 Nayef M. Kazzaz
Nayef M. Kazzaz Gautam Sule
Gautam Sule Jason S. Knight
Jason S. Knight
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 November 2016
Sec. Molecular Innate Immunity
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00453
This article is part of the Research Topic NETosis 2: the excitement continues View all 39 articles
Neutrophil extracellular traps (NETs) are chromatin-derived webs extruded from neutrophils in response to either infection or sterile stimulation with chemicals, cytokines, or microbial products. The vast majority of studies have characterized NET release (also called NETosis) in pure neutrophil cultures in vitro. The situation is surely more complex in vivo as neutrophils constantly sample not only pathogens and soluble mediators but also signals from cellular partners, including platelets and endothelial cells. This complexity is beginning to be explored by studies utilizing in vitro co-culture, as well as animal models of sepsis, infective endocarditis, lung injury, and thrombosis. Indeed, various selectins, integrins, and surface glycoproteins have been implicated in platelet–neutrophil interactions that promote NETosis, albeit with disparate results across studies. NETosis can also clearly be regulated by soluble mediators derived from platelets, such as eicosanoids, chemokines, and alarmins. Beyond platelets, the role of the endothelium in modulating NETosis is being increasingly revealed, with adhesive interactions likely priming neutrophils toward NETosis. The fact that the same selectins and surface glycoproteins may be expressed by both platelets and endothelial cells complicates the interpretation of in vivo data. In summary, we suggest in this review that the engagement of neutrophils with activated cellular partners provides an important in vivo signal or “hit” toward NETosis. Studies should, therefore, increasingly consider the triumvirate of neutrophils, platelets, and the endothelium when exploring NETosis, especially in disease states.
Neutrophil extracellular traps (NETs), first described in 2004, are released by neutrophils via an active process coined NETosis (1, 2). While first characterized for their role in combatting infectious organisms (1), these tangles of chromatin and antimicrobial proteins are now known to play a role in pathogenic autoimmunity and other sterile inflammatory states (3, 4). NETs may place organ systems at risk, including the vasculature (5–7), central nervous system (8), lungs (5), and kidneys (9, 10). Organ failure and thrombotic vessel occlusions are even possible (11–13). Neutrophils, as one of the first responders to inflammatory insults have long been known to interact with other cell types (especially platelets and endothelial cells) with implications for neutrophil recruitment, generation of reactive oxygen species (ROS), and phagocytosis. This cell-to-cell crosstalk may be mediated by either direct cell contact or soluble mediators. In this review, we will focus on the implications of crosstalk for NETosis. Relevant studies have characterized not only in vitro systems (typically with human cells) but also more complex murine models of disease. There is significant heterogeneity between studies, especially in terms of how NETosis is scored and the neutrophil pathways that are considered (which is probably not surprising as a canonical model of NETosis is still not established). Our goal is to highlight the similarities between studies and to point out the discrepancies that necessitate further research. Also, whenever possible, we will try to focus on the implications of these interactions for controlling infection and for regulating inflammation and end-organ damage.
Platelets are megakaryocyte-derived cell bodies that lack nuclei. They circulate in the bloodstream as well-established regulators of the hemostatic system (14). Platelets may be activated by the exposure of subendothelial matrix proteins, such as von Willebrand factor (vWF) and collagen, as might happen with mechanical vessel injury (15). Platelets recognize vWF via a glycoprotein receptor complex, glycoprotein Ib (GPIb)/IX/V (16), with the GPIb subunit playing a particularly key role (17). In parallel, collagen engages a different glycoprotein receptor, GPVI (18). Soluble plasma factors also activate platelets, including fibrinogen (via GPIIb/IIIa) (19) and thrombin (through protease-activated receptors or PARs) (20). When considering research studies, it is important to note that some studies may activate platelets with synthesized activators. An example is thrombin receptor activator peptide (TRAP), which acts as an agonist for all PARs (21), and the more specific TRAP-6, which binds specifically to PAR-1 (22).
These various activating signals lead to platelet aggregation and the release of copious amounts of preformed mediators from platelet granules, such as adenosine diphosphate (ADP) and thromboxane A2 (TXA2) – with the potential for potent local effects and feedforward into further platelet activation (14, 17). Platelet factor 4 (PF4, also known as C–X–C motif ligand 4) is another mediator released by platelets. In addition to functioning as a chemokine for cells, such as neutrophils, PF4 binds and neutralizes negatively charged cell surface glycosaminoglycans, such as heparan sulfate, dermatan sulfate, and chondroitin sulfate, thereby mediating several downstream effects, including platelet aggregation (23). Another soluble mediator that will be discussed in this article is high-mobility group box 1 (HMGB1), a protein “alarmin”/cytokine released by activated platelets (24). Finally, proteins such as P-selectin may be either released locally, or expressed on the platelet surface, thereby regulating the local environment (25, 26). For example, P-selectin has been implicated in platelet aggregation under pulsatile shear stress conditions (27).
While platelets clearly play a key role in stemming blood loss in the event of vessel injury, they also have well-established immunomodulatory properties, potentially acting as sentinels of infectious and inflammatory events (28, 29). In particular, the innate immune receptors toll-like receptor 2 (TLR2) and TLR4 (for Gram-positive and Gram-negative organisms, respectively) are expressed on the platelet surface (30, 31). Activation of these receptors may lead to release of platelet granules (32), PF4 upregulation (33), GPIIb/IIIa conformational changes (34), and ultimately feed forward to thrombin generation (30). Having said that, some studies have found less potent responses. For example, exposure of platelets to triacylated lipoproteins (like Pam3CSK4, a TLR2 agonist) and lipopolysaccharide (LPS, a TLR4 agonist) does not always lead to significant P-selectin release (35).
Platelets interact directly with neutrophils and thereby alter neutrophil function (17). Examples of ligand/receptor pairs that mediate direct platelet/neutrophil interactions include P-selectin/P-selectin glycoprotein ligand 1 (PSGL-1) (36, 37), intercellular adhesion molecule 2 (ICAM-2)/lymphocyte function-associated antigen (LFA-1) (38), and GPIb/macrophage-1 antigen (Mac-1) (17, 39). These interactions clearly support platelet adhesion to leukocytes (40, 41) and, in some cases, have been shown to be of fundamental importance for recruitment of neutrophils to sites of inflammatory insult (40). Furthermore, beyond traditional direct interaction, some molecules (such as GPIIb/IIIa) may be transferred from platelets to neutrophils via microparticles (MP), thereby regulating neutrophil function (an example being nuclear factor kappa B activation) (42).
There is also a key role for platelet-released soluble mediators (ADP, TXA2, etc.) in both perpetuating platelet–neutrophil interplay and activating neutrophils. As an example, ADP (which would presumably be platelet-derived in vivo) induces platelet–neutrophil complexes through a mechanism that may be dependent upon P-selectin, but not PSGL-1 (41). TXA2 augments multiple neutrophil functions, including neutrophil adhesiveness (43), oxidative burst (44), and diapedesis (45). Platelet-derived HMGB1 can engage/activate neutrophil TLRs (46). Beyond TLRs, another well-recognized receptor for HMGB1 is the receptor for advanced glycation end products (RAGE), with engagement by HMGB1 leading to neutrophil recruitment and neutrophil-mediated tissue injury (47). PF4 interacts with neutrophil chondroitin sulfate (48) and (in the presence of co-stimulatory tumor necrosis factor alpha) mediates neutrophil granule release and surface adherence (49). PF4 has also been implicated in neutrophil chemotaxis (50). Neutrophil-activating peptide 2 (NAP-2) released from platelets can regulate neutrophil polarization and motility through CXCR1/2 (51). CCL5 (another chemokine released by platelets) may also play a role in neutrophil infiltration (52).
Platelets are far-and-away the most studied cellular regulators of NETosis. Most model systems have pointed to platelet activation as the first step. This is followed by platelet–neutrophil crosstalk, and ultimately regulation of neutrophil effector function. Studies have employed numerous platelet activators, including LPS, Pam3CSK4, thrombin, collagen, ADP, and TRAP-6 (53–55). These different strategies for activation, beyond anything else, make it challenging to compare studies side-by-side (Table 1).

Table 1. Selected in vitro studies of platelet-stimulated NETosis.
Regarding in vitro studies, platelet–neutrophil interactions have been assessed under static conditions (53, 57), and also with the introduction of shear stress (53–56). It is worth noting that the methodology for quantifying NETosis has varied markedly across studies. Examples include cell-free DNA quantification (53, 55), myeloperoxidase-deoxyribonucleic acid (MPO-DNA) ELISA (5, 55, 60, 61), neutrophil elastase-DNA ELISA (53), neutrophil elastase concentration (57), or direct visualization of NETs by fluorescence microscopy (54). Microscopy samples have been scored by quantifying percent surface area of Sytox green staining (detects extracellular DNA) (54, 58), histone H2Ax percentage surface area (56), or citrullinated histone H3-positive cells per field (62).
We will first describe some notable in vivo studies in the field, which have focused on disease models (Table 2). We will then step through the various stages of platelet–neutrophil interplay, beginning with platelet activation and ending with NETosis (Figure 1).
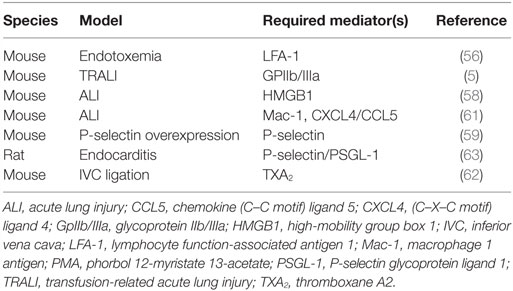
Table 2. Selected in vivo models of platelet-stimulated NETosis.
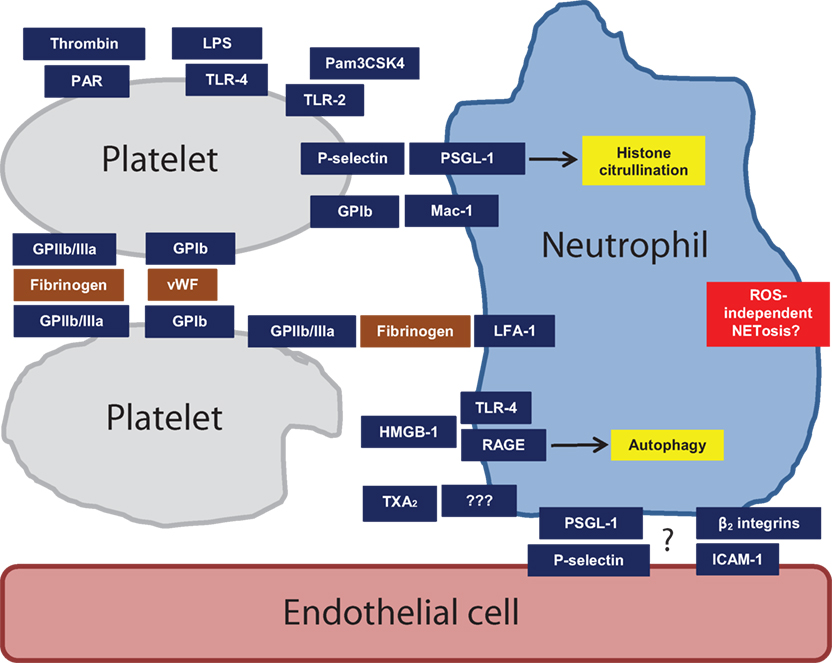
Figure 1. Mechanisms of platelet activation and heterotypic intercellular interactions that may regulate NETosis. Some are more speculative than others, as described in the text. Abbreviations: GPIb, glycoprotein 1b; GPIIb/IIIa, glycoprotein IIb/IIIa; HMGB1, high-mobility group box 1; LFA-1, lymphocyte function-associated antigen 1; LPS, lipopolysaccharide; Mac-1, macrophage-1 antigen; PAR, protease-activated receptor; PSGL-1, P-selectin glycoprotein ligand 1; RAGE, receptor for advanced glycation end products; TLR2, toll-like receptor 2; TLR4, toll-like receptor 4; TXA2, thromboxane A2; vWF, Von Willebrand factor.
One of the first studies to consider the impact of activated platelets on NETosis in vivo utilized a mouse model of endotoxemia (sepsis) induced by intravenous LPS (54, 56). The authors found that LPS triggers the recruitment of neutrophils to liver sinusoids, which then facilitate recruitment of platelets (54) – with platelet recruitment dependent upon neutrophil LFA-1 (56). Importantly, NETosis is only triggered after engagement by the activated platelets (which seem to have been primed by LPS acting through platelet TLR4). This functionality presumably plays a key role in bacterial sequester, but also places the host at risk for significant endothelial damage (54). The authors further mimic these data in vitro, demonstrating that stimulation of platelets through TLR4 enhances both platelet–neutrophil adhesion and NETosis, but without upregulating P-selectin expression or platelet aggregation (54).
Another notable study investigated platelet–neutrophil interplay in the context of transfusion-related acute lung injury (TRALI). TRALI was modeled by treating BALB/c wild-type mice with the combination of LPS and an anti-MHC I monoclonal antibody (5, 64). NETosis was quantified in the lungs by either intravital microscopy or postmortem histological examination (5). Lung NETosis was dependent upon platelet–neutrophil interplay as NETosis was significantly mitigated by inhibiting platelet activation with aspirin (an irreversible inhibitor of platelet TXA2 generation) or a GPIIb/IIIa inhibitor, tirofiban (5). In vitro, TRAP-activated platelets enhanced NETosis (5).
In a murine model of acute lung injury achieved with positive-pressure ventilation, platelet depletion led to depressed NETosis as measured in blood by MPO-DNA ELISA and in the lungs by microscopy (61). A critical role for Mac-1 was demonstrated with blocking antibodies and genetic knockout. By contrast, blocking LFA-1 did not suppress NETosis (61). Beyond integrin signaling, the authors argued that a second hit was also necessary for full neutrophil activation. Indeed, blocking platelet-derived CXCL4/CCL5 chemokine heterodimers reduced lung injury, while also explicitly mitigating NETosis in response to TRAP-activated platelets in vitro (61).
In a model of endocarditis, cultured bacteria from endocarditis patients were infused through carotid catheters into rats (63). By confocal microscopy, a platelet/bacteria layer was demonstrated inside the vegetation film, which was also intermixed with NETs (63). Furthermore, deoxyribonuclease (DNase, an enzyme that degrades DNA) proved to be an effective treatment (63). Platelets were deemed necessary for NETosis in this model, shown by inhibition with aspirin (63). Furthermore, NETosis was inhibited by P-selectin and PSGL-1 blocking antibodies (63).
In a final noteworthy study, the authors were interested in probing mechanisms by which aspirin might mitigate venous thrombosis (62). In a murine model of deep vein thrombosis (achieved by complete inferior vena cava ligation), both aspirin (which reduces the synthesis of TXA2 by platelets) and a selective thromboxane receptor antagonist reduced thrombus size. This was accompanied by a reduction in neutrophil infiltration, as well as deposition of both fibrin and NETs.
If one considers in vitro studies with human neutrophils, then P-selectin has largely been judged dispensable for the ability of stimulated platelets to promote NETosis (53–55). In other species, the story may be different. For example, P-selectin has been implicated as required for thrombin-activated platelets to induce NETosis, as well as histone citrullination (a prerequisite for NETosis); this was demonstrated with cells isolated from knockout mice, and also by antibody-based inhibition (59). In the same study, mice overexpressing soluble P-selectin demonstrated higher neutrophil histone citrullination in vivo. Interestingly, P-selectin overexpression did not seem to regulate baseline NETosis, although accelerated NETosis could be unmasked in these mice with ex vivo stimulation (suggesting the neutrophils had been somehow primed by the overexpression) (59). Additionally, in the aforementioned rat model of infective endocarditis, platelet-induced NETosis was found to be dependent upon P-selectin/PSGL-1 as demonstrated by blocking antibodies (63).
What explains these discrepancies? One simple possibility is species difference (human versus mouse/rat). Another consideration is that P-selectin/PSGL-1 interactions may already be established when neutrophils are purified for in vitro studies, and so blocking antibodies may be less effective in this context (5, 65). As hinted above, the method of platelet stimulation must also be kept in mind, as there was no apparent role for platelet P-selectin in studies in which platelets were stimulated with LPS (54) or TRAP-6 (56), as compared to a positive role in a study using thrombin as the stimulus (59). As P-selectin may serve a priming role in vivo more so than as the primary stimulus (59), and as P-selectin is also well-known to be expressed on endothelial cells (66, 67), intravital studies that can probe these interactions in real time will be important in sorting this out going forward.
There is a suggestion that the β2 integrin Mac-1 is dispensable for platelet-induced NETosis based on in vitro studies with human neutrophils [with either TLR4 agonist (54) or TRAP-6 (5, 55) as the platelet stimulator]. By contrast, a study of acute lung injury demonstrated the requirement of Mac-1 for neutrophil-platelet aggregation as well as NETosis (61). Another interesting study recently revealed that neutrophil Mac-1 is required for crawling on the inflamed endothelium, a process that also requires PSGL-1, albeit without direct PSGL-1/endothelium contact (39). The authors discovered that PSGL-1 instead concentrates in a uropod, which projects into the bloodstream where it receives activating signals from platelets. These PSGL-1-mediated signals then regulate Mac-1 distribution and ultimately crawling (39). This study nicely highlights the potential complexity of platelet–neutrophil interplay in vivo, and how a comprehensive model of neutrophil effector functions (such as NETosis) may not be possible without considering both platelets and the endothelium.
The β2 integrin lymphocyte function-associated antigen 1 (LFA-1) is known to be the key receptor by which neutrophils interact with fibrinogen, an interaction that has been linked to an effective neutrophil oxidative burst (68). Beyond fibrinogen, platelet ICAM-2 may also interact with LFA-1 (38). In vitro studies with human platelets (activated with LPS, TRAP, or Pam3CSK4) have demonstrated that platelet–neutrophil interaction and resulting NETosis can be reversed with blockade of LFA-1 (53, 56), including under conditions of shear stress (56). Similarly, a mouse model of sepsis has supported a key role for LFA-1 in platelet-mediated NETosis, with either genetic deletion or blockade reducing NETosis in liver sinusoids (54, 56). However, in a different study focusing on murine neutrophils, TRAP-activated platelets signaled through neutrophil Mac-1, but not LFA-1, to induce NETosis (61). Differences in species, model, or culture conditions may have contributed to the discrepancies across studies.
An in vitro study has suggested that GPIb (the classic receptor for vWF) is required for platelet-induced NETosis (53), although without a clear understanding of its counterpart on neutrophils. Interestingly, the authors also found that LPS-stimulated platelets increase expression and release of vWF, with blockade of vWF preventing platelet-induced NETosis (53). As GPIb can interact directly with neutrophils through Mac-1 (69, 70), and since vWF is also presented on the surface of endothelial cells, this pathway will need to be further dissected (including in vivo) before definitive conclusions can be drawn (71).
In a mouse model of TRALI, blockade of GPIIb/IIIa (with tirofiban) reduced NETosis in lung tissue (5). This stands in contrast to in vitro human studies, which have not found a role for GPIIb/IIIa in platelet-induced NETosis (53–55). Interestingly, GPIIb/IIIa can be transferred from platelets to neutrophils through platelet-derived MP (42), an observation that could have implications for in vitro and in vivo discrepancies. It may also be that the key role of GPIIb/IIIa is to facilitate platelet–platelet or platelet–endothelial interactions (72–74), which would stand out in in vivo models, more so than the in vitro work.
Platelets stimulated with Pam3CSK4 and TRAP (5, 53) may utilize TXA2 as a means of signaling to promote release of NETs (53). Given that there is no well-characterized receptor for TXA2 on neutrophils, mechanistic details remain to be determined.
PF4 (CXCL4) can play a role in regulating in vitro human NETosis, based on blocking experiments (53), and also direct stimulation of neutrophils with recombinant PF4 (53). In vivo, MKEY (a peptide inhibitor of CXCL4/CCL5 heterodimer formation) reduces NETosis in a model of acute lung injury (61).
Recombinant HMGB1 activates neutrophils to release NETs, dependent upon either neutrophil TLR4 (58) or neutrophil RAGE (55). Human beta defensin-1 (a microbicidal protein found in both neutrophils and platelets) is released by platelets exposed to Staphylococcus aureus alpha toxin, in a manner that then triggers NETosis (57).
It should be noted that neutrophil signaling has not been characterized in most models of platelet-induced NETosis. When Pam3CSK4, LPS, or TRAP were used to stimulate platelets, the resulting NETosis was found to be ROS independent (5, 53, 55). This is in contrast to S. aureus alpha toxin-activated platelets, which promote NETosis in a ROS-dependent manner (57). Platelet HMGB1 seems to leverage neutrophil autophagy to induce NETosis (55). Another study has demonstrated that ERK and PI3K are required for platelet-induced NETosis, when platelets were activated with Pam3CSK4, LPS, or arachidonic acid (53). At this point, the data are too limited to predict whether a consensus signaling pathway will emerge, although there are hints that ROS may not be a critically important factor in a critically important factor in platelet-induced NETosis.
Neutrophils develop in the bone marrow from myeloid precursors, reaching sites of infection or inflammation via the vasculature. This migration of neutrophils from the bloodstream to inflamed tissues is mediated by the interaction of adhesion molecules on the neutrophil surface with their respective ligands on the vascular endothelium. Details regarding this well-coordinated series of events arise from intravital microscopy studies in animals, as well as observations of patients with leukocyte adhesion deficiency (75). As an initial step, neutrophils leverage specific surface ligands in order to tether to P- and E-selectin molecules expressed on activated endothelial cells (selectin ligands potentially expressed on neutrophils include PSGL-1, E-selectin ligand 1, and CD44). Tethering of neutrophils is followed by their rolling along the endothelium (76–80). Rolling neutrophils develop membrane extensions at their rear end (tethers) and front (slings), which stabilize neutrophil rolling and allow the process to proceed despite the high shear stress of flowing blood (81). Subsequently, neutrophils firmly adhere to endothelial cells, mediated by the binding of neutrophil β2 integrins (LFA-1 and Mac-1) to endothelial ligands such as intracellular adhesion molecule 1 (ICAM-1) and ICAM-2 (76, 78, 79, 82). β2 integrins have two main states of activation: the first is an extended (but not open) form with low to intermediate affinity, and the second an extended and open form with high affinity (the form required for firm adhesion). Mechanisms and signaling pathways involved in these transitions have been delineated in great detail, and are reviewed elsewhere (82–84).
Rolling and adhesion may be followed by transmigration, when neutrophils pass between endothelial cells (paracellular) or through endothelial cells (transcellular). While many details remain to be determined, the paracellular process is more prevalent, occurring perhaps 90% of the time (76, 83, 85) and favored by neutrophils expressing Mac-1 (86, 87). By contrast, the transcellular route may be favored by increased endothelial expression of ICAM-1 (88) or by activation of endothelial cells by neutrophils through annexin A1 secretion (89). Beyond the above, adhesion molecules involved in the transmigration process include platelet endothelial cell adhesion molecule 1 (PECAM-1), CD99, ICAM-2, junctional adhesion molecules (JAMs), and cadherins (90). The roles of these adhesion molecules have primarily been demonstrated in mouse models wherein their deletion results in inhibition of transmigration and reduced accumulation of neutrophils in tissues (83, 85, 91).
Within inflamed tissues, neutrophils home via chemokine gradients. Interestingly, recent studies have demonstrated that neutrophils are able to undergo a “reverse transmigration” process such that tissue neutrophils may migrate back to the vascular lumen. Studies in mice have demonstrated that downregulation of JAM-C by neutrophil elastase plays a key role in the process (92). At present, the functional significance of reverse transmigration is not entirely clear. One idea is that the reverse transmigration has a significant downside, as it may contribute to dissemination of a local immune response into a systemic inflammatory phenomenon (93). Alternatively, it may play a role in dampening immune response as observed in zebrafish (94) and, we speculate patients with systemic inflammation (95).
Circulating neutrophils tend to be quiescent in nature, with their activation tightly linked to migration from circulation to tissue. Neutrophil activation can be thought of as a two-step process whereby exposure to one stimulus (priming) ensures a maximum response to a second. So, rolling and adhesion of neutrophils on the endothelium may initiate their activation, but full effector functions only become available to neutrophils once they encounter certain pro-inflammatory chemokines/cytokines or pathogen-derived ligands that can activate other receptors (G protein-coupled receptors and innate pattern-recognition receptors as classic examples). Neutrophils can then rapidly undergo degranulation, activation of their NADPH oxidase pathway for free radical generation, phagocytosis, and even NETosis (96–98). An example comes from studies of P-selectin overexpressing mice in which neutrophils seem to be sensitized to NETosis by excess P-selectin exposure, but do not actually release NETs unless confronted with a second stimulus (59).
Netting neutrophils externalize not just chromatin but also a variety of antimicrobial peptides and proteases that target pathogens. Recent work has demonstrated that these mediators of host defense may also promote tissue damage (12). NETs induce endothelial cell death in a dose-dependent and partially DNA-independent manner (99). Rather than DNA, associated histones and to some extent myeloperoxidase may be most responsible for NET-mediated endothelial cytotoxicity (99). Another study demonstrated the externalization of matrix metalloproteinase-9 (MMP-9) and MMP-25 along with NETs. This externalized MMP-9 activates pro-MMP-2 produced by the endothelium, resulting in cytotoxicity and vessel dysfunction (100).
An interesting in vitro study investigated the implications of co-culture of activated endothelial cells with neutrophils (101). The result was not just increased NETosis by neutrophils, but also increased endothelial cell death (101). The death was attributable to increased IL-8 production by the endothelial cells themselves (101). One can imagine a scenario in vivo in which activated endothelial cells induce NETosis, followed by endothelial cytotoxicity and potentially the release of mediators that feed forward into more NETosis.
It should also be noted that although endothelial cells have not been the explicit focus of most NETs studies, they almost surely play a prominent role in vivo, either through direct regulation of neutrophil activity, or through modulation of other cellular elements, such as platelets (Figure 1). As an example, in the aforementioned sepsis model, liver sinusoids support neutrophil adhesion even in the absence of platelets, perhaps providing certain activating signals to the neutrophils that prime them for subsequent platelet capture (56). One might also point to the TRALI model (5). There, GPIIb/IIIa plays a key role in NETosis beyond anything that has been seen in vitro (53–55) – raising the question of whether additional synergistic signals may emanate from the endothelium in vivo (5). Finally, although studies focusing on platelet–neutrophil interactions in vitro have suggested contradictory roles for P-selectin (53–55), it is worth noting that P-selectin is also present on endothelial cells, which may help explain its more clear-cut role in vivo (59). We expect to see much more on this front in the coming years.
Dendritic cells (DCs) are best known for their role as professional antigen-presenting cells, bridging the gap between innate and adaptive immunity. In recent years, the intersection of neutrophils/NETosis and DCs has been increasingly considered. First, neutrophils are well established to play a role in the recruitment of DCs to sites of inflammation, and promote maturation of DCs via secretion of a variety of soluble mediators, such as CCL3, CCL4, CCL5 (RANTES), CCL20, tumor necrosis factor α, α-defensins, and cathelicidins (102–106). At the same time, in vivo immunization studies have demonstrated that neutrophils can dampen immune responses by competing for antigen with DCs and limiting contact between T cells and DCs (107). So, at least in some contexts, vaccination responses may improve with temporary depletion of neutrophils. In other contexts, NETs seem to do the opposite, quite specifically transferring antigens to DCs, and thereby initiating autoimmune disorders, such as small vessel vasculitis (108).
With further implications for autoimmunity and sterile inflammation, NETs activate plasmacytoid DCs in lupus and atherosclerosis via TLR9. Activated plasmacytoid DCs produce interferons, which in turn prime neutrophils for more NETosis (thereby setting up a positive feedback loop) (109, 110).
Again pointing to different roles in different contexts, DCs may sometimes downregulate NETosis. This has been described in the specific context of human immunodeficiency virus (HIV), which acts through CD209 on DCs to produce interleukin 10 (IL-10). IL-10 then inhibits HIV/TLR7-mediated NETosis (111). Demonstrating at least some specificity, PMA-induced NETosis is not suppressed by IL-10 (111).
MP are small, cell membrane-derived vesicles (112). MP from endothelial cells (113, 114), platelets (115), and red blood cells (116) have all been implicated in activating neutrophils. Furthermore, both platelet-derived (115) and red blood cell-derived (116) MP induce Mac-1 expression on neutrophils and stimulate neutrophil phagocytic activity (115, 116). The role of MP in promoting NETosis was also demonstrated in a paper focusing on preeclampsia, in which placenta syncytiotrophoblast-derived MP seem to promote NETosis (117). In inflammatory bowel disease, MP also appear to activate NETosis (118).
While NETs play a critical role in host defense, excessive formation or persistence of NETs may lead to adverse effects. Thus, clearance of NETs is an important physiological process that helps minimize excessive presentation of both toxic products and potential self-antigens. Degradation of NETs by serum DNase is one mechanism by which NETs are cleared, with impairment of this process leading to a lupus-like syndrome in mice (119). Interestingly, inadequate DNase activity has also been detected in the blood of patients with both lupus (119–121) and autoimmune vasculitis (122). Beyond the enzymatic activity of DNase, macrophages also play a role in the clearance of NETs. DNase processing of NETs prepares them for engulfment by macrophages, with the process further facilitated by the opsonization of NETs by complement C1q (123). Though this process was initially thought to be immunologically silent, recent in vitro studies have demonstrated a potentially complicated response that depends upon macrophage polarization (124). The authors show that M2 macrophages induce a pro-inflammatory response when exposed to NETs (including the release of a variety of pro-inflammatory cytokines/chemokines). By contrast, M1 macrophages initially undergo cell death that leads to their own nuclear decondensation and DNA release. Interestingly, over time, M1 macrophages then degrade this macrophage-derived DNA in a caspase-activated DNase-dependent manner (124). The full implications of this interplay remain unclear in vivo (and in disease states) and will hopefully be elucidated by future studies.
This is a field in which much remains to be defined, as is especially highlighted by the various studies of platelet-induced NETosis. Studies in different systems and by different investigators have revealed surprisingly little mechanistic consensus, which probably points to an involvement of multiple pathways, thereby allowing certain aspects to be revealed by different groups. An obvious barrier is that platelet activation is achieved through different methodology in each study. It would be very interesting to see one group (or preferable a number of groups) take a systematic approach to this question, asking how the method of stimulation influences the specifics of platelet–neutrophil crosstalk. Given the highly regulated crosstalk that exists between the endothelium and neutrophils, endothelial cells surely play an important role in regulating NETosis in vivo – although relatively few studies have specifically probed that role. Studies should, therefore, increasingly consider the triumvirate of neutrophils, platelets, and the endothelium when exploring NETosis, especially in disease states.
NK and GS wrote the review. JK edited and approved the final version for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
NK was supported by Security Forces Hospital Program, Ministry of Interior, Riyadh, Saudi Arabia. JK was supported by awards from NIH–NIAMS (K08AR066569), the Burroughs Wellcome Fund, the Arthritis National Research Foundation, and the Rheumatology Research Foundation.
1. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi:10.1126/science.1092385
2. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ (2009) 16:3–11. doi:10.1038/cdd.2008.150
3. Grayson PC, Kaplan MJ. At the bench: neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J Leukoc Biol (2016) 99:253–64. doi:10.1189/jlb.5BT0615-247R
4. Knight JS, Kaplan MJ. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr Opin Rheumatol (2012) 24:441–50. doi:10.1097/BOR.0b013e3283546703
5. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest (2012) 122(7):2661–71. doi:10.1172/JCI61303
6. Knight JS, Luo W, O’dell AA, Yalavarthi S, Zhao W, Subramanian V, et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res (2014) 114:947–56. doi:10.1161/CIRCRESAHA.114.303312
7. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (2015) 349:316–20. doi:10.1126/science.aaa8064
8. Perez-de-Puig I, Miro-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol (2015) 129:239–57. doi:10.1007/s00401-014-1381-0
9. Yoshida M, Sasaki M, Sugisaki K, Yamaguchi Y, Yamada M. Neutrophil extracellular trap components in fibrinoid necrosis of the kidney with myeloperoxidase-ANCA-associated vasculitis. Clin Kidney J (2013) 6(3):308–12. doi:10.1093/ckj/sft048
10. Knight JS, Zhao W, Luo W, Subramanian V, O’dell AA, Yalavarthi S, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest (2013) 123:2981–93. doi:10.1172/JCI67390
11. Massberg S, Grahl L, Von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med (2010) 16:887–96. doi:10.1038/nm.2184
12. Rao AN, Kazzaz NM, Knight JS. Do neutrophil extracellular traps contribute to the heightened risk of thrombosis in inflammatory diseases? World J Cardiol (2015) 7:829–42. doi:10.4330/wjc.v7.i12.829
13. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Nunez-Alvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol (2015) 67:2990–3003. doi:10.1002/art.39247
14. Rossaint J, Zarbock A. Platelets in leucocyte recruitment and function. Cardiovasc Res (2015) 107:386–95. doi:10.1093/cvr/cvv048
15. Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest (2005) 115:3378–84. doi:10.1172/JCI27196
17. Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev (2007) 21:99–111. doi:10.1016/j.blre.2006.06.001
18. Nieswandt B, Brakebusch C, Bergmeier W, Schulte V, Bouvard D, Mokhtari-Nejad R, et al. Glycoprotein VI but not alpha 2 beta 1 integrin is essential for platelet interaction with collagen. EMBO J (2001) 20:2120–30. doi:10.1093/emboj/20.9.2120
19. Peter K, Schwarz M, Ylanne J, Kohler B, Moser M, Nordt T, et al. Induction of fibrinogen binding and platelet aggregation as a potential intrinsic property of various glycoprotein IIb/IIIa (alpha(IIb)beta(3)) inhibitors. Blood (1998) 92:3240–9.
20. Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature (1998) 394:690–4. doi:10.1038/29325
21. Shankar R, Delamotte CA, Poptic EJ, Dicorleto PE. Thrombin receptor-activating peptides differentially stimulate platelet-derived growth-factor production, monocytic cell-adhesion, and E-selectin expression in human umbilical vein endothelial-cells. J Biol Chem (1994) 269:13936–41.
22. Kaufmann R, Patt S, Kraft R, Zieger M, Henklein P, Neupert G, et al. PAR 1-type thrombin receptors are involved in thrombin-induced calcium signaling in human meningioma cells. J Neurooncol (1999) 42:131–6. doi:10.1023/A:1006246219449
23. Kowalska MA, Rauova L, Poncz M. Role of the platelet chemokine platelet factor 4 (PF4) in hemostasis and thrombosis. Thromb Res (2010) 125:292–6. doi:10.1016/j.thromres.2009.11.023
24. Rouhiainen A, Imai S, Rauvala H, Parkkinen J. Occurrence of amphoterin (HMG1) as an endogenous protein of human platelets that is exported to the cell surface upon platelet activation. Thromb Haemost (2000) 84:1087–94.
25. Brass LF. Thrombin and platelet activation. Chest (2003) 124:18S–25S. doi:10.1378/chest.124.3_suppl.18S
26. Merten M, Thiagarajan P. P-selectin expression on platelets determines size and stability of platelet aggregates. Circulation (2000) 102:1931–6. doi:10.1161/01.CIR.102.16.1931
27. Merten M, Chow T, Hellums JD, Thiagarajan P. A new role for P-selectin in shear-induced platelet aggregation. Circulation (2000) 102:2045–50. doi:10.1161/01.CIR.102.17.2045
28. Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res (2013) 112:1506–19. doi:10.1161/CIRCRESAHA.113.300512
29. Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol (2012) 34:5–30. doi:10.1007/s00281-011-0286-4
30. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi:10.1182/blood-2011-03-343061
31. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity (1999) 11:443–51. doi:10.1016/S1074-7613(00)80119-3
32. Zhang GY, Han JY, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol (2009) 182:7997–8004. doi:10.4049/jimmunol.0802884
33. Rex S, Beaulieu LM, Perlman DH, Vitseva O, Blair PS, McComb ME, et al. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thromb Haemost (2009) 102(1):97–110. doi:10.1160/TH08-08-0513
34. Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, et al. Stimulation of toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res (2009) 104:346–U132. doi:10.1161/CIRCRESAHA.108.185785
35. Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MKB, et al. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost (2005) 94:831–8.
36. Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood (1999) 93:876–85.
37. Yang J, Furie BC, Furie B. The biology of P-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb Haemost (1999) 81:1–7.
38. Diacovo TG, Defougerolles AR, Bainton DF, Springer TA. A functional integrin ligand on the surface of platelets: intercellular adhesion molecule-2. J Clin Invest (1994) 94:1243–51. doi:10.1172/JCI117442
39. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science (2014) 346:1234–8. doi:10.1126/science.1256478
40. Kornerup KN, Salmon GP, Pitchford SC, Liu WL, Page CP. Circulating platelet-neutrophil complexes are important for subsequent neutrophil activation and migration. J Appl Physiol (2010) 109:758–67. doi:10.1152/japplphysiol.01086.2009
41. Mauler M, Seyfert J, Haenel D, Seeba H, Guenther J, Stallmann D, et al. Platelet-neutrophil complex formation – a detailed in vitro analysis of murine and human blood samples. J Leukoc Biol (2015) 99(95):781–9. doi:10.1189/jlb.3TA0315-082R
42. Salanova B, Choi M, Rolle S, Wellner M, Luft FC, Kettritz R. Beta2-integrins and acquired glycoprotein IIb/IIIa (GPIIb/IIIa) receptors cooperate in NF-kappaB activation of human neutrophils. J Biol Chem (2007) 282:27960–9. doi:10.1074/jbc.M704039200
43. Spagnuolo PJ, Ellner JJ, Hassid A, Dunn MJ. Thromboxane-A2 mediates augmented polymorphonuclear leukocyte adhesiveness. J Clin Invest (1980) 66:406–14. doi:10.1172/JCI109870
44. Paterson IS, Klausner JM, Goldman G, Kobzik L, Welbourn R, Valeri CR, et al. Thromboxane mediates the ischemia-induced neutrophil oxidative burst. Surgery (1989) 106:224–9.
45. Goldman G, Welbourn R, Valeri CR, Shepro D, Hechtman HB. Thromboxane-A2 induces leukotriene-B4 synthesis that in turn mediates neutrophil diapedesis via Cd-18 activation. Microvasc Res (1991) 41:367–75. doi:10.1016/0026-2862(91)90035-A
46. Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, et al. High mobility group box 1 protein interacts with multiple toll-like receptors. Am J Physiol Cell Physiol (2006) 290:C917–24. doi:10.1152/ajpcell.00401.2005
47. Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu XR, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest (2015) 125:539–50. doi:10.1172/JCI76887
48. Petersen F, Brandt E, Lindahl U, Spillmann D. Characterization of a neutrophil cell surface glycosaminoglycan that mediates binding of platelet factor 4. J Biol Chem (1999) 274:12376–82. doi:10.1074/jbc.274.18.12376
49. Petersen F, Ludwig A, Flad HD, Brandt E. TNF-alpha renders human neutrophils responsive to platelet factor 4. Comparison of PF-4 and IL-8 reveals different activity profiles of the two chemokines. J Immunol (1996) 156:1954–62.
50. Deuel TF, Senior RM, Chang D, Griffin GL, Heinrikson RL, Kaiser ET. Platelet factor-4 is chemotactic for neutrophils and monocytes. Proc Natl Acad Sci U S A (1981) 78:4584–7. doi:10.1073/pnas.78.7.4584
51. Ghasemzadeh M, Kaplan ZS, Alwis I, Schoenwaelder SM, Ashworth KJ, Westein E, et al. The CXCR1/2 ligand NAP-2 promotes directed intravascular leukocyte migration through platelet thrombi. Blood (2013) 121:4555–66. doi:10.1182/blood-2012-09-459636
52. Grommes J, Alard JE, Drechsler M, Wantha S, Morgelin M, Kuebler WM, et al. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med (2012) 185:628–36. doi:10.1164/rccm.201108-1533OC
53. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol (2016) 99:153–62. doi:10.1189/jlb.3A0415-161R
54. Clark SR, Ma AC, Tavener SA, Mcdonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13:463–9. doi:10.1038/nm1565
55. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost (2014) 12:2074–88. doi:10.1111/jth.12710
56. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe (2012) 12:324–33. doi:10.1016/j.chom.2012.06.011
57. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog (2011) 7:e1002355. doi:10.1371/journal.ppat.1002355
58. Tadie JM, Bae HB, Jiang SN, Park DW, Bell CP, Yang H, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol (2013) 304:L342–9. doi:10.1152/ajplung.00151.2012
59. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood (2015) 126:242–6. doi:10.1182/blood-2015-01-624023
60. Sil P, Yoo D-G, Floyd M, Gingerich A, Rada B. High throughput measurement of extracellular DNA release and quantitative NET formation in human neutrophils in vitro. J. Vis. Exp (2016) (112):e52779. doi:10.3791/52779
61. Rossaint J, Herter JM, Van Aken H, Napirei M, Doring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood (2014) 123:2573–84. doi:10.1182/blood-2013-07-516484
62. Tarantino E, Amadio P, Squellerio I, Porro B, Sandrini L, Turnu L, et al. Role of thromboxane-dependent platelet activation in venous thrombosis: aspirin effects in mouse model. Pharmacol Res (2016) 107:415–25. doi:10.1016/j.phrs.2016.04.001
63. Jung CJ, Yeh CY, Hsu RB, Lee CM, Shun CT, Chia JS. Endocarditis pathogen promotes vegetation formation by inducing intravascular neutrophil extracellular traps through activated platelets. Circulation (2015) 131:571–U414. doi:10.1161/CIRCULATIONAHA.114.011432
64. Xiao W, Chen X, Lin Z, Lu S, Liang Z, Wu X. Binding of divalent H-2Kd/IgG2aFc fusion protein to murine macrophage via Fc-FcR interaction. Cell Mol Immunol (2007) 4:147–51.
65. Barnard MR, Krueger LA, Frelinger AL III, Furman MI, Michelson AD. Whole blood analysis of leukocyte-platelet aggregates. Curr Protoc Cytom (2003) 6:15. doi:10.1002/0471142956.cy0615s24
66. Massberg S, Enders G, Leiderer R, Eisenmenger S, Vestweber D, Krombach F, et al. Platelet-endothelial cell interactions during ischemia/reperfusion: the role of P-selectin. Blood (1998) 92:507–15.
67. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209:819–35. doi:10.1084/jem.20112322
68. Ruf A, Patscheke H. Platelet-induced neutrophil activation: platelet-expressed fibrinogen induces the oxidative burst in neutrophils by an interaction with CD11C/CD18. Br J Haematol (1995) 90:791–6. doi:10.1111/j.1365-2141.1995.tb05197.x
69. Ehlers R, Ustinov V, Chen Z, Zhang X, Rao R, Luscinskas FW, et al. Targeting platelet-leukocyte interactions: identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibalpha. J Exp Med (2003) 198:1077–88. doi:10.1084/jem.20022181
70. Simon DI, Chen Z, Xu H, Li CQ, Dong J, Mcintire LV, et al. Platelet glycoprotein Ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med (2000) 192:193–204. doi:10.1084/jem.192.2.193
71. Petri B, Broermann A, Li H, Khandoga AG, Zarbock A, Krombach F, et al. von Willebrand factor promotes leukocyte extravasation. Blood (2010) 116:4712–9. doi:10.1182/blood-2010-03-276311
72. Bombeli T, Schwartz BR, Harlan JM. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J Exp Med (1998) 187:329–39. doi:10.1084/jem.187.3.329
73. May AE, Kalsch T, Massberg S, Herouy Y, Schmidt R, Gawaz M. Engagement of glycoprotein IIb/IIIa (alpha(IIb)beta(3)) on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells. Circulation (2002) 106:2111–7. doi:10.1161/01.CIR.0000033597.45947.0F
74. Weiss HJ, Hawiger J, Ruggeri ZM, Turitto VT, Thiagarajan P, Hoffmann T. Fibrinogen-independent platelet-adhesion and thrombus formation on subendothelium mediated by glycoprotein Iib-Iiia complex at high shear rate. J Clin Invest (1989) 83:288–97. doi:10.1172/JCI113871
75. Schmidt S, Moser M, Sperandio M. The molecular basis of leukocyte recruitment and its deficiencies. Mol Immunol (2013) 55:49–58. doi:10.1016/j.molimm.2012.11.006
76. Borregaard N. Neutrophils, from marrow to microbes. Immunity (2010) 33:657–70. doi:10.1016/j.immuni.2010.11.011
77. McDonald B, Kubes P. Interactions between CD44 and hyaluronan in leukocyte trafficking. Front Immunol (2015) 6(107):68. doi:10.3389/fimmu.2015.00068
78. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol (2014) 15:602–11. doi:10.1038/ni.2921
79. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med (2011) 17:1381–90. doi:10.1038/nm.2514
80. Yago T, Tsukamoto H, Liu Z, Wang Y, Thompson LF, Mcever RP. Multi-inhibitory effects of A2A adenosine receptor signaling on neutrophil adhesion under flow. J Immunol (2015) 195:3880–9. doi:10.4049/jimmunol.1500775
81. Sundd P, Gutierrez E, Koltsova EK, Kuwano Y, Fukuda S, Pospieszalska MK, et al. ‘Slings’ enable neutrophil rolling at high shear. Nature (2012) 488:399–403. doi:10.1038/nature11248
82. Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol (2009) 27:339–62. doi:10.1146/annurev.immunol.021908.132554
83. Dimasi D, Sun WY, Bonder CS. Neutrophil interactions with the vascular endothelium. Int Immunopharmacol (2013) 17:1167–75. doi:10.1016/j.intimp.2013.05.034
84. Zarbock A, Ley K. Neutrophil adhesion and activation under flow. Microcirculation (2009) 16:31–42. doi:10.1080/10739680802350104
85. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol (2014) 9:181–218. doi:10.1146/annurev-pathol-020712-164023
86. Woodfin A, Voisin MB, Nourshargh S. Recent developments and complexities in neutrophil transmigration. Curr Opin Hematol (2010) 17:9–17. doi:10.1097/MOH.0b013e3283333930
87. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med (2006) 203:2569–75. doi:10.1084/jem.20060925
88. Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood (2005) 106:584–92. doi:10.1182/blood-2004-12-4942
89. Williams SL, Milne IR, Bagley CJ, Gamble JR, Vadas MA, Pitson SM, et al. A proinflammatory role for proteolytically cleaved annexin A1 in neutrophil transendothelial migration. J Immunol (2010) 185:3057–63. doi:10.4049/jimmunol.1000119
90. Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS One (2008) 3:e1649. doi:10.1371/journal.pone.0001649
91. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi:10.1038/nri3399
92. Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, et al. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity (2015) 42:1075–86. doi:10.1016/j.immuni.2015.05.010
93. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol (2011) 12:761–9. doi:10.1038/ni.2062
94. Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol (2006) 80:1281–8. doi:10.1189/jlb.0506346
95. Pillay J, Kamp VM, Van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest (2012) 122:327–36. doi:10.1172/JCI57990
96. Doerfler ME, Danner RL, Shelhamer JH, Parrillo JE. Bacterial lipopolysaccharides prime human neutrophils for enhanced production of leukotriene B4. J Clin Invest (1989) 83:970–7. doi:10.1172/JCI113983
97. Guthrie LA, Mcphail LC, Henson PM, Johnston RB Jr. Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. Evidence for increased activity of the superoxide-producing enzyme. J Exp Med (1984) 160:1656–71. doi:10.1084/jem.160.6.1656
98. Williams MR, Azcutia V, Newton G, Alcaide P, Luscinskas FW. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol (2011) 32:461–9. doi:10.1016/j.it.2011.06.009
99. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One (2012) 7:e32366. doi:10.1371/journal.pone.0032366
100. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis (2015) 74:1417–24. doi:10.1136/annrheumdis-2013-204837
101. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett (2010) 584:3193–7. doi:10.1016/j.febslet.2010.06.006
102. Bennouna S, Bliss SK, Curiel TJ, Denkers EY. Cross-talk in the innate immune system: neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J Immunol (2003) 171:6052–8. doi:10.4049/jimmunol.171.11.6052
103. Bennouna S, Denkers EY. Microbial antigen triggers rapid mobilization of TNF-alpha to the surface of mouse neutrophils transforming them into inducers of high-level dendritic cell TNF-alpha production. J Immunol (2005) 174:4845–51. doi:10.4049/jimmunol.174.8.4845
104. Schuster S, Hurrell B, Tacchini-Cottier F. Crosstalk between neutrophils and dendritic cells: a context-dependent process. J Leukoc Biol (2013) 94:671–5. doi:10.1189/jlb.1012540
105. van Gisbergen KP, Sanchez-Hernandez M, Geijtenbeek TB, Van Kooyk Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J Exp Med (2005) 201:1281–92. doi:10.1084/jem.20041276
106. Yang D, De La Rosa G, Tewary P, Oppenheim JJ. Alarmins link neutrophils and dendritic cells. Trends Immunol (2009) 30:531–7. doi:10.1016/j.it.2009.07.004
107. Yang CW, Strong BS, Miller MJ, Unanue ER. Neutrophils influence the level of antigen presentation during the immune response to protein antigens in adjuvants. J Immunol (2010) 185:2927–34. doi:10.4049/jimmunol.1001289
108. Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood (2012) 120:3007–18. doi:10.1182/blood-2012-03-416156
109. Doring Y, Manthey HD, Drechsler M, Lievens D, Megens RT, Soehnlein O, et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation (2012) 125:1673–83. doi:10.1161/CIRCULATIONAHA.111.046755
110. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra19. doi:10.1126/scitranslmed.3001180
111. Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe (2012) 12:109–16. doi:10.1016/j.chom.2012.05.015
112. Cognasse F, Hamzeh-Cognasse H, Laradi S, Chou ML, Seghatchian J, Burnouf T, et al. The role of microparticles in inflammation and transfusion: a concise review. Transfus Apher Sci (2015) 53:159–67. doi:10.1016/j.transci.2015.10.013
113. Arteaga RB, Chirinos JA, Soriano AO, Jy W, Horstman L, Jimenez JJ, et al. Endothelial microparticles and platelet and leukocyte activation in patients with the metabolic syndrome. Am J Cardiol (2006) 98:70–4. doi:10.1016/j.amjcard.2006.01.054
114. Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E, et al. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol (2005) 45:1467–71. doi:10.1016/j.jacc.2004.12.075
115. Jy W, Mao WW, Horstman L, Tao J, Ahn YS. Platelet microparticles bind, activate and aggregate neutrophils in vitro. Blood Cells Mol Dis (1995) 21:217–31; discussion 231a. doi:10.1006/bcmd.1995.0025
116. Belizaire RM, Prakash PS, Richter JR, Robinson BR, Edwards MJ, Caldwell CC, et al. Microparticles from stored red blood cells activate neutrophils and cause lung injury after hemorrhage and resuscitation. J Am Coll Surg (2012) 214:648–55; discussion 656–57. doi:10.1016/j.jamcollsurg.2011.12.032
117. Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol (2005) 66:1146–54. doi:10.1016/j.humimm.2005.11.003
118. He Z, Si Y, Jiang T, Ma R, Zhang Y, Cao M, et al. Phosphotidylserine exposure and neutrophil extracellular traps enhance procoagulant activity in patients with inflammatory bowel disease. Thromb Haemost (2016) 115:738–51. doi:10.1160/TH15-09-0710
119. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet (2000) 25:177–81. doi:10.1038/76032
120. Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol (2012) 188:3522–31. doi:10.4049/jimmunol.1102404
121. Hakkim A, Furnrohr BG, Amann K, Laube B, Abu Abed U, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A (2010) 107:9813–8. doi:10.1073/pnas.0909927107
122. Nakazawa D, Shida H, Tomaru U, Yoshida M, Nishio S, Atsumi T, et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol (2014) 25:990–7. doi:10.1681/ASN.2013060606
123. Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol (2013) 191:2647–56. doi:10.4049/jimmunol.1300436
Keywords: neutrophil extracellular traps, platelets, endothelium, selectins, integrins
Citation: Kazzaz NM, Sule G and Knight JS (2016) Intercellular Interactions as Regulators of NETosis. Front. Immunol. 7:453. doi: 10.3389/fimmu.2016.00453
Received: 29 July 2016; Accepted: 11 October 2016;
Published: 14 November 2016
Edited by:
Marko Radic, The University of Tennessee Health Science Center, USACopyright: © 2016 Kazzaz, Sule and Knight. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason S. Knight, anNrbmlnaHRAdW1pY2guZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.