 Krittalak Chakrabandhu
Krittalak Chakrabandhu Anne-Odile Hueber
Anne-Odile Hueber
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 17 October 2016
Sec. Immunological Tolerance and Regulation
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00429
This article is part of the Research Topic Ying and Yang of members of TNF superfamily: Friends or Foes in immune-mediated diseases and cancer View all 9 articles
The Fas/FasL system is known, first and foremost, as a potent apoptosis activator. While its proapoptotic features have been studied extensively, evidence that the Fas/FasL system can elicit non-death signals has also accumulated. These non-death signals can promote survival, proliferation, migration, and invasion of cells. The key molecular mechanism that determines the shift from cell death to non-death signals had remained unclear until the recent identification of the tyrosine phosphorylation in the death domain of Fas as the reversible signaling switch. In this review, we present the connection between the recent findings regarding the control of Fas multi-signals and the context-dependent signaling choices. This information can help explain variable roles of Fas signaling pathway in different pathologies.
Fas (TNFRSF6/CD95) belongs to the tumor necrosis factor receptor superfamily. When bound to Fas ligand (FasL) or agonistic antibodies, Fas can recruit Fas-associated death domain-containing protein (FADD), procaspase-8, procaspase-10, and cellular FLICE inhibitory proteins (c-FLIPs). This leads to the formation of the death-inducing signaling complex (DISC), the caspase cascade and ultimately apoptosis (1, 2). Apoptosis mediated by the Fas/FasL system is essential for shutting down chronic immune responses (3–5) and preventing autoimmunity and cancer (6). The downregulation of Fas in some cancers prompted the opinion that it was a tumor suppressor. However, while Fas is often downregulated in cancer, it rarely is completely lost (7). Moreover, Fas also mediates cell survival, proliferation, and motility, which can promote autoimmunity, cancer growth, and metastasis (7–12).
Current Fas-targeting therapies aim to activate or inhibit Fas signaling (13, 14). However, without understanding when and why Fas assumes different roles in different pathological contexts, these therapies face a major challenge.
Physiologically, the presence of different FasL forms is an important extrinsic factor that can influence Fas signaling modes. Membrane-bound FasL is essential for activating Fas-mediated apoptosis and thus instrumental in the safeguard against autoimmunity and cancer. Meanwhile, excess soluble FasL (sFasL) may promote autoimmunity and tumorigenesis through non-apoptotic activities (6). However, knowing the different functions of FasL does not sufficiently describe how Fas ultimately takes the apoptotic or non-apoptotic role.
Fas possesses the protein-interacting domain, death domain (DD) (15, 16). Fas multi-signaling requires an efficient molecular switch in DD allowing different signaling complex formations. This agrees with the observation that most disease-causing mutations are in DD (17, 18). This review discusses the regulation of Fas multi-signaling at DD level by tyrosine phosphorylation and implications in human pathologies.
Unbound human Fas (hFas) DD comprises six α-helices (19) (Figure 1A). Fas DD can interact with many adaptors, including FADD (20–23). Resolved structures of Fas/FADD complex have identified some amino acids whose mutations could have pathological consequences (24–26). However, challenges remain in determining DD-mediated complex structure since the full-length receptor and the post-translational modifications, such as DD tyrosine phosphorylation, have yet to be taken into account.
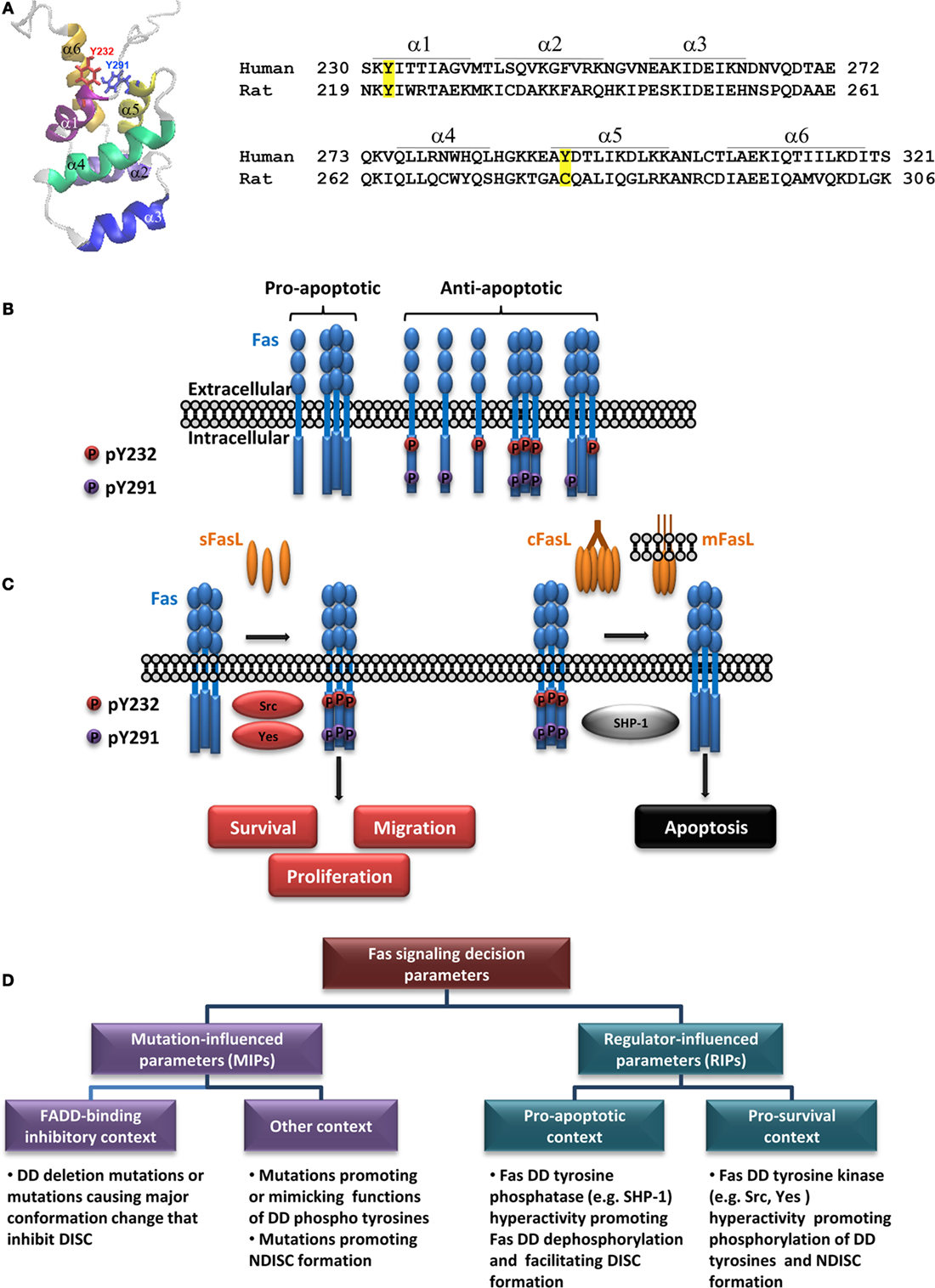
Figure 1. Regulation of Fas multi-signaling by death domain tyrosine phosphorylation and pathologies. (A) Fas death domain structural model demonstrating six antiparallel α-helices (PDB: 1DDF) (19) with the side chains of Y232 and Y291 indicated (left) and amino acid sequences of human Fas and rat Fas encompassing the death domain with positions of each helix indicated (right; α, α-helix). Tyrosine phosphorylation sites in hFas and corresponding residues in rat Fas are highlighted. Amino acid numbering is according to UniProt entries P25445 and Q63199. (B) A diagram depicting different states of Fas, with respect to its ability to transmit apoptotic signal, as affected by its death domain phosphorylation [adapted from Ref. (27)]. The proapoptotic state is allowed when both DD tyrosines are dephosphorylated. The dominant proapoptotic state takes place when Y232 and/or Y291 is phosphorylated (some examples of possible dominant-negative scenarios are given). (C) A simplified illustration of Fas multi-signaling regulation by tyrosine phosphorylation switch. The non-death signaling triggered by activators such as soluble FasL (sFasL) is mediated by Src or Yes phosphorylation of the death domain tyrosines. The death signaling triggered by activators such as cross-linked FasL (cFasL) or membrane FasL (mFasL) is permitted by the dephosphorylation of the death domain tyrosines by SHP-1. (D) A diagram outlining pathologically relevant parameters leading to contexts that define the role of Fas signaling in human diseases (see text).
While structural studies shed light into the DISC formation, an important question remained: “What gives the cue for Fas DD to form the DISC or a non-death-inducing signaling complex (NDISC)?” Because tyrosine Y232 and Y291 in hFas DD are phosphorylable (24), tyrosine phosphorylation is a prime candidate for the mechanism that determines when and which signaling complex is formed. Earlier studies reported that the Y291 of overexpressed hFas inhibited survival signals in mouse neutrophil (25). In the rat, whose Fas lacks the equivalent of Y291 of hFas (Figure 1A), Fas tyrosine phosphorylation associated with apoptosis in hepatocytes (26, 28). For nearly 20 years after the first report of hFas phosphotyrosine (pY), whether Y232 and/or Y291 phosphorylation was a switch for hFas multi-signaling had remained unclear, and the functions of each DD tyrosine had also remained unknown. The lag was due to the lack of practical pY mimetics for functional analyses and site-specific hFas pY detection.
Our recent study based on the analysis of evolution-guided Fas pY proxies and site-specific pY detection has revealed that DD tyrosine phosphorylation of hFas is the reversible antiapoptotic/pro-survival multi-signaling switch, namely, the DD tyrosine phosphorylation turns off the proapoptotic signal and turns on the pro-survival signals by dominantly inhibiting the DISC formation and apoptosis (Figure 1B) while promoting FasL-induced cell proliferation and migration (Figure 1C). We have also shown that pY232 and pY291 are regulated distinctly in some cancers (27).
That Fas DD pY dominantly inhibits Fas apoptotic signal and activates pro-survival signals invites a reassessment of our view about how Fas exerts its action in pathologies, such as cancer. A current opinion suggests that Fas signaling requires at least one wild-type FAS allele and that the signal transition from non-apoptotic to apoptotic signaling occurs when the Fas signal strength, exhibited by wild-type Fas protein, exceeds a threshold. This opinion is based on two reasons: (1) a mutated FAS allele that causes the loss of apoptotic function is often considered completely non-functional and (2) when Fas mutations are detected, tumors rarely have the loss of heterozygosity (18).
The threshold-based switch notion suggests that apoptotic signal requires two wild-type FAS alleles (strong signal) to reach its high threshold, while the threshold for the non-apoptotic signal is so low that it is attainable with one wild-type FAS allele (29). Based on the recent findings, the intermolecular and intramolecular “death-off” dominant inhibitory function of DD pY and its activating function for survival signals (27) suggest that the DD tyrosine phosphorylation is a highly efficient “on-off” multi-signaling switch. This information extends our views on Fas multi-signaling in diseases from threshold-based signaling switch to cover the concept that the apoptotic signal requires conditions that favor double dephosphorylation of the DD tyrosines, and the pro-survival signal is achievable in conditions that favor the phosphorylation of least one DD tyrosine.
Src-family kinases (SFKs), including Src, Yes, Fyn, Blk, Yrk, Fgr, Hck, Lck, and Lyn, are protein tyrosine kinases that are preferentially expressed in different tissues (30, 31). Data from rodent models indirectly implied the role of Fyn and Yes as positive regulators of Fas-mediated apoptosis (32–36). Although, while some SFKs might play a proapoptotic role, they may not directly participate in Fas tyrosine phosphorylation. For example, the activation of human eosinophils led to a transient Fas tyrosine phosphorylation, followed by Lyn activation, which occurred concomitantly with Fas dephosphorylation (37). In fact, the phosphorylation of Fas by SFKs in cells had not been demonstrated till recently.
Studies of hFas in human colorectal cancer (CRC) cells have shown that Src and Yes play an important antiapoptotic and pro-survival roles in hFas signaling by phosphorylating hFas at Y232 and Y291 (27). The phosphorylation of Fas DD by Src and Yes leads to an inhibition of apoptosis and the enhanced cancer cell proliferation and migration, which are consistent with the oncogenic roles of these SFKs often reported in human cancers (38). The findings that (1) the levels of pY232 and pY291 increase in several types of cancer, including breast, ovarian, and colon cancers and (2) pY232 and pY291 levels appear to correlate with CRC progression (27) are in line with observations that the elevated Src and Yes levels correlate with advanced stages and metastatic potential of tumors and poor prognosis (39–42). In human glioblastoma multiforme (GBM), the Fas–Yes interaction and subsequent activation of PI3K/Akt pathway mediate glioblastoma invasion, and the Yes expression and phosphorylation of SFKs are present along with increased FasL expression in the tumor/host interaction zone in tumors of GBM patients (43). Additionally, Fas–Yes association leads to the activation of PI3K/Akt pathway and cell migration in human triple-negative breast cancer model (44). These observations support the role of SFKs in the Fas phosphorylation and tumor malignancy.
A point to keep in mind is the context under consideration. The roles of SFKs in Fas signaling and even the identity of the SFKs involved may differ appreciably in different tissues, disorders, or disease stages since expression profiles of kinases can vary significantly from one setting to another. For instance, while Src and Yes are key regulators of hFas phosphorylation in some solid tumors, this may not hold true for some hematopoietic malignancies where other oncogenic SFKs, such as Lck or Fgr are prominently present. Additionally, divergence in terms of regulatory specificity exists among model systems. For example, a non-conservative tyrosine phosphorylation site in Fas DD among primates and rodents (27) suggests diverse roles and identities of kinases that regulate Fas phosphorylation in different species. Therefore, extrapolating the regulation of Fas tyrosine phosphorylation switch from one species to another is likely to be inappropriate. Thus far, current information supports the notion that oncogenic SFKs, such as Src and Yes, are responsible for the tyrosine phosphorylation of DD of hFas and hence are positive regulators of Fas survival signals and negative regulators of Fas apoptotic signal.
Src homology domain 2 (SH2)-containing tyrosine phosphatase-1 (SHP-1), a protein tyrosine phosphatase, is predominantly present in hematopoietic cells and to a lesser extent in other cell types, including epithelial cells (45–48). Human SHP-1 appears to be a positive regulator of Fas-mediated apoptosis (27, 49) and a negative regulator of survival (25, 50), proliferation (51), and epithelial–mesenchymal transition (52). SHP-1 binding to hFas requires Y291 of Fas DD (25). In human CRC cells, it functionally opposes the effects of Src and Yes by dephosphorylating both pY232 and pY291, switching Fas from the antiapoptotic state to the proapoptotic/anti-proliferative state (Figure 1C) (27).
Notably, the rodent models demonstrate that the roles of SHP-1 depend on contexts, including tissues, diseases, and species. For example, Fas-mediated apoptosis was defective in the lymphoid organs but not in hepatocytes and thymocytes from SHP-1-deficient mice (49, 53). In mouse B cells, SHP-1 negatively regulates the DISC formation through its phosphatase activity on Vav1 (54). In rat granulocytes, SHP-1-CEACAM1 binding is important for downregulating FasL-induced apoptosis (53). The fact that rat Fas lacks the tyrosine shown to be the SHP-1-binding site in hFas (25) implies its distinct requirement for interacting with SHP-1 that may explain the different roles of rodent SHP-1 in Fas signaling. Like SFKs, identifying phosphatases and evaluating their roles in the regulation of Fas phosphorylation and signaling require a careful consideration of the contexts since the details of the signaling regulation can differ appreciably among species, tissues, disorders, or disease stages.
Besides its cognate ligand and agonistic antibodies, other activators, including anticancer drugs and cytokines, can also activate Fas signaling (55–59). As we continue to unveil the control of pY-based mechanism of Fas multi-signaling switch, the roles of other molecules that may directly or indirectly influence the phosphorylation process of hFas DD, and the downstream Fas signaling will be clarified. For example, besides regulating the phosphorylation and non-death signaling of Fas (27), Yes also links Fas to EGFR and the PI3K/Akt pathways (44, 60), and thus PP2A (61). These actors, among others, are likely to participate in the mode of Fas signaling, at least indirectly. Further studies into the cross-talks between Fas and such actors, which are also drug targets (62–65), will not only further reinforce our understanding of context-dependent Fas signaling in human diseases but also aid in the design of efficient combinatorial therapies against diseases in which Fas is involved (66–68).
The involvement of tyrosine phosphorylation in Fas signaling has been well appreciated. In fact, the pY-based survival/apoptotic switch system also applies to other actors in Fas signaling network, with an important example being Caspase-8.
Human Caspase-8 (hCaspase-8) has at least three tyrosine phosphorylation sites. Phosphorylation of Y310, Y397, and/or Y465 (Y293, Y380, and/or Y488, respectively, in isoform B) suppresses Fas-mediated apoptosis (69–72). Additionally, prosurvival activators such as EGF can induce the phosphorylation of Caspase-8, which mediates its interaction with PI3K and cell adhesion and motility (73) and protects the cells from Fas-mediated apoptosis (69). The Caspase-8-mediated cell migration depends on pY380 but not the Caspase-8 activity or the death effector domain (74), thus separating non-apoptotic function of Caspase-8 from that of the receptor-mediated DISC formation path. Caspase-8 tyrosine phosphorylation may also participate in a positive feedback loop of caspase-8-induced Src activation and further promotes survival pathways (75). Additionally, pY310 of Caspase-8 promotes its interaction with SHP-1, which dephosphorylates Caspase-8, permitting its proapoptotic function (71).
Notably, Caspase-8 in rodents, including mouse, rat, guinea pig, and Chinese hamster lacks tyrosines at the positions equivalent to Y310 and Y380 in hCaspase-8. In mouse and guinea pig, it also lacks the tyrosine at the position equivalent to Y465 in hCaspase-8. Therefore, Caspase-8 multi-signaling regulation in rodents is likely to differ significantly from that of human, particularly regarding the dependence on tyrosine phosphorylation switch. Similar to Fas, this highlights the importance of careful consideration when applying the observed regulation and functions of Caspase-8 from one species to another. The lack of a tyrosine at the position equivalent to Y310 (SHP-1-binding site) of hCaspase-8 in rodent SHP-1 coincides with the observation that it participates in Fas signaling differently from human SHP-1. Thus, it is quite probable that, in different species, distinct sets of kinases and phosphatases regulate Fas multimode signaling system. This observation, along with other species-based variations in immune-related signaling (76, 77), serves as a reminder that, while the value of animal models is appreciable, particular attention should be given to the species-dependent details especially when designing targeted therapies. Notably, although the functional outcome among species may seem the same, the underlying mechanism might be very different.
That the SFK/SHP-1 phosphorylation-dephosphorylation mechanism applies to both Fas and Caspase-8, the main DISC components, based on information from human cells, suggests at least one common “survival-ON/death-OFF” switch system in the Fas signaling network and can help explain some observation of Fas-mediated outcome in human pathologies. For instance, hyperactivated Src and/or downregulation of SHP-1, such as in some cancers, may favor Fas DD and Caspase-8 tyrosine phosphorylation and thus the antiapoptotic/survival mode of Fas signaling.
Interrupted apoptotic/non-apoptotic balance begets pathologies. Too little or too much of Fas-mediated apoptosis, as well as the rampant non-apoptotic activity of Fas can cause autoimmune diseases and cancer (3, 4, 12, 17, 43, 78, 79). Thus, several Fas-targeting therapeutics have been designed based on various strategies and are currently in different stages of development and clinical trials (Table 1). Understanding the apoptotic/non-apoptotic switch of Fas, which helps explain its pathological roles can help identify appropriate therapeutic choices. That said, how the apoptotic/non-apoptotic switch is set depends not only on modifications of the FAS gene but also on an intricate signaling network involving not only different forms of its ligand but also direct and indirect regulators, which varies according to genetic background. In the clinical aspect, parameters governing Fas multi-signaling may be divided into two main groups: Fas mutation-influenced parameters (MIPs) and regulator-influenced parameters (RIPs) (Figure 1D). Following examples illustrate the MIPs and RIPs in pathologies and their implications in context-dependent therapeutic design.
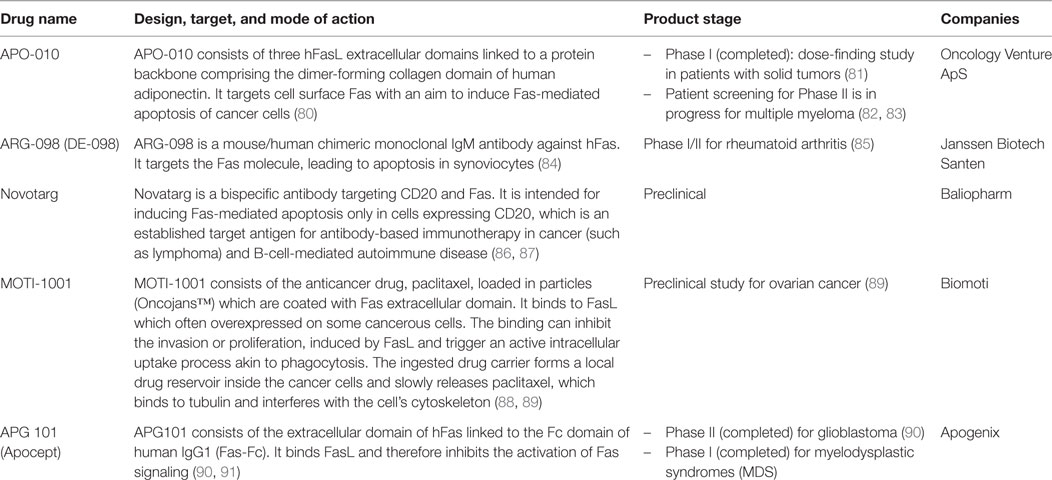
Table 1. Examples of Fas/FasL-targeting drugs.
Autoimmune lymphoproliferative syndrome (ALPS) is often affected by MIPs since Fas mutations, mostly in DD, are the common cause of the disease (17). Some Fas mutations cause defects in the binding to FADD, inhibiting the DISC formation and apoptosis. Other mutations may promote the NDISC formation, which may depend on the regulation of DD tyrosine phosphorylation. As such, ALPS cases that fall into different contexts of MIPs can respond differently to the same intervention. Thus, therapeutic approaches tailored for ALPS in context-dependent manners are desirable.
Systemic lupus erythematosus (SLE) illustrates multi-faceted MIPs and RIPs that involve both the downregulation of apoptosis and the upregulation of migration mediated by Fas. MIPs in SLE include FAS gene polymorphisms and mRNA editing. Fas gene polymorphisms have been associated with susceptibility to SLE (92), although, the consequence of the polymorphisms on the protein’s function is unclear. Recently, a FAS mRNA mutation caused by an mRNA editing has been reported in SLE patients (93). While the FAS gene mutation is absent in the genomic DNA, the mRNA editing leads to the production of an apoptosis-defective truncated Fas protein with a frameshift at the end of DD especially in T cells of the SLE patients (93). Meanwhile, RIPs in SLE include the involvement of Lyn and the expression levels of soluble Fas (sFas) and sFasL. The loss of function of Lyn, which is implicated in the development of SLE (94), can also dampen Fas-mediated apoptosis (37). Elevated levels of sFas and sFasL of a significant number of SLE patients (60, 95) are also associated with the disease flare (96). The sFasL is implicated in the trafficking of T helper 17 lymphocytes to inflamed organs (12). Meanwhile, it is unclear whether the elevated level of sFas in SLE patient prevents Fas-mediated apoptosis, as previously suggested (95) or counteracts the effect of elevated sFasL level in SLE patients. How these RIPs influence the Fas signaling switch is yet to be clarified. And as such, different SLE cases influenced by the different context of MIPS and RIPs will have to be handled accordingly.
The progression of GBM, where Fas mutations are uncommon (97), involves RIPs that promote non-apoptotic signaling of Fas, such as SFK and FasL expression. An elevated SFK activity in GBM (98) can promote NDISC and GBM cell invasion (43). FasL also contributes toward the invasive phenotype of glioblastoma cells (99). In GBM cases, where an SFK that promotes Fas DD phosphorylation (e.g., Src or Yes) is hyperactivated, the intervention should thus be aimed at inhibiting NDISC that can promote cancer progression. Such interventions may be achieved by preventing FasL from binding to Fas and thus preventing the Fas activation and/or by specifically inhibit the activities of Src and Yes and thus preventing Fas DD phosphorylation. In agreement with this view, a drug that prevents FasL from activating Fas non-apoptotic signals shows promise in reducing GBM progression (90). One may expect that integrating this approach with inhibiting Src and Yes (100), can further improve therapeutic success for GBM.
A hyper-apoptotic signaling is significant in myelodysplastic syndromes (MDS) (101), where RIPs, such as overexpression of Fas (79) and SHP-1 (102) are prominent. When SHP-1 (the suppressor of Fas DD phosphorylation) is upregulated, inhibiting the rampant apoptosis, such as by blocking FasL or SHP-1, should be beneficial. In agreement with this view, preventing FasL from activating Fas apoptotic signal in MDS seems promising (91). It can also be envisaged that combining this approach with SHP-1 inhibition may further improve therapeutic success for MDS.
Overall, the findings discussed here emphasize the importance of examining Fas multi-signaling while considering different contexts that can influence Fas signaling switch. A context-oriented understanding of Fas multi-signaling will allow the efficient design of Fas-related therapeutic strategies.
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by institutional funds from the Centre National de la Recherche Scientifique (CNRS) and the Institut National de la Santé et de la Recherche Médicale (INSERM), and by grants from the Institut National du Cancer (INCa; PLBIO09-317); the University of Nice, the Agence Nationale de la Recherche (ANR-10-BLAN-1226; ANR-11-LABX-0028-01).
1. Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J (1995) 14(22):5579–88.
2. Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ (2012) 19(1):36–41. doi:10.1038/cdd.2011.155
3. Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity (2008) 28(2):197–205. doi:10.1016/j.immuni.2007.12.017
4. Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK III, Wu T, Li QZ, et al. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity (2008) 28(2):206–17. doi:10.1016/j.immuni.2007.12.015
5. Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity (2008) 28(2):218–30. doi:10.1016/j.immuni.2007.12.014
6. O’Reilly LA, Tai L, Lee L, Kruse EA, Grabow S, Fairlie WD, et al. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature (2009) 461(7264):659–63. doi:10.1038/nature08402
7. Peter ME, Hadji A, Murmann AE, Brockway S, Putzbach W, Pattanayak A, et al. The role of CD95 and CD95 ligand in cancer. Cell Death Differ (2015) 22(4):549–59. doi:10.1038/cdd.2015.25
8. Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, et al. The CD95 receptor: apoptosis revisited. Cell (2007) 129(3):447–50. doi:10.1016/j.cell.2007.04.031
9. Fouque A, Debure L, Legembre P. The CD95/CD95L signaling pathway: a role in carcinogenesis. Biochim Biophys Acta (2014) 1846(1):130–41. doi:10.1016/j.bbcan.2014.04.007
10. Chakrabandhu K, Huault S, Hueber AO. Distinctive molecular signaling in triple-negative breast cancer cell death triggered by hexadecylphosphocholine (miltefosine). FEBS Lett (2008) 582(30):4176–84. doi:10.1016/j.febslet.2008.11.019
11. Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C, et al. CD95 promotes tumour growth. Nature (2010) 465(7297):492–6. doi:10.1038/nature09075
12. Poissonnier A, Sanseau D, Le Gallo M, Malleter M, Levoin N, Viel R, et al. CD95-mediated calcium signaling promotes T helper 17 trafficking to inflamed organs in lupus-prone mice. Immunity (2016) 45(1):209–23. doi:10.1016/j.immuni.2016.06.028
13. National Institutes of Health. A Phase I Dose Finding Study of APO010 in Patients with Solid Tumors (AP1001) NCT00437736. National Institutes of Health (2015). Available from: http://clinicaltrials.gov/ct2/show/NCT00437736?term=apo010&rank=1
14. Bendszus M, Debus J, Wick W, Kobyakov G, Martens T, Heese O, et al. APG101_CD_002: a phase II, randomized, open-label, multicenter study of weekly APG101 plus reirradiation versus reirradiation in the treatment of patients with recurrent glioblastoma. J Clin Oncol (2012) 30(Suppl 15):abstr2034. doi:10.1200/JCO.2009.26.61
15. Tartaglia LA, Ayres TM, Wong GH, Goeddel DV. A novel domain within the 55 kd TNF receptor signals cell death. Cell (1993) 74(5):845–53. doi:10.1016/0092-8674(93)90464-2
16. Weber CH, Vincenz C. The death domain superfamily: a tale of two interfaces? Trends Biochem Sci (2001) 26(8):475–81. doi:10.1016/S0968-0004(01)01905-3
17. Price S, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood (2014) 123(13):1989–99. doi:10.1182/blood-2013-10-535393
18. Peter ME, Legembre P, Barnhart BC. Does CD95 have tumor promoting activities? Biochim Biophys Acta (2005) 1755(1):25–36. doi:10.1016/j.bbcan.2005.01.001
19. Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature (1996) 384(6610):638–41. doi:10.1038/384638a0
20. Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell (1995) 81(4):505–12. doi:10.1016/0092-8674(95)90071-3
21. Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell (1995) 81(4):513–23. doi:10.1016/0092-8674(95)90072-1
22. Yang X, Khosravi-Far R, Chang HY, Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell (1997) 89(7):1067–76. doi:10.1016/S0092-8674(00)80294-9
23. Ahn EY, Lim ST, Cook WJ, McDonald JM. Calmodulin binding to the Fas death domain. Regulation by Fas activation. J Biol Chem (2004) 279(7):5661–6. doi:10.1074/jbc.M311040200
24. Gradl G, Grandison P, Lindridge E, Wang Y, Watson J, Rudert F. The CD95 (Fas/APO-1) receptor is phosphorylated in vitro and in vivo and constitutively associates with several cellular proteins. Apoptosis (1996) 1:131–40. doi:10.1007/BF01321019
25. Daigle I, Yousefi S, Colonna M, Green DR, Simon HU. Death receptors bind SHP-1 and block cytokine-induced anti-apoptotic signaling in neutrophils. Nat Med (2002) 8(1):61–7. doi:10.1038/nm0102-61
26. Reinehr R, Schliess F, Häussinger D. Hyperosmolarity and CD95L trigger CD95/EGF receptor association and tyrosine phosphorylation of CD95 as prerequisites for CD95 membrane trafficking and DISC formation. FASEB J (2003) 17(6):731–3. doi:10.1096/fj.02-0915fje
27. Chakrabandhu K, Huault S, Durivault J, Lang K, Ta Ngoc L, Bole A, et al. An evolution-guided analysis reveals a multi-signaling regulation of Fas by tyrosine phosphorylation and its implication in human cancers. PLoS Biol (2016) 14(3):e1002401. doi:10.1371/journal.pbio.1002401
28. Reinehr R, Graf D, Haussinger D. Bile salt-induced hepatocyte apoptosis involves epidermal growth factor receptor-dependent CD95 tyrosine phosphorylation. Gastroenterology (2003) 125(3):839–53. doi:10.1016/S0016-5085(03)01055-2
29. Legembre P, Barnhart BC, Zheng L, Vijayan S, Straus SE, Puck J, et al. Induction of apoptosis and activation of NF-kappaB by CD95 require different signalling thresholds. EMBO Rep (2004) 5(11):1084–9. doi:10.1038/sj.embor.7400280
30. Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol (1997) 13:513–609. doi:10.1146/annurev.cellbio.13.1.513
31. Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene (2004) 23(48):7906–9. doi:10.1038/sj.onc.1208160
32. Atkinson EA, Ostergaard H, Kane K, Pinkoski MJ, Caputo A, Olszowy MW, et al. A physical interaction between the cell death protein Fas and the tyrosine kinase p59fynT. J Biol Chem (1996) 271(11):5968–71. doi:10.1074/jbc.271.11.5968
33. Grant SG, O’Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science (1992) 258(5090):1903–10. doi:10.1126/science.1361685
34. Reinehr R, Becker S, Eberle A, Grether-Beck S, Häussinger D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J Biol Chem (2005) 280(29):27179–94. doi:10.1074/jbc.M414361200
35. Reinehr R, Becker S, Höngen A, Haüssinger D. The Src family kinase yes triggers hyperosmotic activation of the epidermal growth factor receptor and CD95. J Biol Chem (2004) 279(23):23977–87. doi:10.1074/jbc.M401519200
36. Reinehr R, Becker S, Wettstein M, Häussinger D. Involvement of the Src family kinase yes in bile salt-induced apoptosis. Gastroenterology (2004) 127(5):1540–57. doi:10.1053/j.gastro.2004.08.056
37. Simon HU, Yousefi S, Dibbert B, Hebestreit H, Weber M, Branch DR, et al. Role for tyrosine phosphorylation and Lyn tyrosine kinase in fas receptor-mediated apoptosis in eosinophils. Blood (1998) 92(2):547–57.
38. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature (2001) 411(6835):355–65. doi:10.1038/35077225
39. Aligayer H, Boyd DD, Heiss MM, Abdalla EK, Curley SA, Gallick GE. Activation of Src kinase in primary colorectal carcinoma: an indicator of poor clinical prognosis. Cancer (2002) 94(2):344–51. doi:10.1002/cncr.10221
40. Cartwright CA, Meisler AI, Eckhart W. Activation of the pp60c-src protein kinase is an early event in colonic carcinogenesis. Proc Natl Acad Sci U S A (1990) 87(2):558–62. doi:10.1073/pnas.87.2.558
41. Park J, Meisler AI, Cartwright CA. c-Yes tyrosine kinase activity in human colon carcinoma. Oncogene (1993) 8(10):2627–35.
42. Pena SV, Melhem MF, Meisler AI, Cartwright CA. Elevated c-yes tyrosine kinase activity in premalignant lesions of the colon. Gastroenterology (1995) 108(1):117–24. doi:10.1016/0016-5085(95)90015-2
43. Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O, et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell (2008) 13(3):235–48. doi:10.1016/j.ccr.2008.02.003
44. Malleter M, Tauzin S, Bessede A, Castellano R, Goubard A, Godey F, et al. CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res (2013) 73(22):6711–21. doi:10.1158/0008-5472.CAN-13-1794
45. Banville D, Stocco R, Shen SH. Human protein tyrosine phosphatase 1C (PTPN6) gene structure: alternate promoter usage and exon skipping generate multiple transcripts. Genomics (1995) 27(1):165–73. doi:10.1006/geno.1995.1020
46. Evren S, Wan S, Ma XZ, Fahim S, Mody N, Sakac D, et al. Characterization of SHP-1 protein tyrosine phosphatase transcripts, protein isoforms and phosphatase activity in epithelial cancer cells. Genomics (2013) 102(5–6):491–9. doi:10.1016/j.ygeno.2013.10.001
47. Keilhack H, Muller M, Bohmer SA, Frank C, Weidner KM, Birchmeier W, et al. Negative regulation of Ros receptor tyrosine kinase signaling. An epithelial function of the SH2 domain protein tyrosine phosphatase SHP-1. J Cell Biol (2001) 152(2):325–34. doi:10.1083/jcb.152.2.325
48. Plutzky J, Neel BG, Rosenberg RD, Eddy RL, Byers MG, Jani-Sait S, et al. Chromosomal localization of an SH2-containing tyrosine phosphatase (PTPN6). Genomics (1992) 13(3):869–72. doi:10.1016/0888-7543(92)90172-O
49. Su X, Zhou T, Wang Z, Yang P, Jope RS, Mountz JD. Defective expression of hematopoietic cell protein tyrosine phosphatase (HCP) in lymphoid cells blocks Fas-mediated apoptosis. Immunity (1995) 2(4):353–62. doi:10.1016/1074-7613(95)90143-4
50. Yousefi S, Simon HU. SHP-1: a regulator of neutrophil apoptosis. Semin Immunol (2003) 15(3):195–9. doi:10.1016/S1044-5323(03)00033-2
51. Duchesne C, Charland S, Asselin C, Nahmias C, Rivard N. Negative regulation of beta-catenin signaling by tyrosine phosphatase SHP-1 in intestinal epithelial cells. J Biol Chem (2003) 278(16):14274–83. doi:10.1074/jbc.M300425200
52. Fan LC, Shiau CW, Tai WT, Hung MH, Chu PY, Hsieh FS, et al. SHP-1 is a negative regulator of epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene (2015) 34(41):5252–63. doi:10.1038/onc.2014.445
53. Singer BB, Klaile E, Scheffrahn I, Muller MM, Kammerer R, Reutter W, et al. CEACAM1 (CD66a) mediates delay of spontaneous and Fas ligand-induced apoptosis in granulocytes. Eur J Immunol (2005) 35(6):1949–59. doi:10.1002/eji.200425691
54. Koncz G, Kerekes K, Chakrabandhu K, Hueber AO. Regulating Vav1 phosphorylation by the SHP-1 tyrosine phosphatase is a fine-tuning mechanism for the negative regulation of DISC formation and Fas-mediated cell death signaling. Cell Death Differ (2008) 15(3):494–503. doi:10.1038/sj.cdd.4402282
55. Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med (1998) 188(11):2033–45. doi:10.1084/jem.188.11.2033
56. Codony-Servat J, Garcia-Albeniz X, Pericay C, Alonso V, Escudero P, Fernandez-Martos C, et al. Soluble FAS in the prediction of benefit from cetuximab and irinotecan for patients with advanced colorectal cancer. Med Oncol (2013) 30(1):428. doi:10.1007/s12032-012-0428-0
57. Nadal C, Maurel J, Gallego R, Castells A, Longaron R, Marmol M, et al. FAS/FAS ligand ratio: a marker of oxaliplatin-based intrinsic and acquired resistance in advanced colorectal cancer. Clin Cancer Res (2005) 11(13):4770–4. doi:10.1158/1078-0432.CCR-04-2119
58. Yildiz R, Benekli M, Buyukberber S, Kaya AO, Ozturk B, Yaman E, et al. The effect of bevacizumab on serum soluble FAS/FASL and TRAIL and its receptors (DR4 and DR5) in metastatic colorectal cancer. J Cancer Res Clin Oncol (2010) 136(10):1471–6. doi:10.1007/s00432-010-0803-1
59. Esser MT, Dinglasan RD, Krishnamurthy B, Gullo CA, Graham MB, Braciale VL. IL-2 induces Fas ligand/Fas (CD95L/CD95) cytotoxicity in CD8+ and CD4+ T lymphocyte clones. J Immunol (1997) 158(12):5612–8.
60. Tauzin S, Chaigne-Delalande B, Selva E, Khadra N, Daburon S, Contin-Bordes C, et al. The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PLoS Biol (2011) 9(6):e1001090. doi:10.1371/journal.pbio.1001090
61. Ruvolo PP. The broken “off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin (2016) 6:87–99. doi:10.1016/j.bbacli.2016.08.002
62. Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature (2015) 526(7572):263–7. doi:10.1038/nature14969
63. Lee MS, Kopetz S. Current and future approaches to target the epidermal growth factor receptor and its downstream signaling in metastatic colorectal cancer. Clin Colorectal Cancer (2015) 14(4):203–18. doi:10.1016/j.clcc.2015.05.006
64. Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol (2016) 27(10):13021. doi:10.1111/bcp.13021
65. Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol (2013) 14(6):e229–38. doi:10.1016/S1470-2045(12)70558-2
66. Gao Q, Lei T, Ye F. Therapeutic targeting of EGFR-activated metabolic pathways in glioblastoma. Expert Opin Investig Drugs (2013) 22(8):1023–40. doi:10.1517/13543784.2013.806484
67. Park MA, Mitchell C, Zhang G, Yacoub A, Allegood J, Haussinger D, et al. Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca(2+)-de novo ceramide-PP2A-reactive oxygen species-dependent signaling pathway. Cancer Res (2010) 70(15):6313–24. doi:10.1158/0008-5472.CAN-10-0999
68. West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, et al. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res (2013) 73(24):7265–76. doi:10.1158/0008-5472.CAN-13-0890
69. Cursi S, Rufini A, Stagni V, Condò I, Matafora V, Bachi A, et al. Src kinase phosphorylates Caspase-8 on Tyr380: a novel mechanism of apoptosis suppression. EMBO J (2006) 25(9):1895–905. doi:10.1038/sj.emboj.7601085
70. Powley IR, Hughes MA, Cain K, MacFarlane M. Caspase-8 tyrosine-380 phosphorylation inhibits CD95 DISC function by preventing procaspase-8 maturation and cycling within the complex. Oncogene (2016) 25(10):99. doi:10.1038/onc.2016.99
71. Jia SH, Parodo J, Kapus A, Rotstein OD, Marshall JC. Dynamic regulation of neutrophil survival through tyrosine phosphorylation or dephosphorylation of caspase-8. J Biol Chem (2008) 283(9):5402–13. doi:10.1074/jbc.M706462200
72. Zonta F, Pagano MA, Trentin L, Tibaldi E, Frezzato F, Gattazzo C, et al. Lyn-mediated procaspase 8 dimerization blocks apoptotic signaling in B-cell chronic lymphocytic leukemia. Blood (2014) 123(6):875–83. doi:10.1182/blood-2013-02-485540
73. Senft J, Helfer B, Frisch SM. Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res (2007) 67(24):11505–9. doi:10.1158/0008-5472.CAN-07-5755
74. Barbero S, Barila D, Mielgo A, Stagni V, Clair K, Stupack D. Identification of a critical tyrosine residue in caspase 8 that promotes cell migration. J Biol Chem (2008) 283(19):13031–4. doi:10.1074/jbc.M800549200
75. Tsang JL, Jia SH, Parodo J, Plant P, Lodyga M, Charbonney E, et al. Tyrosine phosphorylation of caspase-8 abrogates its apoptotic activity and promotes activation of c-Src. PLoS One (2016) 11(4):e0153946. doi:10.1371/journal.pone.0153946
76. Gibbons DL, Spencer J. Mouse and human intestinal immunity: same ballpark, different players; different rules, same score. Mucosal Immunol (2011) 4(2):148–57. doi:10.1038/mi.2010.85
77. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A (2013) 110(9):3507–12. doi:10.1073/pnas.1222878110
78. Rapetti L, Chavele KM, Evans CM, Ehrenstein MR. B cell resistance to Fas-mediated apoptosis contributes to their ineffective control by regulatory T cells in rheumatoid arthritis. Ann Rheum Dis (2015) 74(1):294–302. doi:10.1136/annrheumdis-2013-204049
79. Claessens YE, Bouscary D, Dupont JM, Picard F, Melle J, Gisselbrecht S, et al. In vitro proliferation and differentiation of erythroid progenitors from patients with myelodysplastic syndromes: evidence for Fas-dependent apoptosis. Blood (2002) 99(5):1594–601. doi:10.1182/blood.V99.5.1594
80. Holler N, Tardivel A, Kovacsovics-Bankowski M, Hertig S, Gaide O, Martinon F, et al. Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol Cell Biol (2003) 23(4):1428–40. doi:10.1128/MCB.23.4.1428-1440.2003
81. ClinicalTrials.gov. A Phase I Dose Finding Study of APO010 in Patients with Solid Tumors (AP1001). ClinicalTrials.gov (2016). Available from: https://clinicaltrials.gov/ct2/show/NCT00437736
82. Oncology Venture. Additional Two Centers Started Screening of Multiple Myeloma Patients in OV’s APO010 Study. Oncology Venture Sweden AB (2016). Available from: https://press.aktietorget.se/oncologyventure/79683/551324.pdf
83. Oncology Venture. Oncology Venture’s Immuno-Oncology Drug APO010 for Multiple Myeloma Receives EUROSTARS Grant of a Total Value of 12,7 mNOK from Norway 2016. (2016). Available from: https://press.aktietorget.se/oncologyventure/79121/529816.pdf
84. Odani-Kawabata N, Takai-Imamura M, Katsuta O, Nakamura H, Nishioka K, Funahashi K, et al. ARG098, a novel anti-human Fas antibody, suppresses synovial hyperplasia and prevents cartilage destruction in a severe combined immunodeficient-HuRAg mouse model. BMC Musculoskelet Disord (2010) 11(221):221. doi:10.1186/1471-2474-11-221
85. Matsubara T, Okuda K, Chiba J, Takayama A, Inoue H, Sakurai T, et al. FRI0207 A phase I/II clinical trial of intra-articular administration of ARG098, an anti-FAS IGM monoclonal antibody, in knee joint synovitis of japanese patients with rheumatoid arthritis. Ann Rheum Dis (2013) 71(Suppl 3):384. doi:10.1136/annrheumdis-2012-eular.2664
86. Jung G, Grosse-Hovest L, Krammer PH, Rammensee HG. Target cell-restricted triggering of the CD95 (APO-1/Fas) death receptor with bispecific antibody fragments. Cancer Res (2001) 61(5):1846–8.
87. Herrmann T, Grosse-Hovest L, Otz T, Krammer PH, Rammensee HG, Jung G. Construction of optimized bispecific antibodies for selective activation of the death receptor CD95. Cancer Res (2008) 68(4):1221–7. doi:10.1158/0008-5472.CAN-07-6175
88. Ateh DD, Leinster VH, Lambert SR, Shah A, Khan A, Walklin HJ, et al. The intracellular uptake of CD95 modified paclitaxel-loaded poly(lactic-co-glycolic acid) microparticles. Biomaterials (2011) 32(33):8538–47. doi:10.1016/j.biomaterials.2011.07.060
89. Biomoti. (2016). Available from: http://www.biomoti.com/technology/
90. Wick W, Fricke H, Junge K, Kobyakov G, Martens T, Heese O, et al. A phase II, randomized, study of weekly APG101+reirradiation versus reirradiation in progressive glioblastoma. Clin Cancer Res (2014) 20(24):6304–13. doi:10.1158/1078-0432.CCR-14-0951-T
91. Raimbault A, Pierre-Eugene C, Rouquette A, Deudon C, Willems L, Chapuis N, et al. APG101 efficiently rescues erythropoiesis in lower risk myelodysplastic syndromes with severe impairment of hematopoiesis. Oncotarget (2016) 7(12):14898–911. doi:10.18632/oncotarget.7469
92. Lee YH, Song GG. Associations between the FAS -670 A/G, -1377 G/A, and FASL -844 T/C polymorphisms and susceptibility to systemic lupus erythematosus: a meta-analysis. Clin Exp Rheumatol (2016) 34(4):634–40.
93. Wu J, Xie F, Qian K, Gibson AW, Edberg JC, Kimberly RP. FAS mRNA editing in human systemic lupus erythematosus. Hum Mutat (2011) 32(11):1268–77. doi:10.1002/humu.21565
94. Ban T, Sato GR, Nishiyama A, Akiyama A, Takasuna M, Umehara M, et al. Lyn kinase suppresses the transcriptional activity of IRF5 in the TLR-MyD88 pathway to restrain the development of autoimmunity. Immunity (2016) 45(2):319–32. doi:10.1016/j.immuni.2016.07.015
95. Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, et al. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science (1994) 263(5154):1759–62. doi:10.1126/science.7510905
96. Munroe ME, Vista ES, Guthridge JM, Thompson LF, Merrill JT, James JA. Proinflammatory adaptive cytokine and shed tumor necrosis factor receptor levels are elevated preceding systemic lupus erythematosus disease flare. Arthritis Rheumatol (2014) 66(7):1888–99. doi:10.1002/art.38573
97. Fults D, Pedone CA, Thompson GE, Uchiyama CM, Gumpper KL, Iliev D, et al. Microsatellite deletion mapping on chromosome 10q and mutation analysis of MMAC1, FAS, and MXI1 in human glioblastoma multiforme. Int J Oncol (1998) 12(4):905–10.
98. Stettner MR, Wang W, Nabors LB, Bharara S, Flynn DC, Grammer JR, et al. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res (2005) 65(13):5535–43. doi:10.1158/0008-5472.CAN-04-3688
99. Merz C, Strecker A, Sykora J, Hill O, Fricke H, Angel P, et al. Neutralization of the CD95 ligand by APG101 inhibits invasion of glioma cells in vitro. Anticancer Drugs (2015) 26(7):716–27. doi:10.1097/CAD.0000000000000237
100. Ahluwalia MS, de Groot J, Liu WM, Gladson CL. Targeting SRC in glioblastoma tumors and brain metastases: rationale and preclinical studies. Cancer Lett (2010) 298(2):139–49. doi:10.1016/j.canlet.2010.08.014
101. Lambert C, Wu Y, Aanei C. Bone marrow immunity and myelodysplasia. Front Oncol (2016) 6(172):172. doi:10.3389/fonc.2016.00172
Keywords: apoptosis, Fas/CD95, survival signals, tyrosine phosphorylation, disease
Citation: Chakrabandhu K and Hueber A-O (2016) Fas Versatile Signaling and Beyond: Pivotal Role of Tyrosine Phosphorylation in Context-Dependent Signaling and Diseases. Front. Immunol. 7:429. doi: 10.3389/fimmu.2016.00429
Received: 10 July 2016; Accepted: 30 September 2016;
Published: 17 October 2016
Edited by:
Abdel Hamad, Johns Hopkins University, USAReviewed by:
Aslam Khan, University of Missouri, USACopyright: © 2016 Chakrabandhu and Hueber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krittalak Chakrabandhu, Y2hha3JhYmFAdW5pY2UuZnI=;
Anne-Odile Hueber, aHVlYmVyQHVuaWNlLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.