoxicants including reactive oxygen species (ROS), tumor necrosis factor α (TNF-α), interleukins, and chemoattractants for cytotoxic neutrophils, which will impair the function and viability of the neighboring cells. The “gut–liver axis” theory provides a number of potential therapeutic targets for ALD treatment, which have been evaluated or are under evaluation in clinical trials (10, 11). Furthermore, KCs are exceptionally plastic cells that can polarize to specific activation states and express different functions in different microenvironment. Two extremes of macrophage polarization have been designated as M1 (classically activation) and M2 (alternative activation). M1-polarized KCs can produce a large amount of pro-inflammatory cytokines, such as TNF-α, while M2-polarized KCs exhibit high expression of anti-inflammatory mediators, such as interleukin 10 (IL-10) (4, 12). Results of recent studies suggest that pharmacological intervention targeting M2 KCs polarization may represent an attractive strategy for ALD treatment (5). In this review, we discuss the critical roles of KCs in the pathogenesis of both chronic and acute ALD, KCs polarization in ALD, and potential therapeutic targets for ALD treatment.
Critical Roles of KCs in the Pathogenesis of Chronic ALD
Considerable evidence has demonstrated the critical roles of KCs in the development of chronic ALD. Chronic alcohol exposure can lead to intestinal hyperpermeability, resulting in the elevation of circulating endotoxin/LPS levels. LPS translocates from gut to liver, leading to the activation of KCs. Activated KCs produce a large amount of ROS, pro-inflammatory cytokines, and chemokines and induce the infiltration of other inflammatory cells, which finally cause liver injury (Figure 1).
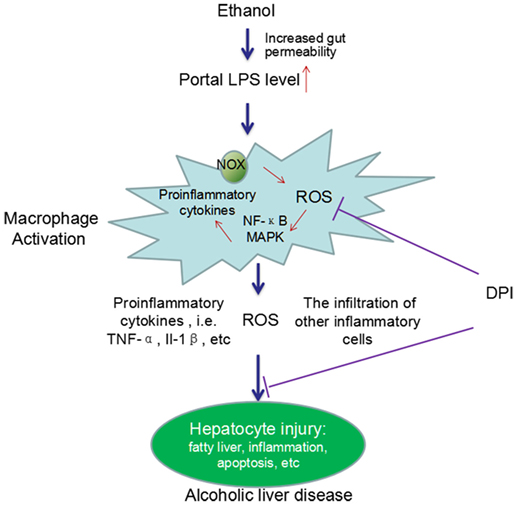
Figure 1. A schema graph for the critical roles of KCs in the pathogenesis of ALD. Chronic ethanol exposure increases the gut permeability, resulting in translocation of gut endotoxin/LPS to liver. In liver, LPS leads to KCs activation via activating NOX and the TLR-4 pathway. Activated KCs produce a large amount of ROS, pro-inflammatory cytokines, and chemokines and induce the infiltration of other inflammatory cells. The ROS, pro-inflammatory cytokines, and the infiltration of other inflammatory cells finally cause liver injury. DPI, a NOX inhibitor, can significantly block ethanol-induced oxidative stress and the liver injury. In addition, chronic ethanol exposure can also sensitize the LPS-induced toxicity by increasing the half-life and cell surface receptor number of TNF-α, the expression of TLR-related co-receptors, and forming oxidative stress-related pro-inflammatory adducts, such as MAA adducts (MDA reacts with acetaldehyde and proteins to form hybrid protein adducts).
Evidence Demonstrates that KCs Activation by Gut-Derived Endotoxin/LPS Plays Pivotal Roles in the Pathogenesis of Chronic ALD
The number of KCs in portal tract of liver was increased in ALD patients as well as in chronic ethanol-intoxicated animals (13, 14). Parallelly, the levels of many pro-inflammatory cytokines and chemokines secreted by KCs in chronic ethanol-intoxicated animals were significantly increased (15, 16). Furthermore, KCs elimination by gadolinium chloride almost completely prevented chronic ethanol-induced fat accumulation, inflammation, and necrosis scores (17–19). Now, it is clear that chronic ethanol-induced activation of KCs is attributed to gut-sourced LPS, which is a major component of outer membrane of Gram-negative bacteria and passes through the intestinal epithelial barrier in trance amounts under normal condition (20, 21). Chronic ethanol exposure can increase translocation of LPS from gut to liver by enhancing the intestinal permeability and altering the gut microflora. It has been demonstrated that chronic ethanol exposure can induce hemorrhagic lesions and pronounced alteration in the ultrastructure of enterocytes in small intestine of animals and human beings, leading to the increased permeability of intestinal mucosa to macromolecules (22–25). Additionally, chronic ethanol consumption may alter gut microflora, favoring the overgrowth of Gram-negative bacteria and thus increasing the source of LPS (26, 27). Elevation of serum LPS levels was observed in chronic ethanol-feeding rats and also in ALD patients (28). Suppressing LPS-producing bacteria by probiotics significantly reduced the serum LPS level and attenuated liver injury (29). Furthermore, animal studies showed that intestinal sterilization by antibiotics or LPS receptors deficiency significantly suppressed chronic ethanol-induced liver injury (30–32). These studies clearly demonstrate that the activation of KCs by gut-derived LPS plays causal roles in the pathogenesis of chronic ALD.
Signaling studies reveal that LPS can activate the toll-like receptor 4 (TLR-4) in KCs by incorporating in an activation complex involving LPS-binding protein (LBP), cluster of differentiation 14 (CD14) and myeloid differentiation factor 2 (MD-2) (21, 33). LPS is transferred by LBP (a shuttle protein) to CD14 and then binds with TLR-4/MD-2 receptor complex (34, 35). TLR-4 undergoes oligomerization and triggers myeloid differentiation primary response gene 88 (MyD88)- and toll-interleukin-1 receptor domain-containing adaptor inducing interferon-β (TRIF)-dependent production of pro-inflammatory cytokines, and type I interferon (IFN), respectively (33, 36). In the MyD88-dependent scenario, MyD88 recruits downstream adaptors including IL-1 receptor-associated kinase-4 (IRAK-4), IRAK-1, and TNF receptor-associated factor 6 (TRAF-6), leading to the activation of transforming growth factor β-activated kinase 1 (TAK-1) (33, 36, 37). TAK-1 can activate IκB kinase (IKK) and mitogen-activated protein kinase (MAPK) (38). Activated IKK phosphorylates IκB, resulting in the degradation of IκB proteins and the subsequent nuclear translocation of active NF-κB dimmers (39), while MAPK activates the early growth response 1 (Egr-1) and activation protein 1 (AP-1) (33, 40–42). On the other scenario, TRIF initiates a signaling pathway which activates interferon regulatory factor 3 (IRF-3) transcription factor and the late-phase activation of NF-κB and MAPK, leading to the expression of type 1 IFN and IFN-inducible chemokines (Figure 2).
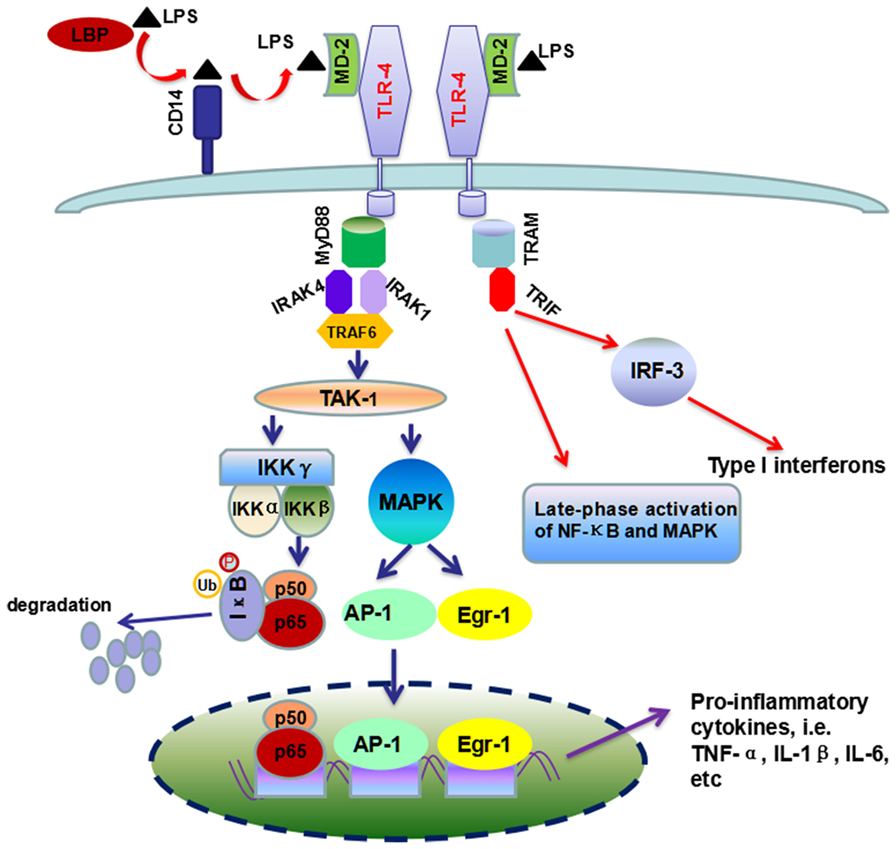
Figure 2. Overview of TLR-4/LPS signaling pathway activated by LPS in ALD. Chronic ethanol exposure leads to increased translocation of LPS to liver. In liver, LBP (the shuttle protein) transfers LPS to CD14, which facilitates the binding of LPS to TLR-4/MD-2 complex. TLR4 undergoes dimerization and transduces signal by two different pathways, i.e., MyD88-dependent and TRIF-dependent pathways. The former pathway included the recruitment of IRAK4, IRAK1, and TRAF-6, which ultimately leads to the production of pro-inflammatory cytokines by the activation of NF-κB and MAPK. In the second scenario, TRIF triggers a signaling pathway which controls the production of type I interferon and some other cytokines, as well as the late-phase activation of NF-κB and MAPK.
Chronic ethanol exposure led to increased DNA binding activity of NF-κB, and KCs were considered as the major cell type for NF-κB activation in liver (43). Importantly, NF-κB activation preceded the histopathological changes of liver in ALD rats and inactivation of NF-κB by delivery of IκB superrepressor gene with adenovirus significantly reduced chronic ethanol-induced liver injury, indicating the causal roles of NF-κB activation in the pathogenesis of ALD (44, 45). NF-κB may mediate liver injury by enhancing the production of TNF-α, as TNF-α antibodies and TNF receptor 1 (TNFR-1) knockout all significantly reduced chronic ethanol-induced liver injury (46, 47). Interestingly, the activation of NF-κB and subsequent TNF-α production could be suppressed by antioxidants or delivery of superoxide dismutase (SOD), which suggests that ROS may act as signaling molecules for NF-κB activation (48). NADPH oxidase (NOX) and cytochrome P4502E1 (CYP2E1) have been suggested to be major sources of ROS in ALD (49–52). NOX inhibitor (diphenyleneiodonium sulfate, DPI) and loss of p47phox subunit of NOX all significantly blunted the ethanol-induced oxidative stress, NF-κB activity, and TNF-α expression in the liver (53). These results strongly support the hypothesis that ROS from NOX in KCs promotes the NF-κB activation and TNF-α production (54).
Hepatocytes May Be Sensitized to Chronic Alcohol-Induced Injury
Alcohol may increase the sensitivity of hepatocytes to liver injury by increasing LPS-induced production of TNF-α, modifying cell surface receptors of TNF-α and forming reactive aldehydes adducts.
First, chronic alcohol might increase the sensitivity of KCs to secrete TNF-α and other pro-inflammatory cytokines (55). In vivo studies showed that chronic ethanol feeding enhanced LPS-stimulated TNF-α production and liver injury, which could be attenuated by TNF-α synthesis inhibitor (56, 57). In vitro studies using isolated KCs suggested that KCs from ethanol-fed rats secreted more TNF-α compared with those isolated from control animals (58–60). Further mechanisms investigation revealed that chronic alcohol exposure increased the half-life of TNF-α mRNA by activating p38MAPK, extracellular signal regulated kinases 1 and 2 (ERK1/2) and Egr-1 in KCs (58, 59, 61). It has been demonstrated that ERK1/2 plays critical roles in mediating the expression of TNF-α, interleukin 1β (IL-1β), etc., as specific inhibitor of ERK1/2 treatment led to the reduction of mRNA levels of these cytokines (40). As DPI could abrogate LPS-induced production of TNF-α as well as the activation of ERK1/2, it is possible that ERK1/2 is an important target of NOX (60). Second, chronic alcohol exposure could markedly elevate the affinity and capacity of binding sites of TNFR on hepatoctyes, and increase the mRNA and/or protein levels of LBP, CD14, and MD-2 in liver tissues, which may sensitize the liver to LPS-induced injury (59, 62–65). Third, chronic alcohol exposure can induce oxidative stress and lipid peroxidation in liver, resulting in overproduction of reactive aldehydes, including malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), which exhibit reactivity with proteins. It has been demonstrated that MDA can react with acetaldehyde and proteins to form hybrid protein adducts, designated as MAA adducts (66). In vitro exposure of KCs to MAA adducts induced pro-inflammatory and profibrogenic response, and MAA could synergistically interact with LPS to increase cytokines and chemokines expression (67).
CYP2E1 Potentiates Liver Injury Induced by LPS-Activated KCs
Cytochrome P4502E1, a member of the hemo-containing cytochrome P450 superfamily, has been suggested to play important roles in the pathogenesis of ALD. In addition to hepatocytes, CYP2E1 is also expressed in the KCs and in other tissues, including intestine and adipose (68, 69). CYP2E1 is an inducible enzyme and is the major component of the microsomal ethanol-oxidizing system (MEOS) (70–72). The activity of hepatic CYP2E1 in rats exposed to ethanol (5%, w/v)-containing Lieber-DeCarli diet was significantly increased by threefold to fivefold (73). The central roles of CYP2E1 in the pathogenesis of ALD have been highlighted by series of studies in which specific CYP2E1 inhibitors [such as diallyl sulfide and clomethiazole (CMZ)] or genetic knockout of CYP2E1 significantly attenuated chronic ethanol-induced liver injury (3, 49, 74–77). The important role of CYP2E1 in the development of ALD has been attributed to CYP2E1-mediated production of ROS (78–81).
Interestingly, LPS/TNF-α and CYP2E1, the two contributors to ALD, are not exclusive of each other, as a number of studies have revealed interactions between CYP2E1 and LPS/TNF-α (20, 73). First, activation of CYP2E1 in hepatocytes sensitizes the hepatocytes to LPS/TNF-α toxicity (82). CYP2E1 overexpression converted the hepatocyte TNF-α response from proliferation to apoptotic and necrotic cell death via activating JNK, which can be suppressed by antioxidants (83). Combination exposure to ethanol and TNF-α resulted in more serious cytotoxicity to CYP2E1-expressing HepG2 cells than the wild-type HepG2 cells (84). In vivo studies showed that chronic alcohol feeding enhanced LPS-stimulated TNF-α production and liver injury in wild-type mice, while CYP2E1−/− mice appeared to be resistant to LPS-induced hepatotoxicity (49, 56, 57). In addition, chronic alcohol exposure can lead to CYP2E1 activation in small intestine as well as in KCs (85, 86). Studies regarding the intestinal CYP2E1 showed that intestinal CYP2E1 might promote ethanol-induced intestinal hyperpermeability via a mechanism involving induction of oxidative stress and upregulation of circadian clock proteins CLOCK and PER2 (52, 85, 87). In KCs, CYP2E1 could induce more TNF-α secretion after stimulation by LPS, which was significantly blunted by CYP2E1 inhibitor, CMZ (86). Take together, CYP2E1 may play multiple roles in the pathogenesis of ALD, such as increasing ROS production, enhancing intestinal permeability, and promoting the TLR-4/LPS signaling to produce more TNF-α (Figure 3).
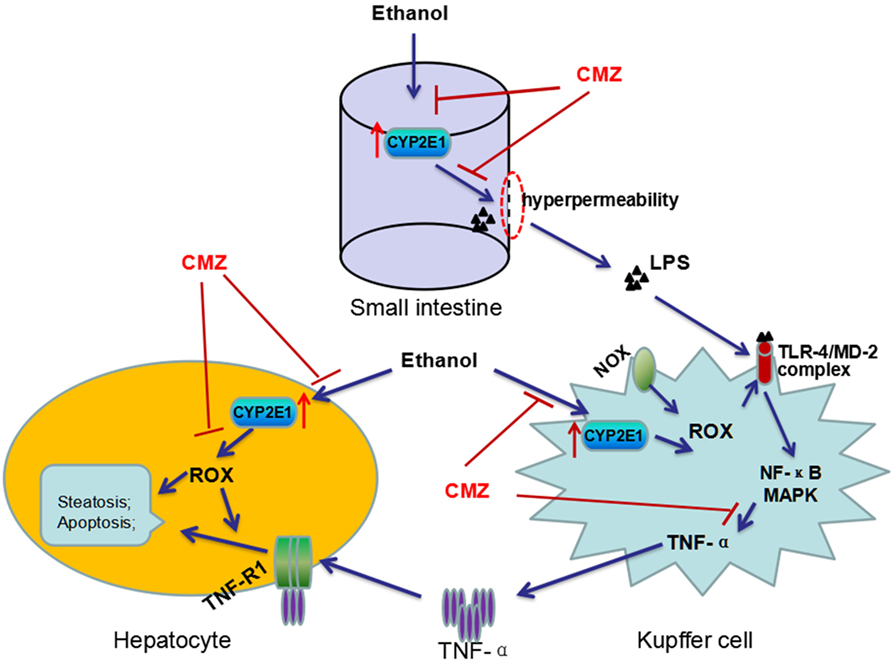
Figure 3. Interactions between CYP2E1 and LPS/TNF-α. CYP2E1 has been demonstrated to be expressed in hepatocytes, KCs, and also in small intestines. Intestinal CYP2E1 activation promotes ethanol-induced gut hyperpermeability, while CYP2E1 in KCs potentiates LPS-induced production of TNF-α. Furthermore, CYP2E1 can sensitize the hepatocytes to LPS/TNF-α toxicity from proliferation to apoptotic and necrotic cell death. All these adverse effects of CYP2E1 can be suppressed by CMZ, a specific inhibitor of CYP2E1.
The Roles of Inflammasome and IL-1β in the Pathogenesis of ALD
Inflammasomes are intracellular multiprotein complexes in both parenchymal and non-parenchymal cells activated upon cellular infection or stress that trigger the activation of caspase-1 and release of pro-inflammatory cytokines (88–90). Inflammasome activates procaspase-1 generating IL-1β, which causes inflammation in an autocrine fashion via IL-1 receptor (11). IL-1β, produced in inactive pro-IL-1β, is an important pro-inflammatory mediator in ALD. Inflammatory stimuli can induce the expression of inactive pro-IL-1β, and also can drive the formation of inflammasome leading to the cleavage of pro-IL-1β into the bioactive IL-1β (88, 91). It has been found that the serum levels of IL-1β in ALD patients and chronic ethanol-treated rats were significantly increased compared with controls (92, 93). In a mouse model of alcoholic hepatitis (AH), Petrasek et al. found that inflammasome was activated in KCs of ALD mice, which promoted caspase-1-mediated activation of IL-1β (91). Importantly, in vivo intervention with a recombinant IL-1 receptor antagonist (IL-1Ra) blocked IL-1β signaling and markedly attenuated ethanol-induced inflammation, steatosis, and liver damage (91). Results of these studies suggest that inflammasome activation is a component of the liver pathophysiology in ALD (89, 94, 95).
Roles of KCs in Acute Ethanol/Binge Drinking-Induced Liver Injury
Biphasic Effect of Acute Ethanol/Binge Drinking on the Activation of KCs
A binge drinking is defined as consumption of five and four drinks for men and women, respectively, in 2 h to produce a blood ethanol level over 80 mg/dl (96). Binge drinking is on the rise at an alarming rate worldwide (97–99). Although acute and chronic ALD may share similar mechanisms, the deleterious effects of alcohol may be affected by the doses and duration of ethanol (100). In contrast to the increased inflammation induced by chronic ethanol consumption, acute/binge drinking and regular moderate drinking has been reported to exert anti-inflammatory effects (96, 101). Numerous studies have demonstrated that acute/binge drinking (2–7 g/kg body weight, by introperitoneal injection or by gavage) could significantly suppress bacteria, LPS, and TLR ligand-induced increase of TNF-α levels in serum, bronchoalveolar lavage (BAL), and peritoneal lavage fluid (56, 102–111). For example, Nelson et al. found that acute ethanol (5.5 g/kg body weight, by introperitoneal injection), but not chronic ethanol consumption, markedly inhibited LPS-induced increase of TNF-α level in serum and BAL, and also suppressed systemic and intrapulmonary polymorphonuclear leukocyte aggregation (103). Honchel et al. found that acute ethanol (2 g/kg body weight, by gavage) suppressed LPS-induced TNF-α production, while chronic ethanol led to further elevation of serum TNF-α and significant liver injury (56). The anti-inflammatory effect of binge drinking was also examined in in vitro studies. Acute ethanol intoxication significantly suppressed LPS-induced elevation of inducible nitric oxide synthase (iNOS) mRNA in KCs and reduced LPS-induced serum TNF-α activity and ROS release (104, 112). Results of these studies seem to suggest that acute/binge drinking induce inactivation and tolerance of KCs to LPS, which is paradoxical to the effects of chronic ethanol exposure. However, time-effects studies revealed that acute/binge drinking might cause transient tolerance of KCs to LPS, followed by the enhanced sensitivity to LPS. For example, the study by Enomoto et al. found that KCs isolated from rats exposed to acute ethanol (4 g/kg body weight) 2 h ago exhibited decreased release of TNF-α, while KCs isolated from rats exposed to acute ethanol 24 h ago displayed enhanced secretion of TNF-α (113). Similarly, the study by Yamashina et al. revealed that ethanol administration at 1 h before LPS exposure alleviated LPS-induced liver injury, while ethanol administration at 21 h before LPS aggregated liver injury. This biphasic effect of ethanol was correlated with the expression of IRAK, NF-κB, and the TNF-α levels (114).
Evidence Suggests that KCs Play Crucial Roles in Acute Ethanol/Binge Drinking-Induced Liver Injury
Although limited, several studies have suggested that KCs may play crucial roles in acute ethanol/binge drinking-induced liver injury (52, 100, 115, 116). Binge drinking (6 g/kg body weight) could also lead to transient increase of the serum endotoxin/LPS levels (115, 116). Interestingly, endotoxin-neutralizing protein abrogated binge drinking-induced increase of hepatic TNF-α level, oxidative stress, and liver injury (115). Using a more stringent binge drinking mice model (mice exposed to three doses of ethanol, 6 g/kg body weight each, at 12 h intervals), wild-type mice in ethanol group developed hepatic steatosis and significant increase of serum endotoxin/LPS level as well as hepatic enterobacteria level (52). As binge drinking-induced TNF-α production and liver injury could be effectively attenuated by antioxidant (N-acetylcysteine) and specific inhibitor of CYP2E1 (chlormethiazole), it could be concluded that oxidative stress might mediate endotoxin/LPS-induced TNF-α production in acute ethanol-induced liver injury (52, 115). Furthermore, probiotics, which could maintain the intestinal integrity, significantly suppressed acute ethanol-induced liver injury (117). Results of these studies suggest that the “gut–liver axis” plays crucial roles in acute/binge drinking-induced liver injury.
To test the roles of gut microbiota in the pathogenesis of acute ALD, the sensitivity to binge drinking (3 g/kg body weight)-induced liver injury was compared between germ-free and conventional C57BL/6 mice (118). Unexpectedly, germ-free mice were found to be more susceptible to binge drinking-induced hepatic fat accumulation, elevation of serum aminotransferase activity, and hepatic inflammation compared with the conventional mice. The germ-free animals exhibited higher basal levels of hepatic fat content and CYP2E1 protein level compared with the conventional mice, which might explain the increased sensitivity to binge drinking-induced liver injury (118). As complete loss of intestinal microbiota seems to affect the metabolic homeostasis of mice liver, it may be not appropriate to use these germ-free animals to test the “gut–liver axis” theory (119).
The Polarization of KCs and the Pathogenesis of ALD
The Polarization of KCs in ALD
Macrophages including KCs are exceptionally plastic cells which can polarize to specific activation state and express different functions in response to microenvironmental signals. Two well-established polarized phenotypes are often referred to as classically activated macrophage (M1 polarization) and alternatively activated macrophage (M2 polarization) (120, 121). The nomenclature of M1/M2 polarization is derived from the cytokines that are associated with these macrophage phenotypes as these cytokines, namely interferon γ (IFN-γ) or interleukin-4 (IL-4), are linked with T helper 1 (Th1)- and Th2-type immune responses, respectively (120, 122). M1-polarized macrophages are characterized by increased expression of pro-inflammatory cytokines, including TNF-α and iNOS, while M2-polarized macrophages exhibit low expression of pro-inflammatory cytokines, but increased expression of anti-inflammatory mediators, such as IL-10 (4, 12). Inflammatory stimuli such as microbes, damaged tissues, and activated lymphocytes, can induce macrophages to acquire the pro-inflammatory M1 polarization (123). The inflammation driven by the M1 macrophages is counterbalanced by the anti-inflammatory M2-polarized macrophages which can promote the inflammation resolution and tissue repair (124) (Figure 4).
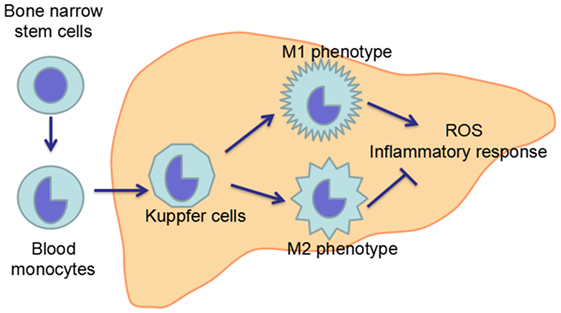
Figure 4. Maturation and polarization of KCs. KCs, the resident macrophage in the liver, originate from the precursor cells in the bone marrow, which give rise to blood monocytes. Blood monocytes migrate into liver and develop in to liver macrophage, namely KCs. In the liver, KCs can polarize in two ways: classical activation/M1 polarization and alternative activation/M2 polarization, which exhibit pro-inflammatory and anti-inflammatory effects, respectively. The imbalance between M1 and M2 polarization of KCs contributes to the pathogenesis of ALD.
Both chronic ethanol exposure and acute/binge drinking can lead to increased hepatic translocation of LPS, which is a strong inducer of M1 activation of KCs (125, 126). Chronic ethanol feeding has been demonstrated to result in increase of both M1 markers genes [iNOS, tumor necrosis factor α (TNFA) and monocyte chemoattractant protein-1 (MCP1)] and M2 markers genes [Arginase 1 (Arg1), mannose receptor C type 2 (Mrc2)] in mice liver, but the induction of M1 marker genes was more profound than the M2 marker genes (127, 128). The imbalance between M1 and M2 polarization leads to the overproduction of pro-inflammatory cytokines, which finally induce hepatocytes injury. Although there is no direct study about the polarization of KCs in acute/binge drinking animal models, it could be speculated that KCs may also undergo M1 polarization after acute ethanol exposure.
KCs Polarization toward the Alternative State (M2 Phenotype) May Be an Attractive Strategy for ALD Treatment
As macrophages of M1 and M2 phenotypes are not stably differentiated subsets, pharmacological interventions targeting M2 polarization during early stages of ALD may represent an attractive strategy to limit ethanol-induced inflammation and hepatocytes injury (5). Indeed, regulation of M1/M2 balance in KCs by agonist of cannabinoid CB2 receptor (a G-protein-coupled receptor predominantly expressed by cells of the immune system) significantly suppressed chronic ethanol-induced hepatic steatosis (127). In a recent study, Wan et al. found that KCs in ALD-resistant BALB/c mice displayed preponderant M2 polarization compared to KCs in ALD-sensitive C57BL6/J mice, and ongoing alcohol drinkers with minimal hepatic injury displayed higher hepatic expression of M2 genes as compared to patients with severe liver lesions (128). Mechanisms studies showed that M2-polarized KCs promoted death of selective M1-polarized KCs by producing IL-10 and triggered hepatocytes senescence (128, 129). Furthermore, many studies have demonstrated that ω-3 polyunsaturated fatty acids (PUFAs) such as docosahexaenoic acid (DHA) and DHA-rich fish oil could attenuate ALD (130–132). As unsaturated fatty acids and saturated fatty acids could induce macrophages M2 activation and M1 activation, respectively (133–135), it would be possible that these PUFAs may confer protection against ALD by modulating the polarization of KCs.
Potential Therapeutic Targets for ALD Treatment Related with Gut Endotoxin/LPS–Liver KCs Axis
The critical roles of KCs and related inflammatory cascade in the pathogenesis of ALD make it a promising target in pharmaceutical drug developments for ALD treatment. Several potentially therapeutic drugs, including antibiotics, probiotics, anti-TNF-α, and anti-IL-1β agents, have been evaluated or are under evaluation for the efficacy in ALD treatment in animal studies and/or clinical trials. Specially, several anti-TNF-α agents, including pentoxifylline, infliximab, and etanercept, have been tested for the treatment of severe AH. Unfortunately, although animal studies usually showed promising effects, clinical trials have brought conflicting results.
Antibiotics
Theoretically, antibiotics can block the development of ALD, as gut-derived endotoxin/LPS initiates the TLR-4 signaling pathway, leading to the production of pro-inflammatory cytokines. Indeed, animal studies showed that antibiotics (polymyxin B and neomycin) completely prevented the elevation of aspartate aminotransferase activity and significantly reduced the average hepatic pathological scores in ethanol-exposed rats (30). Rifaximin, a non-absorbed antibiotic with few side effects and little evidence for resistance, has been evaluated for the treatment of alcoholic cirrhosis in clinical trials (136). One study found that rifaximin (1200 mg/day) could improve thrombocytopenia in patients with alcoholic cirrhosis (137). Portal hypertension is crucial in the transition from the compensated phase to the decompensated phase of cirrhosis, as it is associated with the most severe complication of cirrhosis, including hepatorenal syndrome (138, 139). Results of two studies showed that intestinal decontamination with rifaximin (1200 mg/day) for 4 weeks led to significant decrease of hepatic venous pressure gradient (HVPG), cardiac output, and plasma rennin activity, accompanied with significant increase of glomerular filtration rate and natriuresis (140, 141). A study suggested that long-term rifaximin administration (1200 mg/day, followed up for 5 years) might be associated with reduced risk of developing complications of portal hypertension and improved survival in patients with alcohol-related decompensated cirrhosis (142). Results of these studies suggest that rifaximin may be beneficial for the reduction in portal pressure and improvement of renal function in decompensated alcoholic cirrhotic patients. These results are quite promising, and are needed to be further evaluated by well-designed randomized-controlled trials with larger sample size.
Probiotics
Probiotics are defined as “live microorganism which can confer health benefits on the host when administered in adequate amounts” (143). Probiotics are non-pathogenic beneficial flora that act to regulate and maintain a stable intestinal environment and promote micro-ecological balance (144). The beneficial effects of probiotics against ALD have been gaining increased interest in recent years. Many probiotics such as Lactobacillus rhamnosus and probiotic mixtures VSL#3 have been demonstrated to protect against both chronic and acute ALD in animal studies (117, 145–152). The mechanisms included the improvement of intestinal integrity, reduction of TLR-4 expression, and activation of AMP-activated protein kinase (AMPK) (117, 149, 151, 152). A pilot prospective, randomized, clinical trial showed that probiotics supplementation could restore bowel flora and improve liver enzymes in ALD patients (153). A randomized-controlled multicenter study showed that 7 days of oral supplementation with probiotics restored the bowel flora and improved the LPS in patients with AH (29). Results of these studies provide a scientific rationale to further test probiotics for treatment and/or prevention of ALD in humans.
Anti-TNF-α Agents
Several anti-TNF-α agents have gained great interest due to the important roles of TNF-α in the pathogenesis of AH. Three anti-TNF-α agents have been investigated for AH treatment, i.e., pentoxifylline, infliximab, and etanercept. However, the clinical use of these agents is yet not recommended due to the poor clinical outcomes observed in the largest clinical trials.
Pentoxifylline is a non-selective phosphodiesterase inhibitor, which can inhibit endotoxin-induced production of TNF-α (154). A pilot study showed that pentoxifylline (400 mg × 3 times/day for 28 days) significantly reduced the mortality of severe AH (46 vs. 25%, p = 0.037); while another study showed pentoxifylline treatment significantly improved the renal and hepatic functions accompanied with a trend of decreasing mortality as compared with the placebo group (155, 156). However, the studies comparing the efficacy between pentoxifylline and corticosteroid (the first-line treatment for severe AH) have produced conflicting results. One study found that pentoxifylline was superior to corticosteroid in reducing the mortality and hepatorenal syndrome (157), while another showed that the efficacy of the pentoxifylline was not statistically equivalent to that of corticosteroid (158). In addition, the combination of pentoxifylline and corticosteroid did not significantly improve the 28-day and 6-month survival of severe AH patients when compared with treatment with corticosteroid alone (159). A recently published multicenter, double-blind, randomized trials with a 2-by-2 factorial design showed that pentoxifylline did not improve survival of AH patients (160). Due to these conflicting results, several meta-analyses have been performed, and the results showed that pentoxifylline could not improve the short-term survival of AH patients compared with placebo; however, combination of pentoxifylline and corticosteroid could reduce the incidences of hepatorenal syndrome and the infection risk compared with corticosteroid therapy alone (161–163).
Infliximab is a monoclonal human/mouse anti-TNF-α antibody which can bind to TNF-α with high affinity when administered by intravenous infusion. In contrast to the efficacy observed in the treatment of rheumatoid arthritis and Crohn’s disease, the efficacy of infliximab in the treatment of AH is controversial. Animals study showed that infliximab at 1 mg/kg body weight exhibited immunomodulatory and anti-inflammatory effects, whereas infliximab at 10 mg/kg body weight significantly decreased lipid accumulation in experimental AH model (164). An earlier pilot clinical trial found that serum bilirubin levels, Maddrey scores, neutrophil counts, and C-reactive protein levels fell significantly within the first month in patients receiving single dose of infliximab (5 mg/kg body weight) (165). A randomized-controlled clinical trial in 20 biopsy-proven AH patients showed that combination of single dose of infliximab (5 mg/kg body weight) and prednisone (40 mg/day for 28 days) improved the Maddrey scores and laboratory parameters, while prednisone treatment alone had no significant alteration on these parameters (166). However, another multicenter double-blind randomized-controlled trial of infliximab associated with prednisolone in 36 AH patients found that the survival rate and frequency of severe infections within 2 months was higher in infliximab and prednisolone group than in the placebo and prednisolone group (167). The study was stopped by the follow-up committee and the sponsor at the end of 2 months, as more deaths occurred in infliximab co-treatment groups. Another study showed that a single dose of infliximab (5 mg/kg, i.v.) was associated with significant improvement in parameters of severity, but failed to suppress the infection (168).
Etanercept is a genetically engineered, soluble, systemic TNF-α blocker that competitively binds to and neutralizes both soluble and transmembrane forms of TNF-α (169). A pilot study examining the effects of etanercept in 13 patients with moderate to severe AH showed that the survival rate of 1 month was 92% (12/13), although 23% of patients (3/13) develop serious adverse effects including infection, hepatorenal decompensation, and gastrointestinal bleeding (170). However, a multicenter, randomized, double-blind, placebo-controlled study in 48 patients with moderate to severe AH (defined as model for end-stage liver disease score ≥15) found that etanercept (given six times over 3 weeks) did not significantly affect the 1-month survival rates compared with placebo treatment, while the 6-month mortality rate was significantly higher in the etanercept group compared with the placebo group (57.7 vs. 22.7%, p = 0.017) (171).
IL-1β Signaling Inhibitors
The key role of inflammasome activation in ALD progression has been confirmed using experimental mouse models. Activation of inflammasome leads to the production of pro-inflammatory cytokine IL-1β, which has been demonstrated to play critical roles in alcoholic steatohepatitis (95). IL-1β signals through IL-1R, leading to inflammatory cascade. IL-1Ra is a naturally occurring cytokine that binds to IL-1R to regulate the actions of IL-1β and control inflammation (172). Animal studies have shown that recombinant IL-1Ra could significantly attenuate ethanol-induced liver inflammation and injury (91, 172). As anti-TNF-α agents did not bring convincing results in some clinical trials, IL-1Ra has gained great interest, and a clinical trial is currently underway to test the efficacy of IL-1Ra (Anakinra) in the treatment of AH (173).
Other Drugs Potentially Used for ALD Treatment
In addition to the above referred drugs for ALD treatment, the gut–liver axis also provides many other potentially therapeutic targets, including intestinal permeability maintaining agents, LPS antibodies, TLR-4 antagonist, and caspase inhibitors. The efficiency of these drugs has been examining in several ongoing clinic trials (11).
Conclusion and Remarks
The causal roles of KCs in the pathogenesis of ALD have been highlighted, and the underlying molecular mechanisms have been revealed. KCs are activated in both chronic and acute ALD, which is driven by gut-sourced endotoxin/LPS. LPS mediates activation of TLR-4 signaling pathway in classically activated KCs (M1 phenotypes), leading to the overproduction of ROS, pro-inflammatory cytokines, and chemokines. The increased release of pro-inflammatory cytokines and the infiltration of other inflammatory cells such as the neutrophils finally cause liver injury.
The “gut–liver axis” suggests that suppression of KCs activation (such as suppressing the production of LPS, reducing the combination of LPS with TLRs, etc.) and elimination of cytotoxic products secreted by KCs (such as using TNF-α antibodies and IL-1Ra) may be beneficial to the treatment of ALD. Several drugs (such as rifaximin, pentoxifylline, and infliximab) have been evaluated or are under evaluation for ALD treatment in randomized clinical trials. Although animal studies usually showed beneficial effects, conflicting results were obtained from studies about the anti-TNF-α agents in AH treatment, which might be related with differences in the study design, the enrollment criteria and the baseline characteristics of AH patients, and the ethnicity of study population. For example, although the same threshold of AH (Maddrey scores ≥ 32) was used in all the trials, the baseline characteristics of patients such as the age and rate of encephalopathy in these trials were not consistent, which might influence the accuracy of the results as these characteristics could influence mortality (160, 174, 175). Importantly, the study by Petrasek et al. suggests the therapeutic potential of IL-1β inhibitors for the treatment of ALD. It is urgent to investigate whether IL-1β inhibitors are beneficial for AH patients, as severe AH is associated with high mortality and lack of effective treatments (172, 176). Furthermore, the study by Wan et al. provided solid evidence for the protective roles of M2 KCs against early stage of ALD (128). Thus, screening pharmacological regulators for KCs toward M2 polarization may provide additional therapeutic agents. The combination of these potentially therapeutic drugs with other agents (such as hepatoprotective agents, including zinc, melatonin, silymarin, etc.) may bring encouraging results.
Although the roles of KCs in the pathogenesis of ALD have been clearly demonstrated, there are still some questions that need to be addressed. First, the autophagic response of KCs in ALD remains unclear. Autophagy, an intracellular self-digestion process, can regulate lipid metabolism in hepatocytes and also have anti-inflammatory effects (177, 178). Activation of autophagy could reduce alcoholic and metabolic steatosis by enhancing the decomposition of lipid and suppress LPS-induced inflammation and liver injury (178–181). Although ethanol exposure induces autophagy in the mice/rat livers and in cultured hepatocytes, no studies investigated the autophagic response of KCs in ALD (182, 183). It has been demonstrated that LPS can induce autophagy in human and murine macrophages (184, 185). Therefore, it appears to be reasonable that ethanol may also lead to the activation of autophagy in KCs as ethanol could increase LPS translocation from gut to liver. The autophagic response of KCs and its roles in ALD are needed to be investigated. Second, the relationships between autophagy and polarization in KCs are needed to be elucidated. One previous study showed that KCs from mice with Atg 5 deficiency in macrophages exhibited increased M1 polarization and decreased M2 polarization, suggesting that autophagy has a critical regulatory function in macrophage polarization (178). It should also be necessary to explore whether KCs polarization regulator can influence autophagy activity. Third, the highly heterogeneous KCs can be classified into different subpopulations. For example, two F4/80+ KCs subsets have been revealed, a CD68+ subset with phagocytic activity and a CD11b+ subset with cytokine-producing capacity (6, 186). These different KC subsets may play different roles in the pathogenesis of ALD, which need to be further studied.
In addition to ALD, KCs activation may be also involved in the pathogenesis of alcohol dependence, which is characterized by an individual’s continued drinking despite negative consequences related to alcohol use (187–190). It was found that intestinal permeability and blood LPS were largely increased in alcohol-dependent subjects and the pro-inflammatory cytokines level was positively correlated with craving (191). Evidences from postmortem alcoholic brain showed that chronic ethanol could increase degradation of tight junctions and extracellular matrix in human brain (192). Additionally, animal study demonstrated that TNF-α produced by KCs could transfer to the brain through TNF-R to induce the synthesis of additional TNF-α, creating a persistent and self-propelling neuroinflammation in the brain (193). Results of these studies suggest that the cytokines produced by KCs may cross the blood–brain barrier (BBB) via diffusion or active transport, or by BBB alterations (189, 190). Further studies about the roles of KCs in the pathogenesis of alcohol dependence may reveal new therapeutic targets for pathologic drinking behaviors.
Author Contributions
TZ and K-QX designed the study. TZ, C-LZ, MX, RY, and K-QX contributed to the literature search, interpretation, writing, and proofreading of the manuscript. TZ generated the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the National Science Foundation of China (NSFC Grant No. 81473004, 81102153) and the Young Scholars Program of Shandong University (Grant No. 2015WLJH52).
Abbreviations
4-HNE, 4-hydroxynonenal; AH, alcoholic hepatitis; ALD, alcoholic liver disease; AMPK, AMP-activated protein kinase; AP-1, activation protein 1; BAL, bronchoalveolar lavage; CD14, cluster of differentiation 14; CYP2E1, cytochrome P4502E1; CMZ, clomethiazole; DHA, docosahexaenoic acid; DPI, diphenyleneiodonium sulfate; Egr-1, early growth response 1; ERK1/2, extracellular signal regulated kinases 1 and 2; IFN, interferon; IKK, IκB kinase; IL-1β, interleukin 1β; IL-10, interleukin 10; IL-1Ra, IL-1 receptor antagonist; iNOS, inducible nitric oxide synthase; IRAK-4, IL-1 receptor-associated kinase-4; IRF-3, interferon regulatory factor 3; KCs, Kupffer cells; LPS, lipopolysaccharide; LBP, LPS-binding protein; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; MD-2, myeloid differentiation factor 2; MEOS, microsomal ethanol-oxidizing system; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor kappa B; NOX, NADPH oxidase; PUFAs, polyunsaturated fatty acids; ROS, reactive oxygen species; SOD, superoxide dismutase; TAK-1, transforming growth factor β-activated kinase 1; TNF-α, tumor necrosis factor α; TNFR, tumor necrosis factor receptor 1; TLR-4, toll-like receptor 4; TRIF, toll-interleukin-1 receptor domain-containing adaptor inducing interferon-β; TRAF-6, TNF receptor-associated factor 6.
References
1. Wu XQ, Yang Y, Li WX, Cheng YH, Li XF, Huang C, et al. Telomerase reverse transcriptase acts in a feedback loop with NF-kappaB pathway to regulate macrophage polarization in alcoholic liver disease. Sci Rep (2016) 6:18685. doi:10.1038/srep18685
2. Zeng T, Zhang CL, Song FY, Zhao XL, Yu LH, Zhu ZP, et al. The activation of HO-1/Nrf-2 contributes to the protective effects of diallyl disulfide (DADS) against ethanol-induced oxidative stress. Biochim Biophys Acta (2013) 1830(10):4848–59. doi:10.1016/j.bbagen.2013.06.028
3. Chen YY, Zhang CL, Zhao XL, Xie KQ, Zeng T. Inhibition of cytochrome P4502E1 by chlormethiazole attenuated acute ethanol-induced fatty liver. Chem Biol Interact (2014) 222C:18–26. doi:10.1016/j.cbi.2014.08.009
4. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol (2013) 3(2):785–97. doi:10.1002/cphy.c120026
5. Smith K. Liver disease: Kupffer cells regulate the progression of ALD and NAFLD. Nat Rev Gastroenterol Hepatol (2013) 10(9):503. doi:10.1038/nrgastro.2013.140
6. Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol (2014) 96(4):657–65. doi:10.1189/jlb.6A0114-004RR
8. McCuskey RS, McCuskey PA. Fine structure and function of Kupffer cells. J Electron Microsc Tech (1990) 14(3):237–46. doi:10.1002/jemt.1060140305
9. Diehl AM. Nonalcoholic steatosis and steatohepatitis IV. Nonalcoholic fatty liver disease abnormalities in macrophage function and cytokines. Am J Physiol Gastrointest Liver Physiol (2002) 282(1):G1–5. doi:10.1152/ajpgi.00384.2001
10. Saberi B, Dadabhai AS, Jang YY, Gurakar A, Mezey E. Current management of alcoholic hepatitis and future therapies. J Clin Transl Hepatol (2016) 4:113–22. doi:10.14218/JCTH.2016.00006
11. Singal AK, Shah VH. Therapeutic strategies for the treatment of alcoholic hepatitis. Semin Liver Dis (2016) 36(1):56–68. doi:10.1055/s-0036-1571297
12. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5(12):953–64. doi:10.1038/nri1733
13. Karakucuk I, Dilly SA, Maxwell JD. Portal tract macrophages are increased in alcoholic liver disease. Histopathology (1989) 14(3):245–53. doi:10.1111/j.1365-2559.1989.tb02143.x
14. Shiratori Y, Teraoka H, Matano S, Matsumoto K, Kamii K, Tanaka M. Kupffer cell function in chronic ethanol-fed rats. Liver (1989) 9(6):351–9. doi:10.1111/j.1600-0676.1989.tb00423.x
15. Kamimura S, Tsukamoto H. Cytokine gene expression by Kupffer cells in experimental alcoholic liver disease. Hepatology (1995) 22(4 Pt 1):1304–9. doi:10.1002/hep.1840220441
16. Lin HZ, Yang SQ, Zeldin G, Diehl AM. Chronic ethanol consumption induces the production of tumor necrosis factor-alpha and related cytokines in liver and adipose tissue. Alcohol Clin Exp Res (1998) 22(5 Suppl):231S–7S. doi:10.1111/j.1530-0277.1998.tb04008.x
17. Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology (1994) 20(2):453–60. doi:10.1002/hep.1840200227
18. Knecht KT, Adachi Y, Bradford BU, Iimuro Y, Kadiiska M, Xuang QH, et al. Free radical adducts in the bile of rats treated chronically with intragastric alcohol: inhibition by destruction of Kupffer cells. Mol Pharmacol (1995) 47(5):1028–34.
19. Koop DR, Klopfenstein B, Iimuro Y, Thurman RG. Gadolinium chloride blocks alcohol-dependent liver toxicity in rats treated chronically with intragastric alcohol despite the induction of CYP2E1. Mol Pharmacol (1997) 51(6):944–50.
20. Cederbaum AI, Yang L, Wang X, Wu D. CYP2E1 sensitizes the liver to LPS- and TNF alpha-induced toxicity via elevated oxidative and nitrosative stress and activation of ASK-1 and JNK mitogen-activated kinases. Int J Hepatol (2012) 2012:582790. doi:10.1155/2012/582790
21. Hersoug LG, Moller P, Loft S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: implications for inflammation and obesity. Obes Rev (2016) 17(4):297–312. doi:10.1111/obr.12370
22. Worthington BS, Meserole L, Syrotuck JA. Effect of daily ethanol ingestion on intestinal permeability to macromolecules. Am J Dig Dis (1978) 23(1):23–32. doi:10.1007/BF01072571
23. Parlesak A, Schafer C, Schutz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol (2000) 32(5):742–7. doi:10.1016/S0168-8278(00)80242-1
24. Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M, et al. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol (2009) 50(3):538–47. doi:10.1016/j.jhep.2008.10.028
25. Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res (2009) 33(10):1836–46. doi:10.1111/j.1530-0277.2009.01022.x
26. Bode JC, Bode C, Heidelbach R, Durr HK, Martini GA. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology (1984) 31(1):30–4.
27. Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol (1998) 275(4 Pt 1):G605–11.
28. Nanji AA, Khettry U, Sadrzadeh SM, Yamanaka T. Severity of liver injury in experimental alcoholic liver disease. Correlation with plasma endotoxin, prostaglandin E2, leukotriene B4, and thromboxane B2. Am J Pathol (1993) 142(2):367–73.
29. Han SH, Suk KT, Kim DJ, Kim MY, Baik SK, Kim YD, et al. Effects of probiotics (cultured Lactobacillus subtilis/Streptococcus faecium) in the treatment of alcoholic hepatitis: randomized-controlled multicenter study. Eur J Gastroenterol Hepatol (2015) 27(11):1300–6. doi:10.1097/MEG.0000000000000458
30. Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology (1995) 108(1):218–24. doi:10.1016/0016-5085(95)90027-6
31. Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology (2001) 34(1):101–8. doi:10.1053/jhep.2001.25350
32. Yin M, Bradford BU, Wheeler MD, Uesugi T, Froh M, Goyert SM, et al. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol (2001) 166(7):4737–42. doi:10.4049/jimmunol.166.7.4737
33. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine (2008) 42(2):145–51. doi:10.1016/j.cyto.2008.01.006
34. Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes Infect (2004) 6(15):1361–7. doi:10.1016/j.micinf.2004.08.015
35. Miyake K. Innate immune sensing of pathogens and danger signals by cell surface toll-like receptors. Semin Immunol (2007) 19(1):3–10. doi:10.1016/j.smim.2006.12.002
36. Plociennikowska A, Hromada-Judycka A, Borzecka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci (2015) 72(3):557–81. doi:10.1007/s00018-014-1762-5
37. Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol (2005) 3(1):36–46. doi:10.1038/nrmicro1068
38. Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol (2005) 6(11):1087–95. doi:10.1038/ni1255
39. Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science (1988) 242(4878):540–6. doi:10.1126/science.3140380
40. Scherle PA, Jones EA, Favata MF, Daulerio AJ, Covington MB, Nurnberg SA, et al. Inhibition of MAP kinase kinase prevents cytokine and prostaglandin E2 production in lipopolysaccharide-stimulated monocytes. J Immunol (1998) 161(10):5681–6.
41. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature (2001) 410(6824):37–40. doi:10.1038/35065000
42. Guha M, O’Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, et al. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood (2001) 98(5):1429–39. doi:10.1182/blood.V98.5.1429
43. Nanji AA, Jokelainen K, Rahemtulla A, Miao L, Fogt F, Matsumoto H, et al. Activation of nuclear factor kappa B and cytokine imbalance in experimental alcoholic liver disease in the rat. Hepatology (1999) 30(4):934–43. doi:10.1002/hep.510300402
44. Jokelainen K, Reinke LA, Nanji AA. Nf-kappab activation is associated with free radical generation and endotoxemia and precedes pathological liver injury in experimental alcoholic liver disease. Cytokine (2001) 16(1):36–9. doi:10.1006/cyto.2001.0930
45. Uesugi T, Froh M, Arteel GE, Bradford BU, Gabele E, Wheeler MD, et al. Delivery of IkappaB superrepressor gene with adenovirus reduces early alcohol-induced liver injury in rats. Hepatology (2001) 34(6):1149–57. doi:10.1053/jhep.2001.29400
46. Iimuro Y, Gallucci RM, Luster MI, Kono H, Thurman RG. Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology (1997) 26(6):1530–7. doi:10.1002/hep.510260621
47. Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology (1999) 117(4):942–52. doi:10.1016/S0016-5085(99)70354-9
48. Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med (2001) 31(12):1544–9. doi:10.1016/S0891-5849(01)00748-1
49. Lu Y, Zhuge J, Wang X, Bai J, Cederbaum AI. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology (2008) 47(5):1483–94. doi:10.1002/hep.22222
50. Zeng T, Xie KQ. Ethanol and liver: recent advances in the mechanisms of ethanol-induced hepatosteatosis. Arch Toxicol (2009) 83(12):1075–81. doi:10.1007/s00204-009-0457-4
51. Cederbaum AI. Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig Dis (2010) 28(6):802–11. doi:10.1159/000324289
52. Abdelmegeed MA, Banerjee A, Jang S, Yoo SH, Yun JW, Gonzalez FJ, et al. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic Biol Med (2013) 65:1238–45. doi:10.1016/j.freeradbiomed.2013.09.009
53. Kono H, Rusyn I, Uesugi T, Yamashina S, Connor HD, Dikalova A, et al. Diphenyleneiodonium sulfate, an NADPH oxidase inhibitor, prevents early alcohol-induced liver injury in the rat. Am J Physiol Gastrointest Liver Physiol (2001) 280(5):G1005–12.
54. Kono H, Rusyn I, Yin M, Gabele E, Yamashina S, Dikalova A, et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest (2000) 106(7):867–72. doi:10.1172/JCI9020
55. Bode C, Bode JC. Activation of the innate immune system and alcoholic liver disease: effects of ethanol per se or enhanced intestinal translocation of bacterial toxins induced by ethanol? Alcohol Clin Exp Res (2005) 29(11 Suppl):166S–71S. doi:10.1097/01.alc.0000189280.19073.28
56. Honchel R, Ray MB, Marsano L, Cohen D, Lee E, Shedlofsky S, et al. Tumor necrosis factor in alcohol enhanced endotoxin liver injury. Alcohol Clin Exp Res (1992) 16(4):665–9. doi:10.1111/j.1530-0277.1992.tb00656.x
57. Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology (2000) 32(5):1008–17. doi:10.1053/jhep.2000.19621
58. Kishore R, McMullen MR, Nagy LE. Stabilization of tumor necrosis factor alpha mRNA by chronic ethanol: role of A + U-rich elements and p38 mitogen-activated protein kinase signaling pathway. J Biol Chem (2001) 276(45):41930–7. doi:10.1074/jbc.M107181200
59. Kishore R, Hill JR, McMullen MR, Frenkel J, Nagy LE. ERK1/2 and Egr-1 contribute to increased TNF-alpha production in rat Kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol (2002) 282(1):G6–15. doi:10.1152/ajpgi.00328.2001
60. Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol (2006) 79(6):1348–56. doi:10.1189/jlb.1005613
61. Nagy LE. Stabilization of tumor necrosis factor-alpha mRNA in macrophages in response to chronic ethanol exposure. Alcohol (2004) 33(3):229–33. doi:10.1016/j.alcohol.2004.09.002
62. Deaciuc IV, D’Souza NB, Spitzer JJ. Tumor necrosis factor-alpha cell-surface receptors of liver parenchymal and nonparenchymal cells during acute and chronic alcohol administration to rats. Alcohol Clin Exp Res (1995) 19(2):332–8. doi:10.1111/j.1530-0277.1995.tb01511.x
63. Jarvelainen HA, Fang C, Ingelman-Sundberg M, Lindros KO. Effect of chronic coadministration of endotoxin and ethanol on rat liver pathology and proinflammatory and anti-inflammatory cytokines. Hepatology (1999) 29(5):1503–10. doi:10.1002/hep.510290508
64. Lukkari TA, Jarvelainen HA, Oinonen T, Kettunen E, Lindros KO. Short-term ethanol exposure increases the expression of Kupffer cell CD14 receptor and lipopolysaccharide binding protein in rat liver. Alcohol Alcohol (1999) 34(3):311–9. doi:10.1093/alcalc/34.3.311
65. Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology (2008) 48(4):1224–31. doi:10.1002/hep.22470
66. Tuma DJ. Role of malondialdehyde-acetaldehyde adducts in liver injury. Free Radic Biol Med (2002) 32(4):303–8. doi:10.1016/S0891-5849(01)00742-0
67. Duryee MJ, Klassen LW, Freeman TL, Willis MS, Tuma DJ, Thiele GM. Lipopolysaccharide is a cofactor for malondialdehyde-acetaldehyde adduct-mediated cytokine/chemokine release by rat sinusoidal liver endothelial and Kupffer cells. Alcohol Clin Exp Res (2004) 28(12):1931–8. doi:10.1097/01.ALC.0000148115.90045.C5
68. Nakamura Y, Yokoyama H, Okamura Y, Ohgo H, Fukuda M, Horie Y, et al. Evidence for ethanol oxidation by Kupffer cells. Alcohol Clin Exp Res (1999) 23(4 Suppl):92S–5S. doi:10.1111/j.1530-0277.1999.tb04542.x
69. Yoshinari K, Sato T, Okino N, Sugatani J, Miwa M. Expression and induction of cytochromes p450 in rat white adipose tissue. J Pharmacol Exp Ther (2004) 311(1):147–54. doi:10.1124/jpet.104.067066
70. Takahashi T, Lasker JM, Rosman AS, Lieber CS. Induction of cytochrome P-4502E1 in the human liver by ethanol is caused by a corresponding increase in encoding messenger RNA. Hepatology (1993) 17(2):236–45. doi:10.1016/0270-9139(93)90083-Y
71. Lieber CS. Microsomal ethanol-oxidizing system (MEOS): the first 30 years (1968-1998) – a review. Alcohol Clin Exp Res (1999) 23(6):991–1007. doi:10.1097/00000374-199906000-00006
72. Lieber CS. The discovery of the microsomal ethanol oxidizing system and its physiologic and pathologic role. Drug Metab Rev (2004) 36(3–4):511–29. doi:10.1081/DMR-200033441
73. Cederbaum AI, Lu Y, Wang X, Wu D. Synergistic toxic interactions between CYP2E1, LPS/TNFalpha, and JNK/p38 MAP kinase and their implications in alcohol-induced liver injury. Adv Exp Med Biol (2015) 815:145–72. doi:10.1007/978-3-319-09614-8_9
74. Bradford BU, Kono H, Isayama F, Kosyk O, Wheeler MD, Akiyama TE, et al. Cytochrome P450 CYP2E1, but not nicotinamide adenine dinucleotide phosphate oxidase, is required for ethanol-induced oxidative DNA damage in rodent liver. Hepatology (2005) 41(2):336–44. doi:10.1002/hep.20532
75. Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radic Biol Med (2010) 49(9):1406–16. doi:10.1016/j.freeradbiomed.2010.07.026
76. Ronis MJ, Korourian S, Blackburn ML, Badeaux J, Badger TM. The role of ethanol metabolism in development of alcoholic steatohepatitis in the rat. Alcohol (2010) 44(2):157–69. doi:10.1016/j.alcohol.2009.11.002
77. Zeng T, Zhang CL, Song FY, Zhao XL, Xie KQ. CMZ reversed chronic ethanol-induced disturbance of PPAR-alpha possibly by suppressing oxidative stress and PGC-1alpha acetylation, and activating the MAPK and GSK3beta pathway. PLoS One (2014) 9(6):e98658. doi:10.1371/journal.pone.0098658
78. Wu D, Cederbaum AI. Ethanol cytotoxicity to a transfected HepG2 cell line expressing human cytochrome P4502E1. J Biol Chem (1996) 271(39):23914–9. doi:10.1074/jbc.271.39.23914
79. Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med (2008) 44(5):723–38. doi:10.1016/j.freeradbiomed.2007.11.004
80. Yang L, Wu D, Wang X, Cederbaum AI. Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free Radic Biol Med (2012) 53(5):1170–80. doi:10.1016/j.freeradbiomed.2012.06.029
81. Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J Hepatol (2013) 58(2):395–8. doi:10.1016/j.jhep.2012.08.018
82. Tsukamoto H, Takei Y, McClain CJ, Joshi-Barve S, Hill D, Schmidt J, et al. How is the liver primed or sensitized for alcoholic liver disease? Alcohol Clin Exp Res (2001) 25(5 Suppl ISBRA):171S–81S. doi:10.1111/j.1530-0277.2001.tb02393.x
83. Liu H, Jones BE, Bradham C, Czaja MJ. Increased cytochrome P-450 2E1 expression sensitizes hepatocytes to c-Jun-mediated cell death from TNF-alpha. Am J Physiol Gastrointest Liver Physiol (2002) 282(2):G257–66. doi:10.1152/ajpgi.00304.2001
84. Pastorino JG, Hoek JB. Ethanol potentiates tumor necrosis factor-alpha cytotoxicity in hepatoma cells and primary rat hepatocytes by promoting induction of the mitochondrial permeability transition. Hepatology (2000) 31(5):1141–52. doi:10.1053/he.2000.7013
85. Forsyth CB, Voigt RM, Shaikh M, Tang Y, Cederbaum AI, Turek FW, et al. Role for intestinal CYP2E1 in alcohol-induced circadian gene-mediated intestinal hyperpermeability. Am J Physiol Gastrointest Liver Physiol (2013) 305(2):G185–95. doi:10.1152/ajpgi.00354.2012
86. Ye Q, Wang X, Wang Q, Xia M, Zhu Y, Lian F, et al. Cytochrome P4502E1 inhibitor, chlormethiazole, decreases lipopolysaccharide-induced inflammation in rat Kupffer cells with ethanol treatment. Hepatol Res (2013) 43:1115–23. doi:10.1111/hepr.12063
87. Forsyth CB, Voigt RM, Keshavarzian A. Intestinal CYP2E1: a mediator of alcohol-induced gut leakiness. Redox Biol (2014) 3:40–6. doi:10.1016/j.redox.2014.10.002
88. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi:10.1016/j.cell.2010.01.040
89. Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol (2012) 57(3):642–54. doi:10.1016/j.jhep.2012.03.035
90. Wang HJ, Gao B, Zakhari S, Nagy LE. Inflammation in alcoholic liver disease. Annu Rev Nutr (2012) 32:343–68. doi:10.1146/annurev-nutr-072610-145138
91. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest (2012) 122(10):3476–89. doi:10.1172/JCI60777
92. Tilg H, Wilmer A, Vogel W, Herold M, Nolchen B, Judmaier G, et al. Serum levels of cytokines in chronic liver diseases. Gastroenterology (1992) 103(1):264–74. doi:10.1016/0016-5085(92)91122-K
93. Valles SL, Blanco AM, Azorin I, Guasch R, Pascual M, Gomez-Lechon MJ, et al. Chronic ethanol consumption enhances interleukin-1-mediated signal transduction in rat liver and in cultured hepatocytes. Alcohol Clin Exp Res (2003) 27(12):1979–86. doi:10.1097/01.ALC.0000099261.87880.21
94. Nagy LE. The role of innate immunity in alcoholic liver disease. Alcohol Res (2015) 37(2):237–50.
95. Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol (2015) 12(7):387–400. doi:10.1038/nrgastro.2015.94
96. Shukla SD, Pruett SB, Szabo G, Arteel GE. Binge ethanol and liver: new molecular developments. Alcohol Clin Exp Res (2013) 37(4):550–7. doi:10.1111/acer.12011
97. Reuben A. Alcohol and the liver. Curr Opin Gastroenterol (2007) 23(3):283–91. doi:10.1097/MOG.0b013e3280f27582
98. Mathurin P, Deltenre P. Effect of binge drinking on the liver: an alarming public health issue? Gut (2009) 58(5):613–7. doi:10.1136/gut.2007.145573
99. Patrick ME, Schulenberg JE. Prevalence and predictors of adolescent alcohol use and binge drinking in the United States. Alcohol Res (2013) 35(2):193–200.
100. Kanuri G, Weber S, Volynets V, Spruss A, Bischoff SC, Bergheim I. Cinnamon extract protects against acute alcohol-induced liver steatosis in mice. J Nutr (2009) 139(3):482–7. doi:10.3945/jn.108.100495
101. Pruett SB, Zheng Q, Fan R, Matthews K, Schwab C. Acute exposure to ethanol affects toll-like receptor signaling and subsequent responses: an overview of recent studies. Alcohol (2004) 33(3):235–9. doi:10.1016/j.alcohol.2004.08.003
102. D’Souza NB, Bagby GJ, Nelson S, Lang CH, Spitzer JJ. Acute alcohol infusion suppresses endotoxin-induced serum tumor necrosis factor. Alcohol Clin Exp Res (1989) 13(2):295–8. doi:10.1111/j.1530-0277.1989.tb00329.x
103. Nelson S, Bagby GJ, Bainton BG, Summer WR. The effects of acute and chronic alcoholism on tumor necrosis factor and the inflammatory response. J Infect Dis (1989) 160(3):422–9. doi:10.1093/infdis/160.3.422
104. Bautista AP, Spitzer JJ. Cross-tolerance between acute alcohol intoxication and endotoxemia. Alcohol Clin Exp Res (1996) 20(8):1395–400. doi:10.1111/j.1530-0277.1996.tb01139.x
105. Vinson RB, Carroll JL, Pruett SB. Mechanism of suppressed neutrophil mobilization in a mouse model for binge drinking: role of glucocorticoids. Am J Physiol (1998) 275(4 Pt 2):R1049–57.
106. Boe DM, Nelson S, Zhang P, Bagby GJ. Acute ethanol intoxication suppresses lung chemokine production following infection with Streptococcus pneumoniae. J Infect Dis (2001) 184(9):1134–42. doi:10.1086/323661
107. Pruett SB, Fan R, Zheng Q. Acute ethanol administration profoundly alters poly I:C-induced cytokine expression in mice by a mechanism that is not dependent on corticosterone. Life Sci (2003) 72(16):1825–39. doi:10.1016/S0024-3205(02)02507-9
108. Pruett SB, Schwab C, Zheng Q, Fan R. Suppression of innate immunity by acute ethanol administration: a global perspective and a new mechanism beginning with inhibition of signaling through TLR3. J Immunol (2004) 173(4):2715–24. doi:10.4049/jimmunol.173.4.2715
109. Pruett SB, Zheng Q, Fan R, Matthews K, Schwab C. Ethanol suppresses cytokine responses induced through toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol (2004) 33(2):147–55. doi:10.1016/j.alcohol.2004.08.001
110. Dai Q, Pruett SB. Ethanol suppresses LPS-induced toll-like receptor 4 clustering, reorganization of the actin cytoskeleton, and associated TNF-alpha production. Alcohol Clin Exp Res (2006) 30(8):1436–44. doi:10.1111/j.1530-0277.2006.00172.x
111. Pruett SB, Fan R. Ethanol inhibits LPS-induced signaling and modulates cytokine production in peritoneal macrophages in vivo in a model for binge drinking. BMC Immunol (2009) 10:49. doi:10.1186/1471-2172-10-49
112. Spolarics Z, Spitzer JJ, Wang JF, Xie J, Kolls J, Greenberg S. Alcohol administration attenuates LPS-induced expression of inducible nitric oxide synthase in Kupffer and hepatic endothelial cells. Biochem Biophys Res Commun (1993) 197(2):606–11. doi:10.1006/bbrc.1993.2522
113. Enomoto N, Ikejima K, Bradford B, Rivera C, Kono H, Brenner DA, et al. Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxin. Gastroenterology (1998) 115(2):443–51. doi:10.1016/S0016-5085(98)70211-2
114. Yamashina S, Wheeler MD, Rusyn I, Ikejima K, Sato N, Thurman RG. Tolerance and sensitization to endotoxin in Kupffer cells caused by acute ethanol involve interleukin-1 receptor-associated kinase. Biochem Biophys Res Commun (2000) 277(3):686–90. doi:10.1006/bbrc.2000.3738
115. Zhou Z, Wang L, Song Z, Lambert JC, McClain CJ, Kang YJ. A critical involvement of oxidative stress in acute alcohol-induced hepatic TNF-alpha production. Am J Pathol (2003) 163(3):1137–46. doi:10.1016/S0002-9440(10)63473-6
116. Donohue TM Jr, Osna NA, Trambly CS, Whitaker NP, Thomes PG, Todero SL, et al. Early growth response-1 contributes to steatosis development after acute ethanol administration. Alcohol Clin Exp Res (2012) 36(5):759–67. doi:10.1111/j.1530-0277.2011.01681.x
117. Wang Y, Liu Y, Sidhu A, Ma Z, McClain C, Feng W. Lactobacillus rhamnosus GG culture supernatant ameliorates acute alcohol-induced intestinal permeability and liver injury. Am J Physiol Gastrointest Liver Physiol (2012) 303(1):G32–41. doi:10.1152/ajpgi.00024.2012
118. Chen P, Miyamoto Y, Mazagova M, Lee KC, Eckmann L, Schnabl B. Microbiota protects mice against acute alcohol-induced liver injury. Alcohol Clin Exp Res (2015) 39(12):2313–23. doi:10.1111/acer.12900
119. Zeng T, Zhao YY, Xie KQ. Does intestinal microbiota protect mice against acute/binge drinking-induced liver injury? Alcohol Clin Exp Res (2016) 40(8):1788–90. doi:10.1111/acer.13130
120. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol (2011) 11(11):750–61. doi:10.1038/nri3088
121. Ju C, Mandrekar P. Macrophages and alcohol-related liver inflammation. Alcohol Res (2015) 37(2):251–62.
122. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
123. Villa A, Rizzi N, Vegeto E, Ciana P, Maggi A. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci Rep (2015) 5:15224. doi:10.1038/srep15224
124. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122(3):787–95. doi:10.1172/JCI59643
125. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi:10.2741/2692
126. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi:10.1016/j.immuni.2014.06.008
127. Louvet A, Teixeira-Clerc F, Chobert MN, Deveaux V, Pavoine C, Zimmer A, et al. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology (2011) 54(4):1217–26. doi:10.1002/hep.24524
128. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology (2014) 59(1):130–42. doi:10.1002/hep.26607
129. Wan J, Benkdane M, Alons E, Lotersztajn S, Pavoine C. M2 Kupffer cells promote hepatocyte senescence: an IL-6-dependent protective mechanism against alcoholic liver disease. Am J Pathol (2014) 184(6):1763–72. doi:10.1016/j.ajpath.2014.02.014
130. Song BJ, Moon KH, Olsson NU, Salem N Jr. Prevention of alcoholic fatty liver and mitochondrial dysfunction in the rat by long-chain polyunsaturated fatty acids. J Hepatol (2008) 49(2):262–73. doi:10.1016/j.jhep.2008.04.023
131. Wada S, Yamazaki T, Kawano Y, Miura S, Ezaki O. Fish oil fed prior to ethanol administration prevents acute ethanol-induced fatty liver in mice. J Hepatol (2008) 49(3):441–50. doi:10.1016/j.jhep.2008.04.026
132. Huang LL, Wan JB, Wang B, He CW, Ma H, Li TW, et al. Suppression of acute ethanol-induced hepatic steatosis by docosahexaenoic acid is associated with downregulation of stearoyl-CoA desaturase 1 and inflammatory cytokines. Prostaglandins Leukot Essent Fatty Acids (2013) 88(5):347–53. doi:10.1016/j.plefa.2013.02.002
133. Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through toll-like receptor 4. J Biol Chem (2001) 276(20):16683–9. doi:10.1074/jbc.M011695200
134. Tang T, Sui Y, Lian M, Li Z, Hua J. Pro-inflammatory activated Kupffer cells by lipids induce hepatic NKT cells deficiency through activation-induced cell death. PLoS One (2013) 8(12):e81949. doi:10.1371/journal.pone.0081949
135. Luo WJ, Sui YH, Lian M, Hua J. Effects of different fatty acids on macrophage M1/M2 polarization. Chin J Gastroenterol (2015) 20(1):24–8.
136. Malaguarnera G, Giordano M, Nunnari G, Bertino G, Malaguarnera M. Gut microbiota in alcoholic liver disease: pathogenetic role and therapeutic perspectives. World J Gastroenterol (2014) 20(44):16639–48. doi:10.3748/wjg.v20.i44.16639
137. Kalambokis GN, Mouzaki A, Rodi M, Tsianos EV. Rifaximin improves thrombocytopenia in patients with alcoholic cirrhosis in association with reduction of endotoxaemia. Liver Int (2012) 32(3):467–75. doi:10.1111/j.1478-3231.2011.02650.x
138. de Franchis R, Primignani M. Natural history of portal hypertension in patients with cirrhosis. Clin Liver Dis (2001) 5(3):645–63. doi:10.1016/S1089-3261(05)70186-0
139. Ripoll C, Groszmann R, Garcia-Tsao G, Grace N, Burroughs A, Planas R, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology (2007) 133(2):481–8. doi:10.1053/j.gastro.2007.05.024
140. Vlachogiannakos J, Saveriadis AS, Viazis N, Theodoropoulos I, Foudoulis K, Manolakopoulos S, et al. Intestinal decontamination improves liver haemodynamics in patients with alcohol-related decompensated cirrhosis. Aliment Pharmacol Ther (2009) 29(9):992–9. doi:10.1111/j.1365-2036.2009.03958.x
141. Kalambokis GN, Mouzaki A, Rodi M, Pappas K, Fotopoulos A, Xourgia X, et al. Rifaximin improves systemic hemodynamics and renal function in patients with alcohol-related cirrhosis and ascites. Clin Gastroenterol Hepatol (2012) 10(7):815–8. doi:10.1016/j.cgh.2012.02.025
142. Vlachogiannakos J, Viazis N, Vasianopoulou P, Vafiadis I, Karamanolis DG, Ladas SD. Long-term administration of rifaximin improves the prognosis of patients with decompensated alcoholic cirrhosis. J Gastroenterol Hepatol (2013) 28(3):450–5. doi:10.1111/jgh.12070
143. Imani Fooladi AA, Mahmoodzadeh Hosseini H, Nourani MR, Khani S, Alavian SM. Probiotic as a novel treatment strategy against liver disease. Hepat Mon (2013) 13(2):e7521. doi:10.5812/hepatmon.7521
144. Fuller R. Probiotics in man and animals. J Appl Bacteriol (1989) 66(5):365–78. doi:10.1111/j.1365-2672.1989.tb05105.x
145. Segawa S, Wakita Y, Hirata H, Watari J. Oral administration of heat-killed Lactobacillus brevis SBC8803 ameliorates alcoholic liver disease in ethanol-containing diet-fed C57BL/6N mice. Int J Food Microbiol (2008) 128(2):371–7. doi:10.1016/j.ijfoodmicro.2008.09.023
146. Chang B, Sang L, Wang Y, Tong J, Zhang D, Wang B. The protective effect of VSL#3 on intestinal permeability in a rat model of alcoholic intestinal injury. BMC Gastroenterol (2013) 13:151. doi:10.1186/1471-230X-13-151
147. Wang Y, Liu Y, Kirpich I, Ma Z, Wang C, Zhang M, et al. Lactobacillus rhamnosus GG reduces hepatic TNFalpha production and inflammation in chronic alcohol-induced liver injury. J Nutr Biochem (2013) 24(9):1609–15. doi:10.1016/j.jnutbio.2013.02.001
148. Singh AK, Pandey SK, Naresh Kumar G. Pyrroloquinoline quinone-secreting probiotic Escherichia coli Nissle 1917 ameliorates ethanol-induced oxidative damage and hyperlipidemia in rats. Alcohol Clin Exp Res (2014) 38(7):2127–37. doi:10.1111/acer.12456
149. Hong M, Kim SW, Han SH, Kim DJ, Suk KT, Kim YS, et al. Probiotics (Lactobacillus rhamnosus R0011 and acidophilus R0052) reduce the expression of toll-like receptor 4 in mice with alcoholic liver disease. PLoS One (2015) 10(2):e0117451. doi:10.1371/journal.pone.0117451
150. Tian F, Chi F, Wang G, Liu X, Zhang Q, Chen Y, et al. Lactobacillus rhamnosus CCFM1107 treatment ameliorates alcohol-induced liver injury in a mouse model of chronic alcohol feeding. J Microbiol (2015) 53(12):856–63. doi:10.1007/s12275-015-5239-5
151. Zhang M, Wang C, Zhao H, Zhao C, Chen Y, Wang Y, et al. Enhanced AMPK phosphorylation contributes to the beneficial effects of Lactobacillus rhamnosus GG supernatant on chronic-alcohol-induced fatty liver disease. J Nutr Biochem (2015) 26(4):337–44. doi:10.1016/j.jnutbio.2014.10.016
152. Zhao H, Zhao C, Dong Y, Zhang M, Wang Y, Li F, et al. Inhibition of miR122a by Lactobacillus rhamnosus GG culture supernatant increases intestinal occludin expression and protects mice from alcoholic liver disease. Toxicol Lett (2015) 234(3):194–200. doi:10.1016/j.toxlet.2015.03.002
153. Kirpich IA, Solovieva NV, Leikhter SN, Shidakova NA, Lebedeva OV, Sidorov PI, et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: a pilot study. Alcohol (2008) 42(8):675–82. doi:10.1016/j.alcohol.2008.08.006
154. Doherty GM, Jensen JC, Alexander HR, Buresh CM, Norton JA. Pentoxifylline suppression of tumor necrosis factor gene transcription. Surgery (1991) 110(2):192–8.
155. Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology (2000) 119(6):1637–48. doi:10.1053/gast.2000.20189
156. Sidhu SS, Goyal O, Singla M, Bhatia KL, Chhina RS, Sood A. Pentoxifylline in severe alcoholic hepatitis: a prospective, randomised trial. J Assoc Physicians India (2012) 60:20–2.
157. De BK, Gangopadhyay S, Dutta D, Baksi SD, Pani A, Ghosh P. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: a randomized controlled trial. World J Gastroenterol (2009) 15(13):1613–9. doi:10.3748/wjg.15.1613
158. Park SH, Kim DJ, Kim YS, Yim HJ, Tak WY, Lee HJ, et al. Pentoxifylline vs. corticosteroid to treat severe alcoholic hepatitis: a randomised, non-inferiority, open trial. J Hepatol (2014) 61(4):792–8. doi:10.1016/j.jhep.2014.05.014
159. Sidhu SS, Goyal O, Singla P, Gupta D, Sood A, Chhina RS, et al. Corticosteroid plus pentoxifylline is not better than corticosteroid alone for improving survival in severe alcoholic hepatitis (COPE trial). Dig Dis Sci (2012) 57(6):1664–71. doi:10.1007/s10620-012-2097-4
160. Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med (2015) 372(17):1619–28. doi:10.1056/NEJMoa1412278
161. Singh S, Murad MH, Chandar AK, Bongiorno CM, Singal AK, Atkinson SR, et al. Comparative effectiveness of pharmacological interventions for severe alcoholic hepatitis: a systematic review and network meta-analysis. Gastroenterology (2015) 149(4):958.e–70.e. doi:10.1053/j.gastro.2015.06.006
162. Lee YS, Kim HJ, Kim JH, Yoo YJ, Kim TS, Kang SH, et al. Treatment of severe alcoholic hepatitis with corticosteroid, pentoxifylline, or dual therapy: a systematic review and meta-analysis. J Clin Gastroenterol (2016). doi:10.1097/MCG.0000000000000674
163. Njei B, Do A, McCarty TR, Fortune BE. Corticosteroids versus pentoxifylline for severe alcoholic hepatitis: a sequential analysis of randomized controlled trials. J Clin Gastroenterol (2016) 50(10):871–81. doi:10.1097/MCG.0000000000000585
164. Kirvel P, Goretskaia MV, Lukivskaia O, Naruta EE, Belokonskaia EB, Buko VU. [Hepatoprotective and immunomodulatory effects of infliximab on experimental alcoholic steatohepatitis]. Eksp Klin Farmakol (2012) 75(7):36–9.
165. Tilg H, Jalan R, Kaser A, Davies NA, Offner FA, Hodges SJ, et al. Anti-tumor necrosis factor-alpha monoclonal antibody therapy in severe alcoholic hepatitis. J Hepatol (2003) 38(4):419–25. doi:10.1016/S0168-8278(02)00442-7
166. Spahr L, Rubbia-Brandt L, Frossard JL, Giostra E, Rougemont AL, Pugin J, et al. Combination of steroids with infliximab or placebo in severe alcoholic hepatitis: a randomized controlled pilot study. J Hepatol (2002) 37(4):448–55. doi:10.1016/S0168-8278(02)00230-1
167. Naveau S, Chollet-Martin S, Dharancy S, Mathurin P, Jouet P, Piquet MA, et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology (2004) 39(5):1390–7. doi:10.1002/hep.20206
168. Sharma P, Kumar A, Sharma BC, Sarin SK. Infliximab monotherapy for severe alcoholic hepatitis and predictors of survival: an open label trial. J Hepatol (2009) 50(3):584–91. doi:10.1016/j.jhep.2008.10.024
169. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther (2008) 117(2):244–79. doi:10.1016/j.pharmthera.2007.10.001
170. Menon KV, Stadheim L, Kamath PS, Wiesner RH, Gores GJ, Peine CJ, et al. A pilot study of the safety and tolerability of etanercept in patients with alcoholic hepatitis. Am J Gastroenterol (2004) 99(2):255–60. doi:10.1111/j.1572-0241.2004.04034.x
171. Boetticher NC, Peine CJ, Kwo P, Abrams GA, Patel T, Aqel B, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology (2008) 135(6):1953–60. doi:10.1053/j.gastro.2008.08.057
172. Mathews S, Gao B. Therapeutic potential of interleukin 1 inhibitors in the treatment of alcoholic liver disease. Hepatology (2013) 57(5):2078–80. doi:10.1002/hep.26336
173. Mitchell M. Efficacy Study of Anakinra, Pentoxyfilline, and Zinc Compared to Methylprednisone in Severe Acute Alcoholic Hepatitis. (2016). Available from: https://clinicaltrials.gov/ct2/show/NCT01809132
174. Nguyen-Khac E, Thevenot T, Piquet MA, Benferhat S, Goria O, Chatelain D, et al. Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis. N Engl J Med (2011) 365(19):1781–9. doi:10.1056/NEJMoa1101214
175. Mathurin P, Louvet A, Duhamel A, Nahon P, Carbonell N, Boursier J, et al. Prednisolone with vs without pentoxifylline and survival of patients with severe alcoholic hepatitis: a randomized clinical trial. JAMA (2013) 310(10):1033–41. doi:10.1001/jama.2013.276300
176. Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med (2009) 360(26):2758–69. doi:10.1056/NEJMra0805786
177. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature (2009) 458(7242):1131–5. doi:10.1038/nature07976
178. Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy (2015) 11(2):271–84. doi:10.1080/15548627.2015.1009787
179. Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology (2010) 139(5):1740–52. doi:10.1053/j.gastro.2010.07.041
180. Lin CW, Zhang H, Li M, Xiong X, Chen X, Dong XC, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol (2013) 58(5):993–9. doi:10.1016/j.jhep.2013.01.011
181. Pun NT, Subedi A, Kim MJ, Park PH. Globular adiponectin causes tolerance to LPS-induced TNF-alpha expression via autophagy induction in RAW 264.7 macrophages: involvement of SIRT1/FoxO3A axis. PLoS One (2015) 10(5):e0124636. doi:10.1371/journal.pone.0124636
182. Zeng T, Zhang CL, Song FY, Zhao XL, Yu LH, Zhu ZP, et al. PI3K/Akt pathway activation was involved in acute ethanol-induced fatty liver in mice. Toxicology (2012) 296(1–3):56–66. doi:10.1016/j.tox.2012.03.005
183. Eid N, Ito Y, Otsuki Y. Ethanol-induced hepatic autophagy: friend or foe? World J Hepatol (2015) 7(9):1154–6. doi:10.4254/wjh.v7.i9.1154
184. Xu Y, Liu XD, Gong X, Eissa NT. Signaling pathway of autophagy associated with innate immunity. Autophagy (2008) 4(1):110–2. doi:10.4161/auto.5225
185. Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol (2009) 10(5):461–70. doi:10.1038/ni.1726
186. Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S, et al. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol (2010) 53(5):903–10. doi:10.1016/j.jhep.2010.04.037
187. Achur RN, Freeman WM, Vrana KE. Circulating cytokines as biomarkers of alcohol abuse and alcoholism. J Neuroimmune Pharmacol (2010) 5(1):83–91. doi:10.1007/s11481-009-9185-z
188. Gonzalez-Reimers E, Santolaria-Fernandez F, Martin-Gonzalez MC, Fernandez-Rodriguez CM, Quintero-Platt G. Alcoholism: a systemic proinflammatory condition. World J Gastroenterol (2014) 20(40):14660–71. doi:10.3748/wjg.v20.i40.14660
189. Crews FT, Vetreno RP. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology (Berl) (2016) 233(9):1543–57. doi:10.1007/s00213-015-3906-1
190. Montesinos J, Alfonso-Loeches S, Guerri C. Impact of the innate immune response in the actions of ethanol on the central nervous system. Alcohol Clin Exp Res (2016) 40(11):2260–70. doi:10.1111/acer.13208
191. Leclercq S, Cani PD, Neyrinck AM, Starkel P, Jamar F, Mikolajczak M, et al. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav Immun (2012) 26(6):911–8. doi:10.1016/j.bbi.2012.04.001
192. Rubio-Araiz A, Porcu F, Perez-Hernandez M, Garcia-Gutierrez MS, Aracil-Fernandez MA, Gutierrez-Lopez MD, et al. Disruption of blood-brain barrier integrity in postmortem alcoholic brain: preclinical evidence of TLR4 involvement from a binge-like drinking model. Addict Biol (2016). doi:10.1111/adb.12376
Keywords: Kupffer cells, alcoholic liver disease, lipopolysaccharide, polarization, tumor necrosis factor α, cytochrome P4502E1
Citation: Zeng T, Zhang C-L, Xiao M, Yang R and Xie K-Q (2016) Critical Roles of Kupffer Cells in the Pathogenesis of Alcoholic Liver Disease: From Basic Science to Clinical Trials. Front. Immunol. 7:538. doi: 10.3389/fimmu.2016.00538
Received: 21 September 2016; Accepted: 15 November 2016;
Published: 29 November 2016
Edited by:
Kai Fang, University of California Los Angeles, USAReviewed by:
Alexander Neumeister, Mitsubishi Tanabe Pharma Development America, USAJian Li, Beijing University of Chinese Medicine, China
Steven Dooley, University of Heidelberg, Germany
Brian Duncan Tait, Australian Red Cross Blood Service, Australia
Copyright: © 2016 Zeng, Zhang, Xiao, Yang and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Zeng, zengtao@sdu.edu.cn;
Ke-Qin Xie, keqinx@sdu.edu.cn