 Tabasum Sidiq
Tabasum Sidiq Sayuri Yoshihama
Sayuri Yoshihama Isaac Downs
Isaac Downs Koichi S. Kobayashi
Koichi S. Kobayashi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 September 2016
Sec. Mucosal Immunity
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00367
This article is part of the Research Topic Understanding Crohn's disease: immunity, genes and microbes View all 13 articles
The human intestinal tract harbors large bacterial community consisting of commensal, symbiotic, and pathogenic strains, which are constantly interacting with the intestinal immune system. This interaction elicits a non-pathological basal level of immune responses and contributes to shaping both the intestinal immune system and bacterial community. Recent studies on human microbiota are revealing the critical role of intestinal bacterial community in the pathogenesis of both systemic and intestinal diseases, including Crohn’s disease (CD). NOD2 plays a key role in the regulation of microbiota in the small intestine. NOD2 is highly expressed in ileal Paneth cells that provide critical mechanism for the regulation of ileal microbiota through the secretion of anti-bacterial compounds. Genome mapping of CD patients revealed that loss of function mutations in NOD2 are associated with ileal CD. Genome-wide association studies further demonstrated that NOD2 is one of the most critical genetic factor linked to ileal CD. The bacterial community in the ileum is indeed dysregulated in Nod2-deficient mice. Nod2-deficient ileal epithelia exhibit impaired ability of killing bacteria. Thus, altered interactions between ileal microbiota and mucosal immunity through NOD2 mutations play significant roles in the disease susceptibility and pathogenesis in CD patients, thereby depicting NOD2 as a critical regulator of ileal microbiota and CD.
NLR [Nucleotide binding domain (NBD)-leucine rich repeats (LRR) containing or nucleotide-binding oligomerization domain (NOD)-like receptor] proteins play an important role in shaping the immune system. Humans have 22 NLR protein members and mice have 34 members whereby abundance may vary in relation to cell types (1–4). While certain NLR protein members are critical for the early embryogenesis (5, 6) or upregulation of major histocompatibility complex (MHC) molecules (4, 7–10), other group of NLR proteins have important roles in recognizing pathogen and damage-associated molecular patterns (PAMPs and DAMPs, respectively) in the cytoplasm and eliciting innate immune responses (1, 11–16). NLRs have a tripartite structure consisting of an amino-terminal effector domain, a central NBD, and a carboxy-terminal LRR (17). Most NLR proteins contain distinct protein–protein association domains, such as caspase recruitment domain (CARD), pyrin domain (PYD), or a baculovirus inhibitor repeat (BIR). These variations of the amino-terminal domain are used to categorize NLRs into major subfamilies, which include NLRCs (NLRs containing CARD), NLRPs (NLRs containing Pyrin), and others (1, 3, 11, 14, 18–20). The central NBD, found in all NLRs, is involved in oligomerization and activation (1, 3). The carboxy-terminal LRRs may be used for the recognition of PAMPs and DAMPs by many NLRs, except NLRP4 and NAIP (Neuronal Inhibitor Apoptosis Protein) that do not use LRRs for ligand recognition (1, 3, 20–24).
NOD2 is a 110 kDa cytosolic protein (1040 amino acids) with two CARD domains, thereby is a part of the NLRC subfamily (13, 15). Known as a major genetic risk factor for Crohn’s disease (CD), NOD2 gene is located on human chromosome 16p21. Upstream of the transcription start site of NOD2, are two NF-κB-binding sites within the promoter at 26 and 301 bp as well as vitamin D receptor-binding sites within the NOD2 gene (25–27). NOD2 is highly expressed in myeloid cells, such as dendritic cells and macrophages, but expression is low in T cells (27–30). Furthermore, NOD2 is highly expressed in Paneth cells in the ileum with a lesser degree in epithelial cells from the intestines, oral cavity, and lungs (28, 29, 31–33). NOD2 expression is induced by bacterial components (e.g., LPS), short-chain fatty acids (e.g., butyrate), hormonal vitamin D (1,25-dihydroxyvitamin D3), and pro-inflammatory cytokines (e.g., TNF-α) (25, 27, 28, 34–39). The expression of NOD2 and downstream kinase, receptor-interacting serine/threonine-protein kinase 2 (RIP2) in the intestine is largely dependent on the presence of gut microbiota as the expression of these two genes is significantly low in germ-free mice and can be restored by colonization of single strains of bacteria or microflora from mice under SPF conditions (28, 31, 35).
It has been shown that NOD2 recognizes muramyl dipeptide (MDP), N-acetylmuramyl-l-alanyl-d-isoglutamine (40–42). LRRs at C-terminus are responsible for direct recognition of MDP, although affinity is not strong for in vitro association assays (13, 15). MDP is derived from peptidoglycan found mainly in Gram-positive bacteria and to a lesser extent in Gram-negative bacteria. MDP can be transported into the cytoplasm of mammalian cells from endo/phagosome through transporters, such as solute carrier family protein 5 (SLC15A4) (43–45). Both MDP recognition and NOD2 oligomerization are enhanced when ATP binds to the Walker A motif in the NBD, known to regulate the activity of most NLRs (13, 15).
NOD2 activation promotes oligomerization and recruitment of RIP2 by CARD-CARD homophilic interactions (Figure 1) (36, 46–48). E3 ligases [TNF receptor associated factor 6 (TRAF6), TNF receptor associated factor 2 (TRAF2), cellular inhibitor of apoptosis protein (cIAP2)] are recruited to this NOD2-RIP2 platform to ubiquitinate NEMO (NF-κB essential modulator) at amino acid R285 and R399 (42, 47, 49, 50). Whereas NOD2 is subsequently ubiquitinated and undergoes proteasomal degradation (51), TGFβ-activated kinase 1 (TAK1) and ubiquitinated NEMO form a complex within proximity to activate IκB kinase (IKK) that initiates the NF-κB signaling pathway (37, 46, 50). Once phosphorylated by IKK, IκB-α is targeted to proteasomal degradation that permits free NF-κB to translocate into the nucleus (35, 42, 46, 47, 50) (Figure 1).
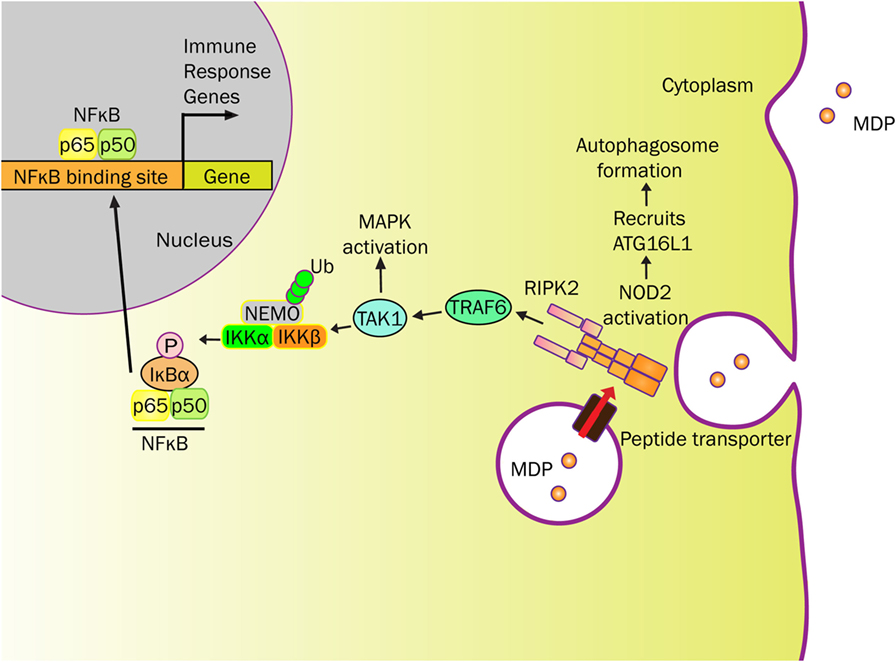
Figure 1. NOD2 senses MDP and elicits immune responses. MDP-derived from bacterial cell wall can be transported into the cytosol by endo/phagosome through peptide transporters (e.g., SLC15A4). MDP activates NOD2 to initiate RIPK2 recruitment resulting in platform formation. This cytosolic NOD2-initiated structure localizes E3 ligases (e.g., TRAF6) to ubiquitinate NEMO, for which NEMO and TAK1 forms a complex near IKK. In addition to activating the mitogen-activated protein kinase (MAPK) pathway, TAK1 phosphorylates IKK to initiate the NF-κB signaling pathway. IκB-α is phosphorylated by IKK, which targets IκB-α to proteasome degradation. No longer restricted, free NF-κB translocates into the nucleus and activates various immune response genes. NOD2 also recruits ATG16L1 near the plasma membrane to promote autophagosome formation, known to degrade antigenic structures. The release of cytokines and chemokines, initiated by NOD2, recruits immune cells to influence Th1 and Th2 responses.
In addition to NF-κB activation, the mitogen-activated protein kinases (MAPK), such as p38 and ERK, are activated in a TRAF6-dependent manner (42, 50) (Figure 1). Activation of these signaling cascades upon NOD2 stimulation induces the production of a variety of cytokines, chemokines, and antimicrobial peptides (TNF-α, IL-6, IFN-γ, IL-1β, IL-10, IL-8/CXCL8, α-defensin) depending on cell types (30, 52). NOD2 signal cascade is enhanced by toll-like receptor (TLR) agonists through NF-κB. Along with MDP, this relationship further highlights the impact of microbial components on the NOD2 pathway (1, 30, 53).
Through the innate immune system, NOD2 provides a defensive strategy to protect the hosts against bacterial infection. Various epithelial cells throughout the human body not only create a physical barrier but also produce antimicrobial peptides to regulate bacterial colonization (29, 31, 54). In addition to NOD2 gene, mutations in ATG16L1 (Autophagy-related 16 Like 1) gene are a risk factor for CD (55). ATG16L1 is a component of a large protein complex essential for autophagy, a mechanism that degrades intracellular components using the lysosome. As NOD2 is capable of recruiting ATG16L1 to the plasma membrane at the bacterial entry site, NOD2 mutants failed to recruit ATG16L1, ultimately impairing autophagosomal encapsulation of invading bacteria in dendritic cells (56, 57). Therefore, both ATGL16 and NOD2 have interrelated roles for regulating the microbial invasion that has yet to be studied in Paneth cells (Figure 1). However, in Paneth cells, NOD2 recruitment onto dense vesicles helps coordinate cargo-sorting events that support host defenses (58). Therefore, NOD2 function and related responses regulate interactions between innate immunity and the intestinal microbiota.
As noted above, NOD2 and TLR signaling cooperate to enhance immune responses. This synergy of NOD2 and TLR activation affects the polarization of T cell adaptive immunity (Th1 vs. Th2 immune responses) (42, 52, 59, 60). While it is well documented that the NOD2 pathway cooperates with signals through NOD1 or various TLRs (e.g., TLR3, TLR4, TLR9) to enhance Th1 immune responses, Th2 responses are also induced through the cooperation of NOD2 with TLR2 agonists (35, 52, 61, 62). Regarding humoral immunity, NOD2 is capable of influencing antigen-specific immunoglobulin production based on studies using Nod2-deficient mice (42, 52). Thus, NOD2 expression impacts immune responses as well as bacterial killing and colonization in the ileum (25, 31, 34, 35, 52, 61–63). In addition to bacterial components, dietary fiber as well as vitamin D-containing products also induces expression of NOD2 to further increase the regulation of the microbiota (26, 28, 35).
Crohn’s disease is a chronic, relapsing inflammatory disorder of the gastrointestinal tract, most commonly involving the ileum and colon. CD patients typically suffer from frequent and chronically relapsing flares with diarrhea, abdominal pain, rectal bleeding, and malnutrition. Histologically, CD is characterized by transmural infiltration of lymphocytes and macrophages with granuloma. Because of the clinical and histological features, most CD patients require lifelong treatment, such as medication and surgery. For efficient treatment of CD, it is important to identify patients who carry risk factors for complications of the disease and initiate appropriate therapy at early phase of the clinical course (64).
In the healthy human intestine, the intestinal epithelium provides an effective barrier against luminal bacteria with the help of epithelial tight junction and the mucus layer (Figure 2). However, in patients with inflammatory bowel diseases, this barrier function is often compromised due to genetic, environmental, microbial, and immunological factors (65, 66). Dysfunctional mucosal barrier allows the penetration of bacterial products, leading to direct interaction with immune cells (Figure 2). Innate immune cells, such as macrophages and dendritic cells produce cytokines, such as TNF-α, IL-1β, IL-6, IL-12, IL-23, and chemokines upon recognition of microbes and microbial products. Activated dendritic cells promote the differentiation of CD4+ T cells into pro-inflammatory T cell subsets, such as Th1 or Th17 effector T cells, which secrete additional cytokines and chemokines (65, 66). Recently identified novel family of innate immune cells termed innate lymphoid cells (ILCs), in particular the type 1 ILC (ILC1) subset, produce high amount of pro-inflammatory cytokines, such as IFN-γ and TNF-α (67–69). High levels of chemokines produced by these innate and adaptive immune cells further recruit lymphocytes, creating a vicious positive feedback cycle of inflammation in CD.
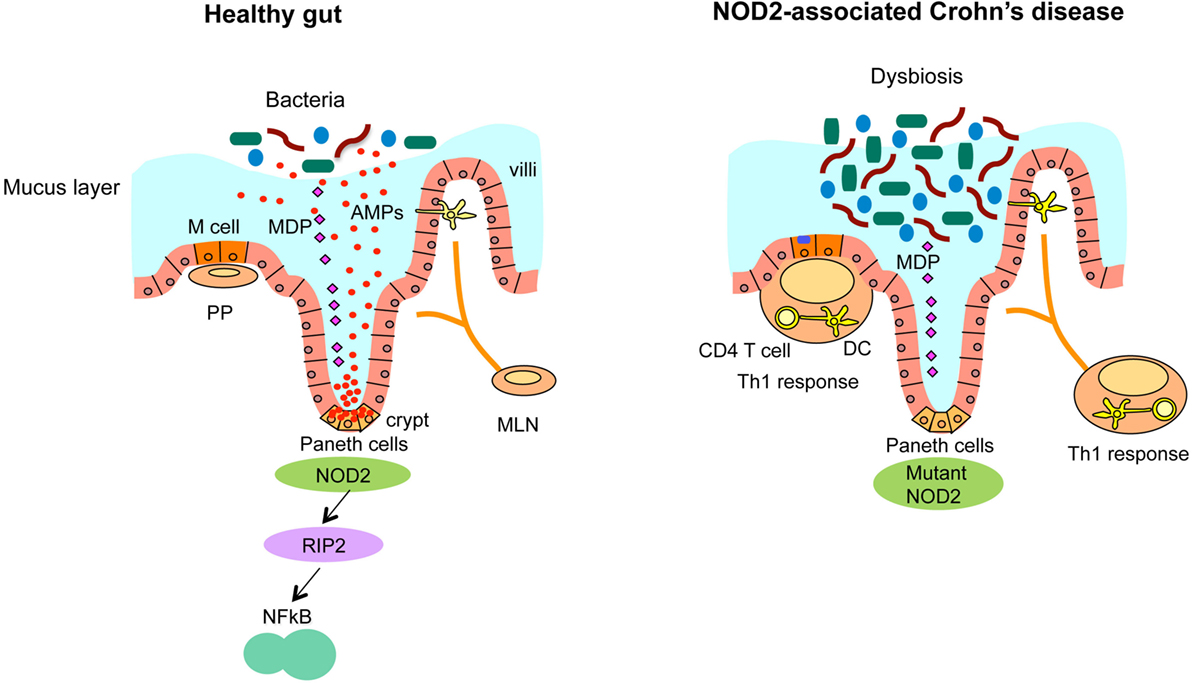
Figure 2. NOD2-associated dysregulated microbiota leads to the susceptibility of CD. Ileum in healthy individuals (left panel). NOD2 senses microbiota-derived MDP and activates NF-κB through the downstream kinase receptor-interacting protein 2 (RIP2), which is critical for the bacterial killing activity of Paneth cells via the secretion of anti-bacterial compounds. Ileum with loss of NOD2 function (right panel). In CD, that is associated with NOD2 mutations, CD-associated NOD2 mutants disturb many characteristics of gut immune homeostasis, including reduced MDP sensing, and impaired antimicrobial responses in Paneth cells, leading to defective bacterial clearance. This dysbiosis caused by impaired Paneth cell function is characterized by increased load of bacteria and abnormalities of Peyer’s patches (PP) and mesenteric lymph nodes (MLN) that stimulate the mucosal immune system to induce Th1 immune response, leading to chronic inflammation.
Recent Genome-wide association studies (GWAS) revealed 163 susceptibility loci for IBD (inflammatory bowel disease), 30 of them being specific to CD (70). Among them, NOD2 was the first gene identified as a risk factor for ileal CD (71, 72), discovered by the genetic mapping study of the CD susceptibility locus (73). One of the cell types that express NOD2 at a high level is the Paneth cell, most of which are located in the terminal ileum (32). Paneth cells play an important role in the innate regulation of gut microbiota by synthesizing and secreting antimicrobial peptides or proteins. Upon the stimulation with bacterial products, such as the NOD2 ligand, MDP, Paneth cells secrete antimicrobial peptides, including lysozyme, secretory phospholipase A2 (sPLA2), and human α-defensins 5 and 6 (HD5 and HD6) in the intestinal lumen (74). Therefore, NOD2 mutations lead to dysregulation of host–microbe interactions, which increases the susceptibility to abnormal ileal inflammation. In addition to this microbial dysbiosis, other genetic or environmental factors trigger the development of ileal CD.
A meta-analysis showed the risk for CD development was increased to 17.1-fold in NOD2 homozygotes or compound heterozygotes, and 2.4-fold in simple NOD2 heterozygotes (Table 1) (75). Three main variants or polymorphisms in NOD2 gene (i) a frame shift mutation at position 1007 (1007fs); (ii) a glycine to arginine conversion at amino acid residue 908 (G908R); and (iii) an arginine to tryptophan conversion at amino acid residue 702 (R702W) were highly associated with susceptibility to CD (71, 72, 76). All three mutations are located within or close to the LRR domain that recognizes MDP. Indeed, patients with ileal CD showed reduced levels of HD5 and HD6 derived from Paneth cells (77, 78). Additionally, the production of α-defensins is particularly reduced in patients with non-functional NOD2 mutations (79, 80). These findings showed that CD-related mutations in NOD2 are loss of function and individuals carrying NOD2 mutations are at high risk for ileal CD.
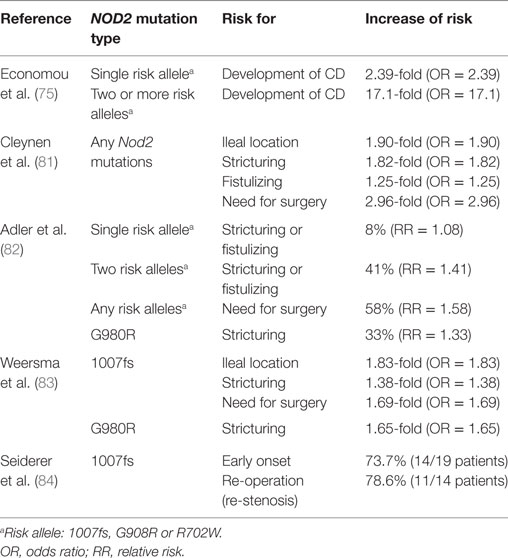
Table 1. NOD2 mutations in CD patients.
Furthermore, many studies have suggested that NOD2 mutations increase the risk of complications in CD (Table 1). It was shown that any of the NOD2 mutations are independent predictive factor for stricturing [odds ratio (OR) = 1.82], fistulizing (OR = 1.25), and need for surgery (OR = 2.96) (81). A meta-analysis reported that the risk for complicated disease, such as stricturing or fistulizing, was increased by 8% [relative risk (RR) = 1.08] in NOD2 heterozygotes and 41% (RR = 1.41) in NOD2 homozygotes or compound heterozygotes (82). Likewise, the risk of surgery was increased by 58% (RR = 1.58) with any of the NOD2 mutations (82). In terms of each risk allele, p.1007fs mutation is the strongest disease predictive factor. p.1007fs was associated with stricturing (OR = 1.38) and the necessity of an operation (OR = 1.69) (83). The homozygous carriers of p.1007fs were significantly younger at the time of diagnosis (under the age of 26 years). Fourteen out of 19 homozygous carriers of p.1007fs (73.7%) required surgery and 11 out of 14 patients (78.6%) underwent the re-operation due to re-stenosis (84). The mutation of p.G980R is also associated with disease complications [RR = 1.33 (82), OR = 1.65 (83)]. Interestingly, multiple studies indicated that association of NOD2 mutations with CD were found in European and Ashkenazi Jewish ancestry population, but not in CD patient cohort of Asian ancestry, such as Japanese, Chinese, Korean, or Indian, indicating the impact of ethnic genetic background on the CD susceptibility caused by NOD2 mutations (85–88).
Taken together, NOD2 mutation and the susceptibility of ileal CD are strongly related via altered interaction between ileal microbiota and mucosal immunity. Carrying the NOD2 mutation is the strong risk factor for ileal CD with multiple complications.
The surfaces of the body, particularly the gastrointestinal tract, are exposed to a large number of diverse microbes that are collectively referred to as the microbiota. Composition of the microbiota can be significantly affected by the genetic background of hosts along with several other factors, such as diet, age, stress, and diseases (89). While commensal bacteria colonize all mucosal surfaces and the skin, the intestinal tract harbors the largest bacterial load (up to 1014 total bacteria) (90, 91). It is becoming increasingly clear that the close symbiotic relationship between host and an adaptable microbiota forms a vital part of the intestinal homeostasis (92, 93). The host microbiota plays important roles in several processes such short-chain fatty acid production (94), the development of intestinal epithelium (95, 96), protection against invading pathogens, and, importantly, the development of mature immune system (96). The intestinal microbiota provides both inflammatory and anti-inflammatory products, which modulate the immune responses (97, 98) and dysbiosis in the microbiota is considered pivotal to the origin of many diseases. It is well known that the disease outcome can be influenced by the host microbiota either due to the increased colonization of pathogenic strains or the introduction of new bacterial species. This is well-exemplified in IBD patients who typically exhibit imbalances in the microbiota composition. Moreover, reducing exposure to intestinal bacteria in these patients has been shown to frequently alleviate inflammation (99, 100).
NOD2 plays an important role in mediating host–microbe interactions in the intestine. Interestingly, NOD2 has been shown to contribute to the homeostatic maintenance of the gut microbiota. Abnormal NOD2 function associated with LRR polymorphisms leads to an inflammatory response mediated by innate immune functions (42, 71, 72, 101). NOD2 controls the expression and secretion of antimicrobial peptides thereby acting as an important regulator of the ileal commensal microbiota in mice (31). Nod2-deficient mice exhibit reduced bactericidal activity, higher loads of commensal bacteria and increased susceptibility to colonization by pathogenic bacteria (31, 102), and are, thus, prone to bacterial infections and intestinal inflammation (30, 42). NOD2 function, therefore, may be pivotal in understanding the balance between the host immune system and commensal bacteria.
NOD2 is critical for regulating the bacterial flora in the ileum through the secretion of anti-bacterial compounds (31, 42, 103–105) (Figure 2). Earlier studies in Nod2-deficient mice indicated the role of NOD2 in the homeostasis of gut epithelium, possibly linking it to the development of CD (42). Furthermore, increased NOD2 expression is observed at the sites for α-defensin secretion, such as intestinal crypts in mice (42) and Paneth cells in human (32, 33). Nod2-deficient mice harbor larger loads of bacteria in the ileum due to impaired Paneth cell function, compared to littermate controls even in the same cage (31). Nod2-deficient mice are susceptible to Th1-dominant granulomatous inflammation in the ileum, which is induced by an opportunistic pathogen Helicobacter hepaticus and ileal dysbiosis (2, 30). Furthermore, NOD2 expression in the intestine is regulated by signals from the microbiota, given that germ-free mice had lower NOD2 expression that was reversible upon monocolonization with commensal bacteria (31). Zhang et al. found that NOD2-mediated lysozyme trafficking in Paneth cells is directed by commensal bacteria, which is important for regulating intestinal infection (58). Two different steps, selective lysozyme trafficking and final secretion (106) are separately regulated by commensals implicating a new mechanism by which symbiotic bacteria direct a lysozyme-trafficking event to promote host defense against pathogens.
Several studies have shown the role of NOD2 in host protection against pathogens in a number of bacterial infectious models including Listeria monocytogenes (42), Staphylococcus aureus (107), Chlamydophila pneumoniae (108), Streptococcus pneumoniae (109), and Mycobacterium tuberculosis (110). Nod2-deficient mice were susceptible to L. monocytogenes infection via oral but not systemic routes, suggesting that NOD2 might have a non-superfluous role in intestinal antimicrobial responses (11).
The discovery that NOD2 mutations are strongly associated with CD highlighted the importance of NOD2 in the regulation of antimicrobial responses. Loss of function mutations in NOD2, rendering it unresponsive to peptidoglycan component, MDP, have been implicated in the development of CD (41, 111, 112). Interestingly, patients with mutant variants of NOD2 have altered microbiota in the ileum (32, 33, 113–117). There was a significant decrease in the relative frequencies of Clostridia and an increase in the Actinobacteria and Proteobacteria in CD patients compared to healthy controls (117).
Apart from having role in the bactericidal ability of Paneth cells, NOD2 also shapes the microbial community profiles via local production of reactive oxygen species (118–120). Adult Nod2-deficient mice display a substantially altered microbial community structure and a significantly elevated bacterial load in their feces and terminal ileum compared to their wild-type counterparts (114). Interestingly, these findings were also present in weaning-age mice, indicating a subtle influence of NOD2 on the early development and composition of the intestinal microbiota. Increased load of the Bacteroidetes and Firmicutes phyla as well as the genus Bacteroides was observed in the terminal ileum of CD patients with homozygosity in NOD2 mutations (114). In addition, altered frequencies of Faecalibacterium and Escherichia have also been associated with NOD2 risk alleles in CD patients. Nod2-deficient mice harbor increased bacterial load in the feces and ileum along with increased abundance of Bacteroidetes and Firmicutes in comparison to wild-type mice (31, 114, 121). These studies are supported by the observations of Petnicki-Ocwieja et al., who used the most strict experimental conditions taking into account the usage of littermates and bacterial quantification by qPCR, with the prominent difference observed only in ileum (31).
Several abnormalities in the small-intestinal epithelium of Nod2-deficient mice were investigated which included the inflammatory gene expression and goblet cell dysfunction (102). Ramanan et al. demonstrated that these abnormalities were associated with excessive interferon-γ production by intraepithelial lymphocytes (IELs) and Myd88-dependent signaling, and were dependent on the expansion of a common member of the intestinal microbiota, Bacteroides vulgatus. Although this study implies NOD2 prevents harmful immune responses by controlling the expansion of a pro-inflammatory member of the microbiota (102), the composition of the gut microbiota may vary among mice from different laboratories, which may lead to the variations in the experimental outcome (31, 114, 121).
The constant exposure of the intestinal tissue to gut microorganisms maintains the mucosa in a state of physiological inflammation, which balances tolerogenic and pro-inflammatory type responses to maintain homeostasis. In line with the idea that NOD2 signaling might indirectly regulate T cell populations, results presented in a recent study indicated that there are dysfunctional and reduced numbers of IELs in Nod2-deficient mice (122). These effects of NOD2 deficiency could hamper the integrity of intestinal epithelium and lead to an altered immune response to the resident microbiota. A recent study showed that there is an increased sensitivity of Nod2 as well as Rip2-deficient mice to DSS-induced colitis and colonic adenocarcinoma as a result of dysbiosis, and that the effect can be transmitted to wild-type mice through the microbiota (123). However, such differences in the composition of gut microbiota as observed in naive mice deficient in NOD2 signaling components is debatable as many other studies could not arrive at the same conclusion (124–126), depicting that, in accordance with the study in TLR-deficient mice (127), changes in the relative abundance of certain bacterial groups in Nod2-deficient mice emulate caging conditions and familial transmission rather than their NOD2 genotype. In regard to this discrepancy, the question of whether NOD2 deficiency can cause different composition of intestinal bacteria requires additional study. Likewise, different reports in human studies suggest a reduced abundance of certain bacteria, including Faecalibacterium prausnitzii and other butyrate-producing organisms (128) in patients with colitis, but whether these changes occur before the onset of inflammation is an important question in the field. Continuing studies, such as these will help to determine whether dysbiosis occurs before disease onset and whether particular microbial profiles are indeed early biomarkers of disease susceptibility.
Studies conducted in healthy SPF mice support the observation in which failure of NOD2 to induce secretion of antimicrobial compounds allows for increase of certain bacterial groups. Studies by Petnicki-Ocwieja et al. (31) reported increased loads of Bacteroides and Firmicutes groups in the ileum of Nod2-deficient mice compared to wild-type/heterozygous mice from the same litter. However, no such difference was observed in the feces of these mice. This observation was attributed to the decreased bactericidal activity of crypt-secreted factors, such as α-defensins (42). Similarly, increased abundance of Bacteroides was also reported in the ileum of Rip2-deficient mice compared to wild-type littermates (31). However, Rehman et al. (114) reported immense abundance of Bacteroidetes in the feces of adult Nod2-deficient mice, whereas greater load of Firmicutes was observed only in the terminal ileum of these mice. Mondot et al. (121) reported increased colonic abundance of the Rikenellaceae, Bacteroidaceae and Prevotellaceae families in Nod2-deficient mice compared to wild-type mice with no specific housing conditions. In conclusion, these studies indicate the increased bacterial load in the ileum of Nod2-deficient mice although the variations observed in these studies can be attributed to different housing conditions.
Microbial dysbiosis in CD is likely to be shaped by the chronic inflammatory state of the intestine. Abnormal microbial composition in association with the detrimental genetic background increases the predisposition of Nod2-deficient mice to inflammatory diseases. Several studies on the microbiota in Nod2-deficient mice reveal its altered diversity as well as richness (114, 121). These studies emphasize the importance of NOD2 in regulating the intestinal microbiota composition and the subsequent outcome of CD.
Previous studies indicate an important role of NOD2 in the development of GALT (Gut associated lymphoid tissues) during the bacterial colonization of gut (129). In Nod2-deficient mice, high proportion of CD4+ T cells, increased inflammatory cytokine levels, and higher antigen and bacterial permeability rates (124) were observed, indicating an increased stimulation of Peyer’s patches (PP) by resident gut microflora. All these changes observed in these mice were suppressed after treatment with oral antibiotics. Therefore, apart from influencing the development of the GALT, NOD2 has a role in modulating the immune response by limiting the development of a Th1 immune response toward bacteria. The results described above support the observation that activation of NOD2 in dendritic cells controls their ability to induce a polarized Th1 response in CD4+ T cells (130), thus elucidating the role of NOD2 in hematopoietic cells. However, this cannot explain why the phenotype of NOD2 mutation is only associated with ileal lesion. Also, the Th1 immune response phenotype was not observed in other studies unless colonized with Helicobacter species (30).
Amendola et al. reported that the absence of spontaneous colonic inflammation in Nod2-deficient mice can be attributed to altered gut permeability caused by the high mucosal cytokine production and increased activity of MLCK. MLCK, myosin light chain kinase, is a factor that likely causes the increased gut permeability in Nod2-deficient mice due to its effects on tight junctions (131). NOD2 deficiency, therefore, causes the permeability changes that increase the exposure of dendritic cells to factors, such as TLR ligands that in turn influence Treg cell development and subsequent changes in the microbiota. These results are in agreement with the studies in IBD patients where increased gut permeability and an altered microbiota are associated with NOD2 polymorphisms with no inflammation (116, 117, 132, 133). Recently, it was found that NOD2 is expressed in Lgr5+ stem cells using culture of intestinal crypt organoids, indicating that NOD2 may contribute to epithelial homeostasis by regulating stem cell function in addition to bacterial killing activity of Paneth cells (134).
Autophagy has emerged as a pivotal component of the innate immune response to intracellular bacteria and is also involved in the delivery of microbial ligands to intracellular compartments containing TLRs to trigger an antimicrobial response. Three groups revealed that autophagy is activated by NOD2 to augment intracellular bacterial killing (56, 57, 135). The studies by Travassos et al. (57) demonstrated that bacterial sensing by NOD proteins is linked to the induction of autophagy, thus providing a functional association between NOD2 and ATG16L1, two of the most important genes associated with CD. Cooney et al. demonstrated that bacterial killing and MHC class II-dependent antigen presentation in primary human dendritic cells are increased by MDP-activated autophagy and this process involves ATG16L1, NOD2, and RIP2, but is independent of NLRP3 (56). Dendritic cells from CD patients expressing CD-associated NOD2 or ATG16L1 risk variants have the defect in autophagy induction, bacterial trafficking, and antigen presentation. Hence, NOD2 polymorphism along with the defective lysosomal degradation, and impaired induction of antigen-specific CD4+ T cells would lead to bacterial persistence and thereby the inflammatory responses in CD (56). Furthermore, a recent study showed that ATG16L1- or NOD2-deficient DCs have impaired capability to induce Tregs upon stimulation with immunomodulatory molecules from the commensal Bacteroides fragilis, indicating the immunosuppressive role of NOD2 and ATG16L1 in intestinal dendritic cells (136). However, this suppressive function does not explain the critical role of NOD2 and ATG16L1 in Paneth cells and ileum specific inflammation (137).
Nod2 deficiency in mice, thus, results in increased intestinal bacterial load and greater susceptibility to pathogenic bacterial colonization (31), which could result from either Paneth cell dysfunction, defects in intestinal autophagic responses, or more likely, a combination of the two. Thus, in addition to Paneth cell defects, ATG16L1 and NOD2 risk variants affect the intestinal epithelial cell antimicrobial responses, probably changing the gut microbiome and enhancing CD susceptibility (135).
Although more studies are needed to fully understand the CD pathogenesis caused by NOD2 mutations, recent studies have shown the important roles of NOD2 in the interactions between microbiota and the intestinal immune system. In the studies using Nod2-deficient mice, it was shown that NOD2 is critical for the function of bactericidal activity of ileal crypts and the regulation of ileal microbiota. Therefore, it is tempting to speculate that ileal CD caused by NOD2 mutations is mainly due to the dysfunction of Paneth cells. Under physiological condition, microbiota in the ileum is tightly regulated by bacterial killing activity of Paneth cells in healthy individuals. This regulates both pathogenic and non-pathogenic bacteria as well as bacterial antigens that constitutively stimulate the intestinal immune system. An increased load of microbiota alone is likely not sufficient to induce CD pathogenesis, since inflammatory responses in mucosal immunity are tightly regulated by various mechanisms. However, in the presence of other genetic, environmental, and immunological factors, dysbiosis of bacteria may increase susceptibility to ileal inflammation by enhancing stimulation of the intestinal immune system beyond the controlled physiological threshold, leading to pathological changes and, finally, to chronic inflammation. Although the studies of Paneth cells and microbiota in Nod2-deficient mice and CD patients support this scenario, further research is required to fully elucidate the molecular mechanism of CD. Additional studies on CD-associated microbiota and animal models of ileal CD may provide a greater insight to develop better treatment and management methods of CD patients.
All authors listed have made substantial, direct, and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from the NIH (R01DK074738), Broad Foundation (IBD-0328), Texas A&M Clinical Science & Translational Research Institute Grant Program and Center for Translational Environmental Health Research Pilot Project Program to KK.
1. Biswas A, Kobayashi KS. Regulation of intestinal microbiota by the NLR protein family. Int Immunol (2013) 25(4):207–14. doi:10.1093/intimm/dxs116
2. Biswas A, Petnicki-Ocwieja T, Kobayashi KS. Nod2: a key regulator linking microbiota to intestinal mucosal immunity. J Mol Med (Berl) (2012) 90(1):15–24. doi:10.1007/s00109-011-0802-y
3. Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol (2012) 12(12):813–20. doi:10.1038/nri3339
4. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A (2010) 107(31):13794–9. doi:10.1073/pnas.1008684107
5. Pisani LF, Ramelli P, Lazzari B, Braglia S, Ceciliani F, Mariani P. Characterization of maternal antigen that embryos require (MATER/NLRP5) gene and protein in pig somatic tissues and germ cells. J Reprod Dev (2010) 56(1):41–8. doi:10.1262/jrd.09-098A
6. Tong ZB, Bondy CA, Zhou J, Nelson LM. A human homologue of mouse mater, a maternal effect gene essential for early embryonic development. Hum Reprod (2002) 17(4):903–11. doi:10.1093/humrep/17.4.903
7. Chang CH, Guerder S, Hong SC, van Ewijk W, Flavell RA. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity (1996) 4(2):167–78. doi:10.1016/S1074-7613(00)80681-0
8. Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, et al. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J Immunol (2012) 188(10):4951–8. doi:10.4049/jimmunol.1103160
9. Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell (1993) 75(1):135–46. doi:10.1016/S0092-8674(05)80090-X
10. Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q, et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res (2012) 22(5):836–47. doi:10.1038/cr.2012.56
11. Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the antimicrobial response by NLR proteins. Immunity (2011) 34(5):665–79. doi:10.1016/j.immuni.2011.05.007
12. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145(5):745–57. doi:10.1016/j.cell.2011.04.022
13. Grimes CL, Ariyananda Lde Z, Melnyk JE, O’Shea EK. The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J Am Chem Soc (2012) 134(33):13535–7. doi:10.1021/ja303883c
14. Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol (2009) 21(4):317–37. doi:10.1093/intimm/dxp017
15. Mo J, Boyle JP, Howard CB, Monie TP, Davis BK, Duncan JA. Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J Biol Chem (2012) 287(27):23057–67. doi:10.1074/jbc.M112.344283
16. Wilmanski JM, Petnicki-Ocwieja T, Kobayashi KS. NLR proteins: integral members of innate immunity and mediators of inflammatory diseases. J Leukoc Biol (2008) 83(1):13–30. doi:10.1189/jlb.0607402
17. Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol (2008) 8(5):372–9. doi:10.1038/nri2296
18. Fairbrother WJ, Gordon NC, Humke EW, O’Rourke KM, Starovasnik MA, Yin JP, et al. The PYRIN domain: a member of the death domain-fold superfamily. Protein Sci (2001) 10(9):1911–8. doi:10.1110/ps.13801
19. Meissner TB, Li A, Kobayashi KS. NLRC5: a newly discovered MHC class I transactivator (CITA). Microbes Infect (2012) 14(6):477–84. doi:10.1016/j.micinf.2011.12.007
20. Meissner TB, Li A, Liu YJ, Gagnon E, Kobayashi KS. The nucleotide-binding domain of NLRC5 is critical for nuclear import and transactivation activity. Biochem Biophys Res Commun (2012) 418(4):786–91. doi:10.1016/j.bbrc.2012.01.104
21. Ludigs K, Seguin-Estevez Q, Lemeille S, Ferrero I, Rota G, Chelbi S, et al. NLRC5 exclusively transactivates MHC class I and related genes through a distinctive SXY module. PLoS Genet (2015) 11(3):e1005088. doi:10.1371/journal.pgen.1005088
22. Scholl T, Mahanta SK, Strominger JL. Specific complex formation between the type II bare lymphocyte syndrome-associated transactivators CIITA and RFX5. Proc Natl Acad Sci U S A (1997) 94(12):6330–4. doi:10.1073/pnas.94.12.6330
23. Cui J, Li Y, Zhu L, Liu D, Songyang Z, Wang HY, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol (2012) 13(4):387–95. doi:10.1038/ni.2239
24. Tenthorey JL, Kofoed EM, Daugherty MD, Malik HS, Vance RE. Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol Cell (2014) 54(1):17–29. doi:10.1016/j.molcel.2014.02.018
25. Rosenstiel P, Fantini M, Brautigam K, Kuhbacher T, Waetzig GH, Seegert D, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology (2003) 124(4):1001–9. doi:10.1053/gast.2003.50157
26. Wang TT, Dabbas B, Laperriere D, Bitton AJ, Soualhine H, Tavera-Mendoza LE, et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease. J Biol Chem (2010) 285(4):2227–31. doi:10.1074/jbc.C109.071225
27. Ikeuchi T, Nakamura T, Fukumoto S, Takada H. A vitamin D3 analog augmented interleukin-8 production by human monocytic cells in response to various microbe-related synthetic ligands, especially NOD2 agonistic muramyldipeptide. Int Immunopharmacol (2013) 15(1):15–22. doi:10.1016/j.intimp.2012.10.027
28. Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem (2002) 277(44):41701–5. doi:10.1074/jbc.M206473200
29. Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol (2007) 44(12):3100–11. doi:10.1016/j.molimm.2007.02.007
30. Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL, et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci U S A (2010) 107(33):14739–44. doi:10.1073/pnas.1003363107
31. Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A (2009) 106(37):15813–8. doi:10.1073/pnas.0907722106
32. Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, et al. Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology (2003) 125(1):47–57. doi:10.1016/S0016-5085(03)00661-9
33. Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut (2003) 52(11):1591–7. doi:10.1136/gut.52.11.1591
34. Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology (2003) 124(4):993–1000. doi:10.1053/gast.2003.50153
35. Leung CH, Lam W, Ma DL, Gullen EA, Cheng YC. Butyrate mediates nucleotide-binding and oligomerisation domain (NOD) 2-dependent mucosal immune responses against peptidoglycan. Eur J Immunol (2009) 39(12):3529–37. doi:10.1002/eji.200939454
36. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol (2014) 14(1):9–23. doi:10.1038/nri3565
37. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature (2001) 412(6844):346–51. doi:10.1038/35085597
38. Muhlbauer M, Cheely AW, Yenugu S, Jobin C. Regulation and functional impact of lipopolysaccharide induced Nod2 gene expression in the murine epididymal epithelial cell line PC1. Immunology (2008) 124(2):256–64. doi:10.1111/j.1365-2567.2007.02763.x
39. Dalmasso G, Nguyen HT, Yan Y, Charrier-Hisamuddin L, Sitaraman SV, Merlin D. Butyrate transcriptionally enhances peptide transporter PepT1 expression and activity. PLoS One (2008) 3(6):e2476. doi:10.1371/journal.pone.0002476
40. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278(11):8869–72. doi:10.1074/jbc.C200651200
41. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem (2003) 278(8):5509–12. doi:10.1074/jbc.C200673200
42. Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science (2005) 307(5710):731–4. doi:10.1126/science.1104911
43. Lee J, Tattoli I, Wojtal KA, Vavricka SR, Philpott DJ, Girardin SE. pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J Biol Chem (2009) 284(35):23818–29. doi:10.1074/jbc.M109.033670
44. Nakamura N, Lill JR, Phung Q, Jiang Z, Bakalarski C, de Maziere A, et al. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature (2014) 509(7499):240–4. doi:10.1038/nature13133
45. Vavricka SR, Musch MW, Chang JE, Nakagawa Y, Phanvijhitsiri K, Waypa TS, et al. hPepT1 transports muramyl dipeptide, activating NF-kappaB and stimulating IL-8 secretion in human colonic Caco2/bbe cells. Gastroenterology (2004) 127(5):1401–9. doi:10.1053/j.gastro.2004.07.024
46. Abbott DW, Yang Y, Hutti JE, Madhavarapu S, Kelliher MA, Cantley LC. Coordinated regulation of toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol Cell Biol (2007) 27(17):6012–25. doi:10.1128/MCB.00270-07
47. Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J (2008) 27(2):373–83. doi:10.1038/sj.emboj.7601962
48. LeBlanc PM, Yeretssian G, Rutherford N, Doiron K, Nadiri A, Zhu L, et al. Caspase-12 modulates NOD signaling and regulates antimicrobial peptide production and mucosal immunity. Cell Host Microbe (2008) 3(3):146–57. doi:10.1016/j.chom.2008.02.004
49. Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol (2004) 14(24):2217–27. doi:10.1016/j.cub.2004.12.032
50. Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y. TRAF6 autoubiquitination-independent activation of the NFkappaB and MAPK pathways in response to IL-1 and RANKL. PLoS One (2008) 3(12):e4064. doi:10.1371/journal.pone.0004064
51. Lee KH, Biswas A, Liu YJ, Kobayashi KS. Proteasomal degradation of Nod2 protein mediates tolerance to bacterial cell wall components. J Biol Chem (2012) 287(47):39800–11. doi:10.1074/jbc.M112.410027
52. Magalhaes JG, Fritz JH, Le Bourhis L, Sellge G, Travassos LH, Selvanantham T, et al. Nod2-dependent Th2 polarization of antigen-specific immunity. J Immunol (2008) 181(11):7925–35. doi:10.4049/jimmunol.181.11.7925
53. Fritz JH, Girardin SE, Fitting C, Werts C, Mengin-Lecreulx D, Caroff M, et al. Synergistic stimulation of human monocytes and dendritic cells by toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur J Immunol (2005) 35(8):2459–70. doi:10.1002/eji.200526286
54. Qiu HN, Wong CK, Chu IM, Hu S, Lam CW. Muramyl dipeptide mediated activation of human bronchial epithelial cells interacting with basophils: a novel mechanism of airway inflammation. Clin Exp Immunol (2013) 172(1):81–94. doi:10.1111/cei.12031
55. Naser SA, Arce M, Khaja A, Fernandez M, Naser N, Elwasila S, et al. Role of ATG16L, NOD2 and IL23R in Crohn’s disease pathogenesis. World J Gastroenterol (2012) 18(5):412–24. doi:10.3748/wjg.v18.i5.412
56. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med (2010) 16(1):90–7. doi:10.1038/nm.2069
57. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11(1):55–62. doi:10.1038/ni.1823
58. Zhang Q, Pan Y, Yan R, Zeng B, Wang H, Zhang X, et al. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol (2015) 16(9):918–26. doi:10.1038/ni.3233
59. Hedegaard CJ, Enevold C, Sellebjerg F, Bendtzen K, Nielsen CH. Variation in NOD2 augments Th2- and Th17 responses to myelin basic protein in multiple sclerosis. PLoS One (2011) 6(5):e20253. doi:10.1371/journal.pone.0020253
60. Netea MG, Kullberg BJ, de Jong DJ, Franke B, Sprong T, Naber TH, et al. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn’s disease. Eur J Immunol (2004) 34(7):2052–9. doi:10.1002/eji.200425229
61. Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect Immun (2005) 73(12):7967–76. doi:10.1128/IAI.73.12.7967-7976.2005
62. van Heel DA, Ghosh S, Hunt KA, Mathew CG, Forbes A, Jewell DP, et al. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn’s disease. Gut (2005) 54(11):1553–7. doi:10.1136/gut.2005.065888
63. van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet (2005) 365(9473):1794–6. doi:10.1016/S0140-6736(05)66582-8
64. Dignass A, Van Assche G, Lindsay JO, Lemann M, Soderholm J, Colombel JF, et al. The second European evidence-based consensus on the diagnosis and management of Crohn’s disease: current management. J Crohns Colitis (2010) 4(1):28–62. doi:10.1016/j.crohns.2009.12.002
65. Podolsky DK. Inflammatory bowel disease. N Engl J Med (2002) 347(6):417–29. doi:10.1056/NEJMra020831
66. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med (2009) 361(21):2066–78. doi:10.1056/NEJMra0804647
67. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science (2012) 336(6086):1321–5. doi:10.1126/science.1222551
68. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14(3):221–9. doi:10.1038/ni.2534
69. Peters CP, Mjosberg JM, Bernink JH, Spits H. Innate lymphoid cells in inflammatory bowel diseases. Immunol Lett (2016) 172:124–31. doi:10.1016/j.imlet.2015.10.004
70. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491(7422):119–24. doi:10.1038/nature11582
71. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411(6837):603–6. doi:10.1038/35079114
72. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411(6837):599–603. doi:10.1038/35079107
73. Hugot JP, Laurent-Puig P, Gower-Rousseau C, Olson JM, Lee JC, Beaugerie L, et al. Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature (1996) 379(6568):821–3. doi:10.1038/379821a0
74. Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A (2005) 102(50):18129–34. doi:10.1073/pnas.0505256102
75. Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol (2004) 99(12):2393–404. doi:10.1111/j.1572-0241.2004.40304.x
76. Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet (2002) 70(4):845–57. doi:10.1086/339432
77. Simms LA, Doecke JD, Walsh MD, Huang N, Fowler EV, Radford-Smith GL. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut (2008) 57(7):903–10. doi:10.1136/gut.2007.142588
78. Perminow G, Beisner J, Koslowski M, Lyckander LG, Stange E, Vatn MH, et al. Defective Paneth cell-mediated host defense in pediatric ileal Crohn’s disease. Am J Gastroenterol (2010) 105(2):452–9. doi:10.1038/ajg.2009.643
79. Wehkamp J, Schmid M, Fellermann K, Stange EF. Defensin deficiency, intestinal microbes, and the clinical phenotypes of Crohn’s disease. J Leukoc Biol (2005) 77(4):460–5. doi:10.1189/jlb.0904543
80. Elphick D, Liddell S, Mahida YR. Impaired luminal processing of human defensin-5 in Crohn’s disease: persistence in a complex with chymotrypsinogen and trypsin. Am J Pathol (2008) 172(3):702–13. doi:10.2353/ajpath.2008.070755
81. Cleynen I, Gonzalez JR, Figueroa C, Franke A, McGovern D, Bortlik M, et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: results from the IBDchip European project. Gut (2013) 62(11):1556–65. doi:10.1136/gutjnl-2011-300777
82. Adler J, Rangwalla SC, Dwamena BA, Higgins PD. The prognostic power of the NOD2 genotype for complicated Crohn’s disease: a meta-analysis. Am J Gastroenterol (2011) 106(4):699–712. doi:10.1038/ajg.2011.19
83. Weersma RK, Stokkers PC, van Bodegraven AA, van Hogezand RA, Verspaget HW, de Jong DJ, et al. Molecular prediction of disease risk and severity in a large Dutch Crohn’s disease cohort. Gut (2009) 58(3):388–95. doi:10.1136/gut.2007.144865
84. Seiderer J, Schnitzler F, Brand S, Staudinger T, Pfennig S, Herrmann K, et al. Homozygosity for the CARD15 frameshift mutation 1007fs is predictive of early onset of Crohn’s disease with ileal stenosis, entero-enteral fistulas, and frequent need for surgical intervention with high risk of re-stenosis. Scand J Gastroenterol (2006) 41(12):1421–32. doi:10.1080/00365520600703900
85. Pugazhendhi S, Santhanam S, Venkataraman J, Creveaux I, Ramakrishna BS. NOD2 gene mutations associate weakly with ulcerative colitis but not with Crohn’s disease in Indian patients with inflammatory bowel disease. Gene (2013) 512(2):309–13. doi:10.1016/j.gene.2012.10.015
86. Yamazaki K, Takazoe M, Tanaka T, Kazumori T, Nakamura Y. Absence of mutation in the NOD2/CARD15 gene among 483 Japanese patients with Crohn’s disease. J Hum Genet (2002) 47(9):469–72. doi:10.1007/s100380200067
87. Long WY, Chen L, Zhang CL, Nong RM, Lin MJ, Zhan LL, et al. Association between NOD2/CARD15 gene polymorphisms and Crohn’s disease in Chinese Zhuang patients. World J Gastroenterol (2014) 20(16):4737–44. doi:10.3748/wjg.v20.i16.4737
88. Croucher PJ, Mascheretti S, Hampe J, Huse K, Frenzel H, Stoll M, et al. Haplotype structure and association to Crohn’s disease of CARD15 mutations in two ethnically divergent populations. Eur J Hum Genet (2003) 11(1):6–16. doi:10.1038/sj.ejhg.5200897
89. Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev (2010) 90(3):859–904. doi:10.1152/physrev.00045.2009
90. Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol (2012) 30:759–95. doi:10.1146/annurev-immunol-020711-074937
91. Grice EA, Segre JA. The human microbiome: our second genome. Annu Rev Genomics Hum Genet (2012) 13:151–70. doi:10.1146/annurev-genom-090711-163814
92. Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr (2002) 22:283–307. doi:10.1146/annurev.nutr.22.011602.092259
93. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature (2012) 489(7415):231–41. doi:10.1038/nature11551
94. Sommer F, Backhed F. The gut microbiota – masters of host development and physiology. Nat Rev Microbiol (2013) 11(4):227–38. doi:10.1038/nrmicro2974
95. Falk PG, Hooper LV, Midtvedt T, Gordon JI. Creating and maintaining the gastrointestinal ecosystem: what we know and need to know from gnotobiology. Microbiol Mol Biol Rev (1998) 62(4):1157–70.
96. Wu HJ, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes (2012) 3(1):4–14. doi:10.4161/gmic.19320
97. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139(3):485–98. doi:10.1016/j.cell.2009.09.033
98. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504(7480):446–50. doi:10.1038/nature12721
99. Packey CD, Sartor RB. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis (2009) 22(3):292–301. doi:10.1097/QCO.0b013e32832a8a5d
100. Feller M, Huwiler K, Schoepfer A, Shang A, Furrer H, Egger M. Long-term antibiotic treatment for Crohn’s disease: systematic review and meta-analysis of placebo-controlled trials. Clin Infect Dis (2010) 50(4):473–80. doi:10.1086/649923
101. Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science (2005) 307(5710):734–8. doi:10.1126/science.1103685
102. Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity (2014) 41(2):311–24. doi:10.1016/j.immuni.2014.06.015
103. Bevins CL, Stange EF, Wehkamp J. Decreased Paneth cell defensin expression in ileal Crohn’s disease is independent of inflammation, but linked to the NOD2 1007fs genotype. Gut (2008) 57:903–10; discussion 883–4.
104. Wehkamp J, Stange EF. NOD2 mutation and mice: no Crohn’s disease but many lessons to learn. Trends Mol Med (2005) 11(7):307–9. doi:10.1016/j.molmed.2005.06.003
105. Wehkamp J, Wang G, Kubler I, Nuding S, Gregorieff A, Schnabel A, et al. The Paneth cell alpha-defensin deficiency of ileal Crohn’s disease is linked to Wnt/Tcf-4. J Immunol (2007) 179(5):3109–18. doi:10.4049/jimmunol.179.5.3109
106. Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol (2000) 1(2):113–8. doi:10.1038/77783
107. Deshmukh HS, Hamburger JB, Ahn SH, McCafferty DG, Yang SR, Fowler VG Jr. Critical role of NOD2 in regulating the immune response to Staphylococcus aureus. Infect Immun (2009) 77(4):1376–82. doi:10.1128/IAI.00940-08
108. Shimada K, Chen S, Dempsey PW, Sorrentino R, Alsabeh R, Slepenkin AV, et al. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog (2009) 5(4):e1000379. doi:10.1371/journal.ppat.1000379
109. Opitz B, Puschel A, Schmeck B, Hocke AC, Rosseau S, Hammerschmidt S, et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem (2004) 279(35):36426–32. doi:10.1074/jbc.M403861200
110. Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, et al. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol (2008) 181(10):7157–65. doi:10.4049/jimmunol.181.10.7157
111. Hampe J, Cuthbert A, Croucher PJ, Mirza MM, Mascheretti S, Fisher S, et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet (2001) 357(9272):1925–8. doi:10.1016/S0140-6736(00)05063-7
112. Kim YG, Shaw MH, Warner N, Park JH, Chen F, Ogura Y, et al. Cutting edge: Crohn’s disease-associated Nod2 mutation limits production of proinflammatory cytokines to protect the host from Enterococcus faecalis-induced lethality. J Immunol (2011) 187(6):2849–52. doi:10.4049/jimmunol.1001854
113. Gasche C, Grundtner P. Genotypes and phenotypes in Crohn’s disease: do they help in clinical management? Gut (2005) 54(1):162–7. doi:10.1136/gut.2003.035600
114. Rehman A, Sina C, Gavrilova O, Hasler R, Ott S, Baines JF, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut (2011) 60(10):1354–62. doi:10.1136/gut.2010.216259
115. Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, Huang H, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med (2014) 6(12):107. doi:10.1186/s13073-014-0107-1
116. Li E, Hamm CM, Gulati AS, Sartor RB, Chen H, Wu X, et al. Inflammatory bowel diseases phenotype, C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PLoS One (2012) 7(6):e26284. doi:10.1371/journal.pone.0026284
117. Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis (2011) 17(1):179–84. doi:10.1002/ibd.21339
118. Lipinski S, Till A, Sina C, Arlt A, Grasberger H, Schreiber S, et al. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J Cell Sci (2009) 122(Pt 19):3522–30. doi:10.1242/jcs.050690
119. Barnich N, Hisamatsu T, Aguirre JE, Xavier R, Reinecker HC, Podolsky DK. GRIM-19 interacts with nucleotide oligomerization domain 2 and serves as downstream effector of anti-bacterial function in intestinal epithelial cells. J Biol Chem (2005) 280(19):19021–6. doi:10.1074/jbc.M413776200
120. Lupfer CR, Anand PK, Liu Z, Stokes KL, Vogel P, Lamkanfi M, et al. Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical NLRP3 inflammasome during enteric pathogen infection. PLoS Pathog (2014) 10(9):e1004410. doi:10.1371/journal.ppat.1004410
121. Mondot S, Barreau F, Al Nabhani Z, Dussaillant M, Le Roux K, Dore J, et al. Altered gut microbiota composition in immune-impaired Nod2(-/-) mice. Gut (2012) 61(4):634–5. doi:10.1136/gutjnl-2011-300478
122. Jiang W, Wang X, Zeng B, Liu L, Tardivel A, Wei H, et al. Recognition of gut microbiota by NOD2 is essential for the homeostasis of intestinal intraepithelial lymphocytes. J Exp Med (2013) 210(11):2465–76. doi:10.1084/jem.20122490
123. Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest (2013) 123(2):700–11. doi:10.1172/JCI62236
124. Robertson SJ, Zhou JY, Geddes K, Rubino SJ, Cho JH, Girardin SE, et al. Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes (2013) 4(3):222–31. doi:10.4161/gmic.24373
125. Shanahan MT, Carroll IM, Grossniklaus E, White A, von Furstenberg RJ, Barner R, et al. Mouse Paneth cell antimicrobial function is independent of Nod2. Gut (2014) 63(6):903–10. doi:10.1136/gutjnl-2012-304190
126. Natividad JM, Petit V, Huang X, de Palma G, Jury J, Sanz Y, et al. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1-/-; Nod2-/- mice. Inflamm Bowel Dis (2012) 18(8):1434–46. doi:10.1002/ibd.22848
127. Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med (2012) 209(8):1445–56. doi:10.1084/jem.20120504
128. Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol (2012) 9(10):599–608. doi:10.1038/nrgastro.2012.152
129. Barreau F, Meinzer U, Chareyre F, Berrebi D, Niwa-Kawakita M, Dussaillant M, et al. CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS One (2007) 2(6):e523. doi:10.1371/journal.pone.0000523
130. Butler M, Chaudhary R, van Heel DA, Playford RJ, Ghosh S. NOD2 activity modulates the phenotype of LPS-stimulated dendritic cells to promote the development of T-helper type 2-like lymphocytes – possible implications for NOD2-associated Crohn’s disease. J Crohns Colitis (2007) 1(2):106–15. doi:10.1016/j.crohns.2007.08.006
131. Amendola A, Butera A, Sanchez M, Strober W, Boirivant M. Nod2 deficiency is associated with an increased mucosal immunoregulatory response to commensal microorganisms. Mucosal Immunol (2014) 7(2):391–404. doi:10.1038/mi.2013.58
132. Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, et al. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut (2006) 55(3):342–7. doi:10.1136/gut.2005.065557
133. D’Inca R, Annese V, di Leo V, Latiano A, Quaino V, Abazia C, et al. Increased intestinal permeability and NOD2 variants in familial and sporadic Crohn’s disease. Aliment Pharmacol Ther (2006) 23(10):1455–61. doi:10.1111/j.1365-2036.2006.02916.x
134. Nigro G, Rossi R, Commere PH, Jay P, Sansonetti PJ. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe (2014) 15(6):792–8. doi:10.1016/j.chom.2014.05.003
135. Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology (2010) 139(5):e1–2. doi:10.1053/j.gastro.2010.07.006
136. Chu H, Khosravi A, Kusumawardhani IP, Kwon AH, Vasconcelos AC, Cunha LD, et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science (2016) 352(6289):1116–20. doi:10.1126/science.aad9948
Keywords: NOD2, ileal Crohn’s disease, Paneth cells, microbiota
Citation: Sidiq T, Yoshihama S, Downs I and Kobayashi KS (2016) Nod2: A Critical Regulator of Ileal Microbiota and Crohn’s Disease. Front. Immunol. 7:367. doi: 10.3389/fimmu.2016.00367
Received: 27 July 2016; Accepted: 06 September 2016;
Published: 20 September 2016
Edited by:
Amélia M. Sarmento, Fernando Pessoa University, PortugalReviewed by:
Dario S. Zamboni, University of São Paulo, BrazilCopyright: © 2016 Sidiq, Yoshihama, Downs and Kobayashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Koichi S. Kobayashi, a29iYXlhc2hpQG1lZGljaW5lLnRhbWhzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.